CompositionPrincipes actifs
Le rozanolixizumab est un anticorps monoclonal recombinant et humanisé de type immunoglobuline G 4P (IgG4P) ciblant le récepteur FcRn (Fraction Fc du récepteur néonatal) produit à partir de cellules d'ovaire de hamster chinois (CHO) par une technique d'ADN recombinant.
Un ml contient 140 mg de rozanolixizumab.
Excipients
L-histidine, chlorhydrate de L-histidine monohydraté, L-proline, polysorbate 80 et eau pour préparations injectables.
Indications/Possibilités d’emploiRystiggo est indiqué en association au traitement standard pour le traitement de la myasthénie auto-immune généralisée (MAg) chez les patients adultes présentant des anticorps anti-récepteurs de l'acétylcholine (R-Ach) ou des anticorps anti-tyrosine kinase spécifique du muscle (MuSK) (voir la rubrique Efficacité clinique).
Posologie/Mode d’emploiLe traitement doit être instauré et supervisé par des médecins expérimentés dans la prise en charge de patients atteints de troubles neuromusculaires ou neuro-inflammatoires.
Posologie
Afin d'assurer la traçabilité des médicaments biotechnologiques, il convient de documenter pour chaque traitement le nom commercial et le numéro de lot.
Un cycle de traitement correspond à 1 dose par semaine administrée par voie sous-cutanée pendant 6 semaines.
Le tableau suivant indique la dose hebdomadaire totale recommandée de rozanolixizumab en fonction du poids corporel du patient:
|
Poids corporel
|
≥35 à <50 kg
|
≥50 à < 70 kg
|
≥70 à < 100 kg
|
≥100 kg
| |
Dose hebdomadaire (mg)
|
280 mg
|
420 mg
|
560 mg
|
840 mg
| |
Dose hebdomadaire (ml)
|
2 ml
|
3 ml
|
4 ml
|
6 ml
| |
Nombre de flacons nécessaires*
|
1
|
2
|
2
|
3
|
* Chaque flacon contient un volume excédentaire pour l'amorçage de la ligne de perfusion, voir « Mode d'administration ».
Il n'existe aucune donnée sur l'utilisation du rozanolixizumab chez les patients de moins de 35 kg et de plus de 155 kg. Les patients présentant une MAg de classe IVb et V selon la Myasthenia Gravis Foundation of America (MGFA) n'ont pas été étudiés dans le cadre du programme d'étude clinique de Rystiggo.
Les cycles de traitement ultérieurs doivent être administrés en fonction de l'évaluation clinique.
La fréquence des cycles de traitement peut varier en fonction des patients. En cas d'aggravation des symptômes rendant nécessaire un traitement supplémentaire (définie comme une augmentation d'au moins 3 points du score sur l'échelle Quantitative-Myasthenia Gravis (QMG) et/ou de 2 points sur l'échelle Myasthenia gravis-Activities of Daily Living (MG-ADL) depuis la fin du dernier cycle de traitement), les patients peuvent recevoir un cycle de traitement supplémentaire de 6 semaines. Dans le programme de développement clinique, la plupart des participants avaient des intervalles sans traitement de 4 à 13 semaines entre les cycles. L'intervalle minimum entre deux cycles de traitement est de quatre semaines (c.-à-d. au moins 28 jours après la dernière dose du cycle précédent).
Le bénéfice du traitement doit être évalué régulièrement. Si un patient ne répond pas à deux cycles de traitements consécutifs de 6 semaines (la réponse étant définie comme une amélioration de moins de 3 points sur l'échelle QMG et/ou de moins de 2 points sur l'échelle MG-ADL par rapport au début du cycle respectif), le traitement par Rystiggo ne doit pas être poursuivi.
Si une perfusion programmée est manquée, Rystiggo peut être administré jusqu'à 4 jours après la date programmée. Par la suite, le schéma posologique initial doit être repris jusqu'à la fin du cycle de traitement.
Instructions posologiques particulières
Patients présentant des troubles de la fonction hépatique
Aucune donnée n'est disponible chez les patients présentant une insuffisance hépatique. Aucun ajustement posologique n'est considéré nécessaire, car il est peu probable que la pharmacocinétique du rozanolixizumab soit modifiée par une insuffisance hépatique (voir la rubrique Pharmacocinétique).
Patients présentant des troubles de la fonction rénale
Les données relatives à la sécurité d'emploi et à l'efficacité chez les patients atteints d'insuffisance rénale légère à modérée (DFGe >45 ml/min/1,73 m2) sont limitées. Aucune donnée n'est disponible chez les patients présentant une insuffisance rénale sévère. Aucun ajustement posologique n'est considéré nécessaire, car il est peu probable que la pharmacocinétique du rozanolixizumab soit modifiée par une insuffisance rénale (voir la rubrique Pharmacocinétique).
Patients âgés
Aucun ajustement posologique n'est nécessaire (voir la rubrique Pharmacocinétique). Il n'existe pas de données sur la sécurité et l'efficacité chez les patients >85 ans. Pour le groupe d'âge ≥65 à 85 ans, les données sont limitées (voir rubriques Mises en garde et précautions et Effets indésirables).
Enfants et adolescents
La sécurité et l'efficacité du rozanolixizumab chez les enfants et adolescents âgés de moins de 18 ans n'ont pas été établies. Aucune donnée n'est disponible. Rystiggo n'est pas autorisé pour une utilisation dans la population pédiatrique.
Mode d'administration
Pour perfusion sous-cutanée à l'aide d'une pompe.
Des pompes à perfusion, seringues et sets de perfusion appropriés à l'administration sous-cutanée de médicaments doivent être utilisés. Dans la mesure où chaque flacon contient un volume excédentaire pour l'amorçage de la ligne de perfusion, il est recommandé d'utiliser des pompes dont le volume à administrer peut être prédéfini.
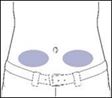
Il est recommandé d'administrer le rozanolixizumab par voie sous-cutanée, de préférence dans la partie inférieure droite ou inférieure gauche de l'abdomen, sous le nombril. Aucun autre site de perfusion n'a été étudié dans le cadre du programme de développement clinique. Les perfusions ne doivent pas être administrées dans des zones où la peau est sensible, érythémateuse ou indurée.
Lors de chaque administration du premier cycle de traitement et de l'administration de la première dose du deuxième cycle de traitement par rozanolixizumab, un traitement approprié pour les réactions liées à l'injection et les réactions d'hypersensibilité doit être immédiatement disponible (voir la rubrique Mises en garde et précautions).
Débit de perfusion
Le rozanolixizumab est administré au moyen d'une pompe à perfusion à un débit constant allant jusqu'à 20 ml/h.
Lire attentivement les Instructions d'utilisation avant l'administration du rozanolixizumab.
Contre-indicationsHypersensibilité à la substance active ou à l'un des excipients mentionnés à la rubrique Composition.
Mises en garde et précautionsSurveillance clinique
Dans le programme d'étude clinique, l'efficacité et la sécurité des traitements cycliques répétés à la dose autorisée de Rystiggo ont été étudiées uniquement chez un nombre limité de patients. Rystiggo est un traitement cyclique axé sur les symptômes.
Compte tenu de différences individuelles imprévisibles dans la réponse clinique, les patients doivent être étroitement surveillés. Au cours du programme d'étude clinique, 13,8 % des patients ont présenté une aggravation de la maladie sous-jacente jusqu'à la crise myasthénique (2,1 %), malgré le traitement cyclique répété par Rystiggo au dosage d'environ (≈) 7 mg/kg ou ≈ 10 mg/kg (voir rubriques Efficacité clinique et Effets indésirables).
Il n'existe pas de donnée sur l'efficacité et la sécurité de Rystiggo dans le traitement de la crise myasthénique.
Poids corporel <50kg
Au cours du programme d'étude clinique, seul un nombre très limité de patients pesant moins de 50 kg a été étudié. Une preuve fiable de l'efficacité du traitement cyclique répété par rozanolixizumab n'est pas démontrée à ce jour (voir la rubrique Efficacité clinique).
Patients âgés
Au cours du programme d'étude clinique, seul un nombre limité de patients âgés de ≥65 à 85 ans a été étudié. L'effet d'un traitement cyclique répété a été moindre et moins robuste dans ce groupe d'âge que chez les patients <65 ans. Simultanément, ces patients âgés présentaient un profil d'effets secondaires moins favorable: ainsi, dans le cadre d'une étude en double aveugle et d'une étude d'extension en ouvert, 39,6 % des patients âgés (≥65 ans) ont présenté des effets indésirables graves, contre 30,7 % dans le groupe <65 ans. Une crise myasthénique est survenue chez 6,3 % des patients ≥65 ans et chez 0,7 % des patients <65 ans (voir la rubrique Efficacité clinique).
Crise myasthénique
Le traitement par rozanolixizumab chez les patients présentant une crise myasthénique imminente ou manifeste n'a pas été étudié. Le rozanolixizumab n'est pas autorisé dans le traitement de la crise myasthénique imminente ou manifeste. En cas de crise myasthénique imminente ou manifeste, il convient de tenir compte de l'interaction entre le rozanolixizumab et les traitements établis de la crise myasthénique, car le rozanolixizumab peut diminuer l'efficacité de ces traitements (voir la rubrique Interactions).
Méningite aseptique
Des cas de méningite aseptique médicamenteuse ont été rapportés après un traitement par rozanolixizumab (voir la rubrique Effets indésirables). En cas de symptômes compatibles avec une méningite aseptique, un bilan diagnostique et un traitement doivent être mis en place conformément aux normes de soins.
Hypogammaglobulinémie
En raison de son mécanisme d'action, Rystiggo peut entraîner une diminution marquée du taux d'immunoglobuline G (IgG) (voir rubrique Effets indésirables). Dans le programme de développement clinique, le traitement par Rystiggo était provisoirement interrompu si le taux d'IgG sérique totale tombait sous 1 g/l et ce, indépendamment de l'éventuelle présence simultanée d'une infection. Dès que le taux d'IgG remontait à ≥2 g/l, le traitement par Rystiggo pouvait être repris. En cas d'infection persistante ou récurrente non grave avec un taux d'IgG sérique totale ≥1 et <2 g/l, le traitement par Rystiggo était également suspendu temporairement jusqu'à ce que l'infection disparaisse et que l'IgG remonte à ≥2 g/L.
Infections
Dans la mesure où le rozanolixizumab entraîne une réduction transitoire du taux d'IgG, le risque d'infections pourrait augmenter (voir rubrique Propriétés/Effets). Le traitement par rozanolixizumab ne doit pas être instauré chez les patients présentant une infection active cliniquement importante tant que l'infection n'a pas disparu ou n'a pas été convenablement traitée. Pendant le traitement par rozanolixizumab, les signes et symptômes cliniques d'infection doivent être surveillés. En cas d'apparition d'une infection active cliniquement importante, le traitement par rozanolixizumab doit être interrompu jusqu'à la résolution de l'infection.
Vaccination
L'immunisation par des vaccins pendant le traitement par rozanolixizumab n'a pas été étudiée. La sécurité de l'immunisation par des vaccins vivants ou vivants atténués ainsi que la réponse à l'immunisation vaccinale sont inconnues. Tous les vaccins doivent être administrés conformément aux recommandations vaccinales et au moins 4 semaines avant le début du traitement. Pour les patients sous traitement, la vaccination par des vaccins vivants ou vivants atténués n'est pas recommandée. Pour tous les autres vaccins, ils doivent avoir lieu au moins 2 semaines après la dernière perfusion d'un cycle de traitement et 4 semaines avant le début du cycle suivant de rozanolixizumab.
Hypersensibilité
Des réactions à la perfusion telles que des éruptions cutanées ou un angiœdème peuvent survenir (voir rubrique Effets indésirables). Dans le programme d'études cliniques, ces réactions étaient légères à modérées. Les patients doivent être surveillés pendant le traitement par rozanolixizumab et pendant les 15 minutes qui suivent la fin de l'administration, afin de détecter les signes cliniques et les symptômes de réactions d'hypersensibilité. En cas de survenue d'une réaction d'hypersensibilité pendant l'administration (voir rubrique Effets indésirables), la perfusion de rozanolixizumab doit être interrompue et des mesures appropriées doivent être appliquées si nécessaire. Après résolution, l'administration pourra être reprise.
Immunogénicité
Dans les données regroupées sur le traitement cyclique du programme de phase III, après 1 cycle de traitement de 6 doses hebdomadaires de rozanolixizumab, 26,9 % (42/156) des patients ont développé des anticorps anti-médicament et 10,3 % (16/156) ont eu des anticorps classés comme neutralisants. Lors de la reprise du traitement, la proportion de patients ayant développé des anticorps anti-médicament et des anticorps neutralisants est passée à 61,4 % (35/57) et 43,9 % (25/57) respectivement, après 5 cycles de traitement. L'apparition d'anticorps neutralisants a été associée à une diminution de 24 % de l'exposition plasmatique globale au rozanolixizumab. L'immunogénicité n'a pas eu d'impact significatif sur l'efficacité et la sécurité (voir rubrique Propriétés/Effets). Les participants à l'étude présentant des anticorps anti-médicament ont toutefois rapporté plus de deux fois plus souvent certains effets indésirables que les participants sans anticorps anti-médicament (voir rubrique Effets indésirables).
Proline (excipient)
Ce médicament contient 29 mg de proline par ml.
L'utilisation chez les patients atteints d'hyperprolinémie doit être limitée dans les cas où aucun autre traitement n'est disponible.
InteractionsAucune étude d'interaction n'a été réalisée. Dans la mesure où le rozanolixizumab interfère avec le mécanisme de recyclage par le FcRn des immunoglobulines G (IgG), les concentrations sériques de médicaments à base d'IgG (par ex., les anticorps monoclonaux et l'immunoglobuline intraveineuse [IgIV]) et de protéines de fusion Fc-peptide devraient être réduites en cas d'administration concomitante. Dans les 2 semaines suivant une perfusion de rozanolixizumab, un effet cliniquement significatif du rozanolixizumab sur la pharmacocinétique ou l'efficacité de ces médicaments est peu probable. Il est recommandé d'instaurer ces traitements 2 semaines après l'administration du rozanolixizumab et de surveiller l'éventuelle diminution d'efficacité de ces médicaments en cas de co-administration.
Les interactions avec les médicaments à forte liaison protéique ou avec les substrats, inducteurs, inhibiteurs ou transporteurs du cytochrome-P450 sont peu probables.
Le traitement par immunoglobulines i.v. (intraveineux) ou s.c. (sous-cutané), échanges plasmatiques/plasmaphérèse et immunoadsorption pourrait réduire les taux circulants de rozanolixizumab.
Grossesse, allaitementGrossesse
Les données limitées ne permettent pas de tirer de conclusions sur l'utilisation du rozanolixizumab chez la femme enceinte. Dans les études menées chez l'animal, des effets sur les avortements spontanés ont été observés. En outre, les portées de femelles traitées présentaient de très faibles taux d'IgG à la naissance, comme attendu en raison du mode d'action pharmacologique du rozanolixizumab. Chez l'animal, le rozanolixizumab n'a pas montré d'effets sur le développement fœtal, la parturition ou le développement postnatal (voir la rubrique Données préclinique). Le traitement des femmes enceintes par rozanolixizumab ne doit être envisagé que si le bénéfice clinique l'emporte sur les risques.
Étant donné que le rozanolixizumab devrait réduire les taux maternels d'IgG et inhiber le transfert d'IgG maternelles au fœtus, une réduction de la protection passive du nouveau-né est présumée. Par conséquent, les risques et les bénéfices de l'administration de vaccins vivants/vivants atténués à des nourrissons exposés au rozanolixizumab in utero doivent être pris en compte au cours des deux premiers mois suivant la naissance (voir rubrique Mises en garde et précautions - Vaccination).
Allaitement
On ne sait pas si le rozanolixizumab est excrété dans le lait maternel. On sait que les IgG maternelles sont excrétées dans le lait maternel pendant les premiers jours qui suivent la naissance, les concentrations diminuant rapidement par la suite jusqu'à devenir faibles; par conséquent, un risque pour les nouveau-nés/nourrissons ne peut être exclu pendant cette courte période. Après cette phase précoce, la décision d'arrêter l'utilisation du rozanolixizumab ou de renoncer à l'allaitement doit être prise en mettant en balance les avantages potentiels de l'allaitement, les besoins cliniques de la mère pour le traitement par rozanolixizumab et les effets nocifs potentiels du rozanolixizumab sur le nourrisson allaité.
Fertilité
L'effet du rozanolixizumab sur la fertilité humaine n'est pas connu. Les études effectuées chez l'animal n'ont pas mis en évidence d'effets délétères sur la fertilité (voir la rubrique Données précliniques).
Effet sur l’aptitude à la conduite et l’utilisation de machinesL'effet du rozanolixizumab sur l'aptitude à la conduite et l'utilisation de machines n'a pas été étudié. Les considérations théoriques suggèrent l'absence d'influence ou une influence négligeable du rozanolixizumab sur l'aptitude à la conduite ou l'utilisation de machines.
Effets indésirablesRésumé du profil de sécurité
Dans une étude en double aveugle, contrôlée contre placebo, menée auprès de patients atteints de myasthénie auto-immune généralisée, 133 patients ont été traités par rozanolixizumab. Les effets indésirables les plus fréquemment rapportés dans cette étude en double aveugle et dans une étude d'extension en ouvert (188 patients) étaient les céphalées (51,6 %), la diarrhée (33,5 %), les infections des voies respiratoires supérieures (25,5 %), la fièvre (20,7 %), les nausées (17,6 %), les réactions au site de perfusion/d'injection (12,2 %) et l'arthralgie (12,2 %).
Liste des effets indésirables
Les effets indésirables signalés dans les études cliniques sur la myasthénie auto-immune généralisée sont répertoriés par classe de systèmes d'organes MedDRA selon la convention suivante. Au sein de chaque classe de systèmes d'organes, les effets indésirables sont classés par fréquence, les plus fréquents étant indiqués en premier.
Les effets indésirables sont présentés par catégorie de fréquence selon la convention suivante: très fréquent (≥1/10); fréquent (≥1/100 à < 1/10); occasionnel (≥1/1000 à < 1/100); rare (≥1/10 000 à < 1/1000); très rare (< 1/10 000), fréquence inconnue (ne peut être estimée sur la base des données disponibles).
Tableau 1: Liste des effets indésirables
|
Classe de système d'organes MedDRA
|
Catégorie de fréquence
|
Effets indésirables
| |
Affections hématologiques et du système lymphatique
|
Très fréquent
|
Baisse du taux d'IgG (11,2 %)
| |
Infections et infestations
|
Très fréquent
|
Infections des voies respiratoires supérieures (25,5 %)1
| |
Fréquent
|
Infections virales2
Infections des voies respiratoires inférieures3
Infections virales à herpès4
| |
Affections du système nerveux
|
Très fréquent
|
Céphalées (51,6 %)5
| |
Rare
|
Méningite aseptique
| |
Affections respiratoires, thoraciques et médiastinales
|
Fréquent
|
Douleurs oropharyngées
| |
Affections gastro-intestinales
|
Très fréquent
|
Diarrhée (33,5 %),
Douleurs abdominales (16,5 %)6
Nausées (17,6 %)
| |
Fréquent
|
Vomissements
| |
Affections de la peau et du tissu sous-cutané
|
Fréquent
|
Éruption cutanée7,
| |
Occasionnel
|
Angiœdème8
| |
Affections musculosquelettiques et du tissu conjonctif
|
Très fréquent
|
Arthralgie (12,2 %)
| |
Fréquent
|
Myalgie
Douleur à la nuque
Spasmes musculaires
| |
Troubles généraux et anomalies au site d'administration
|
Très fréquent
|
Fièvre (20,7 %)
Réactions au site d'injection/perfusion (12,2 %)9
| |
Fréquent
|
Syndrome grippal
Douleur thoracique
|
1 Comprend la sinusite aiguë et chronique, la laryngite, la rhino-pharyngite, la pharyngite, la rhinite, la sinusite, l'amygdalite, l'infection des voies respiratoires supérieures
2 Comprend la gastro-entérite virale, les infections virales des voies respiratoires, l'infection virale, l'infection virale des voies respiratoires supérieures, mais pas les infections au COVID-19
3 Comprend la bronchite, l'infection des voies respiratoires inférieures, la pneumonie
4 Comprend les infections à herpès simplex, herpèsvirus, herpès zoster, herpès simplex ophtalmique, herpès oral
5 Comprend les céphalées et la migraine
6 Comprend les douleurs abdominales, les douleurs abdominales hautes et basses, la douleur gastro-intestinale, les troubles abdominaux
7 Comprend l'éruption cutanée, l'éruption papuleuse et l'éruption érythémateuse
8 Comprend le gonflement de la langue
9 Comprend, mais ne se limite pas à, l'hématome au site d'injection, l'éruption cutanée au site d'injection, la réaction au site d'injection, l'érythème au site d'injection, le prurit au site d'injection, l'érythème au site de perfusion, la réaction au site de perfusion.
Description d'effets indésirables spécifiques et informations complémentaires
Céphalées
Dans l'étude MG0003, les céphalées constituaient la réaction la plus fréquemment rapportée chez 58 (43,6 %) et 13 (19,4 %) des patients traités par le rozanolixizumab et par placebo. Les céphalées sont survenues le plus fréquemment après la première perfusion de rozanolixizumab et dans les 1 à 4 jours suivant la perfusion. Les céphalées n'étaient pas sévères, mais généralement légères ou modérées, et aucune augmentation de leur incidence n'a été observée avec un traitement cyclique répété.
Immunogénicité
La détection des anticorps anti-médicaments (ADAs) dépend en grande partie de la sensibilité et de la spécificité des tests utilisés. De plus, l'incidence observée de la positivité aux anticorps (y compris aux anticorps neutralisants) peut être influencée par plusieurs facteurs, tels que la méthode de test, la manipulation des échantillons, le moment du prélèvement de l'échantillon, les traitements concomitants et la maladie sous-jacente. C'est pourquoi il peut être trompeur de comparer l'incidence des anticorps anti-rozanolixizumab à celle des anticorps contre d'autres médicaments.
Dans les données regroupées sur le traitement cyclique du programme de phase III, après 1 cycle de traitement de 6 doses hebdomadaires de rozanolixizumab 26,9 % (42/156) des patients ont développé des anticorps anti-médicaments et 10,3 % (16/156) des anticorps classés comme neutralisants. Lors de la reprise du traitement, la proportion de patients ayant développé des anticorps anti-médicament et des anticorps neutralisants est passée à 61,4 % (35/57) et 43,9 % (25/57) respectivement, après 5 cycles de traitement. L'apparition d'anticorps neutralisants a été associée à une diminution de 24 % de l'exposition plasmatique globale au rozanolixizumab.
La réduction par le rozanolixizumab de la concentration totale en IgG chez les patients présentant des anticorps neutralisants n'était pas différente de celle des patients ne présentant pas d'anticorps anti-médicament.
L'immunogénicité n'a pas eu d'impact significatif sur l'efficacité et la sécurité globales.
La fréquence de certains effets indésirables (douleurs abdominales hautes, infections des voies respiratoires supérieures, baisse du nombre de granulocytes neutrophiles, hypertension, dys/paresthésie, détresse respiratoire, dyslipidémie) était toutefois au moins deux fois plus élevée chez les participants présentant des anticorps anti-médicaments que chez les participants qui n'en présentaient pas. Un lien de causalité direct entre l'apparition de ces effets indésirables et les anticorps anti-médicaments n'a pas été démontré jusqu'à présent. Les patients sans anticorps anti-médicament étaient plus fréquemment traités par immunosuppresseurs, corticostéroïdes systémiques et agents chimiothérapeutiques.
L'annonce d'effets secondaires présumés après l'autorisation est d'une grande importance. Elle permet un suivi continu du rapport bénéfice-risque du médicament. Les professionnels de santé sont tenus de déclarer toute suspicion d'effet secondaire nouveau ou grave via le portail d'annonce en ligne ElViS (Electronic Vigilance System). Vous trouverez des informations à ce sujet sur www.swissmedic.ch.
SurdosageSignes et symptômes
Il n'existe aucune donnée sur les symptômes associés à un surdosage. Une dose sous-cutanée unique pouvant atteindre 20 mg/kg (2162 mg) et des doses sous-cutanées hebdomadaires d'env. 10 mg/kg (1120 mg) pendant un maximum de 52 semaines ont été administrées conformément au protocole dans des études cliniques sans toxicité limitant la dose.
Traitement
En cas de surdosage, il est recommandé que les patients soient étroitement surveillés afin de déceler tout effet indésirable, et des mesures de soutien appropriées doivent être immédiatement mises en place.
Propriétés/EffetsCode ATC
L04AG16
Mécanisme d'action
Le rozanolixizumab est un anticorps monoclonal humanisé de type IgG4, qui diminue la concentration sérique d'IgG en inhibant la liaison de l'IgG au récepteur Fc néonatal (FcRn), un récepteur qui, dans des conditions physiologiques, protège l'IgG contre la dégradation intracellulaire et recycle l'IgG à la surface cellulaire.
Par le même mécanisme, le rozanolixizumab diminue la concentration d'auto-anticorps IgG pathogènes associés à la MAg. Les données cliniques sur le rozanolixizumab n'ont pas identifié d'impact cliniquement pertinent sur les taux d'albumine, qui se lie à un site différent sur le FcRn.
Pharmacodynamique
L'administration sous-cutanée hebdomadaire de rozanolixizumab a entraîné une réduction rapide et durable des concentrations sériques d'IgG totales, avec une diminution significative de 45 % des IgG par rapport à l'inclusion à 1 semaine, et une diminution maximale de 73 % à environ 3 semaines. Après l'arrêt de l'administration, les concentrations d'IgG sont revenues aux taux de référence en 8 semaines environ. Des changements similaires ont été observés dans toutes les sous-classes d'IgG.
Efficacité clinique
Études de phase-III
La sécurité et l'efficacité du rozanolixizumab ont été étudiées au cours de trois études de phase III chez des patients atteints de myasthénie auto-immune généralisée. Ces patients étaient âgés d'au moins 18 ans, avec un poids corporel ≥35 kg, avaient reçu un diagnostic de myasthénie auto-immune généralisée, présentaient des auto-anticorps contre les récepteurs de l'acétylcholine (R-Ach) ou de la protéine MuSK, étaient atteints d'une myasthénie auto-immune de classe II à IVa selon la Myasthenia Gravis Foundation of America (MGFA), avec un score MG-Activities of Daily Living (MG-ADL) d'au moins 3 (avec un score ≥3 points pour les symptômes non oculaires), un score Quantitative Myasthenia Gravis (QMG) d'au moins 11 et étaient éligibles à un traitement supplémentaire par plasmaphérèse (échange plasmatique (PLEX)) ou immunoglobuline intraveineuse (IVIg).
Les patients ont été exclus de la participation à l'étude:
·s'ils étaient atteints d'une infection active cliniquement pertinente ou d'infections graves, d'infections mycobactériennes, d'une hépatite B, d'une hépatite C, d'infections par le VIH
·s'ils présentaient une immunodéficience primaire ou un déficit en IgA à l'anamnèse, ou avaient subi une splénectomie ou une transplantation (transplantation d'organe solide ou greffe de cellules souches hématopoïétiques / greffe de moelle osseuse)
·s'ils présentaient des antécédents de thymectomie au cours des 6 derniers mois précédant le début du traitement ou de thymome à n'importe quel moment ayant nécessité une chimiothérapie et/ou une radiothérapie, ou une maladie néoplasique active ou une maladie néoplasique datant de moins de 5 ans
·s'ils avaient été traités par échange plasmatique (PLEX), IgIV dans le mois précédant le traitement ou par anticorps monoclonaux dans les 3 à 6 mois avant le début du traitement
·s'ils présentaient un taux sérique d'IgG totales ≤5,5 g/l ou une numération absolue des neutrophiles <1500 cellules/mm3
La sécurité et l'efficacité du rozanolixizumab ont été évaluées chez des patients atteints de myasthénie auto-immune généralisée dans l'étude pivot de phase III MG0003. La sécurité d'emploi, la tolérance et l'efficacité à long terme du rozanolixizumab ont été évaluées dans 2 études d'extension de phase III en ouvert (OLE [open-label extension]). Dans cette étude OLE MG0007, le rozanolixizumab était administré en cycles de traitement de 6 semaines en fonction des besoins cliniques pendant une période de 2 ans maximum (voir ci-dessous).
Étude MG0003
L'étude MG0003 a évalué 200 patients, pendant un maximum de 18 semaines, au cours de laquelle les patients ont été randomisés pour recevoir des doses de rozanolixizumab dépendant du poids, équivalent à environ (≈) 7 mg/kg ou ≈10 mg/kg ou un placebo. Le traitement consistait en une dose hebdomadaire pendant une période de 6 semaines, suivie d'une période d'observation de 8 semaines.
L'efficacité du rozanolixizumab était évaluée par rapport à son impact sur les résultats des questionnaires MG-ADL, MG-C, QMG et d'une série d'autres questionnaires patients, y compris sur le score MG Symptoms PRO. Le critère d'évaluation principal était la variation par rapport à l'inclusion jusqu'au jour 43 du score total de MG-ADL. Les critères d'évaluation secondaires de l'efficacité incluaient la variation entre l'inclusion et le jour 43 du score MG-C et du score QMG ainsi que le score de réponse à l'échelle MG-ADL au jour 43.
En règle générale, les données démographiques des patients ainsi que les caractéristiques de la maladie à l'inclusion étaient équilibrées entre les groupes de traitement. La majorité des patients dans le groupe rozanolixizumab à la dose de ≈7 mg/kg étaient des femmes (59,1 %), âgées de moins de 65 ans (74,2 %), d'origine caucasienne (62,1 %) ou asiatique (13,5 %), et qui présentaient une myasthénie auto-immune généralisée de classe II ou III selon les critères de la MGFA (95,4 %). L'âge médian au moment du diagnostic de la MG était de 44,0 ans et la durée médiane depuis le diagnostic était de 5,3 ans. La répartition des auto-anticorps chez les patients était la suivante: 7,6 % d'anticorps anti-MuSK et 90,9 % d'anticorps anti-R-Ach. Dans l'ensemble, 95,5 % des patients recevaient au moment de l'inclusion au moins un médicament contre la myasthénie auto-immune généralisée qui a été maintenu pendant l'étude, dont 83,3 % d'inhibiteurs de l'acétylcholinestérase, 63,6 % de corticostéroïdes, 47,0 % d'immunosuppresseurs à des doses stables. Dans les groupes rozanolixizumab et placebo, le score total médian MG-ADL était de 8,0 et le score total médian QMG de 15,0 au début de l'étude.
Les résultats pour les critères d'évaluation principal et secondaires d'efficacité sont présentés dans le Tableau 2 ci-dessous.
Une amélioration statistiquement et cliniquement significative des symptômes de la myasthénie généralisée a été observée au jour 43 d'après les scores MG-ADL, MG-C et QMG, dans les deux groupes de traitement par rozanolixizumab par rapport au groupe placebo.
Tableau 2: Variation des résultats d'efficacité entre l'inclusion et le jour 43
|
|
Placebo
(N = 67)
|
Rozanolixizumab
≈ 7 mg/kg
(N = 66)
| |
MG-ADL
| |
Moyenne à l'inclusion
|
8,4
|
8,4
| |
Variation par rapport à l'inclusion
Moyenne des MC (ES)
|
-0,784 (0,488)
|
-3,370 (0,486)
| |
Différence par rapport au placebo
|
NA
|
-2,586
| |
IC à 95% pour la différence
|
NA
|
-4,091, -1,249
| |
Valeur p pour la différence
|
NA
|
< 0,001
| |
MG-C
| |
Moyenne à l'inclusion
|
15,6
|
15,9
| |
Variation par rapport à l'inclusion
Moyenne des MC (ES)
|
-2,029 (0,917)
|
-5,930 (0,916)
| |
Différence par rapport au placebo
|
NA
|
-3,901
| |
IC à 95% pour la différence
|
NA
|
-6,634, -1,245
| |
Valeur p pour la différence
|
NA
|
< 0,001
| |
QMG
| |
Moyenne à l'inclusion
|
15,8
|
15,4
| |
Variation par rapport à l'inclusion
Moyenne des MC (ES)
|
-1,915 (0,682)
|
-5,398 (0,679)
| |
Différence par rapport au placebo
|
NA
|
-3,483
| |
IC à 95% pour la différence
|
NA
|
-5,614, -1,584
| |
Valeur p pour la différence
|
NA
|
< 0,001
|
≈=dose approximative; IC = intervalle de confiance; N=nombre total de patients dans le groupe de traitement; n=nombre de patients; MC = moindre carré; ES = erreur type (standard error).
Un patient répondeur MG-ADL était défini comme un patient qui présentait, à tout moment du traitement et de la période d'observation, une amélioration de son score d'au moins 2 points par rapport à l'inclusion. Au jour 43, le taux de répondeurs MG-ADL dans le groupe sous rozanolixizumab à ≈7 mg/kg était de (46 [71,9 %]), était plus de deux fois plus élevé que dans le groupe placebo (20 [31,3 %]). Le taux de répondeurs QMG au jour 43 était de 39,1 % dans le groupe placebo, contre 54,7 % dans le groupe rozanolixizumab à ≈7 mg/kg.
Un patient répondeur QMG devait présenter à tout moment du traitement et de la période d'observation une amélioration d'au moins 3 points par rapport à l'inclusion.
Le traitement par rozanolixizumab était lié à une amélioration rapide des scores MG-ADL-, MG-C- et QMG, observable chez les patients dès une semaine après administration de la première dose. Les plus grandes améliorations ont été rapportées entre le jour 36 et le jour 43 dans le groupe rozanolixizumab ≈7 mg/kg.
Études OLE
L'étude d'extension en ouvert (OLE, open-label extension) MG0007 étudiait l'efficacité de cycles de traitement répétés par rozanolixizumab de 6 semaines. Les patients dont l'état se détériorait selon l'investigateur (augmentation d'au moins 2 points de l'évaluation sur l'échelle MG-ADL ou de 3 points sur l'échelle QMG), pouvaient recevoir un nouveau cycle de traitement. Au total, 25 patients ont reçu plus de 3 cycles de traitement à ≈7 mg/kg de rozanolixizumab. Certains patients ont reçu jusqu'à 15 cycles. Les patients qui répondaient au traitement répété à ≈7 mg/kg de rozanolixizumab ont montré une réponse stable aux cycles de traitement suivants.
Dans le groupe qui recevait exclusivement la dose autorisée de rozanolixizumab, entre les cycles 1 et 4, les taux de réponse des patients se situaient entre 55,3 % et 87,5 % selon l'échelle MG-ADL et entre 57,9 % et 71,4 % selon l'échelle QMG.
Efficacité chez les patients âgés
L'étude MG0003 étudiait également l'efficacité du rozanolixizumab par rapport au placebo chez les patients âgés (≥65 ans). Dans le groupe recevant exclusivement ≈7 mg/kg de rozanolixizumab, 17 patients étaient âgés de ≥65 ans. Par rapport aux patients recevant le placebo, la différence d'amélioration du score MG-ADL au jour 43 par rapport à l'inclusion était de -2,287 (ES 1.257) dans le groupe ≥65 ans pour -2,577 (ES 0.524) dans le groupe d'âge <65 ans.
Dans l'étude OLE MG0007, 14 patients âgés (≥65 ans) ont été traités par cycles répétés de rozanolixizumab à la dose autorisée (14 patients ont reçu 1 cycle, 12 patients 2 cycles, 7 patients 3 cycles, ≤5 patients ont reçu 4 cycles et plus). L'amélioration moyenne du score MG-ADL au jour 43 par rapport à l'inclusion était moins marquée à chaque cycle respectif que dans le groupe <65 ans (cycle 1: -2,5 (SD 3,6) vs. -3,4 (SD 3,2), cycle 2: -1,6 (SD 3,8) vs. -2,7 (SD 2,5), cycle 3: -1,7 (SD 3,1) vs. -3,3 (SD 2,6) points).
Efficacité chez les participants positifs aux auto-anticorps anti-MuSK et anti-R-Ach
Une analyse de sous-groupes a été effectuée par auto-anticorps spécifiques de la myasthénie généralisée (MuSK+ et R-Ach+).
Au jour 43, lors du traitement cyclique répété, les taux de réponses dans les cycles 1 à 4 aux critères MG-ADL, MG-C et QMG étaient de >40 % pour les participants positifs aux anticorps anti-R-Ach (28 participants au cycle 1, 27 participants au cycle 2, 21 participants au cycle 3 et 15 participants au cycle 4) et de >50 % chez les participants positifs aux anticorps anti-MuSK (4 participants respectivement aux cycles 1 à 4).
Ces données concernent les participants traités exclusivement par rozanolixizumab à la dose autorisée de ≈7 mg/kg de poids corporel.
Efficacité chez les patients pesant <50 kg
Le nombre de participants à l'étude OLE MG0007 pesant <50 kg et ayant reçu un traitement exclusif de cycles répétés à ≈7 mg/kg de rozanolixizumab était réduit (cycles 1-5: jusqu'à 4 participants par cycle). Dans ce groupe, les taux de répondeurs MG-ADL variaient de 0 à 100% sur les cycles 1 à 5, et la variation moyenne du score MG-ADL n'a pas atteint la limite de la pertinence clinique dans 3 des 5 cycles (« clinical meaningfulness », définie comme au moins ≥2 points au jour 43 par rapport à l'inclusion dans le cycle respectif).
Population pédiatrique
Pour plus d'informations sur l'utilisation du médicament en pédiatrie, voir la rubrique Posologie/Mode d'emploi.
PharmacocinétiqueAbsorption
Après l'administration sous-cutanée de rozanolixizumab, les concentrations plasmatiques maximales sont atteintes après environ 2 jours. La biodisponibilité absolue du rozanolixizumab après l'administration sous-cutanée était d'environ 70 %, telle qu'estimée par l'analyse pharmacocinétique de population.
Distribution
Le volume apparent de distribution du rozanolixizumab est d'environ 7 l selon l'estimation de l'analyse pharmacocinétique de population.
Métabolisme
Le rozanolixizumab devrait être dégradé en petits peptides et acides aminés via les voies cataboliques de manière similaire aux IgG endogènes.
Élimination
La clairance linéaire apparente pour la substance active libre est d'environ 0,9 l/jour. La demi-vie du rozanolixizumab dépend de la concentration et ne peut être calculée. Les concentrations plasmatiques de rozanolixizumab sont indétectables une semaine après l'administration.
Linéarité/non-linéarité
Le rozanolixizumab a présenté une pharmacocinétique non linéaire typique d'un anticorps monoclonal subissant une élimination du médicament à médiation par la cible.
À l'état d'équilibre, selon les prévisions, les concentrations plasmatiques maximales et l'aire sous la courbe (ASC) de la concentration en fonction du temps devaient être 3 fois et 4 fois plus élevées aux doses dépendant du poids de ≈10 mg/kg par rapport à ≈7 mg/kg, respectivement.
Cinétique pour certains groupes de patients
Trouble de la fonction hépatique ou rénale
Aucune étude spécifique n'a été menée chez les patients présentant une insuffisance rénale ou hépatique. Il est cependant peu probable que la pharmacocinétique du rozanolixizumab soit modifiée par une insuffisance rénale ou hépatique. Dans le cadre d'une analyse pharmacocinétique de population, les fonctions rénales (débit de filtration glomérulaire [DFGe] estimé à 38-161 ml/min/1,73 m2) ou les bilans biochimiques et hépatiques (ALAT, ASAT, phosphatase alcaline et bilirubine) n'ont montré aucune influence significative sur la clairance linéaire apparente du rozanolixizumab.
Âge, sexe ou groupe ethnique
Une analyse pharmacocinétique de population n'a montré aucun effet cliniquement significatif de l'âge (18 à 89 ans), du sexe ou de l'appartenance ethnique sur la pharmacocinétique du rozanolixizumab.
Données précliniquesToxicité en cas d'administration répétée
Les données non cliniques issues des études conventionnelles de toxicologie en administration répétée n'ont pas révélé de risque particulier pour l'homme. L'administration à des singes cynomolgus et rhésus a entraîné la réduction attendue des IgG.
La réponse thymo-dépendante (T-cell Dependent Antibody Response, TDAR) durant la phase de traitement a induit des taux d'IgM normaux et une faible réponse des IgG en raison de leur dégradation accélérée. Cependant, la vaccination de rappel après la clairance du rozanolixizumab a entraîné une réponse normale des IgM et des IgG.
Carcinogénicité
Aucune étude de carcinogénicité n'a été menée avec le rozanolixizumab.
Génotoxicité
Le rozanolixizumab étant un anticorps monoclonal, aucune étude de génotoxicité n'a été menée.
Toxicité sur la reproduction
Dans une étude sur des singes cynomolgus traités, l'incidence des avortements spontanés à partir du 20e jour de gestation était plus élevée dans le groupe rozanolixizumab que dans le groupe contrôle (principalement entre le 20e et le 5e jour de gestation). On n'a observé aucun effet du rozanolixizumab sur les développements embryo-fœtal et postnatal ni sur la parturition. Les portées des mères traitées présentaient de très faibles taux d'IgG à la naissance, conformément aux prévisions de la pharmacologie. Le taux d'IgG est revenu aux valeurs de contrôle ou supérieures dans les 60 jours. Il n'y a eu aucun impact sur la fonction immunitaire des petits des mères traitées, selon l'évaluation d'un test d'évaluation de la réponse thymo-dépendante (T-cell Dependent Antibody Response, TDAR.)
Altération de la fertilité
Aucune étude de fertilité (mâle ou femelle) n'a été menée.
Une étude de toxicité à doses répétées de rozanolixizumab pendant 26 semaines chez des singes n'a montré aucune modification liée au traitement au niveau des organes reproducteurs des animaux sexuellement matures. Une évaluation du cycle menstruel et des paramètres de la reproduction masculine (poids de l'éjaculat, nombre, motilité et morphologie des spermatozoïdes) n'a montré aucune modification liée au traitement.
Remarques particulièresIncompatibilités
En l'absence d'études de compatibilité, ce médicament ne doit pas être mélangé avec d'autres médicaments pour perfusion.
Stabilité
Le médicament ne doit pas être utilisé au-delà de la date figurant après la mention «EXP» sur l'emballage.
Remarques particulières concernant le stockage
Conserver au réfrigérateur (2-8 °C).
Ne pas congeler.
Conserver le flacon dans son carton pour le protéger de la lumière.
Conserver hors de portée des enfants.
Numéro d’autorisation69227.
PrésentationSolution de 2 ml dans un flacon (verre de type I) muni d'un bouchon en élastomère, scellé par un opercule serti et un capuchon amovible.
Taille d'emballage: 1 flacon [A].
Titulaire de l’autorisationUCB-Pharma AG, Bulle
Mise à jour de l’informationOctobre 2024
Remarques concernant la manipulation
Spécificités des matériaux
La solution injectable de rozanolixizumab peut être administrée à l'aide de seringues en polypropylène et de sets de perfusion contenant du polyéthylène (PE), du polyéthylène basse densité (LDPE), du polyester, du chlorure de polyvinyle (PVC sans DEHP), du polycarbonate (PC), de l'éthylène polypropylène fluoré (FEP), de l'uréthane/acrylate, du polyuréthane, du méta-acrylonitrile butadiène styrène (MABS), de la silicone ou de la cyclohexanone. Ne pas utiliser de dispositifs d'administration étiquetés comme contenant du di(2-éthylhexyl)phtalate (DEHP).
Afin d'éviter d'éventuelles interruptions dans l'administration de Rystiggo, les critères suivants doivent être respectés:
·Les limites d'alarme d'occlusion de la pompe à seringue doivent être paramétrées sur le réglage maximal.
·Une longueur de tubulure d'administration de 61 cm ou plus courte est recommandée.
·Un set de perfusion doté d'une aiguille de calibre 26 gauge ou plus doit être utilisé.
Lire attentivement les instructions avant toute administration de rozanolixizumab.
Le rozanolixizumab ne contient pas de conservateur, et chaque flacon est destiné exclusivement à un usage unique. Tout produit non utilisé ou déchet résiduel dans le flacon ne doit pas être réutilisé mais être éliminé conformément aux exigences locales
Les informations suivantes sont destinées uniquement aux professionnels de la santé.
Instructions d'utilisation destinées aux professionnels de santé
Manipulation de Rystiggo au moyen d'une technique de perfusion assistée par dispositif par ex., une pompe à perfusion
Voie sous-cutanée uniquement.
Le nombre de flacons (2 ml par flacon) à utiliser dépend du poids corporel du patient. Pour administrer la dose de 280 mg aux patients pesant ≥35 kg à <50 kg, 2 ml sont nécessaires. Pour administrer la dose de 420 mg aux patients pesant ≥50 kg à <70 kg, 3 ml sont nécessaires. Pour administrer la dose de 560 mg aux patients pesant ≥70 kg à <100 kg, 4 ml sont nécessaires. Pour administrer la dose de 840 mg aux patients pesant ≥100 kg, 6 ml sont nécessaires.
Lisez TOUTES les instructions ci-dessous avant d'administrer le rozanolixizumab.
1. Retirez Rystiggo de la boîte:
·Laissez le flacon atteindre la température ambiante. Cela peut prendre un minimum de 30 minutes et 120 minutes au maximum. Ne pas utiliser de dispositifs de chauffage.
·Vérifiez chaque flacon avant utilisation:
·Date d'expiration: Ne pas utiliser au-delà de la date de péremption.
·Couleur: la solution doit être incolore à jaune pâle/brunâtre, limpide à légèrement opalescente. Ne pas utiliser le flacon si le liquide semble trouble, contient des particules étrangères ou a changé de couleur.
·Capuchon: ne pas utiliser si le bouchon de protection du flacon est manquant ou défectueux.
2. Rassemblez tous les éléments:
·Rassemblez tous les éléments pour la perfusion. En plus du ou des flacon(s), rassemblez les éléments suivants, qui ne sont pas fournis: seringue, aiguille(s) de seringue, lingette imbibée d'alcool, set de perfusion, ruban adhésif ou pansement transparent, pompe à perfusion et conteneur pour objets tranchants.
3. Utilisez une technique aseptique lors de la préparation et de l'administration de ce produit.
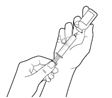
4. Préparez Rystiggo pour la perfusion
·Utilisez des aiguilles de transfert pour remplir la seringue
·Utilisez des canules mélangeuses pour remplir la seringue.
·Retirez le capuchon de protection du flacon et nettoyez le bouchon du flacon avec une lingette imbibée d'alcool. Laissez sécher.
·Prélevez tout le contenu du flacon dans la seringue. Une petite quantité restera dans le flacon et doit être jetée.
·Pour plusieurs flacons, utilisez une aiguille neuve et répétez les étapes précédentes.
·Retirez l'aiguille de la seringue et fixez le set de perfusion à la seringue.
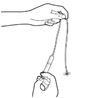
5. Préparez la perfusion
·Suivez les instructions fournies avec la pompe à perfusion afin de préparer la pompe et d'amorcer la ligne de perfusion. Administrez immédiatement après l'amorçage du set de perfusion.
·Chaque flacon contient un volume excédentaire (pour permettre l'amorçage de la ligne de perfusion); par conséquent, paramétrez la pompe au préalable pour administrer le volume prescrit. Pour les pompes qui ne peuvent pas être paramétrées à l'avance, après l'amorçage de la ligne de perfusion, ajustez le volume à administrer en éliminant tout volume excédentaire.
6. Préparez le site de perfusion
·Sélectionnez une zone de perfusion: partie inférieure droite ou inférieure gauche de l'abdomen, sous le nombril. Ne jamais perfuser dans les zones où la peau est sensible, rouge ou dure, ou présente des ecchymoses. Évitez de perfuser dans des cicatrices ou des vergetures.
·Nettoyez le site de perfusion à l'aide d'une lingette imbibée d'alcool. Laissez sécher.
7. Insérez l'aiguille du set de perfusion
·Saisissez un pli cutané abdominal entre deux doigts.
·Insérez l'aiguille du set de perfusion dans le tissu sous-cutané.
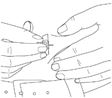
8. Fixez l'aiguille sur la peau
·Si nécessaire, utilisez du ruban adhésif ou un pansement transparent pour maintenir l'aiguille en place.

9. Démarrez la perfusion
·Suivez les instructions du fabricant pour l'utilisation de la pompe
10. Fin de la perfusion
·Lorsque la perfusion est terminée, ne pas rincer la ligne de perfusion car le volume de perfusion a été ajusté en tenant compte des pertes dans la ligne.
·Retirez l'aiguille du site de perfusion.
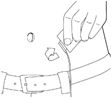
11. Nettoyez
·Jetez tous les éléments contenant le produit restant, c'est à dire, les flacons partiellement utilisés, le kit de perfusion ainsi que tout matériel d'administration dans un récipient pour objets tranchants
|