CompositionPrincipes actifs
Sparsentan
Excipients
Noyau du comprimé: cellulose microcristalline silicifiée, lactose, carboxyméthylamidon sodique (type A), silice colloïdale anhydre, stéarate de magnésium
Pelliculage du comprimé: poly(alcool vinylique), macrogol 3350, talc, dioxyde de titane (E171)
Comprimés pelliculés de 200 mg: 1 comprimé pelliculé contient 42 mg de lactose et au maximum 0,9 mg de sodium.
Comprimés pelliculés de 400 mg: 1 comprimé pelliculé contient 84 mg de lactose et au maximum 1,8 mg de sodium.
Indications/Possibilités d’emploiFilspari est indiqué dans le traitement des patients adultes atteints d’une néphropathie à immunoglobulines A (NIgA) primitive et présentant une protéinurie ≥ 1,0 g/jour (ou un rapport protéinurie/créatininurie ≥ 0,75 g/g) (voir « Efficacité clinique »).
En raison d’une documentation incomplète au moment de l’examen de la demande, cette indication est autorisée pour une durée limitée (art. 9a LPTh). L’autorisation à durée limitée est impérativement liée à la satisfaction de charges en temps opportun. Une fois ces charges satisfaites, l’autorisation à durée limitée pourra être transformée en autorisation sans charge spécifique.
Posologie/Mode d’emploiPosologie habituelle
Il convient d’instaurer le traitement par Filspari à une dose de 200 mg une fois par jour pendant 14 jours, puis d’augmenter la dose en fonction de la tolérance pour atteindre une dose d’entretien de 400 mg une fois par jour.
Pour la titration de la dose initiale de 200 mg une fois par jour jusqu’à la dose d’entretien de 400 mg une fois par jour, des comprimés pelliculés de 200 mg et de 400 mg sont disponibles pour atteindre la dose d’entretien.
Chez les patients présentant des problèmes de tolérance (pression artérielle systolique [PAS] ≤ 100 mmHg, pression artérielle diastolique ≤ 60 mmHg, aggravation d’un œdème ou hyperkaliémie), il est recommandé d’ajuster les médicaments concomitants, puis de réduire temporairement la dose de Filspari ou de l’arrêter (voir « Mises en garde et précautions » et « Efficacité clinique »).
Lors de la reprise du traitement par Filspari après une interruption, il faut envisager de reproduire le schéma posologique initial. En cas d’hypotension persistante ou de modifications de la fonction hépatique, l’interruption du traitement, précédée ou non d’une réduction de la dose de Filspari, peut être envisagée (voir « Mises en garde et précautions »).
Doses oubliées
En cas d’oubli d’une dose, celle-ci ne doit pas être prise et la dose suivante doit être prise à l’heure prévue. Aucune dose double ou supplémentaire ne doit être prise.
Patients âgés
Aucun ajustement posologique n’est recommandé chez les patients âgés (voir « Pharmacocinétique »). Chez les patients âgés, il convient d’instaurer le traitement par Filspari à une dose de 200 mg une fois par jour pendant 14 jours. L’augmentation de la dose jusqu’à une dose d’entretien de 400 mg une fois par jour doit se faire avec prudence, en fonction de la tolérance (voir « Mises en garde et précautions »).
Patients présentant des troubles de la fonction hépatique
Sur la base des données pharmacocinétiques, aucun ajustement posologique de Filspari n’est nécessaire chez les patients présentant une altération légère ou modérée (classe A ou classe B de Child-Pugh) de la fonction hépatique (voir « Pharmacocinétique »).
L’expérience clinique est limitée chez les patients présentant une altération modérée de la fonction hépatique. Par conséquent, Filspari doit être utilisé avec prudence chez ces patients (voir « Mises en garde et précautions »).
Filspari n’a pas été étudié chez les patients présentant une altération sévère (classe C de Child-Pugh) de la fonction hépatique. Son utilisation n’est donc pas recommandée chez ces patients.
L’expérience clinique est limitée chez les patients présentant des taux d’aspartate-aminotransférase (ASAT)/alanine-aminotransférase (ALAT) atteignant plus de deux fois la limite supérieure de la normale (LSN). Par conséquent, il convient de renoncer à instaurer un traitement par Filspari chez les patients présentant des taux d’ASAT/ALAT > 2 × LSN (voir « Mises en garde et précautions »).
Patients présentant des troubles de la fonction rénale
Aucune adaptation posologique n’est nécessaire chez les patients présentant une altération légère (insuffisance rénale chronique [IRC] de stade 2; débit de filtration glomérulaire estimé [DFGe] de 60 à 89 ml/min/1,73 m2) ou modérée (IRC de stade 3a et 3b; DFGe de 30 à 59 ml/min/1,73 m2) de la fonction rénale. Sur la base des données pharmacocinétiques, aucune adaptation posologique ne peut être recommandée pour les patients atteints d’une altération sévère (IRC de stade 4; DFGe < 30 ml/min/1,73 m2) de la fonction rénale (voir « Pharmacocinétique »). L’expérience clinique étant limitée chez les patients présentant une altération sévère de la fonction rénale, l’utilisation de Filspari n’est pas recommandée chez ces derniers (voir « Mises en garde et précautions »).
Filspari n’a pas été étudié chez les patients qui ont bénéficié d’une transplantation rénale. Par conséquent, Filspari doit être utilisé avec prudence chez ces patients.
Filspari n’a pas été étudié chez les patients dialysés. L’instauration d’un traitement par Filspari n’est pas recommandée chez ces patients.
Enfants et adolescents
La sécurité et l’efficacité de Filspari n’ont pas encore été établies chez les enfants et les adolescents de moins de 18 ans. Aucune donnée n’est disponible.
Mode d’emploi
Administration par voie orale.
Il est recommandé d’avaler les comprimés entiers avec de l’eau pour éviter leur goût amer. Filspari peut être pris avec ou sans nourriture.
Contre-indications·Hypersensibilité au principe actif ou à l’un des autres composants (voir « Composition »).
·Grossesse (voir « Mises en garde et précautions » et « Grossesse, allaitement »)
·Administration concomitante d’antagonistes des récepteurs de l’angiotensine (ARA), d’antagonistes des récepteurs de l’endothéline (ARE) ou d’inhibiteurs de la rénine (voir « Mises en garde et précautions » et « Interactions »).
Mises en garde et précautionsFemmes en âge de procréer
Le traitement par Filspari ne peut être instauré chez les femmes en âge de procréer qu’une fois que l’absence de grossesse a été confirmée et qu’une contraception efficace est utilisée (voir « Contreindications » et « Grossesse, allaitement »).
Hypotension
Des cas d’hypotension ont été associés à l’utilisation d’inhibiteurs du système rénine-angiotensine-aldostérone (SRAA), dont Filspari. Une hypotension peut survenir pendant le traitement par Filspari et a été plus fréquemment rapportée chez les patients âgés (voir « Effets indésirables »).
Chez les patients présentant un risque d’hypotension, il convient d’envisager l’arrêt ou l’ajustement des autres médicaments antihypertenseurs ainsi que le maintien d’un statut volémique approprié. Si une hypotension apparaît malgré l’arrêt ou la diminution des autres médicaments antihypertenseurs, il convient d’envisager une réduction de la dose ou une interruption du traitement par Filspari. Une réaction hypotensive transitoire ne constitue pas une contre-indication à la poursuite du traitement par Filspari. Le traitement peut être repris dès la stabilisation de la pression artérielle.
Si l’hypotension persiste malgré l’arrêt ou la diminution des médicaments antihypertenseurs, il convient de ramener la dose de Filspari à la dose initiale jusqu’à la stabilisation de la pression artérielle. Une interruption du traitement par Filspari doit être envisagée si les symptômes d’hypotension persistent après deux semaines de réduction de la dose. Filspari doit être utilisé avec prudence chez les patients présentant une pression artérielle systolique ≤ 100 mmHg (voir « Posologie/Mode d’emploi »). La dose de Filspari ne doit pas être augmentée chez les patients dont la pression artérielle systolique est ≤ 100 mmHg (voir « Posologie/Mode d’emploi »).
Troubles de la fonction rénale
Une augmentation transitoire de la créatinine sérique a été associée aux inhibiteurs du SRAA, dont Filspari. Une augmentation transitoire de la créatinine sérique peut survenir, en particulier au début du traitement par Filspari (voir « Effets indésirables »). Chez les patients à risque, les taux sériques de créatinine et de potassium doivent être surveillés régulièrement. Il convient également de faire preuve de prudence lors de l’utilisation de Filspari chez les patients présentant une sténose bilatérale de l’artère rénale.
L’expérience clinique étant limitée chez les patients présentant un DFGe < 30 ml/min/1,73 m2, l’utilisation de Filspari n’est pas recommandée chez ces patients (voir « Posologie/Mode d’emploi »).
Rétention hydrique
Des cas de rétention hydrique ont été associés à des médicaments antagonistes du récepteur de type A de l’endothéline (ETAR), dont le sparsentan. Pendant le traitement par Filspari, une rétention hydrique peut survenir (voir « Effets indésirables »).
Si une rétention hydrique apparaît pendant le traitement par Filspari, il est recommandé d’administrer un diurétique ou d’augmenter la dose de diurétique en cours avant de modifier la dose de Filspari. Chez les patients chez lesquels une rétention hydrique a été constatée avant le début du traitement par Filspari, un traitement diurétique peut être envisagé.
Filspari n’a pas été étudié chez les patients présentant une insuffisance cardiaque. Par conséquent, il doit être utilisé avec prudence chez les patients insuffisants cardiaques.
Fonction hépatique
Des élévations de l’ALAT et de l’ASAT supérieures ou égales à 3 × LSN ont été observées lors de l’utilisation de Filspari (voir « Effets indésirables »).
Aucune élévation concomitante de la bilirubine > 2 × LSN ni aucun cas d’insuffisance hépatique n’a été observé chez les patients traités par Filspari. Par conséquent, pour réduire le risque d’hépatotoxicité sévère potentielle, les taux sériques d’aminotransférases et la bilirubine totale doivent être contrôlés avant le début du traitement, puis faire l’objet d’une surveillance tous les trois mois.
Les patients doivent être surveillés pour déceler tout signe de lésion hépatique. Si les patients développent une élévation durable, inexpliquée et cliniquement significative de l’ALAT et/ou de l’ASAT, ou si de telles élévations s’accompagnent d’une augmentation de la bilirubine > 2 × LSN, ou si l’élévation de l’ALAT et/ou de l’ASAT est associée à des signes ou symptômes de lésion hépatique (par exemple un ictère), le traitement par Filspari doit être arrêté.
Une nouvelle administration de Filspari ne peut être envisagée qu’une fois que les taux d’enzymes hépatiques et de bilirubine sont revenus aux valeurs initiales avant le traitement, et seulement chez les patients ne présentant pas de symptômes cliniques d’hépatotoxicité. Filspari ne doit pas être utilisé chez les patients présentant un taux élevé d’aminotransférases (> 2 × LSN) avant le début du traitement (voir « Posologie/Mode d’emploi »).
L’expérience clinique est limitée chez les patients présentant une altération modérée de la fonction hépatique. Par conséquent, il convient de faire preuve de prudence lors de l’utilisation de Filspari chez ces patients (voir « Posologie/Mode d’emploi »).
Double inhibition du système rénine-angiotensine-aldostérone (SRAA)
Il existe des preuves que l’utilisation concomitante d’inhibiteurs de l’enzyme de conversion de l’angiotensine (IEC), d’antagonistes des récepteurs de l’angiotensine II (ARA II) ou d’aliskirène augmente le risque d’hypotension, d’hyperkaliémie et d’altération de la fonction rénale (y compris d’insuffisance rénale aiguë). La double inhibition du SRAA par l’utilisation combinée d’IEC et d’ARA II (correspondant en partie au mécanisme d’action du sparsentan) ou d’inhibiteurs de la rénine n’est donc pas recommandée (voir « Interactions » et « Efficacité clinique »). Si un traitement par double inhibition est considéré comme absolument nécessaire, il doit se faire uniquement sous la supervision d’un spécialiste et sous réserve d’une surveillance étroite et fréquente de la fonction rénale, des électrolytes et de la pression artérielle.
Hyperkaliémie
Le traitement ne doit pas être instauré chez les patients dont le taux de potassium sérique est > 5,5 mmol/l. Comme avec d’autres médicaments qui affectent le système rénine-angiotensine-aldostérone, une hyperkaliémie peut survenir pendant le traitement par Filspari, en particulier en présence d’une altération de la fonction rénale et/ou d’une insuffisance cardiaque. Chez les patients à risque, une surveillance étroite de la kaliémie est recommandée. En cas d’apparition d’une hyperkaliémie cliniquement significative, il est recommandé d’ajuster les médicaments concomitants, de réduire temporairement la dose ou d’arrêter le traitement. Si le taux de potassium sérique est > 5,5 mmol/l, l’arrêt du traitement devra être envisagé.
Les patients présentant une intolérance au galactose, un déficit total en lactase ou un syndrome de malabsorption du glucose et du galactose (maladies héréditaires rares) ne devraient pas prendre ce médicament.
Ce médicament contient moins de 1 mmol (23 mg) de sodium par comprimé, c.-à-d. qu’il est essentiellement « sans sodium ».
InteractionsUtilisation concomitante d’ARA, d’ARE et d’inhibiteurs de la rénine
L’utilisation concomitante du sparsentan avec des ARE, comme le bosentan, l’ambrisentan, le macitentan, le sitaxentan, avec des ARA, comme l’irbésartan, le losartan, le valsartan, le candésartan, le telmisartan, ou avec des inhibiteurs de la rénine, comme l’aliskirène, est contre-indiquée (voir « Contre-indications »).
Utilisation concomitante d’IEC et d’inhibiteurs du récepteur des minéralocorticoïdes (aldostérone)
L’utilisation concomitante du sparsentan avec des inhibiteurs du récepteur des minéralocorticoïdes (aldostérone), comme la spironolactone et la finérénone, peut être associée à un risque accru d’hyperkaliémie.
Il n’existe aucune donnée concernant l’association du sparsentan avec des IEC comme l’énalapril ou le lisinopril. Les études cliniques ont montré que la double inhibition du système rénine-angiotensine-aldostérone (SRAA) par l’utilisation combinée d’IEC, d’antagonistes des récepteurs de l’angiotensine II ou d’aliskirène est associée à des événements indésirables plus fréquents tels que l’hypotension, l’hyperkaliémie et l’altération de la fonction rénale (y compris l’insuffisance rénale aiguë) par rapport à l’utilisation d’un seul agent agissant sur le SRAA (voir « Pharmacocinétique »).
L’utilisation du sparsentan en association avec des IEC, comme l’énalapril ou le lisinopril, doit se faire avec prudence et sous la surveillance de la pression artérielle, de la kaliémie et de la fonction rénale (voir « Mises en garde et précautions »).
Utilisation concomitante de préparations de potassium et de diurétiques épargneurs de potassium
Une hyperkaliémie pouvant apparaître chez les patients traités par des médicaments antagonistes du récepteur de type 1 de l’angiotensine II (AT1R) (voir « Effets indésirables »), l’utilisation concomitante de préparations de potassium, de diurétiques épargneurs de potassium, comme la spironolactone, l’éplérénone, le triamtérène ou l’amiloride, ou de substituts de sel contenant du potassium peut majorer le risque d’hyperkaliémie et n’est donc pas recommandée.
Effets d’autres médicaments sur le sparsentan
Le sparsentan est principalement métabolisé par le cytochrome P450 (CYP)3A.
Inhibiteurs puissants et modérés du CYP3A
L’administration concomitante de sparsentan et d’itraconazole (inhibiteur puissant du CYP3A) a multiplié par 1,3 la Cmax du sparsentan et par 2,7 son ASC0-inf. L’administration concomitante d’un inhibiteur puissant du CYP3A, comme le bocéprévir, le télaprévir, la clarithromycine, l’indinavir, le lopinavir/ritonavir, l’itraconazole, la néfazodone ou le ritonavir, ou la consommation concomitante de pamplemousse ou de jus de pamplemousse n’est pas recommandée.
L’administration concomitante de sparsentan et de ciclosporine (inhibiteur modéré du CYP3A) a multiplié par 1,4 la Cmax du sparsentan et par 1,7 son ASC0-inf. L’administration concomitante d’un inhibiteur modéré du CYP3A, comme le conivaptan, le fluconazole ou le nelfinavir, doit se faire avec prudence.
Inducteurs du CYP3A
Le sparsentan est un substrat du CYP3A. L’administration concomitante d’un inducteur modéré ou puissant du CYP3A, comme la rifampicine, l’éfavirenz, la dexaméthasone, la carbamazépine, la phénytoïne ou le phénobarbital, diminue l’exposition au sparsentan, ce qui pourrait altérer son efficacité. Par conséquent, l’administration concomitante d’un inducteur modéré ou puissant du CYP3A n’est pas recommandée.
Médicaments réduisant l’acidité gastrique
D’après une analyse pharmacocinétique (PK) de population, l’administration concomitante d’un médicament réduisant l’acidité gastrique pendant le traitement par le sparsentan n’aurait aucun impact significatif sur la variabilité de la PK du sparsentan. Les médicaments qui modifient le pH gastrique, comme les antiacides, les inhibiteurs de la pompe à protons et les agonistes des récepteurs de l’histamine de type 2, peuvent être utilisés en association avec le sparsentan.
Effet du sparsentan sur d’autres médicaments
In vitro, le sparsentan a inhibé et induit le CYP3A et a induit le CYP2B6, le CYP2C9 ainsi que le CYP2C19.
L’administration concomitante de sparsentan à l’état d’équilibre et de midazolam, un substrat du CYP3A4, n’a eu aucun effet sur l’exposition systémique au midazolam. L’administration concomitante de sparsentan à l’état d’équilibre et de bupropion, un substrat du CYP2B6, a divisé par 1,5 à la fois la Cmax et l’ASC0-inf du bupropion.
Aucun ajustement posologique n’est nécessaire lors de l’utilisation concomitante de sparsentan à l’état d’équilibre et d’un substrat du CYP3A4 ou du CYP2B6.
Aucune étude clinique n’a évalué l’importance de l’induction du CYP2C9 et du CYP2C19 par le sparsentan. Il convient de faire preuve de prudence lors de l’administration concomitante de sparsentan et d’un substrat du CYP2C9, comme la warfarine S, la phénytoïne et l’ibuprofène, ou d’un substrat du CYP2C19, comme l’oméprazole et la phénytoïne.
Aucune étude clinique n’a évalué l’importance de l’induction du CYP3A4 après une dose unique de sparsentan. Le sparsentan étant un inhibiteur du CYP3A4, il peut, lors de son instauration, avoir un effet sur la PK des médicaments qui sont des substrats du CYP3A4. Ainsi, il convient de faire preuve de prudence lors de l’instauration d’un traitement concomitant par sparsentan avec un substrat du CYP3A4, comme l’alfentanil, le conivaptan, l’indinavir, la ciclosporine et le tacrolimus.
In vitro, le sparsentan est un inhibiteur de la P-gp, de la BCRP, de l’OATP1B3 et de l’OAT3 à des concentrations pertinentes.
Aucune étude clinique n’a évalué l’importance de l’inhibition de la P-gp par le sparsentan.
Il convient de faire preuve de prudence lors de l’administration concomitante de sparsentan et de substrats inhibant la P-gp, s’il est établi que l’inhibition de la P-gp a un effet significatif sur l’absorption.
L’administration concomitante de sparsentan et de pitavastatine (un substrat de l’OATP1B1, de l’OATP1B3 et de la BCRP) a divisé par 1,2 la Cmax de la pitavastatine et par 1,4 son ASC0-inf. Aucun ajustement posologique n’est nécessaire lors de l’utilisation concomitante de sparsentan et d’un substrat de l’OATP1B1, de l’OATP1B3 ou de la BCRP.
Aucune étude clinique n’a été menée pour étudier l’effet du sparsentan sur un substrat sensible de l’OAT3. Cependant, à une dose de 800 mg, le sparsentan ne semble pas affecter le biomarqueur 6βhydroxycortisol (substrat de l’OAT3), ce qui semble indiquer que l’effet clinique est très probablement limité.
Grossesse/AllaitementFemmes en âge de procréer
Le traitement par Filspari ne peut être instauré chez les femmes en âge de procréer qu’une fois l’absence de grossesse confirmée. Les femmes en âge de procréer doivent utiliser une contraception efficace pendant le traitement et jusqu’à un mois après son arrêt.
Grossesse
À ce jour, il n’existe pas de données ou il existe des données très limitées sur l’utilisation de Filspari chez la femme enceinte.
Les études effectuées chez l’animal ont mis en évidence une toxicité pour la reproduction (voir « Données précliniques »).
Filspari est contre-indiqué pendant la grossesse (voir « Contre-indications »).
Allaitement
Les données physico-chimiques suggèrent une excrétion du sparsentan dans le lait maternel chez l’être humain. Un risque pour les nouveau-nés/nourrissons ne peut être exclu. Filspari ne doit pas être utilisé pendant l’allaitement.
Fertilité
Il n’existe aucune donnée sur les effets du sparsentan sur la fertilité humaine. Les données chez l’animal n’ont pas mis en évidence d’altération de la fertilité chez les mâles ou les femelles (voir « Donnes précliniques »).
Effet sur l’aptitude à la conduite et l’utilisation de machinesFilspari peut avoir une légère influence sur l’aptitude à la conduite et l’utilisation de machines.
Les effets de Filspari sur l’aptitude à conduire des véhicules et la capacité à utiliser des machines n’ont pas été étudiés. Il convient toutefois de tenir compte du fait que des étourdissements peuvent apparaître pendant le traitement par Filspari (voir « Effets indésirables »). Il doit être conseillé aux patients présentant des étourdissements de ne pas conduire de véhicules et de ne pas utiliser de machines jusqu’à ce que les symptômes aient disparu.
Effets indésirablesRésumé du profil de sécurité
Les effets indésirables les plus fréquemment rapportés ont été l’hypotension (9 %), l’hyperkaliémie (7 %), les étourdissements (7 %) et les œdèmes périphériques (5 %). L’effet indésirable grave le plus fréquemment rapporté était l’insuffisance rénale aiguë (1 %).
Liste des effets indésirables
Des données de sécurité ont été obtenues à partir de 27 études cliniques dans le cadre desquelles plus de 500 patients présentant une maladie rénale chronique, y compris une NIgA, ont reçu Filspari (voir « Efficacité clinique »).
Les effets indésirables sont rangés par classe de systèmes d’organes de MedDRA et par fréquence selon la convention suivante:
« très fréquents » (≥ 1/10)
« fréquents » (≥ 1/100, < 1/10)
« occasionnels » (≥ 1/1000, < 1/100)
« rares » (≥ 1 /10000, < 1/1000)
« très rares » (< 1/10000)
Tableau 1: Effets indésirables observés lors des études cliniques
|
Classe de systèmes d’organes
|
Fréquents
|
Occasionnels
| |
Affections hématologiques et du système lymphatique
|
-
|
Anémie
| |
Troubles du métabolisme et de la nutrition
|
Hyperkaliémie
|
-
| |
Affections du système nerveux
|
Étourdissements
Céphalée
|
-
| |
Affections vasculaires
|
Hypotension
Hypotension orthostatique
|
-
| |
Affections du rein et des voies urinaires
|
Insuffisance rénale
Insuffisance rénale aiguë
|
-
| |
Troubles généraux et anomalies au site d’administration
|
Œdème périphérique
Fatigue
|
-
| |
Investigations
|
Créatinine sanguine augmentée
Transaminases augmentéesa
|
-
|
a Le terme « transaminases augmentées » inclut les termes préférentiels suivants: alanine aminotransférase augmentée, aspartate aminotransférase augmentée, gammaglutamyltransférase augmentée, enzymes hépatiques élevées et transaminases augmentées.
Description d’effets indésirables spécifiques
Diminution de l’hémoglobine
Dans l’étude PROTECT, une anémie ou une diminution de l’hémoglobine a été rapportée en tant qu’effet indésirable chez 2 patients (< 1 %) traités par sparsentan, contre 2 patients (< 1 %) traités par irbésartan. Globalement, une hémoglobine ≤ 9 g/dl a été rapportée à tout moment pendant le traitement chez 5 patients (2,5 %) du groupe traité par sparsentan et chez 3 patients (1,5 %) du groupe traité par irbésartan. Il semble que cette diminution soit en partie due à une hémodilution. Aucun arrêt du traitement en raison d’une anémie n’a été rapporté.
Événements indésirables de nature hépatique
Dans l’étude PROTECT, ce sont au total 6 participants (3 %) du groupe sparsentan et 4 participants (2 %) du groupe irbésartan qui ont présenté une élévation des transaminases hépatiques supérieure à 3 fois la limite supérieure de la normale, sans élévation de la bilirubine totale, après avoir reçu le médicament à l’étude pendant respectivement 168 à 407 jours. Tous les événements étaient sans gravité et asymptomatiques, et la majorité d’entre eux étaient d’intensité légère ou modérée. Tous les événements étaient réversibles et d’autres motifs ont été identifiés comme des facteurs de causalité potentiels ou comme des facteurs contribuant potentiellement à l’élévation des transaminases. Dans le groupe sparsentan, le médicament à l’étude a été arrêté chez 3 participants après un rechallenge positif, tandis que le traitement par sparsentan a été réintroduit sans nouvelle élévation des taux d’enzymes hépatiques chez 2 participants.
Insuffisance rénale aiguë (IRA)
Dans l’étude PROTECT, une insuffisance rénale aiguë a été rapportée en tant qu’effet indésirable chez 4 participants (2 %) du groupe sparsentan et 2 participants (1 %) du groupe irbésartan. Quatre patients (2 %) ayant reçu le sparsentan ont présenté une IRA grave, réversible dans tous les cas. Aucune des IRA graves n’a nécessité de dialyse. Dans le groupe sparsentan, le médicament à l’étude a été arrêté chez 3 participants.
Hyperkaliémie
Dans l’étude PROTECT, une hyperkaliémie a été rapportée en tant qu’effet indésirable chez 18 participants (9 %) du groupe sparsentan et 16 participants (8 %) du groupe irbésartan. Chez les participants du groupe sparsentan, tous les événements étaient sans gravité. La majorité était d’intensité légère à modérée et tous étaient réversibles. Aucun arrêt du traitement en raison d’une hyperkaliémie n’a été rapporté. Le risque d’hyperkaliémie est accru chez les patients présentant un DFGe faible.
Hypotension
Dans l’étude PROTECT, une PAS < 100 mmHg ou une réduction de la PAS supérieure à 30 mmHg a été rapportée chez 10 % et 8 % des patients traités par sparsentan, respectivement, contre 9 % et 6 % des patients traités par irbésartan. Parmi les participants traités par sparsentan, seuls 15 (7,4 %) étaient âgés de plus de 65 ans. Une hypotension a été rapportée chez 17 participants (9 %) de moins de 65 ans et chez 5 participants (33 %) âgés de 65 à 74 ans.
L’annonce d’effets secondaires présumés après l’autorisation est d’une grande importance. Elle permet un suivi continu du rapport bénéfice-risque du médicament. Les professionnels de santé sont tenus de déclarer toute suspicion d’effet secondaire nouveau ou grave via le portail d’annonce en ligne ElViS (Electronic Vigilance System). Vous trouverez des informations à ce sujet sur www.swissmedic.ch.
SurdosageFilspari a été administré à des sujets sains à des doses allant jusqu’à 1600 mg/jour, sans signes de toxicité limitant la dose. Les patients qui présentent un surdosage (avec potentiellement des signes et symptômes d’hypotension) doivent être surveillés attentivement et recevoir un traitement symptomatique approprié.
Propriétés/effetsCode ATC
C09XX01
Mécanisme d’action
Le sparsentan est un antagoniste double des récepteurs de l’endothéline et de l’angiotensine.
Il s’agit d’une molécule unique, qui agit en tant qu’antagoniste à forte affinité et à double action à la fois du récepteur de type A de l’endothéline (ETAR) et du récepteur de type 1 de l’angiotensine (AT1R). L’endothéline 1 (via ETAR) et l’angiotensine II (via AT1R) interviennent dans des processus qui conduisent à la progression de la NIgA, tels que les effets hémodynamiques et la prolifération des cellules mésangiales, l’expression et l’activité accrues des médiateurs pro-inflammatoires et profibrotiques, les lésions des podocytes et le stress oxydant. Le sparsentan inhibe l’activation d’ETAR et d’AT1R, réduisant ainsi la protéinurie et ralentissant la progression de la maladie rénale.
Pharmacodynamique
Dans une étude randomisée comprenant un groupe comparateur actif et un groupe placebo et menée auprès de sujets sains, le sparsentan a entraîné un léger allongement de l’intervalle QTcF, avec un effet maximal de 8,8 ms (IC à 90 %: 5,9; 11,8) à une dose de 800 mg et de 8,1 ms (IC à 90 %: 5,2; 11,0) à une dose de 1600 mg. Dans une autre étude menée auprès de sujets sains avec une exposition au sparsentan correspondant à plus de 2 fois la dose maximale recommandée chez l’être humain, aucun allongement significatif de l’intervalle QTcF n’a été observé. L’effet maximal était de 8,3 ms (6,69; 9,90). Il est donc peu probable que le sparsentan ait un effet cliniquement significatif sur l’allongement de l’intervalle QT.
Efficacité clinique
L’efficacité et la sécurité du sparsentan ont été évaluées dans l’étude PROTECT menée auprès de patients atteints d’une NIgA.
L’étude PROTECT est une étude de phase III internationale, multicentrique, randomisée, en double aveugle (110 semaines) et contrôlée contre comparateur actif qui a été menée auprès de patients atteints d’une NIgA. Ont participé à cette étude des patients âgés de ≥ 18 ans, dont 15 patients (8 %) traités par sparsentan âgés de > 65 ans, présentant un DFGe ≥ 30 ml/min/1,73 m2 et une protéinurie totale ≥ 1,0 g/jour. Avant la participation à l’étude, les patients ont reçu la dose maximale tolérée d’un IEC et/ou d’un ARA pendant au moins 3 mois. Le traitement par IEC et/ou par ARA a été arrêté avant l’instauration du sparsentan. Les patients présentant un taux de potassium supérieur à 5,5 mmol/l à l’inclusion ont été exclus.
Au total, 404 patients ont été randomisés et ont reçu soit le sparsentan (n = 202) soit l’irbésartan (n = 202). Le traitement a été instauré à la dose de 200 mg de sparsentan une fois par jour ou de 150 mg d’irbésartan une fois par jour. Après 14 jours, la dose devait être augmentée selon la tolérance jusqu’à la dose recommandée de 400 mg de sparsentan une fois par jour ou de 300 mg d’irbésartan une fois par jour. La tolérance a été définie comme une pression artérielle systolique > 100 mmHg et une pression artérielle diastolique > 60 mmHg après deux semaines et l’absence d’EI (p. ex. aggravation d’un œdème) ou en fonction des résultats d’analyse biologique (p. ex. kaliémie > 5,5 mEq/l [5,5 mmol/l]). Les inhibiteurs du SRAA ou du système endothéline étaient interdits pendant l’étude. D’autres classes d’agents antihypertenseurs étaient autorisées en fonction des besoins, pour atteindre la pression artérielle cible. Un traitement par agents immunosuppresseurs était autorisé pendant l’étude, à la discrétion de l’investigateur.
Les données à l’inclusion en matière de DFGe et de protéinurie étaient comparables entre les groupes de traitement. La population totale présentait un DFGe moyen (ET) de 57 (24) ml/min/1,73 m2 et un rapport protéinurie/créatininurie (UP/C) médian de 1,24 g/g (écart interquartile: 0,83; 1,77). L’âge moyen était de 46 ans (étendue de 18 à 76 ans); 70 % des patients étaient de sexe masculin, 67 % étaient blancs, 28 % étaient asiatiques, 1 % étaient noirs ou afro-américains et 3 % appartenaient à une autre ethnie.
L’analyse principale (intermédiaire) de la protéinurie a été menée 36 semaines après la randomisation d’environ 280 patients, afin de déterminer si l’effet du traitement sur le critère d’évaluation principal de l’efficacité, à savoir la variation du rapport UP/C entre l’inclusion et la semaine 36, était statistiquement significatif. Le critère d’évaluation principal de l’étude, à savoir la variation du rapport UP/C entre l’inclusion et la semaine 36, a été satisfait. La moyenne géométrique du rapport UP/C à la semaine 36 était de 0,62 g/g dans le groupe sparsentan, contre 1,07 g/g dans le groupe irbésartan. La moyenne géométrique des moindres carrés pour la variation en pourcentage du rapport UP/C entre l’inclusion et la semaine 36 était de -49,8 % (intervalle de confiance [IC] à 95 %: -54,98; -43,95) dans le groupe sparsentan, contre -15,1 % (IC à 95 %: -23,72; -5,39) dans le groupe irbésartan (p < 0,0001; Figure 1). Lors de l’analyse finale, le sparsentan a montré un effet antiprotéinurique rapide et durable sur une période de 2 ans, avec une moyenne géométrique pour le rapport UP/C à la semaine 110 de 0,64 g/g dans le groupe sparsentan contre 1,09 g/g dans le groupe irbésartan, ce qui représente une réduction moyenne de 43 % par rapport à l’inclusion (IC à 95 %: -49,75; -34,97), contre seulement 4,4 % avec l’irbésartan (IC à 95 %: -15,84; 8,70). Une amélioration en matière de réduction de la protéinurie a été observée durablement dans le groupe sparsentan après 4 semaines et jusqu’à la semaine 110 (Figure 1).
Figure 1: Évolution en pourcentage du rapport protéinurie/créatininurie par rapport à l’inclusion, par visite (étude PROTECT)
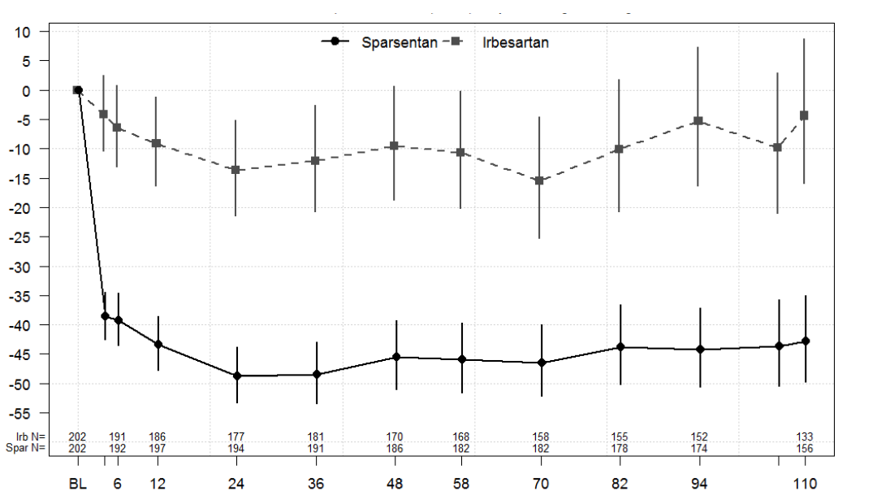
Remarques: la moyenne géométrique des moindres carrés ajustée pour le rapport UP/C par rapport à l’inclusion était basée sur un modèle longitudinal à mesures répétées, stratifié en fonction du DFGe et de la protéinurie à la sélection, avec les valeurs exprimées en pourcentage d’évolution avec leur IC à 95 % respectif. L’analyse inclut les données relatives au rapport UP/C recueillies pendant la phase en double aveugle auprès de tous les patients randomisés ayant reçu au moins une dose de médicament à l’étude. La valeur à l’inclusion a été définie comme la dernière observation non manquante qui a été réalisée avant/pendant l’administration de la première dose.
Abréviations: IC = intervalle de confiance; DFGe = débit de filtration glomérulaire estimé; UP/C = rapport protéinurie/créatininurie.
DFG estimé
Lors de l’analyse confirmatoire, l’amélioration de la pente durable du DFGe à 2 ans (à partir de 6 semaines de traitement) était de 1,1 ml/min/1,73 m2 par an avec le sparsentan par rapport à l’irbésartan (IC à 95 %: -0,07, 2,12; p = 0,037), et l’amélioration correspondante de la pente totale du DFGe à 2 ans (à partir de l’inclusion) était de 1,0 ml/min/1,73 m2 par an (IC à 95 %: -0,03, 1,94; p = 0,058). La variation absolue par rapport à l’inclusion pour le DFGe à 2 ans était de -5,8 ml/min/1,73 m2 (IC à 95 %: -7,38; -4,24) pour le sparsentan, contre -9,5 ml/min/1,73 m2 (IC à 95 %: -11,17; -7,89) pour l’irbésartan.
Informations supplémentaires
Deux études à grande échelle randomisées et contrôlées (ONTARGET [ONgoing Telmisartan Alone and in combination with Ramipril Global Endpoint Trial] et VA NEPHRON-D [The Veterans Affairs Nephropathy in Diabetes]) ont porté sur l’utilisation de l’association d’un IEC et d’un antagoniste des récepteurs de l’angiotensine II. L’étude ONTARGET a été menée auprès de patients présentant des antécédents de maladies cardiovasculaires ou cérébrovasculaires ou de diabète de type 2, et chez lesquels des signes de lésions des organes cibles étaient présents. L’étude VA NEPHRON-D a été menée chez des patients présentant un diabète de type 2 et une néphropathie diabétique. Ces études n’ont pas montré d’effet positif significatif sur les résultats rénaux et/ou cardiovasculaires ni sur la mortalité, tandis qu’un risque accru d’hyperkaliémie, d’insuffisance rénale aiguë et/ou d’hypotension a été observé par rapport à la monothérapie. Compte tenu de leurs propriétés pharmacodynamiques similaires, ces résultats sont aussi pertinents pour les autres IEC et antagonistes des récepteurs de l’angiotensine II. Par conséquent, aucun traitement concomitant par des IEC et des antagonistes des récepteurs de l’angiotensine II ne doit être utilisé chez les patients présentant une néphropathie diabétique. L’étude ALTITUDE (Aliskiren Trial in Type 2 Diabetes Using Cardiovascular and Renal Disease Endpoints) a été conçue pour évaluer les bénéfices de l’ajout d’aliskirène à un traitement standard à base d’IEC ou d’antagoniste des récepteurs de l’angiotensine II chez des patients présentant un diabète de type 2 ainsi qu’une maladie rénale chronique, une maladie cardiovasculaire ou les deux. Il a été mis un terme prématuré à cette étude en raison d’un risque accru d’effets indésirables. Les décès d’origine cardiovasculaire et les accidents vasculaires cérébraux ont été numériquement plus fréquents dans le groupe aliskirène que dans le groupe placebo, et des événements indésirables et des événements indésirables graves d’intérêt (hyperkaliémie, hypotension ou altérations de la fonction rénale) ont également été observés plus fréquemment dans le groupe aliskirène que dans le groupe placebo.
PharmacocinétiqueAbsorption
Après une dose orale unique de 400 mg de sparsentan, le délai médian pour atteindre la concentration plasmatique maximale est d’environ 3 heures.
Après une dose orale unique de 400 mg de sparsentan, les moyennes géométriques de la Cmax et de l’ASC sont respectivement de 6,97 μg/ml et de 83 μg × h/ml. Les concentrations plasmatiques à l’état d’équilibre sont atteintes dans les 7 jours, sans accumulation de l’exposition à la posologie recommandée.
Après une dose quotidienne de 400 mg de sparsentan, les moyennes géométriques de la Cmax et de l’ASC à l’état d’équilibre sont respectivement de 6,47 μg/ml et de 63,6 μg × h/ml.
Interaction avec la nourriture
À des doses allant jusqu’à 400 mg, l’effet d’un repas riche en graisses sur l’exposition au sparsentan n’était pas cliniquement significatif. Le sparsentan peut être pris avec ou sans nourriture.
Distribution
Sur la base d’une analyse pharmacocinétique de population, le volume apparent de distribution à l’état d’équilibre est de 61,4 l.
Le sparsentan se lie fortement (> 99 %) aux protéines plasmatiques humaines, avec une liaison préférentielle à l’albumine et une liaison modérée à l’alpha-1-glycoprotéine acide.
Métabolisme
Le sparsentan est principalement métabolisé par le CYP3A4, avec une contribution mineure du CYP2C8, du CYP2C9 et du CYP3A5. La substance mère est l’entité prédominante dans le plasma humain, représentant environ 90 % de la radioactivité totale dans la circulation. Un métabolite hydroxylé mineur est le seul métabolite présent dans le plasma et représente > 1 % de la radioactivité totale (environ 3 %). La principale voie métabolique du sparsentan est l’oxydation et la désalkylation; 9 métabolites ont été identifiés dans les selles, le plasma et l’urine humains.
Élimination
La clairance du sparsentan est temps-dépendante. Sur la base d’une analyse pharmacocinétique de population, la clairance apparente est de 3,88 l/h et augmente à 5,11 l/h à l’état d’équilibre.
La demi-vie du sparsentan à l’état d’équilibre est estimée à 9,6 heures.
Après une dose unique de 400 mg de sparsentan radiomarqué, 82 % de la radioactivité administrée ont été retrouvés au cours d’une période de recueil de 10 jours: 80 % dans les selles, dont 9 % sous forme inchangée, et 2 % dans les urines, dont une quantité négligeable sous forme inchangée.
Linéarité/non-linéarité
La Cmax et l’ASC du sparsentan augmentent de manière moins que proportionnelle après l’administration de doses uniques de 200 mg à 1600 mg. Le sparsentan a présenté une pharmacocinétique temps-dépendante, sans accumulation de la Cmax et avec une diminution de l’ASC à l’état d’équilibre après une dose quotidienne de 400 mg ou 800 mg.
Cinétique pour certains groupes de patients
Patients âgés
L’analyse pharmacocinétique de population n’a montré aucun effet significatif de l’âge sur l’exposition plasmatique au sparsentan. Aucune adaptation posologique n’est nécessaire chez les patients âgés (voir « Posologie/Mode d’emploi »). Le sparsentan n’a pas été étudié chez les patients âgés de > 75 ans.
Altération de la fonction hépatique
Dans une étude consacrée à l’insuffisance hépatique, l’exposition systémique après une dose unique de 400 mg de sparsentan était similaire chez les patients présentant une altération légère ou modérée (classe A ou classe B de Child-Pugh) de la fonction hépatique à l’inclusion et chez les patients présentant une fonction hépatique normale. Aucun ajustement posologique n’est nécessaire chez les patients présentant une altération légère ou modérée de la fonction hépatique. Il convient toutefois de faire preuve de prudence lors de l’utilisation du sparsentan chez des patients présentant une altération modérée de la fonction hépatique (voir « Posologie/Mode d’emploi » et « Mises en garde et précautions »).
Aucune donnée n’étant disponible concernant les patients présentant une altération sévère (classe C de Child-Pugh) de la fonction hépatique, l’utilisation du sparsentan chez ces derniers n’est pas recommandée (voir « Posologie/Mode d’emploi »).
Altération de la fonction rénale
Selon une analyse pharmacocinétique de population menée chez des patients présentant une insuffisance rénale chronique légère (clairance de la créatinine: 60 à 89 ml/min), modérée (clairance de la créatinine: 30 à 59 ml/min) et sévère (clairance de la créatinine: 15 à 29 ml/min), l’insuffisance rénale n’a pas d’effet cliniquement significatif sur la pharmacocinétique par rapport à une fonction rénale normale (clairance de la créatinine ≥ 90 ml/min). Aucune donnée n’est disponible concernant les patients présentant une insuffisance rénale terminale (clairance de la créatinine < 15 ml/min).
Sur la base des données limitées disponibles, aucun ajustement posologique ne peut être recommandé pour les patients présentant une insuffisance rénale sévère (DFGe < 30 ml/min/1,73 m2, voir « Posologie/Mode d’emploi »). Le sparsentan n’ayant pas été étudié chez les patients dialysés ou présentant une insuffisance rénale sévère, son utilisation chez ces derniers n’est pas recommandée. Le sparsentan n’ayant pas été étudié chez les patients ayant bénéficié d’une transplantation rénale, il convient de faire preuve de prudence lors de son utilisation chez ces derniers (voir « Posologie/Mode d’emploi »).
Autres populations particulières
Les analyses pharmacocinétiques de population indiquent que l’âge, le sexe et l’origine ethnique n’ont pas d’effet cliniquement significatif sur la pharmacocinétique du sparsentan.
Données précliniquesDes études sur le développement embryofœtal menées chez les rats et les lapins ont mis en évidence une toxicité pour le développement chez les deux espèces. Chez les rats, des effets tératogènes dose-dépendants ont été observés, à toutes les doses de sparsentan testées, sous la forme de malformations crâniofaciales, d’anomalies du squelette, d’une augmentation de la létalité embryofœtale et d’une réduction du poids fœtal, et ce à des expositions 8 fois et 13 fois supérieures à l’ASC chez l’homme pour les doses de 800 mg/jour et de 400 mg/jour. Chez les lapins, aucune malformation fœtale ni aucun effet sur la viabilité embryofœtale ou sur la croissance fœtale n’ont été observés. Cependant, une augmentation des modifications du squelette (côtes cervicales surnuméraires) a été observée à une exposition d’environ 0,10 et 0,2 fois l’ASC chez l’être humain pour les doses de 800 mg/jour et 400 mg/jour.
Dans l’étude sur le développement prénatal et postnatal menée chez le rat, une toxicité maternelle incluant le décès a été observée à une exposition ~8 fois et 13 fois supérieure à l’ASC chez l’être humain pour les doses de 800 mg/jour et 400 mg/jour; une toxicité maternelle a également été observée à une exposition ~2 fois et 3 fois supérieure à l’ASC chez l’être humain pour les doses de 800 mg/jour et 400 mg/jour. Une augmentation de la mortalité des petits et une croissance réduite de ces derniers ont été observées à une exposition ~8 fois et 13 fois supérieure à l’ASC chez l’être humain pour les doses de 800 mg/jour et 400 mg/jour, et une croissance réduite a également été observée à une exposition ~2 fois et 3 fois supérieure à l’ASC chez l’être humain pour les doses de 800 mg/jour et 400 mg/jour.
Études sur des animaux juvéniles
Des études menées sur des rats juvéniles ont démontré qu’aucun effet indésirable toxicologique général n’était observé à une posologie allant jusqu’à 10 mg/kg/jour et qu’aucune toxicité pour la reproduction chez les mâles ou les femelles n’était observée jusqu’à 60 mg/kg/jour lorsque l’administration commençait au jour postnatal 14 (ce qui est l’équivalent d’enfants d’un an). Une toxicité vasculaire est apparue à des doses ≥ 3 mg/kg/jour lorsque l’administration commençait au jour postnatal 7 (ce qui est l’équivalent de nouveau-nés).
Remarques particulièresStabilité
Ce médicament ne doit pas être utilisé au-delà de la date figurant après la mention « EXP » sur l’emballage.
Remarques particulières concernant le stockage
Conserver à 15-30 °C.
Tenir hors de portée des enfants.
Remarques concernant la manipulation
Tout médicament non utilisé ou déchet doit être éliminé conformément à la réglementation nationale.
Numéro d’autorisation69241 (Swissmedic)
PrésentationComprimés pelliculés de 200 mg/400 mg: emballages contenant chacun 30 comprimés pelliculés dans un flacon. (B)
Comprimés pelliculés de 400 mg: emballages contenant chacun 30 comprimés pelliculés dans un flacon. (B)
Comprimés pelliculés de 400 mg: 3 emballages contenant chacun 30 comprimés pelliculés dans un flacon. (B)
Titulaire de l’autorisationVifor (International) Inc., Saint-Gall
Mise à jour de l’informationMai 2024
|