CompositionPrincipes actifs
Omalizumabum
Excipients
Solution injectable en stylo prérempli:
Arginini hydrochloridum, Histidini hydrochloridum monohydricum, Histidinum, Polysorbatum 20, Aqua ad iniectabile q.s. ad solutionem
Indications/Possibilités d’emploiAsthme allergique
Xolair est utilisé en association avec d'autres traitements de l'asthme pour améliorer le contrôle de l'asthme chez les adultes et les enfants (âgés de 6 ans et plus) souffrant d'asthme allergique persistant sévère (test cutané positif ou réaction in vitro à un aéroallergène perannuel) présentant une fonction pulmonaire réduite (VEMS < 80%) ainsi que des symptômes fréquents pendant la journée ou des réveils nocturnes et des exacerbations de l'asthme, malgré un traitement quotidien par des corticostéroïdes inhalés à forte dose et un agoniste bêta2 à longue durée d'action.
Polypes nasaux
Xolair (omalizumab) est indiqué dans le traitement des polypes nasaux chez les adultes (âgés de 18 ans et plus) qui ne répondent pas de manière adéquate aux corticostéroïdes intranasaux.
Urticaire chronique spontanée (UCS)
Traitement adjuvant chez l'adulte et l'adolescent (âgé de 12 ans et plus) présentant une urticaire chronique spontanée (UCS) prolongée*, qui ne peut être contrôlée par un traitement antihistaminique H1 et pour lesquels aucune autre maladie sous-jacente n'a été identifiée par un médecin familier de cette pathologie.
(*Les études pivots ont évalué des patients avec UCS durant depuis 6 mois à 66 ans, durée moyenne de 6 ans)
Posologie/Mode d’emploiAsthme allergique
Posologie usuelle
Adultes et enfants à partir de 6 ans
Les stylos préremplis Xolair ne doivent pas être utilisés chez les patients de moins de 12 ans. La seringue préremplie Xolair de 75 mg, la seringue préremplie Xolair de 150 mg ou la poudre et le solvant pour solution injectable Xolair peuvent être utilisés chez les enfants âgés de 6 à 11 ans et souffrant d'asthme allergique.
La dose et la fréquence d'administration appropriées de Xolair sont déterminées en fonction du taux sérique initial d'immunoglobuline E (IgE) (UI/ml), mesuré avant le début du traitement, et du poids corporel (kg). Avant la première administration, il est nécessaire de déterminer le taux d'IgE du patient à l'aide d'un test d'IgE sérique total disponible sur le marché, en vue de déterminer la dose de Xolair. Sur la base de ces mesures, 75 à 600 mg de Xolair sont nécessaires en 1 à 2 injections par administration.
Chez les patients dont le taux d'IgE est inférieur à 76 UI/ml, le bénéfice était moins probable (voir «Pharmacodynamique»). Avant le début du traitement, le médecin prescripteur doit s'assurer que les patients dont le taux d'IgE est inférieur à 76 UI/ml présentent une réactivité in vitro (RAST) claire à un allergène perannuel.
Voir le tableau 1 pour le schéma de conversion et les tableaux 2 et 3 pour la détermination de la dose.
Les patients dont les taux d'IgE de base ou le poids corporel en kilogrammes dépassent des limites des tableaux de dose (≤20 ou > 150 kg de poids corporel) ne doivent pas être traités par Xolair.
|
Tableau 1: Conversion de la dose en nombre de stylos préremplis, en nombre d'injections et en volume total injecté par injection
| |
Dose (mg)
|
Nombre de stylos préremplis
|
Nombre d'injections
|
Volume total
injecté (ml)
| |
75 mga
|
150 mgb
|
300 mgc,*
| |
75
|
1
|
0
|
0
|
1
|
0.5
| |
150
|
0
|
1
|
0
|
1
|
1.0
| |
225
|
1
|
1
|
0
|
2
|
1.5
| |
300
|
0
|
0
|
1
|
1
|
2.0
| |
375
|
1
|
0
|
1
|
2
|
2.5
| |
450
|
0
|
1
|
1
|
2
|
3.0
| |
600
|
0
|
0
|
2
|
2
|
4.0
| |
|
a
0.0.5 ml = volume maximal par stylo prérempli (Xolair 75 mg).
b 1.0 ml = volume maximal par stylo prérempli (Xolair 150 mg).
c 2.0 ml = volume maximal par stylo prérempli (Xolair 300 mg).
|
*Ni la seringue préremplie Xolair 300 mg ni aucun des dosages des stylos préremplis Xolair ne sont destinés à être utilisés par des patients de moins de 12 ans.
Ce tableau donne le nombre minimal d'injections pour les patients. Cependant, d'autres combinaisons de seringue et de stylo sont également possibles pour obtenir la dose souhaitée.
Afin d'assurer la traçabilité des médicaments biotechnologiques, il convient de documenter pour chaque traitement le nom commercial et le numéro de lot.
Durée du traitement
Dans les études cliniques sur l'asthme allergique, une réduction des exacerbations de l'asthme et de l'utilisation d'un traitement de secours ainsi qu'une amélioration des symptômes ont été observées au cours des 16 premières semaines de traitement. 16 semaines après le début du traitement par Xolair, l'efficacité du traitement doit être évaluée par le médecin avant l'administration de nouvelles injections. La décision de poursuivre le traitement par Xolair doit être basée sur une amélioration significative du contrôle général de l'asthme (voir «Pharmacodynamique»).
L'arrêt du traitement entraîne généralement un retour à des taux élevés d'IgE libres et des symptômes associés.
Adaptation posologique
Les taux d'IgE totales sont élevés pendant le traitement et restent élevés jusqu'à un an après l'arrêt du traitement. Par conséquent, une nouvelle mesure des taux d'IgE pendant le traitement par Xolair ne peut pas être utilisée pour ré-estimer la dose de Xolair. Par conséquent, la détermination de la dose après une interruption de moins de 1 an doit être basée sur les taux sériques d'IgE mesurés lors de la détermination de la dose initiale (voir «Propriétés/Effets»). Les taux d'IgE totales ne doivent être réévalués pour la détermination de la dose que si le traitement par Xolair a été interrompu pendant un an ou plus.
La posologie doit être ajustée en cas de modification significative du poids corporel (voir Tableaux 2 et 3).
|
Tableau 2: ADMINISTRATION TOUTES LES 4 SEMAINES Asthme allergique et polypes nasaux. Dose de Xolair (mg par dose) administrée par voie sous-cutanée toutes les 4 semaines
| |
|
Poids corporel (kg)
| |
Valeur IgE initiale (UI/ml)
|
> 20–25
|
> 25–30
|
> 30–40
|
> 40–50
|
> 50–60
|
> 60–70
|
> 70–80
|
> 80–90
|
> 90–125
|
> 125–150
| |
≥30–100
|
75
|
75
|
75
|
150
|
150
|
150
|
150
|
150
|
300
|
300
| |
> 100–200
|
150
|
150
|
150
|
300
|
300
|
300
|
300
|
300
|
450
|
600
| |
> 200–300
|
150
|
150
|
225
|
300
|
300
|
450
|
450
|
450
|
600
|
| |
> 300–400
|
225
|
225
|
300
|
450
|
450
|
450
|
600
|
600
|
|
| |
> 400–500
|
225
|
300
|
450
|
450
|
600
|
600
|
|
|
|
| |
> 500–600
|
300
|
300
|
450
|
600
|
600
|
|
|
|
|
| |
> 600–700
|
300
|
|
450
|
600
|
|
ADMINISTRATION TOUTES LES
2 SEMAINES
VOIR TABLEAU 3
|
|
Tableau 3: ADMINISTRATION TOUTES LES 2 SEMAINES. Asthme allergique et polypes nasaux. Dose de Xolair (mg par dose) administrée par voie sous-cutanée toutes les 2 semaines
| |
|
Poids corporel (kg)
| |
Valeur IgE initiale (UI/ml)
|
> 20-25
|
> 25-30
|
> 30-40
|
> 40-50
|
> 50-60
|
> 60-70
|
> 70-80
|
> 80-90
|
> 90-125
|
> 125-150
| |
≥30–100
|
ADMINISTRATION TOUTES LES 4 SEMAINES, VOIR TABLEAU 2
|
|
|
|
|
| |
> 100–200
|
|
|
|
|
| |
> 200–300
|
|
|
|
|
|
|
|
|
|
375
| |
> 300–400
|
|
|
|
|
|
|
|
|
|
| |
> 400–500
|
|
|
|
|
|
|
375
|
375
|
|
| |
> 500–600
|
|
|
|
|
|
375
|
– Données insuffisantes pour recommander une posologie
| |
> 600–700
|
|
225
|
|
|
375
|
|
La dose maximale recommandée est de 375 mg d'omalizumab toutes les 2 semaines.
Urticaire chronique spontanée (UCS)
Posologie usuelle
La dose initiale recommandée est de 300 mg sous forme d'injection sous-cutanée toutes les 4 semaines. La plupart des patients qui répondent au traitement présentent une amélioration dans les 4 semaines suivant la première dose. Chez les patients qui ne répondent que partiellement au premier traitement, une amélioration supplémentaire des symptômes est possible en poursuivant le traitement.
L'expérience issue des essais cliniques dans cette indication pour une durée de traitement supérieure à 6 mois est limitée.
Le médecin doit régulièrement vérifier la nécessité de poursuivre le traitement.
Les données cliniques montrent que certains patients traités par Xolair 150 mg toutes les 4 semaines peuvent atteindre un contrôle de leurs symptômes. Par conséquent, chez les patients qui, sous Xolair 300 mg en association avec un anthihistaminique H1, sont exempts de démangeaisons et d'urticaire, on peut essayer de réduire la dose à 150 mg toutes les quatre semaines.
Polypes nasaux (à partir de 18 ans)
Dans les études pivots d'efficacité portant sur les polypes nasaux, le même schéma posologique en fonction des IgE et du poids chez les patients âgés de 18 ans et plus et pesant entre 30 kg et 150 kg a été utilisé que celui décrit pour l'asthme allergique. Le schéma posologique recommandé pour le traitement de l'urticaire chronique spontanée n'a pas été étudié pour le traitement de la polypose. Par conséquent, chez les patients atteints de polypes nasaux (âgés de 18 ans et plus et pesant de 30 kg à 150 kg), la même posologie en fonction des IgE et du poids que chez les patients souffrant d'asthme allergique est recommandée (voir ci-dessus).
Instructions posologiques particulières
Patients présentant des troubles de la fonction hépatique et des troubles de la fonction rénale
Il n'existe pas d'étude évaluant l'effet de l'insuffisance hépatique ou rénale sur la pharmacocinétique de Xolair. La clairance de l'omalizumab étant déterminée par le système réticulo-endothélial (SRE) aux doses cliniques, il est peu probable qu'elle soit altérée par un dysfonctionnement hépatique ou rénal. Bien qu'aucun ajustement posologique ne soit recommandé, Xolair doit être utilisé avec prudence chez ces patients (voir «Mises en garde et précautions»).
Patients âgés
Les données sur l'utilisation de Xolair chez les patients âgés de 65 ans et plus sont limitées. Cependant, rien n'indique qu'une posologie différente soit nécessaire chez les patients âgés par rapport aux patients adultes plus jeunes.
Enfants et adolescents
Dans l'asthme allergique, la sécurité et l'efficacité n'ont pas été établies chez les patients de moins de 6 ans. Par conséquent, l'utilisation de Xolair n'est pas recommandée chez ces patients. Xolair en stylo prérempli n'est pas destiné aux patients de moins de 12 ans (voir «Posologie usuelle Adultes et enfants âgés de 6 ans et plus»).
En cas de polypes nasaux, la sécurité et l'efficacité n'ont pas été établies chez les patients de moins de 18 ans.
Urticaire chronique spontanée (UCS)
La sécurité et l'efficacité n'ont pas été établies chez les patients de moins de 12 ans. Par conséquent, l'utilisation de Xolair n'est pas recommandée chez ces patients.
Mode d'administration
Voie sous-cutanée uniquement. Ne pas administrer par voie intraveineuse ou intramusculaire.
Les injections sont administrées par voie sous-cutanée dans la partie supérieure du bras, dans la région deltoïde. Si l'administration dans la région deltoïde n'est pas possible pour une raison quelconque, les injections peuvent être administrées dans la cuisse.
Les patients qui n'ont pas d'antécédents connus d'anaphylaxie peuvent s'injecter eux-mêmes Xolair ou faire faire l'injection par un soignant à partir de la quatrième dose, si le médecin le juge approprié (voir «Mises en garde et précautions»). Le patient ou le soignant doit être formé à la technique d'injection correcte et à la détection des signes et symptômes précoces de réactions allergiques sévères.
Les patients ou les soignants doivent être informés qu'ils doivent injecter la totalité du contenu du stylo prérempli Xolair conformément aux instructions de la notice.
Contre-indicationsHypersensibilité au principe actif ou à l'un des excipients.
Mises en garde et précautionsRéactions allergiques, anaphylaxie
Des réactions allergiques locales ou systémiques, incluant une anaphylaxie mettant en jeu le pronostic vital et un choc anaphylactique, peuvent survenir lors de l'utilisation de l'omalizumab. Ils peuvent également survenir après une période de traitement plus longue. La plupart de ces réactions sont survenues dans les 2 heures suivant la première injection et les injections suivantes de Xolair, mais certaines sont survenues plus de 2 heures et même plus de 24 heures après l'injection. La plupart des réactions anaphylactiques sont survenues lors des 3 premières administrations de Xolair. Les 3 premières administrations doivent donc être réalisées par un professionnel de santé ou sous la surveillance de celui-ci. Des antécédents d'anaphylaxie non liés à l'omalizumab peuvent constituer un facteur de risque d'anaphylaxie suite à l'administration de Xolair. Par conséquent, Xolair doit être administré par un professionnel de santé aux patients ayant des antécédents connus d'anaphylaxie. De plus, des médicaments doivent toujours être immédiatement disponibles pour le traitement d'une réaction anaphylactique après l'administration de Xolair. Les patients doivent être informés que de telles réactions sont possibles et qu'un traitement médical immédiat est nécessaire en cas de réaction allergique. En cas de réactions sévères, le traitement par Xolair doit être arrêté immédiatement (voir «Effets indésirables»).
Une maladie sérique et des symptômes de type maladie sérique, qui sont des réactions allergiques de type III retardées, ont été rarement observés chez des patients traités par des anticorps monoclonaux humanisés, dont l'omalizumab. Le début survenait généralement 1 à 5 jours après la première injection ou une injection ultérieure, même après une longue durée de traitement. Les symptômes évocateurs d'une maladie sérique comprennent une arthrite/arthralgie, un rash (urticaire ou autres formes), de la fièvre et une lymphadénopathie. Les antihistaminiques et les corticoïdes peuvent être utiles pour prévenir ou traiter cette maladie. Les patients doivent être informés de la nécessité de signaler les symptômes évocateurs.
Syndrome de Churg-Strauss et syndrome hyperéosinophilique
Les patients présentant un asthme allergique sévère peuvent rarement présenter un syndrome hyperéosinophilique systémique ou une vascularite granulomateuse à éosinophiles allergique (syndrome de Churg-Strauss, polyangéite granulomateuse à éosinophiles), les deux étant généralement traités par des corticostéroïdes systémiques.
Immunogénicité
Dans de rares cas, les patients peuvent développer des anticorps contre l'omalizumab, comme cela est possible avec tous les anticorps humanisés issus de l'ADN recombinant.
De faibles titres d'anticorps anti-Xolair ont été détectés chez environ 1 patient sur 1843 (< 0.1%) traités par Xolair. Ces données reflètent le pourcentage de patients dont les résultats de test ont été considérés comme positifs à l'aide d'un dosage ELISA pour la recherche d'anticorps contre Xolair et dépendent fortement de la sensibilité et de la spécificité du dosage. De plus, l'incidence observée des réactions positives pour les anticorps lors du dosage peut être influencée par divers facteurs tels que la manipulation des échantillons, le moment du prélèvement des échantillons, d'autres médicaments concomitants et d'autres maladies présentes. Par conséquent, la comparaison de l'incidence des anticorps anti-Xolair avec celle des anticorps vis-à-vis d'autres produits peut induire en erreur.
Affections cérébrovasculaires
Dans les études cliniques contrôlées menées chez des adultes et des adolescents âgés de 12 ans et plus, des événements cérébrovasculaires incluant une attaque ischémique transitoire et un accident vasculaire cérébral ischémique ont été observés plus fréquemment chez les patients traités par Xolair que dans le groupe de contrôle (cf. «Effets indésirables»).
Infections parasitaires (helminthiases)
Les IgE peuvent être impliquées dans la réponse immunologique à certaines infestations par les helminthes. Une étude a été menée au Brésil, dans laquelle des patients vivant dans une région à haut risque d'infestations par des helminthes ont été traités pendant 1 an par l'omalizumab. 53% (36/68) des patients traités par l'omalizumab et 42% (29/69) des patients traités par placebo ont présenté une herlminthiase diagnostiquée par examen des selles. La différence entre les deux groupes pour ce qui est des helminthiases n'est pas satistiquement significative. L'évolution, la sévérité de la maladie et la réponse au traitement de l'infection n'ont pas été modifiées. Le taux d'helminthiase dans le programme clinique, qui n'était pas conçu pour détecter de telles maladies, a été inférieur à 1 cas pour 1000 patients. Cependant, la prudence est recommandée chez les patients à haut risque d'helminthiase, en particulier lorsqu'ils voyagent dans des zones endémiques. Si les patients ne répondent pas au traitement anti-helminthique recommandé, l'arrêt du traitement par Xolair doit être envisagé.
Tumeurs malignes
Au cours des premières études cliniques, un déséquilibre numérique concernant le cancer a été observé dans le groupe avec traitement actif par rapport au groupe témoin. Les différents types de tumeurs, la durée relativement courte du traitement et l'état clinique individuel font qu'un lien de causalité semble peu probable. L'incidence globale observée de tumeurs malignes dans le programme de développement clinique de Xolair a été comparable à celle rapportée dans la population générale. Des études ultérieures ont montré que le risque relatif de tumeurs malignes n'est pas augmenté avec Xolair. Cependant, en l'état actuel des données, on ne peut pas pas exclure totalement la possibilité d'un léger déséquilibre (voir «Effets indésirables»).
Généralités
Xolair n'est pas indiqué dans le traitement des exacerbations aiguës de l'asthme, du bronchospasme aigu ou de l'état de mal asthmatique.
Xolair n'a pas été étudié chez les patients présentant un syndrome d'hyper-IgE, une aspergillose bronchopulmonaire allergique ou dans la prévention des réactions allergiques.
Xolair n'a pas été suffisamment étudié en cas de dermatite atopique, de rhinite allergique ou d'allergie alimentaire.
Le traitement par Xolair n'a pas été étudié chez les patients présentant une maladie auto-immune, une maladie à complexes immuns ou une insuffisance rénale ou hépatique préexistante. La prudence est recommandée lorsque Xolair est utilisé chez ces patients.
L'arrêt brutal des corticostéroïdes systémiques ou inhalés en cas d'asthme allergique ou de polypes nasaux n'est pas recommandé après l'instauration du traitement par Xolair. La réduction des corticoïdes doit avoir lieu sous surveillance médicale et doit être progressive si nécessaire.
InteractionsLes enzymes du cytochrome P450. les pompes à efflux et les mécanismes de liaison aux protéines n'interviennent pas dans la clairance de l'omalizumab; le risque d'interactions médicamenteuses est donc faible. Aucune étude spéciale d'interaction avec des médicaments ou des vaccins n'a été effectuée avec Xolair. D'un point de vue pharmacologique, il n'y a pas de raison de s'attendre à une interaction entre les médicaments habituellement prescrits dans le traitement de l'asthme, des polypes nasaux ou de l'urticaire chronique spontanée (UCS) et l'omalizumab.
Asthme allergique
Dans les études cliniques, Xolair a été généralement utilisé en association avec des corticostéroïdes inhalés ou oraux, des bêta-agonistes inhalés à courte ou à longue durée d'action, des antagonistes des récepteurs des leucotriènes, des théophyllines et des antihistaminiques oraux. Il n'a pas été mis en évidence de modification de la sécurité de Xolair par ces autres médicaments généralement utilisés pour l'asthme.
L'efficacité du traitement par Xolair associé à une immunothérapie spécifique n'a pas été établie.
Polypes nasaux
Lors des études cliniques, Xolair a été utilisé en association avec de la mométasone en pulvérisation nasale conformément au protocole. Les autres médicaments concomitants fréquemment utilisés comprenaient d'autres corticoïdes intranasaux, des bronchodilatateurs, des antihistaminiques, des antagonistes des récepteurs des leucotriènes, des agonistes adrénergiques/sympathomimétiques et des anesthésiques locaux administrés par voie intranasale. Il n'a pas été mis en évidence d'altération de la sécurité de Xolair lors de l'administration concomitante de ces autres médicaments fréquemment utilisés en cas de polypes nasaux.
Urticaire chronique spontanée (UCS)
Dans les études cliniques pourtant sur l'UCS, Xolair a été utilisé en association avec des antihistaminiques (anti-H1, anti-H2) et des antagonistes des récepteurs des leucotriènes (ARLT). Dans les études de phase III Q4881g et Q4882g, tous les patients ont reçu des antihistaminiques H1 en association à Xolair ou au placebo. Dans l'étude de phase III Q4883g, tous les patients ont reçu un ou plusieurs antihistaminiques H1 et/ou antihistaminiques H2 et/ou ARLT en association à Xolair ou au placebo. Il n'a pas été mis en évidence de modification de la sécurité de l'omalizumab par rapport à son profil de sécurité connu pour l'asthme allergique lors de l'administration concomitante avec ces médicaments. De plus, une analyse pharmacocinétique de population n'a montré aucune influence significative des antihistaminiques H2 et des ARLT sur la pharmacocinétique de l'omalizumab (voir «Propriétés/Effets»).
Grossesse, allaitementGrossesse
Il n'existe pas d'études cliniques bien contrôlées avec Xolair chez la femme enceinte. Une étude prospective de registre de grossesses (EXPECT) menée chez 250 femmes enceintes asthmatiques traitées par Xolair a montré que la prévalence des malformations congénitales pertinentes était comparable entre les patientes traitées par Xolair (EXPECT) et les patientes asthmatiques (asthme modéré et sévère) n'ayant pas reçu Xolair (8.1% vs 8.9%). La proportion d'enfants présentant un poids à la naissance < 2.5 kg a été plus élevée sous Xolair (13.7% vs 9.8%), ce qui pourrait également être dû à des différences de sévérité de l'asthme. Dans l'ensemble, le risque de malformations congénitales ne peut pas être évalué de manière définitive sur la base de cette étude en raison de restrictions méthodologiques, y compris un plan non randomisé de l'étude et les éventuelles différences entre la population du registre et le groupe de comparaison (voir «Efficacité clinique»).
On sait que les IgG traversent la barrière placentaire. Les études effectuées chez l'animal n'ont révélé aucune toxicité sur la reproduction (voir «Données précliniques»).
Risque associé à la maladie pour la mère et/ou l'embryon/le fœtus:
Il existe des preuves que, chez les femmes présentant un asthme mal ou modérément contrôlé, le risque de prééclampsie de la mère et d'accouchement prématuré, de faible poids à la naissance et de faible taille du nouveau-né par rapport à l'âge gestationnel est augmenté. C'est pourquoi les femmes enceintes qui sont asthmatiques doivent faire l'objet d'une surveillance étroite et le traitement doit être ajusté, si nécessaire, afin de maintenir un contrôle optimal de l'asthme.
Allaitement
Bien que la présence d'omalizumab dans le lait maternel n'ait pas été étudiée après l'administration de Xolair, les IgG passent dans le lait maternel et on peut en conclure que l'omalizumab sera présent dans le lait maternel. La fréquence des infections observées chez les nourrissons dans l'étude EXPECT a été évaluée comme une mesure indirecte du développement du système immunitaire après exposition pendant la grossesse ou par l'allaitement. La majorité des nourrissons dans la population d'analyse principale (77.5%, 186/240) ont été allaités.
Des événements indésirables graves (EIG) classés comme «infections et infestations» ont été observés chez 11.4% (5/44) des nourrissons non allaités, 10.4% (16/154) des nourrissons exposés à Xolair par l'allaitement et 12.5% (4/32) des nourrissons allaités sans exposition à Xolair. L'étude présente des limites méthodologiques, y compris le plan non randomisé de l'étude.
Les bénéfices de l'allaitement sur le développement et la santé du nourrisson doivent être évalués en tenant compte de la nécessité clinique pour la mère de prendre Xolair et des éventuels effets indésirables de l'omalizumab ou de la maladie maternelle sous-jacente sur l'enfant allaité.
Fertilité
Il n'existe pas de recommandation spécifique pour les femmes en âge de procréer.
Effet sur l’aptitude à la conduite et l’utilisation de machinesAucune étude correspondante n'a été effectuée.
Les patients traités par l'omalizumab doivent être informés que, s'ils présentent des vertiges, une fatigue, des syncopes ou une somnolence, ils ne doivent pas conduire de véhicules ni utiliser Pldes machines.
Effets indésirablesFréquences
«Très fréquent» (≥1/10); «fréquent» (≥1/100 à < 1/10); «occasionnel» (≥1/1000 à < 1/100); «rare» (≥1/10 000 à < 1/1000); «très rare» (< 1/10 000).
Asthme allergique
Dans les essais cliniques menés chez des adultes et des adolescents âgés de 12 ans et plus, les effets indésirables les plus fréquemment observés ont été des céphalées et des réactions au site d'injection, incluant douleur au site d'injection, gonflement, érythème et prurit. Dans les essais cliniques menés chez des patients âgés de 6 ans à moins de 12 ans, les effets indésirables les plus fréquemment observés ont été des céphalées, une pyrexie et des douleurs abdominales hautes. Dans la plupart des cas, ils ont été d'intensité légère à modérée.
Effets indésirables identifiés dans les études cliniques sur l'asthme allergique
Infections et infestations
Occasionnel: Pharyngite.
Rare: Infection parasitaire.
Affections du système immunitaire
Rare: Réactions anaphylactiques et autres réactions allergiques telles que maladie sérique, symptômes pseudo-grippaux tels que fièvre, douleurs articulaires, malaise. Développement d'anticorps antimédicament.
Affections du système nerveux
Fréquent: Céphalées**.
Occasionnel: Vertige, envie de dormir, paresthésie, syncope.
Affections vasculaires
Occasionnel: Hypotension orthostatique, bouffées vasomotrices.
Affections respiratoires, thoraciques et médiastinales
Occasionnel: Toux, bronchospasme allergique.
Rare: Œdème du larynx.
Affections gastro-intestinales
Fréquent: Douleur abdominale haute*.
Occasionnel: Nausées, diarrhée, dyspepsie.
Affections de la peau et du tissu sous-cutané
Occasionnel: Urticaire, rash, prurit, photosensibilité.
Rare: Angiœdème.
Troubles généraux et anomalies au site d'administration
Très fréquent: Pyrexie*.
Fréquent: Douleur, érythème, prurit, gonflement.
Occasionnel: Gain pondéral, fatigue, gonflement des bras, symptômes pseudo-grippaux.
Dans une étude en ouvert chez des adultes sains comparant le stylo prérempli de 300 mg/2 ml à la seringue préremplie de 300 mg/2 ml, des réactions au site d'injection (par exemple induration, douleur, érythème, saignement, gonflement, gêne, ecchymose, hypoesthésie, œdème, prurit) ont été observées chez 24% (16/66) des adultes traités avec le stylo prérempli versus 14% (9/64) des adultes traités avec la seringue préremplie.
Les fréquences des effets indésirables ont été similaires dans le groupe traité et dans le groupe témoin.
* chez les enfants âgés de 6 ans à < 12 ans
** très fréquents chez les enfants âgés de 6 ans à < 12 ans
Polypes nasaux
Résumé du profil de sécurité
Les données présentées ci-dessous sont issues de deux études contrôlées par placebo menées chez des patients âgés de 18 ans et plus. Dans ces études, les patients ont reçu soit 150 à 600 mg de Xolair toutes les 2 ou 4 semaines, soit un placebo. Tous les patients ont reçu de la mométasone en traitement de fond par voie intranasale; le profil de sécurité chez les patients présentant des polypes nasaux et chez les patients atteints d'asthme allergique et d'UCS étaient concordants. Les effets indésirables les plus fréquemment rapportés (> 3%), plus fréquents qu'avec le placebo, sont présentés dans le Tableau 4.
Le tableau 4 présente les effets indésirables survenus au cours des études cliniques dans l'ensemble de la population présentant des polypes nasaux traitée par Xolair, par classe d'organe et par fréquence. Les catégories de fréquence sont définies comme suit: très fréquent (≥1/10), fréquent (≥1/100 à < 1/10), occasionnel (≥1/1000 à < 1/100); rare (≥1/10 000 à < 1/1000) et très rare (< 1/10 000).
|
Tableau 4: Effets indésirables identifiés dans les études cliniques menées chez des patients présentant des polypes nasaux
| |
Effets indésirables
(désignés par le terme préféré MedDRA)
|
Études de l'omalizumab en cas de polypes nasaux 1 et 2
groupées
|
Catégorie de fréquence
| |
Placebo
N = 130
|
Omalizumab
N = 135
| |
Affections du système nerveux
| |
Céphalées
|
7 (5.4%)
|
11 (8.1%)
|
Fréquent
| |
Sensation vertigineuse
|
1 (0.8%)
|
4 (3.0%)
|
Fréquent
| |
Affections musculosquelettiques et du tissu conjonctif
| |
Arthralgie
|
2 (1.5%)
|
4 (3.0%)
|
Fréquent
| |
Affections gastro-intestinales
| |
Douleur abdominale
|
1 (0.8%)
|
4 (3.0%)
|
Fréquent
| |
Troubles généraux et anomalies au site d'administration
| |
Réactions au site d'injection (réactions au site d'injection, réaction liée à l'injection, douleur au site d'injection)
|
2 (1.5%)
|
7 (5.2%)
|
Fréquent
|
Urticaire chronique spontanée (UCS)
Résumé du profil de sécurité
La sécurité et la tolérance de l'omalizumab aux doses de 75 mg, 150 mg et 300 mg toutes les 4 semaines ont été évaluées chez 975 patients atteints d'UCS, dont 242 ont reçu un placebo. 733 patients ont été traités par l'omalizumab pendant un maximum de 12 semaines et 490 patients pendant un maximum de 24 semaines. Respectivement 175 et 412 patients ont été traités pendant jusqu'à 12 semaines et 87 et 333 patients pendant jusqu'à 24 semaines aux doses recommandées de 150 mg ou 300 mg.
Les effets indésirables les plus fréquemment rapportés dans les études cliniques chez les patients adultes et adolescents (âgés de 12 ans et plus) étaient des céphalées et des rhinopharyngites.
|
Tableau 5: Tableau récapitulatif des effets indésirables issus des études de phase III groupées portant sur les doses recommandées de 150 mg et 300 mg (du jour 1 à la semaine 12)
| |
Effets indésirables
(MedDRA)
|
Études groupées de l'omalizumab Q4881g, Q4882g et Q4883g
|
Fréquences
| |
Placebo
N = 242
|
150 mg
N = 175
|
300 mg
N = 412
|
| |
Infections et infestations
| |
Rhinopharyngite
|
17 (7.0%)
|
16 (9.1%)
|
27 (6.6%)
|
Fréquent
| |
Sinusite
|
5 (2.1%)
|
2 (1.1%)
|
20 (4.9%)
|
Fréquent
| |
Inflammation virale des voies respiratoires hautes
|
0
|
4 (2.3%)
|
2 (0.0.5%)
|
Fréquent
| |
Affections du système nerveux
| |
Céphalées
|
7 (2.9%)
|
21 (12.0%)
|
25 (6.1%)
|
Très fréquent
| |
Affections musculosquelettiques et du tissu conjonctif
| |
Douleurs articulaires
|
1 (0.4%)
|
5 (2.9%)
|
12 (2.9%)
|
Fréquent
|
Autres événements rapportés à tout moment pendant la période de traitement du jour 1 à la semaine 24 (études Q4881g et Q4883g) et répondant aux critères d'effets indésirables:
Infections et infestations: Infections des voies aériennes supérieures (placebo 3.1%, 150 mg 3.4%, 300 mg 5.7%), infections des voies urinaires (placebo 1.8%, 150 mg 4.6%, 300 mg 2.4%)
Affections du système nerveux: Céphalées sinusales (placebo 0%, 150 mg 2.3%, 300 mg 0.3%)
Affections musculosquelettiques et du tissu conjonctif: Myalgie (placebo 0%, 150 mg 2.3%, 300 mg 0.9%), douleur dans les membres (placebo 0%, 150 mg 3.4%, 300 mg 0.9%), douleurs musculo-squelettiques (placebo 0%, 150 mg 2.3%, 300 mg 0.9%)
Troubles généraux et anomalies au site d'administration: Fièvre (placebo 1.2%, 150 mg 3.4%, 300 mg 0.9%). Dans les études, des réactions au site d'injection sont survenues plus fréquemment chez les patients traités par l'omalizumab que chez les patients sous placebo (2.7% sous 300 mg, 0.6% sous 150 mg, 0.8% sous placebo). Ces réactions ont inclus: Gonflement, érythème, douleurs, ecchymose, démangeaisons, hémorragie et urticaire.
Dans une étude d'une durée de 48 semaines, 81 patients atteints d'UCS ont reçu 300 mg d'omalizumab toutes les 4 semaines (voir «Efficacité clinique – UCS»). Le profil de sécurité en cas d'utilisation à long terme était similaire à celui observé dans les études portant sur l'UCS et d'une durée maximale de 24 semaines.
Effets indésirables après commercialisation
Les réactions suivantes ont été principalement identifiées grâce à des déclarations spontanées:
Système immunitaire: Des cas d'anaphylaxie et de réactions anaphylactoïdes ont été rapportés aussi bien après l'administration initiale qu'après des administrations ultérieures (voir «Mises en garde et précautions»). Maladie sérique (voir «Mises en garde et précautions»).
Peau: Alopécie.
Sang et système lymphatique: Thrombopénie idiopathique sévère.
Organes respiratoires: Syndrome de Churg-Strauss (polyangéite granulomateuse à éosinophiles).
Système musculo-squelettique: Arthralgies, myalgie, tuméfaction articulaire.
Description de certains effets indésirables
Thrombopénie
Dans les essais cliniques, quelques patients ont présenté des taux de plaquettes en dessous de la limite inférieure de la normale. Aucune de ces modifications n'a été associée à des hémorragies ni à une diminution de l'hémoglobine. Il n'y a pas de signe d'une diminution persistante du taux de plaquettes chez l'homme (patients âgés de plus de 6 ans) comme cela a été observé chez les primates (voir «Données précliniques»). Des cas de thrombopénie ont été rapportés depuis la commercialisation.
Infections parasitaires
Chez les patients allergiques présentant une tendance chronique aux helminthiases, une étude contrôlée par placebo a montré une légère augmentation du taux d'infection sous omalizumab. L'évolution et la sévérité de l'infection, ainsi que la réponse au traitement n'ont pas été modifiées (voir «Mises en garde et précautions»).
Tumeurs malignes
Au cours des premières études cliniques, chez des adultes et des adolescents âgés de 12 ans et plus, un déséquilibre numérique concernant le cancer a été observé dans le groupe avec traitement actif par rapport au groupe témoin. Les cas de cancer ont été occasionnels (< 1/100) dans les groupes de contrôle comme dans les groupes avec traitement actif. Dans une étude observationnelle ultérieure de 5 ans, comparant 5007 patients traités par Xolair et 2829 patients non traités par Xolair, le risque relatif de survenue de tumeurs malignes n'est pas augmenté sous Xolair. Le taux d'incidence de tumeurs malignes primitives pour 1000 années-patients a été respectivement de 16.01 (295/18 426 annéespatients) et de 19.07 (190/9963 années-patients), soit un rapport de fréquence de 0.84 (intervalle de confiance à 95%, 0.62–1.13). Dans une analyse prospective d'études cliniques randomisées, en double aveugle, contrôlées contre placebo, menées chez 4254 patients traités par Xolair et 3178 patients traités par placebo, le traitement par Xolair n'a pas été associé à une augmentation du risque de malignité sur la base des taux d'incidence pour 1000 années-patients de 4.14 (14/3382 annéespatients) chez les patients traités par Xolair et de 4.45 (11/2474 annéespatients) chez les patients du groupe placebo (rapport des fréquences 0.93, intervalle de confiance à 95% 0.39–2.27). Cependant, en l'état actuel des données, on ne peut pas pas exclure totalement la possibilité d'un léger déséquilibre.
Le taux d'incidence global de tumeurs malignes observé au cours des études sur Xolair chez des adultes et des adolescents âgés de 12 ans et plus a été comparable à celui observé dans la population générale.
Dans les études ultérieures, le risque relatif de tumeurs malignes n'a pas augmenté dans le groupe traité par Xolair (voir «Mises en garde et précautions»).
Evénements thromboemboliques artériels (ETA)
Un déséquilibre numérique des ETA a été observé au cours d'essais cliniques contrôlés et d'analyses intermédiaires d'une étude observationnelle. Les ETA incluaient les accidents vasculaires cérébraux, les accidents ischémiques transitoires, l'infarctus myocardique, l'angor instable et les décès de cause cardiovasculaire (incluant décès d'origine inconnue). Dans l'analyse finale de l'étude observationnelle, le taux d'ETA pour 1000 années-patients était de 7.52 (115/15 286 années-patients) chez les patients traités par Xolair et de 5.12 (51/9963 années-patients) chez les patients du groupe de contrôle. Dans une analyse multivariée corrigée selon les facteurs de risque cardiovasculaires présents à l'inclusion, le hazard ratio était de 1.32 (intervalle de confiance à 95%: 0.91–1.91).
Dans une analyse prospective d'études cliniques groupées incluant toutes les études cliniques randomisées en double aveugle contrôlées par placebo d'une durée minimale de 8 semaines, le taux d'ETA pour 1000 années-patients a été de 2.69 (5/1856 années-patients) chez les patients traités par Xolair et de 2.38 (4/1680 années-patients) chez les patients du groupe placebo (rapport des fréquences de 1.13, intervalle de confiance à 95% de 0.24 à 5.71).
Données de laboratoire
Après administration de Xolair, les taux sériques d'IgE totales ont augmenté en raison de la formation de complexes Xolair-IgE (voir «Pharmacocinétique» et «Posologie/Mode d'emploi»).
L'annonce d'effets secondaires présumés après l'autorisation est d'une grande importance. Elle permet un suivi continu du rapport bénéfice-risque du médicament. Les professionnels de santé sont tenus de déclarer toute suspicion d'effet secondaire nouveau ou grave via le portail d'annonce en ligne ElViS (Electronic Vigilance System). Vous trouverez des informations à ce sujet sur www.swissmedic.ch.
SurdosageLa dose maximale tolérée de Xolair n'a pas été déterminée. Des doses intraveineuses uniques allant jusqu'à 4000 mg ont été administrées à des patients sans signes de toxicité limitant la dose. La dose cumulée la plus élevée administrée à un patient était de 44 000 mg (sur une période de 20 semaines) sans effets indésirables aigus associés à cette dose.
Propriétés/EffetsCode ATC
R03DX05
Mécanisme d'action
L'omalizumab est un anticorps monoclonal humanisé recombinant, qui se lie de manière sélective à l'immunoglobuline E (IgE) humaine. Il s'agit d'un anticorps IgG1 kappa, avec un squelette humain, dont la région déterminant la complémentarité est d'origine murine et qui se lie aux IgE humaines.
Pharmacodynamique
Patients présentant un asthme allergique
En se liant aux IgE libres, l'omalizumab empêche la liaison des IgE au récepteur FcεRI à haute affinité (récepteur à IgE à haute affinité). La quantité d'IgE libre disponible pour déclencher la cascade allergique est réduite. Le traitement des patients atopiques par l'omalizumab a réduit le nombre de récepteurs FcεRI présents sur les basophiles. De plus, la sécrétion d'histamine in vitro à partir de basophiles prélevés chez des patients traités par Xolair a été réduite d'environ 90% après stimulation allergénique par rapport aux valeurs avant traitement.
Le traitement par Xolair entraîne une diminution du nombre d'éosinophiles dans le sang et les tissus et une diminution des médiateurs de l'inflammation. Parmi ceux-ci, il y a également des interleukines (IL-4, IL-5 et IL-13). Dans les études cliniques, les taux sériques d'IgE libres ont été réduits de façon dose-dépendante dans l'heure suivant la première dose et maintenus entre les doses.
La réduction moyenne des IgE libres était supérieure à 96% aux doses recommandées. Les taux sériques d'IgE totales (c'est-à-dire liées et non liées) ont augmenté après la première dose en raison de la formation de complexes omalizumab-IgE. Les complexes omalizumab-IgE ont une vitesse d'élimination plus faible que les IgE libres. 16 semaines après la première dose, le taux d'IgE totales était 5 fois plus élevé que le taux avant traitement, avec des dosages standards utilisés pour la détermination. Après l'arrêt du traitement par Xolair, l'augmentation des IgE totales et la réduction des IgE libres ont été réversibles sans effet rebond des IgE après élimination de l'omalizumab. Des taux d'IgE totales similaires à ceux observés avant le traitement n'ont pas été atteints dans l'année suivant l'arrêt du traitement par Xolair.
Les effets de l'omalizumab sur les cellules B porteuses d'IgE et sur la régulation à long terme de la synthèse des IgE spécifiques à l'allergène ne sont pas clairement établis.
Patients atteints de polypes nasaux
Dans les études cliniques menées chez des patients atteints de polypes nasaux, le traitement par Xolair a entraîné une réduction des taux d'IgE sériques libres et une augmentation des taux d'IgE sériques totales, à l'instar de ce qui a été observé chez les patients souffrant d'asthme allergique.
Patients atteints d'urticaire chronique spontanée (UCS)
Il existe plusieurs théories de l'étiologie de l'UCS, dont une qui adopte une origine auto-immune. Chez certains patients atteints d'UCS, des anticorps auto-immuns dirigés contre les IgE et leur récepteur, le FcεRI, ont été isolés dans le sérum. Ces auto-anticorps peuvent activer les granulocytes basophiles ou les mastocytes, déclenchant ainsi la libération d'histamine.
Une hypothèse sur le mécanisme d'action de l'omalizumab dans l'UCS indique que l'omalizumab réduit les taux d'IgE libres dans le sang et donc dans la peau. Ceci entraîne une régulation négative des récepteurs de surface des IgE, ce qui réduit la transmission des signaux en aval via la voie de signalisation FcεRI, ce qui inhibe l'activation cellulaire et la réaction inflammatoire. Par conséquent, la fréquence et la sévérité des symptômes d'UCS sont réduites. Une autre hypothèse est que la réduction des taux d'IgE libres entraîne une désensibilisation rapide et non spécifique des mastocytes cutanés. La régulation négative du FcεRI pourrait aider à maintenir cette réaction.
Dans les études cliniques menées chez des patients atteints d'UCS, le traitement par l'omalizumab a entraîné une réduction dose-dependante du taux d'IgE libres et une augmentation des taux d'IgE totales dans le sérum, à l'instar de ce qui a été observé chez les patients souffrant d'asthme allergique. La suppression maximale des IgE libres a été observée 3 jours après la première dose sous-cutanée. Après administration de doses multiples toutes les 4 semaines, les taux sériques d'IgE libres avant l'administration sont restés stables entre les semaines 12 et 24 de traitement. Le taux sérique d'IgE totales a augmenté après la première dose en raison de la formation de complexes omalizumab-IgE, dont la vitesse d'élimination est plus faible que celle des IgE libres. Après administration de doses répétées de 75 mg à 300 mg une fois toutes les 4 semaines, les taux sériques moyens d'IgE totales avant l'administration ont été deux à trois fois supérieurs à ceux observés avant le début du traitement à la semaine 12 et sont restés stables entre les semaines 12 et 24 de traitement. Après l'arrêt du traitement par Xolair, pendant une période de suivi sans traitement de 16 semaines, les taux d'IgE libres ont augmenté alors que les taux d'IgE totales diminuaient, les deux se rapprochant des valeurs avant traitement.
Efficacité clinique
Asthme allergique
Adultes et adolescents (≥12 ans)
L'efficacité et la sécurité d'emploi de Xolair ont été démontrées dans une étude pivot de 28 semaines contrôlée versus placebo (étude 5) chez 419 patients souffrant d'asthme allergique sévère, âgés de 12 à 79 ans, présentant une insuffisance pulmonaire (volume expiré maximal par seconde: VEMS 40–80%) et dont les symptômes répondaient mal au traitement par > 1000 µg de dipropionate de béclométhasone (ou équivalent) et d'agonistes bêta2 à longue durée d'action. Les patients admis dans l'étude avaient eu plusieurs exacerbations de l'asthme au cours de l'année précédente, nécessitant un traitement systémique par corticostéroïdes, avaient été hospitalisés ou avaient eu recours à un traitement d'urgence pour exacerbation de l'asthme sévère malgré un traitement continu par des corticostéroïdes inhalés à forte dose et des agonistes bêta2 à longue durée d'action. Xolair ou le placebo ont été administrés par voie sous-cutanée comme traitement d'appoint à > 1000 µg de dipropionate de béclométhasone (ou équivalent) et d'agonistes bêta2 à longue durée d'action. Les patients ont également reçu un traitement continu par corticostéroïdes oraux (22%), théophylline (27%) et antagonistes des leucotriènes (35%). Le traitement concomitant de l'asthme n'a pas été modifié au cours de la période de traitement.
Le critère d'évaluation principal est le taux d'exacerbations de l'asthme nécessitant un traitement aigu par corticostéroïdes systémiques. L'omalizumab a réduit le taux d'exacerbations de l'asthme de 19% (p = 0.153). D'autres analyses, qui ont montré une signification statistique (p = 0.05) en faveur de Xolair, ont inclus la réduction des exacerbations sévères (au cours desquelles la fonction pulmonaire du patient était réduite à moins de 60% de la meilleure valeur personnelle et nécessitant des corticostéroïdes systémiques) et la consultation dans un service d'urgences liée à l'asthme (incluant hospitalisation, visites au service des urgences et visites non programmées) et l'amélioration de l'évaluation globale par le médecin de l'efficacité du traitement, de la qualité de vie liée à l'asthme (AQL), des symptômes d'asthme et de la fonction pulmonaire.
Dans une analyse de sous-groupes chez des patients ayant un taux d'IgE de ≥76 UI/ml avant traitement, il est apparu plus probable que Xolair ait un bénéfice cliniquement significatif. Chez ces patients de l'étude 1, Xolair a réduit le nombre d'exacerbations de l'asthme de 40% (p = 0.002). De plus, dans le programme d'étude de Xolair dans l'asthme sévère, un plus grand nombre de patients ont présenté une réponse cliniquement significative dans la population avec un taux d'IgE totale ≥76 UI/ml (voir Tableau 6).
Lors de quatre autres grandes études contrôlées versus placebo, d'une durée de 28 à 52 semaines, incluant 1722 patients adultes et adolescents (études 3, 4, 5, 6), l'efficacité et la tolérance de Xolair ont été évaluées chez des patients atteints d'asthme persistant grave. La plupart des patients n'étaient pas suffisamment contrôlés, mais recevaient moins de médicaments concomitants pour l'asthme que les patients de l'étude 1 ou 2. Les études 3 à 5 comprenaient les exacerbations comme critère d'évaluation principal, alors que l'étude 6 a principalement déterminé l'économie de corticostéroïdes inhalés.
L'étude 2 a démontré la sécurité et l'efficacité de l'omalizumab chez 312 patients atteints d'asthme allergique sévère, correspondant à la population de l'étude 1. Dans cette étude en ouvert, le traitement par Xolair a entraîné une réduction de 61% du taux d'exacerbations de l'asthme cliniquement significatives par rapport au traitement actuel de l'asthme seul.
Dans les études 3, 4 et 5, les patients traités par Xolair avaient respectivement une réduction du taux d'exacerbations de l'asthme de 37.5% (p = 0.027), 40.3% (p < 0.001) et 57.6% (p < 0.001) par rapport au placebo.
Dans l'étude 6, un nombre significativement plus élevé de patients atteints d'asthme allergique sévère ont été capables de réduire leur dose de fluticasone à ≤500 µg/jour sans détérioration du contrôle de l'asthme avec Xolair (60.3%) par rapport au groupe placebo (45.8%, p < 0.05).
|
Tableau 6: Résultats de l'étude
| |
|
Population totale de l'étude
| |
|
Xolair
N = 209
|
Placebo
N = 210
| |
Exacerbations de l'asthme
|
|
| |
Fréquence par 28 semaines
|
0.74
|
0.92
| |
% de réduction, valeur de p pour le rapport des fréquences
|
19.4%, p = 0.153
| |
Exacerbations graves de l'asthme
|
|
| |
Fréquence pour 28 semaines
|
0.24
|
0.48
| |
% de réduction, valeur p pour le rapport des fréquences
|
50.1%, p = 0.002
| |
Visites aux urgences
|
|
| |
Fréquence pour 28 semaines
|
0.24
|
0.43
| |
% de réduction, valeur p pour le rapport des fréquences
|
43.9%, p = 0.038
| |
Évaluation globale du médecin
|
|
| |
% de répondeurs*
|
60.0.5%
|
42.8%
| |
Valeur p**
|
< 0.001
| |
Améliorations AQL***
|
|
| |
% de patients avec une amélioration ≥0.0.5
|
60.8%
|
47.8%
| |
Valeur p
|
0.008
| |
* Amélioration notable ou contrôle complet
** Valeur p pour la distribution générale de l'évaluation
*** Qualité de vie liée à l'asthme
|
Enfants âgés de 6 à < 12 ans
Les données fondamentales de sécurité et d'efficacité de Xolair chez les patients âgés de 6 à < 12 ans proviennent d'une étude multicentrique, randomisée, en double aveugle, contrôlée versus placebo (étude 7).
L'étude 7 était une étude contrôlée versus placebo, menée sur un sous-groupe spécifique (N = 235) de patients selon l'indication actuelle, traités par corticostéroïdes inhalés à haute dose (≥500 µg d'équivalent fluticasone/jour) et par bêta-agonistes de longue durée d'action.
Une exacerbation cliniquement significative a été définie comme une aggravation des symptômes de l'asthme, évaluée cliniquement par l'investigateur, nécessitant un doublement de la dose initiale du corticostéroïde inhalé pendant au moins 3 jours et/ou un traitement d'urgence par corticostéroïde systémique (oral ou intraveineux) pendant au moins 3 jours.
Dans le sous-groupe spécifique de patients recevant de fortes doses de corticoïdes inhalés, le groupe omalizumab a présenté un taux d'exacerbations de l'asthme significativement plus faible que le groupe placebo. Après 24 semaines, l'analyse des différences de taux pour les patients traités par omalizumab a montré l'obtention d'un taux inférieur de 34% (rapport des taux 0.662, p = 0.047) par rapport au placebo. Dans une deuxième période de traitement en double aveugle, d'une durée de 28 semaines, l'analyse des différences de taux pour les patients traités par l'omalizumab a montré l'obtention d'un taux inférieur de 63% (rapport des taux 0.37, p < 0.001) par rapport au placebo.
Au cours du traitement en double aveugle de 52 semaines (comprenant la phase de 24 semaines à dose constante de stéroïde et la phase de 28 semaines à dose adaptée de stéroïde), les différences de taux entre les groupes de traitement ont montré une diminution de 50% (rapport des taux de 0.0.504, p < 0.001) des exacerbations chez les patients traités par l'omalizumab.
À la fin des 52 semaines de traitement, le groupe omalizumab a montré une diminution plus importante de l'utilisation des bêta-agonistes comme traitement de secours que le groupe placebo, même si la différence entre les groupes de traitement n'était pas statistiquement significative. Dans l'évaluation globale de l'efficacité après 52 semaines de traitement en double aveugle, dans le sous-groupe de patients sévèrement malades recevant de fortes doses de corticostéroïdes inhalés et de bêta-agonistes à longue durée d'action, la proportion de patients présentant un «excellent» résultat du traitement a été plus élevée dans le groupe omalizumab que dans le groupe placebo. La proportion de patients avec un résultat «modéré» ou «mauvais» était plus faible dans le groupe omalizumab que dans le groupe placebo. Les différences entre les groupes étaient statistiquement significatives (p < 0.001). Aucune différence n'a été observée entre le groupe omalizumab et le groupe placebo dans les évaluations subjectives de la qualité de vie des patients.
Polypes nasaux
La sécurité et l'efficacité de Xolair ont été évaluées dans deux études cliniques randomisées, multicentriques, en double aveugle, contrôlées contre placebo (Etude 1, N = 138; Etude 2, N = 127) menées chez des patients atteints de rhinosinusite chronique et de polypes nasaux. Les patients ont reçu Xolair ou un placebo par voie sous-cutanée toutes les 2 ou 4 semaines, le dosage et la fréquence d'administration étant donnés aux tableaux 7 et 8 (voir «Posologie/Administration»). De plus, tous les patients ont reçu un traitement de fond par mométasone intranasale pendant toute l'étude. Ni une intervention chirurgicale sinonasale antérieure, ni un traitement systémique antérieur par corticostéroïdes n'étaient nécessaires à l'inclusion dans les études. Les participants ont reçu Xolair ou un placebo pendant 24 semaines, suivies d'une période de suivi de 4 semaines sans traitement. Les données démographiques et les caractéristiques à l'inclusion, y compris les comorbidités allergiques, sont présentées dans le Tableau 7.
|
Tableau 7: Données démographiques et caractéristiques à l'inclusion dans les études sur les polypes nasaux
| |
Paramètres
|
Étude sur les polypes nasaux 1
N = 138
|
Étude sur les polypes nasaux 2
N = 127
| |
Âge moyen en années (ET)
|
51.0 (13.2)
|
50.1 (11.9)
| |
% d'hommes
|
63.8
|
65.4
| |
Patients utilisant des corticostéroïdes systémiques au cours de l'année précédente (%)
|
18.8
|
26.0
| |
Score NPS endoscopique bilatéral moyen* (ET), intervalle: 0-8
|
6.2 (1.0)
|
6.3 (0.9)
| |
Score moyen de congestion nasale (CN)* (ET), intervalle: 0-3
|
2.4 (0.6)
|
2.3 (0.7)
| |
Score moyen de l'odorat* (ET) Intervalle: 0-3
|
2.7 (0.7)
|
2.7 (0.7)
| |
Moyenne du score total SNOT-22* (ET) Intervalle: 0-110
|
60.1 (17.7)
|
59.5 (19.3)
| |
Taux moyen d'éosinophiles dans le sang (cellules/µl) (ET)
|
346.1 (284.1)
|
334.6 (187.6)
| |
Moyenne des IgE totales en UI/ml (ET)
|
160.9 (139.6)
|
190.2 (200.0.5)
| |
Asthme (%)
|
53.6
|
60.6
| |
Léger (%)
|
37.8
|
32.5
| |
Modéré (%)
|
58.1
|
58.4
| |
Sévère (%)
|
4.1
|
9.1
| |
Maladie respiratoire exacerbée par l'aspirine (%)
|
19.6
|
35.4
| |
Rhinite allergique
|
43.5
|
42.5
|
ET = écart type; NPS = score de polypes nasaux; SNOT-22 = questionnaire de 22 questions concernant le test sino-nasal; IgE = immunoglobuline E; UI = unités internationales.
En ce qui concerne le NPS, le SCN et les scores olfactifs, le syndrome post-nasal et l'écoulement nasal ainsi que le score SNOT-22, des valeurs plus élevées indiquent une plus grande sévérité de la maladie.
Les co-critères d'évaluation principaux étaient le score bilatéral de polypes nasaux (NPS) et le score journalier moyen de congestion nasale (SCN) mesurés à la semaine 24. Le NPS a été déterminé par endoscopie à l'inclusion et aux échéances préalablement fixées (intervalle: 0 à 4 par narine) et à partir de ces valeurs, le NPS total a été calculé (intervalle: 0 = meilleure valeur à 8 = plus mauvaise valeur). La congestion nasale a été évaluée quotidiennement à l'aide de l'échelle SCN (intervalle: 0 = meilleure valeur à 3 = plus mauvaise valeur). Les patients devaient avoir un NPS ≥5 et une moyenne hebdomadaire du SCN > 1 malgré l'utilisation de mométasone par voie intranasale, avant la randomisation. Le NPS moyen à l'inclusion était équilibré entre les deux groupes de traitement dans les deux études.
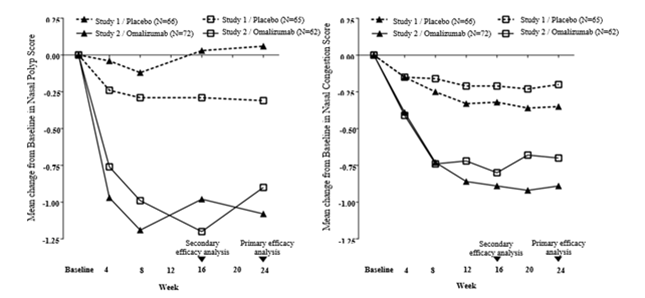
Dans l'étude 1 et l'étude 2 des études sur les polypes nasaux, les patients traités par Xolair ont présenté une amélioration plus importante de manière statistiquement significative du NPS et du SCN hebdomadaire moyen à la semaine 24 par rapport à la valeur initiale que les patients ayant reçu un placebo (voir tableau 8).
Les améliorations plus importantes du NPS et du SCN dans le groupe Xolair par rapport au groupe placebo ont été observées dans les deux études dès la première évaluation à la semaine 4, comme le montre la Figure 1. À la semaine 4, la différence entre les valeurs de la moyenne des moindres carrés (LS) pour la variation par rapport à la valeur initiale du NPS dans le groupe Xolair par rapport au groupe placebo était de -0.92 (IC à 95%: -1.37, -0.48) dans l'étude 1 et -0.0.52 (IC à 95%: -0.94, -0.11) dans l'étude 2. La différence entre les valeurs de la moyenne des moindres carrés (LS) pour la variation par rapport à la valeur initiale à la semaine 4 dans le groupe Xolair par rapport au groupe placebo était de -0.25 (IC à 95%: -0.46, -0.04) dans l'étude 1 et de -0.26 (IC à 95%: -0.45, -0.07) dans l'étude 2. Cependant, les tests statistiques n'étaient pas pré-spécifiés à ce stade.
|
Tableau 8: Variation du score de polypes nasaux et du score de congestion nasale moyen sur 7 jours à la semaine 24 par rapport à la valeur initiale dans les études sur les polypes nasaux 1 et 2
| |
|
Étude sur les polypes nasaux 1
|
Étude sur les polypes nasaux 2
| |
|
Placebo
|
Xolair
|
Placebo
|
Xolair
| |
N
|
66
|
72
|
65
|
62
| |
Score de polypes nasaux
|
|
|
|
| |
Moyenne à l'inclusion
|
6.32
|
6.19
|
6.09
|
6.44
| |
Valeur de la moyenne LS de la variation à la semaine 24 par rapport à l'inclusion
|
0.06
|
-1.08
|
-0.31
|
-0.90
| |
Différence des moyennes LS
vs placebo
|
-1.14
|
-0.0.59
| |
IC à 95% de la différence
|
-1.59, -0.69
|
-1.05, -0.12
| |
Valeur p
|
< 0.0001
|
0.0140
| |
Valeur moyenne du score journalier de congestion nasale sur 7 jours
|
|
|
|
| |
Moyenne à l'inclusion
|
2.46
|
2.40
|
2.29
|
2.26
| |
Valeur moyenne LS de la variation à la semaine 24 par rapport à la valeur initiale
|
-0.35
|
-0.89
|
-0.20
|
-0.70
| |
Différence des moyennes LS
vs placebo
|
-0.0.55
|
-0.0.50
| |
IC à 95% de la différence
|
-0.84, -0.25
|
-0.80. -0.19
| |
Valeur p
|
0.0004
|
0.0017
|
LS = moindres carrés (détermination de la valeur moyenne selon la méthode des moindres carrés)
Figure 1: Variation moyenne par rapport à l'inclusion du score de congestion nasale et du score de polypes nasaux par groupe de traitement dans les études sur les polypes nasaux 1 et 2
Un critère secondaire majeur était l'évaluation de la variation du score total des symptômes nasaux (TNSS, total nasal symptom score) à la semaine 24 par rapport à l'inclusion. Le TNSS rapporté par le patient était un score correspondant à la somme de 4 scores de symptômes quotidiens équivalents pondérés. Il s'agit de: SCN, score de l'odorat, score de rhinorrhée postérieure et score de rhinorrhée antérieure. La fourchette pour le TNSS allait de 0 = meilleure valeur à 12 = plus mauvaise valeur. Une amélioration significative du TNSS quotidien moyen a été observée avec Xolair par rapport au placebo. La différence des moyennes LS pour la variation à la semaine 24 par rapport à la valeur initiale était de -1.91 point (IC à 95%: -2.85, -0.96; p = 0.0001) dans l'étude 1 et -2.09 points (IC à 95%: -3.00, -1.18; p < 0.0001) dans l'étude 2.
Avec Xolair, on a observé une amélioration significative du score SNOT-22 (test sino-nasal), qui combine des questions liées aux symptômes sinonasaux, à la psychologie et à la qualité du sommeil. Le SNOT-22 était compris entre 0 et 110 (0 = meilleure valeur, 110 = plus mauvaise valeur). La différence des moyennes LS pour la variation à la semaine 24 par rapport à la valeur initiale du score SNOT-22 dans le groupe Xolair comparativement au placebo était de -16.12 (IC à 95%: -21.86, -10.38; p < 0.0001) dans l'étude 1 et -15.04 (IC à 95%: -21.26, -8.82; p < 0.0001) dans l'étude 2.
Une amélioration significative des scores UPSIT (test d'identification d'odeur de l'université de Pennsylvanie) quotidiens moyens a de plus été observée sous Xolair par rapport au placebo. Le score UPSIT était compris entre 0 et 40 (0 = plus mauvaise valeur, 40 = meilleure valeur). La différence des moyennes LS pour la variation à la semaine 24 par rapport à la valeur initiale dans le groupe Xolair comparativement au placebo était de 3.81 points (IC à 95%: 1.38, 6.24; p = 0.0024) dans l'étude 1 et de 3.86 points (IC à 95%: 1.57, 6.15; p = 0.0011) dans l'étude 2.
Les effets sur les scores TNSS et SNOT-22 ont été observés dès la première évaluation à la semaine 4 dans les deux études. De plus, l'effet sur le score UPSIT a été observé dès la première évaluation à la semaine 8 dans les deux études.
D'autres analyses des critères d'évaluation secondaires portaient sur les évaluations du NPS et du SCN à la semaine 16. Une amélioration significative du NPS a été observée sous Xolair à la semaine 16 (0 = plus mauvaise valeur, 8 = meilleure valeur) par rapport au placebo. La différence des moyennes LS pour la variation à la semaine 16 par rapport à la valeur initiale dans le groupe Xolair comparativement au placebo était de -1.01 (IC à 95%: -1.43, -0.60; p < 0.0001) dans l'étude 1 et de -0.91 (IC à 95%: -1.39, -0.44; p = 0.0002) dans l'étude 2. Une amélioration significative du SCN a été observée à la semaine 16 sous Xolair par rapport au placebo (0 = meilleure valeur, 3 = plus mauvaise valeur). La différence des valeurs moyennes LS pour la variation du SCN quotidien moyen à la semaine 16 par rapport à la valeur initiale dans le groupe Xolair comparativement au placebo était de -0.57 (IC à 95%: -0.0.83, -0.0.31; p < 0.0.0001) dans l'étude 1 et de -0.0.59 (IC à 95%: -0.0.87, -0.0.30; p < 0.0001) dans l'étude 2.
Urticaire chronique spontanée (UCS)
Le programme de développement clinique de phase III pour l'UCS incluait trois études multicentriques, randomisées, en double aveugle, contrôlées contre placebo, en groupes parallèles: Q4881g, Q4882g et Q4883g.
Les études ont été menées chez des adultes et des adolescents (âgés de 12 ans et plus) atteints d'UCS pendant ≥6 mois (6 mois à 66 ans, moyenne de 6 ans) présentant des poussées continues malgré des antihistaminiques aux doses maximales autorisées (score UAS 7 ≥16/42 pendant ≥8 jours consécutifs).
Les études Q4881g et Q4882g ont permis d'évaluer l'efficacité et la sécurité de l'administration de Xolair 75 mg, 150 mg ou 300 mg toutes les 4 semaines pendant 24 ou 12 semaines avec une période de suivi sans traitement de 16 semaines chez des patients (12 à 75 ans) atteints d'UCS réfractaire malgré un traitement par antihistaminiques H1.
L'étude Q4883g a permis d'évaluer l'efficacité et la sécurité de Xolair 300 mg administré toutes les 4 semaines pendant 24 semaines avec une période de suivi sans traitement de 16 semaines chez des patients (12 à 75 ans) atteints d'UCS réfractaire malgré un traitement par antihistaminiques H1 et/ou H2 et/ou antagonistes des récepteurs des leucotriènes (ARLT).
|
Tableau 9
|
Critères d'évaluation de l'efficacité
| |
Variation du score hebdomadaire de sévérité des démangeaisons (ISS, intervalle 0–21) à la semaine 12 par rapport à l'inclusion
|
Critère d'évaluation principal dans les études Q4881g et Q4882g
Critère d'évaluation secondaire dans l'étude de sécurité Q4883g
| |
Délai avant obtention d'une réponse DMI a (diminution ≥5 points par rapport à l'inclusion) du score ISS évalué chaque semaine jusqu'à la semaine 12
|
Critères d'évaluation secondaires dans les trois études Q4881g, Q4882g et Q4883g
| |
Variation du score d'activité de l'urticaire mesuré sur une période de 7 jours (UAS7 b, intervalle 0-42) à la semaine 12 par rapport à l'inclusion
| |
Proportion de patients avec un score d'activité de l'urticaire mesuré sur une période de 7 jours ≤6 (UAS7 b ≤6) à la semaine 12
| |
Proportion de patients avec un score d'activité de l'urticaire mesuré sur une période de 7 jours = 0 (UAS7 b = 0) à la semaine 12 c
| |
Variation du score hebdomadaire du nombre de papules à la semaine 12 par rapport à l'inclusion
| |
Variation du score total de l'indice dermatologique de qualité de vie (DLQI) à la semaine 12 par rapport à l'inclusion
| |
Proportion de patients ayant des jours sans angiœdème entre la semaine 4 et la semaine 12 d
| |
a
DMI: différence minimale importante (Minimally Important Difference)
b UAS7: composé de la sévérité du prurit et du nombre de papules; somme des scores mesurés sur 7 jours consécutifs
c Analyse post-hoc pour l'étude Q4882g
d La proportion moyenne de jours sans angiœdème entre la semaine 4 et la semaine 12 a été calculée pour l'ensemble de la population de l'étude, y compris les patients sans symptômes d'angiœdème.
|
Dans les études Q4881g et Q4882g, la dose de 75 mg n'a atteint de manière constante ni le critère d'évaluation principal de l'efficacité (variation du score hebdomadaire de sévérité des démangeaisons (ISS) à la semaine 12 par rapport à l'inclusion) ni plusieurs critères d'évaluation secondaires. Par conséquent, cette dose n'a pas été considérée comme efficace et n'est donc pas présentée plus loin.
Le critère principal d'efficacité, la variation du score hebdomadaire de sévérité des démangeaisons à la semaine 12 par rapport à l'inclusion, a été atteint dans les études Q4881g et Q4882g avec les doses de 150 mg et de 300 mg et dans l'étude Q4883g avec la dose de 300 mg (critère d'évaluation secondaire; voir tableau 10).
|
Tableau 10: Variation du score hebdomadaire de sévérité des démangeaisons à la semaine 12 par rapport à l'inclusion, études Q4881g, Q4882g, Q4883g (population mITT*)
| |
|
Placebo
|
Omalizumab
150 mg
|
Omalizumab
300 mg
| |
Étude Q4881g
|
|
|
| |
N
|
80
|
80
|
81
| |
Moyenne (ET)
|
-3.63 (5.22)
|
-6.66 (6.28)
|
-9.40 (5.73)
| |
Différence des moyennes LS vs placebo1
|
-
|
-2.95
|
-5.80
| |
IC à 95% pour la différence
|
-
|
−4.72, −1.18
|
−7.49, −4.10
| |
Valeur p par rapport au placebo2
|
-
|
0.0012
|
< 0.0001
| |
Étude Q4882g
|
|
|
| |
N
|
79
|
82
|
79
| |
Moyenne (ET)
|
-5.14 (5.58)
|
-8.14 (6.44)
|
-9.77 (5.95)
| |
Différence des moyennes LS vs placebo1
|
-
|
-3.04
|
-4.81
| |
IC à 95% pour la différence
|
-
|
−4.85, −1.24
|
−6.49, −3.13
| |
Valeur p par rapport au placebo2
|
-
|
0.0011
|
< 0.0001
| |
Étude Q4883g
|
|
|
| |
N
|
83
|
-
|
252
| |
Moyenne (ET)
|
-4.01 (5.87)
|
-
|
-8.55 (6.01)
| |
Différence des moyennes LS vs placebo1
|
-
|
-
|
-4.52
| |
IC à 95% pour la différence
|
-
|
-
|
−5.97, −3.08
| |
Valeur p par rapport au placebo2
|
-
|
-
|
< 0.0001
| |
* Population en intention de traiter modifiée (mITT): inclut tous les patients randomisés ayant reçu au moins une dose du médicament expérimental
La méthode BOCF (Baseline Observation Carried Forward) a été utilisée pour calculer les données manquantes.
1 La moyenne LS a été calculée en utilisant un modèle ANCOVA. Les facteurs de stratification étaient la valeur initiale du score hebdomadaire de sévérité des démangeaisons (< 13 vs. ≥13) et le poids initial (< 80 kg vs. ≥80 kg).
2 Les valeurs p proviennent du test t ANCOVA.
|
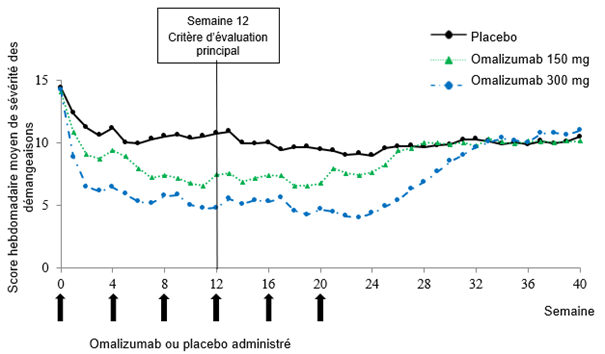
La figure 2 présente la moyenne hebdomadaire de l'ISS au cours du temps dans l'étude Q4881g. Le score hebdomadaire moyen de sévérité des démangeaisons a diminué significativement dans les deux groupes de traitement. L'effet maximal a été atteint approximativement à la semaine 12 et s'est maintenu sur la période de traitement de 24 semaines. Dans les études Q4883g (300 mg sur une période de traitement de 24 semaines) et Q4882g (150 mg ou 300 mg sur une période de traitement de 12 semaines), les résultats étaient similaires à ceux obtenus dans l'étude Q4881g.
Dans les trois études (voir Figure 2 pour l'étude Q4881g), le score hebdomadaire moyen de sévérité des démangeaisons pour les deux doses a augmenté progressivement pendant la période sans traitement de 16 semaines, parallèlement à la réapparition des symptômes. À la fin de la période de suivi, les valeurs moyennes étaient comparables à celles du groupe placebo, mais inférieures aux valeurs moyennes à l'inclusion.
Figure 2: Score hebdomadaire moyen de sévérité des démangeaisons au cours du temps, étude Q4881g (BOCF, population mITT)
BOCF = Baseline Observation Carried Forward; mITT = population en intention de traiter modifiée
Délai avant obtention d'une réponse DMI du score ISS évalué chaque semaine jusqu'à la semaine 12
Dans les études Q4881g et Q4882g, le délai médian d'obtention d'une DMI pour le score hebdomadaire de sévérité des démangeaisons de 5 points a été de 2 semaines chez les patients du groupe de traitement 150 mg (p = 0.0301 dans l'étude Q4881g; p = 0.0101 dans l'étude Q4882g) et de 1 semaine chez les patients du groupe de traitement 300 mg (p < 0.0001), contre 4 semaines chez les patients du groupe placebo. Des résultats comparables ont été observés dans l'étude Q4883g avec un délai médian d'obtention d'une DMI de 2 semaines dans le groupe de traitement 300 mg (p < 0.0001) contre 5 semaines dans le groupe placebo.
Variation de l'UAS7 à la semaine 12 par rapport à l'inclusion
Dans les études de phase III, les groupes de traitement par omalizumab 150 mg et 300 mg ont présenté une différence statistiquement significative pour la variation moyenne du score UAS7 à la semaine 12 par rapport à la valeur initiale comparativement au placebo (Figure 3 pour l'étude Q4881g). La significativité statistique (p < 0.0001) a été atteinte dans les trois études dans le groupe traité par 300 mg et dans les études Q4881g (p = 0.0008) et Q4882g (p = 0.0001) dans le groupe traité par 150 mg.
La figure 3 montre le score UAS7 moyen dans le temps dans l'étude Q4881g, qui a significativement diminué dans les deux groupes de traitement par rapport à la valeur initiale, avec un effet maximal à la semaine 12. L'intensité de cet effet s'est maintenue au cours de la période de traitement de 24 semaines. Dans les études Q4882g (150 mg et 300 mg pendant une période de traitement de 12 semaines) et Q4883g (300 mg pendant une période de traitement de 24 semaines), les résultats ont été comparables à ceux de l'étude Q4881g.
Dans les trois études (voir Figure 3 pour l'étude Q4881g), le score UAS7 dans les deux groupes de traitement par omalizumab a augmenté progressivement pendant la période de suivi sans traitement de 16 semaines, parallèlement à la réapparition des symptômes. A la fin de la période de suivi, les valeurs moyennes étaient comparables à celles du groupe placebo, mais inférieures aux valeurs correspondantes à l'inclusion.
Figure 3: Score UAS7 moyen au cours du temps, étude Q4881g (BOCF, population mITT)

BOCF = Baseline Observation Carried Forward; mITT = Population en intention de traiter modifiée; UAS7 = Score d'activité de l'urticaire mesuré sur une période de 7 jours
Proportion de patients avec un score UAS7 ≤6 à la semaine 12
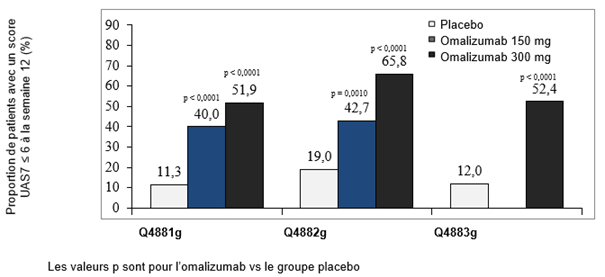
La Figure 4 présente la proportion de patients ayant un score UAS7 ≤6 à la semaine 12. Les taux de réponse étaient tous statistiquement significatifs et étaient compris entre 52% et 66% (dose de 300 mg; p < 0.0001) et entre 40% et 43% (dose de 150 mg; p < 0.001), contre 11–19% dans le groupe placebo.
Figure 4: Proportion de patients avec un score UAS7 ≤6 à la semaine 12, études Q4881g, Q4882g et Q4883g
Proportion de patients avec un score UAS7 = 0 à la semaine 12
La proportion de patients présentant une réponse complète avec un score UAS7 = 0 à la semaine 12 était de 34 à 44% (dose de 300 mg, statistiquement significatif, tous p < 0.0001) et de 15 à 22% (dose de 150 mg), contre 5 à 9% dans le groupe placebo (figure 5).
Figure 5: Proportion de patients avec un score UAS7 = 0 à la semaine 12, études Q4881g, Q4882g et Q4883g

Analyse prospective dans les études Q4881g et Q4883g et analyse post-hoc dans l'étude Q4882g
Variation du score hebdomadaire du nombre de papules à la semaine 12 par rapport à l'inclusion
La variation moyenne du score hebdomadaire du nombre de papules à la semaine 12 par rapport à l'inclusion a été statistiquement significative (p < 0.001) dans les trois études de phase III dans le groupe de traitement 300 mg et a montré une réduction du nombre de papules par rapport au placebo (-11.35 dans l'étude Q4881g, -11.97 dans l'étude Q4882g et -10.46 dans l'étude Q4883g contre -4.37, -5.22 et -4.49 dans les groupes placebo correspondants). Dans le groupe de traitement 150 mg, la variation moyenne était de -7.78 (p = 0.0017) dans l'étude Q4881g et de -9.75 (p < 0.0001) dans l'étude Q4882g.
Proportion de jours sans angiœdème entre les semaines 4 et 12
Dans les trois études de phase III, les groupes traités à 300 mg ont systématiquement atteint la proportion moyenne la plus élevée de jours sans angiœdème entre les semaines 4 et 12 (91–96%). L'augmentation de la proportion de jours sans angiœdème a été statistiquement significative par rapport au placebo (p < 0.001) (fig. 6). Dans le groupe de traitement 150 mg, la proportion moyenne de jours sans angiœdème pendant la même période a été de 89.6% dans l'étude Q4881g et de 91.6% dans l'étude Q4882g. Les valeurs correspondantes pour le placebo dans ces études ont été respectivement de 88.2% et 89.2%. Dans les deux études, les différences entre la dose de 150 mg et le placebo n'ont pas été statistiquement significatives.
Figure 6: Proportion de jours sans angiœdème entre les semaines 4 et 12, études Q4881g, Q4882g et Q4883g
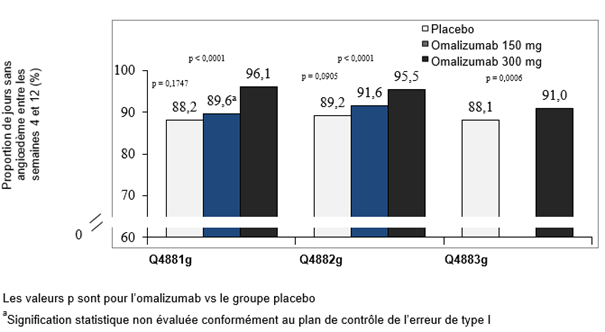
La proportion moyenne de jours sans angioedème entre la semaine 4 et la semaine 12 a été calculée pour l'ensemble de la population de l'étude, y compris les patients sans symptômes d'angioedème.
Variation du score total de l'indice de qualité de vie dermatologique (DLQI) à la semaine 12 par rapport à l'inclusion
Dans les trois études de phase III, la variation moyenne du score DLQI total à la semaine 12 par rapport à l'inclusion était statistiquement significativement plus importante dans le groupe de traitement 300 mg (p < 0.001) que dans le groupe placebo. Dans l'étude Q4882g, le groupe de traitement par l'omalizumab 150 mg a présenté une différence statistiquement significative (p = 0.022) par rapport au placebo (figure 7).
Figure 7: Variation du score total de l'indice de qualité de vie dermatologique (DLQI) à la semaine 12 par rapport à l'inclusion, études Q4881g, Q4882g et Q4883g
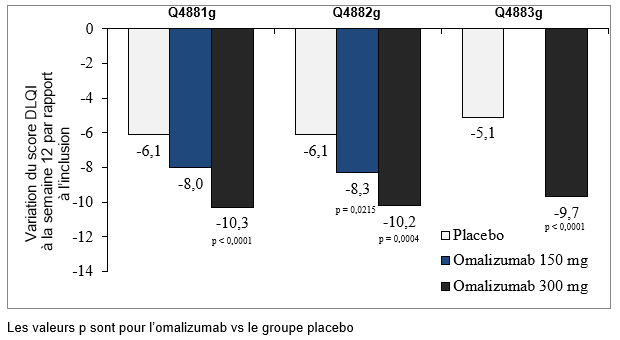
DLQI = Dermatology Life Quality Index, index de qualité de vie lié à la dermatologie
Efficacité après 24 semaines de traitement
Le tableau 11 présente les résultats après 24 semaines de traitement. Les ordres de grandeur de la réponse sont similaires à ceux observés après 12 semaines.
|
Tableau 11: Résultats d'efficacité après 24 semaines de traitement, études Q4881g et Q4883g (population mITT*)
| |
Paramètres de l'étude
|
Semaine
|
Placebo
|
Omalizumab
150 mg
|
Omalizumab
300 mg
| |
Variation du score hebdomadaire de sévérité des démangeaisons (BOCF) par rapport à l'inclusion, moyenne
| |
Étude Q4881g
|
Semaine 24
|
−5.41
|
−6.47
|
−9.84**
| |
Étude Q4883g
|
Semaine 24
|
−4.03
|
NA
|
−8.60**
| |
Variation du score UAS7 (BOCF) par rapport à l'inclusion, moyenne
| |
Étude Q4881g
|
Semaine 24
|
−11.73
|
−14.21
|
−22.11**
| |
Étude Q4883g
|
Semaine 24
|
−8.85
|
NA
|
−19.15**
| |
Proportion de patients avec score UAS7 ≤6, % de patients
| |
Étude Q4881g
|
Semaine 24
|
25.0
|
36.3
|
61.7**
| |
Étude Q4883g
|
Semaine 24
|
16.9
|
NA
|
55.6**
| |
Proportion de patients avec score UAS7 = 0. % de patients
| |
Étude Q4881g
|
Semaine 24
|
12.5
|
20.0
|
48.1**
| |
Étude Q4883g
|
Semaine 24
|
3.6
|
NA
|
42.5**
| |
* Population en intention de traiter modifiée (mITT): inclut tous les patients randomisés ayant reçu au moins une dose du médicament expérimental.
** valeur de p ≤0.0001 dans le test statistique correspondant entre le traitement et le placebo
NA: sans objet (Not Applicable).
BOCF: Baseline Observation Carried Forward.
|
Efficacité après 48 semaines de traitement
Dans une étude d'une durée de 48 semaines, 206 patients atteints d'UCS non contrôlée par un traitement par antihistaminique H1. âgés de 12 à 75 ans, ont participé à une phase de traitement en ouvert par l'omalizumab 300 mg toutes les 4 semaines, d'une durée de 24 semaines, en tant que traitement d'appoint. Les patients qui ont répondu au traitement pendant cette phase en ouvert, ont ensuite reçu à l'aveugle de manière aléatoire, l'omalizumab 300 mg (81 patients) ou un placebo (53 patients), toutes les 4 semaines pendant 24 semaines supplémentaires.
Parmi les patients ayant été traités par l'omalizumab pendant un total de 48 semaines, 21% ont présenté une dégradation clinique (valeur UAS7 de 12 ou plus pendant au moins 2 semaines consécutives après la randomisation entre les semaines 24 et 48), par comparaison à 60.4% des patients traités par le placebo à la semaine 48 (La différence est de -39.4%, p < 0.0001, IC à 95%: -54.5%, -22.5%).
L'étude prospective de registre de grossesses (EXPECT)
Une étude prospective de registre des grossesses (EXPECT) réalisée aux États-Unis de 2006 à 2018 incluait 250 femmes enceintes asthmatiques traitées par Xolair; 246 femmes avaient été traitées par Xolair pendant le premier trimestre de la grossesse et 78.4% (196/250) des femmes avaient été traitées par Xolair au moins une fois dans chacun des 3 trimestres de la grossesse, avec une durée médiane d'exposition de 8.7 mois. Les résultats de EXPECT dans les sous-groupes de mères et de nourrissons concernés ont été comparés aux fréquences liées à l'âge dans une cohorte externe adaptée à la maladie de 1153 femmes enceintes souffrant d'asthme (sans traitement par Xolair), établie à partir de bases de données de santé d'habitants de la province canadienne Québec et nommée Quebec External Comparator Cohort (QECC, cohorte québécoise externe de comparaison).
La prévalence des malformations congénitales pertinentes était de 8.1% chez les nourrissons EXPECT comparés aux nourrissons QECC (n = 223), une prévalence donc comparable à celle des nourrissons QECC (8.9%). Parmi les grossesses de EXPECT, qui ont été comparées aux grossesses de QECC (n = 230), 99.1% ont donné lieu à des naissances vivantes, soit un taux similaire aux 99.3% des grossesses de QECC.
Dans une sous-étude de EXPECT, les taux de plaquettes ont été évalués chez 51 nourrissons nés de femmes exposées à Xolair; les taux ont tous été inclus dans les limites de la normale.
PharmacocinétiqueAbsorption
Après injection sous-cutanée, la biodisponibilité absolue moyenne de l'omalizumab est de 62%. Après administration sous-cutanée unique chez des patients asthmatiques adultes et adolescents, l'omalizumab a été absorbé lentement. Les concentrations sériques maximales ont été atteintes en 7 à 8 jours en moyenne. À des doses supérieures à 0.5 mg/kg, la pharmacocinétique de l'omalizumab est linéaire.
Après administration multiple d'omalizumab, l'ASC (à l'état d'équilibre entre le jour 0 et le jour 14) a été jusqu'à 6 fois plus élevée qu'après la première administration.
Distribution
In vitro, l'omalizumab forme des complexes de poids moléculaire limité avec les IgE. Il n'a pas été observé, in vitro ou in vivo, de complexes qui précipitent, ni la formation de complexes de poids moléculaire supérieur à 1 000 000 Da. Le volume apparent de distribution chez les patients après administration sous-cutanée était de 78 ± 32 ml/kg.
Métabolisme
Aucune donnée pertinente n'est disponible pour l'utilisation.
Élimination
La clairance de l'omalizumab inclut non seulement des processus de clairance des IgG, mais également la clairance par liaison spécifique et formation de complexe avec le ligand cible, l'IgE. L'élimination hépatique des IgG fait intervenir une dégradation dans le système réticulo-endothélial et dans les cellules endothéliales. Les IgG sont également éliminées sous forme inchangée dans la bile. Chez les patients asthmatiques, la demi-vie d'élimination sérique de l'omalizumab est en moyenne de 26 jours, avec une clairance apparente moyenne de 2.4 ± 1.1 ml/kg/jour.
Par ailleurs, à un poids corporel double correspond une clairance apparente double.
Patients atteints de polypes nasaux
Les analyses pharmacocinétiques de population de l'omalizumab ont montré que la pharmacocinétique de l'omalizumab en cas de polypes nasaux était identique à celle pour l'asthme. Des analyses graphiques de covariance ont été réalisées pour évaluer les effets des caractéristiques démographiques et d'autres facteurs sur l'exposition à l'omalizumab et la réponse clinique. Ces analyses montrent qu'aucune adaptation posologique n'est nécessaire en fonction de l'âge (18 à 75 ans) ou du sexe. Les données relatives à la race et à l'origine ethnique disponibles chez les patients atteints de polypes nasaux sont trop limitées pour permettre de déterminer si une adaptation posologique est nécessaire.
Linéarité/non-linéarité
À des doses supérieures à 0.5 mg/kg, la pharmacocinétique de l'omalizumab est linéaire.
Cinétique pour certains groupes de patients
Les analyses pharmacocinétiques en fonction de l'âge, du sexe, de l'origine ethnique ou de l'indice de masse corporelle ont montré qu'aucune adaptation posologique n'était nécessaire chez ces populations de patients (6–76 ans).
Aucune donnée pharmacocinétique ou pharmacodynamique n'est disponible chez les patients présentant une insuffisance rénale ou hépatique (cf. «Mises en garde et précautions»).
Âge, origine ethnique, sexe, poids corporel, indice de masse corporelle, valeur initiale des IgE, auto-anticorps anti-FcεRI, traitements concomitants
Les effets des covariables démographiques et d'autres facteurs sur l'exposition à l'omalizumab ont été évalués à l'aide de méthodes pharmacocinétiques de population. De plus, les effets des covariables ont été évalués par l'analyse du rapport entre les concentrations d'omalizumab et la réponse clinique. Ces analyses suggèrent qu'aucune adaptation posologique n'est nécessaire chez les patients atteints d'UCS en fonction de l'âge (12 à 75 ans), de l'origine ethnique, du sexe, du poids corporel, de l'indice de masse corporelle, de la valeur initiale des IgE, des auto-anticorps anti-FcεRI ou de l'utilisation concomitante d'antihistaminiques H2 ou d'antagonistes des récepteurs des leucotriènes (ARLT).
Données précliniquesL'administration de Xolair à des singes Cynomolgus adultes et juvéniles n'a révélé aucun signe de réaction anaphylactoïde systémique due à une dégranulation mastocytaire.
Dans toutes les études menées chez le singe, des complexes omalizumab-anticorps IgE circulants ont été détectés, mais aucun organe (y compris le rein) n'a présenté de signes de réaction à médiation par un complexe immun après administration d'omalizumab. Les complexes omalizumab-IgE ne peuvent ni lier le complément ni induire une cytotoxicité dépendante du complément.
Des anticorps anti-omalizumab ont été détectés chez quelques singes après administration sous-cutanée ou intraveineuse. Un tel résultat pouvait être attendu après l'administration d'une protéine hétérologue. Quelques animaux n'ont pas pu être évalués en raison de concentrations sériques élevées d'omalizumab, de valeurs élevées d'IgE ou des deux. Cependant, les animaux ont présenté des concentrations sériques élevées d'omalizumab pendant toute la durée du traitement et aucune toxicité apparente due aux anticorps anti-omalizumab n'a été observée.
Les études de distribution tissulaire chez le singe Cynomolgus n'ont pas montré d'absorption spécifique de 125I-omalizumab par un quelconque organe ou tissu.
Dans les études menées chez la souris et le singe, les complexes omalizumab-IgE ont été éliminés par interaction avec les récepteurs Fcγ dans le SRE avec des taux de dégradation généralement plus rapides que la clairance des IgG.
Toxicité à long terme (ou toxicité en administration répétée)
L'administration à long terme d'omalizumab à des doses allant jusqu'à 250 mg/kg (dose clinique maximale admissible pour l'asthme ou l'UCS recommandée de 15 mg/kg) a été bien tolérée chez les singes primates (adultes et juvéniles). Une exception a été une diminution dose-dépendante de la numération plaquettaire chez plusieurs singes primates avec des concentrations sériques généralement supérieures aux niveaux d'exposition maximaux observés chez l'homme dans les études cliniques pivots. Les singes juvéniles étaient plus sensibles aux effets plaquettaires que les singes adultes. De plus, chez le singe Cynomolgus, des saignements aigus et des inflammations ont été observés aux sites d'injection, ce qui est compatible avec une réponse immunitaire localisée à l'administration répétée d'une protéine hétérologue par voie sous-cutanée.
Cancérogénicité
Aucune étude sur la cancérogénicité n'a été effectuée avec l'omalizumab.
Toxicité pour la reproduction
Des études sur la reproduction ont été menées avec l'omalizumab chez le singe Cynomolgus. Des doses sous-cutanées d'omalizumab allant jusqu'à 75 mg/kg/semaine (environ 17 fois l'exposition à la DMRH pendant 4 semaines) pendant l'organogenèse n'ont pas montré de toxicité maternelle, d'embryotoxicité ni de tératogénicité. De plus, aucun effet délétère sur la croissance fœtale ou néonatale n'a été observé lors de l'administration pendant la gestation, l'accouchement ou l'allaitement.
L'excrétion de l'omalizumab dans le lait a été étudiée chez le singe Cynomolgus femelle à des doses sous-cutanées de 75 mg/kg/semaine. Les taux sériques d'omalizumab chez le nouveau-né après une exposition in utero et un allaitement pendant 28 jours étaient compris entre 11% et 94% des taux sériques maternels. Les taux d'omalizumab dans le lait se situaient à 0.15% de la concentration sérique maternelle.
Fertilité
Il n'existe pas de données sur l'effet de l'omalizumab sur la fertilité humaine. Aucune altération de la fertilité mâle ou femelle n'a été observée après administration répétée d'omalizumab à des doses allant jusqu'à 75 mg/kg dans des études de fertilité non cliniques spécifiques, y compris des études d'accouplement.
Remarques particulièresIncompatibilités
Xolair ne doit pas être mélangé à d'autres médicaments.
Stabilité
Ce médicament ne doit pas être utilisé au-delà de la date figurant après la mention «EXP» sur l'emballage.
Remarques particulières concernant le stockage
Conserver au réfrigérateur (2–8 °C).
Ne pas congeler.
Conserver le récipient dans son carton pour le protéger de la lumière (et/ou de l'humidité).
Tenir hors de portée des enfants.
Le stylo prérempli ne doit pas être conservé plus de 48 heures au total à température ambiante (25 °C).
Instructions d'utilisation et de manipulation
Chaque boîte de Xolair contient un stylo prérempli.
Un mode d'emploi détaillé est disponible dans l'information destinée aux patients.
Numéro d’autorisation69272 (Swissmedic)
Présentation1 solution injectable en stylo prérempli à 75 mg d'omalizumab dans 0.5 ml [B]
1 solution injectable en stylo prérempli à 150 mg d'omalizumab dans 1.0 ml [B]
1 solution injectable en stylo prérempli à 300 mg d'omalizumab dans 2.0 ml [B]
Titulaire de l’autorisationNovartis Pharma Schweiz AG, Risch; domicile: 6343 Rotkreuz
Mise à jour de l’informationMars 2025
|