CompositionPrincipes actifs
Inébilizumab.
L'inébilizumab est un anticorps monoclonal humanisé produit dans des cellules ovariennes de hamster chinois par la technologie de l'ADN recombinant.
Excipients
Histidine, chlorhydrate d'histidine monohydraté, chlorure de sodium, tréhalose dihydraté, polysorbate 80 (E433), eau pour préparations injectables.
Ce médicament contien 16,1 mg de sodium par flacon.
Indications/Possibilités d’emploiUplizna est indiqué en monothérapie dans le traitement des troubles du spectre de la neuromyélite optique (TSNMO) chez les patients adultes qui sont séropositifs pour les immunoglobulines G anti-aquaporine-4 (AQP4-IgG) (voir rubrique «Propriétés/Effets»).
Posologie/Mode d’emploiLe traitement doit être initié sous la surveillance d'un médecin expérimenté dans le traitement des TSNMO et ayant accès au matériel médical nécessaire à la prise en charge des effets indésirables sévères potentiels tels que les réactions associées à la perfusion graves.
Le patient doit être surveillé afin de détecter l'apparition de réactions à la perfusion pendant et au moins une heure suivant la fin de la perfusion (voir rubrique «Mises en garde et précautions»).
Évaluations préalables lors de la première dose d'inébilizumab
Avant d'initier le traitement, les analyses suivantes doivent être réalisées:
·Dosage quantitatif des immunoglobulines sériques, numération des lymphocytes B et numération formule sanguine (NFS), y compris formules leucocytaires (voir rubriques «Contre-indications» et «Mises en garde et précautions»)
·Dépistage du virus de l'hépatite B (VHB) (voir rubriques «Contre-indications» et «Mises en garde et précautions»)
·Dépistage du virus de l'hépatite C (VHC) et prise en charge thérapeutique avant le début du traitement par inébilizumab (voir rubrique «Mises en garde et précautions»)
·Détection d'une tuberculose active et d'une infection latente (voir rubriques «Contre-indications» et «Mises en garde et précautions»)
Toutes les vaccinations doivent être administrées conformément aux recommandations vaccinales en vigueur au moins 4 semaines avant d'initier l'inébilizumab pour les vaccins vivants ou vivants atténués (voir rubrique «Mises en garde et précautions»).
S'il est estimé que la perte d'efficacité est due à l'immunogénicité, le médecin doit suivre la numération des lymphocytes B comme mesure directe de l'impact clinique (voir rubrique «Propriétés/Effets»).
Posologie usuelle
Instauration du traitement
La dose de charge recommandée est une perfusion intraveineuse de 300 mg (3 flacons de 100 mg) suivie d'une deuxième perfusion intraveineuse de 300 mg 2 semaines plus tard.
Traitement d'entretien
La dose d'entretien recommandée est une perfusion intraveineuse de 300 mg tous les 6 mois. L'inébilizumab est utilisé en traitement chronique.
Prémédication pour les réactions liées à la perfusion
Évaluation d'une infection
Avant chaque perfusion d'inébilizumab, il convient de déterminer la présence d'une infection cliniquement significative. En cas d'infection, la perfusion d'inébilizumab doit être retardée jusqu'à ce que l'infection soit résolue.
Prémédication requise
Une prémédication par un corticoïde (par ex., méthylprednisolone 80-125 mg intraveineuse ou équivalent) doit être administrée environ 30 minutes avant chaque perfusion d'inébilizumab, ainsi qu'un antihistaminique (par ex., diphénhydramine 25-50 mg par voie orale ou équivalent) et un antipyrétique (par ex., paracétamol 500-650 mg par voie orale ou équivalent) environ 30-60 minutes avant chaque perfusion d'inébilizumab (voir rubrique «Mises en garde et précautions»).
Afin d'assurer la traçabilité des médicaments biotechnologiques, il convient de documenter pour chaque traitement le nom commercial et le numéro de lot.
Instructions posologiques particulières
Patients présentant des troubles de la fonction hépatique et rénale
L'inébilizumab n'a pas été étudié chez les patients atteints d'insuffisance rénale ou hépatique sévère. Cependant, l'adaptation de la posologie selon la fonction rénale ou hépatique n'est pas justifiée car les anticorps monoclonaux de type immunoglobuline (Ig) G ne sont pas principalement éliminés par voie rénale ou hépatique (voir rubrique «Pharmacocinétique»).
Patients âgés
L'inébilizumab a été administré à 6 patients âgés (≥65 ans) dans les études cliniques. Sur la base des données limitées disponibles, aucune adaptation de la posologie n'est considérée comme nécessaire chez les patients de plus de 65 ans (voir rubrique «Pharmacocinétique»).
Enfants et adolescents
La sécurité et l'efficacité de l'inébilizumab chez les enfants et adolescents âgés de 0 à 18 ans n'ont pas encore été établies. Aucune donnée n'est disponible.
Prise retardée
En cas d'oubli d'une perfusion d'inébilizumab, celle-ci doit être administrée dès que possible; ne pas attendre la dose planifiée suivante.
Mode d'administration
Voie intraveineuse.
Les flacons ne doivent pas être secoués.
Les flacons doivent être conservés en position verticale.
La solution préparée doit être administrée par voie intraveineuse à l'aide d'une pompe à perfusion à un débit croissant jusqu'à la fin (environ 90 minutes) par une ligne intraveineuse contenant un filtre en ligne stérile, à faible liaison protéique, de 0,2 ou 0,22 micron, selon le schéma indiqué au Tableau 1.
Tableau 1. Débit de perfusion recommandé pour l'administration une fois dilué dans une poche intraveineuse de 250 ml
|
Temps écoulé (minutes)
|
Débit de perfusion (ml/heure)
| |
0-30
|
42
| |
31-60
|
125
| |
61 - fin
|
333
|
Pour les instructions concernant la dilution du médicament avant administration, voir la rubrique «Remarques concernant la manipulation».
Contre-indications·Hypersensibilité à la/aux substance(s) active(s) ou à l'un des excipients mentionnés à la rubrique «Composition».
·Infection active sévère, y compris infection chronique active telle que l'hépatite B
·Tuberculose latente active ou non traitée
·Antécédents de leucoencéphalopathie multifocale progressive (LEMP)
·Déficit immunitaire sévère
·Affections malignes évolutives
Mises en garde et précautionsRéactions liées à la perfusion et hypersensibilité
L'inébilizumab peut provoquer des réactions liées à la perfusion et des réactions d'hypersensibilité, pouvant inclure céphalée, nausée, somnolence, dyspnée, fièvre, myalgies, rash ou d'autres symptômes. Les réactions liées à la perfusion ont été plus fréquentes lors de la première perfusion, mais ont été observées pendant les perfusions suivantes. Bien que rares, des réactions à la perfusion graves se sont produites dans des essais cliniques sur l'inébilizumab (voir rubrique «Effets indésirables»).
Avant la perfusion
Une prémédication par un corticoïde (par ex., méthylprednisolone 80-125 mg intraveineuse ou équivalent), un antihistaminique (par ex., diphénhydramine 25-50 mg par voie orale ou équivalent) et un antipyrétique (par ex., paracétamol 500-650 mg par voie orale ou équivalent) doit être administrée (voir rubrique «Posologie/Mode d'emploi»). Une cure de corticoïdes oraux de 2 semaines (plus 1 semaine de diminution progressive) a été administrée au début du traitement par inébilizumab dans l'étude pivot (voir rubrique «Propriétés/Effets»).
Pendant la perfusion
Le patient doit être surveillé pour détecter d'éventuelles réactions liées à la perfusion. Les recommandations de prise en charge des réactions à la perfusion dépendent du type et de la gravité de la réaction. En cas de réactions à la perfusion mettant en jeu le pronostic vital, le traitement doit être arrêté immédiatement et définitivement, et un traitement de soutien approprié doit être administré. Pour les réactions à la perfusion moins graves, la prise en charge peut consister à arrêter temporairement la perfusion, à réduire le débit de perfusion et/ou à administrer un traitement symptomatique.
Après la perfusion
Le patient doit être surveillé pour détecter l'apparition de réactions à la perfusion pendant au moins une heure suivant la fin de la perfusion.
Infections
L'inébilizumab entraîne une réduction de la numération de lymphocytes et des taux d'Ig dans le sang périphérique, ce qui est cohérent avec le mécanisme d'action de déplétion en lymphocytes B. Une réduction des numérations des neutrophiles a également été signalée. Par conséquent, l'inébilizumab peut augmenter la susceptibilité aux infections (voir rubrique «Effets indésirables»).
Une numération formule sanguine récente (datant de moins de 6 mois) incluant les formules leucocytaires et les immunoglobulines doit être obtenue avant l'initiation de l'inébilizumab. Des évaluations de la FSC incluant les formules leucocytaires et les immunoglobulines sont également recommandées périodiquement pendant le traitement et après l'arrêt du traitement jusqu'à la repopulation en lymphocytes B. Avant chaque perfusion d'inébilizumab, il convient de déterminer s'il existe une infection cliniquement significative. En cas d'infection, la perfusion d'inébilizumab doit être retardée jusqu'à ce que l'infection soit résolue. Les patients doivent être informés qu'ils doivent signaler rapidement les symptômes d'infection à leur médecin. L'arrêt du traitement doit être envisagé si un patient développe une infection opportuniste grave ou des infections récidivantes si les taux d'Ig indiquent une immunodépression.
Les infections les plus fréquentes rapportées par les patients atteints de TSNMO traités par l'inébilizumab au cours de la période contrôlée randomisée (PCR) et de la période en ouvert (PEO) comprenaient l'infection des voies urinaires (26,2 %), la rhinopharyngite (20,9 %), l'infection des voies aériennes supérieures (15,6 %), la grippe (8,9 %) et la bronchite (6,7 %).
Réactivation du virus de l'hépatite B (VHB)
Un risque de réactivation du VHB a été observé avec d'autres anticorps induisant une déplétion en lymphocytes B. Les patients présentant une infection chronique au VHB ont été exclus des essais cliniques sur l'inébilizumab. Le dépistage du VHB doit être effectué chez tous les patients avant l'instauration d'un traitement par inébilizumab. L'inébilizumab ne doit pas être administré aux patients ayant une hépatite active à VHB positifs pour l'antigène de surface de l'hépatite B (HBsAg) ou pour l'anticorps de noyau de l'hépatite B (anti-HBc). Les patients porteurs chroniques du VHB [HBsAg+] doivent consulter un spécialiste des maladies du foie avant de commencer et pendant le traitement (voir rubrique «Contre-indications»).
Virus de l'hépatite C (VHC)
Les patients positifs pour le VHC ont été exclus des essais cliniques sur l'inébilizumab. Un dépistage initial du VHC est nécessaire pour détecter le virus et commencer le traitement avant d'initier le traitement par inébilizumab.
Tuberculose
Avant d'initier l'inébilizumab, les patients doivent être évalués pour détecter la tuberculose active et l'infection latente. Pour les patients atteints de tuberculose active ou dont le dépistage de la tuberculose est positif et qui n'ont pas reçu de traitement adéquat, il convient de consulter des experts de la maladie infectieuse avant de commencer le traitement par inébilizumab.
Leucoencéphalopathie multifocale progressive (LEMP)
La LEMP est une infection virale opportuniste du cerveau causée par le virus de John Cunningham (JCV), qui survient généralement chez les patients immunodéprimés et peut entraîner la mort ou une invalidité grave. Une infection au JCV entraînant une LEMP a été observée chez des patients traités par d'autres anticorps induisant une déplétion en lymphocytes B.
Dans les essais cliniques sur l'inébilizumab, un patient est décédé des suites de l'apparition de nouvelles lésions cérébrales pour lesquelles un diagnostic définitif n'a pas pu être établi. Cependant, le diagnostic différentiel incluait une poussée de TSNMO atypique, une LEMP ou une encéphalomyélite disséminée aiguë.
Les médecins doivent être attentifs aux symptômes cliniques ou aux résultats de l'imagerie par résonance magnétique (IRM) qui peuvent évoquer une LEMP. Des résultats d'IRM peuvent déjà être disponibles avant l'apparition des signes ou symptômes cliniques. Les symptômes typiques associés à la LEMP sont multiples, se développent sur plusieurs jours ou semaines, et comprennent une faiblesse progressive d'un côté du corps ou une maladresse des membres, des troubles visuels, ainsi que des troubles de la pensée, de la mémoire et de l'orientation qui entraînent une confusion et des modifications de la personnalité.
Au premier signe ou symptôme évocateur de LEMP, le traitement par inébilizumab doit être suspendu jusqu'à ce que la LEMP soit exclue. Une évaluation plus poussée, comprenant la consultation d'un neurologue, une IRM de préférence avec contraste, une analyse du liquide céphalo-rachidien pour la recherche d'ADN du virus JC et des évaluations neurologiques répétées, doit être envisagée. Le traitement par inébilizumab doit être arrêté en cas de LEMP confirmée.
Neutropénie tardive
Des cas de neutropénie d'apparition tardive ont été rapportés (voir rubrique «Effets indésirables»). Bien que certains cas aient été de grade 3, la majorité des cas était de grade 1 ou 2. Des cas de neutropénie d'apparition tardive ont été rapportés au moins 4 semaines après la dernière perfusion d'inébilizumab. Chez les patients présentant des signes et symptômes d'infection, une mesure des neutrophiles sanguins est recommandée.
Traitement des patients sévèrement immunodéprimés
Les patients sévèrement immunodéprimés ne doivent pas être traités tant que cet état persiste (voir rubrique «Contre-indications»).
L'inébilizumab n'a pas été testé avec d'autres immunosuppresseurs. En cas d'association à un autre traitement immunosuppresseur, tenir compte du risque d'augmentation des effets immunosuppresseurs.
Les patients présentant un déficit immunitaire congénital ou acquis connu, y compris une infection à VIH ou une splénectomie, n'ont pas été étudiés.
Vaccinations
Toutes les vaccinations doivent être administrées conformément aux directives de vaccination au moins 4 semaines avant d'initier l'inébilizumab. L'efficacité et la sécurité de la vaccination avec des vaccins vivants ou vivants atténués après un traitement par inébilizumab n'ont pas été étudiées, et la vaccination avec des vaccins vivants ou vivants atténués n'est pas recommandée pendant le traitement et jusqu'à la repopulation en lymphocytes B.
Les nourrissons de mères exposées à l'inébilizumab pendant la grossesse ne doivent pas recevoir de vaccins vivants ou vivants atténués avant qu'il ne soit confirmé que les taux de lymphocytes B sont revenus à la normale chez le nourrisson. La déplétion en lymphocytes B chez ces nourrissons exposés peut augmenter les risques liés aux vaccins vivants ou vivants atténués. Les vaccins non vivants, s'ils sont indiqués, peuvent être administrés avant le retour à la normale de la déplétion en lymphocytes B et des taux d'Ig, mais il doit être envisagé de consulter un spécialiste qualifié afin de déterminer si une réponse immunitaire protectrice s'est développée.
Délai de repopulation en lymphocytes B
Le délai de repopulation en lymphocytes B après l'administration de l'inébilizumab n'est pas connu. La déplétion en lymphocytes B en dessous de la limite inférieure de la normale a été maintenue chez 94 % des patients pendant au moins 6 mois après le traitement.
Grossesse
Par mesure de précaution, il est préférable d'éviter l'utilisation d'inébilizumab pendant la grossesse et chez les femmes en âge de procréer n'utilisant pas de contraception (voir rubrique «Grossesse, Allaitement»). Les patientes doivent être informées que si elles sont enceintes ou prévoient de le devenir pendant le traitement par inébilizumab, elles doivent en informer leur professionnel de santé. Les femmes en âge de procréer doivent utiliser une méthode de contraception efficace (méthodes entraînant un taux de grossesse inférieur à 1 %) pendant le traitement par Uplizna et pendant 6 mois après la dernière administration d'Uplizna.
Tumeurs malignes
Les médicaments immunomodulateurs peuvent augmenter le risque de tumeurs malignes. Sur la base de l'expérience limitée de l'inébilizumab dans le TSNMO (voir rubrique «Effets indésirables»), les données actuelles ne semblent pas suggérer une augmentation du risque de tumeurs malignes. Cependant, le risque possible de développement de tumeurs solides ne peut être exclu à l'heure actuelle.
Teneur en sodium
Ce médicament contient 48,3 mg de sodium par dose, ce qui équivaut à 2 % de l'apport quotidien maximal recommandé par l'OMS de 2 g de sodium par adulte.
InteractionsAucune étude d'interaction n'a été réalisée.
La principale voie d'élimination des anticorps thérapeutiques est la clairance par le système réticulo-endothélial. Les enzymes du cytochrome P450, les pompes d'efflux et les mécanismes de liaison aux protéines ne sont pas impliqués dans la clairance des anticorps thérapeutiques. Par conséquent, le risque potentiel d'interactions pharmacocinétiques entre l'inébilizumab et d'autres médicaments est faible.
Vaccinations
L'efficacité et la sécurité de la vaccination avec des vaccins vivants ou vivants atténués après un traitement par inébilizumab n'ont pas été étudiées. La réponse à la vaccination pourrait être altérée lors de la déplétion en lymphocytes B. Il est recommandé aux patients de compléter leurs vaccinations avant le début du traitement par inébilizumab (voir rubrique «Mises en garde et précautions»).
Immunosuppresseurs
L'inébilizumab a été testé, et est destiné à être utilisé en monothérapie pour cette indication. Aucune donnée n'est disponible sur la sécurité ou l'efficacité de l'association de l'inébilizumab avec d'autres immunosuppresseurs. Dans l'étude pivot, un traitement de 2 semaines par corticoïdes oraux (plus 1 semaine de diminution progressive) a été administré à tous les sujets après la première administration d'inébilizumab.
L'utilisation concomitante d'inébilizumab et d'immunosuppresseurs, y compris de corticoïdes systémiques, peut augmenter le risque d'infection. Les effets de l'inébilizumab sur les lymphocytes B et les immunoglobulines peuvent persister pendant 6 mois ou plus après son administration.
Lors de l'initiation de l'inébilizumab après d'autres traitements immunosuppresseurs à effets immunitaires prolongés ou lors de l'initiation d'autres traitements immunosuppresseurs à effets immunitaires prolongés après l'inébilizumab, la durée et le mode d'action de ces médicaments doivent être pris en compte en raison des effets immunosuppresseurs additifs potentiels (voir rubrique «Propriétés/Effets»).
Grossesse, allaitementFemmes en âge de procréer
Les femmes en âge de procréer doivent utiliser une méthode de contraception efficace (méthodes entraînant un taux de grossesse inférieur à 1 %) pendant le traitement par Uplizna et pendant 6 mois après la dernière administration d'Uplizna.
Grossesse
Il existe des données limitées sur l'utilisation de l'inébilizumab chez la femme enceinte. L'inébilizumab est un anticorps monoclonal IgG1 humanisé et on sait que les immunoglobulines traversent la barrière placentaire. Une déplétion transitoire en lymphocytes B périphériques et une lymphocytopénie ont été rapportées chez des nourrissons nés de mères exposées à d'autres anticorps induisant une déplétion en lymphocytes B pendant la grossesse.
Les études effectuées chez l'animal n'indiquent pas d'effets délétères directs ou indirects en ce qui concerne la toxicité pour la reproduction; elles ont toutefois montré une déplétion en lymphocytes B dans le foie fœtal de la progéniture (voir rubrique «Données précliniques»).
Le traitement par inébilizumab doit être évité pendant la grossesse, sauf si le bénéfice potentiel pour la mère l'emporte sur le risque potentiel pour le fœtus.
En cas d'exposition durant la grossesse, une déplétion en lymphocytes B peut être attendue chez les nouveau-nés compte tenu des propriétés pharmacologiques du produit et des résultats des études sur les animaux (voir rubrique «Données précliniques»). La durée potentielle de la déplétion en lymphocytes B chez les nourrissons exposés à l'inébilizumab in utero, et l'impact de la déplétion en lymphocytes B sur la sécurité et l'efficacité des vaccins, sont inconnus (voir rubriques «Mises en garde et précautions» et «Propriétés/Effets»). Par conséquent, les nouveau-nés doivent être surveillés pour détecter une déplétion en lymphocytes B et les vaccinations par des vaccins à virus vivants, comme le vaccin Bacillus Calmette-Guérin (BCG), doivent être reportées jusqu'à ce que la numération de lymphocytes B du nourrisson soit revenue à la normale (voir rubrique «Mises en garde et précautions»).
Allaitement
L'utilisation de l'inébilizumab chez la femme pendant l'allaitement n'a pas été étudiée. On ignore si l'inébilizumab est excrété dans le lait maternel. Chez l'humain, les anticorps IgG sont excrétés dans le lait maternel pendant les premiers jours suivant la naissance, mais leur taux diminue rapidement jusqu'à des concentrations faibles.
Par conséquent, un risque pour le nourrisson allaité au sein ne peut être exclu pendant cette courte période. Par la suite, Uplizna peut être utilisé pendant l'allaitement si la situation clinique de la mère justifie ce traitement Cependant, si la patiente a été traitée par Uplizna jusqu'aux derniers mois de la grossesse, l'allaitement peut être commencé immédiatement après la naissance.
Fertilité
Il existe peu de données sur l'effet de l'inébilizumab sur la fertilité chez l'homme; cependant, des études chez l'animal ont montré une réduction de la fertilité. La signification clinique de ces résultats non cliniques n'est pas connue (voir rubrique «Données précliniques»).
Effet sur l’aptitude à la conduite et l’utilisation de machinesL'activité pharmacologique et les effets indésirables rapportés à ce jour suggèrent que l'inébilizumab n'a aucun effet ou un effet négligeable sur l'aptitude à conduire des véhicules et à utiliser des machines.
Effets indésirablesRésumé du profil de sécurité
Les effets indésirables les plus fréquemment rapportés par les patients traités par l'inébilizumab ont été l'infection des voies urinaires (26,2 %), la rhinopharyngite (20,9 %), l'infection des voies aériennes supérieures (15,6 %), l'arthralgie (17,3 %) et la dorsalgie (13,8 %) à la fois sur la PCR et la PEO.
Les effets indésirables graves les plus fréquemment rapportés par les patients traités par l'inébilizumab à la fois sur la PCR et la PEO étaient les infections (11,1 %) (y compris les infections des voies urinaires (4,0 %), la pneumonie (1,8 %) et les TSNMO (1,8 %)).
Liste des effets indésirables
Les effets indésirables rapportés lors de l'essai clinique de l'inébilizumab dans les TSNMO sont répertoriés dans le Tableau 2 selon les catégories de fréquence suivantes: très fréquents (≥1/10), fréquents (≥1/100, < 1/10), peu fréquents (≥1/1'000, < 1/100), rares (≥1/10'000, < 1/1'000), très rares (< 1/10'000), fréquence inconnue (ne peut être estimée sur la base des données disponibles).
Tableau 2. Effets indésirables
|
Classe de système d'organe MedDRA
|
Effet indésirable
|
Fréquence
| |
Infections et infestations
|
Infection des voies urinaires (26,2 %),
infection de l'appareil respiratoire (15,6 %), rhinopharingite (20,9 %)
|
Très fréquents
| |
|
Pneumonie, grippe, cellulite, zona, sinusite
|
Fréquents
| |
|
Sepsis,
abcès sous-cutané, bronchiolite
|
Peu fréquents
| |
Affections hématologiques et du système lymphatique
|
Lymphopénie,
Neutropénie,
Neutropénie d'apparition tardive
|
Fréquents
| |
Affections musculosquelettiques et systémiques
|
Arthralgie (17,3 %), dorsalgie (13,8 %)
|
Très fréquents
| |
Investigations
|
Immunoglobulines diminuées (3,8 % - 29,3 %)
|
Très fréquents
| |
Lésions, intoxications et complications liées aux procédures
|
Réaction liée à la perfusion (12,9 %)
|
Très fréquents
|
Description d'effets indésirables spécifiques et informations complémentaires
Réactions liées à la perfusion
L'inébilizumab peut provoquer des réactions liées à la perfusion pouvant inclure céphalées, nausées, somnolence, dyspnée, fièvre, myalgie, rash ou d'autres symptômes. Tous les patients avaient reçu une prémédication. Des réactions à la perfusion ont été observées chez 9,2 % des patients atteints de TSNMO au cours de la première cure d'inébilizumab, contre 10,7 % des patients sous placebo. Les réactions liées à la perfusion étaient plus fréquentes lors de la première perfusion, mais ont été observées lors des perfusions suivantes. La majorité des réactions liées à la perfusion rapportées chez les patients traités par l'inébilizumab étaient de sévérité légère ou modérée.
Infections
Une infection a été signalée par 74,7 % des patients atteints de TSNMO traités par inébilizumab dans l'ensemble de la PCR et de la PEO. Les infections les plus fréquentes étaient les infections des voies urinaires (26,2 %), les rhinopharyngites (20,9 %) et les infections des voies aériennes supérieures (15,6 %), la grippe (8,9 %) et la bronchite (6,7 %). Les infections graves rapportées par plus d'un patient traité par l'inébilizumab ont été l'infection des voies urinaires (4,0 %) et la pneumonie (1,8 %). Voir la rubrique «Mises en garde et précautions» pour les mesures à prendre en cas d'infection.
Infections opportunistes et graves
Au cours de la PCR, aucune infection opportuniste n'est survenue dans l'un ou l'autre des groupes de traitement, et un seul effet indésirable infectieux de grade 4 (pneumonie atypique) est survenu chez un patient traité par l'inébilizumab. Au cours de la PEO, 2 patients traités par inébilizumab (0,9 %) ont présenté une infection opportuniste (dont une n'a pas été confirmée) et 3 patients traités par inébilizumab (1,4 %) ont présenté un effet indésirable infectieux de grade 4. Voir la rubrique «Mises en garde et précautions» pour les mesures à prendre en cas d'infection.
Anomalies biologiques
Immunoglobulines diminuées
En cohérence avec son mécanisme d'action, les taux moyens d'immunoglobulines ont diminué avec l'utilisation de l'inébilizumab. À la fin de la PCR de 6,5 mois, les proportions de patients ayant des taux inférieurs à la limite inférieure de la normale étaient les suivantes: IgA 9,8 % inébilizumab et 3,1 % placebo, IgE 10,6 % inébilizumab et 12,5 % placebo, IgG 3,8 % inébilizumab et 9,4 % placebo, et IgM 29,3 % inébilizumab et 15,6 % placebo. Un effet indésirable isolé d'IgG diminuées a été rapporté (grade 2, au cours de la PEO). La proportion de patients traités par inébilizumab présentant des taux d'IgG inférieurs à la limite inférieure de la normale à la première année était de 7,4 % et à la deuxième année de 9,9 %. Avec une exposition médiane de 3,2 ans, la fréquence de la réduction modérée des IgG (300 à < 500 mg/dl) était de 14,2 % et la fréquence de la réduction sévère des IgG (< 300 mg/dl) était de 3,6 %.
Numération des neutrophiles diminuée
Après 6,5 mois de traitement, des numérations de neutrophiles comprises entre 1,0-1,5 x 109/l (grade 2) ont été observées chez 7,5 % des patients traités par inébilizumab contre 1,8 % des patients traités par placebo. Des numérations des neutrophiles comprises entre 0,5-1,0 x 109/l (grade 3) ont été observées chez 1,7 % des patients traités par inébilizumab contre 0 % des patients traités par placebo. La neutropénie était généralement transitoire et n'était pas associée à des infections graves.
Numération de lymphocytes diminuée
Après 6,5 mois de traitement, une diminution des numérations de lymphocytes a été observée plus fréquemment chez les patients traités par inébilizumab que par placebo: des numérations de lymphocytes comprises entre 500 et < 800/mm3 (grade 2) ont été observées chez 21,4 % des patients traités par inébilizumab contre 12,5 % des patients traités par placebo. Des numérations de lymphocytes comprises entre 200 et < 500/mm3 (grade 3) ont été observées chez 2,9 % des patients traités par inébilizumab contre 1,8 % des patients traités par placebo. Cette observation est cohérente avec le mécanisme d'action de déplétion en lymphocytes B puisque ces derniers constituent un sous-ensemble de la population lymphocytaire.
L'annonce d'effets secondaires présumés après l'autorisation est d'une grande importance. Elle permet un suivi continu du rapport bénéfice-risque du médicament. Les professionnels de santé sont tenus de déclarer toute suspicion d'effet secondaire nouveau ou grave via le portail d'annonce en ligne ElViS (Electronic Vigilance System). Vous trouverez des informations à ce sujet sur www.swissmedic.ch.
SurdosageLa dose d'inébilizumab la plus élevée testée chez les patients atteints de maladies auto-immunes était de 1200 mg, administrée sous forme de deux perfusions intraveineuses de 600 mg à 2 semaines d'intervalle. Les effets indésirables étaient similaires à ceux observés dans l'étude clinique pivot sur l'inébilizumab.
Il n'existe pas d'antidote spécifique en cas de surdosage; la perfusion doit être immédiatement interrompue et le patient doit être mis sous observation pour détecter d'éventuelles réactions liées à la perfusion (voir rubrique «Mises en garde et précautions»). Le patient doit être étroitement surveillé pour déceler tout signe ou symptôme d'effets indésirables et des soins de soutien doivent être instaurés si nécessaire.
Propriétés/EffetsCode ATC
L04AA47
Mécanisme d'action
L'inébilizumab est un anticorps monoclonal qui se lie spécifiquement au CD19, l'antigène de surface cellulaire présent sur les lymphocytes pré-B et B matures, y compris les plasmablastes et certains plasmocytes. Après la liaison de la surface cellulaire aux lymphocytes B, l'inébilizumab favorise la cytotoxicité cellulaire dépendante des anticorps (ADCC) et la phagocytose dépendante des anticorps (ADCP). Les lymphocytes B joueraient un rôle central dans la pathogenèse des TSNMO. Le mécanisme précis par lequel l'inébilizumab exerce ses effets thérapeutiques dans les TSNMO est inconnu, mais on suppose qu'il implique une déplétion en lymphocytes B et peut inclure la suppression de la sécrétion d'anticorps, de la présentation de l'antigène, de l'interaction entre les lymphocytes B et T et de la production de médiateurs inflammatoires.
Pharmacodynamique
La pharmacodynamie de l'inébilizumab a été évaluée à l'aide d'un dosage des lymphocytes B CD20+, car l'inébilizumab peut interférer avec le dosage des lymphocytes B CD19+. Le traitement par inébilizumab diminue le nombre de lymphocytes B CD20+ dans le sang 8 jours après la perfusion. Dans une étude clinique portant sur 174 patients, le nombre de lymphocytes B CD20+ est passé en dessous de la limite inférieure de la normale après 4 semaines chez 100 % des patients traités par inébilizumab et est resté en dessous de la limite inférieure de la normale chez 94 % des patients pendant 28 semaines après le début du traitement. Le délai de réplétion en lymphocytes B après l'administration d'inébilizumab n'est pas connu.
Dans l'étude pivot de patients atteints de TSNMO, la prévalence des anticorps anti-médicaments (ADA) était de 14,7 % à la fin de la PEO; l'incidence globale des ADA apparus au cours du traitement était de 7,1 % (16 sur 225) et la fréquence et le titre des ADA positifs diminuaient au fil du temps avec le traitement par inébilizumab. Le statut ADA-positif n'a pas semblé avoir d'impact cliniquement pertinent sur les paramètres pharmacocinétiques et pharmacodynamiques (lymphocytes B) et n'a pas eu d'impact sur le profil de sécurité à long terme. Il n'y a pas eu d'effet apparent du statut ADA sur le résultat d'efficacité; cependant, l'impact ne peut être pleinement évalué étant donné la faible incidence d'ADA associée au traitement par inébilizumab.
Efficacité clinique
L'efficacité de l'inébilizumab pour le traitement des TSNMO a été étudiée dans un essai clinique randomisé (3:1), en double aveugle, contrôlé par placebo, chez des adultes atteints de TSNMO séropositifs ou séronégatifs pour l'AQP4-IgG. L'étude incluait des patients ayant subi au moins une poussée aiguë de TSNMO au cours de l'année précédente ou au moins 2 poussées au cours des 2 années précédentes ayant nécessité un traitement de secours (par exemple, stéroïdes, échange plasmatique, immunoglobuline intraveineuse), et ayant un score EDSS (Expanded Disability Severity Scale) ≤7,5 (les patients ayant un score de 8,0 étaient éligibles si le patient était raisonnablement capable de participer). Les patients étaient exclus s'ils avaient déjà été traités par des traitements immunosuppresseurs dans un intervalle spécifié pour chacune de ces thérapies. Les traitements immunosuppresseurs de fond pour la prévention des poussées de TSNMO n'étaient pas autorisés. Une cure de corticoïdes oraux de 2 semaines (plus 1 semaine de diminution progressive) a été administrée au début du traitement par inébilizumab dans l'étude pivot.
Les patients ont été traités par des perfusions intraveineuses d'inébilizumab 300 mg le jour 1 et le jour 15, ou par un placebo correspondant, puis ont fait l'objet d'un suivi pendant une période allant jusqu'à 197 jours ou jusqu'à la première poussée confirmée par un comité d'adjudication, appelée période randomisée et contrôlée (PRC). Toutes les poussées potentielles étaient évaluées par un comité d'adjudication (CA) indépendant et soumis à l'aveugle, qui a déterminé si la poussée répondait aux critères définis par le protocole. Les critères de poussée prenaient en compte les poussées dans tous les domaines affectés par les TSNMO (névrite optique, myélite, encéphale et tronc cérébral) et comprenaient des critères basés exclusivement sur des manifestations cliniques substantielles, ainsi que des critères qui majoraient des résultats cliniques plus modestes avec l'utilisation de l'IRM (voir Tableau 3).
Tableau 3. Présentation des critères définis par le protocole pour une poussée de TSNMO
|
Domaine
|
Symptômes représentatifs
|
Résultats cliniques uniquement
|
Résultats cliniques PLUS radiologiques
| |
Nerf optique
|
Vision trouble
Perte de la vue
Douleur oculaire
|
8 critères basés sur les modifications de l'acuité visuelle ou de la malformation pupillaire relative afférente (MPRA)
|
3 critères basés sur les modifications de l'acuité visuelle ou de la MPRA plus présence de résultats d'IRM du nerf optique correspondants
| |
Moelle épinière
|
Douleur profonde ou radiculaire
Paresthésie des extrémités
Faiblesse
Dysfonctionnement du sphincter
Signe de Lhermitte (non isolé)
|
2 critères basés sur les modifications des scores fonctionnels pyramidal, vésical/intestinal ou sensitif
|
2 critères basés sur les modifications des scores fonctionnels pyramidal, vésical/intestinal ou sensitif PLUS résultats d'IRM de la moelle épinière correspondants
| |
Tronc cérébral
|
Nausée
Vomissements réfractaires
Hoquet réfractaire
Autres signes neurologiques (p. ex., vision double, dysarthrie, dysphagie, vertige, paralysie oculomotrice, faiblesse, nystagmus, autre anomalie des nerfs crâniens)
|
Aucun
|
2 critères basés sur les symptômes ou les modifications des scores fonctionnels du tronc cérébral/cérébelleux PLUS résultats d'IRM du tronc cérébral correspondants
| |
Cerveau
|
Encéphalopathie
Dysfonctionnement hypothalamique
|
Aucun
|
1 critère basé sur les modifications des scores fonctionnels cérébral/sensitif/pyramidal PLUS résultats d'IRM cérébrale correspondants
|
Les patients qui ont subi une poussée confirmée par le CA pendant la PCR, ou qui ont terminé la visite du jour 197 sans poussée, ont quitté la PCR et ont eu la possibilité d'intégrer une PEO et d'initier ou de poursuivre le traitement par inébilizumab.
Au total, 230 patients ont été recrutés: 213 patients séropositifs pour l'AQP4-IgG et 17 séronégatifs ont été recrutés; 174 patients ont été traités par inébilizumab et 56 patients ont été traités par placebo durant la PCR de l'étude. Sur les 213 patients séropositifs pour l'AQP4-IgG, 161 ont été traités par l'inébilizumab et 52 ont été traités par placebo pendant la PCR de l'étude. Les caractéristiques à la baseline et les résultats d'efficacité sont présentés pour les patients séropositifs pour l'AQP4-IgG.
Les données démographiques et les caractéristiques de la maladie à l'inclusion étaient équilibrées entre les 2 groupes de traitement (voir Tableau 4).
Tableau 4. Données démographiques et caractéristiques à l'inclusion des patients atteints de TSNMO séropositifs pour l'AQP4-IgG
|
Caractéristique
|
Placebo
N = 52
|
Inébilizumab
N = 161
|
Globalement
N = 213
| |
Âge (années): moyenne (écart type [ET])
|
42,4 (14,3)
|
43,2 (11,6)
|
43,0 (12,3)
| |
Âge ≥65 ans, n (%)
|
4 (7,7)
|
6 (3,7)
|
10 (4,7)
| |
Sexe: Masculin, n (%)
|
3 (5,8)
|
10 (6,2)
|
13 (6,1)
| |
Sexe: Féminin, n (%)
|
49 (94,2)
|
151 (93,8)
|
200 (93,9)
| |
Échelle EDSS (Expanded Disability Status Scale): moyenne (ET)
|
4,35 (1,63)
|
3,81 (1,77)
|
3,94 (1,75)
| |
Durée de la maladie (années): moyenne (ET)
|
2,92 (3,54)
|
2,49 (3,39)
|
2,59 (3,42)
| |
Nombre de rechutes antérieures: ≥2, n (%)
|
39 (75,0)
|
137 (85,1)
|
176 (82,6)
| |
Taux de rechute annualisé: moyenne (ET)
|
1,456 (1,360)
|
1,682 (1,490)
|
1,627 (1,459)
|
Un traitement de secours était instauré si nécessaire en cas de poussées de TSNMO. Tous les patients ont reçu une prémédication avant l'administration du produit expérimental afin de réduire le risque de réactions liées à la perfusion.
Le critère principal d'efficacité était le délai (en jours) de la première poussée de TSNMO confirmée par le CA jusqu'au jour 197 ou avant. Les autres critères d'évaluation secondaires clés comprenaient l'aggravation de l'EDSS par rapport aux valeurs initiales lors de la dernière visite pendant la PCR, le changement par rapport aux valeurs initiales du score binoculaire d'acuité visuelle à faible contraste mesuré par l'échelle des anneaux brisés de Landolt à faible contraste lors de la dernière visite pendant la PCR, le nombre total cumulé de lésions actives à l'IRM (nouvelles lésions rehaussées par le gadolinium ou lésions T2 nouvelles/élargies) pendant la PCR, et le nombre d'hospitalisations liées aux TSNMO. Un patient était considéré comme ayant une aggravation du score EDSS si l'un des critères suivants était rempli: (1) aggravation de 2 points ou plus du score EDSS pour les patients dont le score initial était de 0; (2) aggravation de 1 point ou plus du score EDSS pour les patients dont le score initial était de 1 à 5; (3) aggravation de 0,5 point ou plus du score EDSS pour les patients dont le score initial était de 5,5 ou plus. Bien qu'aucun comparateur n'ait été disponible pendant la PEO, le taux de poussées annualisé a été évalué pendant la période randomisée et celle en ouvert.
Les résultats chez les patients séropositifs pour l'AQP4-IgG sont présentés dans le Tableau 5 et la Figure 1. Dans cette étude, le traitement par inébilizumab a diminué de manière statistiquement significative le risque de poussée de TSNMO confirmée par le CA comparé au placebo (hazard ratio: 0,227, p < 0,0001; réduction de 77,3 % du risque de poussée de TSNMO déterminé par le CA) chez les patients séropositifs pour l'AQP4-IgG. Aucun bénéfice thérapeutique n'a été observé chez les patients séronégatifs pour l'AQP4-IgG.
Dans le groupe inébilizumab, l'aggravation de l'EDSS a été significativement moindre par rapport au groupe placebo (14,9 % versus 34,6 % des patients). Il n'y a pas eu de différence dans le score binoculaire de l'acuité visuelle à faible contraste entre les deux groupes de l'étude. Le nombre cumulé moyen de lésions actives totales à l'IRM (1,7 contre 2,3) et le nombre cumulé moyen d'hospitalisations liées aux TSNMO (1,0 contre 1,4) ont été réduits dans le groupe d'étude inébilizumab.
Tableau 5. Résultats d'efficacité dans l'étude pivot sur les TSNMO séropositifs pour l'AQP4-IgG
|
|
Groupe de traitement
| |
|
Placebo
N = 52
|
Inébilizumab
N = 161
| |
Délai avant la poussée confirmée par le comité d'adjudication (critère d'efficacité principal)
| |
Nombre (%) de patients ayant subi des poussées
|
22 (42,3 %)
|
18 (11,2 %)
| |
Hazard ratio (IC à 95 %)a
|
0,227 (0,1214; 0,4232)
| |
Valeur pa
|
< 0,0001
|
a Méthode de régression de Cox, avec placebo comme groupe de référence.
Figure 1. Courbe de Kaplan-Meier du délai avant la première poussée de TSNMO confirmée par le CA au cours de la PCR chez les patients séropositifs pour l'AQP4-IgG
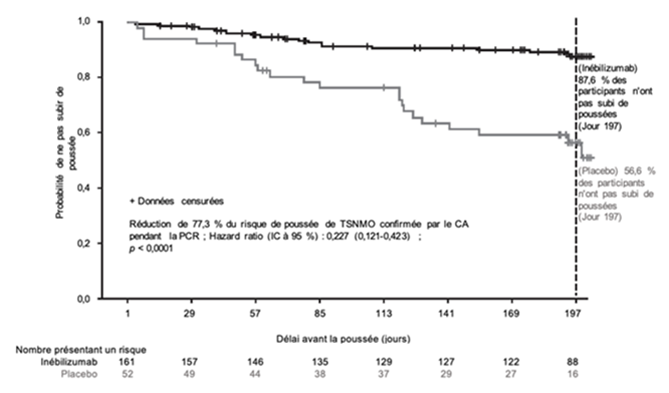
CA comité d'adjudication; AQP4-IgG immunoglobuline G anti-aquaporine-4; IC intervalle de confiance; TSNMO trouble du spectre de neuromyélite optique; PCR période contrôlée randomisée.
Sur les PCR et PEO, le taux annualisé de poussées de TSNMO déterminées par le CA a été analysé à titre de critère d'évaluation secondaire et chez les patients séropositifs pour l'AQP4-IgG traités par l'inébilizumab, le résultat était de 0,09.
PharmacocinétiqueAbsorption
L'inébilizumab est administré sous forme de perfusion intraveineuse.
Distribution
D'après l'analyse pharmacocinétique de population, le volume de distribution central et périphérique typique estimé de l'inébilizumab était respectivement de 2,95 l et 2,57 l.
Métabolisme
L'inébilizumab est un anticorps monoclonal IgG1 humanisé qui est dégradé par des enzymes protéolytiques réparties dans tout l'organisme.
Élimination
Chez les patients adultes atteints de TSNMO, la demi-vie d'élimination terminale a été d'environ 18 jours. D'après l'analyse pharmacocinétique de population, la clairance systémique estimée de l'inébilizumab de la voie d'élimination de premier ordre était de 0,19 l/jour. À de faibles niveaux d'exposition pharmacocinétique, l'inébilizumab était probablement soumis à une clairance médiée par le récepteur (CD19), qui diminuait avec le temps, probablement en raison de la déplétion en lymphocytes B induite par le traitement par inébilizumab.
Cinétique pour certains groupes de patients
Troubles de la fonction hépatique
Aucune étude clinique formelle n'a été réalisée pour étudier l'effet de l'insuffisance hépatique sur l'inébilizumab. Dans les études cliniques, aucun sujet présentant une insuffisance hépatique sévère n'a été exposé à l'inébilizumab. Les anticorps monoclonaux IgG ne sont pas principalement éliminés par la voie hépatique; une modification de la fonction hépatique ne devrait donc pas influencer la clairance de l'inébilizumab. D'après l'analyse pharmacocinétique de population, les biomarqueurs de la fonction hépatique initiaux (AST, ALP et bilirubine) n'avaient aucun effet cliniquement significatif sur la clairance de l'inébilizumab.
Troubles de la fonction rénale
Aucune étude clinique formelle n'a été réalisée pour étudier l'effet de l'insuffisance rénale sur l'inébilizumab. En raison du poids moléculaire important et de la taille hydrodynamique d'un anticorps monoclonal IgG, l'inébilizumab ne devrait pas être filtré par le glomérule. D'après une analyse pharmacocinétique de population, la clairance de l'inébilizumab chez les patients présentant divers degrés d'insuffisance rénale était comparable à celle des patients présentant un débit de filtration glomérulaire estimé normal.
Patients âgés
L'analyse pharmacocinétique de population n'a indiqué aucune incidence de l'âge sur la clairance de l'inébilizumab.
Enfants et adolescents
L'inébilizumab n'a pas été étudié chez les adolescents ou les enfants.
Sexe, race
Une analyse pharmacocinétique de population a indiqué que le sexe et la race n'avaient aucun effet significatif sur la clairance de l'inébilizumab.
Données précliniquesLes données précliniques issues des études conventionnelles de pharmacologie de sécurité, toxicologie en administration répétée, génotoxicité et potentiel carcinogène, n'ont pas révélé de risque particulier pour l'homme.
L'inébilizumab a été évalué dans une étude combinée de fertilité et de développement embryo-fœtal chez des souris huCD19 Tg femelles et mâles à des doses intraveineuses de 3 mg/kg et 30 mg/kg. Il n'y a pas eu d'effet sur le développement embryo-fœtal, cependant, une réduction liée au traitement de l'indice de fertilité a été observée aux deux doses testées. La pertinence de ce résultat pour l'homme n'est pas connue. De plus, une diminution des populations de lymphocytes B au site de développement des lymphocytes B a été observée chez les souris fœtales nées d'animaux traités par l'inébilizumab par rapport à la progéniture des animaux témoins, ce qui suggère que l'inébilizumab traverse le placenta et induit la déplétion en lymphocytes B.
Seuls quelques échantillons toxicocinétiques ont été recueillis dans l'étude combinée sur la fertilité et le développement embryo-fœtal; sur la base de la concentration maximale de la première dose (Cmax), les multiples d'exposition de 3 mg/kg et 30 mg/kg chez les souris femelles huCD19 Tg étaient respectivement 0,4 fois et 4 fois plus élevés pour la dose thérapeutique clinique de 300 mg.
Dans une étude sur le développement pré/postnatal menée chez des souris transgéniques, l'administration d'inébilizumab à la mère du jour 6 de la gestation au jour 20 de la lactation a entraîné une diminution des populations de lymphocytes B chez la progéniture au jour 50 postnatal. Les populations de lymphocytes B de la progéniture sont revenues à la normale au jour 357 postnatal. La réponse immunitaire au néoantigène de la progéniture des animaux traités par l'inébilizumab était diminuée par rapport à la progéniture des animaux témoins, ce qui suggère une altération de la fonction normale des lymphocytes B.
Remarques particulièresIncompatibilités
En l'absence d'études de compatibilité, ce médicament ne doit pas être mélangé à d'autres médicaments.
Stabilité
Le médicament ne doit pas être utilisé au-delà de la date figurant après la mention «EXP» sur l'emballage.
Stabilité après dilution
La solution pour perfusion préparée doit être administrée immédiatement. Si elle n'est pas administrée immédiatement, conserver jusqu'à 24 heures au réfrigérateur entre 2 °C et 8 °C ou 4 heures à température ambiante avant le début de la perfusion.
Remarques particulières concernant le stockage
Conserver au réfrigérateur (2-8°C).
Ne pas congeler.
Conserver le récipient dans son carton pour le protéger de la lumière.
Pour les conditions de conservation du médicament après dilution, voir la rubrique «Stabilité après dilution».
Conserver hors de portée des enfants.
Remarques concernant la manipulation
Préparation de la solution pour perfusion
Avant le début de la perfusion intraveineuse, la solution pour perfusion préparée doit être à température ambiante entre 20 °C et 25 °C.
La solution à diluer doit être inspectée visuellement pour vérifier l'absence de particules et de décoloration. Le flacon doit être jeté si la solution est trouble, décolorée ou si elle contient des particules étrangères discrètes.
·Le flacon ne doit pas être secoué.
·Le flacon doit être conservé en position verticale.
·Se procurer une poche intraveineuse contenant 250 ml de solution injectable de chlorure de sodium 9 mg/ml (0,9 %). Ne pas utiliser d'autres solvants pour diluer l'inébilizumab car leur utilisation n'a pas été testée.
·Prélever 10 ml d'Uplizna dans chacun des 3 flacons contenus dans la boîte et transférer un total de 30 ml dans la poche intraveineuse de 250 ml. Mélanger la solution diluée en retournant délicatement le flacon. Ne pas secouer la solution.
Élimination
Tout médicament non utilisé ou déchet doit être éliminé conformément à la réglementation en vigueur.
Numéro d’autorisation69322 (Swissmedic)
Présentation10 ml de solution à diluer dans un flacon en verre de type 1 avec un bouchon en élastomère et un opercule en aluminium gris brumeux amovible.
Présentation de 3 flacons. (A)
Titulaire de l’autorisationAmgen Switzerland AG, Risch; Domicile: 6343 Rotkreuz
Mise à jour de l’informationOctobre 2023
|