CompositionPrincipes actifs
Éplontersen (un oligonucléotide antisens de synthèse chimiquement modifié)
Excipients
Phosphate monosodique dihydraté (corresp. à max. 0,1 mg de sodium par injection)
Phosphate disodique (anhydre) (corresp. à 0,3 mg de sodium par injection)
Chlorure de sodium (corresp. à 1,9 mg de sodium par injection)
Acide chlorhydrique (pour l'ajustement du pH) (corresp. à max. 0,01 mg de sodium par injection)
Hydroxyde de sodium (pour l'ajustement du pH)
Eau pour préparations injectables ad solutionem pro 0,8 ml
Indications/Possibilités d’emploiWAINZUA est utilisé dans le traitement de la polyneuropathie de stade 1 et 2 associée à une amyloïdose héréditaire à transthyrétine (ATTRv) chez les patients adultes.
Posologie/Mode d’emploiLe traitement doit être prescrit et supervisé par un médecin expérimenté dans le traitement de patients atteints d'amyloïdose.
Posologie
La dose recommandée de WAINZUA est de 45 mg administrés par injection sous-cutanée. Les doses doivent être administrées une fois par mois.
La décision de poursuivre le traitement chez les patients dont la maladie évolue en polyneuropathie de stade 3 doit être prise à la discrétion du médecin sur la base de l'évaluation globale des bénéfices et des risques.
Groupes de patients particuliers
Patients présentant des troubles de la fonction rénale
Aucun ajustement posologique n'est nécessaire chez les patients présentant des troubles légers à modérés de la fonction rénale (débit de filtration glomérulaire estimé [DFGe] ≥45 à < 90 ml/min/1,73 m2) (voir «Pharmacocinétique»). WAINZUA n'a pas été étudié chez les patients présentant un DFGe < 45 ml/min/1,73 m2 ou atteints d'insuffisance rénale terminale et doit être utilisé chez ces patients uniquement si le bénéfice clinique attendu est supérieur au risque potentiel (voir rubrique «Pharmacocinétique»).
Patients présentant des troubles de la fonction hépatique
Aucun ajustement posologique n'est nécessaire chez les patients présentant des troubles légers de la fonction hépatique (voir «Pharmacocinétique»). WAINZUA n'a pas été étudié chez les patients présentant des troubles modérés ou sévères de la fonction hépatique et doit être utilisé chez ces patients uniquement si le bénéfice clinique attendu est supérieur au risque potentiel (voir rubrique «Pharmacocinétique»).
Patients âgés
Aucun ajustement de la posologie n'est nécessaire chez les patients âgés (≥65 ans) (voir «Pharmacocinétique»).
Enfants et adolescents
La sécurité et l'efficacité de WAINZUA pour les enfants et les adolescents âgés de moins de 18 ans ne sont pas établies. On ne dispose d'aucune donnée.
Oubli d'une dose
Si une dose d'éplontersen a été oubliée, la dose suivante doit être administrée dès que possible. L'administration doit être poursuivie à intervalles mensuels à compter de la date de la dernière dose.
Mode d'administration
Administration par voie sous-cutanée uniquement.
La première injection réalisée par le patient ou l'aidant doit être effectuée sous la surveillance d'un professionnel de la santé qualifié. Les patients et/ou les aidants doivent être formés à l'administration sous-cutanée de WAINZUA.
Le stylo prérempli doit être retiré du réfrigérateur au moins 30 minutes avant son utilisation et doit atteindre la température ambiante avant l'injection. Il ne faut pas utiliser d'autres méthodes pour le réchauffer.
Examiner visuellement la solution avant l'emploi. La solution doit être incolore à jaune. Ne pas utiliser si une solution trouble, des particules ou un changement de couleur sont observés avant l'administration.
En cas d'auto-administration, injecter WAINZUA dans l'abdomen ou dans la cuisse. L'arrière du bras peut également être utilisé si l'injection est effectuée par un aidant.
Des instructions complètes concernant l'administration figurent dans le «Mode d'emploi».
Contre-indicationsHypersensibilité au principe actif ou à l'un des excipients selon la composition.
Mises en garde et précautionsBaisse des taux sériques de vitamine A et supplémentation recommandée
En raison de son mécanisme d'action, WAINZUA devrait réduire le taux de vitamine A sérique (rétinol) à un niveau inférieur à la normale (voir «Propriétés/Effets»).
Il faut rechercher la cause de tout symptôme ou signe de carence en vitamine A avant d'instaurer le traitement par WAINZUA.
Les patients qui reçoivent un traitement par WAINZUA doivent prendre un supplément oral à la dose quotidienne recommandée de vitamine A afin de réduire le risque de symptômes oculaires dus à une carence en vitamine A. L'orientation vers un spécialiste en ophtalmologie est recommandée si le patient développe des symptômes oculaires compatibles avec une carence en vitamine A, y compris une vision nocturne réduite ou une cécité nocturne et une sécheresse oculaire persistante.
On ignore si la supplémentation en vitamine A pendant la grossesse est suffisante pour prévenir une carence en vitamine A lorsque les femmes enceintes continuent d'être traitées par WAINZUA (voir «Grossesse/Allaitement»). En raison du mécanisme d'action de l'éplontersen, une augmentation de l'apport en vitamine A au-delà de la dose quotidienne recommandée pendant la grossesse ne corrigerait toutefois probablement pas les taux sériques de rétinol et pourrait être nocive pour la mère et l'enfant à naître.
Excipients revêtant un intérêt particulier
Sodium
Ce médicament contient moins de 1 mmol (23 mg) de sodium par dose, c.-à-d. qu'il est essentiellement «sans sodium».
InteractionsAucune étude clinique formelle en vue d'évaluer les interactions médicamenteuses n'a été réalisée (voir «Pharmacocinétique»).
Grossesse, allaitementFemmes en âge de procréer/Contraception chez la femme
WAINZUA réduit les taux plasmatiques de vitamine A, qui joue un rôle essentiel dans le développement normal du fœtus. On ignore si une supplémentation en vitamine A est suffisante pour réduire le risque pour le fœtus. Une grossesse doit donc être exclue avant d'instaurer le traitement par WAINZUA et les femmes en mesure de procréer doivent utiliser une méthode de contraception efficace.
Si une femme planifie une grossesse, la prise de WAINZUA et de suppléments de vitamine A doit être interrompue, et les taux sériques de vitamine A doivent être surveillés et être revenus à la normale avant de tenter de concevoir. En raison de la longue demi-vie de l'éplontersen (voir «Pharmacocinétique»), une carence en vitamine A peut survenir même après l'arrêt du traitement.
Les femmes en mesure de procréer doivent utiliser une méthode de contraception efficace.
Grossesse
À ce jour, il n'existe pas de données concernant l'emploi de WAINZUA chez la femme enceinte.
L'administration de l'éplontersen ou d'un substitut pharmacologiquement actif spécifique aux rongeurs à des doses jusqu'à 38 fois supérieures à la dose recommandée chez l'humain dans le cadre d'une étude combinée de toxicité sur la fertilité et sur le développement embryofœtal chez la souris n'a révélé aucun effet sur la fertilité des mâles et des femelles ni sur le développement embryofœtal (voir «Données précliniques»).
En raison du risque tératogène potentiel découlant du déséquilibre des taux de vitamine A, WAINZUA ne doit pas être utilisé pendant la grossesse. En cas de grossesse non planifiée, le fœtus et le taux de vitamine A doivent faire l'objet d'une surveillance étroite, en particulier au cours du premier trimestre.
Allaitement
Aucune étude sur la lactation n'a été menée chez l'humain ou l'animal pour évaluer la présence d'éplontersen ou de ses métabolites dans le lait maternel et ses effets sur le nourrisson allaité ou sur la production de lait chez la mère. Un risque pour le nourrisson allaité ne peut pas être exclu. Il faut décider soit d'interrompre l'allaitement, soit d'exclure ou d'arrêter le traitement par WAINZUA, en prenant en compte le bénéfice de l'allaitement pour l'enfant au regard du bénéfice du traitement pour la mère.
Fertilité
Il n'existe pas de données sur les effets de l'éplontersen sur la fertilité humaine.
L'administration de l'éplontersen ou d'un substitut pharmacologiquement actif spécifique aux rongeurs chez la souris à des doses jusqu'à 38 fois supérieures à la dose recommandée chez l'humain n'a révélé aucun effet de l'éplontersen sur la fertilité des mâles ou des femelles.
Effet sur l’aptitude à la conduite et l’utilisation de machinesWAINZUA n'a aucune influence ou a une influence négligeable sur l'aptitude à la conduite ou l'utilisation de machines.
Effets indésirablesRésumé du profil de sécurité
Les données de sécurité décrites ci-dessous proviennent de l'exposition à WAINZUA de 144 patients atteints de polyneuropathie provoquée par ATTRv (ATTRv-PN) qui ont été randomisés pour être traités par WAINZUA et ont reçu au moins une dose de WAINZUA. Parmi ces patients, 141 ont reçu le traitement pendant au moins 6 mois et 137 l'ont reçu pendant au moins 12 mois. La durée moyenne du traitement a été de 541 jours (fourchette: 57 à 582 jours).
Les effets secondaires les plus fréquemment observés lors du traitement par WAINZUA chez ≥5 % des patients étaient les vomissements et la baisse du taux sérique de vitamine A.
Liste des effets indésirables
Les effets indésirables sont rangés par classe de système d'organes (SOC) de la classification MedDRA. Au sein de chacune des SOC, les termes préférentiels sont indiqués par ordre décroissant de fréquence et dans chaque groupe de fréquence, par ordre décroissant de sévérité. Les fréquences de survenue des effets indésirables sont définies de la manière suivante: très fréquent (≥1/10), fréquent (≥1/100 à < 1/10), occasionnel (≥1/1000 à < 1/100), rare (≥1/10 000 à < 1/1000), très rare (< 1/10 000) et fréquence inconnue (ne peut être estimée sur la base des données disponibles).
Tableau 1: Résumé des effets secondaires par catégorie de fréquence
|
Classe de système d'organes
|
Effet secondaire
|
Fréquence
| |
Affections gastro-intestinales
|
Vomissements
|
Fréquent
| |
Troubles généraux et anomalies au site d'administration
|
Érythème au site d'injection
|
Fréquent
| |
Douleurs au site d'injection
|
Fréquent
| |
Prurit au site d'injection
|
Fréquent
| |
Investigations
|
Baisse de la vitamine A
|
Très fréquent
|
Description d'effets indésirables spécifiques
Baisse de la vitamine A
Tous les patients inclus dans l'étude clinique sur l'ATTRv-PN ont été invités à prendre la dose quotidienne recommandée de vitamine A. Tous ceux qui ont été traités par WAINZUA présentaient des taux normaux de vitamine A au début de l'étude et chez 96,5 % d'entre eux, ces taux ont baissé au cours de l'étude et étaient inférieurs à la limite inférieure de la normale (LIN) (voir «Propriétés/Effets»).
Réactions au site d'injection
Des érythèmes au site d'injection, des douleurs au site d'injection et un prurit au site d'injection ont été observés chez respectivement 3,5 %, 3,5 % et 2,1 % des patients atteints d'ATTRv-PN lors du traitement par WAINZUA.
L'annonce d'effets secondaires présumés après l'autorisation est d'une grande importance. Elle permet un suivi continu du rapport bénéfice-risque du médicament. Les professionnels de santé sont tenus de déclarer toute suspicion d'effet secondaire nouveau ou grave via le portail d'annonce en ligne ElViS (Electronic Vigilance System). Vous trouverez des informations à ce sujet sur www.swissmedic.ch.
SurdosageIl n'y a pas de traitement spécifique du surdosage d'éplontersen. En cas de surdosage, un traitement médical de soutien doit être instauré et un médecin doit être consulté.
Propriétés/EffetsCode ATC
N07XX21
Mécanisme d'action
L'éplontersen est un oligonucléotide antisens (OAS) chimérique en conformation «gapmer» modifié par le 2′-O-2-méthoxyéthyle (2′-MOE) et conjugué à la GalNAc, dont l'ossature mixte est composée de liaisons internucléotidiques phosphorothioate (PS) et phosphodiester (PO). La conjugaison de la GalNAc permet l'introduction ciblée de l'OAS dans les hépatocytes. L'éplontersen se lie sélectivement à l'ARN messager (ARNm) de la TTR dans les hépatocytes, ce qui provoque la dégradation de l'ARNm de la TTR, qu'il soit muté ou de type sauvage (normal). Ceci empêche la synthèse de la protéine TTR dans le foie et réduit de manière significative le taux des formes mutée et sauvage de TTR qui sont sécrétées dans la circulation sanguine par le foie.
La TTR est une protéine qui participe au transport de la protéine 4 de liaison au rétinol (RBP4), le principal transporteur de la vitamine A (rétinol). Par conséquent, la réduction du taux plasmatique de TTR devrait entraîner une réduction des taux plasmatiques de rétinol en dessous de la limite inférieure de la normale.
Pharmacodynamique
Dans l'étude clinique au cours de laquelle des patients atteints d'ATTRv-PN ont reçu de l'éplontersen, une réduction des concentrations sériques de TTR a été observée dès la première évaluation (semaine 5) et cette diminution s'est poursuivie jusqu'à la semaine 35. Une réduction durable de la concentration de TTR a été observée pendant toute la phase de traitement (85 semaines). La moyenne (ÉT) du pourcentage de réduction de la concentration sérique de TTR par rapport au taux initial était de 82,1 % (11,7) après 35 semaines, de 83,0 % (10,4) après 65 semaines et de 81,8 % (13,4) après 85 semaines de traitement par l'éplontersen. Une réduction similaire des concentrations sériques de TTR par rapport au taux initial comparativement au placebo (Figure 1, a et b) a été observée indépendamment du sexe, de l'origine ethnique, de l'âge, de la région, du poids corporel, de la présence d'une cardiomyopathie, du traitement antérieur, de la présence de la mutation Val30Met, du stade de la maladie et du diagnostic de cardiomyopathie amyloïde familiale (CAF) au début de l'étude.
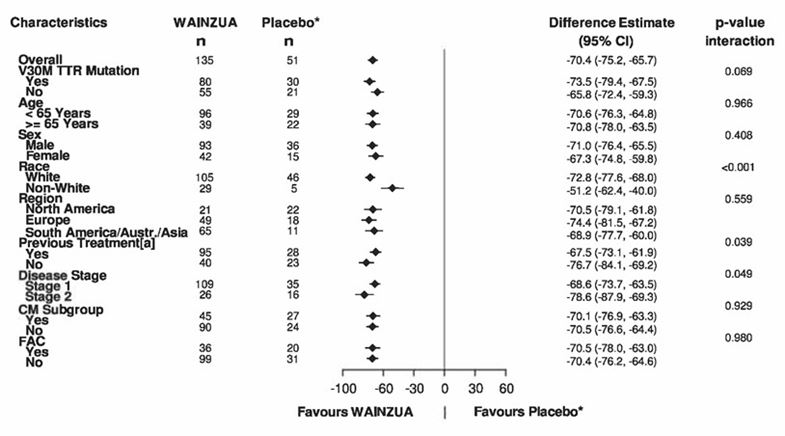
Figure 1: Graphique en forêt représentant la différence entre les traitements exprimée par la MMC du pourcentage de variation de la TTR (g/l) par rapport au taux initial pour les sous-groupes majeurs (étude NEURO-TTRansform) (population d'analyse complète)
a) à la semaine 35

* Le groupe placebo externe provient d'une autre étude contrôlée randomisée (étude NEURO-TTR).
[a] Patients ayant reçu un traitement antérieur par le tafamidis ou le diflunisal.
Se base sur un MMRM ajusté par pondération du score de propension avec les effets des variables catégorielles pour le traitement, le temps, l'interaction traitement/temps et le stade de la maladie, la présence de la mutation Val30Met, le traitement antérieur et avec des covariables fixes pour les valeurs à l'inclusion et l'interaction valeurs à l'inclusion/temps.
Les modèles des sous-groupes comprenaient également les interactions traitement/sous-groupe, temps/sous-groupe et traitement/temps/sous-groupe. Seules les données obtenues jusqu'à la semaine 35 ont été incluses dans l'analyse intermédiaire effectuée à la semaine 35.
Le sous-groupe CM comprend des patients chez lesquels une CAF avait été diagnostiquée à l'inclusion dans l'étude ou des patients chez lesquels une épaisseur du septum interventriculaire (IV) ≥13 mm sans hypertension avait été diagnostiquée au début de l'étude [anamnèse ou diagnostic au cours de l'étude].
Les différences entre les traitements exprimées par la MMC après 35 semaines (WAINZUA – placebo) sont indiquées avec un IC à 95 % (non ajusté).
CAF = cardiomyopathie amyloïde familiale, CM = cardiomyopathie, IC = intervalle de confiance, MMC = moyenne des moindres carrés, MMRM = modèle mixte à mesures répétées, TTR = transthyrétine.
b) à la semaine 65
* Le groupe placebo externe provient d'une autre étude contrôlée randomisée (étude NEURO-TTR).
[a] Patients ayant reçu un traitement antérieur par le tafamidis ou le diflunisal.
Se base sur un MMRM ajusté par pondération du score de propension avec les effets des variables catégorielles pour le traitement, le temps, l'interaction traitement/temps et le stade de la maladie, la présence de la mutation Val30Met, le traitement antérieur et avec des covariables fixes pour les valeurs à l'inclusion et l'interaction valeurs à l'inclusion/temps.
Les modèles des sous-groupes comprenaient également les interactions traitement/sous-groupe, temps/sous-groupe et traitement/temps/sous-groupe.
Le sous-groupe CM comprenait des patients chez lesquels une CAF avait été diagnostiquée à l'inclusion dans l'étude ou des patients chez lesquels une épaisseur du septum interventriculaire ≥13 mm sans hypertension avait été diagnostiquée au début de l'étude [anamnèse ou diagnostic au cours de l'étude].
Les différences entre les traitements exprimées par la MMC après 65 semaines (WAINZUA – placebo) sont indiquées avec un IC à 95 % (non ajusté).
CAF = cardiomyopathie amyloïde familiale, CM = cardiomyopathie, IC = intervalle de confiance, MMC = moyenne des moindres carrés, MMRM = modèle mixte à mesures répétées, TTR = transthyrétine
Électrophysiologie cardiaque
Aucune étude formelle portant sur l'intervalle QTc n'a été réalisée avec WAINZUA. Le risque d'allongement de l'intervalle QTc induit par l'éplontersen a été évalué dans le cadre d'une étude randomisée contrôlée contre placebo menée chez des volontaires sains. Aucun effet cliniquement pertinent sur l'intervalle QT n'a été observé à une dose d'éplontersen correspondant à 2,7 fois la dose recommandée de 45 mg.
Immunogénicité
Au cours de l'étude clinique menée chez des patients atteints d'ATTRv-PN, 58 patients (40,3 %) présentaient des anticorps anti-médicament (AAM) liés au traitement après une durée de traitement de 84 semaines (durée médiane du traitement de 561 jours [80 semaines], fourchette: 57 à 582 jours).
La présence d'anticorps anti-éplontersen n'a eu aucune incidence cliniquement importante sur l'efficacité, la sécurité, la pharmacocinétique ou la pharmacodynamique de WAINZUA.
Efficacité clinique
L'efficacité et la sécurité de WAINZUA ont été évaluées dans le cadre d'une étude randomisée, multicentrique, en ouvert, contrôlée contre groupe témoin externe incluant au total 168 patients atteints d'ATTRv-PN (étude NEURO-TTRansform). Les patients ont été randomisés selon un rapport 6:1 pour recevoir soit une injection sous-cutanée de 45 mg de WAINZUA (N = 144) toutes les 4 semaines, soit 284 mg d'inotersen (N = 24) une fois par semaine en tant que groupe de référence. Parmi les 144 patients qui ont été randomisés pour recevoir l'éplontersen, 140 (97,2 %) ont suivi le traitement jusqu'à la semaine 35, 135 (93,8 %) jusqu'à la semaine 65 et 130 (90,3 %) jusqu'à la semaine 85.
Le groupe témoin externe de patients sous placebo était composé d'une cohorte de patients sous placebo provenant de l'étude pivot sur l'inotersen, une étude clinique multicentrique, randomisée, en double aveugle et contrôlée contre placebo menée chez des patients adultes atteints d'ATTRv-PN (étude NEURO-TTR). Cette cohorte recevait des injections sous-cutanées de placebo une fois par semaine. Les deux études ont utilisé des critères d'inclusion identiques.
Les caractéristiques des groupes éplontersen et placebo externe étaient dans l'ensemble comparables et les déséquilibres potentiels des principales caractéristiques initiales (présence de la mutation Val30Me, stade de la maladie et traitement antérieur) ont été pris en compte dans l'analyse statistique prédéfinie. Les caractéristiques initiales démographiques et spécifiques à la maladie figurent dans le Tableau 2.
Tableau 2 Caractéristiques initiales démographiques et spécifiques à la maladie de l'étude NEURO-TTRansform (population de sécurité)
|
|
Placebo
*(N = 60)
|
WAINZUA
(N = 144)
| |
Âge, ans
|
|
| |
Moyenne (ÉT)
|
59,5 (14,1)
|
53,0 (15,0)
| |
Médiane (min., max.)
|
63 (28, 81)
|
51,5 (24, 82)
| |
< 65, n (%)
|
34 (56,7)
|
100 (69,4)
| |
65-74, n (%)
|
17 (28,3)
|
36 (25,0)
| |
≥75, n (%)
|
9 (15,0)
|
8 (5,6)
| |
Hommes, n (%)
|
41 (68,3)
|
100 (69,4)
| |
Ethnie, n (%)
|
|
| |
Asiatique
|
3 (5,0)
|
22 (15,4)
| |
Noire ou afro-américaine
|
1 (1,7)
|
5 (3,5)
| |
Caucasienne
|
53 (88,3)
|
112 (78,3)
| |
Autre
|
2 (3,3)
|
3 (2,1)
| |
Multiple
|
1 (1,7)
|
1 (0,7)
| |
Appartenance ethnique, n (%)
|
|
| |
m
|
60
|
142
| |
Hispanique ou latino
|
7 (11,7)
|
22 (15,5)
| |
Traitement antérieur par le tafamidis ou le diflunisal, n (%)
|
|
| |
Oui
|
36 (60,0)
|
100 (69,4)
| |
Stade de la maladie de l'ATTRv-PN1, n (%)
|
|
| |
Stade 1
|
42 (70,0)
|
115 (79,9)
| |
Stade 2
|
18 (30,0)
|
29 (20,1)
| |
Score composite mNIS+7, moyenne (ÉT)
|
74,8 (39,0)
|
81,3 (43,4)
| |
Score total au questionnaire Norfolk QoL-DN,
|
|
| |
m
|
59
|
137
| |
Moyenne (ÉT)
|
48,7 (26,8)
|
44,1 (26,6)
| |
Mutation Val30Met de la TTR, n (%)
|
|
| |
Oui2
|
33 (55,0)
|
85 (59,0)
| |
Non3
|
27 (45,0)
|
59 (41,0)
| |
Glu89Gln, Glu109Gln
|
0
|
1 (0,7)
| |
Leu58His, Leu78His
|
3 (5,0)
|
4 (2,8)
| |
Phe64Leu, Phe84Leu
|
3 (5,0)
|
5 (3,5)
| |
Ser50Arg, Ser70Arg
|
1 (1,7)
|
2 (1,4)
| |
Ser77Tyr, Ser97Tyr, S97Y
|
5 (8,3)
|
3 (2,1)
| |
Thr49Ala, Thr69Ala
|
0
|
1 (0,7)
| |
Thr60Ala, Thr80Ala
|
8 (13,3)
|
4 (2,8)
| |
Val122Ile, Val142Ile
|
1 (1,7)
|
4 (2,8)
| |
Autre3
|
6 (10,0)
|
35 (24,3)
| |
Classe NYHA, n (%)
|
|
| |
I
|
40 (66,7)
|
105 (72,9)
| |
II
|
20 (33,3)
|
39 (27,1)
| |
Durée de la maladie à compter du diagnostic de l'ATTRv-PN (mois), moyenne (ÉT)
|
39,3 (40,3)
|
46,8 (58,1)
| |
Durée depuis l'apparition des symptômes de l'ATTRv-PN (mois), moyenne (ÉT)
|
64,0 (52,3)
|
67,7 (50,9)
| |
Diagnostic de cardiomyopathie amyloïde familiale (CAF)4, n (%)
|
|
| |
Critères pour la documentation du diagnostic clinique d'une CAF4, n (%)5
|
22 (36,7)
|
39 (27,1)
| |
Biopsie cardiaque
|
5 (22,7)
|
1 (2,6)
| |
Résultat de l'échocardiographie
|
17 (77,3)
|
24 (61,5)
| |
Autre
|
0
|
24 (61,5)
| |
Durée de la maladie à compter du diagnostic clinique d'une CAF4 recensée dans le CRF (mois), moyenne (ÉT)
|
21,0 (22,5)
|
18,5 (21,4)
| |
Durée depuis l'apparition des symptômes de la CAF4 (mois), moyenne (ÉT)
|
34,1 (29,3)
|
36,3 (63,8)
| |
NT-proBNP (pmol/l), moyenne (ÉT)
|
82,0 (159,2)
|
54,0 (122,6)
| |
Short Form 36 Item Health Survey (SF-36) (forme abrégée du questionnaire sur l'état de santé à 36 questions), score total des composantes physiques, moyenne (ÉT)
|
37,2 (9,8)
|
39,7 (9,3)
| |
Neuropathy Symptoms and Change (NSC) (questionnaire sur les symptômes neuropathiques), score total, moyenne (ÉT)
|
23,0 (12,6)
|
23,1 (12,4)
| |
Score Polyneuropathy Disability (PND) (score des déficiences liées à la polyneuropathie), n (%)
|
|
| |
I
|
23 (38,3)
|
56 (39,2)
| |
II
|
19 (31,7)
|
61 (42,7)
| |
IIIa
|
15 (25,0)
|
16 (11,2)
| |
IIIb
|
3 (5,0)
|
10 (7,0)
| |
Indice de masse corporelle (kg/m2)
|
|
| |
m
|
60
|
138
| |
Moyenne (ÉT)
|
24,2 (4,9)
|
24,4 (4,9)
| |
Médiane (min., max.)
|
23,8 (14,5, 39,8)
|
24,1 (15,4, 35,4)
| |
Indice de masse corporelle modifié (kg/m2 x g/l),
|
|
| |
m
|
60
|
138
| |
Moyenne (ÉT)
|
1049,89 (228,43)
|
1025,78 (235,12)
| |
Médiane (min., max.)
|
1027,55 (668,7, 1710,0)
|
1003,14 (615,7, 1714,0)
| |
Le groupe placebo externe provient d'une autre étude contrôlée randomisée (étude NEURO-TTR).
1 Le stade de la maladie est défini comme suit: stade 1 = pas d'aide à la marche nécessaire et stade 2 = aide à la marche nécessaire.
2 Comprend les génotypes V30M, V50M, V50M-MUTATION, VAL50MET et P.VAL50MET.
3 Se base sur la banque de données cliniques. Les mutations non-Val30Met comprenaient les mutations suivantes: GLU89GLN, LEU58HIS, PHE64LEU, SER50ARG, SER77TYR, THR49ALA, THR60ALA, VAL122LLE et autres mutations (dont la mutation ALA97SER).
4 Cardiomyopathie amyloïde familiale = amyloïdose héréditaire à transthyrétine associée à une cardiomyopathie (ATTRv-CM).
5 Le dénominateur pour le calcul du pourcentage est le nombre de patients présentant un diagnostic de CAF.
Seuls le mois et l'année de la date de la déclaration de consentement ont été recueillis pour calculer la durée de la maladie à compter du diagnostic et à compter de l'apparition des symptômes de l'ATTRv-PN et de la CAF.
CRF = cahier d'observation, ÉT = écart type, m = nombre de patients n'ayant pas de données manquantes si ce nombre est différent de N, N = nombre de patients de la population de sécurité, n = nombre de patients d'un sous-groupe, NT-proBNP = fraction N-terminale du peptide natriurétique de type B.
|
Parmi les 39 patients (27,1%) du groupe éplontersen présentant un diagnostic de cardiomyopathie à TTR à l'entrée dans l'étude, 41,0% étaient dans la classe NYHA I (New York Heart Association) et 59,0% dans la classe NYHA II.
Analyses effectuées à la semaine 35 (analyses intermédiaires)
Les critères principaux d'évaluation de l'efficacité étaient la variation de la concentration sérique de transthyrétine (TTR) (voir Figure 2) et la variation du score composite Modified Neuropathy Impairment Score +7 (mNIS+7) jusqu'à la semaine 35 par rapport à la valeur initiale. Le score composite mNIS+7 constitue une évaluation objective de la neuropathie et correspond à l'agrégation du score NIS et de la composante Modified +7. Dans la version du score composite mNIS+7 utilisée lors de l'étude, le NIS mesure objectivement les déficits de la fonction des nerfs crâniens, de la force musculaire, des réflexes et des sensations, tandis que la composante Modified +7 évalue la réponse de la fréquence cardiaque à la respiration profonde, des tests sensoriels quantitatifs (pression de contact et sensibilité à la chaleur) et l'électrophysiologis des nerfs périphériques.
Dans la version validée du score composite mNIS+7 utilisée lors de l'étude, les valeurs obtenues allaient de -22,3 à 346,3 points, les scores plus élevés indiquant un degré élevé de sévérité de la maladie.
Le critère d'évaluation secondaire était la variation du score total au questionnaire Norfolk Quality of Life – Diabetic Neuropathy (QoL-DN) par rapport à la valeur initiale. Le questionnaire Norfolk QoL-DN est une évaluation effectuée par les patients de l'expérience subjective de la neuropathie dans les domaines suivants: les capacités physiques/la neuropathie des grandes fibres, les activités de la vie quotidienne, les symptômes, la neuropathie des petites fibres et la neuropathie autonome. Les scores obtenus à la version du questionnaire Norfolk QoL-DN utilisée dans le cadre de l'étude allaient de -4 à 136 points, les scores plus élevés indiquant une atteinte plus importante.
Une amélioration statistiquement significative a été mise en évidence avec WAINZUA après 35 semaines par rapport au groupe témoin placebo externe, se traduisant par une réduction de la concentration sérique de TTR avec un pourcentage de variation de -66,43 % (IC à 95 %: --71,39 %, -61,47 %; p < 0,0001) (voir Figure 2). Une amélioration statistiquement significative a été mise en évidence avec WAINZUA après 35 semaines par rapport au groupe témoin placebo externe concernant le score composite mNIS+7, avec une différence de la MMC de -9,0 (IC à 95 %: --13,5, -4,5; p < 0,0001) (voir Figures 3, 4a, 7a). Une amélioration statistiquement significative a été mise en évidence avec WAINZUA après 35 semaines par rapport au groupe témoin placebo externe concernant le score total au questionnaire Norfolk QoL-DN, avec une différence de la MMC de -11,8 (IC à 95 %: --16,8, -6,8; p < 0,0001) (Tableau 3 et Figures 5, 6a, 8a).
Analyses effectuées à la semaine 65/66 (analyses finales)
Les critères d'évaluation co-primaires de l'objectif principal de l'étude lors de l'analyse finale effectuée à la semaine 66 étaient le pourcentage de variation de la concentration sérique de TTR après 65 semaines par rapport à la valeur initiale, la variation du score composite mNIS+7 à la semaine 66 par rapport à la valeur initiale et la variation du score total au questionnaire Norfolk QoL-DN à la semaine 66 par rapport à la valeur initiale. La réduction de la concentration sérique de TTR s'est maintenue jusqu'à la semaine 65. En outre, les résultats obtenus à la semaine 66 concernant le score composite mNIS+7 et le score total au questionnaire Norfolk correspondaient aux résultats obtenus après 35 semaines (voir Tableau 3 et Figures 3, 4b, 5, 6b).
Les critères d'évaluation secondaires étaient la variation du score au questionnaire Neuropathy Symptoms and Change (NSC) à la semaine 66 et à la semaine 35 par rapport à la valeur initiale, la variation du score des composantes physiques (PCS) de la forme abrégée du questionnaire sur l'état de santé à 36 questions (version 2) (SF-36) à la semaine 65 par rapport à la valeur initiale, la variation du score Polyneuropathy Disability (PND) à la semaine 65 par rapport à la valeur initiale et la variation de l'indice de masse corporelle modifié (IMCm) jusqu'à la semaine 65 par rapport à la valeur initiale.
Le NSC est un questionnaire d'auto-évaluation par le patient sur la quantification du type, de la répartition et de la sévérité de la faiblesse musculaire, des symptômes sensoriels, des symptômes douloureux et des symptômes autonomes. Des scores plus élevés indiquent des symptômes plus intenses.
Le SF-36 PCS comprend 4 échelles évaluant la capacité fonctionnelle, les limitations dues à l'état physique, les douleurs physiques et l'état de santé général. Des scores plus élevés indiquent un meilleur état de santé général.
Le PND classifie l'invalidité sur la base de l'évaluation de la mobilité (p.ex. nécessité d'une canne, d'une aide à la marche, d'un fauteuil roulant ou alitement). Des scores plus élevés indiquent une invalidité plus importante.
L'indice de masse corporelle modifié (IMCm) (IMC × albumine sérique) est une méthode acceptable pour l'évaluation de l'état nutritionnel en présence d'ATTR. Des scores plus élevés correspondent à un meilleur état nutritionnel et sont un indicateur de survie plus longue des patients atteints d'ATTRv-PN.
Tous les critères d'évaluation secondaires ont mis en évidence une supériorité statistiquement significative par rapport au placebo externe (voir Tableau 4).
Tableau 3: Effet du traitement concernant les principaux critères d'évaluation et les critères d'évaluation secondaires majeurs (étude NEURO-TTRansform) (population d'analyse complète)
|
Analyse/critère d'évaluation
|
Valeur à l'inclusion, moyenne (ÉT)
|
Variation de la MMC/pourcentage de variation vs valeur à l'inclusion, (ET) [IC à 95 %]
|
Différence de la MMC WAINZUA/placebo externe*
(IC à 95 %)
|
Valeur p
| |
Placebo externe*
|
WAINZUA
|
Placebo externe*
|
WAINZUA
| |
Semaine 35
|
N = 59
|
N = 140
|
N = 59
|
N = 140
|
|
| |
TTR sérique, g/l 1
|
0,15 (0,04)
|
0,23 (0,08)
|
|
|
|
| |
Pourcentage de variation vs valeur à l'inclusion
|
|
|
-14,8 % (2,0)
[-18,73, -10,80]
|
-81,2 % (1,7)
[-84,55, -77,84]
|
-66,4 %
(-71,39, -61,47)
|
p < 0,0001
| |
Score composite mNIS+7 2,3
|
74,1 (39,0)
|
79,6 (42,3)
|
|
|
|
| |
Variation vs valeur à l'inclusion
|
|
|
9,2 (1,9)
[5,54, 12,91]
|
0,2 (1,9)
[-3,46, 3,89]
|
-9,0
(-13,48, -4,54)
|
p < 0,0001
| |
Score total au questionnaire Norfolk QoL-DN2,3
|
48,6 (27,0)
|
43,5 (26,3)
|
|
|
|
| |
Variation vs valeur à l'inclusion
|
|
|
8,7 (2,1)
[4,53, 12,81]
|
-3,1 (2,1)
[-7,19, 0,96]
|
-11,8
(-16,82, -6,76)
|
p < 0,0001
| |
Semaine 65/66
|
N = 59
|
N = 141
|
N = 59
|
N = 141
|
|
| |
TTR sérique, g/l 1
|
0,15 (0,04)
|
0,23 (0,08)
|
|
|
|
| |
Pourcentage de variation vs valeur à l'inclusion
|
|
|
-11,2 % (1,9)
[-15,06, -7,41]
|
-81,7 % (1,6)
[-84,82, -78,48]
|
-70,4 %
(-75,17, -65,66)
|
p < 0,00014
| |
Score composite mNIS+7 1
|
74,1 (39,0)
|
79,8 (42,3)
|
|
|
|
| |
Variation vs valeur à l'inclusion
|
|
|
25,1 (2,4)
[20,23, 29,88]
|
0,3 (2,4)
[-4,46, 5,06]
|
-24,8
(-30,96, -18,56)
|
p < 0,00014
| |
Score total au questionnaire Norfolk QoL-DN 1
|
48,6 (27,0)
|
43,3 (26,2)
|
|
|
|
| |
Variation vs valeur à l'inclusion
|
|
|
14,2 (2,4)
[9,51, 18,97]
|
-5,5 (2,3)
[-10,03, -0,96]
|
-19,7
(-25,63, -13,84)
|
p < 0,00014
|
* Le groupe placebo externe provient d'une autre étude contrôlée randomisée (étude NEURO-TTR).
1 Se base sur un MMRM ajusté par pondération du score de propension avec les effets des variables catégorielles fixes pour le traitement, le temps, l'interaction traitement/temps et le stade de la maladie, la présence de la mutation Val30M, le traitement antérieur et avec des covariables fixes pour la valeur initiale et l'interaction valeur à l'inclusion/temps. Seules les données obtenues jusqu'à la semaine 66 ont été incluses dans l'analyse effectuée après 66 semaines.
2 Se base sur un modèle ANCOVA ajusté en fonction du score de propension avec les effets des variables traitement, stade de la maladie, présence de la mutation Val30Met, traitement antérieur et valeur initiale. Seules les données obtenues jusqu'à la semaine 35 ont été incluses dans l'analyse intermédiaire.
3 La valeur a été générée à l'aide d'un modèle d'imputation selon la méthode de l'imputation multiple chez les participants qui avaient un score mNIS+7 ou Norfolk QoL-D manquant à la semaine 35. Chacun des 500 ensembles de données imputés a été analysé à l'aide d'un modèle ANCOVA simple, et les 500 résultats du modèle ANCOVA ont été regroupés en suivant les règles de Rubin.
4 N'a pas été formellement analysée en raison de résultats statistiquement significatifs à la semaine 35.
L'analyse se basait sur des données saisies jusqu'à 52 jours après l'administration de la dernière dose du médicament à l'étude. Données issues de l'analyse intermédiaire effectuée à la semaine 35 et données de la semaine 65/66 issues de l'analyse effectuée à la semaine 66. Dans la population d'analyse complète, le groupe éplontersen comprenait 140 participants après 35 semaines et 141 participants après 66 semaines. Chez un participant, on ne disposait pas d'évaluation du mNIS+7 ou du Norfolk QoL-DN à la semaine 35, mais d'une évaluation pour au moins l'un de ces paramètres à la semaine 66.
ANCOVA = analyse de la covariance, ÉT = écart type, ET = erreur type, IC = intervalle de confiance, MMC = moyenne des moindres carrés, MMRM = modèle mixte à mesures répétées, mNIS+7 = modified Neuropathy Impairment Score +7, N = nombre de participants dans le groupe, Norfolk QoL-DN = Norfolk Quality of Life – Diabetic Neuropathy Questionnaire, TTR = transthyrétine.
Tableau 4: Analyse hiérarchisée des critères d'évaluation secondaires (étude NEURO-TTRansform)
|
|
|
|
Comparaison WAINZUA vs placebo externe*
| |
Critère d'évaluation secondaire/
groupe de traitement (N)
|
n
|
Variation vs valeur à l'inclusion
MMC (IC à 95 %)
|
Estimation
|
IC à 95 %
|
Valeur p
| |
Variation de la MMC du NSC vs valeur à l'inclusion à la semaine 66
| |
WAINZUA (N = 141)
|
132
|
0,0 (-1,92, 1,86)
|
-8,2
|
-10,65, -5,76
|
< 0,0001
| |
Placebo externe* (N = 59)
|
52
|
8,2 (6,24, 10,12)
| |
Variation de la MMC du NSC vs valeur à l'inclusion à la semaine 35
| |
WAINZUA (N = 141)
|
141
|
0,8 (-0,92, 2,50)
|
-3,9
|
-6,08, -1,80
|
0,0005
| |
Externes Placebo* (N = 59)
|
56
|
4,7 (2,98, 6,48)
| |
Variation de la MMC du SF-36 PCS vs valeur à l'inclusion à la semaine 65
| |
WAINZUA (N = 141)
|
136
|
0,85 (-0,711, 2,412)
|
5,31
|
3,195, 7,416
|
< 0,0001
| |
Placebo externe* (N = 59)
|
50
|
-4,46 (-6,139, -2,770)
| |
Variation de la MMC du score PND vs valeur à l'inclusion à la semaine 65
| |
WAINZUA (N = 141)
|
134
|
0,1 (0,0, 0,2)
|
-0,2
|
-0,4, 0,0
|
0,0241
| |
Placebo externe* (N = 59)
|
51
|
0,3 (0,2, 0,4)
| |
Variation de la MMC de l'IMCm vs valeur à l'inclusion à la semaine 65
| |
WAINZUA (N = 141)
|
130
|
-8,1 (-28,55, 12,42)
|
82,7
|
54,64, 110,76
|
< 0,0001
| |
Placebo externe* (N = 59)
|
49
|
-90,8 (-112,84, -68,69)
|
* Le groupe placebo externe provient d'une autre étude contrôlée randomisée (étude NEURO-TTR).
N = nombre de patients dans la population d'analyse complète à la semaine 66.
n = nombre de patients n'ayant pas de données manquantes pour les covariables à l'inclusion et pour la variation vs valeur à l'inclusion au point temporel correspondant.
L'analyse se basait sur des données saisies jusqu'à 28 jours après l'administration de la dernière dose du médicament à l'étude. L'intervalle de temps pour la visite concernant l'analyse effectuée à la semaine 65 s'étend du jour 419 au jour 479.
Se base sur un modèle mixte à mesures répétées (MMRM) ajusté par pondération du score de propension avec les effets des variables catégorielles fixes pour le traitement, le temps, l'interaction traitement/temps et le stade de la maladie, la présence de la mutation Val30M, le traitement antérieur et avec des covariables fixes pour la valeur initiale et l'interaction valeur à l'inclusion/temps. Seules les données obtenues jusqu'à la semaine 65 ont été incluses dans l'analyse finale après 66 semaines.
IC = intervalle de confiance, IMCm = indice de masse corporelle modifié, MMC = moyenne des moindres carrés, NSC = Neuropathy Symptoms and Change, PCS = score des composantes physiques, PND = Polyneuropathy Disability, SF-36 PCS = Short Form-36 Health Survey Questionnaire Physical Component Score.
Figure 2: Pourcentage de variation de la MMC de la concentration sérique de TTR de l'inclusion à la semaine 65, WAINZUA vs placebo externe* jusqu'à la semaine 65 (étude NEURO-TTRansform) (population d'analyse complète)
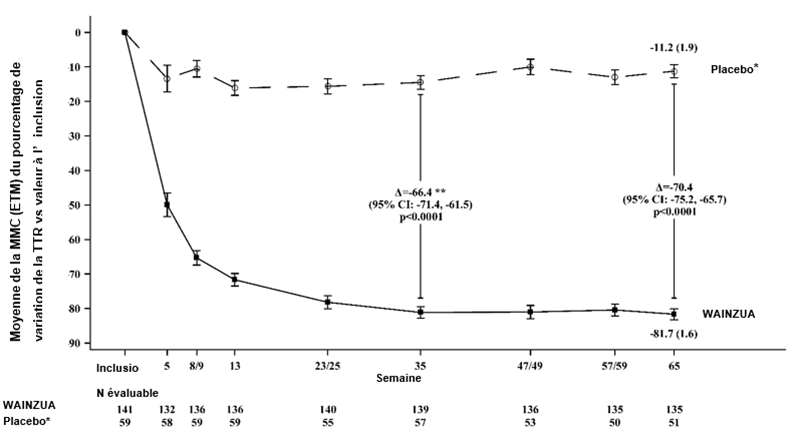
* Le groupe placebo externe provient d'une autre étude contrôlée randomisée (étude NEURO-TTR).
** La différence entre les traitements correspond aux résultats de l'analyse intermédiaire formelle effectuée à la semaine 35. Seules les données obtenues jusqu'à la semaine 35 ont été incluses dans l'analyse intermédiaire réalisée à la semaine 35.
Se base sur un MMRM ajusté par pondération du score de propension avec les effets des variables catégorielles fixes pour le traitement, le temps, l'interaction traitement/temps et le stade de la maladie, la présence de la mutation Val30Met, le traitement antérieur et avec des covariables fixes pour la valeur initiale et l'interaction valeur à l'inclusion/temps.
L'analyse se basait sur des données saisies jusqu'à 28 jours après l'administration de la dernière dose du médicament à l'étude. Les données obtenues jusqu'à la semaine 65 ont été incluses. Le placebo a été évalué à l'inclusion et aux semaines 5, 8, 13, 23, 35, 47, 59 et 65; WAINZUA a été évalué à l'inclusion et aux semaines 5, 9, 13, 25, 35, 49, 57 et 65.
Les différences entre les traitements exprimées par la MMC après 35 semaines et 65 semaines (WAINZUA – placebo) sont indiquées avec un IC à 95 % (non ajusté).
ETM = erreur type de la moyenne, IC = intervalle de confiance, MMC = moyenne des moindres carrés, MMRM = modèle mixte à mesures répétées, TTR = transthyrétine.
Figure 3: Variation de la MMC du score composite mNIS+7 vs inclusion (étude NEURO-TTRansform) (population d'analyse complète)
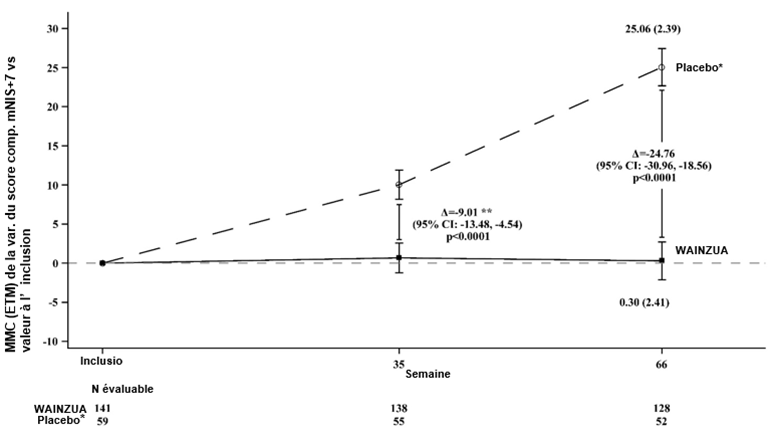
* Le groupe placebo externe provient d'une autre étude contrôlée randomisée (étude NEURO-TTR).
** La différence entre les traitements correspond aux résultats de l'analyse intermédiaire formelle effectuée à la semaine 35. Se base sur une ANCOVA avec IM ajustée par pondération du score de propension avec les effets des variables catégorielles fixes pour le traitement, le stade de la maladie, la présence de la mutation Val30Met, le traitement antérieur et avec des covariables fixes pour la valeur initiale. Seules les données obtenues jusqu'à la semaine 35 ont été incluses dans l'analyse intermédiaire réalisée à la semaine 35.
L'analyse réalisée à la semaine 66 se basait sur un MMRM ajusté par pondération du score de propension avec les effets des variables catégorielles pour le traitement, le temps, l'interaction traitement/temps et le stade de la maladie, la présence de la mutation Val30Met, le traitement antérieur et avec des covariables fixes pour la valeur à l'inclusion et l'interaction valeur à l'inclusion/temps.
L'analyse se basait sur des données saisies jusqu'à 52 jours après l'administration de la dernière dose du médicament à l'étude. Les données obtenues jusqu'à la semaine 66 ont été incluses.
Les différences entre les traitements exprimées par la MMC après 35 semaines et 66 semaines (WAINZUA – placebo) sont indiquées avec un IC à 95 % (non ajusté).
ANCOVA avec IM = analyse de covariance avec la méthode d'imputation multiple, ETM = erreur type de la moyenne, IC = intervalle de confiance, MMC = moyenne des moindres carrés, MMRM = modèle mixte à mesures répétées.
Figure 4: Histogramme de la variation du score composite mNIS+7 vs valeur à l'inclusion (étude NEURO-TTRansform) (population d'analyse de sécurité)
a) À la semaine 35
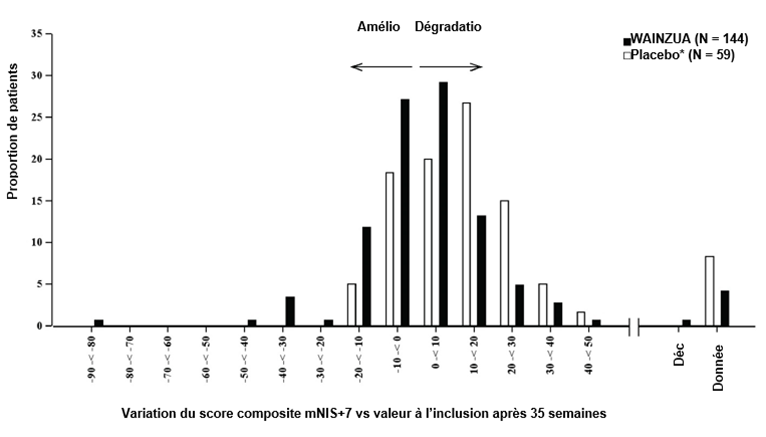
* Le groupe placebo externe provient d'une autre étude contrôlée randomisée (étude NEURO-TTR).
b) À la semaine 66
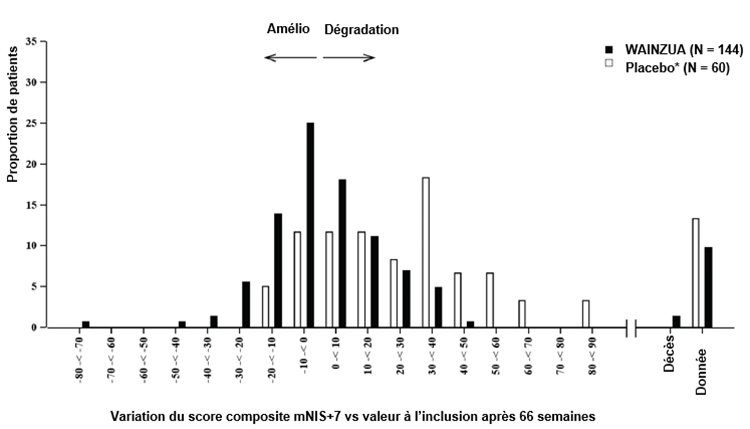
* Le groupe placebo externe provient d'une autre étude contrôlée randomisée (étude NEURO-TTR).
Figure 5: Variation de la MMC du score total au questionnaire Norfolk QoL-DN vs valeur à l'inclusion (étude NEURO-TTRansform)
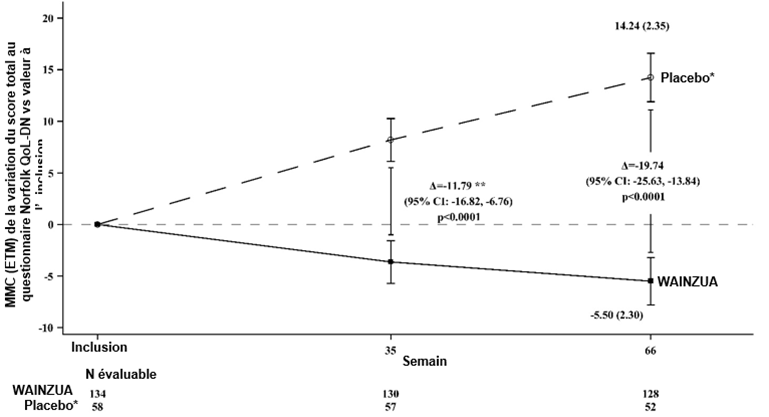
* Le groupe placebo externe provient d'une autre étude contrôlée randomisée (étude NEURO-TTR).
** La différence entre les traitements correspond aux résultats de l'analyse intermédiaire formelle effectuée à la semaine 35. Se base sur une ANCOVA avec IM ajustée par pondération du score de propension avec les effets des variables catégorielles fixes pour le traitement, le stade de la maladie, la présence de la mutation Val30Met, le traitement antérieur et avec des covariables fixes pour la valeur initiale. Seules les données obtenues jusqu'à la semaine 35 ont été incluses dans l'analyse intermédiaire réalisée à la semaine 35.
L'analyse effectuée à la semaine 66 se basait sur un MMRM ajusté par pondération du score de propension avec les effets des variables catégorielles pour le traitement, le temps, l'interaction traitement/temps et le stade de la maladie, la présence de la mutation Val30Met, le traitement antérieur et avec des covariables fixes pour la valeur à l'inclusion et l'interaction valeur à l'inclusion/temps.
L'analyse se basait sur des données saisies jusqu'à 52 jours après l'administration de la dernière dose du médicament à l'étude. Les données obtenues jusqu'à la semaine 66 ont été incluses.
Les différences entre les traitements exprimées par la MMC après 35 semaines et 66 semaines (WAINZUA – placebo) sont indiquées avec un IC à 95 % (non ajusté).
ANCOVA avec IM = analyse de covariance avec la méthode d'imputation multiple, ETM = erreur type de la moyenne, IC = intervalle de confiance, MMC = moyenne des moindres carrés, MMRM = modèle mixte à mesures répétées.
Figure 6: Histogramme de la variation du score total au questionnaire Norfolk QoL-DN vs valeur à l'inclusion (étude NEURO-TTRansform) (population d'analyse de sécurité)
a) À la semaine 35
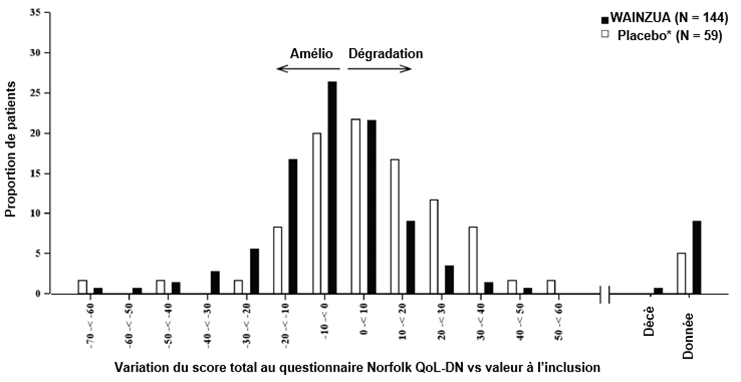
* Le groupe placebo externe provient d'une autre étude contrôlée randomisée (étude NEURO-TTR).
b) À la semaine 66
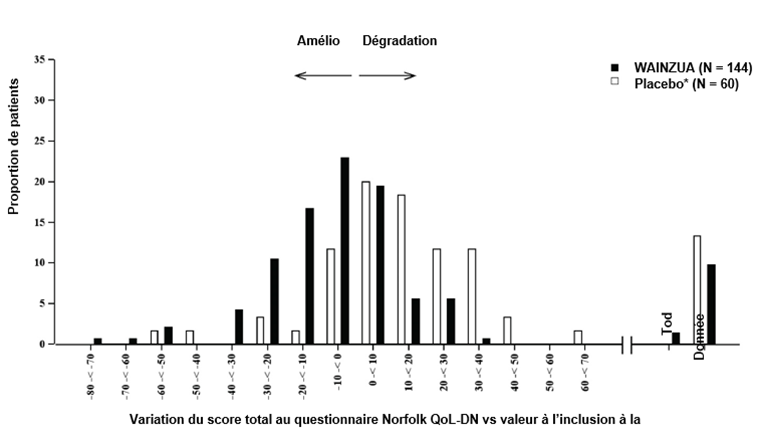
* Le groupe placebo externe provient d'une autre étude contrôlée randomisée (étude NEURO-TTR).
Aussi bien à la semaine 35 qu'à la semaine 65/66, les améliorations observées chez les patients sous WAINZUA étaient similaires en comparaison avec le placebo concernant la réduction de la concentration sérique de TTR, le score composite mNIS+7 et le score total au questionnaire Norfolk QoL-DN dans tous les sous-groupes y compris l'âge, le sexe, l'ethnie, la région, la présence de la mutation Val30Met, la présence d'une cardiomyopathie, le diagnostic de CAF à l'inclusion et le stade de la maladie (Figures 1a et b, 7a et b ainsi que 8a et b).
Figure 7: Graphique en forêt de la différence entre les traitements exprimée par la MMC concernant la variation du score composite mNIS+7 vs valeur à l'inclusion pour les sous-groupes majeurs (étude NEURO-TTRansform) (population d'analyse complète)
a) in Woche 35
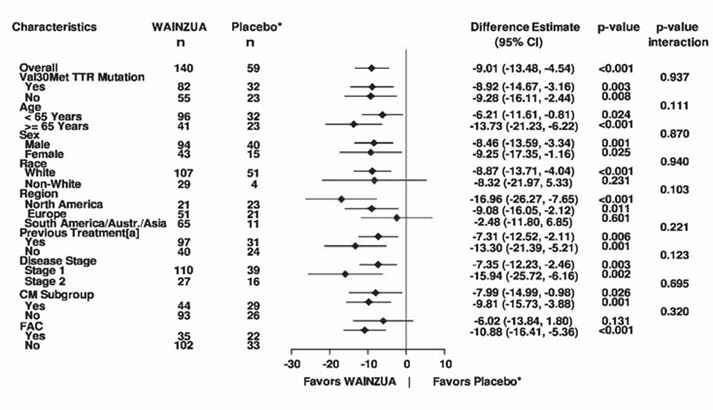
* Le groupe placebo externe provient d'une autre étude contrôlée randomisée (étude NEURO-TTR).
[a] Patients ayant reçu un traitement antérieur par le tafamidis ou le diflunisal.
Le sous-groupe CM comprend des patients chez lesquels une CAF avait été diagnostiquée à l'inclusion dans l'étude ou des patients chez lesquels une épaisseur du septum IV ≥13 mm sans hypertension avait été diagnostiquée au début de l'étude [anamnèse ou diagnostic au cours de l'étude].
La différence de la MMC, l'intervalle de confiance et les valeurs p se basent sur un modèle ANCOVA ajusté en fonction du score de propension avec les effets des variables traitement, facteurs des sous-groupes, stade de la maladie, présence de la mutation Val30Met, traitement antérieur, interaction traitement/sous-groupe et valeur initiale. Les données obtenues jusqu'à la semaine 35 ont été incluses dans l'analyse effectuée à la semaine 35.
b) À la semaine 66

* Le groupe placebo externe provient d'une autre étude contrôlée randomisée (étude NEURO-TTR).
[a] Patients ayant reçu un traitement antérieur par le tafamidis ou le diflunisal.
Se base sur un MMRM ajusté par pondération du score de propension avec les effets des variables catégorielles pour le traitement, le temps, l'interaction traitement/temps et le stade de la maladie, la présence de la mutation Val30Met, le traitement antérieur et avec des covariables fixes pour les valeurs à l'inclusion et l'interaction valeurs à l'inclusion/temps.
Les modèles des sous-groupes comprenaient également les interactions traitement/sous-groupe, temps/sous-groupe et traitement/temps/sous-groupe. Daten bis Woche 66 sind einbezogen.
Le sous-groupe CM comprend des patients chez lesquels une CAF avait été diagnostiquée à l'inclusion dans l'étude ou des patients chez lesquels une épaisseur du septum IV ≥13 mm sans hypertension avait été diagnostiquée au début de l'étude [anamnèse ou diagnostic au cours de l'étude].
Les différences entre les traitements exprimées par la MMC après 66 semaines (WAINZUA – placebo) sont indiquées avec un IC à 95 % (non ajusté).
CAF = cardiomyopathie amyloïde familiale, CM = cardiomyopathie, IC = intervalle de confiance, MMC = moyenne des moindres carrés, MMRM = modèle mixte à mesures répétées.
Figure 8: Graphique en forêt de la différence entre les traitements exprimée par la MMC concernant la variation du score total au questionnaire Norfolk QoL-DN vs valeur à l'inclusion pour les sous-groupes majeurs (étude NEURO-TTRansform) (population d'analyse complète)
a) À la semaine 35

* Le groupe placebo externe provient d'une autre étude contrôlée randomisée (étude NEURO-TTR).
[a] Patients ayant reçu un traitement antérieur par le tafamidis ou le diflunisal.
Le sous-groupe CM comprend des patients chez lesquels une CAF avait été diagnostiquée à l'inclusion dans l'étude ou des patients chez lesquels une épaisseur du septum IV ≥13 mm sans hypertension avait été diagnostiquée au début de l'étude [anamnèse ou diagnostic au cours de l'étude].
La différence de la MMC, l'intervalle de confiance et les valeurs p se basent sur un modèle ANCOVA ajusté en fonction du score de propension avec les effets des variables traitement, facteurs des sous-groupes, stade de la maladie, présence de la mutation Val30Met, traitement antérieur, interaction traitement/sous-groupe et valeur initiale. Seules les données obtenues jusqu'à la semaine 35 ont été incluses dans l'analyse intermédiaire effectuée à la semaine 35.
b) À la semaine 66

* Le groupe placebo externe provient d'une autre étude contrôlée randomisée (étude NEURO-TTR).
[a] Patients ayant reçu un traitement antérieur par le tafamidis ou le diflunisal.
Se base sur un MMRM ajusté par pondération du score de propension avec les effets des variables catégorielles pour le traitement, le temps, l'interaction traitement/temps et le stade de la maladie, la présence de la mutation Val30Met, le traitement antérieur et avec des covariables fixes pour les valeurs à l'inclusion et l'interaction valeurs à l'inclusion/temps.
Les modèles des sous-groupes comprenaient également les interactions traitement/sous-groupe, temps/sous-groupe et traitement/temps/sous-groupe. Les données obtenues jusqu'à la semaine 66 ont été incluses.
Le sous-groupe CM comprend des patients chez lesquels une CAF avait été diagnostiquée à l'inclusion dans l'étude ou des patients chez lesquels une épaisseur du septum IV ≥13 mm sans hypertension avait été diagnostiquée au début de l'étude [anamnèse ou diagnostic au cours de l'étude].
Les différences entre les traitements exprimées par la MMC après 66 semaines (WAINZUA – placebo) sont indiquées avec un IC à 95 % (non ajusté).
CAF = cardiomyopathie amyloïde familiale, CM = cardiomyopathie, IC = intervalle de confiance, MMC = moyenne des moindres carrés, MMRM = modèle mixte à mesures répétées.
Dans une analyse exploratoire des examens de la fonction cardiaque au moyen d'échocardiogrammes en série, une amélioration du rapport E/e' (une mesure de la fonction diastolique du ventricule gauche) a été mise en évidence dans le sous-groupe cardiomyopathie après un traitement de 65 semaines par WAINZUA (différence de la MMC ajustée en fonction du témoin placebo: 3,94 [IC à 95 % 6,46, 1,42]). Des variations en faveur d'un bénéfice de WAINZUA par rapport au placebo à la semaine 66 ont également été observées quant aux critères d'évaluation cardiaques exploratoires prédéfinis, à savoir l'épaisseur de la paroi du ventricule gauche (VG) (différence des MMC -0,04 cm [IC à 95 % -0,12, 0,04]), l'épaisseur du septum interventriculaire (différence de la MMC -0,05 cm [IC à 95 % -0,16, 0,06]) et le NT proBNP, un biomarqueur pronostique d'insuffisance cardiaque (MMC géométrique 0,88 [IC à 95 % 0,68, 1,14]). Malgré ces valeurs observées, un bénéfice clinique en présence d'une cardiomyopathie reste encore à confirmer.
Analyse effectuée à la semaine 85 (analyse de fin de traitement)
Les données de la semaine 85 ne sont pas disponibles pour le groupe placebo externe, car la durée du traitement dans l'étude NEURO-TTR était seulement de 66 semaines.
L'effet observé dans le groupe traité par WAINZUA concernant le score composite mNIS+7 était constant et s'est maintenu jusqu'à la fin du traitement à la semaine 85. La variation moyenne (ÉT) du score composite mNIS+7 vs valeur à l'inclusion était de -0,04 % (16,2) à la semaine 35, de -0,21 % (17,6) à la semaine 66 et de -2,9 % (20,5) à la semaine 85. Le score total moyen au questionnaire Norfolk QoL-DN est resté stable jusqu'à la semaine 85. Dans le groupe éplontersen, la variation moyenne (ÉT) du score au questionnaire Norfolk QoL-DN vs valeur à l'inclusion était de -4,8 (16,5) à la semaine 35, de -7,2 (18,5) à la semaine 66 et de 6,2 (18,0) à la semaine 85.
Les paramètres NSC, PND et IMCm sont restés stables jusqu'à la semaine 85, tandis que le SF-36 continuait à montrer une tendance à l'amélioration.
PharmacocinétiqueLes propriétés pharmacocinétiques (PK) de WAINZUA ont été évaluées après l'administration sous-cutanée d'une dose unique et de doses multiples (une fois toutes les 4 semaines) chez des sujets sains, et de doses multiples (une fois toutes les 4 semaines) chez des patients atteints d'ATTRv-PN.
Absorption
Après une administration sous-cutanée, l'éplontersen est rapidement absorbé dans la circulation systémique, et le temps nécessaire pour atteindre la concentration plasmatique maximale est d'environ 2 heures, d'après les estimations de population.
Distribution
Selon les études animales (chez la souris, le rat et le singe), l'éplontersen est principalement distribué dans le foie et le cortex rénal après son administration sous-cutanée. L'éplontersen se lie fortement aux protéines plasmatiques humaines (> 98 %). L'estimation de population du volume de distribution central apparent est de 12,9 l et celle du volume de distribution périphérique apparent de 11,100 l.
Métabolisme
L'éplontersen est métabolisé dans le foie par des endonucléases et des exonucléases en de courts fragments d'oligonucléotides de taille variable. Aucun métabolite circulant majeur n'a été détecté chez l'humain. Les oligonucléotides thérapeutiques, tels que l'éplontersen, ne sont généralement pas métabolisés par les enzymes du CYP.
Élimination
L'éplontersen est principalement éliminé par métabolisme, puis par excrétion rénale des courts métabolites d'oligonucléotides. La fraction moyenne de l'OAS intact éliminé dans l'urine était inférieure à 1 % de la dose administrée en l'espace de 24 heures. Selon les estimations de population, la demi-vie d'élimination terminale est d'environ 3 semaines.
Linéarité/non-linéarité
Une augmentation de la Cmax et de l'ASC de l'éplontersen légèrement supérieure à une augmentation proportionnelle à la dose a été observée chez des volontaires sains après l'administration de doses sous-cutanées uniques allant de 45 à 120 mg (c.-à-d. 1 à 2,7 fois la dose recommandée).
Les estimations de population des concentrations maximales (Cmax), des concentrations résiduelles (Cmin) et de l'aire sous la courbe (ASCτ) à l'état d'équilibre étaient de 0,218 μg/ml, de 0,000200 μg/ml et de 1,95 μg h/ml respectivement, après l'administration d'une dose de 45 mg toutes les 4 semaines chez des patients atteints d'ATTRv-PN. Aucune accumulation plasmatique d'éplontersen (Cmax et ASC) n'a été observée après son administration répétée (une fois toutes les 4 semaines). Une accumulation d'éplontersen a été observée à la Cmin et l'état d'équilibre est atteint après environ 17 semaines.
Cinétique pour certains groupes de patients
D'après l'analyse pharmacocinétique et pharmacodynamique de population, le poids corporel, le sexe, l'ethnie et la présence de la mutation Val30Met n'ont pas d'incidence d'importance clinique sur l'exposition à l'éplontersen ou sur la réduction de la concentration sérique de TTR à l'état d'équilibre. Dans certains cas, l'évaluation finale n'a été possible qu'en partie, car les covariables étaient limitées par les chiffres globalement faibles.
Patients âgés
Globalement, aucune différence des caractéristiques pharmacocinétiques n'a été observée entre les patients âgés (≥65 ans) et les patients plus jeunes.
Troubles de la fonction rénale
Aucune étude clinique formelle n'a été réalisée en vue d'évaluer l'influence d'une insuffisance rénale sur la pharmacocinétique de l'éplontersen. L'analyse pharmacocinétique et pharmacodynamique de population n'a révélé aucune différence d'importance clinique concernant la pharmacocinétique ou la pharmacodynamique de l'éplontersen en présence d'une insuffisance rénale légère et modérée (DFGe ≥45 à < 90 ml/min). L'éplontersen n'a pas été étudié chez les patients atteints d'insuffisance rénale sévère ou terminale.
Troubles de la fonction hépatique
Aucune étude clinique formelle n'a été réalisée en vue d'évaluer l'influence d'une insuffisance hépatique sur l'éplontersen. L'analyse pharmacocinétique et pharmacodynamique de population n'a révélé aucune différence d'importance clinique concernant la pharmacocinétique ou la pharmacodynamique de l'éplontersen en présence d'une insuffisance hépatique légère (bilirubine totale ≤1 x LSN et ASAT > 1 x LSN, ou bilirubine totale > 1,0 à 1,5 x LSN et tout taux d'ASAT). L'éplontersen n'a pas été étudié chez les patients atteints d'une insuffisance hépatique modérée ou sévèreou chez les patients ayant déjà subi une greffe de foie.
Interactions médicamenteuses
Aucune étude clinique formelle en vue d'évaluer les interactions médicamenteuses n'a été réalisée. Les études in vitro indiquent que l'éplontersen n'est pas un substrat ou un inhibiteur des transporteurs, qu'il n'interagit pas avec les médicaments fortement liés aux protéines plasmatiques et qu'il n'est pas un inhibiteur ou un inducteur des enzymes du CYP. Les oligonucléotides thérapeutiques, tels que l'éplontersen, ne sont généralement pas des substrats des enzymes du CYP. Par conséquent, l'éplontersen ne devrait pas entraîner d'interactions médicamenteuses, ni être affecté par les effets médicamenteux médiés par les transporteurs de médicaments, la liaison aux protéines plasmatiques ou les enzymes du CYP.
Données précliniquesToxicité non clinique/Toxicité en cas d'administration répétée
L'administration répétée d'éplontersen ou d'un substitut spécifique aux rongeurs a entraîné une réduction des concentrations hépatiques d'ARNm de la TTR (réductions allant jusqu'à respectivement ~ 62 % et 82 % chez le singe et la souris) associée à des baisses consécutives des taux plasmatiques de TTR (réduction allant jusqu'à 70 % chez le singe). Aucune anomalie toxicologique pertinente en lien avec cette inhibition pharmacologique de l'expression de la TTR n'a été détectée.
La plupart des anomalies qui ont été observées après une administration répétée pendant une période allant jusqu'à 6 mois chez la souris et 9 mois chez le singe concernaient l'absorption et l'accumulation d'éplontersen et n'ont pas été considérées comme étant nocives. Des anomalies microscopiques dues à l'absorption d'éplontersen ont été observées dans différents types cellulaires de plusieurs organes chez toutes les espèces animales testées, y compris les monocytes/macrophages, l'épithélium tubulaire proximal du rein, les cellules de Kupffer du foie et les infiltrats d'histiocytes dans les ganglions lymphatiques et les sites d'injection.
Dans une étude de toxicité subchronique, une diminution très importante de la numération plaquettaire en raison d'hémorragies spontanées a été constatée chez un singe à la dose maximale testée (24 mg/kg/semaine). Aucun phénomène de ce type n'a été observé chez les singes ayant reçu une dose moyenne de 6 mg/kg/semaine, ce qui correspond à 73 fois l'ASC chez l'humain à la dose thérapeutique d'éplontersen recommandée.
Mutagénicité et carcinogénicité
In vitro et in vivo, l'éplontersen n'a pas montré de potentiel génotoxique et n'était pas carcinogène chez la souris transgénique rasH2.
L'éplontersen n'a pas montré de potentiel génotoxique lors des tests effectués in vitro (mutagénicité bactérienne, aberration chromosomique dans le poumon de hamster chinois) et in vivo (test du micronoyau sur moelle osseuse chez la souris).
Dans une étude de carcinogénicité sous-cutanée effectuée chez la souris transgénique rasH2, l'éplontersen a été administré pendant 26 semaines à des doses de 250, 500 et 1500 mg/kg/mois. Aucun signe de carcinogénicité liée à l'éplontersen n'a été observé après un traitement de 26 semaines chez la souris.
Toxicité sur la reproduction
Toxicité embryofœtale/Toxicité pour le développement/Fertilité
L'administration d'éplontersen chez la souris à des doses jusqu'à 38 fois supérieures à la dose mensuelle recommandée de 45 mg chez l'humain n'a eu aucun effet sur la fertilité ou sur le développement embryofœtal. L'éplontersen n'est pas pharmacologiquement actif chez la souris. L'administration d'un analogue de l'éplontersen spécifique à la souris entraînant une inhibition de > 90 % de l'expression de l'ARNm de la TTR chez la souris n'a cependant pas non plus eu d'effet sur la fertilité ou le développement embryofœtal.
Remarques particulièresIncompatibilités
En l'absence d'études de compatibilité, ce médicament ne doit pas être mélangé à d'autres médicaments.
Stabilité
Ce médicament ne doit pas être utilisé au-delà de la date figurant après la mention «EXP» sur l'emballage.
Remarques particulières concernant le stockage
Conserver au réfrigérateur (2-8°C).
WAINZUA peut être conservé dans son emballage d'origine à l'état non réfrigéré pendant 6 semaines maximum à une température ne dépassant pas 30°C. S'il n'est pas utilisé dans les 6 semaines, il doit être jeté.
Conserver le récipient dans l'emballage d'origine pour le protéger de la lumière.
Ne pas congeler. Ne pas exposer à la chaleur.
Tenir hors de portée des enfants.
Remarques concernant la manipulation
Examiner visuellement WAINZUA avant de l'utiliser. La solution doit être limpide et incolore à jaune. Ne pas administrer WAINZUA si la solution est trouble, si elle contient des particules visibles ou si elle a changé de couleur.
Le stylo prérempli à usage unique doit être éliminé dans un collecteur pour déchets perforants.
Des informations complémentaires et des instructions pour la préparation et l'administration de WAINZUA avec le stylo prérempli figurent dans la notice d'emballage et dans le «Mode d'emploi».
Numéro d’autorisation69332 (Swissmedic)
PrésentationWAINZUA solution injectable: emballage contenant 1 stylo prérempli à usage unique [B]
Titulaire de l’autorisationAstraZeneca AG, 6340 Baar
Mise à jour de l’informationMai 2025
|