CompositionPrincipes actifs
Lébrikizumab.
Le lébrikizumab est produit dans des cellules ovariennes de hamster chinois (cellules CHO) par la technologie de l'ADN recombinant.
Excipients
Histidine, acide acétique 99%, saccharose, polysorbate 20 et eau pour préparations injectables.
Indications/Possibilités d’emploiEbglyss est indiqué pour le traitement de la dermatite atopique modérée à sévère chez les adultes et les adolescents à partir de 12 ans, d'un poids corporel d'au moins 40 kg, lorsque le traitement par des médicaments topiques ne permet pas d'obtenir un contrôle adéquat de la maladie ou n'est pas médicalement recommandé.
Posologie/Mode d’emploiLe traitement doit être instauré par un médecin expérimenté dans le diagnostic et le traitement de la dermatite atopique.
Posologie habituelle
La dose recommandée de lébrikizumab est de 500 mg (deux injections de 250 mg) à la semaine 0 et à la semaine 2, suivie de 250 mg administrés par injection sous-cutanée toutes les deux semaines jusqu'à la semaine 16.
L'arrêt du traitement devra être envisagé chez les patients qui ne présentent aucune réponse clinique après 16 semaines de traitement.
Une fois la réponse clinique obtenue, la dose d'entretien de lébrikizumab recommandée est de 250 mg toutes les quatre semaines.
Le lébrikizumab peut être utilisé avec ou sans corticostéroïdes topiques (CST). Les inhibiteurs de la calcineurine topiques (ICT) peuvent être utilisés, mais ils doivent être limités aux zones sensibles telles que le visage, le cou, les zones intertrigineuses et les parties génitales.
Afin d'assurer la traçabilité des médicaments biotechnologiques, il convient de documenter pour chaque traitement le nom commercial et le numéro de lot.
Dose retardée
En cas d'oubli d'une dose, celle-ci devra être administrée dès que possible. Par la suite, le schéma d'administration devra être repris à la date prévue habituellement.
Instructions posologiques particulières
Patients présentant des troubles de la fonction hépatique ou rénale
Aucun ajustement posologique n'est nécessaire chez les patients présentant une insuffisance hépatique ou rénale (voir «Pharmacocinétique»).
Poids corporel
Aucun ajustement posologique n'est nécessaire en fonction du poids corporel (voir «Pharmacocinétique»).
Patients âgés (≥65 ans)
Aucun ajustement posologique n'est nécessaire chez les patients âgés (voir «Pharmacocinétique»).
Enfants de moins de 12 ans
La sécurité et l'efficacité du lébrikizumab chez les enfants de moins de 12 ans ou chez les adolescents de 12 à 17 ans et pesant moins de 40 kg ne sont pas encore établies. Aucune donnée n'est disponible.
Mode d'administration
Voie sous-cutanée.
Le lébrikizumab est administré par injection sous-cutanée dans la cuisse ou l'abdomen, à l'exception de la zone de 5 centimètres autour du nombril. Si une autre personne administre l'injection, il est également possible de la faire dans le haut du bras.
Pour la dose initiale de 500 mg, deux injections de 250 mg sont administrées consécutivement en choisissant des sites d'injection différents.
Il est recommandé de changer de site d'injection à chaque injection. Le lébrikizumab ne doit pas être injecté dans des zones où la peau est sensible, endommagée ou présente des hématomes ou des cicatrices.
Un patient peut s'auto-injecter le lébrikizumab ou le soignant du patient peut administrer le lébrikizumab si le médecin traitant juge que cela est approprié. Une formation adéquate des patients et/ou des soignants à l'administration du lébrikizumab devra être assurée avant utilisation. Des instructions d'utilisation détaillées sont incluses à la fin de l'information destinée aux patients.
Contre-indicationsHypersensibilité à la substance active ou à l'un des excipients (voir «Composition»).
Mises en garde et précautionsHypersensibilité
En cas de survenue d'une réaction d'hypersensibilité systémique (immédiate ou retardée), l'administration du lébrikizumab doit être interrompue et un traitement approprié doit être instauré.
Conjonctivite
Chez les patients traités par le lébrikizumab et qui développent une conjonctivite non résolue avec un traitement standard doivent effectuer un examen ophtalmologique (voir «Effets indésirables»).
Les patients doivent signaler à leur médecin tout nouveau symptôme oculaire ou tout symptôme qui s'aggrave.
Vaccins
Avant l'instauration du traitement par lébrikizumab, il est recommandé de s'assurer que les patients sont à jour de toutes les vaccinations appropriées selon l'âge, conformément aux recommandations vaccinales en vigueur. Les vaccins vivants et les vaccins vivants atténués ne doivent pas être administrés au cours du traitement par lébrikizumab car la sécurité et l'efficacité cliniques n'ont pas été établies. Les réponses immunitaires aux vaccins non vivants ont été évaluées dans un vaccin combiné diphtérie-tétanos-coqueluche (acellulaire) (TdaP) et un vaccin polysaccharidique contre les méningocoques (voir «Interactions»).
Helminthoses
Les patients présentant des infections connues par des helminthes ont été exclus des études cliniques. Les patients présentant des infections préexistantes par des helminthes doivent être traités avant de commencer le traitement par lébrikizumab. Si des patients sont infectés au cours du traitement par lébrikizumab et ne répondent pas au traitement anti-helminthique, le traitement par lébrikizumab doit être interrompu jusqu'à la guérison de l'infection.
InteractionsVaccins vivants
La sécurité et l'efficacité de l'utilisation concomitante du lébrikizumab et de vaccins vivants ou vivants atténués n'ont pas été étudiées. Les vaccins vivants et les vaccins vivants atténués ne doivent pas être administrés au cours du traitement par lébrikizumab (voir également «Mises en garde et précautions»).
Vaccins non vivants
Les réponses immunitaires aux vaccins non vivants ont été évaluées dans une étude au cours de laquelle des patients adultes atteints de dermatite atopique ont été traités par lébrikizumab 500 mg aux semaines 0 et 2, puis par lébrikizumab 250 mg toutes les deux semaines jusqu'à la semaine 16. Après 12 semaines d'administration de lébrikizumab, les patients ont reçu un vaccin TdaP, vaccin combiné tétanos, diphtérie et coqueluche acellulaire (dépendant des lymphocytes T) et un vaccin polysaccharidique contre les méningocoques (indépendant des lymphocytes T). Les réponses immunitaires à l'anatoxine tétanique et au vaccin polysaccharidique contre les méningocoques du sérogroupe C ont été analysées 4 semaines plus tard. Le traitement concomitant par lébrikizumab n'a pas eu d'impact négatif sur la production d'anticorps avec les deux vaccins non vivants. Dans l'étude, aucune interaction indésirables entre les vaccins non vivants et le lébrikizumab n'a été observée.
Traitements concomitants
Aucune étude n'a été réalisée pour déterminer les interactions pharmacocinétiques des médicaments.
Le risque que le lébrikizumab provoque des interactions médiées par les cytokines avec les enzymes CYP chez les patients atteints de dermatite atopique est considéré comme faible.
Grossesse, allaitementGrossesse
À ce jour, on ne dispose que de peu d'expérience concernant l'utilisation d'Ebglyss chez les femmes enceintes. Les études effectuées chez l'animal n'ont pas mis en évidence d'effets délétères directs ou indirects sur la reproduction (voir «Données précliniques»). Le lébrikizumab ne doit pas être utilisé pendant la grossesse, à moins que le bénéfice potentiel ne dépasse le risque potentiel pour le fœtus.
Allaitement
On ne sait pas si le lébrikizumab est excrété dans le lait maternel ou absorbé par voie systémique après ingestion. Les IgG maternelles étant présentes dans le lait maternel, un risque pour les nouveaux-nés/nourrissons allaités ne peut être exclu. Une décision doit être prise soit d'interrompre l'allaitement, soit d'interrompre le traitement par Ebglyss, en prenant en compte le bénéfice de l'allaitement pour l'enfant au regard du bénéfice du traitement pour la femme.
Fertilité
L'effet d'Ebglyss sur la fertilité humaine n'a pas été étudié. Les études effectuées chez l'animal n'ont montré aucune altération de la fertilité (voir «Données précliniques»).
Effet sur l’aptitude à la conduite et l’utilisation de machinesAucune étude correspondante n'a été effectuée.
Effets indésirablesRésumé du profil de sécurité
Les effets indésirables les plus fréquents sont des conjonctivites (6,9%), des réactions au site d'injection (2,6%), des conjonctivites allergiques (1,8%) et une sécheresse oculaire (1,4%).
Tableau présentant la liste des effets indésirables
Dans toutes les études cliniques menées sur la dermatite atopique, 1720 patients ont été traités par le lébrikizumab, dont 891 patients ont reçu du lébrikizumab pendant au moins un an. Sauf indication contraire, les fréquences sont basées sur les données regroupées de 4 études randomisées, en double aveugle, menées chez des patients atteints de dermatite atopique modérée à sévère au cours desquelles 783 patients ont été traités par lébrikizumab par voie sous-cutanée pendant la phase contrôlée par placebo (16 premières semaines de traitement).
Le tableau 1 présente les effets indésirables observés dans les études cliniques par classe de systèmes d'organes et par fréquence, en utilisant les catégories suivantes: très fréquent (≥1/10); fréquent (≥1/100 à < 1/10); occasionnel (≥1/1000 à <1/100); rare (≥1/10 000 à <1/1000); très rare (<1/10 000).
Tableau 1. Liste des effets indésirables
|
Classe de systèmes d'organes MedDRA
|
Fréquence
|
Effet indésirable
| |
Infections et infestations
|
Fréquent
|
Conjonctivite
| |
|
Occasionnel
|
Zona
| |
Affections hématolo-giques et du système lymphatique
|
Occasionnel
|
Éosinophilie
| |
Affections oculaires
|
Fréquent
|
Conjonctivite allergique
Sécheresse oculaire
| |
|
Occasionnel
|
Kératite
Blépharite
| |
Troubles généraux et anomalies au site d'administration
|
Fréquent
|
Réaction au site d'injection
|
Description d'effets indésirables spécifiques et informations complémentaires
Conjonctivite et événements apparentés
Au cours des 16 premières semaines de traitement, des cas de conjonctivite, de conjonctivite allergique, de blépharite et de kératite ont été rapportés plus fréquemment chez les patients traités par lébrikizumab (respectivement 6,9%, 1,8%, 0,8% et 0,6%) que sous placebo (respectivement 1,8%, 0,7%, 0,2% et 0,3%). La plupart des cas de conjonctivite, de conjonctivite allergique, de blépharite et de kératite étaient d'intensité légère à modérée, se sont améliorés ou ont disparu sans interruption ou arrêt du traitement.
Éosinophilie
Les patients traités par lébrikizumab ont présenté une augmentation moyenne de leur taux à l'inclusion d'éosinophiles, plus importante que les patients ayant reçu le placebo. En général, cette augmentation par rapport à la valeur de référence chez les patients traités par lébrikizumab était transitoire.
Une éosinophilie (>5000 cellules/µl) a été observée chez 0,4% des patients traités par lébrikizumab et chez aucun de ceux ayant reçu le placebo. L'éosinophilie n'a pas entraîné d'arrêt du traitement, et aucun trouble lié aux éosinophiles n'a été rapporté.
Infections
Dans toutes les études cliniques portant sur la dermatite atopique, des infections graves ont été signalées au cours des 16 premières semaines chez 0,4% des patients traités par lébrikizumab et chez 0,5% des patients du groupe placebo.
Réaction au site d'injection
Des réactions au site d'injection (y compris douleur, érythème et rash) ont été rapportées plus fréquemment chez les patients ayant reçu le lébrikizumab (2,6%) que chez ceux ayant reçu le placebo (1,5%). La majorité (95%) des réactions au site d'injection étaient d'intensité légère ou modérée et peu de patients (<0,5%) ont arrêté le traitement par lébrikizumab.
Zona (herpès zoster)
Un zona a été observé chez 0,6% des patients traités par lébrikizumab et chez aucun des patients du groupe placebo. Tous les épisodes de zona rapportés étaient d'intensité légère ou modérée, et aucun n'a entraîné l'arrêt définitif du traitement.
Immunogénicité
Comme pour toutes les protéines thérapeutiques, il existe la possibilité d'une immunogénicité avec le lébrikizumab. L'incidence des anticorps anti-médicaments (ADA) dépend dans une large mesure de la sensibilité et de la spécificité du test. En outre, l'incidence observée de la positivité des anticorps (y compris les anticorps neutralisants) dans un test peut être influencée par plusieurs facteurs, par exemple la méthodologie du test, la manipulation des échantillons, le moment de la collecte des échantillons, les médicaments concomitants et la maladie sous-jacente. Pour cette raison, la comparaison de l'incidence des anticorps dirigés contre le lébrikizumab avec l'incidence des anticorps dirigés contre d'autres médicaments peut induire en erreur.
Au bout de 12 mois de traitement, jusqu'à 2,8% des patients traités par 250 mg de lébrikizumab ont développé des anticorps anti-médicament (ADA) dirigés contre le principe actif. Ils étaient généralement neutralisants et leurs concentrations étaient faibles.
Tolérance à long terme
La tolérance à long terme du lébrikizumab a été évaluée dans 5 études cliniques: dans les deux études en monothérapie (ADvocate 1, ADvocate 2) pendant une durée maximale de 52 semaines, et dans l'étude en association avec les CST (ADhere) avec une étude d'extension à long terme (ADjoin) pendant 56 semaines au total et dans l'étude ADore en monothérapie chez des adolescents pendant également une durée maximale de 52 semaines. Le profil de sécurité du lébrikizumab en monothérapie jusqu'à la semaine 52 incluse ou en association avec des CST jusqu'à la semaine 56 est cohérent avec le profil de sécurité observé jusqu'à la semaine 16 (incluse).
Population pédiatrique
Adolescents âgés de 12 à 17 ans
La tolérance du lébrikizumab a été évaluée chez 372 patients âgés de 12 à 17 ans atteints de dermatite atopique modérée à sévère, dont 270 patients ayant reçu du lébrikizumab pendant au moins un an. Le profil de sécurité du lébrikizumab chez ces patients était comparable à celui des adultes atteints de dermatite atopique.
L'annonce d'effets secondaires présumés après l'autorisation est d'une grande importance. Elle permet un suivi continu du rapport bénéfice-risque du médicament. Les professionnels de santé sont tenus de déclarer toute suspicion d'effet secondaire nouveau ou grave via le portail d'annonce en ligne ElViS (Electronic Vigilance System). Vous trouverez des informations à ce sujet sur www.swissmedic.ch.
SurdosageDans les études cliniques, des doses intraveineuses uniques allant jusqu'à 10 mg/kg et des doses sous-cutanées multiples allant jusqu'à 500 mg ont été administrées à l'être humain lors des essais cliniques, sans observer de toxicité limitant la dose. Il n'y a pas de traitement spécifique en cas de surdosage de lébrikizumab. En cas de surdosage, le patient doit être surveillé afin de détecter tout signe ou symptôme d'effets indésirables et d'instaurer immédiatement un traitement symptomatique approprié.
Propriétés/EffetsCode ATC
D11AH10
Mécanisme d'action
Le lébrikizumab est un anticorps monoclonal d'immunoglobuline (IgG4) qui se lie avec une haute affinité à l'interleukine (IL)-13 et inhibe sélectivement la signalisation de l'IL-13 par la voie du récepteur alpha de l'IL-4 (IL-4Rα) et de l'hétérodimère du récepteur alpha 1 de l'IL-13 (IL-13Rα1), inhibant ainsi les effets de l'IL-13 en aval. L'inhibition de la signalisation de l'IL-13 devrait être bénéfique dans les maladies pour lesquelles l'IL-13 est un facteur clé de la pathogenèse de la maladie. Le lébrikizumab n'empêche pas la liaison de l'IL-13 au récepteur alpha 2 de l'IL-13 (IL-13Rα2 ou récepteur leurre), ce qui permet l'internalisation de l'IL-13 dans la cellule.
Pharmacodynamique
Dans les études cliniques, le lébrikizumab a réduit les taux sériques de périostine, d'immunoglobulines E (IgE) totales, de ligand de chimiokine CC(CCL)17 [chimiokine régulée par le thymus et par activation (TARC)], de CCL18 [[chimiokine régulée par le poumon et par activation (PARC)] et de CCL13 [protéine chimiotactique des monocytes-4 (MCP-4)]. La diminution des médiateurs de l'inflammation de type 2 apporte une preuve indirecte de l'inhibition de la voie de l'IL-13 par le lébrikizumab.
Efficacité clinique
Adultes et adolescents atteints de dermatite atopique
L'efficacité et la sécurité du lébrikizumab en monothérapie (ADvocate 1, ADvocate 2) et en association avec un CST (ADhere) ont été évaluées dans trois études pivot randomisées en double aveugle, contrôlées contre placebo chez 1062 adultes et adolescents (âgés de 12 à 17 ans et pesant ≥40 kg) atteints de dermatite atopique modérée à sévère, définie par un score d'indice de surface et de sévérité de l'eczéma (Eczema Area and Severity Index, EASI) ≥16, un score d'évaluation globale par l'investigateur (Investigator's Global Assessment, IGA) ≥3 et une surface corporelle atteinte (Body Surface Area, BSA) ≥10%. Les patients inclus dans les trois études présentaient auparavant une réponse insuffisante au traitement par voie topique ou bien les traitements par voie topique ne leur étaient pas médicalement recommandés (ce dernier critère ne s'appliquait pas à l'étude ADhere).
Dans les trois études, les patients ont reçu une dose initiale de 500 mg de lébrikizumab (deux injections de 250 mg) aux semaines 0 et 2, puis 250 mg toutes les deux semaines (Q2S) jusqu'à la semaine 16 ou un placebo équivalent selon un rapport de 2:1. Dans ADhere, les patients de l'étude ont également reçu un CST de puissance faible à modérée ou un ICT concomitant sur les lésions actives. Les patients étaient autorisés à recevoir un traitement de secours selon l'appréciation de l'investigateur pour contrôler les symptômes intolérables de la dermatite atopique. Les patients nécessitant un traitement de secours systémique ont arrêté le traitement à l'étude.
Les patients ayant obtenu un score IGA de 0 ou 1 ou une réduction d'au moins 75% de la réponse EASI (EASI 75) sans avoir reçu de traitement de secours étaient randomisés à nouveau en aveugle pour recevoir (i) du lébrikizumab 250 mg toutes les 2 semaines (Q2S), (ii) du lébrikizumab 250 mg toutes les 4 semaines (Q4S) ou (iii) un placebo pendant 52 semaines.
Dans les études ADvocate 1 et ADvocate 2, après avoir terminé l'étude de 52 semaines, et dans l'étude ADhere, après avoir terminé l'étude de 16 semaines, les patients avaient la possibilité de poursuivre le traitement dans une étude distincte d'extension à long terme (ADjoin).
Critères d'évaluation
Dans les trois études, les co-critères d'évaluation primaires étaient le pourcentage de patients présentant un score IGA 0 ou 1 («sans lésion» ou «lésion minime») avec une réduction de ≥2 points par rapport à l'inclusion, et le pourcentage de patients présentant une réduction d'au moins 75% du score EASI (EASI 75) entre l'inclusion et la semaine 16. Les critères d'évaluation secondaires majeurs comprenaient le pourcentage de patients ayant obtenu une réduction d'au moins 90% du score EASI (EASI 90), le pourcentage de patients avec une amélioration par rapport à l'inclusion d'au moins 4 points sur l'échelle d'évaluation numérique de prurit (Pruritus Numerical Rating Scale, Pruritus NRS), le pourcentage de patients avec une amélioration par rapport à l'inclusion d'au moins 4 points sur l'indice de qualité de vie en dermatologie (Dermatology Life Quality Index, DLQI) et l'impact des démangeaisons sur le sommeil (échelle de perte de sommeil). La variation par rapport à l'inclusion du score de mesure de l'eczéma par le patient (Patient Oriented Eczema Measure, POEM) constituait un autre critère d'évaluation secondaire.
Patients
Caractéristiques à l'inclusion
Les caractéristiques démographiques et les données initiales des patients des études ADvocate 1, ADvocate 2 et ADhere sont présentées dans le tableau 2.
Tableau 2. Données démographiques et caractéristiques à l'inclusion par étude
|
|
ADvocate 1 N = 424
|
ADvocate 2 N = 427
|
ADhere N = 211
| |
Âge (moyenne, années)
|
35,5
|
36,2
|
37,2
| |
Adolescents (12 à 17 ans) (%)
|
13,0
|
11,0
|
21,8
| |
Patients âgés (≥65 ans) (%)
|
7,3
|
7,7
|
9,5
| |
Poids (moyenne, kg)
|
77,7
|
76,5
|
76,2
| |
Femmes (%)
|
50,5
|
49,4
|
48,8
| |
Origine ethnique
|
|
|
| |
Blanc (%)
|
68,2
|
59,3
|
61,6
| |
Asiatique (%)
|
16,5
|
28,6
|
14,7
| |
Noir (%)
|
11,6
|
8,2
|
13,3
| |
IGA 3 (DA modérée) (%)
|
59,7
|
63,2
|
69,2
| |
IGA 4 (DA sévère) (%)
|
40,3
|
36,8
|
30,8
| |
Traitement systémique précédent (%)*
|
54,0
|
55,5
|
47,4
| |
EASI (moyenne)
|
29,6
|
29,7
|
27,3
| |
Score NRS de prurit (moyenne)
|
7,3
|
7,1
|
7,1
| |
DLQI (moyenne)
|
15,4
|
15,5
|
14,4
| |
Échelle de perte de sommeil (moyenne)
|
2,3
|
2,2
|
2,0
| |
POEM (moyenne)
|
20,8
|
20,8
|
19,5
|
*Corticostéroïdes, cyclosporine, photothérapie et dupilumab (ADhere uniquement)
Réponse clinique
Études en monothérapie (ADvocate 1 et ADvocate 2) – phase d'induction, semaine 0 à 16
Dans l'étude ADvocate 1 et ADvocate 2, un pourcentage significativement plus élevé de patients du groupe lébrikizumab 250 mg Q2S ont obtenu un score IGA 0 ou 1, avec une amélioration ≥2 points par rapport à l'inclusion, une réponse EASI 75, une réponse EASI 90 et une amélioration de ≥4 points sur le score NRS du prurit à la semaine 16 par rapport au placebo (voir tableau 3).
Tableau 3. Résultats d'efficacité du lébrikizumab en monothérapie à la semaine 16 dans les études ADvocate 1 et ADvocate 2
|
|
ADvocate 1
|
ADvocate 2
| |
|
Semaine 16
| |
|
Placebo
N = 141
|
LEB 250 mg Q2S
N = 283
|
Placebo
N = 146
|
LEB
250 mg Q2S
N = 281
| |
IGA 0 ou 1, %a
|
12,7
|
43,1*
|
10,8
|
33,2*
| |
EASI 75, %b
|
16,2
|
58,8*
|
18,1
|
52,1*
| |
EASI 90, %b
|
9,0
|
38,3*
|
9,5
|
30,7*
| |
NRS du prurit (amélioration ≥4 points), %c
|
13,0
|
45,9*
|
11,5
|
39,8*
|
LEB = lébrikizumab; N = nombre de patients
a Patients présentant un score IGA de 0 ou 1 («sans lésion» ou «lésion minime») avec une réduction >2 points par rapport à l'inclusion sur une échelle IGA de 0 à 4
b Patients présentant une réduction de 75% ou 90% de la réponse EASI entre l'inclusion et la semaine 16.
c Le pourcentage est calculé par rapport au nombre de patients présentant un score NRS de prurit à l'inclusion dans l'étude ≥4.
*p<0,001 par rapport au placebo
Dans les deux études, les patients randomisés dans le groupe lébrikizumab ont été moins nombreux à avoir recours à un traitement de secours (corticostéroïdes topiques, corticostéroïdes systémiques, immunosuppresseurs) que les patients randomisés dans le groupe placebo (14,7% contre 36,6%, respectivement dans les deux études).
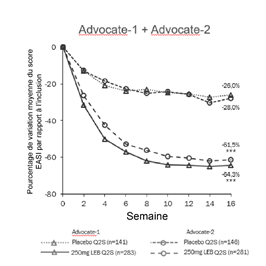
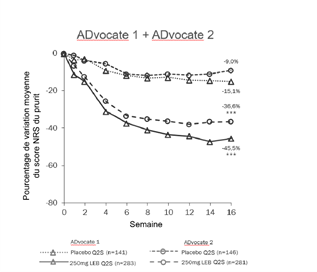
La figure 1 et la figure 2 montrent le pourcentage de variation moyenne de la réponse EASI et du score NRS du prurit entre l'inclusion et la semaine 16.
|
Figure 1. Pourcentage de variation moyenne de la réponse EASI entre l'inclusion et la semaine 16 dans les études ADvocate 1 et 2
|
Figure 2. Pourcentage de variation moyenne du score NRS du prurit entre l'inclusion et la semaine 16 dans les études ADvocate 1 et 2
| |
|
|
|
|
***p<0,001 par rapport au placebo
Les effets du traitement dans les sous-groupes (poids, âge, sexe, origine ethnique, sévérité de la maladie et utilisation antérieure de traitements systémiques) dans les études ADvocate 1 et ADvocate 2 étaient conformes aux résultats de l'ensemble de la population de l'étude pendant la phase d'induction.
Études en monothérapie (ADvocate 1 et ADvocate 2) – Période d'entretien, semaines 16 à 52
Pour évaluer le maintien de la réponse, 157 patients de l'étude ADvocate 1 et 134 patients de l'étude ADvocate 2, traités par le lébrikizumab 250 mg Q2S et ayant atteint un score IGA de 0 ou 1 ou une réponse EASI 75 à la semaine 16 sans traitement de secours topique ou systémique, ont été randomisés une nouvelle fois en aveugle selon un rapport de 2:2:1. Les patients ont reçu en aveugle pendant 36 semaines supplémentaires soit (i) du lébrikizumab 250 mg Q2S, soit (ii) du lébrikizumab 250 mg Q4S, soit (iii) un placebo. La durée totale de l'étude était de 52 semaines (voir tableau 4).
Tableau 4. Résultats d'efficacité du lébrikizumab en monothérapie à la semaine 52 chez les patients répondant au traitement à la semaine 16 dans les études ADvocate 1 et ADvocate 2 (analyse groupée)
|
|
ADvocate 1 et ADvocate 2 (regroupées)
| |
|
Semaine 52
| |
|
Placebod
(retrait du LEB)
N = 60
|
LEB 250 mg
Q2S
N= 113
|
LEB 250 mg
Q4S
N = 118
| |
IGA 0 ou 1, %a
|
47,9
|
71,2*
|
76,9**
| |
EASI 75, %b
|
66,4
|
78,4
|
81,7*
| |
EASI 90, %b
|
41,9
|
64,0*
|
66,4**
| |
NRS du prurit (amélioration ≥4 points), %c
|
66,3
|
84,6
|
84,7
|
a Patients présentant un score IGA 0/1 avec une amélioration ≥2 points par rapport à l'inclusion à la semaine 16, qui présentaient toujours un score IGA 0/1 avec une amélioration ≥2 points à la semaine 52.
b Patients ayant obtenu une réponse EASI 75 à la semaine 16 et qui présentaient toujours une réponse EASI 75 à la semaine 52 ou patients ayant obtenu une réponse EASI 75 à la semaine 16 et qui présentaient une réponse EASI 90 à la semaine 52.
c Le pourcentage est calculé par rapport au nombre de patients présentant un score NRS de prurit à l'inclusion dans l'étude ≥4.
d Patients répondant au lébrikizumab 250 mg Q2S à la semaine 16 (IGA 0 ou 1 ou EASI 75) et à nouveau randomisés dans le groupe placebo.
*p<0,05; **p<0,01 par rapport au placebo.
Étude avec CST concomitant (ADhere)
Dans l'étude ADhere, entre l'inclusion et la semaine 16, un pourcentage significativement plus élevé de patients randomisés dans le groupe lébrikizumab 250 mg Q2S + CST a obtenu un score IGA de 0 ou 1, une réponse EASI 75 et des améliorations ≥4 points du score NRS de prurit, par rapport aux patients recevant le placebo + CST (voir tableau 5).
Tableau 5. Résultats d'efficacité du traitement concomitant avec lébrikizumab et CST à la semaine 16 dans l'étude ADhere
|
|
ADhere
| |
|
Semaine 16
| |
|
Placebo + CST
N = 66
|
LEB
250 mg Q2S + CST
N = 145
| |
IGA 0 ou 1, %a
|
22,1
|
41,2*
| |
EASI 75, %b
|
42,2
|
69,5**
| |
EASI 90, %b
|
21,7
|
41,2**
| |
NRS du prurit (amélioration ≥4 points), %c
|
31,9
|
50,6*
|
a Patients présentant un IGA de 0 ou 1 («sans lésion» ou «lésion minime») avec une réduction de ≥2 points par rapport à la valeur initiale sur une échelle IGA de 0 à 4
b Patients présentant une réduction de l'EASI de 75% ou 90% entre la référence et la semaine 16.
c Le pourcentage est calculé par rapport au nombre de patients présentant un NRS du prurit de référence ≥4.
*p<0,05; **p<0,001 vs Placebo
Dans l'étude ADhere, les patients ayant reçu 250 mg de lébrikizumab Q2S + CTS de la semaine 0 à la semaine 16 ont moins souvent eu recours au CTS que ceux ayant reçu un placebo + CST (1,4% respectivement 4,5%).
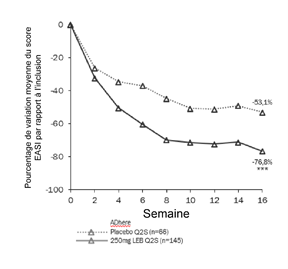
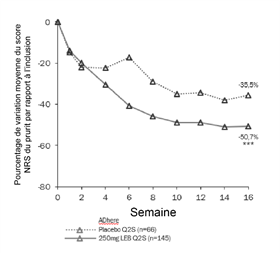
La figure 3 et la figure 4 montrent le pourcentage de variation moyenne du score EASI par rapport à la valeur initiale et le pourcentage de variation moyenne du score NRS du prurit par rapport à la valeur initiale jusqu'à la semaine 16.
|
Figure 3. Pourcentage de variation moyenne des moindres carrés du score EASI par rapport à la valeur initiale de l'étude ADhere
|
Figure 4. Pourcentage de variation moyenne des moindres carrés du score NRS du prurit par rapport à la valeur initiale de l'étude ADhere
| |
|
|
|
|
***p<0,001 par rapport au placebo ***p<0,001 par rapport au placebo
Dans l'étude ADhere, les effets du traitement dans les sous-groupes (poids, âge, sexe, origine ethnique, degré de gravité de la maladie et utilisation préalable de traitements systémiques) étaient conformes aux résultats de l'ensemble de la population étudiée.
Les patients qui étaient répondeurs à la semaine 16 dans l'étude ADhere et qui sont entrés dans l'étude ADjoin étaient traités soit par lébrikizumab 250 mg Q2S, soit par lébrikizumab 250 mg Q4S et ont maintenu leurs réponses jusqu'à 56 semaines (75,4% respectivement 86,8% pour un score IGA de 0 ou 1 et 85,6% respectivement 81,2% pour la réponse EASI 75).
Résultats rapportés par le patient et qualité de vie liée à la santé
Dans les deux études en monothérapie (ADvocate 1 et ADvocate 2), le lébrikizumab 250 mg Q2S a amélioré de manière significative les résultats rapportés par les patients en termes degré de sévérité de la maladie (POEM), de l'impact des démangeaisons sur le sommeil (échelle de perte de sommeil) et de la qualité de vie liée à la santé (DLQI) à la semaine 16 par rapport au placebo. Une proportion significativement plus importante de patients traités par le lébrikizumab ont présenté des améliorations cliniquement significatives du DLQI (défini comme une réduction de ≥4 points par rapport à la référence), du POEM et de l'échelle de perte de sommeil de la référence à la semaine 16 par rapport au groupe placebo (voir tableau 6).
Dans l'étude CST (ADhere), le lébrikizumab 250 mg Q2S + CST a amélioré l'impact des démangeaisons sur le sommeil (échelle de perte de sommeil), le degré de sévérité de la maladie (POEM) rapporté par le patient et la qualité de vie liée à la santé (DLQI) à la semaine 16 par rapport au placebo + CST (voir tableau 6).
Tableau 6. Résultats rapportés par le patient (résultats de la qualité de vie liés à la santé) du lébrikizumab en monothérapie à la semaine 16 dans l'étude ADvocate 1 et ADvocate 2 ou avec traitement concomitant par CST à la semaine 16 dans l'étude ADhere
|
|
ADvocate 1
|
ADvocate 2
|
ADhere
| |
|
Semaine 16
| |
|
Placebo
N = 141
|
LEB 250 mg Q2S
N = 283
|
Placebo
N = 146
|
LEB 250 mg Q2S
N = 281
|
Placebo + CST
N = 66
|
LEB 250 mg Q2S + CST
N = 145
| |
DLQI (adultes) (amélioration ≥4 points), %a
|
33,8
|
75,6**
|
33,6
|
66,3**
|
58,7
|
77,4*
| |
Échelle de perte de sommeil (Sleep-Loss-Scale) (amélioration ≥2 points), %b
|
4,7
|
39.0**
|
8,2
|
28,0**
|
18,4
|
34,5*
| |
POEM; variation de la moyenne des LS par rapport à l'inclusion (± ET)
|
-3,9
(± 0,72)
|
-11,3**
(± 0,47)
|
-3,5
(± 0,77)
|
-9,5**
(± 0,52)
|
-6,24
(± 1,04)
|
-10,23**
(± 0,73)
|
LS = least squares (moindres carrés), ET = écart-type
a Participants avec DLQI ≥4 points par rapport à l'inclusion
b Participants avec ≥2 points dans l'échelle de perte de sommeil par rapport à l'inclusion
*p<0,05; **p<0,001 par rapport au placebo
Adolescents (de 12 à 17 ans)
Dans les études en monothérapie ADvocate 1 et ADvocate 2, l'âge moyen des patients adolescents était de 14,6 ans, le poids moyen de 68,2 kg et de 56,9% étaient des jeunes filles. Dans ces études, 63,7% avaient un score IGA initial de 3 (dermatite atopique modérée), 36,3% avaient un score IGA initial (à l'inclusion) de 4 (dermatite atopique sévère) et 47,1% avaient déjà reçu un traitement systémique. Dans l'étude avec traitement concomitant par CST (ADhere), l'âge moyen des patients adolescents était de 14,6 ans, le poids moyen de 62,2 kg et 50,0% étaient des jeunes filles. Dans cette étude, 76,1% avaient un score IGA initial de 3 (dermatite atopique modérée), 23,9% un score IGA initial de 4 (dermatite atopique sévère) et 23,9% avaient déjà reçu un traitement systémique.
Les résultats d'efficacité à la semaine 16 chez les patients adolescents sont présentés dans le tableau 7.
Tableau 7. Résultats d'efficacité du lébrikizumab en monothérapie dans les études ADvocate 1, ADvocate 2 et du lébrikizumab associé à un CST dans l'étude ADhere à la semaine 16 chez les patients adolescents
|
|
ADvocate 1
|
ADvocate 2
|
ADhere
| |
|
Semaine 16
| |
|
Placebo
N = 18
|
LEB
250 mg Q2S
N = 37
|
Placebo
N = 17
|
LEB
250 mg Q2S
N = 30
|
Placebo + CST
N = 14
|
LEB
250 mg Q2S + CST
N = 32
| |
IGA 0 ou 1, %a
|
22.2
|
48.6
|
5.9
|
44.1**
|
28.6
|
57.3
| |
EASI 75, %a
|
22.2
|
62.2**
|
12.0
|
61.7**
|
57.1
|
88.0*
| |
EASI 90, %a
|
16.7
|
45.9*
|
6.1
|
34.3*
|
28.6
|
55.1
| |
NRS du prurit (amélioration ≥4 points), %b
|
22.8
|
54.3*
|
0.3
|
42.1
|
13.8
|
45.8
| |
a
À la semaine 16, les patients présentant un score IGA de 0 à 1 («sans lésion» ou «lésion minime») avec une réduction ≥2 points par rapport à l'inclusion sur une échelle IGA de 0 à 4 ou une réduction de 75% ou 90% de la réponse EASI entre l'inclusion et la semaine 16.
b Le pourcentage est calculé par rapport au nombre de patients présentant un score NRS du prurit à l'inclusion ≥4.
*p<0,05; **p<0,01 par rapport au placebo.
|
Les patients adolescents traités par lébrikizumab ou lébrikizumab + CST ont obtenu des améliorations cliniquement significatives de la sévérité de la maladie et ont maintenu une réponse jusqu'à la semaine 52. Des données supplémentaires issues de l'étude ADore à bras unique portant sur le lébrikizumab chez 206 adolescents confirment l'efficacité du lébrikizumab chez les patients adolescents jusqu'à 52 semaines de traitement.
PharmacocinétiqueAbsorption
Après injection sous-cutanée de 250 mg de lébrikizumab, les concentrations sériques maximales étaient atteintes environ 7 à 8 jours après l'administration.
Après les doses de charge de 500 mg de la semaine 0 et de la semaine 2, les concentrations sériques à l'état d'équilibre ont été atteintes avec la première dose de 250 mg Q2S à la semaine 4.
D'après une analyse pharmacocinétique (PK) de population, les concentrations minimales à l'état d'équilibre (Cmin,éq) prévues après une administration sous-cutanée de 250 mg de lébrikizumab Q2S et Q4S chez des patients atteints de dermatite atopique (médiane et 5e-95e percentile) ) étaient respectivement de 87 (46-159) µg/ml et 36 (18-68) µg/ml.
La biodisponibilité absolue a été estimée à 86 % d'après une analyse de pharmacocinétique de population. L'emplacement du site d'injection n'avait pas d'influence significative sur l'absorption du lébrikizumab.
Distribution
D'après une analyse de pharmacocinétique de population, le volume de distribution total à l'état d'équilibre était de 5,14 l.
Métabolisme
Aucune étude spécifique du métabolisme n'a été menée car le lébrikizumab est une protéine. Il est attendu que le lébrikizumab se dégrade en petits peptides et en acides aminés individuels par des voies cataboliques de la même manière que les IgG endogènes.
Élimination
Dans l'analyse de pharmacocinétique de population, la clairance était de 0,154 l/jour et était indépendante de la dose. La demi-vie d'élimination moyenne était d'environ 24,5 jours.
Linéarité/non-linéarité
Le lébrikizumab a présenté une pharmacocinétique linéaire avec une augmentation de l'exposition proportionnelle à la dose sur un intervalle de doses allant de 37,5 à 500 mg administrées par injection sous-cutanée chez des patients atteints de DA ou chez des volontaires sains.
Cinétique pour certains groupes de patients
Sexe, âge et origine ethnique
Le sexe, l'âge (de 12 à 93 ans) et l'origine ethnique (64% de Caucasiens, 15% de Noirs/Afro-Américains, 16% d'Asiatiques et 6% d'autres) n'ont pas eu d'effet significative sur la pharmacocinétique du lébrikizumab.
Troubles de la fonction hépatique et rénale
Aucune étude spécifique de pharmacologie clinique n'a été menée pour évaluer l'effet d'une insuffisance hépatique ou rénale sur la pharmacocinétique du lébrikizumab. Le lébrikizumab, en tant qu'anticorps monoclonal, ne devrait pas faire l'objet d'une élimination hépatique ou rénale significative. Les analyses de pharmacocinétique de population ont inclus 54 (3%) sujets avec un taux d'enzymes hépatiques alanine aminotransférase (ALAT) ou aspartate aminotransférase (ASAT) élevé ≥1,5 x LSN au début de l'étude, et 347 (22%) sujets présentant une fonction rénale réduite (taux de filtration glomérulaire entre 30 et 89 ml/min) au début de l'étude. Les analyses de pharmacocinétique de population montrent que les marqueurs de la fonction hépatique ou rénale n'impactent pas la pharmacocinétique du lébrikizumab.
Poids corporel
L'exposition au lébrikizumab était inférieure chez les 10% de patients ayant un poids corporel plus élevé (>100 kg).
Enfants et adolescents
D'après l'analyse de pharmacocinétique de population, les adolescents âgés de 12 à 17 ans atteints de dermatite atopique présentaient des concentrations sériques minimales de lébrikizumab légèrement plus élevées que les adultes, ce qui était lié à leur distribution de poids corporel plus faible.
Données précliniquesLes données précliniques issues des études conventionnelles de toxicité en administration répétée (y compris les critères de pharmacologie de sécurité) et de toxicité sur la reproduction et le développement n'ont pas révélé de risque particulier pour l'homme.
Génotoxicité
Le potentiel mutagène du lébrikizumab n'a pas été évalué; cependant, il n'est pas anticipé que les anticorps monoclonaux altèrent l'ADN ou les chromosomes.
Carcinogénicité
Aucune étude de carcinogénicité n'a été menée avec le lébrikizumab. L'évaluation des preuves disponibles liées à l'inhibition de l'IL-13 et aux données de toxicologie animale avec le lébrikizumab ne suggère pas de potentiel carcinogène pour le lébrikizumab.
Remarques particulièresIncompatibilités
En l'absence d'études de compatibilité, ce médicament ne doit pas être mélangé à d'autres médicaments.
Stabilité
Ce médicament ne doit pas être utilisé au-delà de la date figurant après la mention «EXP» sur l'emballage.
Remarques particulières concernant le stockage
Conserver au réfrigérateur (2-8 °C).
Ne pas congeler.
Conserver dans l'emballage d'origine pour le protéger de la lumière.
Tenir hors de portée des enfants.
Une fois hors du réfrigérateur, ne pas conserver Ebglyss au-dessus de 30°C et l'utiliser ou l'éliminer dans les 7 jours. Ne pas remettre au réfrigérateur après l'avoir conservé hors du réfrigérateur.
Remarques concernant la manipulation
Les instructions détaillées pour la préparation et l'administration d'Ebglyss en seringue préremplie ou en stylo prérempli sont indiquées à la fin de la notice.
La solution doit être limpide à irisée, incolore à légèrement jaunâtre ou légèrement brunâtre et exempte de particules visibles. Si la solution est trouble, présente une modification de la couleur ou contient des particules visibles, elle ne doit pas être utilisée.
Avant l'injection d'Ebglyss, retirer la seringue préremplie ou le stylo prérempli de 250 mg du réfrigérateur et la/le laisser à température ambiante pendant 45 minutes.
La seringue préremplie ou le stylo prérempli ne doit pas être exposé à la chaleur ou à la lumière directe du soleil.
Ne pas secouer.
Tout médicament non utilisé ou déchet doit être éliminé conformément à la réglementation nationale.
Numéro d’autorisation69344, 69460 (Swissmedic)
PrésentationEbglyss, solution injectable en seringue préremplie
1 seringue préremplie [B]
2 seringues préremplies [B]
Ebglyss, solution injectable en stylo prérempli
1 stylo prérempli [B]
2 stylos préremplis [B]
Titulaire de l’autorisationAlmirall AG, 8304 Wallisellen
Mise à jour de l’informationAvril 2024
|