CompositionPrincipes actifs
Paracétamol; Ibuprofène (sous forme d'ibuprofène sodique dihydraté).
Excipients
Chlorhydrate de cystéine monohydraté, phosphate disodique dihydraté, mannitol, acide chlorhydrique (pour l'ajustement du pH), hydroxyde de sodium (pour l'ajustement du pH) et eau pour préparations injectables.
Excipient à effet notoire:
Sodium 42,43 mg pour 100 mL (0,4243 mg/mL). Voir «Mises en garde et précautions».
Indications/Possibilités d’emploiComboval est indiqué chez l'adulte pour le traitement symptomatique de courte durée des douleurs aiguës modérées, lorsqu'un mode d'administration intraveineux est considéré comme cliniquement nécessaire et/ou lorsque les autres voies d'administration sont impossibles.
Posologie/Mode d’emploiPosologie
Pour une administration intraveineuse et une utilisation de courte durée, limitée à un maximum de deux jours. Les effets indésirables peuvent être minimisés en prenant la dose minimale efficace pendant la plus courte durée nécessaire pour contrôler les symptômes (voir «Mises en garde et précautions»).
Adultes (pesant > 50 kg)
Administrer un flacon (100 mL) de Comboval, en perfusion de 15 minutes toutes les 6 heures, à la demande. Ne pas dépasser une dose quotidienne totale de quatre flacons (400 mL), équivalant à 4000 mg (4 g) de paracétamol et 1200 mg d'ibuprofène.
Adultes (pesant ≤50 kg)
Les adultes pesant 50 kg ou moins doivent être traités en fonction de leur poids corporel, à une posologie de 1,5 mL/kg, en perfusion de 15 minutes toutes les 6 heures, à la demande. Cela équivaut à une dose unique maximale de 75 mL (jeter le reliquat de produit restant dans le flacon) et à une dose quotidienne totale de 3000 mg (3 g) de paracétamol et 900 mg d'ibuprofène.
Population pédiatrique
Comboval est contre-indiqué pour les patients de moins de 18 ans (voir «Contre-indications»).
Populations particulières
Sujets âgés
Chez les patients âgés, la sélection de la dose doit être prudente, en commençant généralement à la plus faible dose de l'éventail posologique, compte tenu de la plus grande fréquence des insuffisances hépatique, rénale ou cardiaque, ainsi que des comorbidités ou des autres traitements médicamenteux concomitants.
Les personnes âgées sont à risque majoré de conséquences graves des effets indésirables. Si un AINS est considéré nécessaire, la dose minimale efficace doit être administrée pendant la plus courte durée possible. Le traitement doit être revu à intervalles réguliers et arrêté si aucun bénéfice n'est observé ou en cas d'intolérance. Pendant le traitement par AINS, le patient doit être surveillé régulièrement à la recherche de saignements gastro-intestinaux.
Insuffisance rénale
Des précautions doivent être prises avec la posologie d'ibuprofène chez les patients souffrant d'atteinte rénale. Ce médicament est contre-indiqué chez les patients atteints d'insuffisance rénale sévère (voir «Contre-indications»).
La posologie doit être évaluée au cas par cas. La dose initiale doit être réduite chez les patients atteints d'insuffisance rénale légère à modérée. La dose doit être maintenue aussi faible que possible et prise pendant la durée la plus courte possible requise pour contrôler les symptômes. La fonction rénale doit être surveillée (voir «Contre-indications», «Mises en garde et précautions» et «Propriétés/Effets»).
Insuffisance hépatique
L'usage de paracétamol à des doses supérieures à celles recommandées peut induire une hépatotoxicité, voire une insuffisance hépatique et le décès. Chez les patients présentant des facteurs de risques additionnels d'hépatotoxicité, comme une insuffisance hépatocellulaire, un alcoolisme chronique, une malnutrition chronique (faibles réserves de glutathion dans le foie), ou une déshydratation, la dose quotidienne totale de 2000 mg (2 g) de paracétamol ne doit pas être dépassée.
Ce médicament est contre-indiqué chez les patients souffrant d'insuffisance hépatique sévère (voir «Contre-indications»). Un patient présentant des signes et/ou des symptômes suggérant une atteinte hépatique ou ayant des résultats anormaux aux tests hépatiques doit être évalué à la recherche de signes de développement d'une réaction hépatique plus grave pendant le traitement par ibuprofène, et Comboval doit être arrêté. En cas de survenue de signes cliniques et de symptômes évoquant une maladie hépatique, ou de manifestations systémiques (éosinophilie, rash, etc.), Comboval doit être arrêté.
Mode d'administration
Comboval doit être administré en perfusion intraveineuse de 15 minutes.
Pour prélever la solution, utiliser une aiguille de 0,8 mm (aiguille 21 G) et perforer verticalement le bouchon au point indiqué.
Chez les patients pesant moins de 50 kg pour lesquels un flacon complet (100 mL) n'est pas nécessaire, le volume adéquat doit être perfusé et le reliquat de solution éliminé (voir également «Remarques concernant la manipulation»).
Comme avec toutes les solutions pour perfusion délivrées en flacon de verre, il convient de rappeler qu'une étroite surveillance est requise, en particulier en fin de perfusion, indépendamment de la voie d'administration. Cette surveillance en fin de la perfusion s'applique particulièrement pour la perfusion par voie centrale, afin d'éviter une embolie gazeuse.
Contre-indicationsL'utilisation de ce produit est contre-indiquée dans les cas suivants:
·chez les patients ayant une hypersensibilité connue au paracétamol, à l'ibuprofène, à d'autres AINS ou à l'un des excipients mentionnés (voir «Composition»);
·chez les patients souffrant d'insuffisance cardiaque sévère (classe III - IV NYHA);
·chez les patients présentant un alcoolisme actif, la consommation excessive chronique d'alcool pouvant prédisposer les patients à une hépatotoxicité (en raison du composant paracétamol);
·chez les patients ayant présenté de l'asthme, de l'urticaire ou des réactions allergiques après la prise d'acide acétylsalicylique ou d'autres AINS;
·chez les patients ayant des antécédents d'hémorragie ou de perforation gastro-intestinale associés à un précédent traitement par AINS;
·chez les patients souffrant d'ulcères gastro-duodénaux ou d'hémorragies gastro-intestinales actifs ou passés (deux ou plusieurs épisodes distincts confirmés d'ulcère ou de saignement);
·chez les patients présentant une insuffisance hépatique sévère ou une insuffisance rénale sévère (voir «Mises en garde et précautions»);
·chez les patients présentant une hémorragie cérébrovasculaire ou tout autre type d'hémorragie active;
·chez les patients présentant des troubles de la coagulation sanguine et des affections impliquant une tendance majorée aux saignements;
·déshydratation sévère (secondaire à des vomissements, une diarrhée ou un apport insuffisant en liquide);
·pendant le troisième trimestre de grossesse (voir «Grossesse, Allaitement»);
·chez les patients âgés de moins de 18 ans;
·Traitement postopératoire après une opération de pontage coronarien (ou utilisation d'une machine coeur-poumons).
Mises en garde et précautionsLes effets indésirables peuvent être minimisés en utilisant la dose minimale efficace pendant la plus courte durée requise pour contrôler les symptômes. Ce médicament est destiné à un usage de courte durée; son utilisation n'est pas recommandée au-delà de deux jours.
L'utilisation Comboval avec un AINS concomitant, incluant les inhibiteurs sélectifs de la cyclooxygénase-2, doit être évitée.
Afin d'éviter le risque de surdosage,
·vérifier que les autres médicaments ne contiennent pas de paracétamol ou AINS,
·respecter les doses maximales recommandées (voir «Posologie/Mode d'emploi»).
Événements thrombotiques cardiovasculaires
Les études cliniques suggèrent que l'utilisation d'ibuprofène, en particulier à forte dose (2400 mg/jour), peut être associée à un risque légèrement majoré d'événements thrombotiques artériels (infarctus du myocarde ou accident vasculaire cérébral, par exemple).
Dans l'ensemble, les études épidémiologiques ne suggèrent pas que l'ibuprofène à faible dose (1200 mg/jour) soit associé à un risque majoré d'événements thrombotiques artériels.
Les patients présentant une hypertension non contrôlée, une insuffisance cardiaque congestive (classe NYHA II-III), une cardiopathie ischémique établie, une artériopathie périphérique, et/ou une maladie cérébrovasculaire ne doivent être traités par ibuprofène qu'après une évaluation approfondie, et les fortes doses (2400 mg/jour) doivent être évitées.
Des cas de syndrome de Kounis ont été rapportés chez des patients traités par ibuprofène. Le syndrome de Kounis comprend des symptômes cardiovasculaires suite à une réaction allergique ou d'hypersensibilité avec une constriction des artères coronaires et peut éventuellement mener à un infarctus du myocarde.
Insuffisance hépatique
L'usage de paracétamol à des doses supérieures à celles recommandées peut induire une hépatotoxicité, voire une insuffisance hépatique et le décès. De même, les patients souffrant d'une atteinte hépatique ou ayant un antécédent de maladie hépatique et traités au long cours par ibuprofène ou paracétamol doivent recevoir une surveillance de leur fonction hépatique à intervalles réguliers, dans la mesure où un effet mineur et transitoire de l'ibuprofène sur les enzymes hépatiques a été rapporté. Une réduction de dose est recommandée chez les patients présentant des signes d'altération de la fonction hépatique. Le traitement doit être arrêté chez les patients développant une insuffisance hépatique sévère (voir «Contre-indications»).
Des réactions hépatiques sévères, incluant la jaunisse et des cas d'hépatite fatale, bien que rares, ont été rapportées avec l'ibuprofène comme avec d'autres AINS. En cas de persistance ou d'aggravation des anomalies des tests hépatiques, ou de développement de signes cliniques et de symptômes évoquant une maladie hépatique, ou de survenue de manifestations systémiques (éosinophilie, rash, etc.), l'ibuprofène doit être arrêté. Ces deux médicaments actifs induiraient une hépatotoxicité, voire une insuffisance hépatique, en particulier le paracétamol.
Insuffisance rénale
Le paracétamol peut être utilisé chez les patients souffrant d'insuffisance rénale chronique sans ajustement posologique. Il existe un risque minime de toxicité du paracétamol chez les patients atteints d'insuffisance rénale modérée à sévère. Cependant, compte tenu de la composante ibuprofène de ce produit, une prudence doit être observée lors de l'initiation du traitement chez les patients déshydratés. Les deux principaux métabolites de l'ibuprofène sont excrétés essentiellement dans l'urine et l'atteinte de la fonction rénale peut entraîner leur accumulation. La signification de cette observation n'est pas connue. Les AINS induiraient une néphrotoxicité sous diverses formes: néphropathie interstitielle, syndrome néphrotique et insuffisance rénale. L'atteinte rénale due à la prise d'ibuprofène est généralement réversible. Chez les patients atteints d'insuffisance rénale, cardiaque ou hépatique, ceux prenant des diurétiques et des inhibiteurs de l'enzyme de conversion (IEC), et les personnes âgées, la prudence est requise dans la mesure où l'utilisation des AINS peut entraîner une détérioration de la fonction rénale. La dose doit être maintenue aussi faible que possible et la fonction rénale doit être surveillée chez ces patients. Le traitement doit être arrêté chez les patients qui développent une insuffisance rénale sévère (voir «Contre-indications»).
Utilisation combinée d'inhibiteurs de l'enzyme de conversion (IEC) ou d'antagonistes des récepteurs de l'angiotensine, d'anti-inflammatoires et de diurétiques thiazidiques
L'utilisation concomitante d'un médicament inhibant l'enzyme de conversion (IEC ou antagoniste des récepteurs de l'angiotensine), d'un anti-inflammatoire (AINS ou inhibiteur de la COX-2) et d'un diurétique thiazidique augmente le risque d'insuffisance rénale. Cela inclut les produits à combinaison fixe contenant plus d'une classe de médicament. L'utilisation combinée de ces médicaments doit être associée à une surveillance renforcée de la créatinine sérique, en particulier à l'instauration de la combinaison. L'association de médicaments de ces trois classes doit être utilisée avec prudence en particulier chez les personnes âgées ou chez les patients présentant une insuffisance rénale préexistante.
Sujets âgés
Aucune réduction de la posologie recommandée n'est requise. Cependant, il convient d'être prudent avec l'utilisation de l'ibuprofène, dans la mesure où elle ne doit pas être prise chez les adultes âgés de plus de 65 ans sans tenir compte des comorbidités et des traitements concomitants en raison du risque majoré d'effets indésirables, en particulier d'insuffisance cardiaque, d'ulcère gastro-intestinal et d'insuffisance rénale.
Effets hématologiques
De rares cas de dyscrasies ont été rapportés. Les patients sous traitement au long cours par ibuprofène doivent bénéficier d'une surveillance hématologique régulière.
Réactions anaphylactoïdes
Dans la pratique standard, pendant la perfusion intraveineuse, une surveillance attentive du patient est recommandée, en particulier en début de perfusion afin de détecter toute réaction anaphylactique causée par la substance active ou les excipients.
Les réactions d'hypersensibilité aiguë sévère (choc anaphylactique) sont très rarement observées. Aux premiers signes de réaction d'hypersensibilité après l'administration de Comboval, le traitement doit être arrêté et un traitement symptomatique doit être instauré. Les mesures médicales requises, en fonction des symptômes, doivent être initiées par un personnel spécialisé.
Troubles de la coagulation
Comme d'autres AINS, l'ibuprofène peut inhiber l'agrégation plaquettaire. Il a été démontré que l'ibuprofène prolonge le temps de saignement (mais dans l'intervalle de la normale) chez des sujets normaux. Dans la mesure où cette prolongation des saignements peut être exacerbée chez les patients présentant des troubles hémostatiques sous-jacents, les produits contenant de l'ibuprofène doivent être utilisés avec prudence chez les personnes présentant des anomalies intrinsèques de la coagulation et chez celles sous traitement anticoagulant. Les patients souffrant de troubles de la coagulation ou ceux subissant une chirurgie doivent être surveillés. Une vigilance médicale particulière est requise en cas d'utilisation chez les patients immédiatement après la réalisation d'une chirurgie majeure.
Événements gastro-intestinaux
Des hémorragies gastro-intestinales (GI), des ulcères ou des perforations, pouvant être fatals, ont été rapportés avec tous les AINS, à tout moment pendant le traitement, avec ou sans symptômes précurseurs ou antécédent d'événements GI graves.
Le risque d'hémorragie, d'ulcère ou de perforation GI est supérieur avec des doses croissantes d'AINS chez les patients ayant un antécédent d'ulcère, en particulier s'il est compliqué d'une hémorragie ou d'une perforation (voir «Contre-indications»), et chez les personnes âgées. Ces patients doivent débuter le traitement à la plus faible dose disponible.
Un traitement d'association avec des agents protecteurs (misoprostol ou inhibiteurs de la pompe à protons) doit être envisagé chez ces patients, ainsi que chez ceux nécessitant une faible dose concomitante d'acide acétylsalicylique ou d'autres médicaments susceptibles de majorer le risque gastro-intestinal (voir ci-dessous et «Interactions»). Les patients ayant un antécédent de toxicité GI, en particulier s'ils sont âgés, doivent signaler tout symptôme abdominal inhabituel (en particulier les saignements GI), plus particulièrement au début du traitement.
La prudence doit être recommandée aux patients recevant des médicaments concomitants susceptibles de majorer le risque d'ulcération ou de saignement, tels que les corticoïdes oraux, les anticoagulants comme la warfarine, les inhibiteurs sélectifs de recapture de la sérotonine ou les antiagrégants plaquettaires comme l'acide acétylsalicylique (voir «Interactions»).
En raison de la composante ibuprofène, Comboval doit être administré avec prudence chez les patients ayant un antécédent de maladie GI (rectocolite hémorragique, maladie de Crohn) et chez ceux souffrant de porphyrie.
Les personnes âgées présentent plus souvent des réactions indésirables aux AINS, en particulier d'hémorragies et de perforations gastro-intestinales qui peuvent être fatales (voir «Posologie/Mode d'emploi»).
Ce produit doit être arrêté en cas de signe d'hémorragie ou d'ulcération gastro-intestinale.
Hypertension
Les AINS peuvent induire une hypertension de novo ou l'aggravation d'une hypertension préexistante, et les patients prenant des antihypertenseurs avec des AINS peuvent présenter une altération de la réponse hypotensive. La prudence est recommandée lors de la prescription d'AINS chez les patients hypertendus. La pression artérielle doit être étroitement surveillée lors de l'initiation du traitement par AINS et à intervalles réguliers par la suite.
Insuffisance cardiaque
Des cas de rétention d'eau et d'œdème ont été observés chez certains patients prenant des AINS; en conséquence, la prudence est recommandée chez les patients présentant une rétention d'eau ou une insuffisance cardiaque.
Réactions cutanées sévères
Dans de très rares cas, les AINS peuvent causer des effets indésirables cutanés graves tels qu'une dermatite exfoliative, une nécro-épidermolyse bulleuse aiguë et un syndrome de Stevens-Johnson (SJS), qui peuvent être fatals et survenir sans signe avant-coureur. Des pustuloses exanthématiques aiguës généralisées (PEAG) ont été rapportées associées à des produits contenant de l'ibuprofène. Les patients semblent à plus haut risque de présenter ces réactions au début du traitement, celles-ci survenant dans la majorité des cas au cours du premier mois de traitement.
Les patients doivent être informés des signes et des symptômes de réactions cutanées graves; il doit en outre leur être conseillé de consulter leur médecin à la première apparition d'un rash ou de tout autre signe d'hypersensibilité.
Exceptionnellement, la varicelle peut causer des complications infectieuses graves de la peau et des tissus mous. À ce jour, la contribution des AINS à l'aggravation de ces infections ne peut pas être exclue. En conséquence, il est recommandé d'éviter d'utiliser Comboval en cas de varicelle.
Asthme préexistant
Les produits contenant de l'ibuprofène ne doivent pas être administrés chez les patients présentant un asthme sensible à l'acide acétylsalicylique et doivent être utilisés avec prudence chez les patients ayant un asthme préexistant.
Effets ophtalmologiques
Des effets secondaires ophtalmologiques ont été observés avec les AINS; en conséquence, les patients qui développent des troubles visuels pendant un traitement avec des produits contenant de l'ibuprofène doivent être soumis à un examen ophtalmologique.
Méningite aseptique
Pour les produits contenant de l'ibuprofène, les méningites aseptiques n'ont été rapportées que rarement, généralement mais pas toujours chez des patients souffrant d'un lupus érythémateux systémique (LES) ou d'autres affections du tissu conjonctif.
Interférences potentielles avec les tests de laboratoire
Avec les systèmes analytiques actuels, le paracétamol ne produit pas d'interférence avec les analyses de laboratoire. Cependant, avec certaines méthodes, il existe une possibilité d'interférences biologiques, décrites ci-dessous:
Tests urinaires
Le paracétamol à doses thérapeutiques peut interférer avec la détermination de l'acide 5-hydroxyindole acétique (5-HIAA), entraînant des résultats faux positifs. Les faux positifs peuvent être écartés en évitant la prise de paracétamol plusieurs heures avant et pendant le recueil des échantillons urinaires.
Dissimulation des symptômes d'infections sous-jacentes
Comboval peut masquer des symptômes d'infection, ce qui peut entraîner un retard d'instauration du traitement approprié et partant, l'aggravation de l'issue de l'infection. Cela a été observé dans la pneumonie communautaire bactérienne et dans les complications bactériennes de la varicelle. Lorsque Comboval est administré contre la fièvre ou pour le soulagement de douleurs associées à une infection, la surveillance de l'infection est recommandée. Dans un contexte non hospitalier, le patient doit consulter un médecin en cas de persistance ou d'aggravation des symptômes.
Usage prolongé des antalgiques
En cas d'usage prolongé d'antalgiques, des maux de tête peuvent survenir, qui ne doivent pas être traités par une augmentation de la dose du médicament.
Flucloxacilline
La prudence est recommandée en cas d'administration concomitante de paracétamol et de flucloxacilline en raison d'un risque accru d'acidose métabolique à trou anionique élevé (AMTAE), en particulier chez les patients atteints d'insuffisance rénale sévère, de septicémie, de malnutrition et d'autres sources de déficit en glutathion (par exemple, alcoolisme chronique), ainsi que chez ceux qui utilisent des doses quotidiennes maximales de paracétamol. Une surveillance étroite, incluant la mesure de la 5-oxoproline urinaire, est recommandée.
Précautions spéciales
Il existe des données selon lesquelles les médicaments qui inhibent la synthèse de la cyclo-oxygénase/prostaglandine peuvent causer une altération de la fertilité féminine par un effet sur l'ovulation. Cet effet est réversible à l'arrêt du médicament.
L'ibuprofène doit être utilisé uniquement après une évaluation rigoureuse du rapport bénéfice/risque chez les patients présentant un trouble congénital du métabolisme des porphyrines (porphyrie aiguë intermittente, par exemple).
Avec la consommation concomitante d'alcool, les effets indésirables liés à la substance active, en particulier ceux affectant le tractus gastro-intestinal ou le système nerveux central, peuvent être augmentés avec l'utilisation d'AINS.
La prudence est requise chez les patients présentant certaines affections qui peuvent être aggravées:
·Chez les patients présentant des réactions allergiques à d'autres substances, dans la mesure où il existe également chez eux un risque majoré de survenue de réactions d'hypersensibilité avec la prise de ce médicament.
·Chez les patients souffrant de rhume des foins, de polypes nasaux ou de maladies pulmonaires obstructives chroniques, dans la mesure où ils présentent un risque accru de réactions allergiques. Celles-ci peuvent se manifester sous la forme de crises d'asthme (appelées asthme analgésique), d'œdème de Quincke ou d'urticaire.
Le paracétamol est à utiliser avec précaution en cas:
·d'anorexie, de boulimie ou de cachexie, de malnutrition chronique (faibles réserves en glutathion hépatique)
·de déshydratation, d'hypovolémie
Chez les patients dont les réserves de glutathion sont épuisées, p.ex. en cas de sepsis, le recours au paracétamol peut accroître le risque d'acidose métabolique.
Sodium
Ce médicament contient 42,43 mg de sodium par flacon de 100 mL, ce qui équivaut à 2,12% de l'apport alimentaire quotidien maximal recommandé par l'OMS de 2 g de sodium par adulte.
InteractionsCe médicament ne doit pas être pris avec d'autres médicaments contenant du paracétamol, de l'ibuprofène, de l'acide acétylsalicylique ou des salicylates, ou avec d'autres anti-inflammatoires (AINS), sauf sur prescription d'un médecin.
Ibuprofène:
Comme avec d'autres produits à base d'ibuprofène, les combinaison suivantes avec Comboval doivent être évitées:
·Classe dicoumarol: Les AINS peuvent accroître l'effet des anticoagulants comme la warfarine. Des études expérimentales démontrent que l'ibuprofène renforce les effets de la warfarine sur le temps de saignement. Les AINS et la classe dicoumarol sont métabolisés par la même enzyme, CYP2C9.
·Antiagrégants plaquettaires: Les AINS ne doivent pas être combinés avec des antiagrégants plaquettaires comme la ticlopidine en raison de l'inhibition additive de la fonction plaquettaire (voir ci-dessous).
·Méthotrexate: Les AINS inhibent la sécrétion tubulaire du méthotrexate, et certaines interactions métaboliques avec une clairance réduite du méthotrexate peuvent également en découler. Le risque d'interaction potentielle entre un AINS et le méthotrexate doit également être pris en compte dans le cadre d'un traitement à faible dose par méthotrexate, en particulier chez les patients souffrant d'insuffisance rénale. Lorsqu'un traitement d'association est administré, la fonction rénale doit être surveillée. Il conviendra d'être prudent si un AINS et du méthotrexate sont administrés dans un intervalle de 24 heures, dans la mesure où les taux plasmatiques de méthotrexate peuvent augmenter, induisant une toxicité accrue. En conséquence, dans le traitement à forte dose par méthotrexate, il convient d'éviter systématiquement la prescription d'AINS.
·Acide acétylsalicylique: L'administration concomitante d'ibuprofène et d'acide acétylsalicylique n'est généralement pas recommandée en raison de la possibilité d'augmentation des effets indésirables. Les données expérimentales suggèrent que l'ibuprofène pourrait inhiber compétitivement l'effet de l'acide acétylsalicylique à faible dose sur l'agrégation plaquettaire lorsqu'ils sont administrés concomitamment. Malgré des incertitudes concernant l'extrapolation de ces données en situation clinique, la possibilité que l'utilisation régulière au long cours de l'ibuprofène puisse réduire l'effet cardioprotecteur de l'acide acétylsalicylique à faible dose ne peut être exclue. Aucun effet cliniquement pertinent n'est considéré comme probable en cas de prise occasionnelle d'ibuprofène (voir «Propriétés/Effets»).
·Lithium: L'ibuprofène réduit la clairance rénale du lithium, entraînant une augmentation possible des taux de lithium sérique. Cette association doit être évitée sauf si des contrôles fréquents du lithium sérique peuvent être effectués, avec une possibilité de réduire la dose de lithium.
·Glycosides cardiaques: Les AINS peuvent exacerber l'insuffisance cardiaque, réduire la filtration glomérulaire et augmenter les taux plasmatiques de glycosides cardiaques (digoxine).
·Mifépristone: Une diminution de l'efficacité du médicament peut théoriquement survenir en raison de l'activité antiprostaglandine des anti-inflammatoires non stéroïdiens (AINS) incluant l'acide acétylsalicylique. Selon des données limitées, la co-administration d'AINS le jour de l'administration de prostaglandine n'affecte pas défavorablement les effets du mifépristone ou de la prostaglandine sur la maturation du col ou la contractilité utérine et ne réduit pas l'efficacité clinique sur l'interruption médicale de grossesse.
·Inhibiteurs de l'enzyme de conversion (IEC) et antagonistes des récepteurs de l'angiotensine II (ARA II): Il existe un risque majoré d'insuffisance rénale aiguë, généralement réversible, chez les patients atteints d'insuffisance rénale (par exemple patients déshydratés et/ou âgés) lorsqu'un traitement par IEC ou antagonistes de l'angiotensine II est administré simultanément à un AINS, y compris les inhibiteurs sélectifs de la cyclooxygénase-2. Cette combinaison doit donc être administrée avec prudence chez les patients atteints d'insuffisance rénale, en particulier chez les patients âgés. Les patients doivent être correctement hydratés et un contrôle de la fonction rénale doit être envisagé après l'initiation du traitement d'association, ainsi qu'à intervalles réguliers pendant le traitement (voir «Mises en garde et précautions»).
·Bêta-bloquants: Les AINS neutralisent l'effet hypotenseur des médicaments bloquant les récepteurs bêta-adrénergiques.
·Sulfonylurées: De rares cas d'hypoglycémie ont été rapportés chez des patients sous traitement par sulfonylurée recevant de l'ibuprofène.
·Zidovudine: Des données démontrent un risque majoré d'hémarthroses et d'hématome chez les hémophiles VIH-positifs recevant un traitement concomitant par zidovudine et ibuprofène.
·Quinolones: les données animales indiquent que les AINS peuvent augmenter le risque de convulsions associées aux quinolones. Les patients sous AINS et quinolone peuvent être à risque majoré de convulsions.
·Thiazides, préparations thiazidiques et diurétiques de l'anse: Les AINS peuvent neutraliser l'effet diurétique du furosémide et du bumétanide, possiblement via l'inhibition de la synthèse des prostaglandines. Ils peuvent également neutraliser l'effet hypotenseur des thiazides.
·Diurétiques épargneurs de potassium: L'administration concomitante peut entraîner une hyperkaliémie.
·Aminoglycosides: Les AINS peuvent réduire l'excrétion des aminoglycosides.
·Inhibiteurs sélectifs de recapture de la sérotonine (ISRS): Les ISRS et les AINS induisent chacun un risque majoré de saignement, notamment du tractus gastro-intestinal. Ce risque est accru avec un traitement d'association. Le mécanisme peut potentiellement être lié à une réduction de la capture de la sérotonine dans les plaquettes (voir «Mises en garde et précautions»).
·Cyclosporine: L'administration concomitante d'AINS et de cyclosporine pourrait majorer le risque de néphrotoxicité en raison de la diminution de la synthèse de prostacycline dans le rein. En conséquence, en cas de traitement combiné, la fonction rénale doit être étroitement surveillée.
·Captopril: Les études expérimentales indiquent que l'ibuprofène neutralise les effets du captopril sur l'excrétion de sodium.
·Tacrolimus: L'administration concomitante d'AINS et de tacrolimus serait capable d'augmenter le risque de néphrotoxicité en raison de la diminution de la synthèse de prostacycline dans le rein. En conséquence, en cas de traitement d'association, la fonction rénale doit être étroitement surveillée.
·Corticoïdes: Le traitement concomitant induit un risque accru d'ulcération ou d'hémorragie gastro-intestinale.
·Inhibiteurs du CYP2C9: L'administration concomitante d'ibuprofène et d'inhibiteurs du CYP2C9 peut augmenter l'exposition à l'ibuprofène (substrat du CYP2C9). Dans une étude sur le voriconazole et le fluconazole (inhibiteurs du CYP2C9), une augmentation de l'exposition à l'ibuprofène S(+) d'environ 80 à 100% a été démontrée. Une réduction de la dose d'ibuprofène doit être envisagée lorsque des inhibiteurs puissants du CYP2C9 sont administrés simultanément, en particulier lorsque de l'ibuprofène à forte dose est administré avec le voriconazole ou le fluconazole.
·Phénytoïne: Les taux plasmatiques de phénytoïne peuvent être augmentés dans le traitement concomitant par ibuprofène; par conséquent, le risque de toxicité peut être accru.
·Probénécide et sulfinpyrazone: Les médicaments contenant du probénécide ou de la sulfinpyrazone peuvent retarder l'excrétion de l'ibuprofène.
·Extraits de plantes: Le ginkgo biloba peut augmenter le risque de saignement avec les AINS.
Paracétamol:
·Le probénécide inhibe la liaison du paracétamol à l'acide glucuronique, entraînant ainsi une réduction de la clairance du paracétamol par un facteur de 2 environ. Chez les patients prenant simultanément du probénécide, la dose de paracétamol doit être réduite.
·Des médicaments inducteurs d'enzyme comme certains antiépileptiques (phénytoïne, phénobarbital, carbamazépine) ont diminué l'AUC plasmatique du paracétamol à environ 60% dans les études pharmacocinétiques. D'autres substances ayant des propriétés inductrices enzymatiques (rifampicine, Hypericum) pourraient également entraîner une diminution des concentrations de paracétamol. En outre, le risque d'atteinte hépatique au cours du traitement à la dose maximale recommandée de paracétamol est probablement supérieur chez les patients recevant des inducteurs enzymatiques.
·La zidovudine pourrait affecter le métabolisme du paracétamol et vice versa, ce qui pourrait renforcer la toxicité des deux substances.
·Anticoagulants (warfarine) - la posologie peut devoir être réduite si le paracétamol et l'anticoagulant sont pris pendant une période prolongée.
·Une hépatotoxicité sévère aux doses thérapeutiques ou avec des surdoses modérées de paracétamol a été rapportée chez des patients recevant de l'isoniazide seul ou avec d'autres médicaments contre la tuberculose.
·Le paracétamol peut affecter la pharmacocinétique du chloramphénicol. La surveillance des taux plasmatiques de chloramphénicol est recommandée en cas d'association du paracétamol avec un traitement par injection de chloramphénicol.
·L'éthanol augmente la toxicité du paracétamol, possiblement en induisant une production hépatique de produits hépatotoxiques dérivés du paracétamol.
·Il convient d'être prudent lors de l'utilisation concomitante de paracétamol et de flucloxacilline, car la prise simultanée a été associée à une acidose métabolique à trou anionique élevé (AMTAE), en particulier chez les patients présentant des facteurs de risque (voir «Mises en garde et précautions»).
Effets sur les tests biologiques
La prise de paracétamol peut affecter les analyses d'acide urique utilisant de l'acide phosphotungstique et les dosages de glycémie utilisant la glucose oxydase/peroxydase.
Population pédiatrique
Les études d'interaction n'ont été réalisées que chez l'adulte.
Grossesse, allaitementGrossesse
Il n'existe pas d'expérience sur l'utilisation de ce produit chez l'homme pendant la grossesse. En raison de la composante ibuprofène, Comboval est contre-indiqué pendant le troisième trimestre de grossesse (voir ci-dessous).
Pour l'ibuprofène
L'inhibition de la synthèse des prostaglandines peut affecter défavorablement la grossesse et/ou le développement embryo-fœtal. Les données des études épidémiologiques suggèrent un risque accru de fausse couche, ainsi que de malformation cardiaque et de gastroschisis après l'utilisation d'un inhibiteur de synthèse des prostaglandines en début de grossesse. Le risque absolu de malformation cardiovasculaire était augmenté, passant de moins de 1% à environ 1,5 %. Le risque serait majoré avec la dose et la durée du traitement. Chez l'animal, l'administration d'un inhibiteur de synthèse des prostaglandines a démontré produire une augmentation des pertes pré et post-implantation, ainsi que de la létalité embryo-fœtale. En outre, des incidences accrues de malformations diverses, y compris cardiovasculaires, ont été rapportées chez des animaux recevant un inhibiteur de synthèse des prostaglandines pendant la période organogénétique.
A partir de la 20ème semaine de grossesse, l'utilisation de Comboval peut provoquer un oligohydramnios résultant d'un dysfonctionnement rénal fœtal. Cela peut survenir peu de temps après le début du traitement et est généralement réversible à l'arrêt. De plus, des cas de constriction du canal artériel ont été rapportés, après le traitement au cours du deuxième trimestre, dont la plupart ont disparu après l'arrêt du traitement. Par conséquent, pendant les premier et deuxième trimestres de grossesse, Comboval ne doit être administré que s'il est clairement nécessaire. Si Comboval est utilisé par une femme essayant de concevoir, ou pendant les premier et second trimestres de grossesse, la dose doit être maintenue aussi faible et la durée du traitement aussi courte que possible. Une surveillance prénatale de l'oligohydramnios et de la constriction du canal artériel doit être envisagée après une exposition à Comboval pendant plusieurs jours à partir de la 20ème semaine de gestation. Comboval doit être interrompu en cas d'oligohydramnios ou de constriction du canal artériel.
Au cours du troisième trimestre de grossesse, tous les inhibiteurs de synthèse des prostaglandines peuvent exposer le fœtus aux effets suivants:
·toxicité cardiopulmonaire (constriction / obturation prématurée du canal artériel et hypertension pulmonaire);
·dysfonction rénale (voir ci-dessus);
la mère et le nouveau-né aux effets suivants à la fin de la grossesse:
·prolongation possible du temps de saignement, un effet antiagrégant pouvant survenir même à de très faibles doses;
·inhibition des contractions utérines entraînant un travail retardé ou prolongé.
En conséquence, Comboval est contre-indiqué pendant le troisième trimestre de grossesse (voir «Contre-indications» et «Données précliniques»).
Pour le paracétamol
Un grand nombre de données chez les femmes enceintes utilisant du paracétamol ne suggère aucune toxicité malformative ni foeto/néonatale. Les études épidémiologiques sur le neurodéveloppement chez les enfants exposés au paracétamol in utero révèlent des résultats peu concluants. En cas de nécessité clinique, le paracétamol peut être utilisé pendant la grossesse; cependant, il doit être utilisé à la dose minimale efficace pendant la plus courte durée possible et le moins fréquemment possible.
Allaitement
Le paracétamol est excrété dans le lait maternel, mais pas en quantité cliniquement significative, et les données publiées disponibles ne suggèrent pas de contre-indication de l'allaitement tant que la posologie recommandée n'est pas dépassée.
L'ibuprofène et ses métabolites peuvent passer en très faibles quantités dans le lait maternel. Avec des doses thérapeutiques lors d'un traitement de courte durée, le risque d'effet sur le nouveau-né semble peu probable.
À la lueur des données susmentionnées, il n'est pas nécessaire d'interrompre l'allaitement lors d'un traitement à court terme à la dose recommandée de ce produit.
Fertilité
L'utilisation du produit peut altérer la fertilité féminine et n'est pas recommandée chez les femmes essayant de concevoir. Chez les femmes ayant des difficultés à concevoir ou en cours d'investigations sur l'infertilité, le retrait de ce produit doit être envisagé.
Effet sur l’aptitude à la conduite et l’utilisation de machinesDes effets indésirables comme des vertiges, une somnolence, une fatigue et des troubles visuels sont possibles après la prise d'AINS. Les patients concernés ne doivent pas conduire de véhicule ni utiliser de machines.
Effets indésirablesLes études cliniques sur Comboval et le paracétamol 500 mg/ibuprofène 150 mg comprimés pelliculés n'ont pas démontré d'effets indésirables autres que ceux répertoriés pour le paracétamol seul ou l'ibuprofène seul.
Les effets indésirables sont mentionnés ci-dessous selon les termes préférés MedDRA, par classe de système d'organe et fréquence absolue:
Très fréquents (≥1/10); fréquents (≥1/100 à <1/10); occasionnels (≥1/1 000 à <1/100); rares (≥1/10 000 à <1/1 000); très rares (<1/10 000); Fréquence indéterminée (ne peut être estimée sur la base des données disponibles).
Infections et infestations
Très rare: Une exacerbation des inflammations liées aux infections (développement de fasciite nécrosante) coïncidant avec l'utilisation d'AINS a été décrite.
Affections hématologiques et du système lymphatique
Occasionnel: Diminution de l'hémoglobine et de l'hématocrite. Bien qu'une relation causale n'ait pas été établie, des épisodes hémorragiques (épistaxis, ménorrhagie) ont été rapportés au cours du traitement avec ce médicament.
Très rare: Des troubles hématopoïétiques (agranulocytose, anémie, anémie aplasique, anémie hémolytique, leucopénie, neutropénie, pancytopénie et thrombocytopénie avec ou sans purpura) ont été rapportés après l'administration d'ibuprofène. Cependant, ils n'avaient pas nécessairement de relation causale avec le médicament.
Affections du système immunitaire
Très rare: Des réactions d'hypersensibilité, incluant des éruptions cutanées et une sensibilité croisée avec des sympathomimétiques, ont été rapportées.
Occasionnel : D'autres réactions allergiques ont été rapportées, mais aucune relation causale n'a été établie: Maladie sérique, syndrome de lupus érythémateux, purpura de Henoch-Schönlein, angio-œdème.
Troubles du métabolisme et de la nutrition
Très rare: En cas d'acidose métabolique, la causalité est incertaine dans la mesure où plus d'un médicament a été ingéré. Le cas d'acidose métabolique suivait l'ingestion de 75 grammes de paracétamol, 1,95 grammes d'acide acétylsalicylique et d'une petite quantité de détergent ménager liquide. Le patient présentait en outre un antécédent de crises d'épilepsie qui, selon les auteurs, pourrait avoir contribué à un taux accru de lactate indicateur d'acidose métabolique.
Les effets indésirables métaboliques incluaient l'hypokaliémie. Les effets indésirables métaboliques incluant l'acidose métabolique ont été rapportés après une surdose massive de paracétamol.
Occasionnel : Gynécomastie, réaction hypoglycémique.
Affections du système nerveux
Fréquent: Vertiges, céphalées, nervosité.
Occasionnel: Dépression, insomnie, confusion, instabilité émotionnelle, somnolence, méningite aseptique avec fièvre et coma.
Rare: Paresthésie, hallucinations, rêves anormaux.
Très rare: Stimulation paradoxale, névrite optique, atteinte psychomotrice, effets extrapyramidaux, tremblements et convulsions.
Affections oculaires
Occasionnel: Des amblyopies (vision trouble et/ou altérée, scotomes et/ou altérations de la vision des couleurs) ont été observées, mais elles sont généralement réversibles à l'arrêt du traitement. Tout patient présentant des symptômes oculaires doit effectuer un examen ophtalmologique incluant le champ visuel central.
Affections de l'oreille et du labyrinthe
Très rare: Vertiges.
Fréquent: Acouphènes (pour les médicaments contenant de l'ibuprofène).
Affections cardiaques
Fréquent: Œdème, rétention d'eau; la rétention d'eau répond généralement rapidement à l'arrêt du traitement.
Très rare: Palpitations; tachycardie; arythmies et autres dysrythmies cardiaques ont été rapportées. Fréquence indéterminée: syndrome de Kounis.
Des cas d'hypertension et d'insuffisance cardiaque ont été rapportés en association avec le traitement par AINS.
Affections respiratoires, thoraciques et médiastinales
Occasionnel: Épaississement des sécrétions du tractus respiratoire. Chez les enfants subissant une amygdalectomie, un stridor a été rapporté. Des hypoxémies ont été rapportées.
Très rare: Réactivité respiratoire incluant: asthme, exacerbation de l'asthme, bronchospasme et dyspnée.
Affections gastro-intestinales
Fréquent: Douleurs abdominales, diarrhées, dyspepsie, nausée, inconfort gastrique et vomissements, flatulences, constipation, légères pertes de sang gastro-intestinal pouvant causer une anémie dans des cas exceptionnels.
Occasionnel: Ulcère gastro-duodénal/gastro-intestinal, perforation ou hémorragie gastro-intestinale, avec des symptômes d'hématémèse et méléna parfois fatales, en particulier chez les personnes âgées. Des stomatites ulcéreuses et une exacerbation des colites et de la maladie de Crohn ont été rapportées après l'administration du produit. Moins fréquemment, des gastrites ont été observées et des pancréatites ont été rapportées. Une maladie acido-peptique a été rapportée.
Très rare: Œsophagite, formation d'une sténose intestinale de type diaphragme.
Affections hépatobiliaires
Très rare: Lésion hépatique, en particulier lors d'un traitement au long cours, insuffisance hépatique. Fonction hépatique anormale, hépatite et jaunisse. Dans la surdose, le paracétamol peut causer une insuffisance hépatique aiguë, une insuffisance hépatique, une nécrose hépatique et des lésions hépatiques.
Affections de la peau et du tissu sous-cutané
Fréquent: Rash (notamment de type maculopapulaire), prurit.
Très rare: Alopécie. Hyperhidrose, purpura et photosensibilité. Dermatites exfoliatives. Réactions bulleuses incluant érythème polymorphe, syndrome de Stevens Johnson et nécrolyse épidermique toxique. De très rares cas de réactions cutanées graves ont été rapportés. Dans des cas exceptionnels, des infections sévères de la peau et des complications des tissus mous peuvent survenir au cours de l'infection par la varicelle.
Fréquence indéterminée : Réaction médicamenteuse avec éosinophilie et symptômes systémiques (syndrome DRESS), pustulose exanthématique aiguë généralisée (PEAG), exanthème médicamenteux fixe (FDE).
Affections du rein et des voies urinaires
Occasionnel: Rétention urinaire.
Rare: Lésion du tissu rénal (nécrose papillaire), en particulier au cours du traitement à long terme.
Très rare: Néphrotoxicité sous diverses formes, incluant la néphrite interstitielle, le syndrome néphrotique, et l'insuffisance rénale aiguë et chronique.
Les effets indésirables rénaux sont le plus souvent observés après une surdose, après un usage abusif chronique (souvent avec de multiples antalgiques), ou en association avec une hépatotoxicité liée au paracétamol.
La nécrose tubulaire aiguë survient généralement en association avec une insuffisance hépatique, mais a été observée isolément dans de rares cas. Une augmentation possible du risque de carcinome à cellules rénales a également été associée à l'usage chronique de paracétamol.
Une étude cas-témoin de patients atteints de maladie rénale terminale a suggéré que la consommation à long terme de paracétamol pouvait augmenter significativement le risque de maladie rénale terminale, en particulier chez les patients prenant plus de 1000 mg par jour.
Troubles généraux et anomalies au site d'administration
Occasionnel: Pyrexie
Très rare: Fatigue et malaise.
Lésions, intoxications et complications liées aux procédures
Occasionnel: Des hémorragies post-opératoires après amygdalectomie ont été rapportées.
Investigations
Fréquent: Élévation du taux d'alanine aminotransférase, de gamma-glutamyltransférase et anomalies aux tests de la fonction hépatique avec le paracétamol.
Élévation de la créatininémie et de l'urée sanguine.
Occasionnel: Élévation du taux d'aspartate aminotransférase, des phosphatases alcalines sanguines, de la créatine phosphokinase sanguine, baisse de l'hémoglobine et élévation du nombre de plaquettes.
Rare: élévation des concentrations d'acide urique dans le sang.
Les études cliniques suggèrent que l'utilisation de l'ibuprofène, en particulier à forte dose (2400 mg/jour) peut être associée à un risque légèrement majoré d'événements thrombotiques artériels (infarctus du myocarde ou accident vasculaire cérébral, par exemple) (voir «Mises en garde et précautions»).
L'annonce d'effets secondaires présumés après l'autorisation est d'une grande importance. Elle permet un suivi continu du rapport bénéfice-risque du médicament. Les professionnels de santé sont tenus de déclarer toute suspicion d'effet secondaire nouveau ou grave via le portail d'annonce en ligne ElViS (Electronic Vigilance System). Vous trouverez des informations à ce sujet sur www.swissmedic.ch.
SurdosageSymptômes
Paracétamol
Des lésions du foie, voire une insuffisance hépatique, peuvent survenir après une surdose de paracétamol. Les symptômes de surdose de paracétamol au cours des premières 24 heures sont la pâleur, les nausées, les vomissements, l'anorexie et les douleurs abdominales. L'atteinte hépatique peut devenir patente 12 à 48 heures après l'ingestion. Des anomalies du métabolisme du glucose et une acidose métabolique peuvent survenir. Dans l'intoxication sévère, une insuffisance hépatique peut évoluer vers une encéphalopathie, le coma et le décès. Une insuffisance rénale aiguë avec une nécrose tubulaire aiguë peut se développer en l'absence d'atteinte hépatique sévère. Des arythmies cardiaques ont été rapportées. Une atteinte hépatique est possible chez les adultes ayant pris 10 g ou plus de paracétamol, en raison des quantités excessives d'un métabolite toxique.
Ibuprofène
Les symptômes incluent nausées, douleur abdominale et vomissements, vertiges, convulsion et, dans de rares cas, perte de conscience. Les signes cliniques du surdosage d'ibuprofène pouvant apparaître sont une dépression du système nerveux central et du système respiratoire.
Une utilisation prolongée à des doses supérieures à celles recommandées peut entraîner une hypokaliémie sévère et une acidose tubulaire rénale. Les symptômes peuvent inclure une diminution de la conscience et une faiblesse généralisée.
Dans l'intoxication grave, une acidose métabolique peut survenir.
Traitement
Paracétamol
Un traitement rapide est essentiel dans la prise en charge du surdosage de paracétamol, même en l'absence de symptômes patents, en raison du risque de lésions hépatiques qui se manifestent au bout de quelques heures voire quelques jours. Un traitement médical est recommandé, sans délai, chez tout patient ayant ingéré 7,5 g ou plus de paracétamol au cours des 4 dernières heures. Un lavage gastrique doit être envisagé. Une thérapie spécifique pour faire régresser l'atteinte hépatique, avec un antidote tel que l'acétylcystéine (par voie intraveineuse) ou la méthionine (par voie orale) doit être instaurée dès que possible.
L'efficacité de l'acétylcystéine est optimale lorsqu'elle est administrée au cours des huit premières heures suivant l'ingestion de la surdose; l'effet diminue progressivement entre 8 et 16 heures. Il a généralement été considéré que l'initiation du traitement plus de 15 heures après le surdosage ne produisait aucun bénéfice et pouvait éventuellement aggraver le risque d'encéphalopathie hépatique. Cependant, il est désormais démontré que l'administration tardive est sûre, et les études menées chez des patients traités jusqu'à 36 heures après l'ingestion suggèrent que des résultats bénéfiques peuvent être obtenus au-delà de 15 heures. En outre, l'administration d'acétylcystéine intraveineuse chez des patients ayant déjà développé une insuffisance hépatique fulminante a démontré réduire la morbidité et la mortalité.
Une dose initiale de 150 mg/kg d'acétylcystéine dans 200 mL de glucose à 5% est administrée par voie intraveineuse pendant 15 minutes, suivie d'une perfusion IV de 50 mg/kg dans 500 mL de glucose à 5% sur 4 heures, puis 100 mg/kg dans 1 litre de glucose à 5% sur 16 heures. Le volume de fluides IV doit être modifié chez les enfants.
La méthionine est administrée par voie orale à 2,5 g toutes les 4 heures jusqu'à 10 g. Le traitement par méthionine doit être instauré dans les 10 heures suivant l'ingestion du paracétamol; si tel n'est pas le cas, il sera inefficace et pourrait exacerber l'atteinte hépatique.
Les signes de symptômes graves peuvent ne pas être visibles jusqu'à 4 ou 5 jours suivant le surdosage, et les patients doivent faire l'objet d'une surveillance attentive pendant une durée prolongée.
Ibuprofène
Le traitement doit être symptomatique et de soutien, et inclure le maintien de voies aériennes dégagées ainsi que la surveillance des signes vitaux et cardiaques jusqu'à la stabilisation. Le lavage gastrique n'est recommandé que dans un intervalle de 60 minutes suivant l'ingestion d'une dose engageant le pronostic vital. Dans la mesure où le médicament est acide et excrété dans l'urine, il est théoriquement bénéfique d'administrer un alcalin pour induire une diurèse. Outre les mesures de soutien, l'utilisation de charbon activé par voie orale peut contribuer à réduire l'absorption et la réabsorption des comprimés d'ibuprofène.
Propriétés/EffetsCode ATC
N02BE51 (Autres analgésiques et antipyrétiques, Anilides)
Mécanisme d'action
Bien que le site exact et le mécanisme de l'action analgésique du paracétamol ne soient pas clairement définis, il semble que la substance induise une analgésie par le biais d'une élévation du seuil de la douleur. Le mécanisme potentiel peut impliquer l'inhibition de la voie de l'oxyde nitrique médiée par divers récepteurs des neurotransmetteurs, incluant les récepteurs N-méthyl-D-aspartate et la substance P.
L'ibuprofène est un dérivé de l'acide propionique ayant une activité analgésique, anti-inflammatoire et antipyrétique. Les effets thérapeutiques du médicament en tant qu'AINS résultent de son effet inhibiteur sur l'enzyme cyclo-oxygénase, entraînant une réduction de la synthèse des prostaglandines.
Les données expérimentales suggèrent que l'ibuprofène pourrait inhiber compétitivement l'effet de l'acide acétylsalicylique à faible dose sur l'agrégation plaquettaire lorsqu'ils sont administrés concomitamment.
Pharmacodynamique
Des études pharmacodynamiques ont démontré que lorsque des doses uniques d'ibuprofène à 400 mg étaient prises dans les 8 h précédant ou dans les 30 min suivant la libération immédiate de la dose d'acide acétylsalicylique (81 mg), l'effet de l'acide acétylsalicylique sur la formation de thromboxane ou l'agrégation plaquettaire était diminué. Malgré des incertitudes concernant l'extrapolation de ces données en situation clinique, la possibilité que l'utilisation régulière au long cours de l'ibuprofène puisse réduire l'effet cardioprotecteur de l'acide acétylsalicylique à faible dose ne peut être exclue. Aucun effet cliniquement pertinent n'est considéré comme probable en cas de prise occasionnelle d'ibuprofène (voir «Interactions»).
Efficacité clinique
Études cliniques
Les études cliniques sur Comboval n'ont pas inclus de patients de 65 ans et plus pour déterminer s'ils répondaient différemment des sujets plus jeunes.
Dans une étude d'efficacité de phase III menée chez 276 patients présentant des douleurs légères à modérées après une bunionectomie, Comboval apportait un soulagement de la douleur supérieur au placebo ou à des doses comparables de paracétamol ou d'ibuprofène seuls.
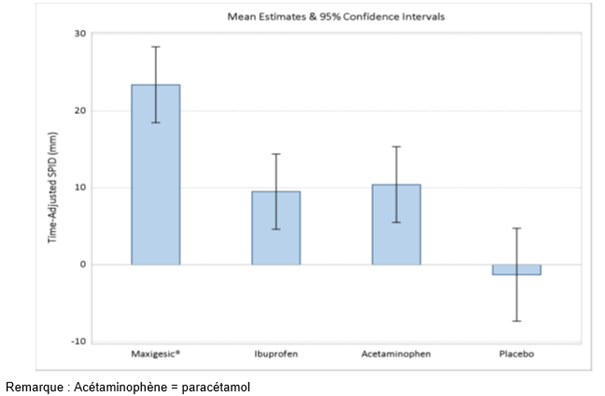
L'analyse de la somme des différences d'intensité de la douleur en fonction du temps (SPID) de 0-48 heures a démontré que Comboval (moyenne = 23,41, ET = 2,50) produisait un soulagement plus efficace de la douleur que le placebo (moyenne = -1,30, ET = 3,07), le paracétamol (moyenne = 10,42, ET = 2,50) ou l'ibuprofène (moyenne = 9,51, ET = 2,49), avec un haut degré de signification statistique (p < 0,001).
Tableau 1: Résumé de la SPID (0-48 heures) en fonction du temps par groupe de traitement
|
|
Comboval
|
Ibuprofène
|
Paracétamol
|
Placebo
| |
N=75
|
N=76
|
N=75
|
N=50
| |
N
|
75
|
76
|
75
|
50
| |
Moyenne (ET)
|
23,41 (2,89)
|
9,51 (2,53)
|
10,42 (2,49)
|
-1,30 (2,08)
| |
Médiane
|
23,10
|
5,40
|
3,45
|
-4,00
| |
Min.; Max.
|
-34,08; 74,17
|
-30,68; 79,98
|
-26,78; 65,43
|
-22,42; 47,50
| |
Estimation de la moyenne (ET)
|
23,41 (2,50)
|
9,51 (2,49)
|
10,42 (2,50)
|
-1,30 (3,07)
| |
Intervalle de confiance 95 %
|
18,48; 28,34
|
4,61; 14,40
|
5,49; 15,35
|
-7,33; 4,74
| |
Estimation de la différence (ET)
|
-
|
13,90 (3,53)
|
12,99 (3,54)
|
24,71 (3,96)
| |
Intervalle de confiance 95 %
|
-
|
6,95; 20,85
|
6,02; 19,96
|
16,92; 32,50
| |
Valeur de p
|
-
|
<0,001
|
<0,001
|
<0,001
|
Figure 1: SPID48 en fonction du temps jusqu'à la première dose du médicament de secours
PharmacocinétiqueAbsorption
Comboval s'administre en perfusion de 15 minutes; le pic de concentration plasmatique de chaque médicament est atteint à la fin de la perfusion. Les deux substances actives dans Comboval atteignent la concentration plasmatique maximum dans le même délai et ont une demi-vie plasmatique similaire (paracétamol 2,39 ± 0,27 heures, ibuprofène 1,88 ± 0,28 heure).
Les paramètres pharmacocinétiques de Comboval, déterminés par une étude menée chez 29 volontaires sains, sont présentés au Tableau 2.
Tableau 2: Moyenne (DS) des paramètres pharmacocinétiques du paracétamol et de l'ibuprofène dans chaque groupe de traitement.
|
|
Traitement (Moyenne ± DS)
| |
Paracétamol
|
Comboval
Perfusion IV, 15 mn
|
Paracétamol IV
Perfusion IV, 15 mn
|
Comboval Demi-dose
Perfusion IV, 15 mn
|
Paracetamol 1000 mg + Ibuprofen 300 mg
Comprimés Comprimé
oral
| |
Cmax (ng/mL)
|
26709,57
± 5814,74
|
26236,06
± 5430,52
|
12880,39
± 2553,15
|
14907,16
± 6255,10
| |
AUC0-t (ng.h/mL)
|
37553,97
± 9816,96
|
35846,20
± 8734,15
|
18327,40
± 4758,34
|
34980,80
± 9430,21
| |
AUC0-∞ (ng.h/mL)
|
39419,95
± 10630,63
|
37651,43
± 9454,60
|
19337,01
± 5146,46
|
37023,82
± 10388,31
| |
Tmax (h)
|
0,25 (fin de la perfusion)
|
0,25 (fin de la perfusion)
|
0,25 (fin de la perfusion)
|
0,73 ± 0,42
| |
t1/2 (h)
|
2,39 ± 0,27
|
2,38 ± 0,25
|
2,44 ± 0,25
|
2,51 ± 0,33
| |
Ibuprofène
|
Comboval Perfusion IV, 15 mn
|
Ibuprofène IV
Perfusion IV, 15 mn
|
Comboval
Demi-dose
Perfusion IV, 15 mn
|
Paracetamol 1000 mg + Ibuprofen 300 mg
Comprimés
Comprimé oral
| |
Cmax (ng/mL)
|
39506,69
± 6874,06
|
40292,97
± 7460,04
|
20352,05
± 3090,87
|
19637,38
± 5178,29
| |
AUC0-t (ng.h/mL)
|
73492,69
± 16509,61
|
72169,59
± 15608,70
|
39642,48
± 9679,16
|
70417,75
± 16260,16
| |
AUC0-∞ (ng.h/mL)
|
74743,31
± 17388,69
|
73410,65
± 16500,76
|
40333,88
± 10240,30
|
72202,48
± 17445,46
| |
Tmax (h)
|
0,25 (fin de la perfusion)
|
0,25 (fin de la perfusion)
|
0,25 (fin de la perfusion)
|
1,49 ± 0,89
| |
t1/2 (h)
|
1,88 ± 0,28
|
1,87 ± 0,27
|
1,88 ± 0,30
|
1,99 ± 0,36
|
Les paramètres pharmacocinétiques étaient similaires après une dose unique de Comboval administrée par voie intraveineuse ou par voie orale, à l'exception de la Cmax de la formulation intraveineuse qui était le double de celle de la formulation orale et, comme attendu, la Tmax suivant l'administration intraveineuse était atteinte beaucoup plus rapidement (en 15 minutes) qu'avec la formulation orale.
Distribution
Le paracétamol est distribué dans la majorité des tissus corporels. L'ibuprofène se lie fortement (90-99%) aux protéines plasmatiques.
Métabolisme
Le paracétamol est fortement métabolisé dans le foie et excrété dans l'urine, essentiellement sous forme de glucuronide inactif et de conjugués de sulfate. Moins de 5% sont excrétés sous forme inchangée. Les métabolites du paracétamol incluent un intermédiaire hydroxylé mineur qui a une activité hépatotoxique. Cet intermédiaire actif est détoxifié par conjugaison avec le glutathion; cependant, il peut s'accumuler après un surdosage de paracétamol et en l'absence de traitement, peut causer des lésions hépatiques sévères et même irréversibles.
L'ibuprofène est fortement métabolisé en composés inactifs dans le foie, essentiellement par glucuronidation.
Dans une étude clinique à dose unique, l'effet de l'ibuprofène sur le métabolisme oxydatif du paracétamol a été évalué chez des volontaires sains à jeun. Les résultats de l'étude indiquaient que l'ibuprofène ne modifiait pas la quantité de paracétamol soumis au métabolisme oxydatif, dans la mesure où la quantité de paracétamol et de ses métabolites (mercapturate-, cystéine-, glucuronide- et sulfate-paracétamol) étaient similaires en administration seuls, sous forme de paracétamol, ou avec l'administration concomitante d'ibuprofène (en combinaison fixe).
Élimination
La demi-vie d'élimination du paracétamol varie d'environ 1 à 3 heures.
Tant les métabolites inactifs qu'une petite quantité d'ibuprofène inchangé sont excrétés rapidement et complètement par les reins, 95% de la dose administrée étant éliminés dans l'urine dans les quatre heures suivant la prise. La demi-vie d'élimination de l'ibuprofène se situe dans l'intervalle de 1,9 à 2,2 heures.
Cinétique pour certains groupes de patients
Aucune donnée disponible.
Données précliniquesDans des études de toxicité à dose unique et à doses répétées menées chez des rats, la co-administration de paracétamol et d'ibuprofène selon un rapport correspondant à celui de Comboval (soit un ratio paracétamol-ibuprofène de 3,3-à-1) et à des niveaux de dose à peu près équivalents à ceux que les patients recevraient lors de l'administration de Comboval à la dose maximale recommandée n'augmentait pas le risque de toxicité GI ou rénale.
L'effet de doses uniques intraveineuses ou périveineuses de Comboval dans une étude sur des irritations locales aiguës chez des lapins mâles a démontré que Comboval est peu susceptible de produire une irritation locale lorsqu'il est administré par voie intraveineuse à la dose recommandée. De plus, lors de la réalisation d'une évaluation de compatibilité sanguine in vitro, aucune hémolyse additionnelle, floculation/précipitation de protéines plasmatiques ou agrégation plaquettaire n'ont été observées avec Comboval comparé au paracétamol IV ou à l'ibuprofène IV seuls.
Ibuprofène
La toxicité subchronique et chronique de l'ibuprofène dans les expériences animales a été observée principalement sous forme de lésions et d'ulcérations dans le tractus gastro-intestinal. Les études in vitro et in vivo n'ont produit aucune preuve cliniquement pertinente de potentiel mutagène de l'ibuprofène. Dans les études menées chez des rats et des souris, aucun signe d'effet carcinogène de l'ibuprofène n'a été retrouvé. L'ibuprofène a causé l'inhibition de l'ovulation chez les lapins ainsi qu'une altération de l'implantation dans diverses espèces animales (lapin, rat, souris). Les études expérimentales ont démontré que l'ibuprofène traverse le placenta. Concernant les doses maternellement toxiques, une incidence accrue de malformations (malformations du septum ventriculaire) a été observée.
Paracétamol
Le paracétamol à des doses hépatotoxiques a démontré un potentiel génotoxique et carcinogène (tumeurs du foie et de la vessie) chez les souris et les rats. Cependant, il est considéré que cette activité génotoxique et cancérogène est associée à des changements dans le métabolisme du paracétamol à fortes doses/concentrations et que cela ne constitue pas un risque en utilisation clinique.
Il n'existe aucune étude conventionnelle disponible utilisant les normes actuellement acceptées pour l'évaluation de la toxicité sur la reproduction et le développement.
Remarques particulièresIncompatibilités
En l'absence d'études de compatibilité, ce médicament ne doit pas être mélangé avec d'autres médicaments.
Influence sur les méthodes de diagnostic
Aucune donnée disponible.
Stabilité
Le médicament ne doit pas être utilisé au-delà de la date figurant après la mention «EXP» sur l'emballage.
Remarques particulières concernant le stockage
Ne pas conserver au-dessus de 25°C. Ne pas conserver au réfrigérateur. Ne pas congeler. Conserver le récipient dans son carton pour le protéger de la lumière. Conserver hors de portée des enfants.
Remarques concernant la manipulation
Inspecter visuellement Comboval pour rechercher la présence éventuelle de particules et d'une décoloration avant l'administration, si la solution et le conditionnement le permettent. En présence de particules opaques, d'une décoloration ou d'autres corps étrangers, la solution ne doit pas être utilisée.
En l'absence d'études de compatibilité, ce médicament ne doit pas être mélangé avec des diluants. Si moins d'un flacon complet est requis pour une dose unique, le volume adéquat doit être perfusé et le reliquat de solution éliminé (voir également «Posologie/Mode d'emploi»).
Comboval doit être utilisé chez un seul patient et une seule fois. Le produit ne contient aucun conservateur antimicrobien. Tout reliquat de solution doit être jeté.
Numéro d’autorisation69568 (Swissmedic)
PrésentationComboval, solution pour perfusion: 10 flacons [B]
Titulaire de l’autorisationLabatec Pharma SA, 1217 Meyrin (Genève)
Mise à jour de l’informationMars 2025
|