CompositionPrincipes actifs
Pomalidomide.
Excipients
Contenu des gélules: lactose monohydraté, crospovidone B, povidone K-30, laurylsulfate de sodium, fumarate de stéaryle sodique.
Enveloppe des gélules: gélatine, bleu brillant FCF (E133), dioxyde de titane (E171), oxyde de fer jaune (E172, uniquement pour les gélules 1 mg, 2 mg et 3 mg), oxyde de fer rouge (E172, uniquement pour les gélules 2 mg).
Encre d'impression: gomme laque, propylène glycol (E1520), oxyde de fer noir (E172), hydroxyde de potassium, solution d'ammoniaque concentrée.
Gélule de 1 mg: contient 50,0 mg de lactose monohydraté et 0,13 mg de sodium.
Gélule de 2 mg: contient 100,0 mg de lactose monohydraté et 0,27 mg de sodium.
Gélule de 3 mg: contient 150,0 mg de lactose monohydraté et 0,40 mg de sodium.
Gélule de 4 mg: contient 200,0 mg de lactose monohydraté et 0,54 mg de sodium.
Indications/Possibilités d’emploiPomalidomid-Teva, en association avec le bortézomib et la dexaméthasone, est indiqué pour le traitement du myélome multiple (MM) chez les patients adultes ayant déjà reçu au moins un traitement antérieur comportant le lénalidomide.
Pomalidomid-Teva, en association avec la dexaméthasone, est indiqué pour le traitement du myélome multiple en rechute et réfractaire chez les patients ayant déjà reçu au moins deux traitements antérieurs (comportant le lénalidomide et le bortézomib) et dont la maladie a progressé pendant le dernier traitement.
Posologie/Mode d’emploiLe traitement doit être initié et surveillé par un hématologue ou un oncologue expérimenté.
Pomalidomid-Teva en association avec le bortézomib et la dexaméthasone (PVd) chez les patients atteints d'un myélome multiple, ayant déjà reçu au moins un traitement antérieur
La dose initiale recommandée de Pomalidomid-Teva est de 4 mg par voie orale une fois par jour aux jours 1 à 14 d'un cycle répété de traitement de 21 jours.
La dose recommandée de bortézomib est de 1,3 mg/m2 et la dose recommandée de dexaméthasone est de 20 mg/jour par voie orale une fois par jour, l'administration devant suivre le schéma posologique indiqué au tableau 1.
La posologie est maintenue ou modifiée en fonction des résultats des examens cliniques et des analyses biologiques. Le traitement doit être interrompu en cas de progression de la maladie.
Tableau 1: Schéma posologique recommandé pour Pomalidomid-Teva en association avec le bortézomib et la dexaméthasone
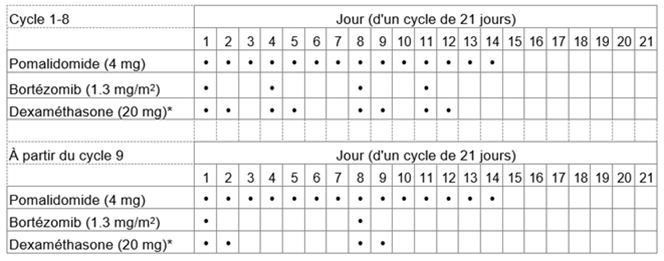
* Pour les patients âgés de >75 ans, voir rubrique «Instructions spéciales pour la posologie».
Pomalidomid-Teva en association avec la dexaméthasone (Pd) chez les patients présentant un myélome multiple en rechute et réfractaire, ayant déjà reçu au moins deux traitements antérieurs
La dose initiale recommandée de Pomalidomid-Teva est de 4 mg par voie orale une fois par jour aux jours 1 à 21 d'un cycle répété de traitement de 28 jours jusqu'à la progression de la maladie. La dose recommandée de dexaméthasone est de 40 mg une fois par jour aux jours 1, 8, 15 et 22 de chaque cycle de traitement de 28 jours.
La posologie est maintenue ou modifiée en fonction des résultats des examens cliniques et des analyses biologiques.
Le schéma posologique est indiqué au tableau 2.
Tableau 2: Schéma posologique recommandé pour Pomalidomid-Teva en association avec la dexaméthasone
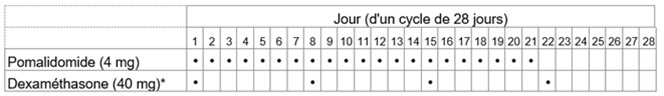
* Pour les patients âgés de >75 ans, voir rubrique «Instructions spéciales pour la posologie».
Ajustement de la posologie
Hématotoxicité
En cas de thrombocytopénie avec chute de la numération plaquettaire <25 x 109/l ou en cas de neutropénie avec chute des polynucléaires neutrophiles (PNN) <0,5 x 109/l ou en cas de neutropénie fébrile (fièvre ≥38,5 °C et PNN <1,0 x 109/l), le traitement par pomalidomide doit être interrompu et suivi d'un contrôle hebdomadaire de la numération formule sanguine complète (et en cas de chute des polynucléaires neutrophiles, l'utilisation de facteurs de croissance (G-CSF) doit être envisagée à l'appréciation du médecin traitant). Après le retour à la normale de la numération des thrombocytes/du taux des polynucléaires neutrophiles, le traitement par le pomalidomide doit être repris à la dose de 3 mg par jour. Pour chaque chute ultérieure (<25 x 109/l et <0,5 × 109/l respectivement), le traitement par le pomalidomide doit être interrompu. Après le retour à la normale de la numération des thrombocytes/du taux des polynucléaires neutrophiles, le traitement par le pomalidomide doit être repris à une dose inférieure de 1 mg à la dose antérieure.
Pomalidomid-Teva en association avec le bortézomib et la dexaméthasone (PVd): pour commencer un nouveau cycle de pomalidomide, le taux des polynucléaires neutrophiles doit être ≥1 × 109/l et la numération des plaquettes ≥50 × 109/l.
Pomalidomid-Teva en association avec la dexaméthasone (Pd): pour commencer un nouveau cycle de pomalidomide, le taux des polynucléaires neutrophiles doit être ≥0.5 x 109/l et la numération des plaquettes ≥50 x 109/l.
Autres effets indésirables de grade 3/4
En cas d'autres effets indésirables de grade 3/4, jugés comme étant liés au pomalidomide, le traitement doit être interrompu et repris à une dose inférieure de 1 mg à la dose antérieure après résolution de l'effet indésirable à un grade ≤2, à l'appréciation du médecin. Si les effets indésirables réapparaissent après réduction de la dose à 1 mg, le médicament doit être arrêté.
Pour des précisions relatives aux ajustements posologiques en raison d'une toxicité au bortézomib, il convient de se référer à l'information destinée aux professionnels du médicament concerné.
Ajustement posologique en cas d'administration concomitante d'inhibiteurs du CYP1A2
Il convient d'éviter l'utilisation concomitante du pomalidomide avec des inhibiteurs puissants du CYP1A2. Envisager d'autres modes de traitement. En cas d'administration concomitante d'inhibiteurs puissants du CYP1A2 (p.ex. ciprofloxacine et fluvoxamine) avec le pomalidomide, la dose de pomalidomide doit être réduite de 50%.
Arrêt de pomalidomide
L'interruption ou l'arrêt du traitement par le pomalidomide doit être envisagé(e) en cas d'éruption cutanée de grade 2 ou 3.
Pomalidomide doit être arrêté en cas d'angioedème, d'anaphylaxie, d'éruption cutanée de grade 4, de dermatite exfoliative ou d'éruption bulleuse, ou de suspicion de syndrome de Stevens-Johnson (SJS), de syndrome de Lyell (TEN) ou d'une réaction médicamenteuse accompagnée d'une éosinophilie et de symptômes systémiques (DRESS). Après l'interruption en raison de ces réactions, le traitement ne doit pas être repris.
Instructions posologiques particulières
Patients présentant des troubles de la fonction hépatique
Chez les patients présentant une insuffisance hépatique légère à modérée (grade A ou B de la classification de Child-Pugh), la dose initiale recommandée est de 3 mg par jour (réduction de la dose de 25%). Chez les patients présentant une insuffisance hépatique sévère (grade C de la classification de Child-Pugh), la dose recommandée est de 2 mg (réduction de la dose de 50%).
Patients présentant des troubles de la fonction rénale
Aucun ajustement posologique du pomalidomide n'est nécessaire chez les patients présentant une insuffisance rénale modérée et chez ceux présentant une insuffisance rénale sévère ne nécessitant pas de dialyse. Chez les patients présentant une insuffisance rénale sévère et nécessitant des dialyses, la dose initiale recommandée est de 3 mg par jour (réduction de la dose de 25%). Les jours d'hémodialyse, le pomalidomide doit être pris après l'hémodialyse.
Patients âgés
Aucune adaptation de la dose de pomalidomide n'est nécessaire.
Pomalidomid-Teva en association avec le bortézomib et la dexaméthasone (PVd) après au moins un traitement antérieur
Chez les patients âgés de >75 ans, la dose de dexaméthasone est de 10 mg une fois par jour aux jours 1, 2, 4, 5, 8, 9, 11 et 12 d'un cycle de 21 jours pour les cycles 1-8, et à partir du cycle 9, de 10 mg une fois par jour aux jours 1, 2, 8 et 9 d'un cycle de 21 jours.
Pomalidomid-Teva en association avec la dexaméthasone (Pd) après au moins 2 traitements antérieurs
Chez les patients âgés de plus de 75 ans, la dose initiale de dexaméthasone est de 20 mg un fois par jour aux jours 1, 8, 15 et 22 de chaque cycle de 28 jours.
Enfants et adolescents
La sécurité et l'efficacité chez les enfants et adolescents ne sont pas démontrées.
Mode d'administration
Pomalidomid-Teva doit être pris chaque jour par voie orale environ à la même heure. Les gélules ne doivent pas être ouvertes, cassées ou mâchées. Les gélules de Pomalidomid-Teva doivent être avalées entières, de préférence avec de l'eau, indépendamment des repas. Si le patient oublie de prendre une dose de Pomalidomid-Teva pendant une journée, il doit prendre la dose normale prescrite à l'heure habituelle le lendemain. La dose ne doit pas être ajustée pour compenser une dose de Pomalidomid-Teva omise le jour précédent.
Pour des précisions relatives à d'autres médicaments utilisés en association avec le pomalidomide, il convient de se référer à l'information destinée aux professionnels du médicament concerné.
Contre-indicationsHypersensibilité au pomalidomide, à l'un des excipients, ou encore au thalidomide et au lénalidomide.
Grossesse.
Femmes en âge de procréer, sauf si toutes les conditions du programme de prévention de la grossesse sont remplies.
Pour des précisions relatives à d'autres médicaments utilisés en association avec le pomalidomide, il convient de se référer à l'information destinée aux professionnels du médicament concerné.
Mises en garde et précautionsLe pomalidomide est un analogue du thalidomide. Le thalidomide a des effets tératogènes connus chez l'homme, lesquels provoquent des anomalies congénitales graves menaçant le pronostic vital de l'enfant à naître. Chez le rat et le lapin, le pomalidomide s'est avéré tératogène lorsqu'il a été administré pendant la phase d'organogenèse majeure (voir «Données précliniques»). Si le pomalidomide est pris pendant la grossesse, il faut s'attendre à un effet tératogène chez l'homme.
Programme de prévention de la grossesse
Programme pour les patientes
Les conditions du programme de prévention de la grossesse doivent être remplies par toutes les patientes, à moins de pouvoir certifier que la patiente est dans l'impossibilité de procréer.
Critères pour l'évaluation du potentiel de procréer
Toute patiente ou conjointe de patient est considérée comme capable de procréer, à moins qu'elle ne remplisse au moins l'une des conditions suivantes:
·Âge ≥50 ans et aménorrhée naturelle depuis ≥1 an*
·Déficience ovarienne prématurée avérée
·Antécédent de salpingo-oophorectomie bilatérale, de ligature des trompes bilatérale ou d'hystérectomie
·Génotype XY, syndrome de Turner, aplasie de l'utérus
* Une aménorrhée après un traitement anticancéreux n'exclut pas la capacité de procréer.
Consultation
Le pomalidomide est contre-indiqué chez les femmes capables de procréer qui ne remplissent pas toutes les conditions ci-dessous:
·La patiente comprend le risque tératogène attendu auquel serait exposé un enfant à naître.
·Elle comprend la nécessité d'une contraception fiable sans interruption commencée 4 semaines avant le début du traitement, poursuivie pendant toute la durée du traitement, pauses de traitement comprises, et pendant les 4 semaines suivant la fin du traitement.
·Même en cas d'aménorrhée, toute patiente en âge de procréer doit respecter strictement toutes les recommandations fournies pour une contraception efficace.
·Elle doit être capable de respecter les exigences d'une méthode de contraception fiable.
·Elle est informée et a compris les conséquences d'une grossesse ainsi que la nécessité de consulter rapidement le médecin si une grossesse est suspectée.
·Elle comprend la nécessité de faire des tests de grossesse toutes les 4 semaines et accepte de s'y soumettre.
·Elle a confirmé avoir compris les risques et mesures de sécurité nécessaires dans le cadre d'un traitement par le pomalidomide.
Chez les femmes en mesure de procréer, le médecin prescripteur doit s'assurer que
·La patiente remplit les conditions indiquées ci-dessus.
·La patiente remplit les conditions spécifiées par le programme de prévention de la grossesse, y compris confirmation d'une compréhension suffisante.
·La patiente a appliqué des mesures contraceptives suffisantes au moins 4 semaines avant le début du traitement, continue à appliquer des mesures contraceptives suffisantes pendant toute la durée du traitement, pauses de traitement comprises, et continuera à les appliquer encore au moins 4 semaines au-delà de la fin du traitement. Chez les patientes nécessitant un traitement immédiat par le pomalidomide, une contraception adéquate, associée à l'utilisation de préservatifs, doit être assurée pendant les 7 jours précédant le début du traitement.
·Un test de grossesse fait avant le traitement soit négatif.
Contraception
Les femmes en mesure de procréer doivent appliquer une méthode de contraception fiable pendant les 4 semaines précédant le début du traitement, pendant toute la durée du traitement, pauses de traitement comprises, et pendant les 4 semaines suivant la fin du traitement. Chez les patientes nécessitant un traitement immédiat par le pomalidomide, une contraception adéquate, associée à l'utilisation de préservatifs, doit être assurée pendant les 7 jours précédant le début du traitement. Si une méthode de contraception fiable n'a pas été utilisée auparavant, la patiente doit être adressée à un service médical de consultation capable de conseiller et informer la patiente de façon approfondie pour le choix d'une méthode contraceptive fiable.
Les méthodes de contraception suivantes peuvent être considérées comme fiables:
- Méthodes indépendantes de la patiente:
·Implant
·Acétate de médroxyprogestérone retard
·Stérilisation
- Méthodes dépendantes de la patiente:
·Renoncement total aux rapports hétérosexuels
·Rapports hétérosexuels uniquement avec un partenaire stérilisé par une vasectomie attestée par deux examens confirmant l'absence de spermatozoïdes.
·Contraceptifs oraux à la progestérone seule.
En raison du risque accru de thromboembolies veineuses sous pomalidomide, les contraceptifs oraux œstro-progestatifs ne sont pas recommandés. Si une patiente utilise déjà des contraceptifs oraux œstro-progestatifs, il convient de considérer le passage à une autre méthode de contraception. Le risque de thromboembolies veineuses persiste pendant 4 à 6 semaines après l'arrêt du contraceptif oral œstro-progestatif. Si aucune autre méthode ne peut être utilisée, on considérera une prévention antithrombotique pendant la durée d'utilisation du contraceptif oral œstro-progestatif. La patiente doit être dûment informée du risque de thromboembolie veineuse.
Les dispositifs intra-utérins (stérilets) sont associés à un risque accru d'infections lors de la mise en place et peuvent provoquer des saignements vaginaux ou règles irrégulières. Ces méthodes ne sont donc pas recommandées.
Test de grossesse
Des tests de grossesse avec une sensibilité d'au moins 25 UI/ml hCG doivent être effectués chez les femmes capables de procréer.
Chaque cas d'une patiente dont le test de grossesse est positif doit, sans attendre, être reporté au Swiss Teratogen Information Service (STIS) à Lausanne, au moyen du formulaire Swissmedic «Annonce d'effets indésirables suspectés d'un médicament (EI)».
- Avant le début du traitement
Un test de grossesse sous contrôle médical doit être réalisé lors de la consultation où le pomalidomide est prescrit ou au cours des trois jours suivant la visite chez le médecin prescripteur. À la date du test, la patiente doit avoir appliqué une méthode contraceptive fiable depuis au moins 4 semaines. Le test doit assurer que la patiente n'est pas enceinte au début du traitement par le pomalidomide.
- Avant le début du traitement chez les patientes nécessitant un traitement immédiat
Une détermination quantitative du taux sérique de hCG doit être faite immédiatement. Après 7 jours d'application d'une méthode de contraception efficace, associée à l'utilisation de préservatifs, le test doit être répété. Si les deux tests confirment que la patiente n'est pas enceinte, le traitement peut être commencé.
- Pendant et après le traitement
Un test de grossesse doit être fait toutes les 4 semaines pendant le traitement, et 4 semaines après la fin du traitement. Ces tests de grossesse doivent être faits pendant les visites chez le médecin pour la prescription du pomalidomide ou dans les trois jours précédant une telle visite.
Les tests de grossesse et la prescription et remise du pomalidomide doivent de préférence avoir lieu le même jour. Le pomalidomide doit être remise au plus tard sous 7 jours à compter de la date de prescription.
Programme chez les patients de sexe masculin
Chez les patients de sexe masculin, les données cliniques attestent du passage de la substance dans le sperme pendant la prise de pomalidomide. Les patients dont la partenaire est en mesure de procréer doivent de ce fait utiliser des préservatifs lors des rapports sexuels pendant le traitement par pomalidomide et au moins pendant les 7 jours après l'arrêt du traitement. Les hommes traités par pomalidomide doivent remplir les conditions suivantes:
·Ils doivent avoir compris le risque tératogène attendu s'ils ont des rapports sexuels avec une femme en mesure de procréer.
·Ils doivent avoir compris et accepté qu'ils doivent utiliser des préservatifs pour tous les rapports sexuels avec une femme en mesure de procréer pendant toute la durée du traitement, pauses de traitement comprises, et pendant les 7 jours suivant la fin du traitement.
Le médecin prescripteur doit assurer que les patients de sexe masculin ont compris et accepté d'utiliser des préservatifs pendant toute la durée du traitement, pauses du traitement compris, et pendant les 7 jours suivant la fin du traitement, s'ils ont des rapports sexuels avec une femme en mesure de procréer.
Les patients ne doivent en aucun cas faire un don de sperme pendant leur traitement par Pomalidomid-Teva ou pendant les 7 jours suivant la fin de ce traitement.
Précautions supplémentaires
Les patients et patientes doivent être instruits de ne jamais donner ce médicament à d'autres personnes et de rapporter les gélules non utilisées à leur médecin ou à leur pharmacien à la fin du traitement.
Les professionnels de santé et les aidants doivent porter des gants jetables pour manipuler la plaquette ou la gélule. Les femmes enceintes ou qui pensent l'être ne doivent pas manipuler la plaquette ou la gélule.
Autres mises en garde et précautions
Effets indésirables hématologiques
Les patients doivent être surveillés pour détecter l'apparition d'effets indésirables hématologiques, en particulier d'une neutropénie. L'hémogramme complet doit être surveillé une fois par semaine pendant les 8 premières semaines, puis une fois par mois. Une modification de dose peut être nécessaire (voir rubrique «Posologie/Mode d'emploi»).
Evénements thrombo-emboliques
On a rapporté des événements thrombo-emboliques veineux (principalement des thromboses veineuses profondes et des embolies pulmonaires). C'est pourquoi, un traitement anticoagulant est conseillé (sauf s'il est contre-indiqué). La décision de mettre en place des mesures prophylactiques anti-thrombotiques devra être prise après une évaluation attentive des facteurs de risque sous-jacents propres à chaque patient.
Affections cardiaques
Des cas d'insuffisance cardiaque, y compris d'insuffisance cardiaque congestive, d'œdème pulmonaire et de fibrillation auriculaire ont été rapportés, surtout chez les patients ayant présenté une affection cardiaque antérieure ou des facteurs de risque cardiaque. Lorsqu'il est envisagé de traiter ces patients par le pomalidomide, la prudence s'impose ainsi qu'une surveillance régulière des signes et symptômes d'une insuffisance cardiaque.
Syndrome de lyse tumorale
Un syndrome de lyse tumorale peut survenir. Les patients ayant le plus grand risque de syndrome de lyse tumorale sont ceux qui ont une charge tumorale élevée avant le traitement. Ces patients doivent faire l'objet d'une surveillance étroite, et des précautions appropriées doivent être prises.
Réactions allergiques et réactions cutanées sévères
Des cas d'angiœdème, d'anaphylaxie et de réactions cutanées sévères, notamment le syndrome de Stevens-Johnson (SJS), le syndrome de Lyell (nécrolyse épidermique toxique - TEN), et une réaction médicamenteuse accompagnée d'une éosinophilie et de symptômes systémiques (DRESS), ont été rapportés. Le DRESS peut se manifester sous forme d'éruption cutanée ou de dermatite exfoliative associée à une éosinophilie, une fièvre et/ou une lymphadénopathie avec complications systémiques comme une hépatite, une néphrite, une pneumopathie inflammatoire, une myocardite et/ou une péricardite. Ces événements peuvent être fatals. L'interruption ou l'arrêt du traitement par le pomalidomide doit être envisagé(e) en cas d'éruption cutanée de grade 2 ou 3. Pomalidomide doit être arrêté en cas d'angiœdème, d'anaphylaxie, d'éruption cutanée de grade 4, de dermatite exfoliative ou d'éruption bulleuse, ou de suspicion d'un SJS, d'une TEN ou d'un DRESS. Après l'interruption en raison de ces réactions, le traitement ne doit pas être repris.
Étourdissements et confusion
Des étourdissements et une confusion ont été rapportés. Il convient d'indiquer aux patients les situations dans lesquelles les étourdissements ou la confusion peuvent constituer un problème.
Cancers secondaires
Des cancers secondaires ont été rapportés chez des patients recevant le pomalidomide. Le médecin doit examiner soigneusement les patients avant et pendant le traitement en utilisant les méthodes habituelles de dépistage des cancers pour surveiller le développement de cancers secondaires et instaurer un traitement le cas échéant.
Troubles de la fonction hépatique
Des élévations importantes des taux d'alanine-aminotransférase et de bilirubine sériques ont été observées chez les patients traités par le pomalidomide (voir rubrique «Effets indésirables»). Il existe également des cas d'hépatite, y compris de réactivation de l'hépatite B, qui ont donné lieu à l'arrêt du pomalidomide. Un contrôle régulier de la fonction hépatique est conseillé.
Infections
Des infections (24,0 à 30,9% de grade 3 ou 4) se sont présentées chez 55,0 à 80,2% des patients qui avaient reçu un traitement d'association avec le pomalidomide dans des études cliniques. Des infections des voies respiratoires supérieures et des pneumonies étaient les infections qui sont survenues le plus fréquemment. Des infections à issue fatale (grade 5) sont survenues chez 2,7 à 4,0% des patients. Chez 2,0 à 2,9% des patients, les infections ont conduit à l'arrêt du pomalidomide.
Les patients présentant des facteurs de risque connus de survenue d'infections doivent être étroitement surveillés. Il convient de conseiller à tous les patients de contacter immédiatement un médecin dès le premier signe d'une infection (p.ex. toux, fièvre, etc.), afin d'en diminuer si possible la sévérité par un traitement précoce.
De rares cas de réactivation de l'hépatite B ont été rapportés à la suite du traitement par le pomalidomide en association avec la dexaméthasone chez des patients présentant des antécédents d'infection par le virus de l'hépatite B (VHB). Certains de ces cas ont évolué vers une insuffisance hépatique aiguë et ont conduit à l'arrêt du traitement par le pomalidomide. La sérologie VHB doit être déterminée avant l'initiation du traitement par le pomalidomide. Chez les patients ayant un résultat positif au dépistage du virus de l'hépatite B, une consultation chez un médecin spécialisé dans le traitement de l'hépatite B est recommandée. La prudence s'impose en cas d'administration de pomalidomide en association avec la dexaméthasone chez des patients préalablement infectés par le VHB. Ces patients doivent être étroitement surveillés tout au long du traitement afin de détecter les signes et symptômes d'infection active par le VHB.
Leucoencéphalopathie multifocale progressive (LMP)
Des cas de leucoencéphalopathie multifocale progressive, y compris ceux ayant un issue fatale, ont été observés en lien avec le pomalidomide. Dans ce contexte, la LMP est survenue quelques mois à années après le début du traitement par pomalidomide.
Les médecins doivent surveiller les patients à intervalles réguliers et considérer la LMP comme un diagnostique différentiel chez les patients présentant une apparition ou aggravation de symptomes neurologiques, des signes ou symptômes cognitifs ou comportementaux. Les patients doivent être invités à informer leurs partenaires ou aidants au sujet de leur traitement car ceux-ci pourraient remarquer des symptomes dont le patient n'est pas conscient.
L'évaluation liée à la LMP doit se reposer une analyse neurologique, une IRM du cerveau ainsi qu'un test de dépistage de l'ADN du virus JC (VJC) dans le liquide céphalo-rachidien à l'aide de la réaction en chaîne de la polymérase (PCR) ou d'une biopsie du cerveau avec test de dépistage du VJC. Un résultat négatif au test PCR du VJC n'exclut pas une LMP. Les résultats de l'IRM peuvent être déjà visibles avant l'apparition des signes et symptomes cliniques. Des cas de LMP qui ont été diagnostiqués sur la base de résultats IRM et la présence de l'ADN du VJC dans le liquide céphalo-rachidien en l'absence de signes ou symptomes cliniques spécifiques à la LMP ont été rapportés. Nombreux de ces patients ont ensuite présentés des symptomes d'une LMP. Par conséquent, une surveillance par IRM des signes d'une LMP peut être utile et tous les résultats suspects doivent mener à une analyse minutieuse pour permettre, le cas échéant, un diagnostic précoce de la LMP. Un suivi et une évaluation supplémentaires peuvent être nécessaires, si aucun autre diagnostic ne peut être posé.
Si une LMP est suspectée, la prise du pomalidomide doit être suspendue jusqu'à ce qu'une LMP soit exclue.
Si une LMP a été confirmée, le pomalidomide doit être interrompu durablement.
Troubles rénaux
Dans des études cliniques, on a observé un taux plus élevé d'effets indésirables hématologiques (anémie et thrombocytopénie) et rénaux (lésions rénales aiguës) (voir aussi «Propriétés/Effets») chez les patients dont la clairance de la créatinine était ≤45 ml/min, et ayant reçu le pomalidomide en association avec le bortézomib et la dexaméthasone. Ces patients doivent être soigneusement surveillés.
Pour des précisions relatives à d'autres médicaments utilisés en association avec le pomalidomide, il convient de se référer à l'information destinée aux professionnels du médicament concerné.
Excipients
Ce médicament contient du lactose. Les patients présentant l'intolérance héréditaire rare au galactose, un déficit total en lactase ou un syndrome de malabsorption du glucose et du galactose ne devraient pas prendre ce médicament.
Ce médicament renferme moins de 1 mmol de sodium (23 mg) par gélule, ce qui signifie qu'il s'agit d'un médicament pratiquement «sans sodium».
InteractionsInteractions éventuelles d'autres médicaments avec le pomalidomide
Le pomalidomide est métabolisé en partie par les CYP1A2 et CYP3A4/5. C'est également un substrat de la glycoprotéine P. L'administration concomitante de pomalidomide avec des médicaments comme par exemple, le kétoconazole, un inhibiteur puissant du CYP3A4/5 et de la P-gp, ou avec la carbamazépine, un inducteur puissant du CYP3A4/5, n'a pas eu d'effets cliniquement pertinents sur l'exposition au pomalidomide.
L'administration concomitante de fluvoxamine, un inhibiteur puissant du CYP1A2, et de pomalidomide en présence de kétoconazole a augmenté de 107% l'exposition moyenne au pomalidomide intervalle de confiance à 90% [91% à 124%] par rapport au pomalidomide plus kétoconazole seuls. L'utilisation concomitante de fluvoxamine (un inhibiteur puissant du CYP1A2) et du pomalidomide a augmenté de 125% l'exposition moyenne au pomalidomide intervalle de confiance à 90% [98% à 157%] par rapport au pomalidomide seul chez les volontaires sains. La dose de pomalidomide doit être réduite de 50% en cas d'administration concomitante d'inhibiteurs puissants du CYP1A2 (p.ex. ciprofloxacine et fluvoxamine) avec le pomalidomide.
Interactions éventuelles du pomalidomide avec d'autres médicaments
Le pomalidomide n'inhibe par les CYP1A2, CYP2A6, CYP2B6, CYP2C8, CYP2C9, CYP2C19, CYP2D6, CYP2E1 ou CYP3A4/5 in vitro. En outre, in vitro, le pomalidomide n'est pas un inducteur des enzymes CYP1A2, CYP2B6, CYP2C9, CYP2C19 et CYP3A4/5.
Le pomalidomide n'est pas un inhibiteur de la glycoprotéine P (P-gp) et n'a montré dans les études in vitro aucun ou un faible effet inhibiteur sur la «Breast Cancer Resistant Protein» (BCRP), les transporteurs «Organic Anion Transporter Protein» (OATP) 1B1, OATP1B3, «Organic Cation Transporter» OAT1 et OAT3 ainsi que sur le transporteur «Organic Anion Transporter Protein» OCT2.
On ne s'attend pas à ce que le pomalidomide provoque des interactions pharmacocinétiques cliniquement pertinentes en raison d'une inhibition ou induction ou d'une inhibition des transporteurs lorsqu'il est administré en association avec des substrats de ces enzymes ou des transporteurs. Le potentiel de telles interactions médicamenteuses ainsi que l'effet possible du pomalidomide sur la pharmacocinétique des contraceptifs oraux, n'ont pas été évalués dans le cadre d'études cliniques.
Grossesse, allaitementGrossesse
Un effet tératogène du pomalidomide est attendu chez l'être humain. Le pomalidomide est contre-indiqué pendant la grossesse et ne doit pas être utilisé chez des femmes en âge de procréer à moins que toutes les conditions relatives à la contraception soient remplies (voir la rubrique «Mises en garde et précautions»).
Allaitement
On ignore si le pomalidomide est excrété dans le lait maternel. Après administration chez des rates allaitantes, le pomalidomide a été détecté dans le lait. Compte tenu du risque d'effets indésirables du pomalidomide chez l'enfant allaité, une décision doit être prise soit d'interrompre l'allaitement soit d'interrompre le traitement en prenant en compte l'importance du traitement par le pomalidomide pour la mère.
Effet sur l’aptitude à la conduite et l’utilisation de machinesAucune étude relative à l'effet du pomalidomide sur l'aptitude à la conduite et l'utilisation de machines n'a été réalisée. Des cas de syncope, fatigue et de vertiges ont été signalés lors de l'utilisation du pomalidomide. C'est pourquoi, la prudence est conseillée aux patients sous pomalidomide s'ils conduisent des véhicules ou utilisent des machines.
Effets indésirablesPomalidomid-Teva en association avec le bortézomib et la dexaméthasone (PVd) chez des patients présentant un myélome multiple, ayant reçu au moins un traitement antérieur
Dans l'étude randomisée CC-4047-MM-007, 278 patients ont reçu le pomalidomide, le bortézomib et la dexaméthasone.
Les maladies du sang et du système lymphatique les plus fréquemment rapportées étaient la neutropénie, la thrombocytopénie et l'anémie. L'effet indésirable le plus fréquemment rapporté était la neuropathie périphérique sensitive. L'effet indésirable sévère le plus fréquemment rapporté était la pneumonie (11,5%).
Pomalidomid-Teva en association avec la dexaméthasone (Pd) chez les patients présentant un myélome multiple en rechute et réfractaire, ayant déjà reçu au moins deux traitements antérieurs
Dans trois études cliniques (CC-4047-MM-003, CC-4047-MM-002 et CC-4047-IFM-2009-02), 455 patients ont été exposés à 4 mg de pomalidomide.
Les effets indésirables les plus fréquemment rapportés étaient neutropénie, anémie, thrombocytopénie, fatigue, fièvre, constipation et diarrhée.
L'effet indésirable grave le plus fréquemment rapporté était la pneumonie (12,5%).
Les fréquences sont définies comme suit: très fréquent (≥1/10); fréquent (<1/10, ≥1/100); occasionnel (<1/100, ≥1/1000); rares (1/1000, ≥1/10'000); très rares (<1/10'000).
Infections et infestations#
Très fréquent: infections des voies respiratoires supérieures (20,9%), pneumonie (19,1%), bronchite (14,0%), infection virale des voies respiratoires supérieures (11,2%).
Fréquent: septicémie, choc septique, colite à Clostridium difficile, bronchopneumonie, grippe, infection des voies urinaires, infection des voies respiratoires, infections des voies respiratoires basses, bronchiolite, infection pulmonaire, sinusite, rhinopharyngite, candidose, candidose orale.
Occasionnel: septicémie neutropénique, herpès zoster*.
Rare: réactivation du virus de l'hépatite B*, leucoencéphalopathie multifocale progressive*.
# = Tous les termes préférentiels de la classe de système/organe des infections (y compris les infections bactériennes, virales et fongiques) sont mentionnés, à l'exception des infections rares d'intérêt sanitaire public.
Tumeurs bénignes, malignes et non précisées (incluant kystes et polypes)
Fréquent: carcinome baso-cellulaire.
Occasionnel: carcinome épidermoïde de la peau*.
Affections hématologiques et du système lymphatique
Très fréquent: neutropénie (47,9%), anémie (44,0%), thrombocytopénie (36,7%), leucopénie (13,0%).
Fréquent: neutropénie fébrile, lymphopénie, diminution du nombre de globules blancs, diminution du taux des polynucléaires neutrophiles, diminution de la numération des thrombocytes.
Occasionnel: pancytopénie*.
Affections du système immunitaire
Occasionnel: réactions allergiques (p.ex. angioedème, urticaire)*.
Rare: anaphylaxie*.
Fréquence inconnue: rejet du greffon après une transplantation d'organe*.
Affections endocriniennes
Rare: hypothyroïdie*.
Troubles du métabolisme et de la nutrition
Très fréquent: hypokaliémie (15,5%), hyperglycémie (14,4%), perte de l'appétit (11,6%).
Fréquent: déshydratation, hyponatrémie, hyperkaliémie, hypercalcémie, hypocalcémie, hypophosphatémie, hypoalbuminémie, hypomagnésiémie.
Affections psychiatriques
Très fréquent: troubles du sommeil (16,2%).
Fréquent: confusion, anxiété, dépression, modification de l'humeur.
Affections du système nerveux
Très fréquent: neuropathie périphérique sensitive (47,8%), vertiges (17,3%), tremblements (10,8%).
Fréquent: neuropathie périphérique, neuropathie périphérique sensitivomotrice, syncope, somnolence, léthargie, diminution du niveau de conscience, céphalées, dysgueusie, paresthésie.
Affections oculaires
Fréquent: vision trouble, cataracte.
Affections de l'oreille et du labyrinthe
Fréquent: vertiges.
Affections cardiaques
Fréquent: insuffisance cardiaque décompensée, fibrillation auriculaire, tachycardie.
Affections vasculaires
Fréquent: thrombose veineuse profonde, hypotension, hypertension.
Affections respiratoires, thoraciques et médiastinales
Très fréquent: toux (20,5%), dyspnée (20,2%).
Fréquent: embolie pulmonaire, toux productive, nez bouché, douleur oropharyngée, dysphonie, saignements de nez, dyspnée à l'effort.
Occasionnel: dyspnée, pneumopathie interstitielle y compris des cas de pneumopathie inflammatoire*.
Affections gastrointestinales
Très fréquent: constipation (36,7%), diarrhée (33,8%), nausées (17,6%), vomissements (11,5%).
Fréquent: douleurs abdominales, douleurs épigastriques, stomatite, sécheresse buccale, ventre ballonné.
Occasionnel: hémorragies gastro-intestinales*.
Affections hépatobiliaires
Fréquent: augmentation de l'alanine-aminotransférase.
Occasionnel: hyperbilirubinémie, élévation des paramètres hépatiques et hépatite*.
Affections de la peau et du tissu sous-cutané
Fréquent: éruption cutanée, prurit, peau sèche, hyperhydrose, sueurs nocturnes.
Très rares: syndrome de Stevens-Johnson*, nécrolyse épidermique toxique*, réaction médicamenteuse accompagnée d'une éosinophilie et de symptômes systémiques (DRESS)*.
Affections musculosquelettiques et systémiques
Très fréquent: dorsalgies (18,7%), faiblesse musculaire (13,7%), douleurs osseuses (13,6%), spasmes musculaires (13,0%).
Fréquent: arthralgie, douleurs de l'appareil locomoteur, douleurs musculo-squelettiques dans le thorax, douleurs dans une des extrémités/douleurs dans les membres.
Affections du rein et des voies urinaires
Fréquent: lésion rénale aiguë, insuffisance rénale, insuffisance rénale aiguë, affection rénale chronique, rétention urinaire, augmentation de la créatininémie.
Affections des organes de reproduction et du sein
Fréquent: douleurs pelviennes.
Troubles généraux et anomalies au site d'administration
Très fréquent: fatigue (37,1%), œdème périphérique (33,8%), fièvre (23,7%), asthénie (18,2%).
Fréquent: aggravation de l'état général, douleurs thoraciques, douleurs, frissons, douleurs thoraciques non cardiaques, œdème.
Occasionnel: syndrome de lyse tumorale*.
Investigations
Fréquent: perte de poids.
Lésions, intoxications et complications liées aux procédures
Fréquent: chute.
* = Données de pharmacovigilance
Population pédiatrique
Les données disponibles issues de deux études non contrôlées ouvertes sont limitées. Il n'y a pas d'indication de nouveaux aspects liés à la sécurité.
L'annonce d'effets secondaires présumés après l'autorisation est d'une grande importance. Elle permet un suivi continu du rapport bénéfice-risque du médicament. Les professionnels de santé sont tenus de déclarer toute suspicion d'effet secondaire nouveau ou grave via le portail d'annonce en ligne ElViS (Electronic Vigilance System). Vous trouverez des informations à ce sujet sur www.swissmedic.ch.
SurdosageDans les études, le pomalidomide a été administré à des doses uniques allant jusqu'à 50 mg chez des volontaires sains et à des doses répétées de 10 mg une fois par jour chez des patients atteints d'un myélome multiple. Il n'a pas été rapporté d'événements indésirables graves liés à un surdosage. Le pomalidomide est éliminé par hémodialyse.
Propriétés/EffetsCode ATC
L04AX06
Mécanisme d'action
Le pomalidomide est un dérivé du thalidomide et se présente sous forme d'un racémate. Il possède des propriétés immunomodulatrices ainsi qu'anti-angiogénétiques.
Le pomalidomide inhibe la prolifération des cellules malignes hématopoïétiques et il inhibe également in vitro la prolifération des lignées cellulaires résistantes au lénalidomide. Il stimule la prolifération des lymphocytes T et la cytotoxicité médiée par les cellules tueuses naturelles (Natural killer - NK), il inhibe la production des cytokines pro-inflammatoires (par ex. TNF-α et IL-6) et bloque la migration et l'adhésion des cellules endothéliales.
Le pomalidomide se lie directement à la protéine cereblon (CRBN), qui fait partie d'un complexe E3-ligase comprenant les protéines DDB1 (DNA damage-binding protein 1), Cullin 4 (CUL4) et Roc1 et pouvant inhiber l'auto-ubiquitination du CRBN dans le complexe. Les E3-ubiquitines ligases sont responsables de la polyubiquitination de différentes protéines substrats et peuvent partiellement expliquer les effets pléiotropiques observés sur les cellules dans le cadre d'un traitement par le pomalidomide.
In vitro, en présence du pomalidomide, les protéines substrats Aiolos et Ikaros font l'objet d'une ubiquitination et ensuite d'une dégradation, avec pour résultat des effets cytotoxiques et immunomodulateurs. In vivo, le traitement par le pomalidomide donne lieu à une diminution du taux d'Ikaros chez les patients atteints d'un myélome multiple en rechute réfractaire au lénalidomide.
Pharmacodynamique
Voir la section sur le mécanisme d'action.
Efficacité clinique
Le pomalidomide en association avec le bortézomib et la dexaméthasone (PVd) à plus faible dose
Une étude clinique multicentrique, randomisée, ouverte, à 2 bras de phase III (CC-4047-MM-007) a évalué l'efficacité et la sécurité d'emploi du pomalidomide en association avec le bortézomib et la dexaméthasone (PVd) à plus faible dose comparé au bortézomib et à la dexaméthasone (Vd) à plus faible dose chez des patients adultes préalablement traités, qui avaient reçu au moins un traitement antérieur, dont un devait être un traitement par le lénalidomide.
Les patients dans le bras PVd ont reçu 4 mg de pomalidomide par voie orale aux jours 1 à 14 d'un cycle de 21 jours. La dexaméthasone à plus faible dose de 20 mg a été administrée une fois par jour aux jours 1, 2, 4, 5, 8, 9, 11 et 12 d'un cycle de 14 jours du cycle 1 jusqu'au cycle 8 inclus et ensuite une fois par jour aux jours 1, 2, 8 et 9 de chaque cycle suivant de 21 jours à partir du cycle 9. Les patients âgés de >75 ans ont reçu 10 mg de dexaméthasone conformément au même schéma thérapeutique que les patients plus jeunes. Le bortézomib (1,3 mg/m2/dose) a été administré aux jours 1, 4, 8 et 11 du cycle de 21 jours, du cycle 1 jusqu'au cycle 8 inclus et ensuite à la même dose aux jours 1 et 8 du cycle de 21 jours à partir du cycle 9. Dans le bras Vd, le bortézomib et la dexaméthasone ont été administrés à la même dose et conformément au même schéma thérapeutique que celui du bras PVd. Le traitement a été poursuivi jusqu'à la progression de la maladie ou la survenue d'une toxicité inacceptable. Lorsqu'il était nécessaire de traiter les symptômes d'une toxicité, les doses avaient été réduites ou le traitement temporairement interrompu ou arrêté.
Au total, 559 patients ont été randomisés: 281 dans le bras PVd et 278 dans le bras Vd. 54% des patients étaient de sexe masculin d'un âge médian de la population totale de 68 ans (min., max.: 27, 89 ans). Environ 70% des patients étaient réfractaires au lénalidomide (71,2% dans le bras PVd, 68,7% dans le bras Vd). Dans l'ensemble, les caractéristiques démographiques et pathologiques étaient généralement bien équilibrées dans les deux bras.
Le critère d'efficacité principal était la survie sans progression (SSP). La SSP était définie comme le délai entre la randomisation et la progression de la maladie ou le décès. La réponse a été évaluée par un comité indépendant d'évaluation de la réponse (IRAC, Independent Response Adjudication Committee) conformément aux critères de réponse de l'International Myeloma Working Group (IMWG) en utilisant l'analyse de la population en intention de traiter comme analyse principale. Le critère d'évaluation secondaire était la survie globale.
Pour la population en ITT, après un suivi médian de 15,9 mois, la médiane de SSP était de 11,20 mois (IC à 95%: 9,66; 13,73) dans le bras PVd. Dans le bras Vd, la médiane de SSP était de 7,10 mois (IC à 95%: 5,88; 8,48). La SSP dans le bras PVd était significativement plus longue que dans le bras Vd: HR 0,61 (IC à 95%: 0,49; 0,77) et suggérait une réduction du risque de progression de la maladie ou de décès de 39% dans le bras PVd.
Chez les patients avec une CrCl initiale <45 ml/min, la médiane de SSP était de 7,16 mois (IC à 95%: 1,94; 10,74) dans le bras PVd (N = 37) comparé à 4,44 mois (IC à 95%: 2,83; 9,49) dans le bras Vd (N = 38); HR = 1,06, IC à 95%: 0,61; 1,83.
D'après l'analyse finale de la survie globale (last patient last visit, 13 mai 2022; suivi médian de 64,5 mois) 197/281 (70,1%) des patients dans le bras PVd étaient décédés et 193/278 (69,4%) dans le bras Vd. La médiane de la survie globale d'après l'évaluation de Kaplan-Meier était de 35,6 mois pour le bras PVd et 31,6 mois pour le bras Vd; HR = 0,94, IC à 95%: 0,77, 1,15, avec un taux global d'événements de 69,8%. Ces différences observées n'étaient pas statistiquement significatives.
Le pomalidomide en association avec la dexaméthasone à faible dose (Pd)
Une étude de phase III (CC-4047-MM-003) et une étude de phase II (CC-4047-MM-002) ont été réalisées pour évaluer l'efficacité et la sécurité du pomalidomide.
La phase II
L'étude MM-002 a été réalisée chez 221 patients présentant un myélome multiple en rechute et réfractaire, qui étaient réfractaires au dernier traitement de leur myélome et qui avaient reçu le lénalidomide et le bortézomib.
Le pomalidomide 4 mg a été administré jusqu'à la progression de la maladie, seul pendant 21 jours d'un cycle de 28 jours ou en association avec de la dexaméthasone (40 mg par semaine, administrés aux jours 1, 8, 15 et 22 de chaque cycle de 28 jours).
Près de 81% des patients étaient réfractaires au lénalidomide, 74% au bortézomib et 64% aux lénalidomide et bortézomib (selon les critères de l'International Myeloma Working Group (IMWG)).
Pour le pomalidomide plus dexaméthasone, la survie médiane sans progression (critère d'évaluation principal) était de 16,6 semaines et pour le pomalidomide en monothérapie, elle était de 10,7 semaines. Le taux de réponse pour le pomalidomide plus dexaméthasone s'élevait à 30,1% contre 9,3% pour le pomalidomide en monothérapie.
La phase III
L'étude de phase III (MM-003) a comparé le traitement par pomalidomide plus dexaméthasone (Pom+LD-dex) avec une monothérapie par la dexaméthasone (HD-dex) chez 455 patients présentant un myélome multiple en rechute et réfractaire qui avaient reçu au moins deux traitements et chez qui tant le lénalidomide que le bortézomib avaient été un échec et dont la maladie avait évolué au cours du dernier traitement.
La médiane de SSP (critère d'évaluation principal) dans le bras POM+LD-dex était de 15,7 semaines (IC à 95%: 13,0-20,1) et de 8 semaines dans le bras HD-dex (IC à 95%: 7,0- 9,0) avec un rapport des risques instantanés (hazard ratio [HR]) de 0,45 (IC à 95%: 0,35-0,59); (p <0,001).
Dans le bras pomalidomide la survie globale médiane n'était pas encore obtenu. La différence entre les deux bras de traitement dans la survie globale était statistiquement significative (hazard ratio de 0,53 (IC à 95%: 0,37-0,74); p <0,001).
PharmacocinétiqueAbsorption
Le pomalidomide atteint une concentration plasmatique maximale (Cmax) en 2 à 3 heures et après administration d'une dose orale unique, son absorption est >70%. La biodisponibilité est à peu près dose-proportionnelle. Sa biodisponibilité n'est pas affectée lorsqu'il est administré avec un repas riche en graisses et en calories. Le pomalidomide peut être administré en dehors des repas.
Distribution
Le volume de distribution à l'état d'équilibre est de 62 à 138 litres. La concentration dans le sperme de volontaires sains s'élevait à environ 67% de la concentration plasmatique. La liaison aux protéines est faible.
Métabolisme
Le pomalidomide est le principal composant retrouvé dans la circulation sanguine (environ 70% de la radioactivité plasmatique). Aucun métabolite n'est présent à une concentration supérieure à 10% de la radioactivité totale dans le plasma.
Les principales voies métaboliques de la radioactivité excrétée sont une hydroxylation suivie d'une glucuroconjugaison ainsi que d'une hydrolyse. In vitro, le CYP1A2 et le CYP3A4 ont été identifiés comme les principales enzymes impliquées dans l'hydroxylation du pomalidomide induite par le CYP, avec des contributions supplémentaires minimes du CYP2C19 et du CYP2D6.
La Cmax du pomalidomide avait augmenté de 14% tandis que l'ASC du pomalidomide avait diminué de 32% chez 14 volontaires sains de sexe masculin qui, après une dose orale unique de 4 mg de pomalidomide, avaient fumé 25 cigarettes par jour pendant une période de 10 jours, par rapport à 13 volontaires sains de sexe masculin non-fumeurs.
Élimination
La demi-vie plasmatique du pomalidomide est d'environ 9,5 heures chez les volontaires sains et d'environ 7,5 heures chez les patients atteints d'un myélome multiple. La clairance corporelle totale (Cl/F) moyenne est d'environ 7 à 10 l/heure.
73% de la dose sont éliminés dans les urines et 15% dans les fèces, environ 2% et 8% étant éliminés sous forme de pomalidomide dans les urines et les fèces.
Cinétique pour certains groupes de patients
Troubles de la fonction hépatique
Les paramètres pharmacocinétiques sont légèrement modifiés chez les patients présentant une insuffisance hépatique (définie selon les critères de Child-Pugh) par rapport aux volontaires sains. Par rapport aux volontaires sains, l'exposition moyenne au pomalidomide est augmentée de 51% (intervalle de confiance à 90% [9% à 110%] ) chez les patients présentant une insuffisance hépatique légère, de 58% (intervalle de confiance à 90% [13% à 119%]) chez les patients présentant une insuffisance hépatique modérée et de 72% (intervalle de confiance à 90%, [24% à 138%]) chez les patients présentant une insuffisance hépatique sévère.
Troubles de la fonction rénale
Les analyses pharmacocinétiques de population ont montré que les paramètres pharmacocinétiques du pomalidomide n'étaient pas significativement modifiés chez les patients présentant une insuffisance rénale (définie par la clairance de la créatinine ou le débit de filtration glomérulaire estimé [DFGe]) par rapport aux patients ayant une fonction rénale normale (ClCr ≥60 ml/minute). L'exposition systémique (ASC) normalisée moyenne au pomalidomide était de 98,2% avec un intervalle de confiance à 90% [77,4% à 120,6%] chez les patients atteints d'insuffisance rénale modérée (DFGe ≥30 et ≤45 ml/minute/1,73 m2) par rapport aux patients ayant une fonction rénale normale. L'exposition systémique (ASC) normalisée moyenne au pomalidomide était de 100,2% avec un intervalle de confiance à 90% [79,7% à 127,0%] chez les patients atteints d'insuffisance rénale sévère ne nécessitant pas de dialyse (ClCr <30 ml/minute ou DFGe <30 ml/minute/1,73 m2) par rapport aux patients ayant une fonction rénale normale. L'exposition systémique (ASC) normalisée moyenne au pomalidomide était augmentée de 35,8% avec un IC à 90% [7,5% à 70,0%] chez les patients atteints d'insuffisance rénale sévère nécessitant des dialyses (ClCr <30 ml/minute nécessitant des dialyses) par rapport aux patients ayant une fonction rénale normale.
Patients âgés
Chez des patients âgés de 61 à 82 ans, les paramètres pharmacocinétiques moyens de l'ASC (0-∞) et de la Cmax étaient en général similaires à ceux de patients plus jeunes.
Enfants et adolescents
Les données actuelles chez les patients pédiatriques ne permettent pas de fournir des recommandations d'utilisation dans cette tranche d'âge.
Données précliniquesGénotoxicité/carcinogénicité
Le pomalidomide n'a pas montré d'effets mutagènes dans les tests de mutation sur cellules bactériennes et cellules de mammifères et n'a pas induit d'aberrations chromosomiques dans des lymphocytes de sang périphérique humains ni de formation de micronoyaux dans les érythrocytes polychromatiques dans la moelle osseuse de rats ayant reçu des doses allant jusqu'à 2'000 mg/kg/jour. Il n'a pas été réalisé d'études de cancérogenèse.
Fertilité et développement embryonnaire précoce
Dans une étude de fertilité et de développement embryonnaire précoce chez le rat, le pomalidomide a été administré chez les mâles et les femelles aux doses de 25, 250 et 1'000 mg/kg/jour. L'examen de l'utérus le 13e jour de gestation a montré une diminution du nombre moyen d'embryons viables et une augmentation des pertes post-implantatoires à toutes les doses. Par conséquent, la DSENO pour ces effets était inférieure à 25 mg/kg/jour (ASC24h: 39'960 ng·h/ml; exposition 99 fois supérieure comparativement à la dose clinique la plus faible testée de 4 mg). Lorsque les mâles traités dans cette étude ont été accouplés avec les femelles non traitées, tous les paramètres utérins ont été comparables à ceux des témoins. Sur la base de ces résultats, les effets observés ont été imputés au traitement des femelles.
Développement embryonnaire et fœtal
Chez le rat et le lapin, le pomalidomide s'est avéré tératogène lorsqu'il a été administré pendant la phase d'organogenèse majeure. Dans l'étude de toxicité sur le développement embryofœtal chez le rat, des malformations ou une absence de vessie, absence de thyroïde et fusion et défaut d'alignement des vertèbres thoraciques et lombaires (corps vertébraux et/ou arcs neuraux) ont été observées à toutes les doses (25, 250 et 1'000 mg/kg/jour). Il n'a pas été mis en évidence de toxicité maternelle dans cette étude. Par conséquent, la DSENO maternelle a été de 1'000 mg/kg/jour et la DSENO en termes de toxicité sur le développement a été <25 mg/kg/jour (l'ASC24h était de 34'340 ng·h/ml le 17e jour de gestation à cette dose la plus faible testée et le rapport d'exposition était de 85 comparativement à une dose clinique de 4 mg). Chez le lapin, le pomalidomide administré à des doses allant de 10 à 250 mg/kg a induit des malformations embryonnaires et fœtales. Une augmentation des anomalies cardiaques a été observée à toutes les doses, avec une incidence significativement plus élevée à la dose de 250 mg/kg/jour. Aux doses de 100 et 250 mg/kg/jour, on a observé une légère augmentation des pertes post-implantatoires et une légère baisse du poids fœtal à la naissance. A la dose de 250 mg/kg/jour, les malformations fœtales consistaient en anomalies des membres (flexion et/ou rotation des membres antérieurs et/ou postérieurs, doigts non attachés ou absents) et malformations osseuses associées (absence d'ossification métacarpienne, défaut d'alignement des phalanges et métacarpes, doigt absent, absence d'ossification des phalanges et tibia court non ossifié ou courbé); dilatation modérée du ventricule latéral du cerveau; position anormale de l'artère sous-clavière droite; absence du lobe intermédiaire du poumon; implantation basse des reins; modifications de la morphologie hépatique; ossification absente ou incomplète du pelvis; augmentation du nombre moyen de côtes thoraciques surnuméraires et diminution du nombre moyen de tarses ossifiés. Une faible réduction de la prise de poids des mères, une diminution significative des triglycérides et une diminution significative des poids absolu et relatif de la rate ont été observées aux doses de 100 et 250 mg/kg/jour. La DSENO maternelle a été de 10 mg/kg/jour et la DSENO sur le développement a été inférieure 10 mg/kg/jour (l'ASC24h était de 418 ng·h/ml le 19e jour de gestation à cette dose la plus faible testée, soit une valeur similaire à celle obtenue avec une dose clinique de 4 mg).
Remarques particulièresStabilité
Le médicament ne doit pas être utilisé au-delà de la date figurant après la mention «EXP» sur l'emballage.
Remarques particulières concernant le stockage
Conserver dans l'emballage d'origine. Ne pas conserver au-dessus de 30°C. Conserver hors de la portée des enfants.
Remarques concernant la manipulation
Comme c'est le cas pour les cytostatiques, la manipulation et l'élimination de Pomalidomid-Teva nécessitent une prudence particulière (voir également la rubrique «Posologie/Mode d'emploi»).
Les professionnels de santé et les aidants qui manipulent le pomalidomide, doivent porter des gants de protection.
Les gélules ne doivent pas être ouvertes ou écrasées. Si la poudre de pomalidomide entre en contact avec la peau, laver immédiatement et abondamment la peau au savon et à l'eau. En cas de contact avec les muqueuses, rincer abondamment à l'eau.
Numéro d’autorisation69576 (Swissmedic).
PrésentationPomalidomid-Teva 1 mg: 14 ou 21 gélules (A)
Pomalidomid-Teva 2 mg: 14 ou 21 gélules (A)
Pomalidomid-Teva 3 mg: 14 ou 21 gélules (A)
Pomalidomid-Teva 4 mg: 14 ou 21 gélules (A)
Titulaire de l’autorisationTeva Pharma AG, Basel.
Mise à jour de l’informationFévrier 2023.
Numéro de version interne: 1.5
|