CompositionPrincipes actifs
Ranibizumab (préparé dans ces cellules de E. coli).
Excipients
α,α-tréhalose dihydraté, L-histidine, chlorhydrate de L-histidine monohydraté, polysorbate 20, eau pour préparations injectables.
Indications/Possibilités d’emploiXimluci est indiqué chez les adultes dans le traitement:
·de la forme exsudative (humide) de la dégénérescence maculaire liée à l'âge (DMLA humide),
·d'une perte de vision due à un œdème maculaire diabétique (OMD),
·de la rétinopathie diabétique non proliférante (RDNP) moyennement sévère à sévère ou de la rétinopathie diabétique proliférante (RDP),
·d'une perte de vision due à un œdème maculaire consécutif à une occlusion de veine rétinienne (occlusion de branche veineuse rétinienne OBVR et occlusion de la veine centrale de la rétine OVCR),
·d'une néovascularisation choroïdienne (NVC) active entraînant une baisse de vision,
·d'une perte de vision due à une néovascularisation choroïdienne (NVC) consécutive à une myopie pathologique (MP).
Ximluci est indiqué chez les prématurés dans le traitement de la rétinopathie du prématuré (RDP):
·RDP dans la zone I (stade de la maladie 1+, 2+, 3 ou 3+), zone II (stade de la maladie 3+) ou AP-RDP (RDP agressive postérieure).
Posologie/Mode d’emploiXimluci ne doit être utilisé que par un ophtalmologue qualifié disposant d'une infrastructure adéquate. Ximluci est injecté dans le corps vitré (voie intravitréenne). Un flacon de Ximluci (pour adultes et prématurés) contient 0,23 ml et est destiné à l'usage unique pour un seul patient.
Posologie usuelle
Posologie chez l'adulte
La dose recommandée de 0,5 mg est administrée sous forme d'injection intravitréenne unique. Cela correspond à un volume d'injection de 0,05 ml. L'intervalle entre deux injections dans le même œil ne doit pas être inférieur à un mois.
Le traitement est initié avec une injection par mois jusqu'à ce que l'acuité visuelle maximale soit atteinte et/ou jusqu'à l'absence de signe d'activité de la maladie. Chez les patients atteints de DMLA humide, d'OMD, de RDNP moyennement sévère à sévère ou de RDP, d'OBVR ou d'OVCR, trois injections mensuelles consécutives ou plus peuvent être nécessaires à l'initiation.
Ensuite, les intervalles de suivi et de traitement doivent être déterminés par le médecin. Le suivi de l'activité de la maladie peut inclure des examens cliniques, des contrôles de l'acuité visuelle et/ou des techniques d'imagerie, comme la tomographie à cohérence optique ou l'angiographie à la fluorescéine.
Les intervalles de suivi et de traitement peuvent être allongés ou raccourcis progressivement en fonction des signes d'activité de la maladie et de l'évolution du traitement.
Si cela est indiqué selon l'avis du médecin, Ximluci peut aussi être utilisé mensuellement dans un œil donné.
Le traitement d'une perte de vision due à une NVC doit être déterminé individuellement pour chaque patient en fonction de l'activité de la maladie. Pour la NVC consécutive à une myopie pathologique (MP) ou pour d'autres NVC ne survenant pas dans le cadre d'une DMLA, on ne dispose d'aucune expérience ayant concerné une durée de traitement de plus d'une année.
Posologie chez le prématuré
La dose recommandée de Ximluci chez le prématuré est de 0,1 mg, administrée sous forme d'injection intravitréenne unique. Cela correspond à un volume d'injection de 0,01 ml. Chez le prématuré, le traitement de la rétinopathie du prématuré (RDP) commence par une dose unique, qui peut être administrée dans les deux yeux le même jour. Au total, jusqu'à trois injections peuvent être administrées dans chaque œil en six mois en présence de signes d'activité de la maladie. Si plusieurs injections sont nécessaires, l'intervalle entre deux injections doit être d'au moins un mois.
Patients présentant des troubles de la fonction hépatique
Aucune étude n'est disponible. Étant donné que la quantité de médicament atteignant la circulation systémique est négligeable, aucune mesure de précaution particulière n'est prévue.
Patients présentant des troubles de la fonction rénale
Aucun ajustement posologique n'est nécessaire pour les patients insuffisants rénaux (voir «Pharmacocinétique»).
Patients âgés (de plus de 65 ans)
Aucun ajustement posologique n'est nécessaire.
Enfants et adolescents
L'utilisation de Ximluci est déconseillée chez les enfants et les adolescents, car les données de sécurité et d'efficacité dans ce groupe de patients sont insuffisantes. Il existe des données limitées chez des patients adolescents âgés de 12 à 17 ans ayant une perte de vision due à une NVC (voir «Propriétés/Effets»).
Mode d'administration
L'administration intravitréenne doit être précédée d'une anamnèse approfondie recherchant d'éventuelles réactions d'hypersensibilité (voir «Mises en garde et précautions»).
L'administration de Ximluci doit avoir lieu sous des conditions d'asepsie (environnement approprié, recouvrement, gants, ustensiles stériles). Une anesthésie adaptée et un microbicide à large spectre topique pour la désinfection de la peau périoculaire, de la paupière et de la surface oculaire doivent être utilisés avant l'injection.
Prélever la totalité du contenu du flacon avec une aiguille filtre de 5 µm montée sur une seringue. Avant l'injection intravitréenne, retirer l'aiguille filtre et la remplacer par une aiguille à injection (13 mm, 30 Gauge). Expulser le contenu jusqu'à ce que l'extrémité du piston atteigne la graduation 0,05 ml (50 µl) sur la seringue. Le contenu d'un flacon est destiné à l'administration d'une dose unique. Jeter la solution non utilisée.
Chez l'adulte, l'aiguille à injection doit être introduite complètement, 3,5–4,0 mm derrière le limbe, en évitant le méridien horizontal et en visant le centre du globe oculaire.
Délivrer alors lentement le volume d'injection; changer de site sur la sclérotique lors des injections suivantes.
Après l'injection, surveiller la pression intraoculaire du patient. La surveillance doit comprendre le contrôle de la circulation sanguine de la papille du nerf optique immédiatement après l'injection, une mesure de la pression intraoculaire dans les 30 minutes, un examen ophtalmoscopique, un examen à la lampe à fente et un examen du fond d'œil après 2–7 jours. Instruire le patient pour qu'il signale immédiatement au médecin les signes éventuels d'une endophtalmie (voir «Mises en garde et précautions»).
Chez le prématuré, l'aiguille à injection doit être introduite 1,0 à 2,0 mm derrière le limbe, en direction du nerf optique. Le volume d'injection de 0,01 ml est ensuite administré.
Contre-indicationsHypersensibilité au ranibizumab ou à l'un des excipients.
Ximluci est contre-indiqué chez les patients atteints d'infections oculaires ou périoculaires, ainsi que chez les patients présentant une inflammation intraoculaire active.
Mises en garde et précautionsRéactions liées aux injections intravitréennes
Une endophtalmie infectieuse et des décollements de la rétine peuvent survenir lors d'injections intravitréennes. C'est pourquoi il faudra utiliser des techniques d'injection aseptiques lors de l'injection de Ximluci. En outre, il faudra surveiller les patients dans les jours qui suivent l'injection afin de déceler une infection à temps et de pouvoir la traiter (voir «Effets indésirables»).
Chez l'adulte, une augmentation transitoire de la pression intraoculaire a été observée dans les 60 minutes suivant l'injection de ranibizumab. Une augmentation prolongée de la pression intraoculaire a aussi été rapportée. C'est pourquoi il faudra surveiller la pression intraoculaire ainsi que la perfusion de l'artère centrale de la rétine et les traiter le cas échéant (voir «Effets indésirables»).
Traitement bilatéral
La sécurité d'emploi et l'efficacité d'un traitement simultané des deux yeux par Ximluci n'ont pas fait l'objet d'études spécialement conçues à cet effet.
Les données limitées à disposition concernant l'utilisation bilatérale de ranibizumab (y compris administration le même jour) ne semblent pas indiquer un risque accru d'événement indésirable systémique par rapport au traitement unilatéral.
Événements thromboemboliques artériels
Il existe un risque potentiel d'événements thromboemboliques artériels dans le cas d'une administration intravitréenne d'inhibiteurs du VEGF (vascular endothelial growth factor). Le risque est éventuellement accru chez les patients présentant un risque connu d'AVC (p.ex. ayant des antécédents d'AVC ou d'accident ischémique transitoire).
Immunogénicité
Dans tous les groupes de traitement, 0–3% des patients qui n'étaient pas encore traités ont montré une réaction immunitaire à ranibizumab. Des titres d'anticorps bas ont été observés chez 1–6% des patients après 12–24 mois lors d'une administration mensuelle. Ces données sur l'immunogénicité reflètent le pourcentage des patients pour qui un test par électrochimioluminescence a donné des résultats positifs. Les données dépendaient fortement de la sensibilité et de la spécificité du test. La signification clinique de la réaction immunitaire à ranibizumab est actuellement incertaine. Des iritis et des inflammations du vitré ont été observées chez les patients montrant les plus hauts titres d'immunoréactivité.
Populations de patients chez lesquelles les données sont limitées
L'emploi de Ximluci chez des patients avec infection systémique active ou autres affections oculaires, telles que décollement de rétine ou trou maculaire n'a pas été étudié jusqu'ici.
Ximluci et photocoagulation au laser
Tant l'administration de ranibizumab à des patients ayant subi dans le passé une photocoagulation au laser que l'emploi simultané de ranibizumab et d'une photocoagulation au laser ont été étudiés. Si une injection de Ximluci est prévue le même jour qu'une photocoagulation au laser, l'injection devra être faite au plus tôt 30 minutes après le traitement au laser.
Traitement concomitant avec d'autres médicaments anti-VEGF (vascular endothelial growth factor)
Ximluci ne peut pas être administré en même temps que d'autres médicaments anti-VEGF (systémiques ou oculaires).
Interruption du traitement par Ximluci chez l'adulte
Dans les cas suivants, la dose doit être suspendue et le traitement ne peut être repris avant la dose prévue suivante:
·Réduction de la meilleure acuité visuelle corrigée (MAVC) de ≥30 niveaux par comparaison à la dernière évaluation de l'acuité visuelle;
·Pression intraoculaire ≥30 mm Hg;
·Déchirure rétinienne;
·Saignement sous-rétinien concernant le centre de la fovéa ou, lorsque la taille du saignement est ≥50%, l'entièreté de la lésion concernée;
·Intervention intraoculaire réalisée au cours des 28 jours précédant ou prévue au cours des 28 jours suivants.
Durée du traitement
Pour la NVC consécutive à une myopie pathologique (MP) ou pour d'autres NVC ne survenant pas dans le cadre d'une DMLA, on ne dispose d'aucune expérience ayant concerné une durée de traitement de plus d'un an.
InteractionsAucune étude spéciale d'interactions n'a été effectuée.
Grossesse, allaitementFemmes en âge de procréer
Les femmes en âge de procréer doivent utiliser des méthodes contraceptives efficaces pendant le traitement et les 30 jours suivants.
Grossesse
Il n'existe pas de données concernant l'emploi du ranibizumab chez la femme enceinte. Des études réalisées sur les animaux n'ont révélé aucune toxicité directe ou indirecte ayant une incidence sur la grossesse, le développement embryonnaire ou le développement fœtal (voir «Données précliniques»). Le ranibizumab inhibe le VEGF-A, un facteur angiogénique important pour la formation de néovaisseaux pendant le développement embryonnaire et fœtal et la formation du placenta. L'exposition systémique au ranibizumab est faible après administration oculaire. Compte tenu de son mécanisme d'action, le ranibizumab doit être considéré comme potentiellement tératogène et embryo-/fœtotoxique et ne doit pas être utilisé pendant la grossesse, sauf en cas de nécessité absolue. Chez les patientes qui envisagent une grossesse, le ranibizumab doit être arrêté 3 mois avant la conception.
Allaitement
Des données limitées indiquent que le ranibizumab est présent dans le lait maternel et qu'il peut supprimer la production du VEGF. L'effet du ranibizumab sur le nourrisson allaité et son effet sur la production de lait/l'excrétion dans le lait maternel ne sont pas connus. Étant donné que de nombreuses substances sont excrétées dans le lait maternel et qu'une résorption et une perturbation de la croissance et du développement de l'enfant sont possibles, l'allaitement pendant le traitement par Ximluci n'est pas recommandé par mesure de précaution. Le bénéfice de l'allaitement pour l'enfant et le bénéfice du traitement pour la femme doivent tous deux être pris en compte.
Fertilité
L'influence de Ximluci sur la fertilité masculine et féminine n'a pas été étudiée.
Effet sur l’aptitude à la conduite et l’utilisation de machinesLe traitement par Ximluci peut entraîner des troubles visuels passagers, ce qui peut altérer l'aptitude à la conduite et à l'utilisation de machines (voir «Effets indésirables»). Les patients présentant de tels signes ne doivent pas conduire de véhicules ou utiliser de machines jusqu'à ce que ces troubles visuels passagers aient disparu.
Effets indésirablesLes 1315 patients au total ayant participé aux trois études cliniques de phase III sur le traitement de la DMLA humide constituent la base de la population soumise à l'évaluation de la sécurité. Tous les patients ont été traités pendant 24 mois au minimum par ranibizumab, et 440 d'entre eux ont reçu la dose recommandée de 0,5 mg.
Les effets indésirables graves liés à la procédure d'injection comprennent: endophtalmie, décollement de rétine rhegmatogène, déchirure de la rétine et cataracte traumatique iatrogène (voir «Mises en garde et précautions»).
Une inflammation intraoculaire et une hypertension intraoculaire ont également été observées chez les patients traités par ranibizumab (voir «Mises en garde et précautions»).
Les effets indésirables mentionnés plus bas sont survenus durant les études contrôlées de phase III FVF2598g (MARINA), FVF2587g (ANCHOR) et FVF3192g (PIER) sur la DMLA humide de façon plus fréquente (au moins 2%) chez les patients avec DMLA humide traités par 0,5 mg de ranibizumab que chez les patients du groupe témoin (traitement simulé (voir «Propriétés/Effets») ou PDT par la vertéporfine). Ils sont donc considérés comme des effets indésirables (EI) potentiels. Les données relatives à la sécurité spécifiées ci-dessous comprennent en outre tous les événements indésirables supposés avoir été au moins potentiellement causés par l'injection elle-même ou par le médicament, et qui sont survenus chez les 440 patients traités par 0,5 mg de ranibizumab contre la DMLA humide dans le cadre du traitement combiné.
Collectif des patients atteints de RD
La sécurité de ranibizumab a été examinée au cours d'une étude clinique de 24 mois dans l'étude de protocole S incluant entre autres 191 patients atteints de rétinopathie diabétique (RD) traités par le ranibizumab. Les événements oculaires et non oculaires observés étaient en accord avec ce à quoi on pourrait s'attendre dans le cas d'une population de patients diabétiques atteints de RD, ou ils étaient similaires aux événements ayant été observés lors d'études cliniques antérieures portant sur ranibizumab en ce qui concerne leur fréquence et leur gravité.
Collectif des patients présentant une NVC
La sécurité de ranibizumab a été examinée au cours d'une étude clinique de 12 mois (MINERVA) à laquelle ont participé 171 patients ayant une perte de vision due à une NVC et traités par le ranibizumab (voir «Propriétés/Effets»). Chez ces patients, le profil de sécurité a correspondu à celui observé dans des études cliniques antérieures menées avec ranibizumab.
Collectif des patients atteints de NVC consécutive à une MP
La sécurité de ranibizumab a été examinée au cours de l'étude clinique de 12 mois RADIANCE. Cette étude portait sur 224 patients présentant une NVC consécutive à une MP, qui ont été traités par le ranibizumab (voir «Propriétés/Effets»). La fréquence et la gravité des événements oculaires et non oculaires qui se sont produits au cours de cette étude ont été comparables à celles observées dans les études portant sur la DMLA humide.
Population atteinte de rétinopathie du prématuré (RDP)
·La sécurité de ranibizumab 0,1 mg a fait l'objet d'une étude clinique de six mois (RAINBOW), portant sur 77 prématurés atteints de RDP et traités par le ranibizumab (voir «Propriétés/Effets»). Les effets indésirables oculaires observés lors de l'étude RAINBOW étaient conformes à ceux qui étaient également survenus chez des adultes pendant le traitement par le ranibizumab 0,5 mg ou qui correspondaient à un événement dans le cadre de la RDP. Les effets indésirables non oculaires dans cette étude clinique correspondaient généralement à ceux que l'on pouvait attendre chez des patients atteints de comorbidités multiples liées à la naissance prématurée. Pour des raisons théoriques, il convient de tenir compte du fait que la maturation d'autres organes pourrait être retardée après un traitement par des anti-VEGF, ce qui pourrait entraîner des complications.
Fréquences: «très fréquents» (≥1/10), «fréquents» (≥1/100 à < 1/10), «occasionnels» (≥1/1000 à < 1/100), «rares» (≥1/10 000 à < 1/1000), «très rares» (< 1/10 000).
Infections et infestations
Très fréquents: rhinopharyngite (12,5–16,4%).
Fréquents: influenza, infections des voies urinaires.
Affections hématologiques et du système lymphatique
Fréquents: anémie.
Affections du système immunitaire
Fréquents: réactions d'hypersensibilité.
Affections psychiatriques
Fréquents: états anxieux.
Affections du système nerveux
Très fréquents: céphalées (8–15%).
Fréquents: accident vasculaire cérébral.
Affections oculaires
Très fréquents: inflammations intraoculaires (10–18%), inflammation du corps vitré (2,3–10%), décollement du corps vitré (18–19%), hémorragie rétinienne (25%), troubles visuels (6,6–10,5%), douleurs oculaires (27–32%), mouches volantes (7–25%), hémorragies conjonctivales (55–72%), irritation oculaire (12–15%), sensation de corps étranger dans les yeux (12–15%), larmoiement accru (10–14%), blépharite (8–11%), prurit (8,5–10%).
Fréquents: dégénérescence rétinienne, troubles de la rétine, décollement de la rétine, déchirures de la rétine, décollement de l'épithélium pigmentaire rétinien, déchirures dans l'épithélium pigmentaire rétinien, défauts visuels, hémorragies et altérations du corps vitré, uvéite, iritis, iridocyclite, cataracte (sous-capsulaire), opacification de la capsule postérieure du cristallin, kératite ponctuée, abrasions de la cornée, trouble de l'humeur aqueuse, vision trouble, saignements au site d'injection, hémorragie oculaire, conjonctivite (allergique), sécrétion oculaire, photopsie, photophobie, troubles oculaires, douleurs et œdème des paupières, hyperémie conjonctivale.
Occasionnels: endophtalmie, hypopion, hyphéma, kératopathie, adhérence de l'iris, nécrolyse et œdème de la cornée, stries dans la cornée, douleurs et irritations au site d'injection, cécité, irritations des paupières.
Affections cardiaques, affections vasculaires
Des événements thromboemboliques artériels selon la définition de l'Antiplatelet Trialists' Collaboration (1994), tels que des décès d'origine vasculaire, des infarctus du myocarde non mortels, des accidents vasculaires cérébraux ischémiques non mortels et des accidents vasculaires cérébraux hémorragiques non mortels, ont été mis en rapport avec la disponibilité systémique d'inhibiteurs très puissants du VEGF. Durant la première année, la proportion d'événements thromboemboliques s'est élevée à 2,3% dans les deux groupes de patients traités par ranibizumab (0,3 et 0,5 mg). En comparaison, elle s'élevait à 1,3% seulement dans le groupe témoin. Durant la deuxième année de l'étude MARINA, la proportion a atteint 3,0% pour les deux groupes de traitement, et 3,2% dans le groupe témoin.
Affections respiratoires, thoraciques et médiastinales
Fréquents: toux.
Affections gastro-intestinales
Fréquents: nausées.
Affections de la peau et du tissu sous-cutané
Occasionnels: réactions allergiques (rash, urticaires, prurit, érythème).
Affections musculo-squelettiques et du tissu conjonctif
Très fréquents: douleurs articulaires (8–12%).
Investigations
Élévation de la pression intraoculaire.
Immunogénicité
Comme avec toutes les protéines thérapeutiques, il existe chez les patients traités par Ximluci un risque de réponse immunitaire. Les données d'immunogénicité reflètent le pourcentage de patients présentant un test positif à la recherche d'anticorps contre le ranibizumab par immunodosage. Les résultats dépendaient beaucoup de la sensibilité et de la spécificité du test.
Dans les études sur la DMLA, 0% à 3% des patients encore non traités de l'ensemble des groupes de traitement ont présenté une réponse immunitaire à ranibizumab. Après des injections mensuelles de ranibizumab durant 12 à 24 mois, environ 1% à 8% des patients avec DMLA néovasculaire étaient porteurs d'anticorps contre le ranibizumab.
Dans les études sur l'OMD, 0% à 2% des patients encore non traités de l'ensemble des groupes de traitement ont présenté une réponse immunitaire à ranibizumab. Après des injections mensuelles de ranibizumab durant 12 mois, environ 2% à 4% des patients avec OMD étaient porteurs d'anticorps contre le ranibizumab.
Dans les études sur l'OVR, 2% à 3% des patients encore non traités de l'ensemble des groupes de traitement ont présenté une réponse immunitaire à ranibizumab. Après des injections mensuelles de ranibizumab durant 12 mois, environ 4% à 5% des patients avec OVR étaient porteurs d'anticorps contre le ranibizumab.
La signification clinique de l'immunoréactivité à ranibizumab n'est pas encore claire à ce jour.
Parmi les données agrégées des études cliniques en double aveugle et randomisées terminées, des infections/inflammations cutanées non oculaires et non graves se sont développées plus fréquemment chez les patients atteints d'OMD sous ranibizumab à 0,5 mg que chez les patients sous traitement de contrôle (1,85/100 années-patient contre 0,27/100 annéespatient). Un lien avec le ranibizumab n'est jusqu'ici pas établi. Il existe toutefois un risque théorique d'un lien entre ces événements et l'inhibition du VEGF.
L'annonce d'effets secondaires présumés après l'autorisation est d'une grande importance. Elle permet un suivi continu du rapport bénéfice-risque du médicament. Les professionnels de santé sont tenus de déclarer toute suspicion d'effet secondaire nouveau ou grave via le portail d'annonce en ligne ElViS (Electronic Vigilance System). Vous trouverez des informations à ce sujet sur www.swissmedic.ch.
SurdosageDes cas de surdosage non intentionnel (injection de volumes supérieurs à la dose recommandée de 0,05 ml de ranibizumab) ont été rapportés lors des études cliniques sur la DMLA humide et durant la surveillance post-marketing.
Signes et symptômes
Les effets indésirables les plus fréquemment enregistrés étaient une élévation de la pression intraoculaire ainsi que des douleurs oculaires.
Traitement
En cas d'administration d'une dose trop élevée, il convient de surveiller la pression intraoculaire et, le cas échéant, de laisser le soin au médecin traitant d'instaurer le traitement adéquat.
Dans des études cliniques, des patients avec DMLA humide et OMD ont reçu des doses allant jusqu'à 2 mg de ranibizumab dans un volume d'injection de 0,05 ml à 0,10 ml. La nature et la fréquence des événements indésirables oculaires et systémiques correspondaient à celles rapportées pour la dose de 0,5 mg (dans 0,05 ml) de ranibizumab.
Propriétés/EffetsCode ATC
S01LA04
Ximluci est un biosimilaire.
Mécanisme d'action
Le principe actif de Ximluci (le ranibizumab) est un fragment d'anticorps monoclonal recombinant humanisé (Fab), dirigé contre le facteur de croissance de l'endothélium vasculaire A (VEGF-A) humain. Le fragment se lie avec une grande affinité au VEGF-A et à ses isoformes. Les isoformes, telles que le VEGF121 et le VEGF165, sont formées par épissage alternatif de l'ARNm et l'isoforme VEGF110 est formée par protéolyse. La liaison du ranibizumab au VEGF-A et à ses isoformes bloque l'activation des récepteurs VEGFR-1 et VEGFR-2 à la surface des cellules endothéliales.
Pharmacodynamique
L'activation des récepteurs VEGFR-1 et VEGFR-2 entraîne une prolifération des cellules endothéliales, une néovascularisation ainsi qu'une hyperperméabilité vasculaire. On suppose que tous ces facteurs contribuent à la progression de la forme néovasculaire de la dégénérescence maculaire liée à l'âge (DMLA), au développement de la NVC, y compris de la NVC consécutive à une myopie pathologique (MP), des œdèmes maculaires diabétiques (OMD) et des occlusions de veines rétiniennes (OVR) induisant la perte de vision.
Efficacité clinique
Traitement de la DMLA humide
La sécurité et l'efficacité cliniques du ranibizumab dans le traitement de la DMLA humide ont été étudiées dans trois études randomisées et en double aveugle, menées au total auprès de 1323 patients (ranibizumab: N = 879, groupe témoin N = 444) avec DMLA néovasculaire. Les traitements simulés de l'étude MARINA et la PDT par Visudyne comme traitement actif de l'étude ANCHOR ont servi de bras de contrôle. Les patients inclus présentaient des lésions d'une taille allant jusqu'à 12 surfaces papillaires et une acuité visuelle dans l'œil étudié allant de 20/40 à 20/320 (selon Snellen). L'âge moyen des patients était de 77 ans. Dans le cadre des études cliniques, les patients ont reçu la consigne de s'appliquer eux-mêmes des gouttes antimicrobiennes dans les yeux (quatre fois par jour, 3 jours avant et après chaque injection).
L'étude MARINA de 24 mois a inclus 716 patients atteints de NVC classique minimale ou de NVC occulte. Ils ont reçu des injections intravitréennes mensuelles de 0,3 mg de ranibizumab (N = 238), de 0,5 mg de ranibizumab (N = 240) ou des injections simulées (N = 238). Durant les 24 mois de la phase de traitement, les patients ont reçu en moyenne 22 traitements sur les 24 possibles. Les résultats de l'étude MARINA observés après 12 mois de traitement ont été pour l'essentiel également confirmés après 24 mois de traitement (1 fois par mois) chez 90% des patients.
L'étude ANCHOR de 24 mois a inclus 423 patients atteints de NVC principalement classique. Ils ont reçu des injections intravitréennes mensuelles de 0,3 mg de ranibizumab et une PDT simulée (N = 143), de 0,5 mg de ranibizumab et une PDT simulée (N = 140) ou des injections intravitréennes simulées et une PDT active par la vertéporfine (N = 143). La première PDT simulée ou active par la vertéporfine a été administrée avec l'injection initiale de ranibizumab. Ensuite, le rythme du traitement était tous les 3 mois lorsque l'œil étudié montrait une poursuite ou une reprise de l'hyperperméabilité vasculaire (angiographie fluorescéinique).
Les résultats sont résumés dans les tableaux suivants:
Tableau 0-1 Étude MARINA: résultats après 12 et 24 mois
|
Variation de l'acuité visuelle (en lettres, ETDRS)
|
Mois
|
Traitement simulé
(n = 238)
|
Ranibizumab
0,5 mg
(n = 240)
|
Différence
(IC 95%)a
| |
Perte de l'acuité visuelle <15 lettres (%)b
|
12 mois
|
62%
|
95%
|
32%
(26%, 39%)
| |
24 mois
|
53%
|
90%
|
37%
(29%, 44%)
| |
Gain de l'acuité visuelle ≥15 lettres (%)b
|
12 mois
|
5%
|
34%
|
29%
(22%, 35%)
| |
24 mois
|
4%
|
33%
|
29%
(23%, 35%)
| |
Variation moyenne de l'acuité visuelleb
(lettres)
|
12 mois
|
-10,5 (16,6)
|
+7,2 (14,4)
|
17,5
(14,8, 20,2)
| |
24 mois
|
-14,9 (18,7)
|
+6,6 (16,5)
|
21,1
(18,1, 24,2)
| |
a
après stratification
b p <0,01
|
Tableau 0-2 Étude ANCHOR: résultats après 12 et 24 mois
|
Variation de l'acuité visuelle
|
Mois
|
PDT par vertéporfine
(n = 143)
|
Ranibizumab 0,5 mg
(n = 140)
|
Différence
(IC 95%)a
| |
Perte de l'acuité visuelle <15 lettres (%)b
|
12 mois
|
64%
|
96%
|
33%
(25%, 41%)
| |
24 mois
|
66%
|
90%
|
25%
(16%, 34%)
| |
Gain de l'acuité visuelle ≥15 lettres (%)b
|
12 mois
|
6%
|
40%
|
35%
(26%, 44%)
| |
24 mois
|
6%
|
41%
|
35%
(26%, 44%)
| |
Variation moyenne de l'acuité visuelleb(lettres)
|
12 mois
|
-9,5 (16,4)
|
+11,3 (14,6)
|
21,1
(17,5, 24,6)
| |
24 mois
|
-9,8 (16,4)
|
+10,7 (16,5)
|
20,7
(16,8, 24,7)
| |
a
après stratification
b p <0,01
|
Dans les deux études MARINA et ANCHOR, l'amélioration de l'acuité visuelle observée avec ranibizumab 0,5 mg à 12 mois s'est traduite par un bénéfice pour le patient. Ce bénéfice a été mesuré à l'aide des trois sous-échelles du National Eye Institute Visual Function Questionnaire (VFQ-25) qui étaient les critères secondaires pré-spécifiés pour l'évaluation de l'efficacité (activités liées à la vision de près, activités liées à la vision de loin et activités indépendantes de la vision). Toutes les différences entre ranibizumab 0,5 mg et les deux groupes témoins ont été statistiquement significatives et cliniquement pertinentes, avec des valeurs de p comprises entre 0,009 et < 0,0001.
L'étude PIER a inclus 184 patients présentant des lésions NVC (avec ou sans composante classique). Ils ont reçu pendant les 3 premiers mois une injection intravitréenne par mois de ranibizumab à 0,3 mg ou de ranibizumab à 0,5 mg ou une injection intravitréenne simulée. D'autres injections de ranibizumab ont eu lieu à intervalles de 3 mois. Après le mois 14, les patients qui avaient reçu une injection simulée pouvaient également être traités par ranibizumab et à partir du mois 19, la fréquence des injections de ranibizumab pouvait être augmentée. Les patients traités par ranibizumab dans PIER ont reçu en moyenne 10 traitements en 24 mois.
Le critère d'évaluation principal de l'efficacité était la variation moyenne de l'acuité visuelle pendant les 12 mois. Après une augmentation initiale de l'acuité visuelle durant l'administration des doses mensuelles, les patients traités par une dose de ranibizumab tous les 3 mois ont perdu de l'acuité visuelle, celle-ci revenant à la valeur initiale au mois 12 et cet effet a été conservé à 24 mois chez la plupart des patients traités par ranibizumab (82%). Les données recueillies chez un nombre limité de patients étant passés à ranibizumab après avoir reçu des injections simulées pendant plus d'un an, indiquent qu'un début précoce du traitement est associé à une meilleure conservation de l'acuité visuelle.
Tableau 0-3 Étude PIER: résultats après 12 mois
|
Variation de l'acuité visuelle
|
Traitement simulé
(n = 63)
|
Ranibizumab
0,5 mg
(n = 61)
|
Différence
(IC 95%)a
| |
Perte de l'acuité visuelle <15 lettres (%)b
|
49
|
90
|
37
(23, 52)
| |
Gain de l'acuité visuelle ≥15 lettres (%)b
|
10
|
13
|
2
(-8, 12)
| |
Variation moyenne de l'acuité visuelleb
|
-16,3 (22,3)
|
-0,2 (13,1)
|
14,7
(8,2, 21,2)
| |
a
après stratification
b p < 0,0001
|
L'étude SAILOR de phase IIIb, multicentrique d'une durée de 1 an, a été réalisée chez des patients atteints de NVC due à une DMLA, aussi bien naïfs de traitement que prétraités. L'objectif primaire de cette étude était d'évaluer l'incidence des effets indésirables oculaires et non oculaires pendant les 12 mois de traitement. 2378 patients ont été randomisés dans 2 groupes qui ont reçu respectivement 0,3 mg ou 0,5 mg de ranibizumab chaque mois pendant 3 mois; le traitement a ensuite été poursuivi en fonction des résultats, à intervalles d'au moins 1 mois.
Au total, il n'y a pas eu de déséquilibre entre les deux groupes quant aux effets indésirables oculaires et non oculaires. Il n'y a pas eu non plus de déséquilibre entre les deux groupes quant au nombre d'AVC. Sous 0,3 mg, 8 patients sur 1169 (0,7%, IC 95%: de 0,3% à 1,3%) en ont été atteints et sous 0,5 mg, 15 patients sur 1209 (1,2%, IC 95%: de 0,7% à 2%). Les patients ayant des facteurs de risque connus, tels qu'antécédents d'AVC ou d'accident ischémique transitoire, présentent vraisemblablement un risque accru d'AVC pendant le traitement par Ximluci.
Traitement de la perte de vision due à un OMD
L'efficacité et la sécurité cliniques de ranibizumab chez les patients avec perte de vision due à un œdème maculaire diabétique (OMD) ont été étudiées dans le cadre de l'étude RESTORE, qui a inclus 345 patients présentant une perte de vision due à un OMD. L'étude comportait trois groupes: dans le groupe 1, les patients (N = 116) ont reçu initialement une injection intravitréenne de ranibizumab 0,5 mg en monothérapie et une photocoagulation au laser simulée. Les sujets du groupe 2 (N = 118) ont reçu initialement du ranibizumab 0,5 mg par injection intravitréenne et une photocoagulation au laser. Les patients du groupe 3 (N = 111) ont bénéficié initialement d'une photocoagulation au laser avec une injection simulée.
Le traitement par le ranibizumab a ensuite été poursuivi sous forme d'injections intravitréennes mensuelles et interrompu en cas de vision stable sous ranibizumab au cours de trois contrôles consécutifs. Après une interruption, le traitement a été repris en cas de détérioration de la vision du patient consécutive à la progression de l'OMD. Les répétitions des photocoagulations au laser ont été effectuées le même jour, au moins 30 minutes avant l'injection de ranibizumab, selon les critères de l'ETDRS.
Les résultats sont résumés dans les tableaux suivants:
Tableau 0-4 Étude RESTORE: résultats à 12 mois
|
Variation de la meilleure vision corrigée
|
Ranibizumab
0,5 mg
(n = 115)
|
Ranibizumab
0,5 mg + laser
(n = 118)
|
Laser
(n = 110)
| |
Variation moyenne de la meilleure acuité visuelle corrigée entre le 1er et le 12e mois en nombre de lettres, versus vision au début de l'étude (déviation standard)a
|
6,1 (6,4)
|
5,9 (7,9)
|
0,8 (8,6)
| |
Variation moyenne de la meilleure acuité visuelle corrigée au 12e mois en nombre de lettres, versus vision au début de l'étude (déviation standard)
|
6,8 (8,3)a
|
6,4 (11,8)b
|
0,9 (11,4)
| |
Augmentation de la meilleure vision corrigée ≥10 lettres (% des patients)
|
37,4c
|
43,2
|
15,5
| |
Augmentation de la meilleure vision corrigée ≥15 lettres (% des patients)
|
22,6d
|
22,9e
|
8,2
| |
a
p <0,0001, b p = 0,0004, c p = 0,0001, d p = 0,0032, e p = 0,0021
|
240 patients, qui avaient précédemment terminé l'étude RESTORE à 12 mois, ont été inclus dans l'étude d'extension de 24 mois multicentrique en ouvert (RESTORE Extension). Ils ont été traités par le ranibizumab 0,5 mg pro re nata (PRN) dans le même œil que celui sélectionné comme œil d'étude dans l'étude centrale. Le traitement était réinstauré mensuellement après diminution de la MAVC due à l'OMD et poursuivi jusqu'à ce qu'une stabilisation de la MAVC soit atteinte. De plus, un traitement par laser était réalisé conformément aux critères ETDRS, si l'investigateur le jugeait nécessaire.
Le nombre moyen d'injections administrées chez les patients traités par le ranibizumab dans l'étude centrale a été de 6,4 pendant la période d'extension de 24 mois. Parmi les 74 patients du bras traité par laser de l'étude centrale, 59 (79%) ont reçu le ranibizumab à un moment donné dans la phase d'extension. En moyenne, ces 59 patients ont reçu 8,1 injections de ranibizumab pendant la période d'extension de 24 mois. La proportion de patients qui n'a pas eu besoin d'un traitement par le ranibizumab durant la phase d'extension a été respectivement de 19%, 25% et 20% pour le groupe préalablement traité par le ranibizumab, le groupe préalablement traité par le ranibizumab + laser, et le groupe préalablement traité par laser seul.
Les résultats sont résumés dans le tableau suivant:
Tableau 0-5 Étude d'extension RESTORE: résultats à 36 mois
|
Paramètre d'évaluation par rapport aux valeurs initiales dans l'étude centrale
|
Au préalable ranibizumab
0,5 mg
n = 83
|
Au préalable ranibizumab
0,5 mg + laser
n = 83
|
Au préalable laser
n = 74*
| |
Variation moyenne de la MAVC par rapport aux valeurs initiales dans l'étude centrale au mois 36 (ET)
|
8,0 (10,09)
|
6,7 (9,59)
|
6,0 (9,35)
| |
Gain ≥10 lettres par rapport aux valeurs initiales dans l'étude centrale ou MAVC ≥84 (%) au mois 36
|
39 (47,0)
|
37 (44,6)
|
31 (41,9)
| |
Gain ≥15 lettres par rapport aux valeurs initiales dans l'étude centrale ou MAVC ≥84 (%) au mois 36
|
23 (27,7)
|
25 (30,1)
|
16 (21,6)
| |
n = nombre de patients pour lesquels les valeurs étaient disponibles, non seulement au début de l'étude (étude centrale, mois 0) mais aussi à la visite du mois 36.
* 59 (79%) des 74 patients avec traitement préalable par laser ont reçu du ranibizumab pendant la phase d'extension.
|
Le profil de sécurité à long terme du ranibizumab observé dans l'étude d'extension de 24 mois est en accord avec le profil de sécurité connu de ranibizumab.
Dans l'étude de phase IIIb (RETAIN), 372 patients présentant une baisse visuelle due à un OMD ont été randomisés pour recevoir des injections intravitréennes:
·de ranibizumab 0,5 mg avec une photocoagulation au laser concomitante selon un protocole «treat-and-extend» (TE) (n = 121),
·de ranibizumab 0,5 mg en monothérapie selon un protocole TE (n = 128) ou
·de ranibizumab 0,5 mg en monothérapie selon un protocole PRN (n = 123).
·Dans tous les groupes, le traitement par le ranibizumab a été initié par des injections intravitréennes mensuelles et poursuivi jusqu'à ce que la MAVC soit stable lors d'au moins trois évaluations mensuelles consécutives. La photocoagulation au laser a été réalisée au début de l'étude, le même jour que la première injection de ranibizumab, puis selon les besoins conformément aux critères ETDRS. Dans le protocole TE, le ranibizumab était ensuite administré à des intervalles prévus de 2 ou maximum 3 mois. Dans le protocole PRN, la MAVC était évaluée mensuellement et le ranibizumab était alors administré au cours de la même visite, si besoin. Dans tous les groupes, le traitement mensuel était réinstauré après une diminution de la MAVC due à la progression de l'OMD et poursuivi jusqu'à ce qu'une stabilisation de la MAVC soit de nouveau atteinte. La durée de l'étude a été de 24 mois.
·Dans l'étude RETAIN, le nombre moyen (médian) d'injections était de 12,4 (12,0) dans le groupe ranibizumab TE + laser, de 12,8 (12,0) dans le groupe ranibizumab TE seul, et de 10,7 (10,0) dans le groupe ranibizumab PRN. Après les trois premières visites mensuelles de traitement, le nombre de visites de traitement nécessaires dans le protocole TE a été de 13 vs 20 requises dans le protocole PRN. Dans les deux schémas de traitement, plus de 70% des patients ont conservé leur MAVC à une fréquence de visite ≥2 mois. L'association du laser n'a pas diminué le nombre moyen d'injections de ranibizumab dans le protocole TE.
Les résultats sont résumés dans le tableau suivant:
Tableau 0-6 Résultats de l'étude RETAIN
|
Résultat exprimé par rapport aux valeurs initiales
|
Ranibizumab TE
0,5 mg + Laser
n = 117
|
Ranibizumab TE
0,5 mg seul
n = 125
|
Ranibizumab PRN
0,5 mg
n = 117
| |
Variation moyenne de la MAVC du mois 1 au mois 12 (ET)
|
5,9 (5,5)b
|
6,1 (5,7)b
|
6,2 (6,0)
| |
Variation moyenne de la MAVC du mois 1 au mois 24 (ET)
|
6,8 (6,0)
|
6,6 (7,1)
|
7,0 (6,4)
| |
Variation de la MAVC au mois 24 (ET)
|
8,3 (8,1)
|
6,5 (10,9)
|
8,1 (8,5)
| |
Gain ≥10 lettres ou MAVC ≥84 (%) au mois 24
|
43,6
|
40,8
|
45,3
| |
Gain ≥15 lettres ou MAVC ≥84 (%) au mois 24
|
25,6
|
28,0
|
30,8
| |
Perte ≥10 lettres au mois 24
|
2,6
|
7,2
|
3,4
| |
Perte ≥15 lettres au mois 24
|
0,9
|
4,0
|
2,6
| |
b
p <0,0001
|
Dans les études dans l'OMD, l'amélioration de la MAVC était accompagnée d'une réduction de la valeur moyenne de l'ECR au cours du temps dans tous les groupes de traitement.
Traitement de la rétinopathie diabétique non proliférante (RDNP) moyennement sévère à sévère ou de la rétinopathie diabétique proliférante (RDP)
La sécurité et l'efficacité cliniques de ranibizumab chez les patients atteints de rétinopathie diabétique proliférante (RDP) ont été évaluées dans une étude de non-infériorité de phase III à groupes parallèles (protocole S), multicentrique, randomisée, contrôlée par traitement actif à laquelle 305 patients (394 yeux étudiés) atteints de RDP avec ou sans OMD (œdème maculaire diabétique) au début de l'étude ont participé et dans laquelle l'injection intravitréenne de 0,5 mg de ranibizumab a été comparée au traitement standard avec photocoagulation panrétinienne (PPR). Au total, 191 yeux (48,5%) ont été randomisés et traités par 0,5 mg de ranibizumab et 203 yeux (51,5%) ont été randomisés et traités par PPR. Au total, 88 yeux (22,3%) présentaient un OMD en situation initiale: 42 (22,0%) et 46 (22,7%) des yeux dans le groupe avec le ranibizumab et la PPR respectivement. Au total, 306 yeux (77,7%) ne présentaient pas d'OMD en situation initiale: 149 (78,0%) et 157 (77,3%) des yeux dans le groupe avec le ranibizumab et la PPR respectivement.
Après 2 ans de traitement, le score de MAVC a été modifié de +2,7 lettres depuis la situation initiale dans le groupe de ranibizumab et de -0,7 lettre depuis la situation initiale dans le groupe de PPR. La différence de 3,5 lettres était comprise dans la marge de non-infériorité, de telle sorte que la non-infériorité de ranibizumab par rapport à la PPR a été confirmée.
La variation de la sévérité de la rétinopathie diabétique a été évaluée au moyen de photos du fond de l'œil par l'utilisation du score de sévérité pour la rétinopathie diabétique (DRSS) de l'étude sur le traitement précoce de la rétinopathie (ETDRS). Dans cette étude, une amélioration du DRSS d'au moins 2 degrés avait eu lieu chez 41,8% des yeux sous traitement par le ranibizumab (n = 189) à 12 mois, comparé à 14,6% chez les yeux traités à la PPR (n = 199).
Dans une méta-analyse de 3 études de phase III randomisées, en double aveugle et contrôlées par traitement actif [D2301 (RESTORE), D2303 (REVEAL) et D2305 (REFINE)], réalisées chez 875 patients atteints d'OMD en tout, 48,4% des 315 patients atteints de RDNP moyenne à sévère ou de RDP (n = 192) traités par le ranibizumab ont montré une amélioration du DRSS d'au moins 2 degrés au 12e mois, comparé à 14,6% des patients traités au laser (n = 123).
Traitement de la perte de vision par œdème maculaire consécutif à une OVR
La sécurité et l'efficacité cliniques de ranibizumab chez les patients avec perte de vision due à un œdème maculaire consécutif à l'occlusion d'une veine rétinienne (OVR) ont été étudiées au cours de deux études BRAVO (N = 397) et CRUISE (N = 392), randomisées, contrôlées, en double aveugle. Dans les deux études, les patients ont reçu soit 0,3 mg de ranibizumab, soit 0,5 mg de ranibizumab, soit un traitement simulé. Dans BRAVO, une photocoagulation au laser était autorisée à tout moment de l'étude à partir du 3e mois dans tous les groupes, en guise de traitement de secours. Les résultats de BRAVO et CRUISE sont résumés dans les tableaux suivants.
Tableau 0-7 Étude BRAVO: résultats à 6 mois et à 12 mois
|
Variation de l'acuité visuelle
|
Traitement simulé
(n = 132)
|
Ranibizumab 0,5 mg
(n = 131)
| |
Variation moyenne de la meilleure acuité visuelle corrigée à 6 mois en nombre de lettres versus acuité visuelle au début de l'étudea
|
+7,3
|
+18,3
| |
Variation moyenne de la meilleure acuité visuelle corrigée au 12e mois en nombre de lettres versus acuité visuelle au début de l'étude
|
+12,1
|
+18,3
| |
Proportion de patients avec un gain de l'acuité visuelle ≥15 lettres (%) au 6e mois
|
28,8%
|
61,1%
| |
Proportion de patients avec un gain de l'acuité visuelle ≥15 lettres (%) au 12e mois
|
43,9%
|
60,3%
| |
Proportion de patients ayant reçu un laser de secours pendant 12 mois
|
61,4%
|
34,4%
| |
a
p <0,0001
|
Tableau 0-8 Étude CRUISE: résultats à 6 mois et à 12 mois
|
Variation de l'acuité visuelle
|
Traitement simulé
(n = 132)
|
Ranibizumab 0,5 mg
(n = 131)
| |
Variation moyenne de la meilleure acuité visuelle corrigée au mois 6, en nombre de lettres, versus début de l'étudea
|
+0,8
|
+14,9
| |
Variation moyenne de la meilleure acuité visuelle corrigée au 12e mois, en nombre de lettres, versus début de l'étude
|
+7,3
|
+13,9
| |
Proportion de patients avec un gain de l'acuité visuelle ≥15 lettres (%) au 6e mois
|
16,9%
|
47,7%
| |
Proportion de patients avec un gain de l'acuité visuelle ≥15 lettres (%) au 12e mois
|
33,1%
|
50,8%
| |
a
p <0,0001
|
Les patients traités par ranibizumab ont présenté dans les deux études (BRAVO et CRUISE) une diminution constante de l'épaisseur centrale de la rétine.
Hormis l'amélioration de l'acuité visuelle sous ranibizumab aux mois 6 et 12, l'étude comportait également un volet d'évaluation de la qualité de vie à ces deux échéances, au moyen du National Eye Institute Visual Function Questionnaire (VFQ-25). Les différences entre le groupe ranibizumab 0,5 mg et le groupe témoin ont donné des valeurs de p à 6 mois situées entre 0,02 et 0,0002.
Les résultats de l'étude d'extension HORIZON de BRAVO et CRUISE ont montré les éléments suivants après 12 mois:
La réduction de la fréquence des traitements dans l'étude HORIZON a eu peu de répercussions sur les patients OBVR qui ont conservé l'amélioration initiale de leur acuité visuelle telle qu'observée dans l'étude BRAVO (+17,5 lettres après 24 mois avec une dose de 0,5 mg et en moyenne 2,4 injections au cours de la deuxième année).
En revanche, la réduction de la fréquence des traitements chez les patients OVCR s'est traduite par une diminution de l'acuité visuelle acquise dans l'étude CRUISE (+12 lettres après 24 mois avec une dose de 0,5 mg et en moyenne 3,8 injections au cours de la deuxième année).
Études E2401 (CRYSTAL) et E2402 (BRIGHTER)
Les études BRIGHTER et CRYSTAL ont évalué la sécurité et l'efficacité cliniques de ranibizumab sur 24 mois chez les patients atteints de troubles de la vision dus à un œdème maculaire consécutif à une OVR. Les études ont inclus des patients atteints d'OBVR (n = 455) et d'OVCR (n = 357). Dans les deux études, les patients ont reçu 0,5 mg de ranibizumab à la demande. BRIGHTER était une étude randomisée à 3 groupes, contrôlée par principe actif, dans laquelle le ranibizumab à 0,5 mg en monothérapie était comparé au ranibizumab en association avec la photocoagulation laser et à la photocoagulation laser seule. Après 6 mois, les participants du groupe laser en monothérapie ont pu recevoir 0,5 mg de ranibizumab. CRYSTAL était une étude à un groupe sur le ranibizumab à 0,5 mg en monothérapie.
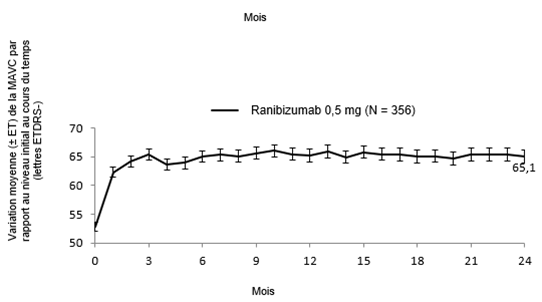
Les principaux résultats fonctionnels et anatomiques des études BRIGHTER et CRYSTAL sont résumés dans le tableau 0-9 et dans les figures 1-0 et 2-0.
Tableau 0-9 Résultats à 6 mois (BRIGHTER) et à 24 mois (BRIGHTER et CRYSTAL)
|
|
BRIGHTER
|
CRYSTAL
| |
|
Ranibizumab 0,5 mg
n = 180
|
Ranibizumab 0,5 mg
+ laser
n = 178
|
Laser*
n = 90
|
Ranibizumab 0,5 mg
n = 356
| |
Variation moyenne de la MAVC à 6 moisb (lettres) (ET)
|
+14,8
(10,7)
|
+14,8
(11,13)
|
+6,0
(14,27)
|
+12,0
(13,95)
| |
Variation moyenne de la MAVC à 24 moisb (lettres) (ET)
|
+15,5
(13,91)
|
+17,3
(12,61)
|
+11,6
(16,09)
|
+12,1
(18,60)
| |
Proportion de patients qui ont atteint une MAVC ≥15 lettres à 24 mois
|
52,8%
|
59,6%
|
43,3%
|
49,2%
| |
Nombre moyen d'injections (ET) (mois 0–23)
|
11,4
(5,81)
|
11,3
(6,02)
|
N/A
|
13,1
(6,39)
| |
* À partir du 6e mois, un traitement avec ranibizumab 0,5 mg était autorisé (24 patients n'ont été traités que par laser).
b: p <0,0001 pour les deux comparaisons dans l'étude BRIGHTER à 6 mois: ranibizumab 0,5 mg comparé à laser et ranibizumab 0,5 mg + laser comparé à laser.
|
Figure 1-0 BRIGHTER: variation moyenne de la MAVC par rapport au niveau initial sur une période de 24 mois
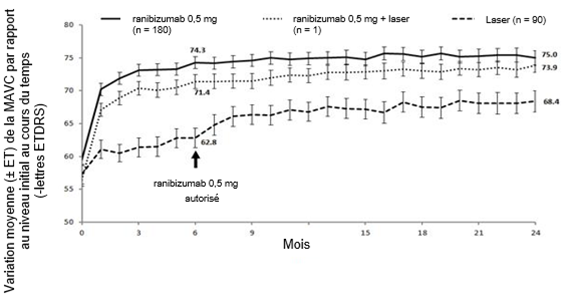
Figure 2-0 CRYSTAL: variation moyenne de la MAVC par rapport au niveau initial sur une période de 24 mois
L'amélioration de la vision s'est révélée similaire chez les patients avec et sans ischémie rétinienne: dans l'étude BRIGHTER, les patients avec ischémie rétinienne (n = 87) ou sans ischémie rétinienne (n = 35) traités par le ranibizumab en monothérapie présentaient, à 24 mois, une variation moyenne par rapport au niveau initial de respectivement +15,4 et +12,9 lettres. Dans l'étude CRYSTAL, les patients avec ischémie rétinienne (n = 107) ou sans ischémie rétinienne (n = 109) présentaient une variation moyenne par rapport au niveau initial de respectivement +11,1 et +12,9 lettres.
Chez les patients malades depuis < 3 mois, une amélioration de l'acuité visuelle de respectivement 13,3 et 10,0 lettres a été observée à 1 mois pour les études BRIGHTER et CRYSTAL, et de respectivement 17,7 et 13,2 lettres à 24 mois. Pour une maladie présente depuis ≥12 mois, elle était respectivement de 4,8 et 7,9 lettres à 1 mois, et de respectivement 8,4 et 8,6 lettres à 24 mois. Une initiation du traitement lors du diagnostic est à envisager.
Le profil de sécurité du ranibizumab, observé dans ces études sur 24 mois, correspond au profil de sécurité de ranibizumab observé dans les études précédentes.
Traitement d'une perte de vision due à une NVC–Étude G2301 (MINERVA)
La sécurité et l'efficacité cliniques de ranibizumab chez des patients ayant une perte de vision due à une NVC consécutive à des étiologies autres qu'une DMLA néovasculaire et une MP ont été évaluées sur la base des données à 12 mois de l'étude G2301 (MINERVA) randomisée, en double aveugle, contrôlée contre une injection simulée. Pour l'analyse, cinq sous-groupes ont été prédéfinis selon l'étiologie (stries angioïdes, choriorétinopathie post-inflammatoire, choriorétinopathie centrale séreuse, choriorétinopathie idiopathique et autres étiologies). 178 patients ont été randomisés selon un ratio 2:1 dans l'un des bras suivants:
Ranibizumab 0,5 mg au début de l'étude, puis schéma posologique individuel selon l'activité de la maladie.
Injection simulée au début de l'étude, puis schéma posologique individuel selon l'activité de la maladie.
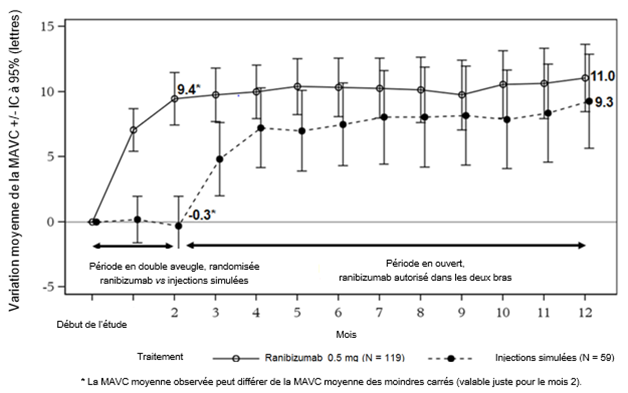
À partir du mois 2, tous les patients ont reçu un traitement par le ranibizumab à la demande. Le critère d'évaluation principal était la variation de la meilleure acuité visuelle corrigée (MAVC) entre le début de l'étude et le mois 2.
Les principaux résultats de MINERVA sont résumés dans les tableaux 0-10 et 0-11 et dans la figure 3-0.
Tableau 0-10 Résultats au mois 2 (MINERVA)
|
|
Ranibizumab 0,5 mg
(n = 119)
|
Injections simulées
(n = 59)
| |
Variation moyenne de la MAVC entre le début de l'étude et le mois 2 (lettres) (moyenne des moindres carrés)a
|
+9,5
|
-0,4
| |
Proportion de patients ayant gagné ≥15 lettres depuis le début de l'étude ou ayant atteint 84 lettres au mois 2
|
31,4%
|
12,3%
| |
Proportion de patients n'ayant pas perdu plus de 15 lettres entre le début de l'étude et le mois 2
|
99,2%
|
94,7%
| |
Diminution de l'épaisseur fovéolaire centrale entre le début de l'étude et le mois 2 (moyenne des moindres carrés)a
|
77 µm
|
-9,8 µm
| |
a
Test unilatéral (p <0,001) avec comparaison avec les injections simulées
|
Figure 3-0: Variation moyenne de la MAVC entre le début de l'étude et le mois 12 au cours du temps (MINERVA)
Lors de la comparaison du ranibizumab avec les injections simulées au mois 2, un effet cohérent a été observé, que ce soit dans la population globale de l'étude ou dans les différents sous-groupes définis selon l'étiologie.
Tableau 0-11 Efficacité du traitement pour la population globale et dans les sous-groupes définis selon l'étiologie au début de l'étude, pour la variable principale au mois 2 (MINERVA)
|
Population globale et sous-groupes stratifiés en fonction de l'étiologie au début de l'étude
|
Efficacité du traitement par rapport aux injections simulées (lettres)
|
Nombre de patients (n) (traitement + injections simulées)
| |
Population globale
|
9,9
|
175*
| |
Stries angioïdes
|
14,6
|
27
| |
Choriorétinopathie post-inflammatoire
|
6,5
|
27
| |
Choriorétinopathie séreuse centrale
|
5,0
|
23
| |
Choriorétinopathie idiopathique
|
11,4
|
62
| |
Autres étiologiesa
|
10,6
|
36
| |
a
Étiologies NVC non comprises dans les autres sous-groupes
*Nombre de patients pour lesquels des données sont disponibles pour l'analyse
|
L'amélioration de la vision s'est accompagnée d'une diminution de l'épaisseur fovéolaire centrale sur une période de 12 mois.
Le nombre moyen d'injections de ranibizumab sur une période de 12 mois dans l'œil étudié a été de 5,8 dans le bras ranibizumab et de 5,4 dans le groupe ayant reçu les injections simulées.
Enfants et adolescents
Cinq patients adolescents âgés de 12 à 17 ans ayant une perte de vision due à une NVC (1x NVC subfovéale lors de drusen papillaires; 1x NVC juxtafovéale et 1x NVC subfovéale lors d'une NVC idiopathique; 2x NVC subfovéale lors d'une maladie de Best) ont reçu un traitement initial par ranibizumab 0,5 mg, puis un schéma thérapeutique individualisé en fonction des signes d'activité de la maladie (p.ex. baisse de l'acuité visuelle, liquide intra/sous-rétinien, hémorragies ou fuites). La variation de la MAVC entre le début de l'étude et le mois 12 s'est améliorée chez les cinq patients et a atteint de + 5 à + 38 lettres. L'amélioration de la vision s'est accompagnée d'une stabilisation ou d'une diminution de l'épaisseur fovéolaire centrale pendant une période de 12 mois (ΔCSFT0-12 mois: de -286 μm à +10 μm). 2 à 5 injections ont été administrées dans l'œil étudié pendant les 12 mois (voir «Posologie/Mode d'emploi»).
Traitement d'une perte de vision due à une NVC consécutive à une MP
La sécurité et l'efficacité cliniques de ranibizumab chez les patients présentant une perte de vision due à une NVC consécutive à une MP ont été évaluées à partir des données portant sur une période de 12 mois provenant de l'étude pivot randomisée, contrôlée, et conduite en double aveugle RADIANCE. Cette étude a été conçue pour évaluer deux schémas posologiques de 0,5 mg de ranibizumab par injection intravitréenne en comparaison à une PDT à la vertéporfine (vPDT, thérapie photodynamique avec Visudyne).
Les 277 patients ont été randomisés dans l'un des bras d'étude suivants:
Groupe I (0,5 mg de ranibizumab, le schéma posologique s'est basé sur des critères de stabilité définis comme une absence de variation de la meilleure acuité visuelle corrigée (MAVC) par rapport à deux examens mensuels précédents);
Groupe II (0,5 mg de ranibizumab, le schéma posologique s'est basé sur des critères d'activité de la maladie définis comme une perte de vision en raison de la présence de liquide intra- ou sous-rétinien ou d'un écoulement actif de liquide suite à la lésion de NVC, avec mise en évidence par une TCO et/ou par une AF);
Groupe III (vPDT; les patients ont pu être traités par le ranibizumab dès le 3e mois).
Au cours des 12 mois de l'étude, les patients du groupe I ont reçu 4,6 injections en moyenne (entre 1 et 11 injections) et ceux du groupe II ont reçu 3,5 injections en moyenne (entre 1 et 12 injections). Dans le groupe II (dans lequel les patients ont reçu le traitement recommandé sur la base de critères déterminés par l'activité de la maladie, voir «Posologie/Mode d'emploi»), 50,9% des patients ont eu besoin d'une ou deux injections, 34,5% des patients de trois à cinq injections et 14,7% des patients de 6 à 12 injections au cours de la période d'étude de 12 mois. 62,9% des patients du groupe II n'ont pas eu besoin d'injections supplémentaires au cours des six derniers mois de l'étude.
Les résultats thérapeutiques les plus importants de l'étude RADIANCE sont résumés dans le tableau 0-12 et dans la figure 0-4.
Tableau 0-12 Résultat du traitement au 3e mois et au 12e mois (RADIANCE)
|
|
Groupe I
0,5 mg de ranibizumab
Stabilité de l'acuité visuelle
(n = 105)
|
Groupe II
0,5 mg de ranibizumab
Activité de la maladie
(n = 116)
|
Groupe III
vPDT*
(n = 55)
| |
3e mois
| |
Variation moyenne de la MAVC entre le 1er et le 3e mois par rapport à la situation initialea (lettres)
|
+10,5
|
+10,6
|
+2,2
| |
Proportion des patients présentant une amélioration de la MAVC:
|
|
|
| |
de ≥10 lettres ou une MAVC globale ≥84 lettres
|
61,9%
|
65,5%
|
27,3%
| |
de ≥15 lettres ou une MAVC globale ≥84 lettres
|
38,1%
|
43,1%
|
14,5%
| |
12e mois
| |
Nombre d'injections jusqu'au 12e mois:
|
|
|
| |
Moyenne
|
4,6
|
3,5
|
N/A
| |
Médiane
|
4,0
|
2,0
|
N/A
| |
Variation moyenne de la MAVC entre le 1er et le 12e mois par rapport à la situation initiale (lettres)
|
+12,8
|
12,5
|
N/A
| |
Proportion des patients présentant une amélioration de la MAVC:
|
|
|
| |
de ≥10 lettres ou une MAVC globale ≥84 lettres
|
69,5%
|
69,0%
|
N/A
| |
de ≥15 lettres ou une MAVC globale ≥84 lettres
|
53,3%
|
51,7
|
N/A
| |
* Contrôle comparatif jusqu'au 3e mois. Les patients randomisés dans le groupe vPDT ont pu recevoir un traitement par le ranibizumab dès le 3e mois (dans le groupe III, 38 patients ont reçu du ranibizumab dès le 3e mois): p< 0,00001 pour la comparaison au groupe témoin vPDT
|
Figure 0-4 Variation moyenne de la MAVC par rapport à la MAVC initiale au cours du temps jusqu'au 12e mois (RADIANCE)
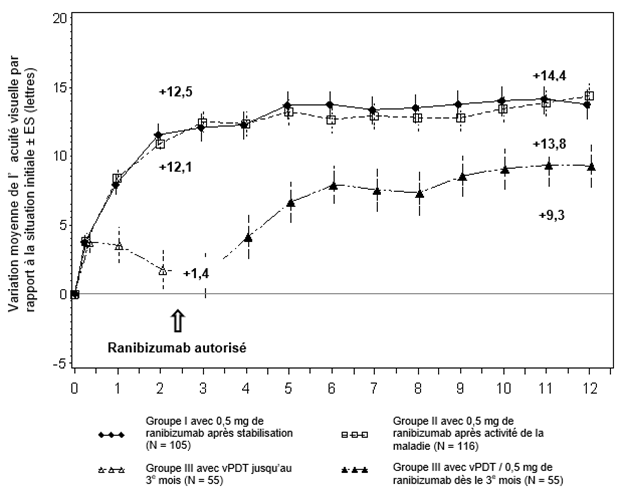
BL = niveau initial; ES = erreur standard de la moyenne.
Figure:
Amélioration moyenne de l'acuité visuelle par rapport au BL ± ES (lettres)
Groupe I avec 0,5 mg de ranibizumab selon stabilisation (N = 105)
Groupe II avec 0,5 mg de ranibizumab selon activité de la maladie (N = 116)
Groupe III avec PDT par Visudyne (N = 55)
Les patients qui ont été randomisés dans le groupe vPDT ont pu recevoir un traitement par le ranibizumab à partir du 3e mois.
L'amélioration de l'acuité visuelle a été accompagnée d'une diminution de l'épaisseur centrale de la rétine.
Dans les bras de traitement par le ranibizumab, un bénéfice subjectivement rapporté par les patients dans le questionnaire VFQ-25 a été constaté par rapport au groupe vPDT (p < 0,05) en ce qui concerne l'amélioration du résultat combiné ainsi que du résultat dans plusieurs sous-échelles (vision globale, activités nécessitant la vision de près, condition psychique et statut d'indépendance fonctionnelle).
Traitement de la RDP chez le prématuré: étude H2301 (RAINBOW)
L'efficacité et la sécurité cliniques de ranibizumab 0,1 mg dans le traitement de la RDP chez le prématuré ont été analysées dans le cadre de l'étude de supériorité randomisée, en ouvert, à trois bras et en groupes parallèles H2301 (RAINBOW) de six mois, conçue pour évaluer l'utilisation du ranibizumab à des doses de 0,1 mg et 0,2 mg, administré par injection intravitréenne, en comparaison avec un traitement au laser. Les patients candidats devaient présenter l'un des signes rétiniens suivants dans les deux yeux:
·Zone I, stade de la maladie 1+, 2+, 3 ou 3+
·Zone II, stade de la maladie 3+
·RDP agressive postérieure (AP-RDP)
Dans le cadre de cette étude, 225 patients ont été randomisés selon un rapport 1:1:1 dans des groupes recevant le ranibizumab par voie intravitréenne à une dose de 0,1 mg (n = 77) ou de 0,2 mg (n = 74) ou un traitement au laser (n = 74).
La réussite du traitement, mesurée par l'absence de RDP active et d'effets indésirables structurels dans les deux yeux 24 semaines après le premier traitement de l'étude, était de 75% dans le groupe ranibizumab 0,1 mg et de 66,2% dans le groupe de traitement au laser. La majorité des patients traités par ranibizumab 0,1 mg (77,6%) ont reçu une seule injection dans chaque œil.
Dans le groupe de traitement ranibizumab 0,1 mg, moins de patients ont changé de mode de traitement en raison d'un manque de réponse que dans le groupe de traitement au laser (16,9% vs 24,3%). Les effets indésirables structurels ont été moins fréquents dans le groupe de traitement ranibizumab 0,1 mg (5 patients, 6,7%) que dans le groupe de traitement au laser (7 patients, 10,1%). En outre, 75% des patients ont obtenu une diminution de la maladie en 8 jours sous ranibizumab 0,1 mg, contre 22,5 jours dans le groupe de traitement au laser.
PharmacocinétiqueAbsorption
L'administration intravitréenne mensuelle de ranibizumab à des patients atteints de DMLA néovasculaire conduit à des concentrations sériques de ranibizumab généralement faibles et à une concentration sérique maximale (Cmax) nettement inférieure à la concentration ayant entraîné une inhibition de l'activité du VEGF de 50% (11–27 ng/ml dans un test de prolifération cellulaire).
Distribution
La concentration sérique maximale (Cmax) se situe généralement dans un intervalle compris entre 0,46 et 1,76 ng/ml et la concentration sérique minimale (Cmin) entre 0,04 et 0,29 ng/ml. La Cmax sérique était proportionnelle à la dose dans l'intervalle posologique compris entre 0,05 et 1,0 mg/œil. La concentration sérique chez les patients avec OMD et OVR était comparable à celle de patients avec DMLA néovasculaire.
Métabolisme
Non applicable.
Élimination
Sur la base de la concentration sérique de ranibizumab, la demi-vie d'élimination moyenne du ranibizumab dans le corps vitré de patients avec DMLA néovasculaire est de 9 jours environ. On suppose que les concentrations sériques du ranibizumab sont 90 000 fois inférieures à celles mesurées dans le corps vitré.
Troubles de la fonction hépatique
Aucune étude spécifique n'est disponible.
Troubles de la fonction rénale
Aucune étude prospective n'a été réalisée pour examiner la pharmacocinétique de Ximluci chez les patients avec une limitation de la fonction rénale. Dans une analyse de pharmacocinétique de population réalisée chez les patients avec DMLA néovasculaire, 68% des patients (n = 136/200) présentaient une limitation de la fonction rénale (pour 46,5% légère, pour 20% modérée et pour 1,5% sévère). Parmi les patients avec OVR examinés, 48,2% (n = 253/525) présentaient une insuffisance rénale (légère dans 36,4% des cas, modérée dans 9,5% des cas et sévère dans 2,3% des cas). La baisse de la clairance systémique du ranibizumab observée était statistiquement non significative.
Population pédiatrique (prématurés atteints de RDP)
Après administration intravitréenne de ranibizumab 0,1 mg dans chaque œil, la concentration sérique de ranibizumab chez les prématurés atteints de RDP était plus élevée que chez les patients adultes atteints de DMLA néovasculaire et traités avec 0,5 mg dans chaque œil. Selon une analyse pharmacocinétique de la population, les différences de Cmax et d'ASCinf étaient environ respectivement 8 et 5 fois supérieures. La demi-vie systémique apparente était d'environ 6 jours. Lors de cette analyse, aucune relation n'a été observée entre la concentration systémique de ranibizumab et celle du VEGF.
Données précliniquesToxicité en cas d'administration répétée
L'administration intravitréenne bilatérale de ranibizumab chez le singe Cynomolgus à des doses variant entre 0,25 mg/œil et 2,0 mg/œil toutes les 2 semaines pendant une période allant jusqu'à 26 semaines a entraîné des effets oculaires dose-dépendants.
Dans la chambre antérieure, une augmentation dose-dépendante des «Flare» et des cellules a été observée, avec un maximum à 2 jours après l'injection. L'intensité de la réaction inflammatoire a généralement régressé avec les injections suivantes ou pendant la phase de récupération, sans être entièrement réversible dans tous les cas. Dans la chambre postérieure, des hémorragies rétiniennes périveineuses focales réversibles sont survenues principalement après la première administration, ainsi que des infiltrations cellulaires vitréennes et des «mouches volantes». Ces dernières ont eu tendance à être dose-dépendantes et ont persisté jusqu'à la fin du traitement. Dans l'étude sur 26 semaines, l'intensité de l'inflammation a augmenté avec le nombre d'injections et le test a dû être arrêté de façon prématurée. Une réversibilité des résultats a été observée pendant la phase de récupération. La formation d'une cataracte a été observée chez certains animaux après une phase relativement longue de forte inflammation, ce qui indique que les modifications du cristallin étaient plutôt secondaires à l'inflammation sévère. Une augmentation transitoire de la pression intraoculaire a été observée après l'injection intravitréenne, indépendamment de la dose.
Les modifications microscopiques au niveau des tissus oculaires ont toutes été liées à l'inflammation et n'ont pas suggéré l'existence de processus dégénératifs dans de quelconques structures oculaires. Dans certains cas, des modifications inflammatoires granulomateuses ont été observées au niveau de la papille du nerf optique. Ces modifications du segment postérieur ont régressé pendant la phase de repos et ont complètement disparu dans certains cas.
Aucun signe de toxicité systémique n'a été observé après une injection intravitréenne induisant des taux sériques plus de 100 fois supérieurs à ceux obtenus lors d'une utilisation thérapeutique normale chez l'être humain. Des anticorps contre le ranibizumab dans le sérum ou dans le corps vitré n'ont pas été observés chez tous les animaux traités.
La documentation préclinique couvre uniquement l'exposition systémique après exposition intravitréenne.
Génotoxicité, carcinogénicité et immunotoxicité
On ne dispose d'aucune donnée sur la mutagénicité, la carcinogénicité et l'immunotoxicité de ranibizumab chez l'animal.
Toxicité sur la reproduction
Lors d'un traitement intravitréen de singes femelles gestantes avec des doses de ranibizumab correspondant à l'exposition systémique maximale, soit 0,9-7 fois l'exposition clinique la plus élevée imaginable, aucun effet embryo-fœtotoxique ou tératogène et aucun effet sur le poids et la structure du placenta n'ont été relevés; le ranibizumab devra cependant être considéré comme potentiellement tératogène et embryo-/fœtotoxique, compte tenu de ses propriétés pharmacologiques.
L'absence d'effets imputés au ranibizumab sur le développement embryo-fœtal est probablement surtout due au fait que le fragment Fab ne passe pas la barrière placentaire. Un cas de présence de ranibizumab dans le sérum d'un fœtus dont la mère avait des taux sériques élevés de cette substance a néanmoins été rapporté; cela suggère que l'anticorps anti-ranibizumab a servi de protéine de transport (avec région Fc) du ranibizumab, diminuant ainsi la clairance sérique chez la mère et permettant le passage à travers la barrière placentaire. Étant donné que les examens sur le développement embryo-fœtal ont été réalisés chez des animaux gestants sains et que la perméabilité du placenta au fragment Fab pourrait être modifiée par une maladie, l'interprétation de ces résultats impose une certaine prudence.
Remarques particulièresIncompatibilités
En l'absence d'étude de tolérance, ce médicament ne doit pas être mélangé à d'autres médicaments.
Stabilité
Le médicament ne doit pas être utilisé au-delà de la date figurant après la mention «EXP» sur l'emballage.
Remarques particulières concernant le stockage
Conserver au réfrigérateur (2-8 °C). Ne pas congeler. Conserver dans l'emballage original à l'abri de la lumière et hors de portée des enfants. Le flacon peut être conservé jusqu'à 48 heures avant emploi à température ambiante (25 °C).
Remarques concernant la manipulation
Pour des raisons microbiologiques, la solution injectable prête à l'emploi provenant d'un flacon doit être utilisée immédiatement après ouverture. La solution non utilisée doit être éliminée (voir «Posologie/Mode d'emploi»). Un flacon est destiné à l'administration d'une dose unique.
Flacon (adultes et prématurés):
Le flacon est stérile. Il ne doit pas être utilisé si l'emballage est endommagé. La stérilité du flacon n'est garantie que si l'emballage et son scellage sont intacts. Il ne doit pas être utilisé si la solution est colorée ou trouble ou si elle contient des particules.
Pour la préparation et la réalisation de l'injection intravitréenne, les éléments à usage unique suivants sont nécessaires:
·aiguille filtre de 5 µm (18 G)
·seringue stérile de 1 ml
·aiguille à injection (13 mm, 30 Gauge)
Ces éléments ne sont pas inclus dans l'emballage qui ne contient que le flacon. Le paquet qui contient le flacon et l'aiguille filtre ne contient ni la seringue stérile de 1 ml, ni l'aiguille à injection.
Veuillez suivre les instructions d'utilisation pour préparer Ximluci en vue d'une administration intravitréenne, voir à la fin de l'information professionnelle.
Numéro d’autorisation69578 (Swissmedic).
Présentation1 flacon de 0,23 ml y compris 1 aiguille filtre. [B]
1 flacon de 0,23 ml. [B]
Titulaire de l’autorisationSpirig HealthCare SA, 4622 Egerkingen
Mise à jour de l’informationAvril 2024
Instructions de préparation et d'utilisation
Instructions de préparation et d'utilisation du flacon (adultes et prématurés)
Veuillez suivre les instructions suivantes pour la préparation de Ximluci en vue d'un usage intravitréen
|
Schéma/Figure
|
Instructions
| |
A.
|
|
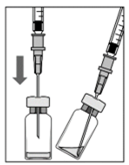
1. Avant l'extraction, retirez la capsule du flacon et nettoyez le septum du flacon (par exemple, avec un tampon imbibé d'alcool à 70%).
2. Dans des conditions stériles, fixez une aiguille-filtre de 5 µm (18 G x 1½ pouce, 1,2 mm x 40 mm) sur une seringue de 1 ml. Insérez l'aiguille-filtre émoussée au centre du bouchon de caoutchouc jusqu'à ce que l'aiguille atteigne le fond du flacon.
3. Aspirez l'entièreté du liquide du flacon en maintenant ce dernier en position verticale, légèrement inclinée pour faciliter une aspiration complète.
| |
B.
|
|
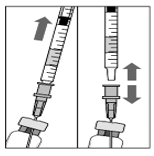
4. Veillez à ce que la tige du piston soit suffisamment rétractée lorsque le flacon est vidé pour vider complètement l'aiguille-filtre.
5. Laissez l'aiguille-filtre émoussée dans l'ampoule et déconnectez la seringue de l'aiguille-filtre émoussée. L'aiguille-filtre sera jetée après le prélèvement du contenu du flacon, elle ne doit pas être utilisée pour l'injection intravitréenne.
| |
C.
|
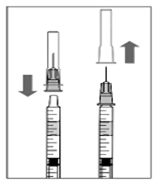
|
6. Dans des conditions aseptiques, fixez une aiguille pour injection (30 G × ½ pouce, 0,3 mm x 13 mm) sur la seringue.
7. Retirez prudemment le capuchon de l'aiguille pour injection, sans séparer l'aiguille de la seringue.
Remarque: lors de l'enlèvement du capuchon de l'aiguille pour injection, le tenir par la coiffe d'insertion.
| |
D.
|
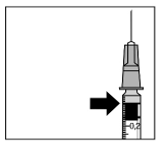
|
8. Éliminez prudemment l'air de la seringue et ajustez la dose au marquage approprié sur la seringue. La dose s'élève à 0,05 ml chez l'adulte. La dose s'élève à 0,01 ml chez le prématuré. La seringue est maintenant prête pour l'injection.
Remarque: ne pas essuyer l'aiguille pour injection. Ne pas tirer le piston vers l'arrière.
| |
|
Après l'injection, ne pas replacer le capuchon sur l'aiguille, ni retirer l'aiguille de la seringue. La seringue usagée, avec l'aiguille, doit être éliminée dans un récipient pour objets tranchants ou conformément à la réglementation locale.
|
|