CompositionPrincipes actifs
Némolizumab produit à partir de cellules d’ovaire de hamster chinois par la technologie de l’ADN recombinant.
Excipients
Poudre: Saccharose, trométamol, chlorhydrate de trométamol (pour l’ajustement du pH), chlorhydrate d’arginine, poloxamère 188
Solvant: Eau pour préparations injectables
Indications/Possibilités d’emploiDermatite atopique (DA)
Nemluvio est indiqué pour le traitement de la dermatite atopique modérée à sévère en association de corticostéroïdes topiques et/ou d’inhibiteurs de la calcineurine chez les adultes et adolescents à partir de 12 ans avec un poids corporel d’au moins 30 kg, dès lors les traitements topiques seuls ne permettent pas un contrôle raisonnable de la maladie (voir les rubriques «Posologie/Mode d’emploi» et «Efficacité clinique»).
Prurigo nodulaire (PN)
Nemluvio est indiqué pour le traitement des adultes atteints de prurigo nodulaire modéré à sévère éligibles à un traitement systémique.
Posologie/Mode d’emploiDermatite atopique (DA)
La posologie de Nemluvio recommandée chez les adultes et adolescents à partir de 12 ans est la suivante:
§Une dose initiale de 60 mg (deux injections de 30 mg), puis 30 mg toutes les 4 semaines (1x/4 sem.).
§Après 16 semaines de traitement, la dose d’entretien recommandée chez les patients qui obtiennent une réponse clinique est de 30 mg toutes les 8 semaines (1x/8 sem.).
Traitements topiques concomitants:
Nemluvio est utilisé en association de corticostéroïdes topiques de puissance faible à modérée et/ou d’inhibiteurs de la calcineurine topiques. Les inhibiteurs de la calcineurine topiques doivent rester réservés aux zones à problème comme le visage et le cou, ainsi qu’aux zones intertrigineuses et génitales En cas d’amélioration suffisante de la maladie, l’utilisation de traitements topiques doit être progressivement réduite, puis arrêtée.
Chez les patients qui ne répondent pas au traitement après une durée de 16 semaines, un arrêt du traitement doit être envisagé.
Prurigo nodulaire (PN)
Chez les patients avec un poids corporel inférieur à 90 kg, il est recommandé d’administrer Nemluvio à une dose initiale de 60 mg (deux injections de 30 mg), puis à une dose de 30 mg toutes les 4 semaines (1x/4 sem.).
Chez les patients avec un poids corporel d’au moins 90 kg, il est recommandé d’administrer Nemluvio à une dose initiale de 60 mg (deux injections de 30 mg), puis à une dose de 60 mg toutes les 4 semaines (1x/4 sem.).
En cas d’absence de réponse au traitement après une durée de 16 semaines, un arrêt du traitement doit être envisagé.
Traçabilité
Afin d’assurer la traçabilité des médicaments biotechnologiques, il convient de documenter pour chaque traitement le nom commercial et le numéro de lot.
Oubli d’une dose (pour toutes les indications)
Si une dose a été oubliée, elle doit être administrée dès que possible, puis le calendrier d’administration doit être repris comme prévu.
Instructions posologiques particulières
Patients âgés (≥65 ans)
Aucun ajustement posologique n’est nécessaire chez les patients âgés (voir la rubrique «Pharmacocinétique»).
Patients présentant des troubles de la fonction hépatique et rénale
Aucun ajustement posologique n’est nécessaire chez les patientes présentant une insuffisance hépatique ou rénale légère ou modérée. Les données sont très limitées chez les patients en insuffisance hépatique ou rénale sévère (voir la rubrique «Pharmacocinétique»).
Enfants et adolescents
La sécurité et l’efficacité de Nemluvio chez les enfants de moins de 12 ans atteints de dermatite atopique modérée à sévère ne sont pas encore établies.
La sécurité et l’efficacité de Nemluvio chez les enfants et adolescents de moins de 18 ans atteints de prurigo nodulaire ne sont pas établies. On ne dispose d’aucune donnée.
Poids corporel
Pour les patients à partir de 12 ans atteints de dermatite atopique, aucun ajustement posologique en fonction du poids corporel n’est recommandé (voir la rubrique «Pharmacocinétique»).
Pour les patients atteints de prurigo nodulaire avec un poids corporel d’au moins 90 kg, la dose recommandée est de 60 mg (deux injections de 30 mg) (voir la rubrique «Pharmacocinétique»).
Mode d’administration
Le traitement doit être instauré par un professionnel de santé expérimenté dans le diagnostic et le traitement des maladies pour lesquelles Nemluvio est indiqué (voir la rubrique «Indications/Possibilités d’emploi»). Nemluvio peut être injecté par le patient lui-même ou par un proche aidant, si le professionnel de santé estime cela pertinent. Avant l’utilisation, le patient et/ou le proche aidant doivent être formés à la préparation et à l’administration de Nemluvio conformément aux instructions d’utilisation figurant dans la notice d’emballage.
Voie sous-cutanée
Nemluvio est injecté par voie sous-cutanée dans la partie avant de la cuisse ou dans l’abdomen, en dehors d’un rayon de 5 cm autour du nombril. Si l’injection est réalisée par une tierce personne, la partie supérieure du bras peut également être choisie comme site d’injection.
Le site d’injection doit changer à chaque injection. Nemluvio ne doit pas être injecté dans des zones cutanées sensibles, enflammées, gonflées ou lésées, ou présentant une ecchymose, une cicatrice ou une plaie ouverte.
Des informations complémentaires sur l’administration de ce médicament sont fournies à la rubrique «Remarques concernant la manipulation».
Contre-indicationsHypersensibilité au principe actif ou à l’un des excipients mentionnés à la rubrique «Composition».
Mises en garde et précautionsRéactions d’hypersensibilité:
En cas de réaction d’hypersensibilité systémique généralisée (immédiate ou retardée), l’utilisation de Nemluvio doit être immédiatement arrêtée et un traitement adapté doit être instauré.
Asthme non contrôlé
Les patients atteints d’asthme non contrôlé ont été exclus des études et on ne dispose d’aucune donnée concernant l’utilisation de Nemluvio dans cette population de patients.
Vaccins
Avant d’instaurer le traitement par Nemluvio, il est recommandé de mettre à jour le statut vaccinal des patients conformément aux recommandations vaccinales actuelles. Les vaccins vivants doivent être évités chez les patients traités par Nemluvio. On ignore si l’administration de vaccins vivants pendant le traitement par Nemluvio peut avoir une influence sur sa sécurité et son efficacité. On ne dispose d’aucune donnée concernant la réponse aux vaccins inactivés.
InteractionsInteractions avec le cytochrome P450
Les effets du némolizumab sur la pharmacocinétique du midazolam (substrat du CYP3A4/5), la warfarine (substrat du CYP2C9), l’oméprazole (substrat du CYP2C19), le métropolol (substrat du CYP2D6) et la caféine (substrat du CYP1A2) ont été évalués dans une étude (SPR.201593) menée sur 14 patients atteints de DA modérée à sévère qui avaient reçu une dose de 60 mg en sous-cutané, puis une dose de 30 mg en sous-cutané toutes les 4 semaines sur une période de 12 semaines.. Aucune altération cliniquement significative de l’exposition aux substrats du CYP450 n’a été observée avant et après plusieurs injections de némolizumab, avec des ratios compris entre 88,24 et 107,81% pour la Cmax et l’ASC. Le némolizumab ne devrait pas avoir d’influence sur la pharmacocinétique de médicaments administrés en concomitance.
Grossesse/AllaitementGrossesse
À ce jour, il existe très peu de données sur l’utilisation du némolizumab chez la femme enceinte. Les études expérimentales animales ne permettent pas de conclure à des effets délétères directs ou indirects en termes de toxicité sur la reproduction (voir «Données précliniques»). Par mesure de précaution, l’utilisation du némolizumab doit être évitée pendant la grossesse.
Allaitement
On ignore si Nemluvio passe dans le lait maternel ou s’il a des effets sur le nourrisson allaité ou sur la production de lait. On ne dispose d’aucune donnée concernant l’excrétion du némolizumab dans le lait maternel. Chez l’être humain, des anticorps anti-IgG sont excrétés dans le lait dans les premiers jours après l’accouchement, leur concentration chutant rapidement pour atteindre des taux faibles. Par conséquent, dans les premiers jours, des anticorps anti-IgG peuvent être transmis au nouveau-né via le lait maternel. Pendant cette courte période, un risque pour l’enfant allaité ne peut pas être exclu.
Fertilité
Dans les études expérimentales animales, aucune dégradation de la fertilité n’a été mise en évidence (voir la rubrique «Données précliniques»).
Effet sur l’aptitude à la conduite et l’utilisation de machinesLes effets de Nemluvio sur l’aptitude à la conduite ou l’utilisation de machines n’ont pas été directement évalués.
Effets indésirablesRésumé du profil de sécurité
Les effets indésirables les plus fréquents en cas de dermatite atopique et de prurigo nodulaire sont les suivants: réactions d’hypersensibilité de type I (1,1%; dont urticaire 1,0% et angio-œdème 0,1%) et réactions au site d’injection (1,2%). D’autres effets indésirables ont été rapportés en cas de prurigo nodulaire, notamment: céphalées (7,0%), dermatite atopique (4,6%), eczéma (3,8%) et eczéma nummulaire (3,5%)
(voir la rubrique «Mises en garde et précautions»).
Liste des effets indésirables
La sécurité d’emploi de Nemluvio a été évaluée dans 3 études poolées, randomisées et contrôlées par placebo menées sur des patients atteints de dermatite atopique (1192 patients sous Nemluvio et 640 patients sous placebo) et deux études randomisées et contrôlées par placebo menées sur des patients atteints de prurigo nodulaire (370 patients sous Nemluvio et 186 patients sous placebo).
Le Tableau 1 récapitule les effets indésirables observés lors des études cliniques, qui sont rangés par classe de système d’organes de la classification MedDRA et par fréquence. Les catégories de fréquence suivantes ont été utilisées: très fréquent (≥1/10), fréquent (≥1/100 à <1/10), occasionnel (≥1/1000 à <1/100), rare (≥1/10’000 à <1/1000), très rare (<1/10’000). Au sein de chaque catégorie de fréquence, les effets indésirables sont mentionnés par degré de gravité décroissant.
Tableau 1: Liste des effets indésirables
|
Classe de système d’organes MedDRA
|
Fréquence
|
Effets indésirables
| |
Affections du système nerveux
|
Fréquent
|
Céphalées (dont céphalées de tension)*
| |
Affections de la peau et du tissu sous-cutané
|
Fréquent
|
Urticaire†
Dermatite atopique*, eczéma*, eczéma nummulaire
| |
Occasionnel
|
Angio-œdème*
| |
Troubles généraux et anomalies au site d’administration
|
Occasionnel
|
Réactions au site d’injection (dont érythème, prurit, douleurs†, irritation†, hématome*)
| |
Rare
|
Œdème au site d’injection†
|
† Effet survenu lors des études sur la dermatite atopique.
* Effet survenu lors des études sur le prurigo nodulaire.
En cas de dermatite atopique, le profil de sécurité de Nemluvio pendant l’étude ouverte (ARCADIA LTE) jusqu’à la semaine 52 correspondait globalement au profil de sécurité observé à la semaine 16.
En cas de prurigo nodulaire, le profil de sécurité de Nemluvio pendant l’étude ouverte (OLYMPIA LTE) jusqu’à la semaine 52 correspondait globalement au profil de sécurité observé à la semaine 16 et à la semaine 24.
Description d’effets indésirables spécifiques et informations complémentaires
Hypersensibilité
Des réactions d’hypersensibilité de type I (réactions médiées par les IgE) ont été rapportées chez des patients traités par Nemluvio dans le contexte d’une dermatite atopique et d’un prurigo nodulaire. Ces réactions comprenaient un urticaire léger et un cas d’angio-œdème (péri-oculaire) léger dans le visage (0,3%), réactions qui n’ont pas conduit à l’arrêt du traitement. Aucun cas de choc anaphylactique ni de maladie sérique n’a été rapporté.
Réactions au site d’injection
L’incidence des réactions au site d’injection pendant la période initiale était faible chez les patients atteints de dermatite atopique traités soit par Nemluvio (1,3% des patients), soit par placebo (1,1% des patients). Pendant la période d’entretien, l’incidence est restée faible avec Nemluvio administré toutes les 8 semaines (0%) et avec le placebo (0,5%).
Chez les patients atteints de prurigo nodulaire, l’incidence des réactions au site d’injection était faible, aussi bien avec Nemluvio (1,1%) qu’avec le placebo (1,6%). Aucune réaction sévère au site d’injection n’est survenue.
Pour les deux indications, aucune réaction ayant mené à l’arrêt du traitement n’a été rapportée.
Céphalées
Chez les patients atteints de prurigo nodulaire, des céphalées ont été rapportées plus fréquemment chez les patients traités par Nemluvio (7,0%) que chez les patients qui recevaient le placebo (3,6%). Des céphalées ont été observées plus fréquemment dans les deux groupes. Dans le groupe sous Nemluvio, les céphalées étaient généralement d’intensité légère à modérée et n’ont pas mené à un arrêt du traitement.
Réactions eczémateuses
Chez les patients atteints de prurigo nodulaire, des réactions eczémateuses (notamment dermatite atopique, eczéma nummulaire ou eczéma) ont été rapportées plus fréquemment chez les patients sous Nemluvio que chez les patients recevant le placebo: dermatite atopique (4,6% patients contre 0,5% des patients), eczéma (3,8% des patients contre 2,2% des patients) et eczéma nummulaire (3,5% des patients contre 0% des patients). Ces réactions eczémateuses étaient d’intensité légère à modérée. La dermatite atopique a conduit à l’arrêt de Nemluvio chez deux patients (0,5%). Aucun événement d’eczéma nummulaire ou d’eczéma n’a conduit à l’abandon de l’étude.
Œdème périphérique et facial
Chez les patients atteints de dermatite atopique et de prurigo nodulaire, des œdèmes périphériques et faciaux ont été rapportés plus fréquemment chez les patients traités par Nemluvio (1,6% et 3,0%) que chez les patients qui recevaient le placebo (0,3% et 1,6%).
Les informations disponibles à ce jour ne permettent pas d’établir un rapport de cause à effet entre Nemluvio et ces œdèmes périphériques et faciaux.
Immunogénicité
Comme pour toutes les protéines thérapeutiques, il existe avec Nemluvio une possibilité d’immunogénicité.
L’incidence des anticorps anti-médicament (AAM) observée dépend fortement de la sensibilité et de la spécificité du test. Les différences entre les procédures d’analyse ne permettent pas d’effectuer des comparaisons valables entre l’incidence d’anticorps anti-médicament dans les études décrites ci-dessous et l’incidence d’anticorps anti-médicament dans d’autres études, y compris celles menées sur le némolizumab.
Lors des études pivots de phase III menées sur la DA (ARCADIA 1, ARCADIA 2) et de l’étude ARCADIA LTE d’une durée allant jusqu’à 128 semaines, l’incidence d’AAM liés au traitement était de 11,2%. Des anticorps neutralisants ont été observés chez 0,5% des patients.
Lors des études pivots de phase III menées sur le PN (OLYMPIA 1, OLYMPIA 2) et de l’étude OLYMPIA LTE d’une durée allant jusqu’à 116 semaines, l’incidence d’AAM liés au traitement était de 12,8%. Des anticorps neutralisants ont été observés chez 3,5% des patients.
Population pédiatrique
Dermatite atopique (DA) – Adolescents (12 à 17 ans)
La sécurité de Nemluvio a été évaluée chez 176 patients pédiatriques âgés de 12 à 17 ans atteints de dermatite atopique modérée et sévère qui étaient inclus dans les études ARCADIA 1 et ARCADIA 2. Le profil de sécurité de Nemluvio chez ces patients jusqu’à la semaine 16 était comparable au profil de sécurité observé chez des adultes atteints de dermatite atopique.
Le profil de sécurité de Nemluvio chez les patients pédiatriques qui faisaient l’objet d’un suivi jusqu’à la semaine 48 était comparable au profil de sécurité observé à la semaine 16. Le profil de sécurité à long terme de Nemluvio chez les patients pédiatriques âgés de 12 à 17 ans correspondait à celui observé chez les adultes atteints de dermatite atopique (ARCADIA LTE).
Remarque concernant l’annonce d’effets secondaires
L’annonce d’effets secondaires présumés après l’autorisation est d’une grande importance. Elle permet un suivi continu du rapport bénéfice-risque du médicament. Les professionnels de santé sont tenus de déclarer toute suspicion d’effet secondaire nouveau ou grave via le portail d’annonce en ligne ElViS (Electronic Vigilance System). Vous trouverez des informations à ce sujet sur www.swissmedic.ch.
SurdosageIl n’y a pas de traitement spécifique du surdosage de Nemluvio. En cas de surdosage, le patient doit faire l’objet d’une surveillance visant à détecter l’apparition de signes et symptômes d’effets indésirables et, si nécessaire, un traitement symptomatique adapté doit être immédiatement instauré.
Propriétés/EffetsCode ATC
D11AH12
Mécanisme d’action
Le némolizumab est un anticorps anti-IgG2 monoclonal humanisé qui inhibe la transduction du signal de l’interleukine-31 (IL-31) en se liant sélectivement au récepteur alpha de l’interleukine-31 (IL-31 RA). L’IL-31 est une cytokine naturelle impliquée dans le prurit, l’inflammation, le dysfonctionnement épidermique et la fibrose. Le némolizumab inhibe les réactions induites par l’IL-31, notamment la libération de chimiokines et de cytokines pro-inflammatoires.
Pharmacodynamique
Efficacité clinique
1) Efficacité clinique et sécurité chez les adultes et adolescents atteints de dermatite atopique
L’efficacité et la sécurité de Nemluvio en cas de traitement de fond topique concomitant ont été évaluées dans deux études pivot randomisées, en double aveugle et contrôlées par placebo (ARCADIA 1 et ARCADIA 2) qui incluaient un total de 1728 participants âgés d’au moins 12 ans, atteints de dermatite atopique modérée à sévère et insuffisamment contrôlée par les traitements topiques. Le degré de sévérité de la maladie était défini par un score IGA (Investigator’s Global Assessment) de 3 (modéré) et 4 (sévère) à l’évaluation globale de la dermatite atopique, un score EASI (Eczema Area and Severity Index) ≥16, une surface corporelle atteinte (BSA) ≥10% et un score PP-NRS (Peak Pruritus Numeric Rating Scale) ≥4.
Les patients inclus dans les études ont reçu soit une première injection sous-cutanée de némolizumab 60 mg, puis des injections de 30 mg toutes les 4 semaines (1x/4 sem.), soit le placebo correspondant. Des corticostéroïdes topiques (CT) et/ou des inhibiteurs de la calcineurine topiques (ICT) de puissance faible et/ou moyenne (selon la classification américaine) étaient administrés en concomitance dans les groupes némolizumab et placebo pendant au moins 14 jours avant la référence et ont été poursuivis pendant l’étude. Selon l’activité de la maladie, ces traitements concomitants pouvaient être diminués progressivement et/ou arrêtés à la discrétion du médecin investigateur.
Après 16 semaines, les patients ayant obtenu une réponse EASI-75 ou un succès selon le score IGA ont intégré la période d’entretien de l’étude pendant 32 semaines supplémentaires afin d’évaluer le maintien de la réponse obtenue à la semaine 16. Les patients répondeurs à Nemluvio ont été randomisés à nouveau pour recevoir soit Nemluvio 30 mg toutes les 8 semaines, soit le placebo toutes 4 semaines (tous les groupes ont continué le traitement par CT/ICT). Les patients randomisés pour recevoir le placebo au cours de la période initiale de traitement et qui avaient obtenu la même réponse clinique à la semaine 16 ont continué à recevoir le placebo toutes les 4 semaines. Les patients non-répondeurs à la semaine 16, les patients ayant présenté une perte de réponse clinique pendant la période d’entretien et les patients ayant terminé la période d’entretien ont eu la possibilité d’être inclus dans l’étude en ouvert (ARCADIA LTE) et de recevoir un traitement par Nemluvio 30 mg toutes les 4 semaines pendant au maximum 200 semaines.
Critères d’évaluation
Dans les études ARCADIA 1 et ARCADIA 2, les critères d’évaluation primaires étaient les suivants :
§Pourcentage de patients présentant un succès selon le score IGA (défini comme un score IGA de 0 [absence d’atteintes] ou 1 [atteintes à peine visibles] et une réduction ≥2 points par rapport à l’inclusion) à la semaine 16
§Pourcentage de patients ayant obtenu une réponse EASI-75 (amélioration ≥75% du score EASI par rapport à l’inclusion) à la semaine 16
Les principaux critères d’évaluation secondaires comprenaient une amélioration du score PP-NRS ≥4 par rapport à la référence aux semaines 1, 2, 4 et 16, un score PP-NRS <2 à la semaine 4 et à la semaine 16, une amélioration du score SD-NRS (Sleep Disturbance Numeric Rating Scale) ≥4 par rapport à la référence à la semaine 16, les patients avec à la fois une réponse EASI-75 et une amélioration du score PP-NRS ≥4 par rapport à la référence à la semaine 16 et les patients avec à la fois un succès selon le score IGA et une amélioration du score PP-NRS ≥4 par rapport à la référence à la semaine 16.
Caractéristiques à l’inclusion
Dans ces études, à l’inclusion, 51,0% des patients étaient des hommes, 79,9% étaient blancs et 15,4% étaient âgés de 12 à 17 ans. À l’inclusion, 70% des patients avaient un score IGA de 3 (DA modérée) et 30% un score IGA de 4 (DA sévère). Le score EASI moyen à l’inclusion était de 27,5, le score PP-NRS hebdomadaire à l’inclusion était de 7,1 (prurit sévère) et le score SD-NRS hebdomadaire à l’inclusion était de 5,8. Globalement, 63,3% des patients avaient reçu auparavant d’autres traitements systémiques pour la dermatite atopique.
Efficacité clinique – ARCADIA 1 et ARCADIA 2 – Adultes et adolescents – Période d’induction, semaine 0 à semaine 16
La supériorité de Nemluvio par rapport au placebo était statistiquement significative en ce qui concerne les co-critères primaires d’évaluation cutanée, à savoir le succès selon le score IGA et la réponse EASI-75 sur 16 semaines (Tableau 2). Les résultats pour les deux co-critères primaires d’évaluation étaient homogènes dans la population atteinte de prurit sévère (score PP-NRS à la référence ≥7).
Tableau 2 – Résultats d’efficacité pour Nemluvio (30 mg 1x/4 sem.) avec traitement concomitant par CT/ICT dans les études ARCADIA 1 et ARCADIA 2 à la semaine 16
|
|
ARCADIA 1
|
ARCADIA 2
| |
Nemluvio + CT/ICT
|
Placebo +
CT/ICT
|
Nemluvio + CT/ICT
|
Placebo +
CT/ICT
| |
Nombre de patients randomisés et traités (PP-NRS à l’inclusion ≥4)
|
620
|
321
|
522
|
265
| |
% de patients avec IGA de 0 ou 1 a
|
35,6#
|
24,6
|
37,7#
|
26,0
| |
% de patients avec EASI-75a
|
43,5*
|
29,0
|
42,1#
|
30,2
|
a Les patients qui ont reçu un traitement de secours ou pour lesquels des données étaient manquantes ont été considérés comme non-répondeurs
* Valeur p <0,0001, #valeur p <0,001La valeur p ajustée par strate est basée sur le test CMH stratifié selon le score PP-NRS et le score IGA à l’inclusion
Figure 1 – Pourcentage de patients présentant un succès selon le score IGA et une réponse EASI-75 entre l’inclusion et la semaine 16 dans les études ARCADIA 1 et ARCADIA 2
Une amélioration significative du prurit chez les patients traités par Nemluvio dans les études ARCADIA 1 et ARCADIA 2 par rapport au placebo, basée une l’amélioration du score PP-NRS ≥4 par rapport à l’inclusion, a été observée à partir de la semaine 1 et s’est maintenue jusqu’à la semaine 16 (Tableau 3). Les résultats étaient homogènes dans la population atteinte de prurit sévère (score PP-NRS initial ≥7).
Tableau 3 – Résultats d’efficacité concernant le prurit pour Nemluvio avec traitement concomitant par CT/ICT dans les études ARCADIA 1 et ARCADIA 2 jusqu’à la semaine 16
|
|
ARCADIA 1
|
ARCADIA 2
| |
Nemluvio + CT/ICT
|
Placebo + CT/ICT
|
Nemluvio + CT/ICT
|
Placebo + CT/ICT
| |
Nombre de patients randomisés et traités (PP-NRS à l’inclusion ≥4)a
|
620
|
321
|
522
|
265
| |
% de patients avec amélioration du PP-NRS ≥4a
| |
À la semaine 1
|
4,7§
|
1,2
|
6,7*
|
0,4
| |
À la semaine 2
|
17,7*
|
3,1
|
16,9*
|
1,9
| |
À la semaine 4
|
27,4*
|
6,5
|
26,1*
|
5,3
| |
À la semaine 16
|
42,7*
|
17,8
|
41,0*
|
18,1
| |
% de patients avec PP-NRS <2a
| |
À la semaine 4
|
16,0*
|
3,7
|
15,9*
|
2,6
| |
À la semaine 16
|
30,6*
|
11,2
|
28,4*
|
11,3
|
a Les patients qui ont reçu un traitement de secours ou pour lesquels des données étaient manquantes ont été considérés comme non-répondeurs
* Valeur p 0,0001, # valeur p <0,001, § valeur p <0,05 La valeur p ajustée par strate est basée sur le test CMH stratifié selon le score PP-NRS et le score IGA à l’inclusion
L’échelle numérique d’évaluation des troubles du sommeil (SD-NRS) est une échelle d’évaluation quotidienne utilisée par les patients pour rapporter la sévérité de la perte de sommeil associée à la dermatite atopique. Une amélioration significative des troubles du sommeil a été observée à la semaine 16 par rapport au placebo (Tableau 4). Les résultats étaient homogènes dans la population atteinte de prurit sévère (score PP-NRS initial ≥7).
Tableau 4 – Efficacité concernant les troubles du sommeil pour Nemluvio avec traitement concomitant par CT/ICT dans les études ARCADIA 1 et ARCADIA 2 jusqu’à la semaine 16
|
|
ARCADIA 1
|
ARCADIA 2
| |
Nemluvio + CT/ICT
|
Placebo + CT/ICT
|
Nemluvio + CT/ICT
|
Placebo + CT/ICT
| |
Nombre de patients randomisés et traités (PP-NRS à l’inclusion ≥4)a
|
620
|
321
|
522
|
265
| |
% de patients avec amélioration du SD-NRS ≥4a
Variation moyenne par rapport à l’inclusion (%)
|
37,9*
-64,6
|
19,9
-38,1
|
33,5*
-59,7
|
16,2
-35,4
|
a Les participants qui ont reçu un traitement de secours ou pour lesquels des données étaient manquantes ont été considérés comme non-répondeurs
* Valeur p <0,0001La valeur p ajustée par strate est basée sur le test CMH stratifié selon le score PP-NRS et le score IGA à l’inclusion
Adolescents atteints de dermatite atopique (12 à 17 ans)
Les résultats d’efficacité des études ARCADIA 1 et ARCADIA 2 à la semaine 16 pour les participants pédiatriques âgés de 12 à 17 ans sont représentés dans le Tableau 5. Les résultats dans la population de patients pédiatriques étaient globalement cohérents avec les résultats dans la population de patients adultes. Les résultats pour les co-critères primaires et les principaux critères d’évaluation secondaires étaient homogènes dans la population atteinte de prurit sévère (score PP-NRS à la référence ≥7).
Tableau 5 – Résultats d’efficacité pour le némolizumab (30 mg 1x/4 sem.) avec traitement concomitant par CT/ICT dans les études ARCADIA 1 et ARCADIA 2 à la semaine 16 chez les patients pédiatriques âgés de 12 à 17 ans
|
|
ARCADIA 1 ET ARCADIA 2
| |
Nemluvio + CT/ICT
|
Nemluvio + CT/ICT
| |
Nombre de patients traités de randomisés (PP-NRS à la référence ≥4)
|
179
|
90
| |
% de patients avec IGA de 0 ou 1 a
|
48,9*
|
34,4
| |
% de patients avec EASI-75a
|
53,4§
|
43,3
|
a Les patients qui ont reçu un traitement de secours ou pour lesquels des données étaient manquantes ont été considérés comme non-répondeurs
≠ valeur p <0,0001, # valeur p <0,001, * valeur p <0,05, ∞ valeur p =0,0591, § valeur p =0,1824
La valeur p stratifiée est basée sur le test CMH stratifié selon le score PP-NRS et le score IGA à l’inclusion
Efficacité clinique – ARCADIA 1 et ARCADIA 2 – Adultes et adolescents – Période d’entretien, semaine 16 à semaine 48
La réponse clinique chez les répondeurs au némolizumab (score IGA 0/1 ou réponse EASI-75 à la semaine 16) a été évaluée entre la semaine 16 et la semaine 48 lors des études ARCADIA 1 et ARCADIA 2. Pour la période de traitement d’entretien, 338 répondeurs au némolizumab ont été de nouveau randomisés pour recevoir du némolizumab 30 mg 1x/8 sem. ou le placebo 1x/4 sem. (arrêt du némolizumab) associé à un traitement concomitant par CT/ICT. À la semaine 48, 60,4% des patients ont obtenu un score IGA de 0/1 et 75,7% une réponse EASI-75 dans le groupe némolizumab 30 mg 1x/8 sem. + CT/ICT, contre respectivement 49,7% et 63,9% dans le groupe placebo 1x/4 sem. + CT/ICT.
2) Efficacité clinique et sécurité chez les adultes atteints de prurigo nodulaire
L’efficacité et la sécurité de Nemluvio en monothérapie ont été évaluées dans deux études pivots randomisées, en double aveugle et contrôlées par placebo (OLYMPIA 1 et OLYMPIA 2) qui incluaient au total 560 patients âgés d’au moins 18 ans et atteints de prurigo nodulaire. La sévérité de la maladie était définie par un score IGA à l’évaluation globale des nodules de prurigo nodulaire sur une échelle de sévérité de 0 à 4. Les patients inclus dans ces deux études présentaient un score IGA ≥3, un prurit sévère défini par une moyenne hebdomadaire du score sur l’échelle d’évaluation numérique du pic de prurit (PP-NRS) ≥7 sur une échelle de 0 à 10, et au moins 20 lésions nodulaires. Les études OLYMPIA 1 et OLYMPIA 2 évaluaient l’effet de Nemluvio en monothérapie sur les signes et symptômes du prurigo nodulaire et ciblaient l’amélioration des lésions cutanées et du prurit sur 16 semaines. L’étude OLYMPIA 1 comprenait une période de traitement de 24 semaines et l’étude OLYMPIA 2 une période de traitement de 16 semaines.
Les participants qui terminaient les études OLYMPIA 1 et OLYMPIA 2 avaient la possibilité d’être inclus dans l’étude en ouvert (OLUMPIA LTE) et de recevoir Nemluvio toutes les 4 semaines pendant une durée maximale de 184 semaines.
Dans le groupe qui recevaient Nemluvio en monothérapie, les patients pesant moins de 90 kg ont reçu des injections sous-cutanées de Nemluvio 60 mg (2 injections de 30 mg) à la semaine 0, puis des injections de 30 mg toutes les 4 semaines, et les patients pesant au moins 90 kg ont reçu des injections sous-cutanées de Nemluvio 60 mg (2 injections de 30 mg) à la semaine 0, puis toutes les 4 semaines.
Critères d’évaluation
Dans les études OLYMPIA 1 et OLYMPIA 2, les deux critères d’évaluation primaires étaient identiques:
§Pourcentage de patients présentant une amélioration ≥4 par rapport à l’inclusion du score sur l’échelle d’évaluation numérique du pic de prurit (NRS) à la semaine 16
§Pourcentage de patients présentant un succès selon le score IGA (défini comme un score IGA de 0 [absence d’atteintes] ou 1 [atteintes à peine visibles] et une amélioration ≥2 points par rapport à l’inclusion) à la semaine 16.
Les principaux critères d’évaluation secondaires comprenaient une amélioration ≥4 du score PP-NRS à la semaine 4 par rapport à l’inclusion, un score PP-NRS <2 à la semaine 4 et à la semaine 16, une amélioration ≥4 du score SD-NRS à la semaine 4 et à la semaine 16 par rapport à l’inclusion.
Caractéristiques à l’inclusion
Dans ces études, à l’inclusion, 59,6% des patients étaient des femmes, 81,4% étaient blancs et 25,4% étaient âgés de plus de 65 ans. Le score PP-NRS moyen hebdomadaire à l’inclusion correspondait à une moyenne (ET) de 8,4 (0,9). À l’inclusion, 58% des patients avaient un score IGA de 3 (PN modéré) et 42% un score IGA de 4 (PN sévère).
Efficacité clinique
Études en monothérapie (OLYMPIA 1 et OLYMPIA 2) – Semaine 0 à semaine 16
Les résultats des études pivots OLYMPIA 1 et OLYMPIA 2 évaluant le traitement par Nemluvio sont présentés dans le Tableau 7 et montrent une amélioration significative chez les patients traités par Nemluvio par rapport au placebo à la fois pour les critères d’évaluation primaires (Figure 2 et Figure 3) et pour les principaux critères d’évaluation secondaires.
Tableau 6 – Résultats d’efficacité pour le némolizumab en monothérapie (1x/4 sem.) dans les études OLYMPIA 1 et OLYMPIA 2
|
|
OLYMPIA 1
|
OLYMPIA 2
| |
Nemluvio
|
Placebo
|
Nemluvio
|
Placebo
| |
Nombre de patients randomisés
|
190
|
96
|
183
|
91
| |
% de patients avec amélioration du PP-NRS ≥4 par rapport à l’inclusiona
| |
Semaine 4
|
41,1*
|
6,3
|
41,0*
|
7,7
| |
Semaine 16
|
58,4*
|
16,7
|
56,3*
|
20,9
| |
% de patients avec IGA de 0 ou 1 à la semaine 16a
|
26,3#
|
7,3
|
37,7*
|
11
| |
% de patients avec PP-NRS <2a
| |
Semaine 4
|
21,6*
|
1,0
|
19,7*
|
2,2
| |
Semaine 16
|
34,2*
|
4,2
|
35,0*
|
7,7
| |
% de patients avec amélioration du SD-NRS ≥4 par rapport à l’inclusiona
| |
Semaine 4
|
31,1*
|
5,2
|
37,2*
|
9,9
| |
Semaine 16
|
50,0*
|
11,5
|
51,9*
|
20,9
|
a Lorsque des patients recevaient un traitement de secours, une stratégie de variable composite était appliquée: les données de base au moment de l’administration du traitement de secours/après le traitement de secours étaient définies comme la pire valeur possible, et la réponse était dérivée de la valeur des données de base. Les patients pour lesquels des résultats étaient manquants ont été considérés comme non-répondeurs
b Non ajusté en fonction de la multiplicité
* Valeur p <0,0001, # valeur p =0,0025, ajustée par strate à l’aide des variables de stratification randomisées (centre d’analyse et poids corporel à l’inclusion [<90 kg, ≥90 kg])
§ Valeur p <0,0001 ajustée par strate vs placebo (ANCOVA MI-MAR)
|
Figure 2 – Pourcentage de patients présentant une amélioration ≥4 du score PP-NRS entre l’inclusion et la semaine 16
| |
| |
Figure 3 – Pourcentage de répondeurs IGA entre l’inclusion et la semaine 16
| |
|
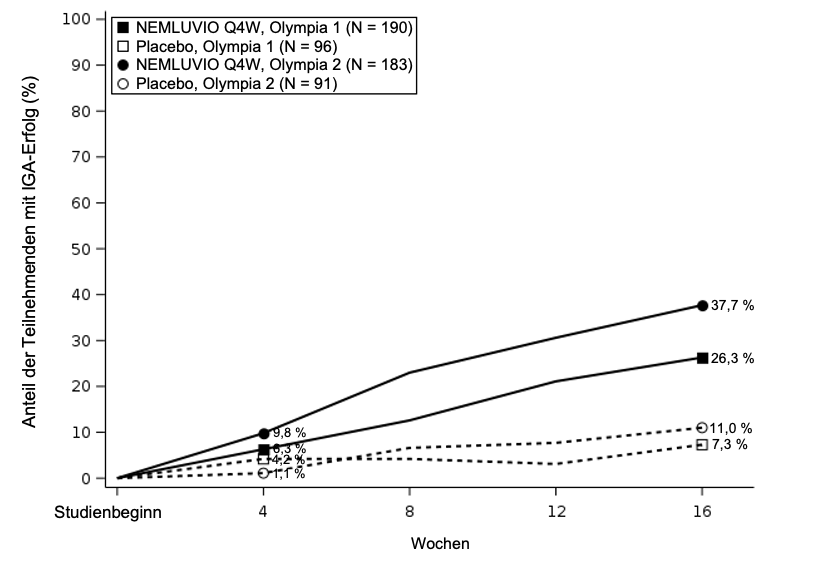
|
PharmacocinétiqueAbsorption
Après une injection sous-cutanée initiale d’une dose de 60 mg dans le cadre d’une étude de phase I (96 patients par bras), la concentration maximale (Cmax) moyenne (ET) de némolizumab était de 7,5 (2,31) µg/ml environ 6 jours après l’administration.
Après plusieurs traitements par Nemluvio chez des patients atteints de dermatite atopique, les concentrations résiduelles moyennes (ET) estimées à l’état d’équilibre du némolizumab étaient de 2,63 (1,27) µg/ml pour la dose de 30 mg 1x/4 sem. et de 0,74 (0,44) µg/ml pour la dose de 30 mg 1x/8 sem.
Après plusieurs traitements par Nemluvio chez des patients atteints de prurigo nodulaire, les concentrations résiduelles moyennes (ET) estimées à l’état d’équilibre du némolizumab étaient de 3,04 (1,23) µg/ml chez les patients d’un poids corporel <90 kg pour la dose de 30 mg 1x/4 sem. et de 3,66 (1,63) µg/ml chez les patients d’un poids corporel ≥90 kg pour la dose de 60 mg 1x/4 sem.
Aussi bien chez les populations atteintes de dermatite atopique et que de prurigo nodulaire, les concentrations de némolizumab à l’état d’équilibre ont été atteintes à la semaine 4 après une dose de charge de 60 mg et à la semaine 12 sans dose de charge.
Distribution
Sur la base d’une analyse de la pharmacocinétique de population, le volume de distribution était de 7,67 l.
Métabolisme
Le némolizumab étant une protéine, aucune étude spécifique du métabolisme n’a été menée. Le némolizumab devrait être métabolisé en petits peptides par les voies cataboliques.
Élimination
Le némolizumab devrait être dégradé de la même manière que les IgG endogènes. Dans l’analyse de la pharmacocinétique de population, la demi-vie d’élimination terminale (ET) du némolizumab a été estimée à 18,9 (4,96) jours et la clairance systémique à 0,263 l/jour.
Linéarité/non-linéarité
La pharmacocinétique du némolizumab après administration d’une dose unique était linéaire, avec une exposition augmentant de manière proportionnelle à la dose entre 0,03 et 3 mg/kg.
Après plusieurs doses, l’exposition systémique au némolizumab a augmenté de manière approximativement proportionnelle à la dose dans toute la plage de doses SC allant jusqu’à 30 mg. Une légère diminution de la biodisponibilité de 9% a été observée avec la dose SC de 60 mg et de 15% avec la dose SC de 90 mg.
Cinétique pour certains groupes de patients
Sexe, âge et origine ethnique
Sur la base d’une analyse de la pharmacocinétique de population, le sexe, l’âge (12 à 85 ans pour la dermatite atopique et 18 à 84 ans pour le prurigo nodulaire) et l’origine ethnique n’ont pas eu d’effet significatif sur la pharmacocinétique du némolizumab.
Troubles de la fonction hépatique
S’agissant d’un anticorps monoclonal, le némolizumab ne devrait pas être éliminé de manière importante par voie hépatique. Aucune étude clinique n’a été réalisée pour évaluer l’effet d’une insuffisance hépatique sur la pharmacocinétique du némolizumab. On a constaté qu’une insuffisance hépatique légère à modérée n’avait aucune influence sur la pharmacocinétique du némolizumab déterminée par l’analyse de la pharmacocinétique de population. Aucune donnée n’est disponible chez les patients atteints d’insuffisance hépatique sévère.
Troubles de la fonction rénale
S’agissant d’un anticorps monoclonal, le némolizumab ne devrait pas être éliminé de manière importante par voie rénale. Aucune étude clinique n’a été réalisée pour évaluer l’effet d’une insuffisance rénale sur la pharmacocinétique du némolizumab. L’analyse de la pharmacocinétique de population n’a pas mis en évidence d’impact cliniquement significatif d’une insuffisance rénale légère ou modérée sur l’exposition systémique au némolizumab. Les données disponibles chez les patients atteints d’insuffisance rénale sévère sont très limitées.
Poids corporel
L’exposition au némolizumab était plus faible chez les patients ayant un poids corporel plus élevé.
Dermatite atopique
La différence d’exposition systémique due au poids corporel n’a eu aucun impact cliniquement significatif sur l’efficacité. Aucun ajustement posologique en fonction du poids corporel n’est nécessaire (voir la rubrique «Posologie/Mode d’emploi»).
Prurigo nodulaire
La variabilité de l’exposition systémique due au poids corporel a eu un impact cliniquement significatif sur l’efficacité sur les lésions cutanées, évaluée par la réponse IGA, mais pas sur l’amélioration du prurit, et requiert un ajustement posologique chez les patients atteints de prurigo nodulaire (voir la rubrique «Posologie/Mode d’emploi).
Population pédiatrique
Dermatite atopique
Dans l’analyse de la PK de population, aucune différence cliniquement significative au niveau de la pharmacocinétique du némolizumab n’a été constatée chez les patients pédiatriques âgés de 12 à 17 ans par rapport aux adultes. Un ajustement posologique dans cette population n’est pas recommandé.
Données précliniquesMutagénicité et carcinogénicité
Le potentiel mutagène du némolizumab n’a pas fait l’objet d’études. Cependant, des anticorps monoclonaux ne devraient pas altérer l’ADN ou les chromosomes.
Aucune étude de carcinogénicité n’a été conduite avec le némolizumab Les résultats des études expérimentales animales ainsi que les informations disponibles concernant l’inhibition de l’IL-31 n’indiquent pas de potentiel cancérogène du némolizumab.
Toxicité sur la reproduction
Aucun effet sur les paramètres de fertilité n’a été observé chez des singes cynomolgus sexuellement matures après un traitement par voie sous-cutanée à long terme par némolizumab. Après l’administration par voie sous-cutanée de némolizumab à des mères entre le début de l’organogenèse et la mise bas, ainsi qu’après l’administration directe chez la progéniture pendant une durée de 26 semaines à partir du jour 35 après la naissance, aucun effet délétère direct ou indirect n’a été observé ni sur la grossesse, ni sur le développement embryonnaire, fœtal ou postnatal. Toutefois, une légère augmentation de l’incidence des décès de la progéniture au début de la période postnatale a été observée à une exposition élevée (43 ou 34 fois supérieure à l’exposition chez l’être humain à la dose maximale recommandée pour le traitement de la DA ou du PN, respectivement) dans le groupe qui recevait du némolizumab à une dose de 25 mg/kg toutes les 2 semaines. Bien que peu probable, un lien entre cette observation et le némolizumab ne peut pas être totalement exclu.
Remarques particulièresLes remarques suivantes s’appliquent à toutes les formes pharmaceutiques disponibles.
Incompatibilités
En l’absence d’études de compatibilité, ce médicament ne doit pas être mélangé à d’autres médicaments.
Influence sur les méthodes de diagnostic
On ne dispose d’aucune donnée.
Stabilité
Ce médicament ne doit pas être utilisé au-delà de la date figurant après la mention «EXP» sur l’emballage. Si nécessaire, le carton contenant le stylo prérempli ou la seringue préremplie peut être conservé à température ambiante (jusqu’à 25°C) en dehors du réfrigérateur pendant une période unique de 90 jours maximum. La date de la première sortie du réfrigérateur doit alors être notée dans l’espace prévu à cet effet sur le carton du stylo prérempli ou de la seringue préremplie. Nemluvio ne doit pas être utilisé après expiration de la date de péremption ou au-delà de 90 jours après la date de la première sortie du réfrigérateur (selon la première éventualité).
Remarques particulières concernant le stockage
Tenir hors de la vue et de la portée des enfants.
Conserver la seringue préremplie ou le stylo prérempli dans l’emballage d’origine pour le protéger de la lumière.
Conserver au réfrigérateur (2-8°C). Ne pas congeler ni faire chauffer le médicament.
Pour les conditions de conservation après reconstitution du médicament, consulter la rubrique «Remarques concernant la manipulation».
Remarques concernant la manipulation
Des instructions complètes sur l’administration de Nemluvio en stylo prérempli ou en seringue préremplie figurent à la fin de la notice d’emballage.
Nemluvio doit être sorti du réfrigérateur 30 à 45 minutes avant sa reconstitution. Une fois les étapes de reconstitution effectuées, Nemluvio doit être utilisé ou éliminé dans un délai de 4 heures.
Nemluvio doit être examiné visuellement avant la reconstitution. Nemluvio est composé d’une poudre blanche et d’un solvant limpide. Nemluvio ne doit pas être utilisé si la poudre n’est pas blanche ou si le solvant est trouble ou contient des particules visibles. Avant administration, il faut contrôler que Nemluvio est limpide et incolore à légèrement jaunâtre, et ne contient pas de particules.
Le stylo prérempli ou la seringue préremplie ne doivent pas être exposés à la chaleur ou aux rayons directs du soleil, ni être agités.
Tout médicament non utilisé ou déchet doit être éliminé conformément aux exigences nationales.
Numéro d’autorisationNemluvio, poudre et solvant pour solution injectable en stylo prérempli: 69707 (Swissmedic)
Nemluvio, poudre et solvant pour solution injectable en seringue préremplie: 69818 (Swissmedic)
PrésentationNemluvio, poudre et solvant pour solution injectable en stylo prérempli
Cartouche à usage unique avec deux compartiments en verre borosilicate de type 1 dans un auto-injecteur avec aiguille inox intégrée.
Conditionnements:
• 1 stylo prérempli [B]
• Conditionnement multiple avec 2 stylos préremplis (2 boîtes de 1) [B]
Nemluvio, poudre et solvant pour solution injectable en seringue préremplie
Seringue préremplie à usage unique avec deux compartiments en verre borosilicate de type 1, conditionnée avec une aiguille 27G (inox) munie d’un dispositif de protection.
Conditionnement:
• 1 seringue préremplie [B]
Titulaire de l’autorisationGalderma SA
Zählerweg 10
CH – 6300 Zoug
Mise à jour de l’informationDécembre 2024
|