CompositionPrincipes actifs
Lutathera: Lutécium (177Lu) oxodotréotide 370 MBq/ml solution pour perfusion à la date et à l'heure de calibration. Le lutécium-177 est préparé à partir de l'ytterbium-176 et est sans porteur ajouté.
Lutathera CA: Lutécium (177Lu) oxodotréotide 370 MBq/ml solution pour perfusion à la date et à l'heure de calibration. Le lutécium-177 est préparé à partir du lutécium-176 et est avec porteur ajouté. La préparation contient l'impureté 177mLutécium.
Excipients
Acide acétique, acétate sodique 0,66 mg/ml, acide gentisique, acide ascorbique, acide pentétique, chlorure de sodium 6,85 mg/ml, hydroxyde de sodium 0,64 mg/ml, eau pour préparations injectables.
Chaque millilitre de solution contient jusqu'à 0,14 mmol (3,24 mg) de sodium.
Indications/Possibilités d’emploiLutathera/Lutathera CA est indiqué chez l'adulte pour le traitement des tumeurs neuroendocrines gastroentéropancréatiques (TNE-GEP) au stade métastatique ou inopérables, progressives, bien différenciées (G1 et G2) et exprimant des récepteurs de la somatostatine.
Posologie/Mode d’emploiLe médicament est réservé à l'usage hospitalier et ne peut être administré que par des médecins spécialisés titulaires du titre de formation postgraduée en médecine nucléaire.
Étant donné la quantité de radioactivité administrée, le patient doit être placé dans une chambre isolée après le traitement. Les mesures de précautions relatives à la radioprotection doivent être respectées. Voir les rubriques «Mises en garde et précautions» ainsi que «Remarques particulières: Remarques concernant la manipulation/Radioprotection».
Avant le début du traitement par Lutathera/Lutathera CA, une imagerie des récepteurs de la somatostatine (scintigraphie ou tomographie par émission de positons [TEP]) doit confirmer la surexpression de ces récepteurs sur les tissus tumoraux avec une fixation par les foyers tumoraux au moins aussi élevée que la fixation hépatique normale.
Posologie usuelle
Adultes
En règle générale, le protocole thérapeutique recommandé chez l'adulte consiste en 4 administrations par voie intraveineuse de 7400 MBq chacune. Un délai de 8 semaines (± 1 semaine) doit être respecté entre chaque perfusion (voir également la rubrique «Ajustement posologique»).
Solution d'acides aminés
Afin de protéger la fonction rénale, une solution d'acides aminés contenant de la L-lysine et de la Larginine doit être administrée par voie intraveineuse pendant 4 heures (voir tableau 1 et tableau 2). La perfusion de la solution d'acides aminés doit être initiée 30 minutes avant le début de l'administration de Lutathera/Lutathera CA.
La perfusion de la solution d'acides aminés et celle de Lutathera/Lutathera CA par des accès veineux distincts dans chacun des bras du patient est la procédure préférée. Si toutefois, en raison de veines difficiles ou des préférences de l'institution/l'hôpital, deux accès veineux ne sont pas possibles, la perfusion de la solution d'acides aminés et celle de Lutathera/Lutathera CA peuvent être réalisées par le même accès via un robinet à trois voies; il convient dans ce cas de surveiller le débit et le maintien de l'ouverture de l'accès. La dose de la solution d'acides aminés ne doit pas être réduite, même en cas d'administration d'une dose réduite de Lutathera/Lutathera CA.
Compte tenu de la grande quantité de solution d'acides aminés et des volumes considérables que les solutions commerciales disponibles nécessitent pour remplir les spécifications mentionnées ci-dessus, la solution en préparation magistrale est considérée comme le produit de choix du fait de son plus faible volume à perfuser et de sa plus faible osmolalité. La solution d'acides aminés peut être préparée à la demande, en tenant compte des bonnes pratiques de préparation de médicaments stériles de l'hôpital et selon la composition spécifiée au tableau 1.
Tableau 1: Composition de la solution d'acides aminés en tant que préparation magistrale
|
Composition
|
Quantité
| |
L-Lysine.HCl
|
25 g (équivalent à 20 g de lysine)
| |
L-Arginine.HCl
|
25 g (équivalent à 20,7 g d'arginine)
| |
Solution de chlorure de sodium à 9 mg/ml (0,9%) pour préparations injectables ou eau pour préparations injectables
|
1 l
|
Le pH de la solution d'acides aminés en tant que préparation magistrale suivant la composition décrite dans le tableau 1 doit être ajusté à 7,4 ± 0,2 en utilisant de l'hydroxyde de sodium (NaOH).
Alternativement, certaines solutions d'acides aminés disponibles dans le commerce peuvent être utilisées si elles sont compatibles avec les spécifications indiquées dans le tableau 2.
Tableau 2: Spécification pour les solutions d'acides aminés du commerce
|
Propriété
|
Spécification
| |
L-Lysine.HCl
|
Entre 18 et 25 g (équivalent à 14,4 à 20 g de L-lysine)
| |
L-Arginine.HCI
|
Entre 18 et 25 g (équivalent à 14,9 à 20,7 g de L-arginine)
| |
Volume
|
1 l à 2 l
| |
Osmolalité
|
< 1200 mOsmol/kg
|
Surveillance du traitement
Des analyses biologiques sont nécessaires avant chaque administration et en cours de traitement afin de réévaluer l'état du patient et d'adapter le protocole thérapeutique si nécessaire (dose, intervalle entre les perfusions, nombre de perfusions).
Les analyses biologiques minimales à réaliser avant chaque perfusion sont les suivantes:
·hématologie (hémoglobine [Hb], numération des globules blancs avec formule sanguine différentielle, nombre de plaquettes);
·fonction rénale (créatinine sérique et clairance de la créatinine selon la formule de Cockcroft-Gault);
·fonction hépatique (alanine aminotransférase [ALAT], aspartate aminotransférase [ASAT], albumine sérique, rapport normalisé international (INR) et bilirubine).
Ces analyses doivent être réalisées au moins une fois dans les 2 à 4 semaines avant l'administration et une fois juste avant l'administration. Il est également recommandé de réaliser ces analyses toutes les 4 semaines pendant au moins 3 mois suivant la dernière perfusion de Lutathera/Lutathera CA et tous les 6 mois par la suite afin de détecter de possibles effets indésirables retardés (voir la rubrique «Effets indésirables»). La posologie peut être modifiée si nécessaire selon les résultats des analyses biologiques.
Ajustement posologique
La prise en charge des effets indésirables sévères ou intolérables du médicament peut nécessiter une interruption temporaire de l'administration (allongement de l'intervalle entre les doses de 8 à 16 semaines maximum), une réduction de la dose ou l'arrêt du traitement par Lutathera/Lutathera CA. (voir tableau 3 et figure 1).
Tableau 3: Ajustements posologiques recommandés en cas d'effets indésirables (EI)
|
EI
|
Sévérité des EI
|
Ajustement posologique
| |
Thrombocytopénie
|
Première apparition de:
Grade 2 (nombre de plaquettes < 75 à 50 × 109/l)
Grade 3 (nombre de plaquettes < 50 à 25 × 109/l)
Grade 4 (nombre de plaquettes < 25 × 109/l)
|
Suspendre la dose jusqu'à obtenir une résolution complète ou partielle (grade 0 à 1).
Reprendre Lutathera/Lutathera CA à la dose de 3700 MBq (100 mCi) pour les patients ayant obtenu une résolution complète ou partielle. Si une dose réduite n'entraîne pas de thrombocytopénie de grade 2, 3 ou 4, administrer Lutathera/Lutathera CA à la dose de 7400 MBq (200 mCi) lors de l'administration suivante.
Arrêter définitivement Lutathera/Lutathera CA en cas de thrombocytopénie de grade ≥2 nécessitant un intervalle entre les doses de plus de 16 semaines.
| |
|
Grade 2, 3 ou 4 récidivant
|
Arrêter définitivement le traitement par Lutathera/Lutathera CA
| |
Anémie et neutropénie
|
Première apparition d'une anémie:
Grade 3 (Hb < 8,0 g/dl); transfusion indiquée
Grade 4 (conséquences engageant le pronostic vital)
Première apparition d'une neutropénie:
Grade 3 (nombre absolu de neutrophiles (NAN) < 1,0 à 0,5 × 109/l)
Grade 4 (NAN < 0,5 × 109/l)
|
Suspendre la dose jusqu'à obtenir une résolution complète ou partielle (grade 0, 1 ou 2).
Reprendre Lutathera/Lutathera CA à la dose de 3700 MBq (100 mCi) pour les patients ayant obtenu une résolution complète ou partielle. Si une dose réduite n'entraîne pas d'anémie ou de neutropénie de grade 3 ou 4, administrer Lutathera/Lutathera CA à la dose de 7400 MBq (200 mCi) lors de l'administration suivante.
Arrêter définitivement Lutathera/Lutathera CA en cas d'anémie ou de neutropénie de grade ≥3 nécessitant un intervalle entre les doses de plus de 16 semaines.
| |
|
Grade 3 ou 4 récidivant
|
Arrêter définitivement le traitement par Lutathera/Lutathera CA.
| |
Toxicité rénale
|
Première apparition de:
·clairance de la créatinine inférieure à 40 ml/min, calculée en utilisant la formule de Cockcroft Gault et le poids corporel réel, ou
·augmentation de 40% de la valeur initiale de la créatinine sérique, ou
·diminution de 40% de la clairance de la créatinine par rapport à la valeur initiale, calculée en utilisant la formule de Cockcroft Gault et le poids corporel réel.
|
Suspendre la dose jusqu'à obtenir une résolution ou un retour aux valeurs initiales.
Reprendre Lutathera/Lutathera CA à la dose de 3700 MBq (100 mCi) pour les patients ayant obtenu une résolution ou un retour aux valeurs initiales. Si une dose réduite n'entraîne pas de toxicité rénale, administrer Lutathera/Lutathera CA à la dose de 7400 MBq (200 mCi) lors de l'administration suivante.
Arrêter définitivement Lutathera/Lutathera CA en cas de toxicité rénale nécessitant un intervalle entre les doses de plus de 16 semaines.
| |
|
Toxicité rénale récidivante
|
Arrêter définitivement Lutathera/Lutathera CA.
| |
Hépatotoxicité
|
Définie comme:
·bilirubinémie > 3 fois la limite supérieure de la normale (grade 3 ou 4), ou
·albuminémie < 30 g/l avec INR > 1,5
|
Suspendre la dose jusqu'à obtenir une résolution ou un retour aux valeurs initiales.
Reprendre Lutathera/Lutathera CA à la dose de 3700 MBq (100 mCi) pour les patients ayant obtenu une résolution complète ou un retour aux valeurs initiales. Si une dose réduite n'entraîne pas d'hépatotoxicité, administrer Lutathera/Lutathera CA à la dose de 7400 MBq (200 mCi) lors de l'administration suivante.
Arrêter définitivement Lutathera/Lutathera CA en cas d'hépatotoxicité nécessitant un intervalle entre les doses de plus de 16 semaines.
| |
|
Hépatotoxicité récidivante
|
Arrêter définitivement Lutathera/Lutathera CA
| |
Tout autre EI de grade 3 ou de grade 4 selon CTCAE*
|
Première apparition d'un
grade 3 ou 4
|
Suspendre la dose jusqu'à obtenir une résolution complète ou partielle (grade 0 à 2).
Reprendre Lutathera/Lutathera CA à la dose de 3700 MBq (100 mCi) pour les patients ayant obtenu une résolution complète ou partielle. Si une dose réduite n'entraîne pas de toxicité de grade 3 ou 4, administrer Lutathera/Lutathera CA à la dose de 7400 MBq (200 mCi) lors de l'administration suivante.
Arrêter définitivement Lutathera/Lutathera CA en cas d'EI de grade ≥3 nécessitant un intervalle entre les doses de plus de 16 semaines.
| |
|
Toxicité de grade 3 ou 4 récidivante
|
Arrêter définitivement Lutathera/Lutathera CA.
| |
Aucun ajustement posologique n'est nécessaire en cas de toxicités hématologiques de grade 3 ou 4 qui sont uniquement dues à une lymphopénie.
*CTCAE: Common Terminology Criteria for Adverse Events (Critères communs de terminologie pour les événements indésirables), National Cancer Institute
|
Figure 1: Schéma d'indications pour les ajustements posologiques

TMD: toxicité modifiant la dose
Parmi les autres raisons d'envisager une interruption temporaire du traitement par Lutathera/Lutathera CA, on relèvera l'apparition d'une maladie intercurrente (par exemple une infection des voies urinaires) qui, selon le médecin, pourrait accroître les risques associés à l'administration de Lutathera/Lutathera CA et qui doit être résolue ou stabilisée pour que le traitement puisse reprendre; et une intervention chirurgicale majeure, auquel cas le traitement par Lutathera/Lutathera CA doit être suspendu pendant 12 semaines après la date de l'intervention.
Populations spécifiques
Patients âgés
Il n'a pas été observé de différence dans les résultats d'essais cliniques en termes de réponse entre les patients âgés et les patients plus jeunes. Cependant, comme le risque de présenter une toxicité hématologique est plus élevé chez les patients âgés (≥70 ans), un suivi rapproché permettant un ajustement rapide de la dose est recommandé dans cette population.
Patients présentant des troubles de la fonction rénale
L'activité à administrer doit être considérée avec attention chez les patients présentant une insuffisance rénale, une augmentation de l'exposition aux rayonnements étant possible. Le profil pharmacocinétique et la sécurité du lutécium (177Lu) oxodotréotide chez les patients présentant une insuffisance rénale sévère ou une insuffisance rénale en phase terminale préexistantes n'ont pas été étudiés. Le traitement par Lutathera/Lutathera CA est contre-indiqué chez les patients présentant une insuffisance rénale sévère avec une clairance de la créatinine < 30 ml/min (voir la rubrique «Contre-indications»). Le traitement par Lutathera/Lutathera CA chez les patients ayant une clairance de la créatinine < 40 ml/min comme valeur initiale (en utilisant la formule de Cockcroft Gault) n'est pas recommandé. Aucun ajustement posologique n'est recommandé chez les patients insuffisants rénaux ayant une clairance de la créatinine ≥40 ml/min préexistante. Cependant, ce médicament étant connu pour être principalement éliminé par les reins, la fonction rénale doit être surveillée plus fréquemment pendant le traitement car ces patients peuvent être exposés à un risque plus élevé de toxicité.
Pour des informations complémentaires sur la prise en charge des patients atteints d'une toxicité rénale, voir les rubriques «Posologie/Mode d'emploi» (tableau 3) et «Mises en garde et précautions».
Patients présentant des troubles de la fonction hépatique
L'activité à administrer doit être considérée avec attention chez les patients présentant une insuffisance hépatique, une augmentation de l'exposition aux rayonnements étant possible. Le profil pharmacocinétique et la sécurité du lutécium (177Lu) oxodotréotide chez les patients présentant une insuffisance hépatique sévères préexistante (bilirubine totale supérieure à trois fois la limite supérieure de la normale, indépendamment du taux d'ASAT) n'a pas été étudié. Par conséquent, le traitement par Lutathera/Lutathera CA n'est pas recommandé chez ces patients. Les patients présentant une insuffisance hépatique préexistante chez lesquels la bilirubine totale est plus de 3 fois supérieure à la limite supérieure de la normale ou dont l'albuminémie est < 30 g/l et l'INR > 1,5 ne doivent être traités par Lutathera/Lutathera CA qu'après une évaluation minutieuse du rapport bénéfice-risque.
Pour la conduite à tenir chez des patients présentant une hépatotoxicité, voir le tableau 3 de la rubrique «Posologie/Mode d'emploi» et la rubrique «Mises en garde et précautions».
Population pédiatrique
Il n'existe pas d'utilisation justifiée de Lutathera/Lutathera CA chez les enfants et les adolescents dans l'indication du traitement des TNE-GEP (à l'exception du neuroblastome,
du neuroganglioblastome et du phéochromocytome). Lutathera/Lutathera CA n'est pas autorisé pour un emploi au sein de la population pédiatrique.
Prémédication
Antiémétiques
Une prémédication avec des antiémétiques doit être administrée en prévoyant un délai de temps suffisant avant le début de la perfusion de la solution d'acides aminés. Veuillez-vous reporter à l'information professionnelle détaillée des antiémétiques pour connaître les instructions relatives à l'administration.
Si pendant la perfusion de la solution d'acides aminés et malgré l'administration antérieure d'un antiémétique de fortes nausées ou vomissements surviennent, un antiémétique d'une autre classe pharmacologique peut être administré.
Médication concomitante avec des analogues de la somatostatine
Avant de débuter le traitement par Lutathera/Lutathera CA: interrompre l'administration d'analogues de la somatostatine à libération prolongée (tel que l'octréotide à libération prolongée [LP]) au moins 4 à 6 semaines avant le début du traitement par Lutathera/Lutathera CA. Si nécessaire, administrer de l'octréotide à courte durée d'action jusqu'à 24 heures avant l'administration de Lutathera/Lutathera CA (voir la rubrique «Interactions»).
Pendant le traitement par Lutathera/Lutathera CA: dans les 4 à 6 semaines précédant chaque perfusion de Lutathera/Lutathera CA, ne pas administrer d'octréotide LP. Afin de contrôler les symptômes de la maladie pendant le traitement par Lutathera/Lutathera CA, de l'octréotide à courte durée d'action peut-être administré au patient, mais l'administration doit être interrompue au moins 24 heures avant chaque perfusion de Lutathera/Lutathera CA.
Après le traitement par Lutathera/Lutathera CA: continuer l'administration d'octréotide LP 30 mg par voie intramusculaire toutes les 4 semaines après la fin du traitement par Lutathera/Lutathera CA, si indiqué cliniquement.
Mode d'administration
Lutathera/Lutathera CA est destiné à être administré par voie intraveineuse. C'est un médicament radiopharmaceutique prêt à l'emploi à usage unique seulement.
Instructions d'administration
La méthode de perfusion pour l'administration de la dose recommandée peut être la méthode par gravité, la méthode par pompe péristaltique ou la méthode par pousse-seringue. Le personnel médical traitant peut utiliser d'autres méthodes connues pour être appropriées et sûres, en particulier lorsqu'une réduction de la dose est nécessaire.
En cas d'utilisation de la méthode par gravité ou de la méthode par pompe péristaltique, Lutathera/Lutathera CA doit être perfusé directement depuis le contenant original. La méthode par pompe péristaltique ou par pousse-seringue doit être utilisée lorsqu'une dose réduite de Lutathera/Lutathera CA est administrée après une modification de la dose en raison d'une réaction indésirable (voir le tableau 3). L'utilisation de la méthode par gravité pour l'administration d'une dose réduite de Lutathera/Lutathera CA peut entrainer l'injection d'une quantité erronée de Lutathera/Lutathera CA lorsque la dose n'est pas ajustée avant l'administration. Au cours de l'administration, les mesures de précautions usuelles relatives à la radioprotection doivent être mises en œuvre quelle que soit la méthode de perfusion (voir la rubrique «Remarques concernant la manipulation/Radioprotection»).
Lutathera/Lutathera CA ne doit pas être injecté en bolus.
Peu de temps après le début de la perfusion, le patient doit être surveillé quant à la radioactivité émise au moyen d'un système étalonné de mesure de la radioactivité, afin de s'assurer que la dose est délivrée. Pendant la perfusion, la radioactivité émise par le patient doit croitre régulièrement alors que la radioactivité émise par le flacon de Lutathera/Lutathera CA doit diminuer.
Il est recommandé de surveiller étroitement les signes vitaux du patient pendant la perfusion.
Le tableau 4 résume les procédures nécessaires au cours d'un cycle de traitement par Lutathera/Lutathera CA.
Tableau 4: Procédure d'administration des antiémétiques, de la solution d'acides aminés et de Lutathera/Lutathera CA
|
Agents administrés
|
Heure de début
(min)
|
Débit de perfusion
(ml/h)
|
Durée
| |
Antiémétiques
|
délai de temps suffisant avant la solution d'acides aminés
|
selon les indications de l'information professionnelle
|
selon les indications de l'information professionnelle
| |
Solution d'acides aminés: soit la préparation magistrale (1 l) soit la solution commerciale (1 à 2 l).
|
0
|
250–500
en fonction du volume
|
4 heures
| |
Lutathera/Lutathera CA avec solution de chlorure de sodium 9 mg/ml (0,9%) pour préparation injectable
|
30
|
Jusqu'à 400
|
30 ± 10 minutes
|
Pour les instructions concernant la manipulation du médicament avant administration, voir la rubrique «Remarques particulières».
Pour les indications concernant la préparation du patient, voir la rubrique «Mises en garde et précautions».
Pour les recommandations en cas d'extravasation, voir la rubrique «Mises en garde et précautions».
Procédure d'administration intraveineuse
Indications pour la perfusion par gravité (avec une pince ou une pompe à perfusion)
1.Introduire une aiguille 20G longue de 2,5 cm (aiguille courte) dans le flacon de Lutathera/Lutathera CA et la relier par un cathéter à 500 ml de solution stérile de chlorure de sodium à 0,9% (qui est utilisée pour le transport de la solution de Lutathera/Lutathera CA pendant la perfusion). Veiller à ce que l'aiguille courte ne touche pas la solution de Lutathera/Lutathera CA contenue dans le flacon. L'aiguille courte ne doit pas être raccordée directement au patient. La solution de chlorure de sodium ne doit pas s'écouler dans le flacon de Lutathera/Lutathera CA avant que la perfusion de Lutathera/Lutathera CA ne soit initiée. La solution de Lutathera/Lutathera CA ne doit pas être injectée directement dans la solution de chlorure de sodium.
2.Introduire une aiguille 18G longue de 9 cm (aiguille longue) dans le flacon de Lutathera/Lutathera CA et veiller à ce que cette aiguille longue touche le fond du flacon de Lutathera/Lutathera CA et y soit fixée pendant toute la durée de la perfusion. Relier l'aiguille longue au patient par un cathéter intraveineux qui est relié à la solution stérile de chlorure de sodium à 0,9% et qui est utilisé pour la perfusion de Lutathera/Lutathera CA au patient.
3.Utiliser une pince à roulette ou une pompe à perfusion pour réguler le flux de la solution de chlorure de sodium via l'aiguille courte dans le flacon de Lutathera/Lutathera CA. La solution de chlorure de sodium, qui arrive dans le flacon par l'aiguille courte, transporte la solution de Lutathera/Lutathera CA du flacon vers le patient par le cathéter intraveineux relié à l'aiguille longue sur une durée totale de 30 ± 10 minutes avec une vitesse de perfusion maximum de 400 ml/h. La perfusion doit commencer avec une vitesse de perfusion plus faible, < 100 ml/h pendant les 5 à 10 premières minutes, la vitesse de perfusion étant ensuite augmentée en fonction de l'état de la veine du patient. La pression dans le flacon doit être maintenue constante tout au long de la perfusion.
4.Pendant la perfusion, s'assurer que le niveau de la solution dans le flacon de Lutathera/Lutathera CA reste constant. Il convient pour cela de procéder à un contrôle visuel direct à plusieurs reprises lorsqu'un récipient blindé transparent est utilisé, ou de manipuler le flacon avec une pince en cas d'utilisation d'un récipient de transport en plomb.
5.L'écoulement de Lutathera/Lutathera CA du flacon vers le patient doit être surveillé pendant toute la durée de la perfusion.
6.La perfusion doit être arrêtée (déconnecter le flacon de la tubulure avec l'aiguille longue et clamper la conduite de solution saline) dès que la radioactivité reste stable pendant au moins cinq minutes.
7.À la suite de la perfusion, fournir au patient par voie intraveineuse 25 ml de solution stérile de chlorure de sodium à 0,9% par le cathéter veineux.
Indications pour la perfusion réalisée à l'aide d'une pompe péristaltique
1.Introduire une aiguille 20G longue de 2,5 cm et avec filtre (aiguille d'aération courte) dans le flacon de Lutathera/Lutathera CA. Veiller à ce que l'aiguille courte ne touche pas la solution de Lutathera/Lutathera CA contenue dans le flacon. L'aiguille courte ne doit pas être raccordée directement au patient ni à la pompe péristaltique.
2.Introduire une aiguille 18G longue de 9 cm (aiguille longue) dans le flacon de Lutathera/Lutathera CA et veiller à ce que cette aiguille longue touche le fond du flacon de Lutathera/Lutathera CA et y soit fixée pendant toute la durée de la perfusion. Raccorder l'aiguille longue par une tubulure appropriée à un robinet à trois voies et une solution stérile de chlorure de sodium à 0,9%.
3.Relier la sortie du robinet à trois voies à la tubulure qui est raccordée à l'entrée de la pompe péristaltique. Suivre pour cela les instructions du fabricant de la pompe.
4.Préremplir la tubulure en ouvrant le robinet à trois voies et en pompant la solution de Lutathera/Lutathera CA dans la tubulure jusqu'à ce qu'elle atteigne la sortie de la vanne.
5.Préremplir le cathéter intraveineux qui est raccordé au patient en ouvrant le robinet à trois voies pour que la solution stérile de chlorure de sodium à 0,9% puisse s'écouler. La solution stérile de chlorure de sodium à 0,9% est pompée jusqu'à ce qu'elle atteigne l'extrémité du cathéter.
6.Raccorder le cathéter intraveineux prérempli au patient et régler le robinet à trois voies de sorte que la solution de Lutathera/Lutathera CA se trouve connectée à la pompe péristaltique.
7.Administrer en perfusion le volume approprié de la solution de Lutathera/Lutathera CA sur une période de 30 ± 10 minutes pour délivrer la radioactivité souhaitée.
8.Une fois que la radioactivité de Lutathera/Lutathera CA souhaitée a été délivrée, arrêter la pompe péristaltique, puis modifier la position du robinet à trois voies de sorte que la pompe péristaltique soit reliée à la solution stérile de chlorure de sodium à 0,9%. Redémarrer la pompe péristaltique et administrer au patient un rinçage intraveineux avec 25 ml de solution stérile de chlorure de sodium à 0,9% via le cathéter veineux.
Indications pour la perfusion avec le pousse-seringue
1.Prélever le volume approprié de solution de Lutathera/Lutathera CA pour administrer la radioactivité souhaitée en utilisant une seringue jetable avec protection et une aiguille jetable 18G stérile, longue de 9 cm (aiguille longue). Pour faciliter le prélèvement de la solution, une aiguille 20G longue de 2,5 cm avec filtre (aiguille d'aération courte) peut être utilisée afin de réduire la résistance du flacon sous pression. Veiller à ce que l'aiguille courte ne touche pas la solution de Lutathera/Lutathera CA contenue dans le flacon.
2.Introduire la seringue dans la pompe blindée et placer un robinet à trois voies entre la seringue et un cathéter intraveineux qui est rempli de solution stérile de chlorure de sodium à 0,9% et qui est utilisé pour l'administration de Lutathera/Lutathera CA au patient.
3.Administrer en perfusion le volume approprié de solution de Lutathera/Lutathera CA sur une période de 30 ± 10 minutes pour délivrer la radioactivité souhaitée.
4.Une fois que la radioactivité de Lutathera/Lutathera CA souhaitée a été délivrée, arrêter le pousse-seringue puis modifier la position du robinet à trois voies de façon à rincer la seringue avec 25 ml de la solution stérile de chlorure de sodium à 0,9%. Redémarrer le pousse-seringue.
5.Une fois le rinçage de la seringue terminé, effectuer un rinçage du cathéter du patient avec 25 ml de solution stérile de chlorure de sodium à 0,9%.
Figure 2: Aperçu des méthodes d'administration

EXPOSITION AUX RAYONNEMENTS
Les analyses dosimétriques effectuées lors des études cliniques avec Lutathera/Lutathera CA ont conduit aux conclusions suivantes:
·L'organe critique est la moelle osseuse. Néanmoins, en utilisant la dose cumulative recommandée de Lutathera CA de 29 600 MBq (4 administrations de 7400 MBq), il n'a pas été observé de corrélation entre la toxicité hématologique et la radioactivité totale administrée ou la dose absorbée par la moelle osseuse ni dans l'étude Erasmus de phase I/II, ni dans l'étude NETTER-1 de phase III.
·Le rein n'est pas un organe critique si une perfusion simultanée d'une solution appropriée d'acides aminés est effectuée.
Globalement, les résultats des analyses dosimétriques effectuées au cours de l'étude NETTER-1 de phase III et au cours de l'étude Erasmus de phase I/II concordent et indiquent que le schéma posologique de Lutathera/Lutathera CA (4 administrations de 7400 MBq) est sans danger.
Tableau 5: Estimation de dose absorbée de lutécium (177Lu) oxodotréotide issue de l'étude NETTER-1 de phase III (Olinda-Output)
|
Organe
|
Dose absorbée par l'organe (mGy/MBq)
(n = 20)
| |
|
Moyenne
|
MS
| |
Glandes surrénales
|
0,037
|
0,016
| |
Cerveau
|
0,027
|
0,016
| |
Seins**
|
0,027
|
0,015
| |
Paroi de la vésicule biliaire
|
0,042
|
0,019
| |
Paroi du gros intestin inférieur
|
0,029
|
0,016
| |
Intestin grêle
|
0,031
|
0,015
| |
Paroi de l'estomac
|
0,031
|
0,015
| |
Paroi du gros intestin supérieur
|
0,032
|
0,015
| |
Paroi du cœur
|
0,032
|
0,015
| |
Reins
|
0,654
|
0,295
| |
Foie*
|
0,199
|
0,226
| |
Poumons
|
0,031
|
0,015
| |
Muscle
|
0,029
|
0,015
| |
Ovaires***
|
0,031
|
0,013
| |
Pancréas
|
0,038
|
0,016
| |
Moelle osseuse rouge
|
0,035
|
0,029
| |
Cellules ostéogéniques
|
0,151
|
0,268
| |
Peau
|
0,027
|
0,015
| |
Rate
|
0,846
|
0,804
| |
Testicules**
|
0,026
|
0,018
| |
Thymus
|
0,028
|
0,015
| |
Thyroïde
|
0,027
|
0,016
| |
Paroi de la vessie
|
0,437
|
0,176
| |
Utérus***
|
0,032
|
0,013
| |
Organisme entier
|
0,052
|
0,027
|
*n = 18 (deux patients ont été exclus car la dose absorbée par le foie était erronée en raison de l'absorption par des métastases hépatiques)
**n = 11 (patients de sexe masculin uniquement)
***n = 9 (patients de sexe féminin uniquement)
La dose de radiation des organes spécifiques, lesquels ne sont pas nécessairement des organes cibles de la thérapie, peut être influencée de manière significative par les changements physiopathologiques induits par la maladie. Ceci doit être pris en compte lors de l'utilisation de ces informations.
Contre-indications·Hypersensibilité au principe actif ou à l'un des excipients mentionnés à la rubrique «composition».
·Grossesse établie ou suspectée, ou lorsque la grossesse n'a pas été exclue (voir la rubrique «Grossesse, allaitement»).
·Insuffisance rénale sévère avec clairance de la créatinine < 30 ml/min.
Mises en garde et précautionsRisques liés à l'exposition aux rayonnements
Lutathera/Lutathera CA contribue à l'exposition globale cumulée du patient aux radiations ionisantes sur le long terme. L'exposition cumulée aux rayonnements sur le long terme peut être associée à un risque accru de cancer.
Myélosuppression
Dans l'étude NETTER-1, les cas de myélosuppression ont été observés plus fréquemment chez les patients recevant Lutathera/Lutathera CA avec de l'octréotide LP que chez les patients recevant de l'octréotide LP à fortes doses (tous grades/grade 3 ou 4): anémie (81%/0) versus (54%/1%); thrombocytopénie (53%/1%) versus (17%/0); et neutropénie (26%/3%) versus (11%/0). Dans l'étude NETTER-1, le délai médian d'obtention de la numération plaquettaire la plus faible (nadir des plaquettes) a été de 5,1 mois après la première dose. Sur les 59 patients ayant développé une thrombocytopénie, une récupération plaquettaire avec un retour au niveau initial ou à la normale a été observée chez 68% d'entre eux. Le délai médian de la récupération plaquettaire était de 2 mois. Quinze des dix-neuf patients pour lesquels une récupération plaquettaire n'a pas été documentée présentaient un taux plaquettaire post-nadir. Parmi ces 15 patients, 5 ont présenté une amélioration atteignant un grade 1, 9 un grade 2, et 1 un grade 3.
Les patients ayant une fonction médullaire réduite, ainsi que ceux ayant déjà reçu une chimiothérapie ou une radiothérapie par faisceau externe, peuvent présenter un risque plus élevé de toxicité hématologique pendant le traitement par Lutathera/Lutathera CA. Le traitement n'est pas recommandé chez les patients dont la fonction hématologique est gravement altérée avant et pendant le traitement par Lutathera/Lutathera CA (par exemple Hb < 4,9 mmol/l ou 8 g/dl, nombre de plaquettes < 75 G/l, ou leucocytes < 2 G/l), sauf si elle est exclusivement attribuée à une lymphopénie.
Une surveillance de la numération globulaire au début du traitement et avant chaque administration de dose de Lutathera/Lutathera CA est nécessaire. En fonction de la sévérité des effets indésirables, il faut suspendre temporairement le traitement, ajuster la posologie ou arrêter définitivement le traitement (voir la rubrique «Posologie/Mode d'emploi: Adaptation du traitement»).
Syndrome myélodysplasique secondaire et leucémie aiguë
Des syndromes myélodysplasiques (SMD) à survenue tardive et des leucémies aiguës (LA) ont été observés après le traitement par Lutathera/Lutathera CA (voir la rubrique «Effets indésirables»). Dans l'étude NETTER-1, après un délai médian de suivi de 76 mois dans la partie principale de l'étude, des cas de syndrome myélodysplasique (SMD) ont été rapportés chez 3 patients (2 patients de la partie principale de l'étude et 1 patient de la partie de dosimétrie de l'étude; 2,3%) ayant reçu Lutathera/Lutathera CA et de l'octréotide LP, mais aucun cas n'a été rapporté chez des patients ayant reçu de l'octréotide LP à fortes doses. Dans l'étude Erasmus, 16 patients (2,0%) ont développé un SMD et 4 (0,5%) une leucémie aiguë. Le délai médian de survenue a été de 29 mois (9 à 45 mois) pour le SMD et de 55 mois (32 à 125 mois) pour la LA. L'étiologie de ces néoplasies myéloïdes secondaires liées au traitement n'est pas clairement établie. Des facteurs comme un âge > 70 ans, une insuffisance rénale, des cytopénies préexistantes, le nombre de traitements antérieurs, une exposition antérieure aux agents chimiothérapeutiques (en particulier les agents alkylants) et une radiothérapie antérieure constitueraient des risques potentiels et/ou des facteurs prédictifs pour le SMD/la LA.
Toxicité rénale
Dans l'étude Erasmus, 8 patients (< 1%) ont développé une insuffisance rénale 3 à 36 mois après l'administration de Lutathera/Lutathera CA. Deux de ces patients présentaient une insuffisance rénale sous-jacente ou des facteurs de risque d'insuffisance rénale (notamment diabète ou hypertension) et nécessitaient une dialyse.
La solution d'acides aminés doit être administrée avant, pendant et après l'administration de Lutathera/Lutathera CA (voir la rubrique «Posologie/Mode d'emploi: Solution d'acides aminés») afin de diminuer la réabsorption du lutécium (177Lu) oxodotréotide à travers les tubules proximaux et diminuer ainsi l'irradiation au niveau des reins. Le patient doit être encouragé à bien s'hydrater et à vider sa vessie le plus fréquemment possible la veille, le jour et le lendemain de l'administration de Lutathera/Lutathera CA. La créatinine sérique et la clairance de la créatinine calculée doivent être surveillées.
En fonction de la sévérité des effets indésirables, il faut suspendre temporairement le traitement, ajuster la posologie ou arrêter définitivement le traitement (voir la rubrique «Posologie/Mode d'emploi: Adaptation du traitement»).
Le risque de toxicité peut être accru chez les patients présentant une insuffisance rénale au départ ou des anomalies des voies rénales ou urinaires.
Pour les patients présentant une clairance de la créatinine < 50 ml/min, un risque accru d'hyperkaliémie transitoire doit également être pris en considération (voir la rubrique «Mises en garde et précautions» concernant la solution d'acides aminés coadministrée pour la protection rénale).
Hépatotoxicité
Dans l'étude Erasmus, il a été rapporté chez 2 patients (0,25%) des hémorragies tumorales hépatiques, un œdème ou une nécrose, et l'un d'entre eux (0,12%) a présenté une congestion et une cholestase intra-hépatique. Lutathera/Lutathera CA étant indiqué chez de nombreux patients présentant des métastases hépatiques, il est ainsi fréquent d'observer chez ces patients une fonction hépatique de base altérée. Une augmentation du risque d'hépatotoxicité due à l'exposition aux rayonnements ionisants peut être observée chez ces patients.
Les concentrations en transaminases, bilirubine et albumine sérique doivent être surveillées pendant le traitement (voir la rubrique «Posologie/Mode d'emploi»).
En fonction de la sévérité des effets indésirables, il faut suspendre temporairement le traitement, ajuster la posologie ou arrêter définitivement le traitement (voir la rubrique «Posologie/Mode d'emploi: Adaptation du traitement»).
Hypersensibilité
Des cas de réactions d'hypersensibilité (y compris d'angio-œdèmes isolés) ont été rapportés après la mise sur le marché chez des patients traités par Lutathera/Lutathera CA (voir la rubrique «Effets indésirables»). En cas de réactions graves d'hypersensibilité, le traitement par Lutathera/Lutathera CA doit être immédiatement interrompu. Des médicaments adaptés et un équipement approprié pour le traitement de ces réactions doivent être disponibles pour une utilisation immédiate.
Crises hormonales neuroendocrines
Des crises hormonales neuroendocrines dues à la sécrétion excessive d'hormones ou de substances bioactives se manifestant par des bouffées de chaleur, des diarrhées, un bronchospasme et de l'hypotension sont survenues chez 2 patients (0,25%) dans l'étude Erasmus, typiquement pendant le traitement par Lutathera/Lutathera CA ou dans les 24 heures suivant son administration. En outre, des cas d'hypercalcémie ont été rapportés chez 2 patients (0,25%). C'est pourquoi l'observation des patients doit être envisagée durant une hospitalisation d'une nuit dans certains cas (p.ex. patients avec un faible contrôle pharmacologique des symptômes).
Les patients doivent faire l'objet d'une observation en cas de survenue de bouffées de chaleur, de diarrhée, d'une hypotension, d'une bronchoconstriction ou de tout autre signe ou symptôme en rapport avec la maladie tumorale neuroendocrine. En fonction des besoins, de fortes doses d'analogues de la somatostatine, des corticoïdes et une solution de réhydratation par voie intraveineuse doivent être administrés.
Nausées et vomissements
Afin de prévenir des nausées et vomissements liés au traitement, une injection intraveineuse d'antiémétiques doit être réalisée en prévoyant un délai de temps suffisant avant le début de la perfusion d'acides aminés (voir la rubrique «Posologie/Mode d'emploi»).
Utilisation concomitante d'analogues de la somatostatine
Une utilisation concomitante d'analogues froids de la somatostatine peut être nécessaire pour le contrôle des symptômes de la maladie (voir la rubrique «Posologie/Mode d'emploi»).
Syndrome de lyse tumorale
Un syndrome de lyse tumorale a été rapporté à la suite d'un traitement par des médicaments contenant du lutécium (177Lu). Les patients ayant des antécédents d'insuffisance rénale et une masse tumorale importante peuvent présenter un risque accru et doivent être traités avec prudence. La fonction rénale ainsi que l'équilibre électrolytique doivent être évalués avant et pendant le traitement.
Règles de radioprotection
Le traitement doit avoir lieu dans un service bénéficiant d'une autorisation de l'OFSP en vue de l'utilisation thérapeutique de sources de rayonnements non scellées.
En conformité avec les bonnes pratiques de radioprotection du service et avec la procédure de prise en charge du patient, l'exposition au rayonnement du patient et du personnel soignant doit être minimisée pendant et après le traitement par Lutathera/Lutathera CA, tout comme il convient de limiter le contact avec d'autres personnes.
Pendant l'administration de Lutathera/Lutathera CA, la personne traitée doit être placée dans une chambre séparée, spécifiquement équipée. L'hospitalisation et la sortie après thérapie avec des substances radioactives doivent être effectuées en accord avec l'ordonnance sur la radioprotection, l'ordonnance du DFI sur l'utilisation des matières radioactives ainsi que les directives du service fédéral de la santé.
Les patients doivent être encouragés à boire des quantités importantes d'eau (par exemple, au moins 1 verre d'eau par heure) la veille de la perfusion, le jour de la perfusion et le jour suivant pour faciliter l'élimination par les urines. Le patient doit également être encouragé à aller à la selle tous les jours et à utiliser un laxatif si nécessaire. Les urines et les selles doivent être éliminées selon les réglementations nationales (ordonnances et directives).
Dans la mesure où la peau du patient n'est pas contaminée, comme en cas de fuite durant la perfusion ou en raison d'une incontinence urinaire, la peau et les vomissures ne devraient pas être contaminées par la radioactivité. Cependant, lors des soins standards ou lors des traitements avec des dispositifs médicaux ou autres instruments qui entrent en contact avec la peau (p.ex. électrocardiogramme [ECG]), il est recommandé de prendre des mesures de protection telles que le port de gants, l'installation du matériel et des électrodes avant le début de la perfusion du produit radiopharmaceutique, le changement du matériel et de l'électrode après la mesure et éventuellement la mesure de la radioactivité des équipements après utilisation.
En accord avec les exigences de l'ordonnance sur la radioprotection, avant la sortie, le médecin spécialiste en médecine nucléaire est tenu d'expliquer au patient, lors d'un entretien individuel, les règles de comportement à adopter vis-à-vis des proches et des tiers en matière de radioprotection ainsi que les précautions générales à respecter pendant ses activités quotidiennes après le traitement afin de limiter autant que possible l'exposition aux radiations des personnes de son entourage.
Après chaque administration, les recommandations générales suivantes ainsi que les procédures et dispositions nationales, locales et institutionnelles doivent être prises en compte:
·le contact rapproché (moins de 1 mètre) avec d'autres personnes doit être limité pendant 7 jours;
·pour les enfants et/ou les femmes enceintes, le contact rapproché (moins de 1 mètre) doit être limité à moins de 15 minutes par jour pendant 7 jours;
·les patients doivent dormir dans une chambre séparée d'autres personnes pendant 7 jours. Les patients doivent dormir dans des chambres séparées des enfants et/ou des femmes enceintes pendant 15 jours.
La radioactivité peut être détectée dans les urines jusqu'à 30 jours après l'administration de Lutathera/Lutathera CA.
Mesures recommandées en cas d'extravasation
Porter des gants jetables et étanches. La perfusion du médicament doit être immédiatement arrêtée et le dispositif d'administration (cathéter, etc.) doit être enlevé. Le médecin spécialiste en médecine nucléaire et la personne responsable de la radioprotection doivent être informés.
Le matériel d'administration doit être conservé afin de mesurer la radioactivité résiduelle et de déterminer l'activité réellement administrée, et éventuellement la dose absorbée. La zone d'extravasion doit être délimitée à l'aide d'un stylo indélébile et une photo doit être prise dans la mesure du possible. Il est également recommandé d'enregistrer le moment de l'extravasation ainsi que l'estimation du volume extravasé.
Pour continuer la perfusion de Lutathera/Lutathera CA, il est obligatoire d'utiliser un nouveau cathéter en le plaçant dans une veine opposée, et aucun autre médicament ne doit être administré du même côté que celui où l'extravasation est survenue.
Afin d'accélérer la dispersion du produit et de prévenir sa stagnation au niveau des tissus, il est recommandé d'augmenter le flux sanguin en élevant le bras concerné. Selon le cas, une aspiration du liquide extravasé, une injection de solution de chlorure de sodium à 9 mg/ml (0,9%) ou l'application de compresses chaudes ou de coussins chauffants sur le site de perfusion afin d'accélérer la vasodilatation peuvent être envisagées.
Les symptômes, notamment l'inflammation et/ou la douleur, doivent être traités. Selon la situation, le médecin spécialiste en médecine nucléaire doit informer le patient des risques liés à une extravasation et lui donner des conseils sur les traitements potentiels ainsi que sur la marche à suivre. La zone d'extravasation doit être surveillée jusqu'à ce que le patient sorte de l'hôpital. En fonction de sa gravité, cet événement doit être déclaré comme un effet indésirable.
Patients souffrants d'incontinence urinaire
Il est recommandé de suivre les patients souffrant d'incontinence urinaire plus fréquemment pendant le traitement. Chez Les patients souffrant d'incontinence urinaire, il convient de prendre des précautions spéciales après l'administration de Lutathera/Lutathera CA pour éviter toute contamination radioactive. Ceci comprend en particulier la manipulation de tout matériel potentiellement contaminé par l'urine.
Patients présentant des métastases cérébrales
Aucune donnée relative à l'efficacité chez les patients présentant des métastases cérébrales n'est disponible. Par conséquent, le rapport bénéfice-risque doit être évalué de manière individuelle pour ces patients.
Néoplasmes malins secondaires
L'exposition aux radiations ionisantes peut induire le développement de cancers et d'anomalies héréditaires. L'irradiation résultant d'une exposition thérapeutique peut conduire à une augmentation de l'incidence des cancers et des mutations génétiques. Dans tous les cas, il est nécessaire de s'assurer que les risques liés à une exposition aux rayonnements sont inférieurs aux risques résultant de la maladie elle-même.
Pour les précautions à prendre dans le respect du risque environnemental, voir la rubrique «Remarques particulières».
Autres patients présentant des facteurs de risque
Les patients présentant une des maladies suivantes ont un risque plus élevé de développer des effets indésirables. Par conséquent, ces patients doivent être suivis plus fréquemment pendant le traitement. En cas de toxicité dose-dépendante, se référer au tableau 3.
·métastases osseuses;
·précédentes thérapies radiométaboliques oncologiques avec des composés marqués au 131I ou toute autre thérapie utilisant des sources radioactives non scellées;
·antécédents d'autres tumeurs malignes, à moins que le patient soit considéré comme étant en rémission depuis au moins 5 ans.
Mises en garde et précautions concernant la solution d'acides aminés pour la protection rénale
Hyperkaliémie associée à la solution d'acides aminés
Une augmentation transitoire des taux de potassium sérique peut se produire chez les patients recevant de l'arginine et de la lysine. Ces taux reviennent généralement à la normale dans les 24 heures qui suivent le début de la perfusion de la solution d'acides aminés. Chez les patients présentant une réduction de la clairance de la créatinine, le risque d'hyperkaliémie transitoire peut être augmenté (voir rubrique «Toxicité rénale»).
Avant chaque administration d'une solution d'acides aminés, le taux de potassium sérique du patient doit être mesuré. En cas d'hyperkaliémie, il convient de vérifier les antécédents du patient en matière d'hyperkaliémie et les médicaments concomitants. L'hyperkaliémie doit être corrigée en conséquence avant de commencer la perfusion.
En cas d'hyperkaliémie préexistante cliniquement significative, un deuxième contrôle avant la perfusion de la solution d'acides aminés doit confirmer que l'hyperkaliémie a été bien corrigée. Le patient doit être surveillé de près pour détecter les signes et symptômes d'hyperkaliémie, par exemple dyspnée, faiblesse, engourdissement, douleurs thoraciques et manifestations cardiaques (troubles de la conduction et arythmies cardiaques). Un électrocardiogramme (ECG) doit être effectué avant la sortie du patient.
Les signes vitaux doivent être surveillés pendant la perfusion, quel que soit le taux de potassium sérique de départ. Les patients doivent être encouragés à boire des quantités importantes d'eau (à savoir au moins 1 verre par heure) la veille de la perfusion, le jour de la perfusion et le lendemain de la perfusion pour rester hydratés et faciliter l'excrétion de l'excès de potassium sérique.
Si des symptômes d'hyperkaliémie se développent pendant la perfusion de la solution d'acides aminés, des mesures correctives adéquates doivent être prises. En cas d'hyperkaliémie symptomatique grave, l'arrêt de la perfusion de la solution d'acides aminés doit être envisagé. Il conviendra de tenir compte du rapport bénéfice-risque entre la protection rénale et l'hyperkaliémie aiguë.
Insuffisance cardiaque
En raison du risque de complications cliniques liées à la surcharge volumique, l'utilisation de l'arginine et de la lysine chez les patients atteints d'insuffisance cardiaque sévère (classe III ou classe IV dans la classification NYHA [New York Heart Association]) requiert une attention particulière. Les patients atteints d'insuffisance cardiaque sévère (classe III ou classe IV dans la classification NYHA) ne doivent être traités qu'après une évaluation minutieuse du rapport bénéfice-risque, en tenant compte du volume et de l'osmolalité de la solution d'acides aminés.
Acidose métabolique
Une acidose métabolique a été observée lors de l'administration de solutions d'acides aminés complexes dans le cadre de protocoles de nutrition parentérale totale (NPT). Les variations de l'équilibre acido-basique modifient l'équilibre entre le potassium extracellulaire et le potassium intracellulaire, et le développement de l'acidose peut être associé à des augmentations rapides du taux de potassium plasmatique.
Teneur en sodium
Ce médicament contient 3,5 mmol (81,1 mg) de sodium par dose, soit 4% de l'apport alimentaire quotidien maximal recommandé par l'OMS pour un adulte, qui est de 2 g de sodium.
InteractionsLa somatostatine et ses analogues se lient de façon compétitive aux récepteurs de la somatostatine et peuvent interférer avec l'efficacité de Lutathera/Lutathera CA. Par conséquent, le traitement avec des analogues de la somatostatine à longue durée d'action doit être arrêté 4 à 6 semaines avant le traitement par Lutathera/Lutathera CA. Si nécessaire, les patients peuvent être traités avec des analogues de la somatostatine à courte durée d'action jusqu'à 24 heures avant la perfusion de Lutathera/Lutathera CA.
Il existe des indications selon lesquelles les glucocorticoïdes peuvent induire une régulation négative de l'expression du récepteur de la somatostatine de sous-type 2 (SSTR2). Par conséquent, il convient de renoncer, par mesure de précaution, à l'administration répétée de doses élevées de glucocorticoïdes pendant le traitement par Lutathera/Lutathera CA. Chez les patients ayant été traités par des glucocorticoïdes sur une longue durée, il convient d'évaluer soigneusement s'il existe une expression suffisante du récepteur de la somatostatine. On ignore si l'utilisation intermittente de glucocorticoïdes pour la prévention des nausées et des vomissements pendant l'administration de Lutathera/Lutathera CA peut entrainer une régulation négative de l'expression des SSTR2. Par mesure de précaution, il convient donc d'éviter d'utiliser des glucocorticoïdes comme traitement antiémétique préventif. Dans le cas où le traitement administré en prévention des nausées et des vomissements avant la perfusion de la solution d'acides aminés s'avère insuffisant, une dose unique de glucocorticoïdes peut être utilisée, mais elle ne doit pas être administrée avant d'initier la perfusion de Lutathera/Lutathera CA ni dans l'heure qui suit la fin de la perfusion.
Grossesse, allaitementFemmes en âge de procréer
Il convient d'exclure toute grossesse avant d'utiliser Lutathera/Lutathera CA en utilisant une méthode de test valide/adéquate.
Contraception chez les hommes et les femmes
Lutathera/Lutathera CA peut être nocif pour le fœtus lorsqu'il est administré à une femme enceinte. Pendant le traitement par Lutathera/Lutathera CA et au minimum au cours des 7 mois suivant la fin du traitement, des mesures adaptées doivent être prises pour prévenir toute grossesse chez les patientes. Pendant le traitement par Lutathera/Lutathera CA et pendant au moins les 4 mois suivant la fin du traitement, des mesures appropriées doivent être prises pour éviter une grossesse chez les partenaires des patients de sexe masculin.
Grossesse
Aucune étude n'a été conduite chez l'animal pour déterminer les effets du lutécium (177Lu) oxodotréotide sur la fonction de reproduction chez la femme et le développement embryo-fœtal.
Les examens utilisant des radionucléides chez la femme enceinte entraînent également l'irradiation du fœtus. L'utilisation de Lutathera/Lutathera CA est contre-indiquée pendant une grossesse confirmée ou suspectée ou si une grossesse n'a pas pu être exclue, en raison du risque associé aux rayonnements ionisants (voir la rubrique «Contre-indications»). Les femmes enceintes doivent être informées du risque pour le fœtus.
Allaitement
Il n'est pas établi si le lutécium (177Lu) oxodotréotide est excrété dans le lait maternel.
Un risque pour l'enfant allaité associé à des radiations ionisantes ne peut être exclu. L'allaitement doit être évité durant le traitement par ce médicament. Si le traitement avec Lutathera/Lutathera CA s'avère nécessaire pendant l'allaitement, l'enfant doit être sevré et l'allaitement doit être interrompu.
Fertilité
Aucune étude n'a été conduite chez l'animal pour déterminer les effets du lutécium (177Lu) oxodotréotide sur la fertilité des deux sexes. Les radiations ionisantes du lutécium (177Lu) oxodotréotide peuvent avoir des effets toxiques sur les gonades femelles et mâles, et entraîner une infertilité temporaire ou définitive. Une consultation génétique est recommandée si le patient ou la patiente souhaite avoir des enfants après le traitement. La cryoconservation de sperme ou d'ovules avant le traitement peut être proposée comme option pour les patients.
Effet sur l’aptitude à la conduite et l’utilisation de machinesLutathera/Lutathera CA n'a aucune influence ou a une influence négligeable sur l'aptitude à la conduite ou l'utilisation de machines. Néanmoins, l'état de santé général du patient et les éventuels effets indésirables liés au traitement doivent être pris en compte avant de conduire des véhicules ou d'utiliser des machines.
Effets indésirablesRésumé du profil de sécurité
Le profil de sécurité général de Lutathera/Lutathera CA a été établi sur des données groupées de patients ayant participé à des essais cliniques (étude NETTER-1 de phase III et les patients néerlandais de l'étude Erasmus de phase I/II) et à des programmes d'usage compassionnel.
Les effets indésirables les plus fréquents chez les patients ayant reçu le traitement par Lutathera/Lutathera CA ont été les nausées et les vomissements, qui sont survenus au début de la perfusion respectivement chez 58,9% et 45,5% des patients. Le lien de causalité entre les nausées et les vomissements est difficile à déterminer en raison des effets émétisants de la perfusion concomitante de solutions d'acides aminés administrée pour protéger les reins.
Compte tenu de la toxicité de Lutathera/Lutathera CA sur la moelle osseuse, les effets indésirables les plus attendus étaient liés à la toxicité hématologique: thrombocytopénie (25%), lymphopénie (22,3%), anémie (13,4%), pancytopénie (10,2%).
La fatigue (27,7%) et la perte d'appétit (13,4%) ont été rapportées comme d'autres effets indésirables très fréquents.
Au moment de l'analyse finale de l'étude NETTER-1, après une durée médiane de suivi de 76 mois dans chaque bras de l'étude, le profil de sécurité d'emploi est resté conforme au profil précédemment décrit.
Tableau récapitulatif des effets indésirables
Les effets indésirables sont présentés dans le tableau 6 par fréquence et par classe de systèmes d'organes (MedDRA). Les fréquences ont été classées selon la convention suivante: très fréquent (≥1/10), fréquent (≥1/100 à < 1/10), occasionnel (≥1/1000 à < 1/100), rare (≥1/10 000 à < 1/1000), très rare (< 1/10 000) et fréquence indéterminée (ne peut être estimée sur la base des données disponibles).
Tableau 6: Fréquence des effets indésirables signalés dans les études cliniques et au cours de la surveillance post-commercialisation
|
Classe de système d'organes MedDRA
|
Très fréquent
|
Fréquent
|
Occasionnel
|
Fréquence inconnue
| |
Infections et infestations
|
|
|
Conjonctivite
Infection des voies respiratoires
Cystite
Pneumonie
Zona herpétique
Zona herpétique ophtalmique
Grippe
Infections staphylococciques
Bactériémies streptococciques
|
| |
Tumeurs bénignes, malignes et non précisées (incl kystes et polypes)
|
|
Cytopénie réfractaire avec dysplasie multilignée (syndrome myélodysplasique)
|
Leucémie myéloïde aiguë
Leucémie aiguë
Leucémie myélomonocytaire chronique
|
| |
Affections hématologiques et du système lymphatique
|
Thrombocytopénie (25%)2
Lymphopénie (22,3%)3
Anémie (13,4%)4
Pancytopénie (10,2%)
|
Leucopénie5
Neutropénie6
|
Cytopénie réfractaire avec dysplasie multilignée
Anémie néphrogénique
Insuffisance de la moelle osseuse
Purpura thrombocytopénique
|
| |
Affections du système immunitaire
|
|
|
Hypersensibilité
|
Angio-œdème12
| |
Affections endocriniennes
|
|
Hypothyroïdie secondaire
|
Hypothyroïdie
Diabète sucré
Crise carcinoïde
Hyperparathyroïdie
|
| |
Troubles du métabolisme et de la nutrition
|
Diminution de l'appétit (13,4%)
|
Hyperglycémie
Déshydratation
Hypomagnésémie
Hyponatrémie
|
Hypoglycémie
Hypernatrémie
Hypophosphatémie
Syndrome de lyse tumorale
Hypercalcémie
Hypocalcémie
Hypoalbuminémie
Acidose métabolique
|
| |
Affections psychiatriques
|
|
Troubles du sommeil
|
Anxiété
Hallucinations
Désorientation
|
| |
Affections du système nerveux
|
|
Étourdissements
Dysgueusie
Céphalées10
Léthargie
Syncope
|
Fourmillements
Encéphalopathie hépatique
Paresthésie
Parosmie
Somnolence
Compression de la moelle épinière
|
| |
Affections oculaires
|
|
|
Affections oculaires
|
| |
Affections de l'oreille et du labyrinthe
|
|
|
Vertiges
|
| |
Affections cardiaques
|
|
Allongement de l'intervalle QT à l'électrocardiogramme
|
Fibrillation auriculaire
Palpitations
Infarctus du myocarde
Angine de poitrine
Choc cardiogénique
|
| |
Affections vasculaires
|
|
Hypertension7
Rougissement
Bouffées de chaleur
Hypotension
|
Vasodilatation
Extrémités froides
Pâleur
Hypotension orthostatique
Phlébite
|
| |
Affections respiratoires, thoraciques et médiastinales
|
|
Dyspnée
|
Douleur oropharyngée
Épanchement pleural
Augmentation des expectorations
Sensation de pression
|
| |
Affections gastro-intestinales
|
Nausées (58,9%)
Vomissements (45,5%)
|
Distension abdominale
Diarrhée
Douleur abdominale
Constipation
Douleur abdominale haute
Dyspepsie
Gastrite
|
Bouche sèche
Flatulence
Ascite
Douleur gastro-intestinale
Stomatite
Hématochézie
Gêne intestinale
Obstruction intestinale
Colite
Pancréatite aiguë
Rectorragie
Méléna
Douleur abdominale basse
Hématémèse
Ascite hémorragique
Iléus
|
| |
Affections hépatobiliaires
|
|
Hyperbilirubinémie9
|
Baisse des enzymes pancréatiques
Atteinte hépatocellulaire
Cholestase
Congestion hépatique
Insuffisance hépatique
|
| |
Affections de la peau et du tissu sous-cutané
|
|
Alopécie
|
Éruption cutanée
Sécheresse cutanée
Gonflement du visage
Hyperhidrose
Prurit généralisé
|
| |
Affections musculo-squelettiques et du tissu conjonctif
|
|
Douleur musculosquelettique8
Spasmes musculaires
|
|
| |
Affections du rein et des voies urinaires
|
|
Insuffisance rénale aiguë
Hématurie
Insuffisance rénale
Protéinurie
|
Leucocyturie
Incontinence urinaire
Diminution du débit de filtration glomérulaire
Trouble rénal
Insuffisance prérénale aiguë
Atteinte rénale
|
| |
Troubles généraux et anomalies au site d'administration
|
Fatigue (27,7%)1
|
Réaction au site d'injection11
Œdème périphérique
Douleur au site d'administration
Frissons
Symptômes grippaux
|
Masse au site d'injection
Inconfort dans la poitrine
Douleur dans la poitrine
Fièvre
Malaise
Douleur
Décès
Sensation anormale
|
| |
Investigations
|
|
Augmentation de la créatinine sanguine [Affections du rein et des voies urinaires]
Augmentation de la GGT* [Affections hépatobiliaires]
Augmentation de l'ALAT** [Affections hépatobiliaires]
Augmentation de l'ASAT*** [Affections hépatobiliaires]
Augmentation de la PAL**** [Affections hépatobiliaires]
|
Baisse du potassium sérique [Affections du rein et des voies urinaires]
Augmentation de l'urée sanguine [Affections du rein et des voies urinaires]
Augmentation de l'hémoglobine glycosylée [Troubles du métabolisme et de la nutrition]
Baisse de l'hématocrite [Affections hématologiques et du système lymphatique]
Protéinurie [Affections du rein et des voies urinaires]
Perte de poids [Troubles généraux et anomalies au site d'administration]
Augmentation des concentrations sériques de créatine-phosphokinase [Affections musculo-squelettiques et du tissu conjonctif]
Augmentation des concentrations sériques de lactate déshydrogénase [Affections musculo-squelettiques et systémiques]
Augmentation des catécholamines dans le sang [Affections endocriniennes]
Augmentation de la protéine C-réactive [Infections et infestations]
|
| |
Lésions, intoxications et complications d'interventions
|
|
|
Fracture de la clavicule
|
| |
Actes médicaux et chirurgicaux
|
|
Transfusion
|
Drainage de la cavité abdominale
Dialyse
Pose d'une sonde gastrique
Pose de stent
Drainage d'abcès
Prélèvement de moelle osseuse
Polypectomie
|
| |
Caractéristiques socio-environnementales
|
|
|
Handicap physique
|
|
1 Inclut asthénie et fatigue
2 Inclut thrombocytopénie et diminution de la numération plaquettaire
3 Inclut lymphopénie et diminution de la numération lymphocytaire
4 Inclut anémie et diminution de l'hémoglobine
5 Inclut leucopénie et diminution du nombre de globules blancs
6 Inclut neutropénie et diminution du nombre de neutrophiles
7 Inclut hypertension et crises hypertensives
8 Inclut arthralgie, douleur au niveau des extrémités, douleur dorsale, douleurs osseuses, douleur du flanc, douleur thoracique musculo-squelettique et douleur de la nuque
9 Inclut augmentation de la bilirubine sérique et hyperbilirubinémie
10 Inclut maux de tête et migraine
11 Inclut réaction au site d'injection, hypersensibilité au site d'injection, induration au site d'injection, gonflement au site d'injection
12 Rapporté dans la période suivant la mise sur le marché
*Augmentation de la gamma-glutamyltransférase
**Alanine aminotransférase
***Aspartate aminotransférase
****Phosphatase alcaline.
Effets indésirables rapportés spontanément (fréquence inconnue)
Les effets indésirables suivants proviennent de rapports spontanés de cas signalés avec Lutathera/Lutathera CA après la mise sur le marché (voir tableau 6). Ces réactions ayant été rapportées spontanément à partir d'une population de taille inconnue, il n'est pas possible d'en évaluer la fréquence avec fiabilité. Pour cette raison, ces effets sont classés comme étant de fréquence inconnue. Les effets indésirables sont répertoriés par classe de systèmes d'organes selon MedDRA. À l'intérieur de chacune des classes de systèmes d'organes, les effets indésirables sont présentés dans l'ordre décroissant de sévérité.
Affections du système immunitaire
Angio-œdème.
L'annonce d'effets secondaires présumés après l'autorisation est d'une grande importance. Elle permet un suivi continu du rapport bénéfice-risque du médicament. Les professionnels de santé sont tenus de déclarer toute suspicion d'effet secondaire nouveau ou grave via le portail d'annonce en ligne ElViS (Electronic Vigilance System). Vous trouverez des informations à ce sujet sur www.swissmedic.ch.
SurdosageAucun cas de surdosage n'a été rapporté à ce jour.
En cas de surdosage, une augmentation de la fréquence des effets indésirables liés à la radiotoxicité est attendue.
En cas de surexposition aux rayonnements lors de l'administration de Lutathera/Lutathera CA, la dose absorbée par le patient doit être réduite autant que possible en augmentant l'élimination du radionucléide de l'organisme par une diurèse forcée et des mictions fréquentes ainsi que par une hydratation renforcée pendant les 48 premières heures suivant la perfusion. Il convient d'estimer la dose efficace administrée.
Il est recommandé d'effectuer les analyses suivantes toutes les semaines pendant les 10 semaines qui suivent le surdosage:
·surveillance hématologique: numération des globules blancs, plaquettes et hémoglobine;
·surveillance biochimique: créatinine sérique et glycémie.
Propriétés/EffetsCode ATC
V10XX04
Propriétés physiques
Lutathera/Lutathera CA avec lutécium (177Lu) se désintègre en hafnium (177Hf) stable avec une demi-vie de 6,647 jours en émettant majoritairement des rayonnements β d'une énergie maximale de 0,498 MeV. L'énergie bêta moyenne est de 0,13 MeV. Des rayonnements photoniques (γ) de 0,113 MeV (6,2%) et 0,208 MeV (11%) sont également émis.
Lutathera CA est préparé en utilisant du 176Lu qui contient une petite quantité de l'isomère nucléaire lutécium métastable (177mLu). L'isomère 177mLu a une demi-vie de 160,44 jours. L'isomère 177mLu se désintègre partiellement (22,8%) par transition isomérique avec émission gamma et conversion électronique à l'état fondamental de Lu-177, et partiellement (77,2%) par émission bêta (40,8 keV) en hafnium-177 métastable (177mHf) qui se désintègre immédiatement par de multiples émissions gamma et conversion électronique en 177Hf stable. Tout déchet radioactif résultant de l'utilisation du lutécium (177Lu) oxodotréotide doit tenir compte de la présence et de la quantité de cet isomère particulier pour une élimination appropriée.
Mécanisme d'action
Le lutécium (177Lu) oxodotréotide a une haute affinité pour les récepteurs de la somatostatine de sous-type 2 (sst2). Il se fixe spécifiquement aux cellules malignes qui surexpriment les récepteurs sst2.
Le lutécium-177 est un radionucléide émetteur β moins dont la distance maximum de pénétration dans les tissus est d'environ 2,2 mm (moyenne de pénétration de 0,67 mm), ce qui est provoque la mort des cellules tumorales ciblées tout en ayant un effet limité sur les cellules voisines saines.
Pharmacodynamique
À la concentration utilisée (environ 10 μg/ml au total pour les formes libre et radiomarquée), le peptide oxodotréotide n'exerce aucun effet pharmacodynamique cliniquement significatif.
Efficacité clinique
Étude NETTER-1
L'étude NETTER-1 de phase III était une étude randomisée, multicentrique, ouverte, contrôlée versus comparateur, comparant le traitement par Lutathera CA (4 doses de 7400 MBq chacune, une dose toutes les 8 semaines [± 1 semaine]) administré en concomitance avec une solution d'acides aminés et avec les meilleurs soins de soutien possibles (octréotide 30 mg à libération prolongée [LP] après chaque dose de Lutathera CA et toutes les 4 semaines après la fin du traitement par Lutathera CA pour le contrôle des symptômes, remplacé par de l'octréotide à courte durée d'action durant les 4 à 6 semaines précédant l'administration de Lutathera CA) à une dose élevée d'octréotide LP (60 mg toutes les 4 semaines) chez les patients présentant des tumeurs carcinoïdes de l'intestin moyen, inopérables, progressives, surexprimant des récepteurs de la somatostatine. Le critère d'évaluation principal de l'étude était la survie sans progression (SSP) évaluée par les critères d'évaluation de la réponse des tumeurs solides (critères RECIST V1.1), basée sur une évaluation radiologique indépendante en aveugle. Les critères d'évaluation secondaires de l'efficacité incluaient le taux de réponse objective (TRO), la survie globale (SG), le temps avant la progression de la tumeur (TPT), la sécurité et la tolérance du médicament ainsi que la qualité de vie liée à la santé (QdVLS).
Au moment de l'analyse principale, 229 patients ont été randomisés pour recevoir soit Lutathera CA (n = 116) soit la dose élevée de 60 mg de l'octréotide LP (n = 113). La randomisation était stratifiée par le score à la scintigraphie Octreoscan® (grades 2, 3 et 4) et la plus longue durée à dose constante d'octréotide reçue par le patient avant la randomisation (soit ≤6 ou > 6 mois). Les critères démographiques ainsi que les caractéristiques des patients et des pathologies étaient bien équilibrés entre les deux bras, avec un âge médian de 64 ans et un taux de 82,1% de patients blancs par rapport à la population totale.
Les résultats de l'analyse finale per protocole (date limite le 24 juillet 2015) sont présentés dans le tableau 7.
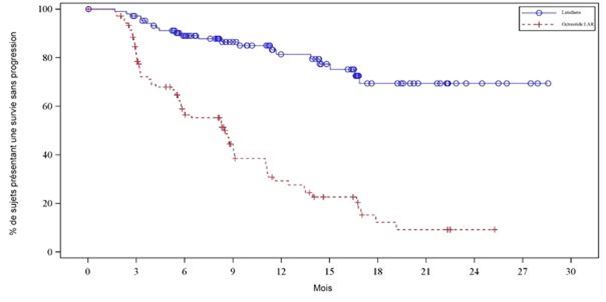
Tableau 7: SSP observée dans le cadre de l'étude NETTER-1 de phase III chez les patients atteints de tumeurs carcinoïdes progressives de l'intestin moyen – (ensemble d'analyse complet (EAC), n = 229)
|
|
Traitement
| |
|
Lutathera CA et octréotide LP
|
Dose élevée d'octréotide LP
| |
N
|
116
|
113
| |
Patients présentant des événements
|
21
|
70
| |
Patients exclus
|
95
|
43
| |
Médiane en mois (IC à 95%)
|
Non atteint
|
8,5 (5,8; 9,1)
| |
Valeur p du test du log-rank
|
< 0,0001
| |
Hazard ratio (IC à 95%)
|
0,177 (0,108; 0,289)
|
N = nombre de patients, IC = indice de confiance, LP: libération prolongée.
Le graphique de Kaplan-Meier de la SSP pour l'ensemble d'analyse complet (EAC) est présenté à la figure 3.
Figure 3: Courbes de Kaplan-Meier de la SSP pour les patients atteints de tumeurs carcinoïdes progressives de l'intestin moyen – (étude NETTER-1 de phase III; EAC, n = 229)
Les résultats de SG de l'analyse intermédiaire (date limite le 24 juillet 2015) et de l'analyse finale (date limite le 18 janvier 2021) sont présentés dans le tableau 8.
Au moment de l'analyse finale de la SG, qui a eu lieu 5 ans après la randomisation du dernier patient (n = 231, date limite le 24 juillet 2015), la durée médiane de suivi dans chaque bras de l'étude était de 76 mois. Les résultats finaux pour la SG n'ont pas atteint le niveau de significativité statistique.
Dans le bras recevant la dose élevée d'octréotide LP, 22,8% des patients avaient reçu un traitement subséquent par ligand radiomarqué (dont lutécium (177Lu) oxodotréotide) dans les 24 mois suivant la randomisation et 36% avant la date limite finale pour la SG, ce qui, avec d'autres facteurs, a pu avoir une influence sur la SG dans ce sous-groupe de patients.
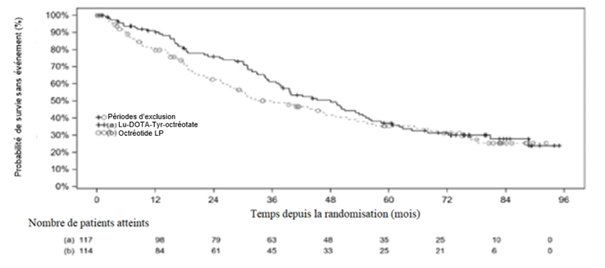
Tableau 8: Résultats de l'étude NETTER-1 de phase III pour la survie globale des patients atteints de tumeurs carcinoïdes progressives de l'intestin moyen (EAC)
|
|
LUTATHERA CA et octréotide LP
|
Dose élevée d'octréotide LP
| |
Analyse intermédiaire de la survie globale (24 juillet 2015) – N = 229*
| |
Décès (%)
|
17 (14,7%)
|
31 (27,4%)
| |
Médiane en mois (IC à 95%)
|
NA (NE; NE)
|
27,4 (20,1; NE)
| |
Hazard ratioa,b (IC à 99,9915%)
|
0,46 (0,14; 1,51)
| |
Analyse finale de la survie globale (18 janvier 2021) – N = 231**
| |
Décès (%)
|
73 (62,4%)
|
69 (60,5%)
| |
Médiane en mois (IC à 95%)
|
48,0 (37,4; 55,2)
|
36,3 (25,9; 51,7)
| |
Hazard ratioa,b,c (IC à 95%)
|
0,84 (0,60; 1,17)
| |
Analyse finale de la survie globale par temps de survie moyen restreint (RMST = Restricted Mean Survival Time) après 60 mois (18 janvier 2021) – N = 231**
| |
Décès (%)
|
65 (55,6)
|
63 (55,3)
| |
RMST (IC à 95%)
|
41,2 (37,6; 44,9)
|
36,1 (31,9; 40,4)
| |
Différence (IC à 95%)
|
5,1 (-0,5; 10,7)
| |
a:
Hazard ratio fondé sur un modèle de Cox non stratifié
b: Statistiquement non significatif selon les critères de significativité prédéfinis
c: HR fondé sur les risques non proportionnels
*: L'analyse a été réalisée chez 116 patients du groupe Lutathera CA et 113 patients du groupe recevant la dose élevée d'octréotide LP (N = 229).
**: L'analyse a été réalisée chez 117 patients du groupe Lutathera CA et 114 patients du groupe recevant la dose élevée d'octréotide LP (N = 231).
NA = non atteint
NE = non évaluable
|
La courbe de Kaplan-Meier de la SG pour l'ensemble d'analyse complet (EAC) à la date limite du 18 janvier 2021 est représentée à la figure 4.
Figure 4: Courbes de Kaplan-Meier de la SG pour les patients atteints de tumeurs carcinoïdes progressives de l'intestin moyen - date limite du 18 janvier 2021 (étude NETTER-1 de phase III; EAC, n = 231)
En présence de risques non proportionnels, au moment de l'analyse finale de la SG, une analyse supplémentaire de la sensibilité (Restricted Mean Survival Time) a été réalisée pour estimer l'effet du traitement de manière plus approfondie (voir tableau 8).
La qualité de vie liée à la santé (QdVLS) a été évaluée à l'aide du questionnaire de qualité de vie de l'Organisation européenne pour la recherche et le traitement du cancer (EORTC QLQ-C30) (instrument générique) et de son module tumeur neuroendocrine (EORTC QLQ-GI.NET-21).
Les résultats indiquent une amélioration de la qualité de vie globale liée à la santé jusqu'à la semaine 84 pour les patients du bras de traitement par Lutathera CA par rapport aux patients du bras de traitement par dose élevée d'octréotide LP.
Tumeurs neuroendocrines gastroentéropancréatiques (TNE-GEP) exprimant les récepteurs de la somatostatine
L'efficacité de Lutathera CA chez des patients atteints de tumeurs neuroendocrines (TNE) gastroentéropancréatiques (GEP) et bronchiques exprimant les récepteurs de la somatostatine a été évaluée dans l'étude clinique Erasmus monocentrique, non comparative, ouverte, dans laquelle le protocole thérapeutique de Lutathera CA consistait en 4 administrations par voie intraveineuse de 7400 MBq chacune, en association avec une solution d'acides aminés. Lutathera CA était initialement administré dans le cadre d'un programme compassionnel suivant un protocole général de radiothérapie à récepteur peptidique, mené dans un seul centre aux Pays-Bas. Un protocole ultérieur spécifique à Lutathera CA a été rédigé huit ans après l'initiation de ce programme, permettant une collecte rétrospective des données même en l'absence de description spécifique de l'effectif total et de l'hypothèse testée. Au total, 360 patients présentant à l'initiation du traitement une tumeur neuroendocrine gastroentéropancréatique et bronchique (de l'intestin moyen 183, du pancréas 133, des bronches 19, de l'intestin postérieur 13, de l'intestin antérieur autre que bronchique et pancréatique 12) ont été suivis à long terme. La moyenne d'âge était de 60 ans dont 51% d'hommes, 99,4% présentaient une fixation tumorale à l'Octreoscan ≥2 (5,6% (2)/62,8% (3)/31,1% (4)), 71,4% avaient un indice de Karnofsky ≥90 et 52% une médication concomitante avec des analogues de la somatostatine. Le taux de réponse objective déterminée par l'investigateur en tant que critère majeur d'efficacité, était de 45% (IC à 95%: 40; 50). La médiane de la durée de réponse était de 16,3 mois (IC à 95%: 12,2; 17,8). Le taux de réponse objective le plus élevé a été observé chez les patients atteints de tumeurs neuroendocrines pancréatiques (61%, IC à 95%: 52; 69) et le taux de réponse le plus faible chez les patients atteints de tumeurs neuroendocrines de l'intestin moyen (33%, IC à 95%: 27; 41).
PharmacocinétiqueAbsorption
Le médicament est administré par voie intraveineuse, il est donc immédiatement et complètement biodisponible.
Distribution
Afin de déterminer l'étendue de la fixation aux protéines plasmatiques du composé non radioactif (lutécium (175Lu) oxodotréotide), une analyse a été effectuée sur du plasma humain et a mis en évidence qu'environ 50% du composé est lié aux protéines plasmatiques.
Il n'a pas été observé de transchélation du lutécium à partir du lutécium (175Lu) oxodotréotide sur les protéines sériques.
Fixation aux organes
Dans les quatre heures suivant l'administration, la distribution du lutécium (177Lu) oxodotréotide montre une fixation rapide dans les reins, les lésions tumorales, le foie et la rate et, chez certains patients, dans l'hypophyse et la thyroïde. L'administration concomitante d'une solution d'acides aminés diminue la fixation rénale, favorisant ainsi l'élimination du produit radioactif (voir «Mises en garde et précautions»). Les études de biodistribution montrent que le lutécium (177Lu) oxodotréotide est rapidement éliminé de la circulation sanguine.
Métabolisme
Biotransformation
L'analyse d'échantillons d'urine de 20 patients inclus dans l'étude ancillaire de dosimétrie, pharmacocinétique et ECG de l'étude NETTER-1 de phase III a démontré que le lutécium (177Lu) oxodotréotide est peu métabolisé et est excrété principalement sous forme inchangée par les reins.
Les analyses HPLC des échantillons d'urine collectés jusqu'à 48 heures après la perfusion ont montré un lutécium (177Lu) oxodotréotide inchangé à presque 100% dans la plupart des échantillons analysés (la valeur la plus faible étant supérieure à 92%), indiquant que le produit est éliminé dans les urines sous forme inchangée majoritairement.
Ces résultats confirment ceux précédemment observés dans l'étude Erasmus de phase I/II, au cours de laquelle l'analyse HPLC des échantillons d'urine collectés sur un patient 1 heure après l'administration de 1,85 MBq de lutécium (177Lu) oxodotréotide a mis en évidence que la majeure partie (91%) était excrétée sous forme inchangée.
Ces résultats sont corroborés par des données in vitro du métabolisme dans les hépatocytes humains, dans lesquels aucune dégradation métabolique du lutécium (175Lu) oxodotréotide n'a été observée.
Élimination
Sur la base des données collectées lors des études Erasmus de phase I/II et NETTER-1 de phase III, l'élimination du lutécium (177Lu) oxodotréotide est principalement rénale: environ 60% du médicament sont éliminés dans les urines dans les 24 premières heures et environ 65% dans les 48 premières heures qui suivent l'administration.
Évaluation in vitro du potentiel d'interaction
L'absence d'inhibition ou d'induction significative des enzymes CYP450 humaines, l'absence d'interaction spécifique avec la glycoprotéine P (transporteur d'efflux) ainsi qu'avec les transporteurs OAT1, OAT3, OCT1, OCT2, OATP1B1, OATP1B3, et BCRP dans les études précliniques suggèrent que Lutathera/Lutathera CA présente une faible probabilité de causer d'autres interactions médicamenteuses significatives.
Données précliniquesLes études toxicologiques réalisées chez le rat ont démontré qu'une injection intraveineuse unique de jusqu'à 4550 MBq/kg était bien tolérée et aucun décès n'a été observé. Lorsqu'il a été testé en une seule injection intraveineuse sur des rats et des chiens avec des doses respectivement de 20 000 µg/kg (rats) et 3200 µg/kg (chiens), le composé froid (lutécium non radioactif (175Lu) oxodotréotide) a été bien toléré chez les deux espèces et aucun décès n'a été observé. Aucune toxicité n'a été observée lors de l'administration répétée de quatre doses de composé froid une fois toutes les 2 semaines, respectivement de 1250 µg/kg chez le rat et de 80 µg/kg chez le chien. Ce médicament n'est pas destiné à une administration régulière ou continue.
Les études de mutagénicité et de carcinogénicité à long terme n'ont pas été réalisées.
Des données précliniques obtenues avec le composé froid (lutécium non radioactif (175Lu) oxodotréotide) issues des études conventionnelles sur la pharmacologie de sécurité, la toxicité en administration répétée et la génotoxicité, n'ont pas révélé de risque particulier pour l'homme.
Remarques particulièresIncompatibilités
Ce médicament ne peut être mélangé qu'aux médicaments mentionnés sous la rubrique «Posologie/Mode d'emploi».
Stabilité
Ce médicament doit être conservé au maximum 72 heures à partir de la date et de l'heure de calibration.
Remarques particulières concernant le stockage
Ne pas conserver au-dessus de 25 °C. Ne pas congeler.
Conserver dans l'emballage d'origine pour protéger des radiations ionisantes (blindage de plomb).
Le médicament ne doit pas être utilisé au-delà de la date figurant après la mention «EXP» sur l'emballage.
La conservation des produits radiopharmaceutiques doit être effectuée conformément aux réglementations nationales relatives aux produits radioactifs.
Remarques concernant la manipulation/Radioprotection
L'utilisation de substances radioactives chez l'Homme est réglementée par l'Ordonnance sur la radioprotection. Une autorisation délivrée par l'Office fédéral de la santé publique est nécessaire pour pouvoir utiliser des substances radioactives.
Lutathera/Lutathera CA doit être réceptionné, utilisé et administré uniquement par des personnes autorisées dans des services agréés. La réception, le stockage, l'utilisation, le transfert et l'élimination de Lutathera/Lutathera CA sont soumis aux réglementations en matière de radioprotection et autorisations appropriées des autorités compétentes.
Lutathera/Lutathera CA doit être utilisé avec les mesures de sécurité appropriées pour minimiser l'exposition au rayonnement. Les précautions d'asepsie appropriées doivent être respectées.
La solution doit être inspectée visuellement avant utilisation pour vérifier l'absence de particules, de coloration et d'impuretés. Seules les solutions limpides et exemptes de particules peuvent être utilisées. Le flacon doit être éliminé si des particules/colorations/impuretés sont présentes Dans un but de radioprotection, l'inspection visuelle de la solution doit être effectuée derrière un écran blindé.
L'emballage doit être examiné quant aux dommages et la présence d'une contamination radioactive doit être établie au moyen d'un appareil de mesure de la radioactivité approprié. Si l'intégrité du flacon ou du récipient en plomb est compromise, le produit ne doit pas être utilisé. Lutathera/Lutathera CA ne doit pas être injecté directement dans une autre solution pour injection intraveineuse.
La quantité de radioactivité dans le flacon doit être mesurée à l'aide d'un activimètre étalonné avant et après chaque perfusion afin de confirmer que la quantité réelle de radioactivité qui sera administrée est identique à la quantité prévue au moment de la perfusion.
Les procédures d'administration doivent être effectuées de manière à minimiser le risque de contamination du médicament et l'irradiation des opérateurs. Un blindage adéquat est obligatoire et il est nécessaire de porter des gants imperméables lors de la manipulation du médicament.
L'administration des produits radiopharmaceutiques présente des risques pour l'entourage du patient en raison de l'irradiation externe ou de la contamination par les liquides corporels comme les urines, les selles, les vomissements, etc. Des mesures de protection adaptées contre les radiations doivent par conséquent être prises.
Il est conseillé au personnel de santé de limiter le temps de contact rapproché avec les patients ayant reçu Lutathera/Lutathera CA. L'utilisation d'écran de surveillance pour suivre les patients est recommandée. Étant donné la longue demi-vie du 177Lu, il est d'autant plus important d'éviter les contaminations internes. Il est obligatoire de porter des gants protecteurs de haute qualité (latex/nitrile) pour éviter tout contact direct avec le produit radiopharmaceutique (flacon/seringue). Afin de minimiser l'exposition aux radiations, les principes de temps, de distance et de protection doivent être toujours appliqués (en réduisant la manipulation du flacon et en utilisant le matériel fourni par le fabricant).
L'administration peut induire une exposition environnementale significative aux rayonnements.
Ceci pourrait concerner la famille proche des individus subissant le traitement ou la population générale. Par conséquent, les exigences de radioprotection doivent être respectées (voir la rubrique «Mises en garde et précautions»). Afin d'éviter toute contamination, des précautions adaptées concernant l'activité éliminée par les patients doivent être prises en conformité avec les réglementations nationales.
Elimination des déchets
Le produit radioactif non utilisé ou les déchets ne doivent être éliminés que conformément aux prescriptions suisses de radioprotection en vigueur.
Lutathera
Le lutécium-177 de Lutathera est préparé en utilisant l'isotope stable ytterbium-176 («sans porteur ajouté»).
Lutathera CA
Le lutécium-177 de Lutathera CA est préparé en utilisant l'isotope stable lutécium-176 («avec porteur ajouté») et nécessite une attention particulière pour l'élimination des déchets en raison de la présence de lutécium-177 métastable (177mLu).
Numéro d’autorisation66580, 69776 (Swissmedic)
PrésentationFlacon en verre incolore et transparent de type I, fermé par un bouchon en caoutchouc de bromobutyle et scellé par une capsule en aluminium.
Chaque flacon contient un volume variant de 20,5 à 25,0 ml de solution correspondant à une activité de 7400 MBq à la date et à l'heure de perfusion.
Le flacon est placé dans un emballage en plomb assurant le blindage.
Catégorie de remise A.
Titulaire de l’autorisationNovartis Pharma Schweiz AG, Risch; domicile 6343 Rotkreuz
Mise à jour de l’informationJuin 2024
|