CompositionPrincipes actifs
Sotatercept.
Excipients
Acidum citricum monohydricum (E330), trinatrii citras dihydricus (E331), polysorbatum 80 (E433), saccharum.
La solution reconstituée contient 0,55 mg de sodium par ml.
Indications/Possibilités d’emploiWinrevair est indiqué, en association avec un traitement standard de l'hypertension artérielle pulmonaire (HTAP), dans le traitement de l'HTAP chez les adultes en classe fonctionnelle (CF) II à III de l'OMS afin d'améliorer la capacité physique et de ralentir la progression de la maladie (voir «Efficacité clinique»).
L'efficacité a été montrée dans une population présentant une HTAP incluant les étiologies suivantes: HTAP idiopathique, HTAP héritable, HTAP associée à une connectivite, HTAP induite par des médicaments ou des toxines ou HTAP associée à une cardiopathie congénitale avec shunts réparés (voir «Efficacité clinique»).
Posologie/Mode d’emploiLe traitement par Winrevair doit être initié et supervisé par un médecin expérimenté dans le diagnostic et le traitement de l'HTAP.
Posologie
Dose initiale recommandée chez l'adulte
Winrevair est administré une fois toutes les 3 semaines sous forme d'injection sous-cutanée (s.c.). La dose à administrer dépend du poids corporel du patient. La dose initiale de Winrevair est de 0,3 mg/kg (voir Tableau 1).
Le taux d'hémoglobine (Hb) et le nombre de plaquettes doivent être déterminés avant la première dose de Winrevair. Chez les patients présentant un nombre de plaquettes <50 000/mm3 (<50,0 x 109/l), il ne faut pas initier de traitement par Winrevair (voir «Posologie/Mode d'emploi», «Ajustements posologiques chez l'adulte en raison d'une augmentation du taux d'hémoglobine ou d'une diminution du nombre de plaquettes»). Après le début du traitement, des augmentations rapides (de plus de 2 g/dl) du taux d'Hb ont été observées.
Tableau 1: Volume à injecter pour une dose de 0,3 mg/kg
|
Plage de poids corporel du patient (kg)
|
Volume à injecter (ml)
|
Type de kit
| |
30,0–40,8
|
0,2
|
Kit de 45 mg (avec 1 flacon de 45 mg)
| |
40,9–57,4
|
0,3
| |
57,5–74,1
|
0,4
| |
74,2–90,8
|
0,5
| |
90,9–107,4
|
0,6
| |
107,5–124,1
|
0,7
| |
124,2–140,8
|
0,8
| |
140,9–157,4
|
0,9
| |
157,5–174,1
|
1,0
|
Kit de 60 mg (avec 1 flacon de 60 mg)
| |
174,2–180,0
|
1,1
|
Posologie cible recommandée chez l'adulte
La dose cible de Winrevair est de 0,7 mg/kg (voir Tableau 2) toutes les 3 semaines.
Avant d'augmenter la dose cible, il convient de déterminer et de vérifier le taux d'hémoglobine (Hb) et le nombre de plaquettes. Le traitement doit être poursuivi à la dose de 0,7 mg/kg toutes les 3 semaines, sauf si des ajustements posologiques sont nécessaires (voir «Posologie/Mode d'emploi», «Ajustements posologiques chez l'adulte en raison d'une augmentation du taux d'hémoglobine ou d'une diminution du nombre de plaquettes»).
Tableau 2: Volume à injecter pour une dose de 0,7 mg/kg
|
Plage de poids corporel du patient (kg)
|
Volume à injecter (ml)
|
Type de kit
| |
30,0–31,7
|
0,4
|
Kit de 45 mg (avec 1 flacon de 45 mg)
| |
31,8–38,9
|
0,5
| |
39,0–46,0
|
0,6
| |
46,1–53,2
|
0,7
| |
53,3–60,3
|
0,8
| |
60,4–67,4
|
0,9
| |
67,5–74,6
|
1,0
|
Kit de 60 mg (avec 1 flacon de 60 mg)
| |
74,7–81,7
|
1,1
| |
81,8–88,9
|
1,2
| |
89,0–96,0
|
1,3
|
Kit de 90 mg (avec 2 flacons de 45 mg)
| |
96,1–103,2
|
1,4
| |
103,3–110,3
|
1,5
| |
110,4–117,4
|
1,6
| |
117,5–124,6
|
1,7
| |
124,7–131,7
|
1,8
| |
131,8–138,9
|
1,9
|
Kit de 120 mg (avec 2 flacons de 60 mg)
| |
139,0–146,0
|
2,0
| |
146,1–153,2
|
2,1
| |
153,3–160,3
|
2,2
| |
160,4–167,4
|
2,3
| |
167,5 et plus
|
2,4
|
Ajustements posologiques chez l'adulte en raison d'une augmentation du taux d'hémoglobine ou d'une diminution du nombre de plaquettes
Des augmentations de l'Hb supérieures à 2 g/dl au-dessus de la limite supérieure de la normale (LSN) et des diminutions du nombre de plaquettes à un niveau <50 000/mm3 (<50,0 x 109/l) ont été observées. Il convient de contrôler les taux d'Hb et le nombre de plaquettes avant chaque dose lors de l'administration des cinq premières doses ou plus longtemps si les taux ne sont pas stables. Ensuite, il faut contrôler régulièrement les taux d'Hb et le nombre de plaquettes. Afin de pouvoir décider si un ajustement de la dose est adapté, il faut prendre en compte le rapport bénéfice-risque pour chaque patient (voir «Mises en garde et précautions», «Érythrocytose», «Thrombopénie sévère»).
Si l'une des situations suivantes se présente, il faut repousser le traitement de trois semaines:
·Augmentation de l'Hb >2,0 g/dl depuis la dose précédente et taux d'Hb supérieur à la LSN
·Augmentation de l'Hb >4,0 g/dl par rapport à la valeur initiale
·Augmentation de l'Hb >2,0 g/dl au-dessus de la LSN
·Diminution du nombre de plaquettes à <50 000/mm3 (<50,0 x 109/l)
En cas d'interruption du traitement >9 semaines, il convient de le reprendre tout d'abord à la dose de 0,3 mg/kg.
Oubli d'une dose, surdosage et sous-dosage
En cas d'oubli d'une dose de Winrevair, celle-ci doit être administrée dès que possible. Si la dose de Winrevair oubliée n'est pas administrée dans les 3 jours suivant la date prévue, ajuster le schéma posologique afin de maintenir des intervalles de 3 semaines entre les administrations. En cas de surdosage ou de sous-dosage, il faudra envisager, le cas échéant, une nouvelle formation du patient ou de son aidant ou aidante au sujet de l'administration correcte du médicament. En cas de surdosage, il faut surveiller l'apparition d'une érythrocytose chez le patient (voir «Surdosage»).
Groupes de patients particuliers
Patients présentant des troubles de la fonction hépatique
Winrevair n'a pas été étudié chez les patients présentant des troubles de la fonction hépatique (classe Child-Pugh A à C). On suppose que les troubles de la fonction hépatique ne devraient pas influencer le métabolisme du sotatercept dans la mesure où il est métabolisé par catabolisme intracellulaire (voir «Pharmacocinétique»).
Patients présentant des troubles de la fonction rénale
Les troubles de la fonction rénale ne nécessitent aucun ajustement de la dose de Winrevair. Le sotatercept n'a pas été étudié chez des patients présentant une HTAP et des troubles graves de la fonction rénale (DFGe <30 ml/min/1,73 m2) (voir «Pharmacocinétique»).
Patients âgés
Aucun ajustement de la dose de Winrevair spécifique à l'âge n'est nécessaire (voir «Pharmacocinétique»).
Enfants et adolescents
La sécurité et l'efficacité de Winrevair n'ont pas été étudiées chez les patients de moins de 18 ans.
Mode d'administration
Winrevair doit être reconstitué avant utilisation et administré par injection sous-cutanée au niveau de l'abdomen (à une distance d'au moins 5 cm du nombril), du haut du bras ou de la cuisse.
Le kit Winrevair est destiné à être utilisé sous la supervision d'un professionnel de santé. Les patients et leurs aidants peuvent administrer Winrevair à condition d'être en mesure de le faire et de recevoir une formation sur la reconstitution, la préparation, la posologie et l'injection de Winrevair, puis l'aide d'un professionnel de santé. Des instructions détaillées sur la manière appropriée de préparer et d'administrer Winrevair sont fournies aux paragraphes «Remarques particulières/Remarques concernant la manipulation» et dans le Mode d'emploi du kit.
Lors des rendez-vous de suivi, il convient de vérifier si le patient ou son aidant peut préparer et administrer correctement Winrevair:
·en cas de modification de la posologie ou si le patient a besoin d'un autre kit
·si le patient commence à présenter une érythrocytose (voir «Mises en garde et précautions», «Érythrocytose»)
Choix du kit adapté de produits
Si, du fait de son poids corporel, un patient a besoin de deux flacons de 45 mg ou de deux flacons de 60 mg contenant la poudre lyophilisée, il faut utiliser un kit comprenant deux flacons et pas deux kits individuels comprenant chacun un flacon. Le kit comprenant 2 flacons contient des instructions pour combiner le contenu des deux flacons, ce qui permet de mesurer plus aisément la dose correcte. De plus, cela permet d'éviter de faire plusieurs injections (voir «Remarques particulières/Remarques concernant la manipulation»).
Afin d'assurer la traçabilité des médicaments issus de la biotechnologie, il est recommandé de consigner le nom du produit et le numéro de lot à chaque traitement.
Contre-indicationsHypersensibilité au principe actif ou à l'un des excipients mentionnés à la rubrique «Composition».
Mises en garde et précautionsÉrythrocytose
Chez les patients traités par Winrevair, on a observé des augmentations des taux d'Hb (>2 g/dl à <4 g/dl au-dessus de la LSN chez ~15% des participants à l'étude). Une érythrocytose sévère peut faire augmenter le risque d'événements thrombo-emboliques ou d'un syndrome d'hyperviscosité. Surveillez le taux d'Hb avant chaque dose lors de l'administration des cinq premières doses ou plus longtemps si les taux ne sont pas stables, puis effectuez des contrôles à intervalles réguliers pour déterminer si un ajustement de la dose est nécessaire (voir «Posologie/Mode d'emploi», «Ajustements posologiques chez l'adulte en raison d'une augmentation du taux d'hémoglobine ou d'une diminution du nombre de plaquettes» et «Effets indésirables»).
Thrombopénie sévère
Chez certains patients utilisant Winrevair, on a observé une diminution du nombre de plaquettes et une thrombopénie sévère (nombre de plaquettes <50 000/mm3 [<50,0 x 109/l]). Les thrombopénies sont survenues plus fréquemment chez les patients recevant également une perfusion de prostacycline.
Ne commencez pas le traitement si le nombre de plaquettes est <50 000/mm3 (<50 x 109/l) (voir «Posologie/Mode d'emploi»).
Surveillez le nombre de plaquettes avant chaque dose lors de l'administration des cinq premières doses ou plus longtemps si les taux ne sont pas stables, puis effectuez des contrôles à intervalles réguliers pour déterminer si un ajustement de la dose est nécessaire (voir «Posologie/Mode d'emploi», «Ajustements posologiques chez l'adulte en raison d'une augmentation du taux d'hémoglobine ou d'une diminution du nombre de plaquettes» et «Effets indésirables»).
Saignements graves
Dans les études cliniques, des événements hémorragiques graves (p.ex. hémorragies gastro-intestinales ou intracrâniennes) ont été signalés chez 4% des patients sous Winrevair et 1% des patients sous placebo. Les patients présentant des événements hémorragiques graves étaient plus susceptibles d'être sous traitement de fond avec une prostacycline et/ou sous antithrombotiques ou d'avoir un nombre de plaquettes bas. Veuillez informer les patients au sujet des signes et des symptômes de saignements. Veuillez évaluer et traiter les saignements de manière appropriée. N'administrez pas Winrevair si le patient souffre d'un événement hémorragique grave (voir «Mises en garde et précautions/Thrombopénie sévère» et «Effets indésirables»).
Toxicité embryofœtale
Compte tenu des résultats des études de reproduction chez l'animal, il est possible que Winrevair cause des dommages au fœtus en cas d'utilisation lors de la grossesse. Il faut expliquer le risque pour le fœtus aux femmes enceintes. Il faut donner des instructions aux femmes en âge de procréer afin qu'elles utilisent une méthode de contraception efficace pendant le traitement par Winrevair et pendant au moins quatre mois après la prise de la dernière dose (voir «Grossesse, Allaitement», «Grossesse» et «Données précliniques/Toxicité sur la reproduction»).
Altération de la fertilité
Compte tenu des résultats des études expérimentales chez l'animal, il est possible que Winrevair altère la fertilité des femmes et des hommes. Il faut expliquer les effets potentiels sur la fertilité aux patients (voir «Grossesse, Allaitement/Fertilité» et «Données précliniques/Toxicité sur la reproduction»).
Sodium
Ce médicament contient moins de 1 mmol (23 mg) de sodium par dose, c'est-à-dire qu'il est essentiellement «sans sodium».
InteractionsAucune étude d'interaction n'a été menée.
Grossesse, allaitementFemmes en âge de procréer/Contraception chez les femmes
Chez les femmes en âge de procréer, il est recommandé d'effectuer un test de grossesse avant le traitement par Winrevair. Pendant le traitement par Winrevair, les femmes en âge de procréer doivent utiliser une méthode de contraception efficace pendant ≥ quatre mois après la prise de la dernière dose (fin du traitement) (voir «Données précliniques»).
Grossesse
Il n'existe pas de données sur l'utilisation du sotatercept chez la femme enceinte. Les études expérimentales chez l'animal ont montré une toxicité sur la reproduction (voir «Données précliniques»).
L'utilisation de Winrevair n'est pas recommandée pendant la grossesse, ainsi que chez les femmes en âge de procréer qui n'utilisent pas de contraception.
Considérations cliniques
Chez les femmes enceintes présentant une HTAP, il y a un risque d'insuffisance cardiaque, d'accouchement prématuré et de décès de la mère ou du fœtus.
Allaitement
On ignore si le sotatercept ou ses métabolites passent dans le lait maternel. Il n'est pas possible d'exclure un risque pour le nouveau-né ou le nourrisson.
Il est recommandé d'interrompre l'allaitement au cours du traitement par Winrevair et pendant 4 mois après la dernière dose du traitement.
Fertilité
Compte tenu des résultats des études expérimentales chez l'animal, il est possible que le sotatercept altère la fertilité des femmes et des hommes (voir «Données précliniques»).
Effet sur l’aptitude à la conduite et l’utilisation de machinesLe sotatercept n'a aucune influence ou a une influence négligeable sur l'aptitude à la conduite ou l'utilisation de machines.
Effets indésirablesRésumé du profil de sécurité
La sécurité de Winrevair a été étudiée dans l'étude pivot STELLAR. Il s'agissait d'une étude contrôlée contre placebo sur le long terme et incluant 323 patients souffrant d'HTAP (voir «Propriétés/Effets/Efficacité clinique»). Après la fin de la phase de traitement de 24 semaines, les patients ont poursuivi leur traitement dans une étude à long terme en double aveugle tout en conservant leur traitement en cours et jusqu'à ce que tous les patients aient terminé la première phase de traitement. La durée médiane du traitement par Winrevair était de 252 jours. L'incidence totale des interruptions de traitement en raison d'effets indésirables était de 4% dans le groupe Winrevair et de 7% dans le groupe placebo. Il n'y a pas eu d'effets indésirables spécifiques menant à l'interruption du traitement qui sont survenus plus fréquemment et à une fréquence supérieure à 1% dans le groupe Winrevair.
Parmi les effets indésirables observés, les événements graves sont survenus de manière occasionnelle (<1,0%) (voir «Description d'effets indésirables sélectionnés»). Les effets indésirables les plus fréquemment signalés étaient les céphalées (24,5%), l'épistaxis (22,1%) et les télangiectasies (16,6%), les diarrhées (15,3%), les étourdissements (14,7%), les éruptions (12,3%) et la thrombopénie (10,4%).
Liste des effets indésirables
Les effets indésirables associés à Winrevair qui ont été observés dans l'étude STELLAR sont présentés dans le tableau ci-dessous par classe de systèmes d'organes selon la classification MedDRA et par fréquence. Les fréquences sont définies comme suit: très fréquents (≥1/10), fréquents (≥1/100, <1/10), occasionnels (≥1/1000, <1/100), rares (≥1/10'000, <1/1000), très rares (<1/10'000).
Tableau 3: Effets indésirables
|
Classes de systèmes d’organes
|
Fréquence
|
Effet indésirable
| |
Affections hématologiques et du système lymphatique
|
Très fréquents
|
Thrombopénie1,2
| |
Fréquents
|
Hémoglobine augmentée1,2
| |
Affections du système nerveux
|
Très fréquents
|
Étourdissement
Céphalées
| |
Affections respiratoires, thoraciques et médiastinales
|
Très fréquents
|
Épistaxis
| |
Affections gastro-intestinales
|
Très fréquents
|
Diarrhées
| |
Affections de la peau et du tissu sous-cutané
|
Très fréquents
|
Télangiectasie2
Éruption3
| |
Fréquents
|
Érythème3
| |
Investigations
|
Fréquents
|
Pression artérielle augmentée2
|
1 Voir «Mises en garde et précautions»
2 Voir «Description d'effets indésirables sélectionnés»
3 Termes de haut niveau de MedDRA (HLT, High Level Terms)
Description d'effets indésirables spécifiques et informations complémentaires
Les réactions indésirables cliniquement significatives suivantes sont décrites à un autre endroit de l'information professionnelle:
·Érythrocytose (voir «Mises en garde et précautions/Érythrocytose»)
·Thrombopénie sévère (voir «Mises en garde et précautions/Thrombopénie sévère»)
·Saignements graves (voir «Mises en garde et précautions/Saignements graves»)
·Toxicité embryo-fœtale (voir «Mises en garde et précautions/Toxicité embryo-fœtale»)
·Altération de la fertilité (voir «Mises en garde et précautions/Altération de la fertilité»)
Thrombopénie
La plupart des événements de thrombopénie (thrombopénie et numération plaquettaire diminuée) étaient non graves, légers, réversibles et ne s'accompagnaient pas d'une interruption du traitement. Une forte diminution du nombre de plaquettes (<50'000/mm3 [<50,0 x 109/l]) est survenue chez 1,8% des patients sous Winrevair.
Télangiectasie
Les événements de télangiectasie étaient non graves et leur gravité n'a pas augmenté au fil du temps. Chez tous les patients sous Winrevair, la durée médiane jusqu'à leur survenue était de 47,1 semaines. Une interruption du traitement due à une télangiectasie a eu lieu chez 1% des patients du groupe Winrevair et 0% des patients du groupe placebo. La télangiectasie n'a pas été associée à des épisodes hémorragiques graves.
Pression artérielle augmentée
Les événements de pression artérielle augmentée (hypertension, pression artérielle diastolique augmentée, pression artérielle augmentée) n'étaient pas graves et aucun événement sévère n'a été signalé. Chez les patients ayant reçu Winrevair, la pression artérielle systolique moyenne au bout de 24 semaines avait augmenté de 2,2 mmHg et la pression artérielle diastolique de 4,9 mmHg par rapport à la valeur initiale. Chez les patients sous placebo, la pression artérielle systolique moyenne avait diminué de 1,6 mmHg et la pression artérielle diastolique de 0,6 mmHg par rapport à la valeur initiale.
Données de sécurité à long terme
Les données de sécurité à long terme sont issues d'une étude clinique de phase II (PULSAR), qui comprenait une phase de traitement en double aveugle et contrôlée contre placebo de 24 semaines, puis une phase de prolongation en ouvert de 30 mois (n = 104). Pour la plupart, ces patients ont ensuite participé à une étude de suivi à long terme.
Dans l'étude PULSAR et dans l'étude de suivi à long terme, la durée moyenne de l'exposition à Winrevair était de 151 semaines et l'exposition maximale de 218 semaines. Le profil de sécurité était généralement comparable à celui observé dans l'étude pivot STELLAR. Toutefois, dans la phase de traitement de PULSAR en double aveugle et contrôlée contre placebo, aucune télangiectasie n'a été signalée. La télangiectasie a été signalée pour la première fois lors de la phase de prolongation en ouvert, et ce chez 27% des patients à l'issue de l'étude. Ainsi, la durée médiane jusqu'à sa survenue était de 106 semaines.
Immunogénicité
La fréquence d'anticorps anti-médicament observée dépend fortement de la sensibilité et de la spécificité du test. Du fait des différences entre les méthodes de test, il est impossible de comparer de manière pertinente la fréquence des anticorps anti-médicaments dans l'étude décrite ci-dessous et la fréquence des anticorps anti-médicaments dans d'autres études. Cela est aussi valable pour les études sur Winrevair ou d'autres produits contenant du sotatercept.
Lors de la phase de traitement de 24 semaines de l'étude pivot (STELLAR), 44/163 (27%) patients traités par sotatercept ont développé des anticorps contre ce médicament. Sur ces 44 patients, 12 (27%) ont obtenu un résultat positif au test des anticorps neutralisants contre le sotatercept. Le titre médian des anticorps dirigés contre le sotatercept était de 30 (intervalle <20 à 640), c'est-à-dire qu'il était généralement faible.
Pendant la phase de traitement de 24 semaines, aucun effet clinique des anticorps dirigés contre le sotatercept n'a été constaté sur la pharmacocinétique, la pharmacodynamique, la sécurité et l'efficacité du sotatercept.
Patients âgés
Hormis pour les événements hémorragiques (un groupe combiné d'événements indésirables d'intérêt clinique), il n'y a pas eu de différences concernant la sécurité entre les patients <65 ans et les patients ≥65 ans. Les événements hémorragiques étaient plus fréquents sous Winrevair dans le sous-groupe plus âgé. Toutefois, il n'y avait pas de déséquilibre notable par rapport à certains événements hémorragiques entre les différents sous-groupes d'âge.
L'annonce d'effets secondaires présumés après l'autorisation est d'une grande importance. Elle permet un suivi continu du rapport bénéfice-risque du médicament. Les professionnels de santé sont tenus de déclarer toute suspicion d'effet secondaire nouveau ou grave via le portail d'annonce en ligne ElViS (Electronic Vigilance System). Vous trouverez des informations à ce sujet sur www.swissmedic.ch.
SurdosageChez les sujets sains, l'administration de Winrevair à la dose de 1 mg/kg a entraîné une augmentation d'Hb associée à une hypertension; ces deux phénomènes se sont améliorés après une phlébotomie. En cas de surdosage, il convient de surveiller étroitement le patient à la recherche d'une augmentation du taux d'hémoglobine et de la pression artérielle et, le cas échéant, de mettre en place des mesures de soutien. Winrevair n'est pas dialysable par hémodialyse.
Propriétés/EffetsCode ATC
C02KX06
Mécanisme d'action
Le sotatercept, protéine de fusion recombinante du récepteur de l'activine de type IIA-Fc (ActRIIA-Fc), est un inhibiteur de la voie de signalisation de l'activine, qui se lie à l'activine A et d'autres ligands de la superfamille des TGF-β. En conséquence, le sotatercept améliore l'équilibre entre la signalisation pro-proliférative (médiée par ActRIIA/Smad2/3) et antiproliférative (médiée par BMPRII/Smad1/5/8) pour moduler la prolifération vasculaire.
Pharmacodynamique
Une étude clinique de phase II a évalué les résistances vasculaires pulmonaires (RVP) chez des patients atteints d'HTAP après 24 semaines de traitement par le sotatercept. La diminution des RVP par rapport à l'inclusion était significativement plus importante dans les groupes sotatercept 0,7 mg/kg et 0,3 mg/kg que dans le groupe placebo. La différence moyenne des moindres carrés (MC) ajustée au placebo par rapport à l'inclusion était de -269,4 dynes•s/cm5 (IC à 95%: -365,8; -173,0) pour le groupe sotatercept 0,7 mg/kg et de -151,1 dynes•s/cm5 (IC à 95%: -249,6; -52,6) pour le groupe sotatercept 0,3 mg/kg. Dans l'étude STELLAR, la diminution des RVP par rapport à l'inclusion était également significativement plus importante dans le groupe sotatercept 0,7 mg/kg que dans le groupe placebo (voir «Efficacité clinique»).
Dans des modèles d'HTAP chez le rat, un analogue du sotatercept a réduit l'expression des marqueurs pro-inflammatoires au niveau de la paroi artérielle pulmonaire, a réduit le recrutement de leucocytes, a inhibé la prolifération des cellules endothéliales et des cellules musculaires lisses et a favorisé leur apoptose dans le système vasculaire affecté. Ces modifications cellulaires étaient associées à un amincissement de la paroi des vaisseaux, à une inversion du remodelage artériel et ventriculaire droit et à une amélioration de l'hémodynamique.
Efficacité clinique
L'efficacité de Winrevair a été évaluée chez des patients adultes atteints d'HTAP dans l'étude STELLAR. STELLAR était une étude clinique mondiale, en double aveugle, contrôlée contre placebo, multicentrique et en groupes parallèles, dans laquelle 323 adultes atteints d'HTAP (groupe 1 de l'OMS, classe fonctionnelle II ou III) ont été randomisés selon un rapport de 1/1 pour recevoir Winrevair (dose cible de 0,7 mg/kg) (n = 163) ou le placebo (n = 160) administré par voie sous-cutanée une fois toutes les 3 semaines.
Les caractéristiques initiales démographiques et cliniques étaient généralement comparables entre le groupe Winrevair et le groupe placebo. Les personnes participant à l'étude étaient des adultes d'un âge médian de 48,0 ans (intervalle:18 à 82 ans); leur poids médian était de 68 kg (intervalle: 38,0 à 141,3 kg); 89,2% de ces personnes étaient blanches et 79,3% n'étaient ni hispaniques, ni latino-américaines; 79,3% étaient des femmes. Les étiologies les plus fréquentes de l'HTAP étaient: HTAP idiopathique (58,5%), HTAP héritable (18,3%) et HTAP associée à une connectivite (14,9%). La durée médiane entre le diagnostic d'HTAP et la sélection était de 8,76 ans. La plupart des participants recevaient un traitement de fond en trithérapie (61,3%) ou en bithérapie (34,7%). Plus d'un tiers (39,9%) recevaient des perfusions de prostacycline. Le pourcentage de participants ayant une HTAP et se trouvant en classe fonctionnelle II de l'OMS (48,6%) ou en classe fonctionnelle III de l'OMS (51,4%) était similaire dans les deux groupes. L'étude STELLAR a exclu les patients avec un diagnostic d'HTAP associée au virus de l'immunodéficience humaine (VIH), d'HTAP associée à une hypertension portale, d'HTAP associée à une schistosomiase et de maladie veino-occlusive pulmonaire.
Le critère principal d'évaluation de l'efficacité était la comparaison entre la distance parcourue au test de marche de 6 minutes (6MWD) à l'inclusion et à la semaine 24. Dans le groupe traité par Winrevair, la variation médiane de la distance parcourue au 6MWD ajustée au placebo entre l'inclusion et la semaine 24 était de 40,8 mètres (IC à 95%: 27,5; 54,1; p<0,001). La variation médiane de la distance parcourue au 6MWD ajustée au placebo entre l'inclusion et la semaine 24 a également été évaluée dans des sous-groupes (voir Figure 1).
Figure 1: Variation de la distance parcourue au test de marche de 6 minutes (en mètres) entre l'inclusion et la semaine 24 dans les sous-groupes
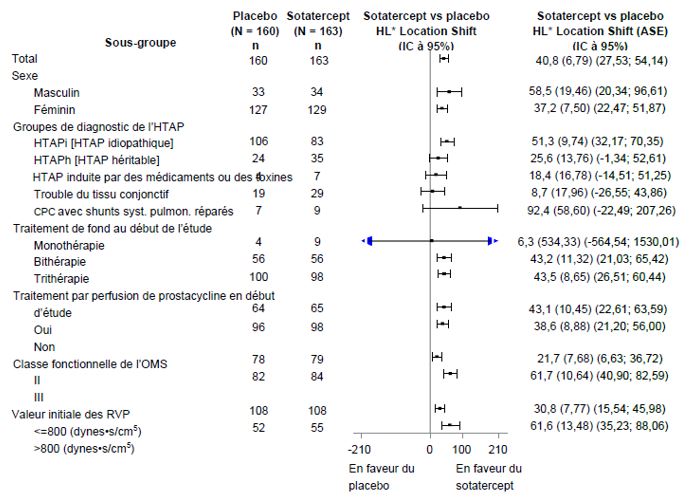
CPC = cardiopathie congénitale
* Estimation du changement de localisation de Hodges-Lehmann par rapport au placebo (médiane de toutes les différences appariées). ASE= asymptomatic standard error (erreur type asymptomatique). Pour les participants qui sont décédés, une valeur allant jusqu’à -2000 mètres a été attribuée à la variation de la distance parcourue au 6MWD à la semaine 24 par rapport à la valeur initiale, afin d’obtenir le plus mauvais rang. Pour les participants dont les données sont manquantes en raison d’une aggravation clinique non fatale, une valeur allant jusqu’à -1000 mètres a été attribuée à la variation de la distance parcourue au 6MWD à la semaine 24 par rapport à la valeur initiale, afin d’obtenir le deuxième plus mauvais rang.
L'amélioration clinique était un critère d'évaluation prédéfini, qui a été mesuré grâce au pourcentage de patients qui remplissaient les trois critères suivants à la semaine 24 par rapport à la situation initiale: amélioration de la distance parcourue au 6MWD (augmentation ≥30 m), amélioration du taux de peptides natriurétiques de type pro-B N-terminal (NT-proBNP) (réduction du NT-proBNP de ≥30% ou maintien/atteinte d'un taux de NT-proBNP <300 ng/l, et amélioration de la classe fonctionnelle de l'OMS ou maintien en classe fonctionnelle II. La progression de la maladie a été mesurée en tant que délai jusqu'au décès ou jusqu'à la survenue du premier événement d'aggravation clinique. Événements d'aggravation clinique: inscription sur liste d'attente de transplantation pulmonaire et/ou cardiaque liée à une aggravation, nécessité de débuter un traitement de secours par un traitement de référence de l'HTAP approuvé ou nécessité d'augmenter la dose de la perfusion de prostacycline de ≥10%, nécessité d'une septostomie atriale, hospitalisation liée à une aggravation de l'HTAP (≥24 h), ou aggravation de l'HTAP (dégradation de la classe fonctionnelle de l'OMS et diminution de la distance parcourue au 6MWD de ≥15% par rapport à l'inclusion, ces deux événements pouvant se produire simultanément ou à des moments différents). Les événements d'aggravation clinique et les cas de décès ont été recensés jusqu'à ce que le dernier patient termine les visites en semaines 24 (données obtenues jusqu'à la date limite; durée médiane d'exposition de 33,6 semaines).
Chez les patients traités par Winrevair, par rapport aux patients du groupe placebo, on a observé une amélioration clinique statistiquement significative, une amélioration de la classe fonctionnelle de l'OMS et une progression retardée de la maladie comprenant un risque moindre de décès et d'hospitalisation (voir Tableau 4 et Figure 2).
En semaine 24, 38,9% des patients sous sotatercept ont présenté une amélioration du MCI contre 10,1% dans le groupe placebo (p<0,001). La différence médiane de traitement des RVP entre le groupe sotatercept et le groupe placebo était de -234,6 dynes*s/cm5 (IC à 95%: -288,4; -180,8; p<0,001). La différence médiane de traitement des NT-proBNP entre le groupe sotatercept et le groupe placebo était de -441,6 pg/ml (IC à 95%: -573,54; -309,61; p<0,001). Une amélioration de la classe fonctionnelle par rapport à la valeur initiale est survenue chez 29% des patients du groupe sotatercept contre 13,8% dans le groupe placebo (p<0,001).
Tableau 4: Décès ou événements d'aggravation clinique
|
|
Placebo
(N = 160)
|
Winrevair
(N = 163)
| |
Nombre total de patients qui sont décédés ou qui ont souffert d'au moins un événement d'aggravation clinique, n (%)
|
42 (26,3)
|
9 (5,5)
| |
Évaluation du décès ou de la survenue du premier événement d'aggravation clinique*, n (%)
|
|
| |
Décès
|
6 (3,8)
|
2 (1,2)
| |
Inscription sur liste d'attente de transplantation pulmonaire et/ou cardiaque liée
à une aggravation
|
1 (0,6)
|
1 (0,6)
| |
Nécessité de débuter un traitement de secours par un traitement de référence
de l'HTAP approuvé ou nécessité d'augmenter la dose de la perfusion de
prostacycline de ≥10%
|
17 (10,6)
|
2 (1,2)
| |
Nécessité d'une septostomie atriale
|
0 (0,0)
|
0 (0,0)
| |
Hospitalisation spécifiquement liée à l'HTAP (≥24 h)
|
7 (4,4)
|
0 (0,0)
| |
Aggravation de l'HTAP†
|
15 (9,4)
|
4 (2,5)
| |
* Pour un même patient, plus d'une évaluation peut être documentée pour le premier événement d'aggravation clinique. Chez 3 patients sous placebo et 0 patient sous sotatercept, plus d'une évaluation a été documentée pour leur premier événement d'aggravation clinique.
† La détérioration de l'HTAP est définie par la survenue des deux événements suivants à tout moment, même s'ils se sont produits à des moments différents, par rapport à leurs valeurs à l'inclusion: (a) aggravation de la classe fonctionnelle de l'OMS (II à III, III à IV, II à IV, etc.); et (b) diminution de la distance parcourue au 6MWD de ≥15% (confirmée par deux tests de marche de 6 minutes à au moins 4 heures d'intervalle, mais sans dépasser une semaine).
N = nombre de patients dans la population FAS; n = nombre de patients dans la catégorie. Les pourcentages sont calculés sous forme de (n/N)•100.
|
Figure 2: Courbe de Kaplan-Meier du délai jusqu'au décès ou jusqu'à la survenue du premier événement d'aggravation clinique
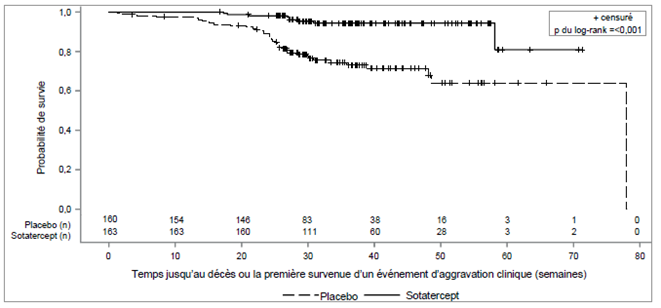
n = nombre de sujets à risque
PharmacocinétiqueChez les patients atteints d'HTAP, la moyenne géométrique (coefficient de variation en % [CV %]) de l'AUC à l'état d'équilibre et de la concentration maximale (Cmax) à l'état d'équilibre, à la dose de 0,7 mg/kg toutes les 3 semaines (Q3S), était respectivement de 171,3 μg•d/ml (34,2%) et 9,7 μg/ml (CV de 30%). Après l'administration de doses uniques de 0,1 mg/kg à 1,0 mg/kg par voie sous-cutanée, l'AUC et la Cmax du sotatercept augmentent de manière proportionnelle à la dose. L'état d'équilibre est atteint après environ 15 semaines en cas d'administrations multiples Q3S. Le rapport d'accumulation de l'AUC du sotatercept est d'environ 2,2.
Absorption
La formulation sous-cutanée a une biodisponibilité absolue d'environ 66%. La concentration maximale du sotatercept est atteinte en un temps médian (Tmax) d'environ 7 jours (intervalle de 2 à 8 jours) après des doses sous-cutanées multiples (0,1 mg/kg toutes les 4 semaines) chez les femmes ménopausées.
Distribution
Le volume de distribution centrale (CV %) du sotatercept est d'environ 3,6 l (24,7%). Le volume de distribution périphérique (CV %) est d'environ 1,7 l (73,3%).
Métabolisme
Le sotatercept est catabolisé par des processus généraux de dégradation des protéines.
Élimination
La clairance du sotatercept est d'environ 0,18 l/jour. La moyenne géométrique de la demi-vie terminale (CV %) est d'environ 21 jours (33,8%).
Cinétique pour certains groupes de patients
Aucune différence cliniquement significative de la pharmacocinétique (PK) du sotatercept n'a été observée en fonction de l'âge (18 à 81 ans), du sexe ou de l'origine ethnique.
La clairance (CL) et le volume de distribution centrale (Vc) du sotatercept ont augmenté avec le poids corporel. Le schéma posologique recommandé en fonction du poids entraîne des expositions cohérentes du sotatercept, quel que soit le poids corporel.
Troubles de la fonction hépatique
Les troubles de la fonction hépatique (déterminés par la classification de Child-Pugh) ne devraient pas influencer le métabolisme du sotatercept puisqu'il est métabolisé par catabolisme intracellulaire. Le sotatercept n'a pas été étudié chez les patients atteints d'HTAP présentant des troubles de la fonction hépatique (classe Child-Pugh A à C).
Troubles de la fonction rénale
La PK du sotatercept était comparable chez les patients atteints d'HTAP présentant des troubles légers à modérés de la fonction rénale (DFGe compris entre 30 et 89 ml/min/1,73 m2) et ceux ayant une fonction rénale normale (DFGe ≥90 ml/min/1,73 m2). De plus, la PK du sotatercept était comparable entre les patients atteints d'insuffisance rénale terminale (IRT) sans HTAP et les patients présentant une fonction rénale normale.
Winrevair n'est pas dialysable par hémodialyse. Aucun ajustement posologique n'est recommandé chez les patients présentant des troubles de la fonction rénale. Le sotatercept n'a pas été étudié chez les patients atteints d'HTAP présentant des troubles sévères de la fonction rénale (DFGe <30 ml/min/1,73 m2).
Données précliniquesToxicité en cas d'administration répétée
Chez les rats et les singes, les études de toxicité par voie sous-cutanée les plus longues ont duré respectivement 3 mois et 9 mois. Chez les rats recevant des doses hebdomadaires de 0,3, 3 et 30 mg/kg pendant 3 mois, les effets indésirables observés étaient une dégénérescence du canal déférent/testiculaire, une congestion/nécrose de la glande surrénale ainsi qu'une glomérulonéphrite membranoproliférative et une néphrite tubulo-interstitielle au niveau des reins. Les modifications surrénales et rénales étaient partiellement réversibles après une période de récupération d'un mois. Chez des singes recevant des doses de 1, 2,6 et 10 mg/kg une fois toutes les 4 semaines et de 10 mg/kg une fois toutes les 2 semaines, les effets toxiques comprenaient une glomérulonéphrite et une néphrite tubulo-interstitielle au niveau des reins. Les modifications rénales chez les singes étaient partiellement réversibles après une période de récupération de 3 mois. Chez les singes, des infiltrats inflammatoires dans le plexus choroïde sont apparus en cas d'exposition clinique. À la dose sans effet nocif observé (NOAEL) chez les rats et les singes, les expositions au sotatercept étaient ≤2 fois l'exposition clinique à la dose humaine maximale recommandée (MRHD).
Génotoxicité et carcinogénicité
Aucune étude de carcinogénicité ou de mutagénicité n'a été réalisée sur le sotatercept.
Toxicité sur la reproduction
Dans une étude de fertilité et de développement embryonnaire précoce chez des rates, le sotatercept a été administré par voie sous-cutanée une fois par semaine à des doses de 5, 15 et 50 mg/kg, en commençant 2 semaines avant l'accouplement jusqu'au jour 7 de la gestation. Aux doses ≥15 mg/kg (exposition ≥9 fois la MRHD, sur la base de l'AUC estimée), les taux de gestation étaient diminués et des augmentations des pertes pré-implantatoires et post-implantatoires, ainsi que des réductions de la taille des portées vivantes, ont été observées. L'augmentation de la durée du cycle a été observée à la dose de 50 mg/kg uniquement (21 fois la MRHD, sur la base de l'AUC estimée).
Dans une étude de fertilité chez les rats, le sotatercept a été administré par voie sous-cutanée une fois par semaine à des doses de 0,3, 3 et 30 mg/kg pendant 13 semaines (en commençant 10 semaines avant l'accouplement). Un sous-groupe d'animaux a été examiné après une période de récupération de 13 semaines. À une dose ≥0,3 mg/kg (0,5 fois la MRHD, sur la base de l'AUC estimée), des modifications histologiques irréversibles ont été observées au niveau des canaux déférents, des testicules et des épididymes. Une diminution réversible de la fertilité a été observée à la dose de 30 mg/kg (20 fois la MRHD, sur la base de l'AUC estimée).
Dans les études de toxicité sur le développement embryo-fœtal, le sotatercept a été administré par voie sous-cutanée à des animaux gravides pendant la période d'organogenèse. Le sotatercept a été administré à des rates aux jours 6 et 13 de la gestation à des doses de 5, 15 ou 50 mg/kg et à des lapines aux jours 7 et 14 de la gestation à des doses de 0,5, 1,5 ou 5 mg/kg. Les effets observés chez les deux espèces ont été notamment des réductions des nombres de fœtus vivants et des poids corporels des fœtus, des retards d'ossification et des augmentations des résorptions et des pertes post-implantatoires. Chez les rates et les lapines, ces effets ont été observés à des expositions (sur la base de l'aire sous la courbe [AUC]) d'environ 4 fois et 0,6 fois la MRHD respectivement. Chez les rates uniquement, des variations squelettiques (augmentation du nombre de côtes surnuméraires et modifications du nombre de vertèbres thoraciques ou lombaires) sont survenues à une exposition 15 fois supérieure à l'exposition humaine à la MRHD.
Dans une étude de développement prénatal et postnatal chez les rates, le sotatercept a été administré par voie sous-cutanée à des doses de 1,5 et 5 mg/kg aux jours 6 et 13 de la gestation ou à des doses de 1,5, 5 ou 10 mg/kg pendant l'allaitement aux jours 1, 8 et 15. Aucun effet indésirable n'a été observé chez les petits de la première génération (F1) dont les mères étaient traitées par le sotatercept pendant la gestation à des expositions estimées jusqu'à 2 fois la MRHD. Chez les petits F1 dont les mères étaient traitées pendant l'allaitement, les diminutions du poids des petits étaient corrélées à des retards de maturation sexuelle à des expositions estimées (sur la base de l'AUC) à ≥2 fois la MRHD.
Remarques particulièresIncompatibilités
Ce médicament ne peut être mélangé qu'aux médicaments mentionnés sous «Remarques concernant la manipulation».
Stabilité
Le médicament ne doit pas être utilisé au-delà de la date figurant après la mention «EXP» sur le récipient.
Pour des raisons microbiologiques, le médicament doit être utilisé immédiatement ou dans un délai de 4 heures après reconstitution.
Si le médicament n'est pas utilisé immédiatement, le délai d'utilisation et les conditions de stockage avant l'utilisation relèvent de la responsabilité de l'utilisateur.
Remarques particulières concernant le stockage
Conserver au réfrigérateur (2–8°C).
Ne pas congeler.
Conserver dans l'emballage d'origine à l'abri de la lumière.
Pour connaître les conditions de conservation après la reconstitution du médicament, voir «Stabilité».
Conserver hors de portée des enfants.
Remarques concernant la manipulation
Présentation sous forme de kit
Veuillez consulter le mode d'emploi pour obtenir des conseils détaillés sur la manière appropriée de préparer et d'administrer Winrevair.
Remarques concernant la reconstitution
·Sortez le kit d'injection du réfrigérateur et attendez 15 minutes pour que la ou les seringues préremplies et le médicament arrivent à température ambiante avant la préparation.
·Sur le flacon, vérifiez que le produit n'est pas périmé. La poudre doit être blanche à blanc cassé et peut ressembler à un bloc entier ou en morceaux.
·Retirez le capuchon du flacon de Winrevair contenant la poudre lyophilisée et nettoyez le bouchon en caoutchouc avec un tampon alcoolisé.
·Placez l'adaptateur pour flacons sur le flacon.
·Vérifiez si la seringue préremplie est endommagée ou présente des fuites et qu'il n'y a pas de particules visibles dans l'eau stérile qu'elle contient.
·Cassez le bouchon de la seringue préremplie et connectez la seringue à l'adaptateur pour flacons.
·Injectez toute l'eau stérile de la seringue fixée au flacon dans le flacon contenant la poudre lyophilisée. La concentration finale obtenue est de 50 mg/ml. Les flacons contiennent un volume excédentaire de sotatercept pour obtenir, après reconstitution avec 1 ml ou 1,3 ml, un volume nominal prélevable de 45 mg/0,9 ml ou de 60 mg/1,2 ml.
·Agitez délicatement le flacon pour reconstituer le médicament en poudre. NE secouez PAS le flacon et NE le bougez PAS violemment.
·Laissez reposer le flacon pendant 3 minutes jusqu'à ce que les bulles disparaissent.
·Effectuez une inspection visuelle de la solution reconstituée. Si Winrevair a bien été mélangé, la solution doit être limpide à opalescente et incolore à légèrement jaune-brunâtre et ne contenir ni grumeaux ni poudre.
·Retirez la seringue de l'adaptateur pour flacons et jetez la seringue vide dans un conteneur pour objets pointus et tranchants.
·Si un kit contenant deux flacons a été prescrit, répétez les étapes de ce paragraphe avec le deuxième flacon.
·Utilisez la solution reconstituée le plus rapidement possible, aux plus tard 4 heures après la reconstitution.
Préparation de la seringue
·Essuyez l'adaptateur pour flacons avec un tampon alcoolisé.
·Retirez la seringue doseuse de son emballage et fixez la seringue à l'adaptateur pour flacons.
·Retournez la seringue et le flacon et prélevez le volume d'injection correspondant en fonction du poids du patient.
·Si deux flacons doivent être utilisés pour atteindre la dose nécessaire, prélevez tout le contenu du premier flacon et transférez-le complètement et lentement dans le deuxième flacon.
·Retournez la seringue et le flacon à l'envers et prélevez la quantité nécessaire de médicament.
·Si nécessaire, enfoncez le piston pour retirer l'excès de médicament ou l'air de la seringue.
·Séparez la seringue du flacon et mettez l'aiguille en place.
Remarques concernant l'administration
Winrevair s'administre par injection sous-cutanée.
·Choisissez un site d'injection situé sur le ventre (à au moins 5 cm du nombril), sur la cuisse ou sur le haut du bras et nettoyez-le avec un tampon alcoolisé. Pour chaque injection, choisissez une nouvelle zone, qui ne comporte pas de cicatrice, d'irritation ou d'hématome.
·Si c'est le patient ou son aidant qui procède à l'administration, seuls le ventre et la cuisse doivent être utilisés comme sites d'injection (voir le mode d'emploi).
·Effectuez l'injection sous-cutanée.
·Jetez la seringue vide dans un conteneur pour objets pointus et tranchants. Ne réutilisez pas la seringue.
Numéro d’autorisation69787 (Swissmedic)
PrésentationFlacon en verre de type I, fermé par un bouchon en caoutchouc bromobutyle et une capsule en aluminium munie d'un capuchon flip-off de couleur citron vert en polypropylène et seringue préremplie (corps de la seringue en verre de type I, fermé par un bouchon en caoutchouc bromobutyle) avec 1 ml d'eau pour préparations injectables pour les kits contenant 45 mg/flacon ou 1,3 ml d'eau pour préparations injectables pour les kits contenant 60 mg/flacon.
Winrevair 45 mg Poudre et solvant pour préparation d'une solution injectable
·Emballage contenant 1 flacon de poudre, 1 seringue préremplie de solvant, 1 seringue doseuse graduée au dixième de millilitre (0,1 ml), 1 adaptateur pour flacons (13 mm), 1 aiguille d'injection et 4 tampons alcoolisés. [B]
·Emballage contenant 2 flacons de poudre, 2 seringues préremplies de solvant, 1 seringue doseuse graduée au dixième de millilitre (0,1 ml), 2 adaptateurs pour flacons (13 mm), 1 aiguille d'injection et 8 tampons alcoolisés. [B]
Winrevair 60 mg Poudre et solvant pour préparation d'une solution injectable
·Emballage contenant 1 flacon de poudre, 1 seringue préremplie de solvant, 1 seringue doseuse graduée au dixième de millilitre (0,1 ml), 1 adaptateur pour flacons (13 mm), 1 aiguille d'injection et 4 tampons alcoolisés. [B]
·Emballage contenant 2 flacons de poudre, 2 seringues préremplies de solvant, 1 seringue doseuse graduée au dixième de millilitre (0,1 ml), 2 adaptateurs pour flacons (13 mm), 1 aiguille d'injection et 8 tampons alcoolisés. [B]
Titulaire de l’autorisationMSD MERCK SHARP & DOHME AG, Lucerne
Mise à jour de l’informationAoût 2024
MK7962-kit-CCDS082023-OMA+CCDS042024+CCDS062024/RCN000025703-000027094-000027270-CH
|