Composition
Principes actifs
Facteur de coagulation XIII (FXIII) humain.
Excipients
Poudre: albumine humaine, glucose, chlorure de sodium (correspond à ≤0,76 mmol ou 17,48 mg de sodium par flacon de Fibrogammin 250 UI et à ≤3,8 mmol ou 87,4 mg de sodium par flacon de Fibrogammin 1250 UI), hydroxyde de sodium (pour ajustement du pH).
Solvant: eau pour injection.
Indications/Possibilités d’emploi
Fibrogammin est indiqué pour
·la prophylaxie en cas de déficit congénital en FXIII et
·le traitement périopératoire d'hémorragies chirurgicales en cas de déficit congénital en FXIII.
Fibrogammin est également indiqué
·en cas de diathèses hémorragiques complètement ou partiellement dues à un déficit acquis en FXIII.
·pour la thérapie de soutien lors de troubles de la cicatrisation, spécialement lors d'Ulcus cruris, après interventions chirurgicales majeures ou lors de lésions.
Posologie/Mode d’emploi
Posologie
1 ml correspond à 62,5 UI et 100 UI correspondent à 1,6 ml.
Important: La posologie et la durée du traitement doivent toujours être adaptées au cas particulier, en fonction de l'efficacité clinique observée.
La posologie doit être ajustée pour chaque cas en fonction du poids corporel, des paramètres de laboratoire et du tableau clinique du patient.
Posologie dans la prophylaxie de routine
Dose initiale
·40 unités internationales (UI) par kg de poids corporel.
·Le débit d'injection ne doit pas excéder 4 ml par minute
Posologie ultérieure
·La posologie doit être basée sur le dernier taux résiduel d'activité du FXIII. L'intervalle est de 28 jours (4 semaines) pour maintenir un taux résiduel d'activité du FXIII d'environ 5 à 20%.
·Les ajustements posologiques recommandés de ± 5 UI par kg doivent tenir compte des taux résiduels d'activité du FXIII indiqués dans le tableau I et du tableau clinique du patient.
·Les ajustements posologiques devraient être faits sur la base d'un test sensible spécial qui est utilisé pour déterminer le taux de FXIII. Un exemple d'ajustement posologique à l'aide du dosage de l'activité du FXIII par le test standard Berichrom® est montré dans le tableau 1.
Tableau 1: Ajustement posologique à l'aide du dosage de l'activité du FXIII par test Berichrom®
|
Taux résiduel d'activité du FXIII (%) |
Changement de posologie |
|
Un taux résiduel de <5% |
Augmentation de 5 unités par kg |
|
Taux résiduel de 5% à 20% |
Aucun changement |
|
Deux taux résiduels de >20% |
Diminution de 5 unités par kg |
|
Un taux résiduel de >25% |
Diminution de 5 unités par kg |
La puissance exprimée en unités est déterminée à l'aide du dosage de l'activité du FXIII par le test Berichrom® et se rapporte au standard international actuel du facteur de coagulation FXIII plasmatique. Une unité est par conséquent équivalente à une unité internationale.
Prophylaxie périopératoire (cette recommandation est basée sur des données cliniques clairsemées dans des interventions en général mineures)
Après la dernière prophylaxie de routine du patient, lorsqu'une opération est programmée:
·entre 21 et 28 jours après – une dose prophylactique complète est administrée juste avant l'opération. La dose prophylactique suivante doit être administrée après 28 jours.
·Entre 8 et 21 jours après – une dose supplémentaire (complète ou partielle) peut être administrée avant l'opération. La posologie devrait être ajustée en fonction du taux d'activité du FXIII et du tableau clinique du patient et être adaptée à la demi-vie de Fibrogammin.
·Dans les 7 jours après la dernière dose – une administration supplémentaire peut ne pas être nécessaire.
Les ajustements posologiques peuvent s'écarter de ces recommandations et devraient toujours être ajustés en fonction du taux de FXIII et du tableau clinique du patient. Tous les patients devraient être surveillés étroitement pendant et après l'opération. Il est par conséquent recommandé de surveiller l'augmentation de l'activité du FXIII au moyen d'un dosage du FXIII. Dans les cas d'interventions majeures et d'hémorragies sévères, des valeurs normales approchantes (individus sains: 70%-140%) sont souhaitables.
Déficit acquis en facteur XIII
Pour le traitement de diathèses hémorragiques, il faut administrer par jour au moins 15-20 unités internationales (UI) par kg de poids corporel jusqu'à l'atténuation des symptômes ou bien jusqu'à la régularisation spontanée du taux de FXIII.
Traitement de soutien dans les cas d'anomalies de la cicatrisation
10 unités internationales (UI) par kg de poids corporel le jour de l'opération et une fois par jour pendant les 3 jours suivants. Chez les patients à risque, la dose individuelle peut être augmentée jusqu'à 15-20 UI/kg de poids corporel.
Patients âgés
La posologie et l'utilisation chez les patients âgés (>65 ans) n'ont pas été étudiées.
Enfants et adolescents
La posologie et l'utilisation chez l'enfant et l'adolescent sont basées sur le poids corporel et suivent par conséquent les mêmes directives que chez l'adulte. La dose et la fréquence de l'administration devraient être adaptées pour chaque individu en fonction de l'effet clinique et du taux de facteur XIII (voir également «Propriétés/Effets»).
Mode d'administration
La solution reconstituée doit être limpide ou légèrement opalescente. Avant l'emploi, la préparation doit être portée à la température ambiante ou à la température corporelle et injectée ou perfusée lentement par voie intraveineuse distincte à un débit sans inconfort pour le patient. Le débit d'injection ou de perfusion ne doit pas excéder 4 ml par minute.
Une surveillance du patient est nécessaire en raison de possibles réactions immédiates. En cas de réaction susceptible d'être liée à l'administration de Fibrogammin, il convient de baisser la vitesse de perfusion ou – selon l'état clinique du patient – d'interrompre la perfusion.
La dissolution de Fibrogammin s'effectue comme décrit dans la rubrique «Remarques particulières» sous «Remarques concernant la manipulation».
Afin de garantir la traçabilité des médicaments biologiques, il est recommandé de documenter le nom commercial et le numéro de lot lors de chaque traitement.
Contre-indications
Hypersensibilité au principe actif ou à l'un des composants selon la composition.
Mises en garde et précautions
Contre-indications relatives:
L'administration à titre prophylactique d'antihistaminiques ou de corticostéroïdes peut être indiquée chez les patients avec antécédent allergique connu à la préparation (avec des symptômes tels que l'urticaire généralisée, rougeur de la peau, hypotension ou dyspnée).
Des réactions d'hypersensibilité sont possibles. Si des réactions d'hypersensibilité (telles qu'éruptions papuleuses, urticaire généralisée, angine de poitrine, stridor, hypotension et anaphylaxie) se présentent, cesser immédiatement l'apport de Fibrogammin (par ex. en interrompant la perfusion) et instaurer un traitement adéquat. Suivre les directives médicales actuelles pour le traitement d'un choc.
Chez des patients à antécédents d'embolie et d'évènements thrombotiques veineux et artériels, la prudence est de mise, car le facteur XIII a un effet stabilisant sur la fibrine. Il peut apparaître une stabilisation du thrombus, qui augmente le risque d'une occlusion vasculaire. Le risque d'un évènement thromboembolique peut être accru chez ces patients.
Immunogénicité
La formation d'anticorps neutralisants (inhibiteurs) contre le FXIII est une complication connue dans le traitement par Fibrogammin. Il convient par conséquent de surveiller les patients afin de déceler un éventuel développement d'inhibiteurs. La présence d'inhibiteurs peut se manifester par une réponse insuffisante au traitement. En cas de présence suspectée d'inhibiteurs, en cas de taux de FXIII plasmatique non atteints ou si des hémorragies surviennent au cours du traitement prophylactique, un dosage de la concentration d'inhibiteurs de FXIII s'impose.
Remarque pour les patients suivant un régime pauvre en sel:
Fibrogammin 250 UI contient ≤0,76 mmol (17,48 mg) de sodium par flacon, ce qui correspond à 0,87 % de la dose journalière de 2 g recommandée par l'OMS.
Fibrogammin 1250 UI contient ≤3,8 mmol (87,4 mg) de sodium par flacon , ce qui correspond à 4,37 % de la dose journalière de 2 g recommandée par l'OMS.
Sécurité virale
Fibrogammin est produit à partir de plasma humain. Les mesures habituelles de prévention du risque de transmission d'agents infectieux par les médicaments préparés à partir de sang ou de plasma humain comprennent la sélection clinique des donneurs, la recherche des marqueurs spécifiques d'infection sur chaque don et sur les pools de plasma et l'inclusion dans le procédé de fabrication d'étapes efficaces pour l'inactivation/l'élimination virale. Cependant, lorsque des médicaments préparés à partir de sang ou de plasma humain sont administrés, le risque de transmission d'agents infectieux ne peut pas être totalement exclu. Ceci s'applique également aux virus inconnus ou émergents ou à d'autres types d'agents infectieux. Les mesures mises en place sont considérées comme efficaces vis-à-vis des virus enveloppés tels que le virus de l'immunodéficience humaine (VIH), le virus de l'hépatite B (VHB) et le virus de l'hépatite C (VHC) ainsi que des virus non enveloppés tels que le virus de l'hépatite A et le parvovirus B19.
Il est impératif de recommander la vaccination contre l'hépatite A et B aux patients recevant régulièrement des produits sanguins ou plasmatiques d'origine humaine (Fibrogammin inclus).
Interactions
Aucune étude visant à déterminer des interactions n'a été effectuée.
Grossesse, allaitement
Grossesse
Aucune étude de toxicité pour la reproduction n'a été effectuée (voir «Données précliniques»).
Des données limitées concernant l'utilisation clinique de Fibrogammin pendant la grossesse n'ont montré aucun effet négatif sur le déroulement de la grossesse ou la phase de développement pendant ou après la naissance.
Après évaluation attentive, Fibrogammin peut donc être administré pendant la grossesse et l'allaitement.
Allaitement
Il n'existe pas de données concernant l'excrétion de Fibrogammin dans le lait maternel. En raison de la taille de sa molécule, il n'y a cependant pas lieu de suspecter une excrétion dans le lait et, au vu de sa structure protéique, l'absorption de molécules fonctionnelles après prise orale par le nourrisson est elle aussi improbable. Fibrogammin peut donc être administré pendant l'allaitement.
Fertilité
Il n'existe pas de données concernant les effets de Fibrogammin sur la faculté reproductrice.
Effet sur l’aptitude à la conduite et l’utilisation de machines
Aucune étude relative aux effets sur la conduite de véhicules et l'aptitude à l'utilisation de machines n'a été effectuée.
Effets indésirables
Les effets indésirables suivants sont basés sur l'analyse de données post-marketing et sont classés selon la fréquence d'apparition et la classification des systèmes d'organes MedDRA.
Pour la fréquence, les définitions suivantes ont été adoptées:
Très fréquent: ≥1/10
Fréquent: ≥1/100 et <1/10
Occasionnel: ≥1/1 000 et <1/100
Rare: ≥1/10 000 et <1/1 000
Très rare: <1/10 000
Affections du système immunitaire:
Rare: Réactions d'hypersensibilité (telles qu'urticaire généralisée, rougeurs, hypotension, troubles respiratoires).
Très rare: Apparition d'inhibiteurs anti-FXIII.
Troubles généraux et anomalies au site d'administration:
Rare: Élévation de la température.
Des informations sur la sécurité virale sont présentées au chapitre «Mises en garde et précautions».
Population pédiatrique
Dans des études cliniques, le profil de sécurité pour les enfants et les adolescents correspond à celui des adultes.
L'annonce d'effets secondaires présumés après l'autorisation est d'une grande importance. Elle permet un suivi continu du rapport bénéfice-risque du médicament. Les professionnels de santé sont tenus de déclarer toute suspicion d'effet secondaire nouveau ou grave via le portail d'annonce en ligne ElViS (Electronic Vigilance System). Vous trouverez des informations à ce sujet sur www.swissmedic.ch.
Surdosage
Aucune conséquence due à un surdosage de Fibrogammin n'est connue à ce jour.
Propriétés/Effets
Code ATC
B02BD07
Groupe pharmacothérapeutique: antihémorragiques
Mécanisme d'action/Pharmacodynamique
Fibrogammin est un concentré purifié de facteur XIII de coagulation. La préparation est produite à partir de lots de pools de plasma humain provenant de donneurs sains. L'information sur le dépistage des dons et des pools de plasma se trouve au chapitre «Mises en garde et précautions».
Le procédé de fabrication de Fibrogammin comporte plusieurs étapes contribuant à l'élimination/l'inactivation des virus. Parmi celles-ci, on compte une étape de chromatographie échangeuse d'ions ainsi qu'un traitement thermique/pasteurisation et une filtration virale.
Comme le facteur XIII joint les groupes aminés de lysine aux groupes aminés de glutamine par réactions enzymatiques, il est aussi capable de relier les molécules de fibrine (activité transamidase). Cela a pour conséquence de stabiliser les caillots et d'accélérer une infiltration des fibroblastes dans le caillot, de façon à produire une accélération de la cicatrisation.
Efficacité clinique
L'efficacité clinique et la sécurité ont été examinées dans l'étude 3001, une étude prospective ouverte multicentrique, menée sur 25 patients masculins et 16 patientes, âgés de 1 à 42 ans. 18 patients (44%) avaient moins de 18 ans, dont 2 enfants en bas âge (5%), 8 enfants et 8 adolescents. Dans le cadre de l'étude, les 41 patients ont reçu au total 533 perfusions, tous les patients (100%) ayant reçu au moins 13 perfusions. La dose unitaire moyenne était de 41,2 U/kg et le volume moyen était de 35,9 ml par dose. En moyenne, 33 584 U ont été administrées au total. À ce dosage, aucune hémorragie spontanée nécessitant une prise en charge thérapeutique n'est survenue.
Population pédiatrique
Dans des études cliniques auxquelles participaient également des enfants et des adolescents de <18 ans à déficit congénital en FXIII, il a été montré qu'avec une administration prophylactique de Fibrogammin tous les 28 jours, il était possible d'atteindre un taux résiduel d'activité du FXIII d'environ 5% à 20%.
Pharmacocinétique
Absorption
Non pertinent.
Distribution
Le produit est appliqué par voie intraveineuse et est disponible immédiatement à une dose qui correspond à la concentration plasmatique.
Métabolisme
Données manquantes.
Élimination
La demi-vie biologique du facteur XIII chez des patients à déficit congénital a été déterminée à 6,6 ± 2,29 jours (médiane ± ET). Fibrogammin est éliminé comme le facteur de coagulation XIII endogène.
Enfants et adolescents
Chez l'enfant, le produit a une demi-vie plus courte et une clairance plus rapide que chez l'adulte. Toutefois, étant donné que la posologie est calculée individuellement en fonction du poids corporel, indépendamment du groupe d'âge, et adaptée au taux résiduel d'activité du FXIII, il n'est pas nécessaire d'ajuster la posologie en fonction de l'âge.
Données précliniques
Les protéines contenues dans Fibrogammin proviennent du plasma humain et ont par conséquent entraîné une forte activité antigénique lors de l'essai sur l'animal après administration parentérale. Les études réalisées sur des animaux de laboratoire avec administration unique et administrations répétées n'ont pas révélé de potentiel toxicologique de Fibrogammin. Aucune étude relative à la reproduction ou au développement embryo-fœtal n'a été réalisée.
Remarques particulières
Remarque pour les diabétiques: Fibrogammin contient du glucose (96 mg par 1000 UI). Avec un dosage de 10 UI/kg de poids corporel, un patient de 75 kg reçoit 72 mg de glucose, ce qui correspond à 252 mg de glucose pour une dose maximale journalière de 35 UI/kg de poids corporel.
Incompatibilités
Fibrogammin ne peut pas être mélangé à d'autres médicaments, solvants ou diluants et doit être administré via une ligne d'injection/de perfusion séparée.
Stabilité
Ce médicament ne doit pas être utilisé au-delà de la date figurant après la mention «EXP» sur l'emballage.
Stabilité après ouverture
Si le produit reconstitué n'est pas utilisé immédiatement, il ne doit pas être stocké à température ambiante (≤25 °C) pendant plus de 4 heures.
La stabilité physico-chimique a été démontrée pendant cette période. Cependant, la préparation doit être utilisée immédiatement pour des raisons microbiologiques. Le produit reconstitué ne doit pas être conservé au réfrigérateur ou congelé.
Remarques particulières concernant le stockage
Conserver au réfrigérateur (2-8 °C) dans l'emballage original fermé pour protéger le contenu de la lumière. Ne pas surgeler. Tenir les médicaments hors de portée des enfants!
Remarques concernant la manipulation
La solution doit être préparée comme décrit sous «Mise en solution».
Remarques générales
·La solution doit être limpide ou légèrement opalescente. Après filtration/inspiration dans la seringue (voir «Mise en solution» ci-dessous) et avant utilisation, le produit reconstitué doit être inspecté visuellement à la recherche de particules ou d'une coloration.
·La préparation et l'administration doivent se faire dans des conditions aseptiques.
·Des solutions nettement troubles ou des solutions contenant des résidus (précipités/particules) ne doivent pas être utilisées.
·Il est important d'éviter toute entrée de sang dans la seringue remplie de produit, car il existe un risque de coagulation du sang dans la seringue et de formation de caillots de fibrine qui pourraient alors être administrés au patient.
·La solution doit être administrée via une voie distincte.
Mise en solution
Avant l'ouverture de l'emballage du Mix2Vial, retirer les capuchons amovibles des flacons de solvant et de produit, puis nettoyer les bouchons avec une solution antiseptique et laisser sécher.
|
|
|
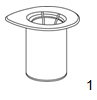
1. Retirer l'opercule en papier du dispositif Mix2Vial. Ne pas extraire le dispositif Mix2Vial de son blister! |
|
|
|
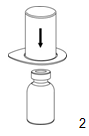
2. Poser et tenir le flacon de solvant sur une surface plane et propre. Saisir le dispositif Mix2Vial avec son blister et enfoncer la pointe de l'adapteur bleu verticalement dans le bouchon du flacon de solvant. |
|
|
|
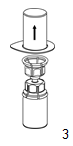
3. Retirer avec précaution l'emballage du dispositif Mix2Vial en tenant le blister par le bord scellé et en le tirant verticalement vers le haut. Ce faisant, il est important que seul le blister soit retiré et non le dispositif Mix2Vial. |
|
|
|
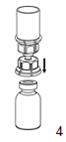
4. Poser le flacon de produit sur une surface dure. Retourner le flacon de solvant avec le dispositif Mix2Vial fixé dessus et enfoncer la pointe de l'adaptateur transparent verticalement dans le bouchon du flacon de produit. Le solvant est transféré automatiquement dans le flacon de produit. |
|
|
|
5. En maintenant la partie produit reconstitué d'un côté et la partie solvant de l'autre, séparer les flacons avec précaution en dévissant le dispositif dans le sens inverse des aiguilles d'une montre. Éliminer la partie solvant avec l'adaptateur bleu. |
|
|
|
6. Agiter délicatement le flacon de produit avec l'adapteur transparent jusqu'à ce que le produit soit entièrement dissout. Ne pas secouer le flacon. |
|
|
|
7. Aspirer de l'air dans une seringue stérile vide. Tout en maintenant le flacon de produit reconstitué en position verticale, visser la seringue sur l'embout Luer-Lock du dispositif Mix2Vial en tournant dans le sens des aiguilles d'une montre et injecter l'air dans le flacon de produit. |
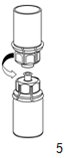
Aspiration de la solution dans la seringue et administration
|
|
|
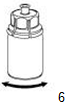
8. Maintenir le piston de la seringue enfoncé et retourner l'ensemble. Prélever ensuite le contenu du flacon en tirant doucement sur le piston. |
|
|
|
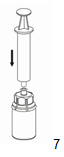
9. Une fois la solution entièrement transférée dans la seringue, tenir le corps de la seringue fermement (en maintenant le piston dans sa position) et déconnecter la seringue de l'adaptateur transparent Mix2Vial en dévissant dans le sens inverse des aiguilles d'une montre. |
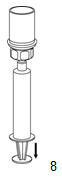
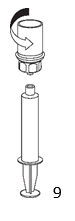
Si plusieurs flacons de Fibrogammin sont nécessaires pour une perfusion unique, il est possible de collecter le contenu de plusieurs flacons dans un dispositif de perfusion disponible dans le commerce.
Voir les informations sous le chapitre «Posologie/Mode d'emploi – Mode d'administration».
Les solutions non utilisées doivent être éliminées selon la réglementation en vigueur.
Numéro d’autorisation
00671 (Swissmedic)
Présentation
Présentation avec 250 UI (B)
1 flacon perforable contenant la poudre
1 flacon perforable contenant 4 ml d'eau pour injection
1 dispositif de transfert avec filtre 20/20
Présentation avec 1250 UI (B)
1 flacon perforable contenant la poudre
1 flacon perforable contenant 20 ml d'eau pour injection
1 dispositif de transfert avec filtre 20/20
Titulaire de l’autorisation
CSL Behring AG, Berne
Mise à jour de l’information
Janvier 2024