Composition
Principes actifs
Rotigotine
Excipients
·Couche de support: aluminée, feuille de polyester siliconée, revêtue d'une couche de pigment sur la face externe.
·Matrice auto-adhésive: adhésif silicone, povidone K90, métabisulfite de sodium (E223), palmitate d'ascorbyle (E304) et alpha-tocophérol (E307).
·Feuille protectrice: feuille de polyester transparente, recouverte de polymère fluoré.
Indications/Possibilités d’emploi
Syndrome des jambes sans repos
Neupro est indiqué dans le traitement symptomatique du syndrome des jambes sans repos (SJSR) idiopathique d'intensité modérée à sévère chez l'adulte.
Maladie de Parkinson
Neupro est utilisé en monothérapie (c.-à-d. sans lévodopa) pour le traitement symptomatique à un stade précoce de la maladie de Parkinson ou en association avec la lévodopa, quand, au cours de l'évolution de la maladie, jusqu'aux stades avancés, l'efficacité de la lévodopa s'atténue ou devient irrégulière et que des fluctuations de l'efficacité thérapeutique apparaissent (fluctuations de type fin de dose ou effet «on-off»).
L'instauration et la surveillance du traitement, en particulier à un stade avancé, doivent être effectuées par un médecin disposant d'une expérience dans le domaine du diagnostic et du traitement de la maladie de Parkinson idiopathique, conformément aux directives actuelles.
Posologie/Mode d’emploi
Posologie
Les recommandations en matière de posologie sont exprimées en termes de dose nominale.
Syndrome des jambes sans repos
Le traitement doit débuter à une dose quotidienne unique de 1 mg/24 h. En fonction de la réponse individuelle du patient, la dose peut être augmentée de 1 mg/24 h chaque semaine sans dépasser la dose maximale de 3 mg/24 h. La nécessité de poursuivre le traitement devra être reconsidérée tous les 6 mois.
Maladie de Parkinson
Posologie au stade précoce de la maladie idiopathique
Le traitement est initié par une dose journalière unique de 2 mg/24 h, que l'on augmente par paliers hebdomadaires de 2 mg/24 h jusqu'à atteindre la dose efficace, au maximum de 8 mg/24 h.
Une dose de 4 mg/24 h peut suffire chez certains patients. Chez la plupart des patients, la dose efficace est atteinte en 3 ou 4 semaines avec des doses de 6 mg/24 h ou de 8 mg/24 h.
La dose maximale est de 8 mg/24 h par jour.
Posologie au stade avancé de la maladie de Parkinson avec fluctuations
Le traitement est initié par une dose journalière unique de 4 mg/24 h, que l'on augmente par paliers hebdomadaires de 2 mg/24 h jusqu'à atteindre une dose cliniquement efficace et tolérée. La dose maximale est de 16 mg/24 h.
Une dose de 4 mg/24 h ou de 6 mg/24 h peut suffire chez certains patients.
Chez la plupart des patients, la dose efficace est atteinte avec une dose de 8 mg/24 h.
Pour des doses supérieures à 8 mg/24 h, on peut utiliser plusieurs patchs transdermiques simultanément. Un patch transdermique 6 mg/24 h et un patch transdermique 4 mg/24 h peuvent être combinés pour atteindre une dose de 10 mg/24 h.
Neupro est appliqué une fois par jour. Le patch transdermique doit être appliqué à peu près à la même heure chaque jour. Celui-ci reste sur la peau pendant 24 heures et doit ensuite être remplacé par un patch transdermique neuf que l'on appliquera sur un site différent.
Si le patient oublie d'appliquer le timbre à l'heure habituelle de la journée ou si le timbre se détache, un nouveau timbre doit être appliqué pour le reste de la journée.
Neupro étant administré par voie transdermique, aucun effet causé par des aliments ou une maladie gastro-intestinale n'est attendu, si bien que le patch peut être appliqué indépendamment de l'heure des repas.
Substitution d'autres agonistes dopaminergiques par Neupro
Le pramipexole ou le ropinirole peuvent être remplacés par Neupro après une nuit, soit après 12 heures, la cabergoline après 24 heures.
Arrêt du traitement
Syndrome des jambes sans repos
Neupro doit être arrêté progressivement. La dose quotidienne doit être réduite par paliers de 1 mg/24 h en réduisant de préférence la dose tous les deux jours jusqu'à l'arrêt total de Neupro (voir aussi «Mises en garde et précautions, Syndrome malin des neuroleptiques et Syndrome de sevrage des agonistes dopaminergiques»). En suivant cette procédure, aucun effet rebond (aggravation des symptômes au-delà de leur intensité initiale) après arrêt du traitement n'a été constaté.
Maladie de Parkinson
Si un arrêt du traitement s'impose, Neupro doit être arrêté progressivement. La dose quotidienne doit être réduite par paliers de 2 mg/24 h en réduisant de préférence la dose tous les deux jours jusqu'à l'arrêt total de Neupro (voir aussi «Mises en garde et précautions, Syndrome malin des neuroleptiques et Syndrome de sevrage des agonistes dopaminergiques»).
Instructions spéciales pour le dosage
Vu que le traitement par Neupro est initié à faible dose et augmenté progressivement en fonction de la tolérance clinique jusqu'à atteindre un effet thérapeutique optimal, aucun ajustement de la dose en fonction du sexe, du poids ou de l'âge n'est nécessaire (voir «Pharmacocinétique, cinétique dans des groupes particuliers de patients»).
Patients présentant des troubles de la fonction hépatique
Un ajustement de la dose n'est pas nécessaire pour les patients souffrant d'insuffisance hépatique légère à modérée. Neupro n'a pas été étudié chez les patients souffrant d'insuffisance hépatique sévère et est contre-indiqué chez les patients atteints d'insuffisance hépatique sévère (voir «Contre-indications»).
Une réduction de la dose peut s'avérer nécessaire en cas d'aggravation de l'insuffisance hépatique.
Patients présentant des troubles de la fonction rénale
Un ajustement de la dose n'est pas nécessaire pour les patients souffrant d'insuffisance rénale légère à sévère, y compris pour les patients dialysés.
Une accumulation inattendue de rotigotine peut également survenir en cas d'aggravation aiguë de la fonction rénale (voir rubrique «Pharmacocinétique, cinétique dans des groupes particuliers de patients»).
Enfants et adolescents
On ne dispose pas de données sur l'efficacité et la sécurité chez les enfants et les adolescents.
Mode d'administration
Neupro est destiné à un usage transdermique.
Le patch transdermique doit être appliqué sur une peau propre, sèche, intacte et saine, sur l'abdomen, la cuisse, la hanche, le flanc, l'épaule et/ou le bras. Pour prévenir autant que possible les risques d'irritation, il est recommandé de changer de site d'application chaque jour (p.ex. en alternant entre les côtés droit et gauche et entre le haut et le bas du corps). Éviter de répéter l'application sur un même endroit en l'espace de deux semaines. Le changement du site d'application peut entraîner des fluctuations des concentrations plasmatiques. Il n'existe cependant aucun indice d'une influence significative sur le résultat clinique (cf. «Pharmacocinétique»). Le patch ne doit pas être appliqué à des endroits couverts par des vêtements trop serrés car il pourrait se décoller sous l'effet du frottement. S'il est prévu d'appliquer le patch transdermique sur une région pileuse, celle-ci doit être rasée au moins trois jours avant l'application.
Neupro ne doit pas être appliqué sur une peau rougie, irritée ou blessée.
On s'abstiendra d'appliquer des crèmes, lotions, poudres ou autres produits topiques sur les régions de la peau destinées à l'application de Neupro car cela pourrait diminuer l'adhérence du patch transdermique.
Chaque patch transdermique est emballé dans un sachet-dose individuel et doit être appliqué immédiatement après ouverture du sachet-dose. Retirer l'un des deux volets de la feuille protectrice et appliquer la face adhésive du patch transdermique sur la peau en appuyant fermement. Replier ensuite le patch transdermique et enlever l'autre volet de la feuille protectrice. Ne pas toucher la face adhésive du patch transdermique. Avec la paume de la main, appuyer fermement sur le patch transdermique pendant 30 secondes pour qu'il adhère bien.
Le patch transdermique doit être décollé lentement et avec précaution pour éviter d'irriter inutilement la peau.
Ne jamais exposer directement le patch transdermique aux rayons du soleil.
Le patient trouvera des instructions d'emploi détaillées dans la notice d'emballage.
Contre-indications
Hypersensibilité connue au principe actif ou à l'un des excipients selon la composition.
Imagerie par résonance magnétique (IRM) ou cardioversion. La couche de support de Neupro contient de l'aluminium. Pour éviter des brûlures cutanées, Neupro doit être retiré si le patient doit subir un examen d'imagerie par résonance magnétique (IRM) ou une cardioversion. Insuffisance hépatique sévère.
Mises en garde et précautions
Chez un patient parkinsonien qui serait insuffisamment stabilisé par le traitement à la rotigotine, le passage à un autre agoniste de la dopamine peut procurer un bénéfice supplémentaire (voir «Propriétés/Effets, mécanisme d'action et pharmacodynamique»).
Attaques de sommeil subites et somnolence
Comme d'autres agonistes de la dopamine, Neupro a été associé à des accès de somnolence et à des attaques de sommeil subites (endormissement soudain), notamment chez des patients atteints de la maladie de Parkinson. Des cas d'endormissement soudain au cours des activités quotidiennes, parfois sans prodromes visibles, ont été rapportés. Le médecin prescripteur devra régulièrement réévaluer ses patients quant à leurs épisodes de somnolence ou d'assoupissement, car il arrive que le patient n'identifie ces problèmes que lorsqu'on l'interroge directement à ce sujet. Le cas échéant, une réduction de la dose ou un arrêt du traitement devra être envisagé avec circonspection.
Les patients doivent être rendus attentifs aux impératifs de prudence lors de la conduite de véhicules ou de l'utilisation de machines pendant le traitement par Neupro. Les patients qui ont déjà éprouvé de la somnolence et/ou connu une attaque de sommeil subite doivent s'abstenir de conduire des véhicules ou d'utiliser des machines (voir aussi «Effets sur l'aptitude à la conduite et l'utilisation de machines»).
Hypotension orthostatique
Les agonistes de la dopamine sont connus pour altérer la régulation systémique de la tension artérielle, ce qui se manifeste par de l'hypotension posturale/orthostatique. Ces événements ont aussi été observés chez des patients traités par Neupro, mais leur incidence était similaire à celle constatée sous placebo.
Compte tenu du risque général d'hypotension orthostatique associé à un traitement dopaminergique, il est conseillé de surveiller la tension artérielle, particulièrement en début de traitement.
Syncopes
Des cas de syncope ont été observés durant les études cliniques avec la rotigotine, cependant, leur fréquence était comparable à celle enregistrée chez les patients sous placebo. Étant donné que ces études excluaient les patients atteints d'une maladie cardiovasculaire cliniquement pertinente, il faut demander aux patients atteints d'une maladie cardiovasculaire grave s'ils présentent des symptômes de syncope et de présyncope.
Syndrome malin des neuroleptiques
Des symptômes évoquant un syndrome malin des neuroleptiques ont été décrits suite à un arrêt brutal du traitement dopaminergique. Il est par conséquent recommandé de réduire le traitement progressivement (voir «Posologie/Mode d'emploi», «Arrêt du traitement»).
Syndrome de sevrage des agonistes dopaminergiques
Des symptômes suggérant un syndrome de sevrage des agonistes dopaminergiques (par exemple, douleur, fatigue, dépression, transpiration et anxiété) ont été rapportés lors d'un arrêt soudain du traitement dopaminergique. Il est donc recommandé de réduire progressivement le traitement et d'éviter un arrêt brutal (voir «Posologie/Mode d'emploi», «Arrêt du traitement»).
Troubles psychiques et comportementaux
Des troubles psychiques et comportementaux ont été rapportés. Ces phénomènes peuvent se manifester de différentes manières, telles que: idées paranoïdes, idées délirantes, hallucinations, confusion, comportements psychotiques, désorientation, agressivité, agitation, délire.
Troubles compulsifs et autres troubles apparentés
Les patients devraient être surveillés régulièrement concernant l'apparition et/ou le développement de troubles compulsifs et de troubles apparentés, y compris le syndrome de dysrégulation dopaminergique. Les patients ainsi que leur entourage devraient être informés que les traitements avec des antagonistes de la dopamine – y compris la rotigotine – peuvent mener à des troubles compulsifs, y compris addiction au jeu, une prise de nourriture ou des dépenses excessives, hypersexualité, augmentation de la libido et actions répétitives dépourvues de sens (martelage). Chez certains patients, un syndrome de dysrégulation dopaminergique a été observé sous traitement par rotigotine.
Si de tels symptômes apparaissent, une réduction de la dose ou un arrêt progressif du traitement doivent être envisagés.
Complications fibrotiques
Des cas de fibrose rétropéritonéale, d'infiltration pulmonaire, d'épanchement pleural, d'épaississement de la plèvre, de péricardite et de valvulopathie cardiaque ont été décrits chez certains patients traités par des agonistes dopaminergiques dérivés de l'ergot. Bien que ces complications puissent disparaître avec l'arrêt du médicament, elles ne sont pas toujours complètement réversibles.
On pense que ces effets indésirables sont associés à la structure ergoline de ces substances, mais on ignore si d'autres agonistes dopaminergiques non dérivés de l'ergot peuvent avoir les mêmes effets.
Neuroleptiques
Il ne faut pas administrer de neuroleptiques antiémétiques aux patients traités par des agonistes de la dopamine (voir «Interactions»).
Contrôles ophtalmologiques
Un suivi ophtalmologique est recommandé à intervalles réguliers ou en cas d'anomalies de la vision.
Augmentation
Un phénomène d'augmentation peut se produire chez les patients atteints du syndrome des jambes sans repos. Ce phénomène d'augmentation fait référence à une apparition plus précoce des symptômes le soir (voire l'après- midi), à une sévérité accrue des symptômes et à une extension des symptômes à d'autres parties du corps.
Dans les études cliniques à long terme sur la rotigotine, la plupart des cas d'augmentation sont survenus au cours de la première et de la deuxième année du traitement. Des doses plus élevées que celles permises pour le SJSR devraient être évitées, car cela peut entraîner une hausse du taux d'augmentation.
Sur la base de deux études de suivi en ouvert d'une durée de 1 an, les symptômes reflétant une augmentation cliniquement pertinente ou non, peuvent atteindre une fréquence de 9,4%. Par contre, selon deux études d'une durée de 6 mois, en double aveugle contrôlées versus placebo, un phénomène d'augmentation cliniquement significatif a été observé chez 1,5% des patients traités par rotigotine versus 0,5% des patients sous placebo. Dans deux études en ouvert ayant un suivi complémentaire de 12 mois, le taux a été de 2,9%. Aucun de ces patients n'a arrêté le traitement en raison du phénomène d'augmentation. Les données émanant d'une étude ouverte portant sur le traitement pendant 5 ans ont montré une augmentation chez 11,9% des patients traités par une posologie autorisée pour le SJSR (1 à 3 mg/24 h); chez 5,1%, l'augmentation a été jugée comme étant cliniquement significative. La plupart des cas d'augmentation sont survenus au cours de la première et de la seconde année de traitement. La posologie de 4 mg/24 h également autorisée dans cette étude était associée à un taux d'augmentation accru. Une posologie de 4 mg/24 h n'est pas autorisée pour le traitement du SJSR (voir «Posologie/Mode d'emploi»).
Application de chaleur
Ne pas exposer la zone où se trouve le patch transdermique à des sources de chaleur externes (coussins chauffants et autres sources de chaleur telles que sauna, bain chaud), car la part absorbée du principe actif pourrait s'en trouver augmentée. Ne pas exposer directement le patch transdermique aux rayons du soleil.
Réactions au site d'application
Des réactions cutanées peuvent se produire au site d'application et sont généralement d'intensité bénigne à modérée. Il est recommandé de changer chaque jour le site d'application (p.ex. en passant du côté droit au côté gauche et en alternant entre le haut et le bas du corps). Éviter de réutiliser le même site en l'espace de 14 jours. Le risque/bénéfice pour le patient devra être évalué en cas de réaction au site d'application durant plus de quelques jours ou persistante, d'augmentation de la sévérité de ces réactions ou d'extension de la réaction cutanée au-delà du site d'application.
Dans les cas où le système transdermique aura provoqué une éruption ou une irritation cutanée, la zone touchée de la peau ne devra pas être exposée au rayonnement solaire direct jusqu'à la guérison, car une exposition pourrait entraîner une modification de la pigmentation de la peau.
Neupro doit être arrêté en cas de réaction cutanée généralisée (p.ex. éruption allergique – y c. éruption érythémateuse, maculaire ou papulaire – ou prurit) associée à l'utilisation du patch transdermique.
Effets indésirables dopaminergiques
L'incidence de certains effets indésirables dopaminergiques (tels que par exemple les hallucinations, la dyskinésie et les oedèmes périphériques) est généralement plus importante lors de l'administration en association avec la L-dopa chez les patients souffrant de maladie de Parkinson. Il convient d'en tenir compte lors de la prescription de rotigotine (voir «Interactions»).
Syndrome de dysrégulation dopaminergique (DDS)
Un syndrome de dysrégulation de la dopamine (SDD) a été observé chez certains patients recevant Neupro. Il s'agit d'un symptome de dépendance qui conduit à une surutilisation de ce médicament ou d'autres médicaments dopaminergiques à des doses croissantes. Avant de commencer le traitement, les patients et les soignants doivent être avertis du risque potentiel de développer un DDS (voir également «Effets indésirables»).
Oedème périphérique
Lors des études cliniques menées chez des patients parkinsoniens, la fréquence des oedèmes périphériques évaluée à 6 mois à environ 4%, s'est maintenue pendant toute la durée d'observation, et cela jusqu'à 36 mois. Des oedèmes périphériques ont été observés lors des études sur les patients avec SJSR.
Sensibilité aux sulfites
Neupro contient du métabisulfite de sodium, un sulfite susceptible d'entraîner des réactions de type allergique y compris symptômes anaphylactiques avec engagement du pronostic vital ou épisodes asthmatiformes moins sévères, chez certaines personnes prédisposées.
La prudence est recommandée lors du changement de traitement pour une autre forme posologique et / ou un autre médicament contenant la même substance active. Le patient doit être correctement surveillé.
Interactions
La rotigotine étant un agoniste de la dopamine, il y a lieu de supposer que d'autres antagonistes de la dopamine tels que les neuroleptiques (p.ex. phénothiazines, butyrophénones, thioxanthènes) ou le métoclopramide réduisent l'efficacité de Neupro. Leur administration concomitante doit donc être évitée (voir «Mises en garde et précautions»).
Comme d'autres agonistes de la dopamine, Neupro peut renforcer les effets indésirables dopaminergiques de la Levodopa et déclencher ou aggraver des dyskinésies existantes.
La pharmacocinétique de la rotigotine n'est pas influencée par l'administration concomitante de Levodopa ou de carbidopa; de même, la rotigotine n'a eu aucun effet sur la pharmacocinétique de la Levodopa ou de la carbidopa.
En raison d'effets additifs potentiels, la prudence est de rigueur chez les patients prenant des médicaments sédatifs ou d'autres substances déprimant le système nerveux central (SNC) (p.ex. benzodiazépines, antipsychotiques, antidépresseurs) ou de l'alcool en association avec la rotigotine.
L'administration concomitante d'oméprazole (inhibiteur du CYP2C19) à des doses de 40 mg/jour n'a eu aucun effet sur la pharmacocinétique et sur le métabolisme de la rotigotine chez des volontaires sains.
L'utilisation concomitante de dompéridone et de rotigotine n'a eu aucun effet sur la pharmacocinétique de la rotigotine.
Des études in vitro montrent que différentes isoformes CYP peuvent catalyser le métabolisme de la rotigotine.
Sur la base de ces essais, on peut supposer que pour des doses thérapeutiques de rotigotine il n'existe aucun risque d'inhibition du CYP1A2, du CYP2C9 et du CYP3A4 et un risque minime d'inhibition du CYP2C19 et du CYP2D6.
Dans l'hépatocyte humain il n'y a pas in vitro d'indication d'induction du CYP1A2, du CYP2B6, du CYP2C9, du CYP2C19 et du CYP3A4.
L'administration simultanée de rotigotine (jusqu'à 4 mg/24 h.) et de cimétidine (400 mg 2× par jour), un inhibiteur du CYP1A2, du CYP2C19, du CYP2D6 et du CYP3A4, n'a pas modifié chez des sujets sains la pharmacocinétique à l'état d'équilibre de la rotigotine.
L'administration concomitante d'inducteurs enzymatiques (p.ex. rifampicine, phénobarbital, carbamazépine, phénytoïne, millepertuis/hypericum perforatum) n'a pas été étudiée.
L'administration concomitante de rotigotine (3 mg/24 h) n'a pas affecté la pharmacodynamique et la pharmacocinétique des contraceptifs oraux (0,03 mg d'éthinylestradiol, 0,15 mg de lévonorgestrel).
Les interactions avec d'autres formes de contraception hormonale n'ont pas été étudiées.
Grossesse, allaitement
Grossesse
On ne dispose pas de données cliniques sur l'exposition de la femme enceinte à Neupro. Les études expérimentales chez l'animal n'ont pas mis en évidence d'effets tératogènes (voir «Données précliniques»).
Neupro ne doit être utilisé pendant la grossesse qu'en cas d'absolue nécessité.
Allaitement
On doit s'attendre à une inhibition de la lactation, la rotigotine diminuant la sécrétion de prolactine chez l'être humain.
Des études chez le rat ont montré que la rotigotine et/ou ses métabolites passent dans le lait maternel. En l'absence de données chez l'être humain, Neupro ne doit pas être pris pendant l'allaitement. Si l'application de Neupro s'impose néanmoins, l'allaitement doit être arrêté.
Effet sur l’aptitude à la conduite et l’utilisation de machines
La rotigotine peut avoir une influence importante sur l'aptitude à conduire des véhicules et à utiliser des machines.
Les patients traités par la rotigotine et qui présentent des accès de somnolence et/ou des attaques de sommeil subites doivent être avertis qu'ils ne doivent ni conduire ni exercer une quelconque activité dans laquelle une baisse de la vigilance pourrait comporter un risque de blessure grave, voire mortelle, pour eux-mêmes ou pour autrui (p.ex. utilisation de machines), et ce jusqu'à disparition de ce type d'épisodes (voir aussi «Mises en garde et précautions»). Les patients doivent être informés de ces problèmes et s'abstenir de telles activités tant que l'on ne possède pas d'expériences suffisantes sur ces effets handicapants.
Effets indésirables
Généralités
Des effets indésirables dopaminergiques tels que nausées et vomissements sont possibles en début de traitement. Ces effets ont généralement une intensité faible à modérée et sont passagers, même quand le traitement est poursuivi.
Syndrome des jambes sans repos
L'analyse de l'ensemble des études cliniques contrôlées contre placebo, qui incluaient au total 748 patients traités par Neupro et 214 patients traités par placebo, montre que 65,5% des patients traités avec Neupro et 33,2% des patients sous placebo ont rapporté au moins une réaction indésirable.
D'autres effets indésirables décrits chez plus de 10% des patients traités par le patch transdermique Neupro sont: nausées (19,3%), réactions à l'endroit de l'application (34,2%), états asthéniques (11,2%) et céphalées (16,7%).
Lors des études où une alternance des sites d'application était pratiquée conformément aux instructions figurant dans l'information professionnelle et l'information aux patients, 34,2% des 748 patients utilisant Neupro patch transdermique ont présenté des réactions au site d'application. La majorité de ces réactions étaient légères à modérées, se limitaient aux zones d'application et n'ont motivé un abandon du traitement que chez 7,2% des patients traités par Neupro.
Taux d'arrêt
Le taux d'arrêt de traitement a été étudié au cours de 3 études cliniques ayant eu jusqu'à 3 ans de durée. Le pourcentage de patients ayant arrêté l'étude a été de 25–38% durant la première année, 10% la seconde année, et 11% la troisième année. Une évaluation périodique de l'efficacité doit être réalisée, accompagnée de l'évaluation de la sécurité, y compris le phénomène d'augmentation.
Maladie de Parkinson
L'analyse de l'ensemble des études cliniques contrôlées par placebo, qui incluaient au total 1'307 patients traités par Neupro et 607 patients traités par placebo, montre que 72,5% des patients traités avec Neupro et 57,8% des patients sous placebo ont rapporté au moins une réaction indésirable.
D'autres effets indésirables décrits chez plus de 10% des patients traités par le patch transdermique Neupro sont: nausées (29,7%), vomissements (10,6%), réaction à l'endroit de l'application (31%), somnolence (19,4%), vertiges (14,5%) et céphalées (10,4%).
Lors des études où une alternance des sites d'application était pratiquée conformément aux instructions figurant dans l'information professionnelle et l'information aux patients, 35,7% des 830 patients utilisant Neupro patch transdermique ont présenté des réactions au site d'application. La majorité des réactions aux zones d'application étaient légères à modérées, se limitaient aux zones d'application et ont motivé un abandon du traitement chez 4,3% des patients traités par Neupro.
Liste des effets indésirables
Syndrome des jambes sans repos
La liste suivante présente les effets indésirables observés lors des études poolées mentionnées ci-dessus et contrôlées contre placebo, réalisées chez des patients atteints de syndrome des jambes sans repos. Au sein de chaque groupe de fréquence, les effets indésirables sont présentés par ordre décroissant de gravité.
Nombre de patients avec des effets indésirables attendus: Nombre de patients susceptibles de présenter cette réaction en utilisant les catégories suivantes: «très fréquent» (≥1/10), «fréquent» (<1/10, ≥1/100), «occasionnel» (<1/100, ≥1/1000), «rare» (<1/1000, ≥1/10'000), «très rare» (<1/10'000) et «inconnu» (fréquence non quantifiable sur la base des données à disposition).
D'autres effets indésirables ont été observés chez les patients atteints de la maladie de Parkinson. Voir la liste des effets indésirables sous la rubrique Maladie de Parkinson.
Classe système/organe selon MedDRA
Affections du système immunitaire
Fréquent: Hypersensibilité pouvant inclure des angio-œdème, des oedèmes de la langue ou des lèvres.
Affections psychiatriques
Fréquent: Attaques d'endormissement soudain, apnée soudaine du sommeil, troubles du désir sexuel¹ (y compris hypersexualité, augmentation de la libido), insomnie, troubles du sommeil, rêves anormaux, troubles du contrôle des pulsionsa,b (y compris jeu pathologique, comportement compulsif/stéréotypé, hyperphagie/trouble de l'alimentation).
Occasionnel: Troubles obsessionnels compulsifs.
Rare: Comportement agressif/agression, désorientationb.
Affections du système nerveux
Très fréquent: Céphalées.
Fréquent: Somnolence.
Affections vasculaires
Fréquent: Hypertension.
Occasionnel: Hypotension orthostatique.
Affections gastro-intestinales
Très fréquent: Nausées.
Fréquent: Vomissements, dyspepsie.
Affections de la peau et du tissu sous-cutané
Fréquent: Prurit.
Affections musculosquelettiques et du tissu conjonctif
Inconnu: augmentation des taux de créatine phosphokinase (CPK)b
Troubles généraux et anomalies au site d'administration
Très fréquent: Réactions au site d'application et d'administrationa (y compris érythème, prurit, irritation, rash, dermatite, vésicules, douleur, eczéma, inflammation, oedème, dépigmentation, papules, exfoliation, urticaire, hypersensibilité), états asthéniquesa (incl. fatigue, asthénie, malaise).
Fréquent: Irritabilité, oedèmes périphériques.
Effets indésirables après commercialisation
Comportement d'achat compulsif, syndrome de dérégulation dopaminergique, rhabdomyolyse.
a Terme de haut niveau.
b Données issues d'études mixtes, en double aveugle, contrôlées versus placebo
Maladie de Parkinson
La liste suivante reprend les effets indésirables des études poolées susmentionnées, réalisées sur des patients atteint de la maladie de Parkinson. Les effets indésirables sont présentés en ordre de gravité décroissant dans chaque classe système/organe.
Classe système/organe selon MedDRA
Affections du système immunitaire
Occasionnel: Hypersensibilité pouvant inclure des angio-œdème, des oedèmes de la langue ou des lèvres.
Affections psychiatriques
Fréquent: Troubles de la perceptiona (incl. hallucinations, hallucination visuelle, hallucination auditive, illusion), insomnie, trouble du sommeil, cauchemar, rêves anormaux, troubles du contrôle des pulsionsa,b (y compris jeu pathologique, comportement compulsif/stéréotypé).
Occasionnel: Attaques de sommeil/endormissement soudain, paranoia, troubles du désir sexuela (incl. hypersexualité, augmentation de la libido), état confusionnel, désorientationb, agitationb.
Rare: Troubles psychotiques, trouble obsessionnel compulsif, comportement agressif, agression, idées délirantesb, délireb.
Affections du système nerveux
Très fréquent: Somnolence, sensations vertigineuses, céphalées.
Fréquent: Troubles de la conscience non spécifiéesa (incl. syncope, syncope vasovagale, perte de conscience), dyskinésiesa, sensations vertigineuses posturales, léthargie.
Rare: Convulsions.
Affections oculaires
Occasionnel: Vision trouble, détérioration de la vision, photopsie.
Affections de l'oreille et du labyrinthe
Fréquent: Vertiges.
Affections cardiaques
Fréquent: Palpitations.
Occasionnel: Fibrillation auriculaire.
Rare: Tachycardie supraventriculaire.
Affections vasculaires
Fréquent: Hypotension orthostatique, hypertension.
Occasionnel: Hypotension.
Affections respiratoires, thoraciques et médiastinales
Fréquent: Singultus.
Affections gastro-intestinales
Très fréquent: Nausées, vomissementsa.
Fréquent: Constipation, sécheresse buccale, dyspepsie.
Occasionnel: Douleurs abdominales.
Affections hépatobiliaires
Inconnu: Élévation des enzymes hépatiques (y c. ASAT, ALAT, GGT).
Affections de la peau et du tissu sous-cutané
Fréquent: Érythèmea, hyperhidrosea, prurit.
Occasionnel: Prurit généralisé, irritation, cutanée, dermatite de contact.
Rare: Rash généralisé.
Affections musculosquelettiques, du tissu conjonctif et des os
Inconnu: augmentation des taux de créatine phosphokinase (CPK)b
Affections des organes de reproduction et du sein
Occasionnel: Dysfonction érectile.
Troubles généraux et anomalies au site d'administration
Très fréquent: Réactions au site d'application et d'instillation¹ (y compris érythème, prurit, irritation, rash, dermatite, vésicules, douleur, eczéma, inflammation, oedème, dépigmentation, papules, exfoliation, urticaire, hypersensibilité).
Fréquent: Oedème périphérique, état asthénique¹ (y c. fatigue, asthénie, malaise).
Rare: Irritabilité.
Examens
Fréquent: Perte de poids.
Occasionnel: Prise de pois, augmentation du rythme cardiaque.
Lésions, intoxication et complications liées aux procédures
Fréquent: Chutes.
a Terme de haut niveau.
b Données issues d'études mixtes, en double aveugle, contrôlées versus placebo
Effets indésirables après commercialisation
Hyperphagie et trouble alimentaire, comportement d'achat compulsif, syndrome de dysrégulation dopaminergique, diarrhée, rhabdomyolyse, sydrome de la tête tombante (Dropped Head Syndrome).
Description de certains effets indésirables
Endormissement soudain et somnolence
Neupro a été associé à une somnolence, y compris une somnolence excessive pendant la journée et à des accès de sommeil d'apparition brutale. Des cas isolés d'«endormissement brutal» sont survenus pendant la conduite et ont provoqué des accidents de la circulation (voir également «Mises en garde et précautions» et «Effets sur l'aptitude à la conduite et l'utilisation de machines»).
Syndrome de dysrégulation dopaminergique
Le syndrome de dysrégulation dopaminergique (SDD) est un symptome de dépendance qui a été observé chez certains patients traités par Neupro. Les patients concernés présentent un abus compulsif de médicaments dopaminergiques avec l'utilisation de doses croissantes et plus élevées que nécessaires pour un contrôle adéquat des symptômes liés au mouvement. Dans certains cas, cela peut entraîner une dyskinésie sévère et des symptômes psychiatriques (voir également «Mises en garde et précautions»).
Troubles compulsifs
Chez des patients traités par agonistes dopaminergiques, y compris Neupro, des signes de jeu pathologique, d'augmentation de la libido et d'hypersexualité, des dépenses ou achat compulsifs, une consommation excessive de nourriture et une alimentation compulsive ont été rapportés (voir «Mises en garde et précautions»).
L'annonce d'effets secondaires présumés après l'autorisation est d'une grande importance. Elle permet un suivi continu du rapport bénéfice-risque du médicament. Les professionnels de santé sont tenus de déclarer toute suspicion d'effet secondaire nouveau ou grave via le portail d'annonce en ligne ElViS (Electronic Vigilance System). Vous trouverez des informations à ce sujet sur www.swissmedic.ch.
Surdosage
Aucun cas de surdosage n'a été rapporté dans les études cliniques. Les effets indésirables auxquels il faudrait s'attendre sont ceux liés au profil pharmacodynamique d'un agoniste de la dopamine, comme par exemple nausées, vomissements, hypotension, mouvements involontaires, hallucinations, confusion, convulsions et autres signes de stimulation dopaminergique centrale.
Traitement ou prise en charge d'un surdosage
Il n'existe pas d'antidote connu à un surdosage d'agonistes de la dopamine. En cas de suspicion de surdosage, il faut envisager le retrait du ou des patch(s) transdermique(s). Les taux de rotigotine diminuent rapidement après le retrait du patch transdermique.
Le patient doit faire l'objet d'une surveillance étroite, y compris de la fréquence cardiaque, du rythme cardiaque et de la tension artérielle.
Le traitement d'un surdosage peut nécessiter des mesures générales de soutien afin de préserver les fonctions vitales.
Il ne faut pas s'attendre à une quelconque utilité de la dialyse, étant donné que la rotigotine ne s'élimine pas par dialyse.
Lorsqu'un arrêt de la rotigotine est nécessaire, celui-ci aura lieu progressivement afin de prévenir un syndrome malin des neuroleptiques et un syndrome de sevrage des agonistes dopaminergiques (voir «Posologie/Mode d'emploi, arrêt du traitement», «Mises en garde et précautions, Syndrome malin des neuroleptiques et Syndrome de sevrage des agonistes dopaminergiques»).
Propriétés/Effets
Code ATC
N04BC09
Mécanisme d'action
La rotigotine est un agoniste dopaminergique D3/D2/D1 de type non ergoline destiné au traitement symptomatique de la maladie de Parkinson idiopathique et du syndrome des jambes sans repos idiopathique.
Conformément à l'activité fonctionnelle au niveau des différents sous-types de récepteurs et de leur répartition dans le cerveau, la rotigotine est décrite comme un agoniste des récepteurs D2 et D3, ayant également une efficacité au niveau des récepteurs D1, D4 et D5. Au niveau des récepteurs non dopaminergiques, la rotigotine montre un antagonisme au niveau des récepteurs alpha2B et un agonisme au niveau du récepteur 5HT-1A, par contre aucune activité au niveau des récepteurs 5HT2B.
On suppose que l'effet bénéfique de la rotigotine sur la maladie de Parkinson est lié à l'activation des récepteurs D3, D2 et D1 au niveau du noyau caudé et du putamen dans le cerveau.
Le mécanisme d'action précis de la rotigotine dans le traitement du SJSR n'est pas connu. On suppose que la rotigotine exerce principalement son effet par le biais des récepteurs dopaminergiques.
Pharmacodynamique
N'est pas applicable.
Efficacité clinique
Études cliniques relatives au syndrome des jambes sans repos
L'efficacité de Neupro a été évaluée au cours de 5 études contrôlées versus placebo sur plus de 1'400 patients souffrant du syndrome des jambes sans repos (SJSR). Son efficacité a été démontrée au cours d'études contrôlées chez des patients traités jusqu'à 29 semaines. L'effet s'est maintenu sur une période de 6 mois.
Les critères principaux d'efficacité ont été les changements par rapport aux valeurs initiales sur l'échelle IRLS (International Restless Legs Syndrom) et de l'item n° 1 de la CGI (gravité de la maladie). Pour les deux critères principaux, des différences statistiquement significatives ont été observées pour les doses de 1 mg/24 h, de 2 mg/24 h et de 3 mg/24 h comparé au placebo. Après un traitement d'entretien de 6 mois chez des patients souffrant de SJSR modéré à sévère, le score IRLS initial a été amélioré de 30,7 à 20,7 dans le groupe placebo et de 30,2 à 13,8 dans le groupe Neupro. La différence moyenne ajustée a été de –6,5 points (IC 95% –8,7; –4,4, p <0,0001).
Le taux de répondeurs sur l'item 1 de la CGI (fortement amélioré, très fortement amélioré) ont été de 43,0% et 67,5% respectivement pour le placebo et Neupro (différence 24,5% IC 95%; 14,2%; 34,8%, p <0,0001).
Dans une étude contrôlée versus placebo d'une durée de 7 semaines, les paramètres polysomnographiques ont été étudiés. Neupro a significativement réduit l'indice des mouvements périodiques des membres (PLMI: Periodic Limb Movement Index) de 50,9 à 7,7 contre 37,4 à 32,7 dans le groupe placebo (p <0,0001).
Études cliniques relatives à la maladie de Parkinson
L'efficacité de Neupro dans le traitement symptomatique de la maladie de Parkinson idiopathique a été évaluée dans le cadre de quatre études menées en groupes parallèles, randomisées en double aveugle et contrôlées contre placebo. D'autres travaux ont porté sur l'efficacité de la rigotine concernant certains aspects spécifiques de la maladie de Parkinson.
Deux études pivots ont porté sur l'efficacité de Neupro en monothérapie dans le traitement symptomatique de la maladie de Parkinson idiopathique au stade précoce. Les patients inclus dans ces études ne recevaient aucun traitement par un agoniste de la dopamine et la durée d'un éventuel traitement antérieur par la Levodopa était au moins inférieure à 6 mois avant l'inclusion dans l'étude.
L'évaluation primaire du résultat s'est faite sur la base du score des deux composantes Activités de la vie quotidienne (ADL, partie II) et Motricité (partie III) de l'échelle UPDRS «Unified Parkinson's Disease Rating Scale».
L'efficacité a été déterminée par la réponse des patients au traitement, mesurée à l'amélioration du taux de répondeurs et de la somme absolue en nombre de points des scores ADL et Examen moteur combinés (parties II + III de l'échelle UPDRS).
Dans l'une des études en double aveugle, 177 patients ont reçu de la rotigotine et 96 ont reçu un placebo. Partant d'une dose initiale de 2 mg/24 h, la dose individuelle optimale de rotigotine ou de placebo a été obtenue au terme d'une augmentation par paliers hebdomadaires de 2 mg/24 h jusqu'à une dose maximale de 6 mg/24 h. Les patients de chaque groupe de traitement ont continué de recevoir leur dose optimale pendant 6 mois.
À la fin du traitement d'entretien, 91% des sujets du groupe rotigotine avaient pour dose optimale la dose maximale autorisée dans cette étude, à savoir 6 mg/24 h. Une amélioration de 20% a été observée chez 48% des sujets recevant de la rotigotine et chez 19% des sujets recevant un placebo (différence: 29%, IC95% 18% – 39%, p <0,0001). L'amélioration moyenne du score UPDRS (parties II + III) par la rotigotine était de –3,98 points (valeur initiale: 29,9 points), tandis qu'on observait une péjoration de 1,31 point (valeur initiale: 30,0 points) dans le groupe traité par placebo. La différence, de 5,28 points, était statistiquement significative (p <0,0001).
Dans une seconde étude en double aveugle, 213 patients ont reçu de la rotigotine, 227 ont reçu du ropinirole et 117 un placebo. Partant d'une dose initiale de 2 mg/24 h, la dose individuelle optimale de rotigotine a été obtenue au terme d'une augmentation de 2 mg/24 h par paliers hebdomadaires sur 4 semaines jusqu'à une dose maximale de 8 mg/24 h. Chez les patients du groupe ropinirole, la dose optimale (au maximum 24 mg/jour) a été atteinte en 13 semaines. Les patients de chaque groupe de traitement ont ensuite été maintenus sous ce traitement d'entretien pendant 6 mois.
À la fin du traitement d'entretien (étude n° 17 dans le graphique ci-dessous), la dose optimale correspondait à la dose maximale autorisée, à savoir 8 mg/24 h, chez 92% des sujets du groupe rotigotine. Une amélioration de 20% a été observée chez 52% des sujets recevant de la rotigotine, 68% des sujets recevant du ropinirole et 30% des sujets sous placebo (différence rotigotine vs placebo: 21,7%, IC95% 11,1% – 32,4%; différence ropinirole vs placebo: 38,4%, IC95% 28,1% – 48,6%; différence ropinirole vs rotigotine: 16,6%, IC95% 7,6% – 25,7%). L'amélioration moyenne du score UPDRS (parties II+III) était de 6,83 points pour le bras rotigotine (valeur initiale: 33,2 points), de 10,78 points pour le bras ropinirole (valeur initiale: 32,2 points) et de 2,33 points pour le bras placebo (valeur initiale: 31,3 points). Toutes les différences entre traitements actifs et placebo étaient statistiquement significatives. Cette étude n'a pas permis de démontrer la non-infériorité de la rigotine par rapport au ropinirole.
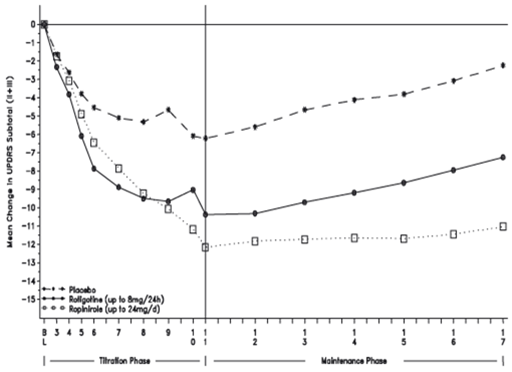
Une étude ouverte multicentrique et multinationale effectuée ultérieurement a porté sur la tolérance d'un changement de traitement consistant à remplacer pendant la nuit le ropinirole, le pramipexole ou la cabergoline par un patch de rotigotine. L'efficacité de cette substitution sur les symptômes de la maladie de Parkinson idiopathique faisait également l'objet de ce travail. 116 patients traités jusqu'ici par voie orale ont reçu une dose de rigotine pouvant atteindre 8 mg/24 h. Parmi eux, 47 étaient traités au ropinirole à une dose de 9 mg par jour maximum, 47 au pramipexole à 2 mg par jour maximum et 22 à la cabergoline à 3 mg par jour maximum. La substitution à la rigotine s'est révélée réalisable, la posologie n'ayant dû être légèrement ajustée (2 mg/24 h en moyenne) que chez 2 des patients traités au ropinirole, 5 des patients traités au pramipexole et 4 des patients traités à la cabergoline. Des améliorations du score UPDRS, parties I à IV, ont été observées. Il n'y avait pas de modification du profil de sécurité par rapport aux études antérieures.
Deux études pivot supplémentaires ont été réalisées chez des patients sous traitement concomitant par la lévodopa. Le critère principal d'évaluation du résultat était la réduction du temps «off» (en heures). L'efficacité a été déterminée par la réponse du sujet au traitement en termes de répondeur et d'amélioration absolue du temps «off».
Lors d'une étude en double aveugle, 113 patients ont reçu de la rotigotine à une dose maximale de 8 mg/24 h, 109 patients ont reçu de la rotigotine à une dose maximale de 12 mg/24 h et 119 patients ont reçu un placebo.
La dose optimale de rotigotine ou de placebo a été obtenue par une augmentation hebdomadaire de 2 mg/24 h en débutant à 4 mg/24 h.
Les patients de chaque groupe de traitement ont été maintenus à leur dose optimale pendant 6 mois. À la fin du traitement d'entretien, on a constaté une amélioration d'au moins 30% chez 57% et 55% des sujets traités à la rotigotine aux doses respectives de 8 mg/24 h et 12 mg/24 h et chez 34% des sujets recevant un placebo (différence: 22% et 21% respectivement, IC95% 10% – 35% et 8% – 33% respectivement, p <0,001 pour les deux groupes rotigotine). Avec la rotigotine, les réductions moyennes du temps «off» ont été respectivement de 2,7 et 2,1 heures, tandis que dans le groupe placebo il a été observé une réduction de 0,9 heures. Les différences étaient statistiquement significatives (p <0,001 et p= 0,003 respectivement).
Au cours d'une seconde étude en double aveugle, 201 patients ont reçu de la rotigotine, 200 ont reçu du pramipexole et 100 patients ont reçu un placebo. La dose optimale de rotigotine a été obtenue par une augmentation hebdomadaire de 2 mg/24 h en débutant à 4 mg/24 h jusqu'à une dose maximale de 16 mg/24 h. Les patients du groupe pramipexole ont reçu 0,375 mg la première semaine, 0,75 mg la deuxième semaine et la dose a ensuite été augmentée de 0,75 mg par semaine jusqu'à la dose individuelle optimale, avec un maximum de 4,5 mg/jour. Les patients de chaque groupe de traitement ont été maintenus à leur dose optimale pendant 4 mois.
À la fin du traitement d'entretien, on a constaté une amélioration d'au moins 30% de la période «off» absolue chez 60% des sujets traités par la rotigotine, chez 67% des sujets traités par pramipexole et chez 35% des sujets recevant un placebo (différence rotigotine versus placebo: 25%, IC95% 13%; 36%, différence pramipexole versus placebo: 32%, IC95% 21%; 43%, différence pramipexole versus rotigotine: 7%, IC95% –2%; 17%). La non-infériorité par rapport au pramipexole n'a ainsi pas été mise en évidence.
La réduction moyenne du temps «off» a été de 2,5 heures dans le groupe rotigotine, de 2,8 heures dans le groupe pramipexole et de 0,9 heures dans le groupe placebo. Toutes les différences entre traitements actifs et placebo ont été statistiquement significatives.
Une autre étude multinationale a été menée en double aveugle sur 287 patients atteints de la maladie de Parkinson au stade précoce ou au stade avancé accompagnée d'un contrôle insuffisant de la mobilité matinale. 81,5% des patients ont été en outre traités par la lévodopa. 190 patients ont reçu de la rotigotine et 97 un placebo. Les patients ont été titrés sur une période de 8 semaines en commençant par une dose de 2 mg/24 h par paliers de 2 mg/24 h par semaine à la dose optimale de rotigotine ou de placebo jusqu'à la dose maximale de 16 mg/24 h et ces doses d'entretien ont été maintenues pendant 4 semaines. Les paramètres cibles co-primaires étaient la mobilité matinale, mesurée par la Unified Parkinson's Disease Rating Scale (UPDRS III) et les troubles du sommeil nocturne, mesurés par la Parkinson's Disease Sleep Scale (PDSS-2) modifiée. À la fin de la phase d'entretien, la valeur UPDRS III des patients traités par la rotigotine (initialement 29,6) s'était améliorée de 7,0 points et la valeur des patients du groupe placebo (initialement 32,0) s'était améliorée de 3,9 points. L'augmentation de la valeur totale de la PDSS-2 dans le groupe rotigotine était de 5,9 points (initialement 19,3) et de 1,9 points dans le groupe placebo (initialement 20,5). Les différences de traitements pour les variables co-primaires étaient statistiquement significatives (p= 0,0002 et p <0,0001).
Pharmacocinétique
Absorption
Après application, la rotigotine est libérée en continu par le patch transdermique et est absorbée par la peau. Lorsque le patch transdermique est appliqué une fois par jour et porté pendant 24 heures, les concentrations à l'équilibre sont atteintes un à deux jours après le début de l'application. La rotigotine présente un profil pharmacocinétique proportionnel à la dose sur une plage posologique de 1 mg/jour (5 cm²) à un maximum de 16 mg/jour (2× 40 cm²).
Environ 45% du principe actif contenu dans le patch transdermique est libéré dans la peau en 24 heures. La biodisponibilité absolue après application transdermique est d'environ 37%.
L'alternance des sites d'application du patch transdermique peut engendrer des variations du taux plasmatique d'un jour à l'autre. Les différences de biodisponibilité de la rotigotine varient de 2% (diff. entre bras et flanc) à 46% (diff. entre épaule et cuisse). Néanmoins, rien n'indique que ces variations ont un impact significatif sur le résultat clinique.
Distribution
La liaison in vitro de la rotigotine aux protéines plasmatiques est de l'ordre de 92%.
Le volume de distribution apparent chez l'être humain est d'environ 84 l/kg.
Métabolisme
La rotigotine est en grande partie métabolisée.
La rotigotine est métabolisée par N-désalkylation et par conjugaison directe et secondaire. Les résultats in vitro indiquent que différentes isoformes du CYP sont capables de catalyser la N-désalkylation de la rotigotine. Les principaux métabolites de la rotigotine sont des sulfates et des glucoronates conjugués de la substance mère, ainsi que des métabolites N-désalkylés qui sont biologiquement inactifs. Les données disponibles sur les métabolites sont incomplètes.
Élimination
Environ 71% de la dose de rotigotine sont éliminés avec les urines sous forme de conjugués inactifs, et une plus faible proportion, environ 23%, est excrétée par voie fécale.
La clairance de la rotigotine après administration transdermique est de l'ordre de 10 l/min et sa demi-vie d'élimination totale est de 5 à 7 heures. Le profil pharmacocinétique montre une élimination biphasique avec une demi-vie initiale d'environ 2 à 3 heures.
Neupro étant administré par voie transdermique, aucun effet causé par des aliments ou une maladie gastro-intestinale n'est attendu.
Cinétique pour certains groupes de patients
Comme le traitement par Neupro débute à faible dose et est augmenté progressivement en fonction de la tolérance clinique pour obtenir un effet thérapeutique optimal, aucun ajustement de la dose en fonction du sexe, du poids ou de l'âge n'est nécessaire (voir «Posologie/Mode d'emploi»).
Troubles de la fonction hépatique
Chez les patients atteints d'insuffisance hépatique modérée, aucune augmentation significative des taux plasmatiques de rotigotine n'a été observée. Neupro n'a pas été étudié chez les patients souffrant d'insuffisance hépatique sévère et est contre-indiqué chez ces patients (voir «Contre-indications»).
Troubles de la fonction rénale
Chez les patients atteints d'insuffisance rénale légère à sévère, aucune augmentation significative des taux plasmatiques de rotigotine n'a été observée.
Les concentrations plasmatiques de conjugués de la rotigotine et de ses métabolites désalkylés augmentent en cas d'atteinte de la fonction rénale. Néanmoins, une contribution de ces métabolites aux effets cliniques est peu probable (voir «Posologie/Mode d'emploi, instructions posologiques particulières»).
Données précliniques
Lors d'études de toxicité de doses répétées et à long terme, les principaux effets étaient associés aux effets pharmacodynamiques des agonistes de la dopamine et à la réduction subséquente de la sécrétion de prolactine.
Comme on l'a rapporté pour d'autres agonistes de la dopamine, une liaison de la rotigotine aux tissus contenant de la mélanine (yeux) était mesurable chez le rat pigmenté et le singe après administration d'une dose unique de rotigotine, mais celle-ci a progressivement disparu au cours de la période d'observation de deux semaines.
Dans une étude de 3 mois chez des rats albinos, une dégénérescence rétinienne a été observée à l'examen microscopique par transmission chez les animaux ayant reçu une dose (en mg/m²) équivalente à 2,8 fois la dose maximale recommandée chez l'être humain. Les effets étaient plus marqués chez les rats femelles. Aucune étude supplémentaire n'a eu lieu pour évaluer plus en détail cette pathologie spécifique. Chez aucune des espèces animales testées, on n'a observé de dégénérescence rétinienne lors de l'évaluation histopathologique de routine des yeux dans le cadre des études toxicologiques. On ignore la signification de ces observations pour l'être humain.
Mutagénicité
La rotigotine n'a pas induit de mutations génétiques au test d'Ames, mais a montré des effets lors de l'analyse du lymphome de souris in vitro avec activation métabolique, ainsi que des effets moins prononcés en l'absence d'activation métabolique. Cet effet mutagène, qui pourrait être imputable à un effet clastogène de la rotigotine, n'a toutefois été confirmé ni par le test du micronoyau in vivo chez la souris, ni par le test de synthèse d'ADN non programmée (test UDS) chez le rat. Cet effet étant plus ou moins parallèle à une diminution globale de la croissance relative des cellules, il pourrait être lié à un effet cytotoxique du principe actif. Par conséquent, on ne sait quelle importance accorder à ce seul test de mutagénicité positif in vitro.
Cancérogénicité
Dans une étude de carcinogenèse, des rats mâles ont développé des tumeurs, ainsi qu'une hyperplasie, au niveau des cellules de Leydig. Des tumeurs malignes ont été observées principalement au niveau de l'utérus chez les femelles ayant reçu des doses moyennes et élevées de rotigotine. Ces altérations sont des effets bien connus des agonistes de la dopamine chez le rat après un traitement à vie et sont considérées comme dépourvues de signification pour l'être humain.
Toxicité de reproduction
Les effets potentiels de Neupro ont été examinés chez le rat, le lapin et la souris. Neupro s'est avéré non tératogène chez les trois espèces animales, mais a montré des effets embryotoxiques chez le rat et la souris à des doses toxiques pour la mère. Neupro n'a pas eu d'effet sur la fertilité des mâles chez le rat, mais a clairement réduit la fertilité des femelles chez le rat et la souris en raison de ses effets sur les concentrations de prolactine, particulièrement marqués chez les rongeurs.
Remarques particulières
Incompatibilités
Pour ne pas diminuer l'adhérence du patch transdermique, on évitera d'appliquer des crèmes, lotions, poudres ou autres produits topiques sur les régions de la peau destinées à l'application de Neupro.
Influence sur les méthodes de diagnostic
La couche de support de Neupro contient de l'aluminium. Pour éviter des brûlures cutanées, Neupro doit être retiré si le patient doit subir un examen d'imagerie par résonance magnétique (IRM) ou une cardioversion.
Stabilité
Le médicament ne doit pas être utilisé au-delà de la date figurant après la mention «EXP» sur l'emballage.
Remarques particulières concernant le stockage
Ne pas conserver au-dessus de 30 °C.
Conserver le récipient dans l'emballage d'origine et dans son sachet non ouvert.
Conserver hors de portée des enfants.
Remarques concernant la manipulation
Sachets-dose scellés.
Le patch Neupro ne doit pas être coupé en morceaux.
Il doit être appliqué immédiatement après avoir été retiré du sachet qui le protège.
Après usage, le patch transdermique contient encore des quantités non négligeables de principe actif. Après retrait, le patch transdermique usagé doit être plié en deux, le côté adhésif vers l'intérieur, de telle sorte que la matrice ne soit pas exposée, puis replacé dans le sachet d'origine et éliminé hors de portée des enfants. Tout patch transdermique usagé ou non utilisé doit être éliminé conformément à la réglementation en vigueur ou restitué au pharmacien.
Numéro d’autorisation
57417 (Swissmedic).
Présentation
Neupro 1 mg/24 h: 7 et 28 patchs (B)
Neupro 2 mg/24 h: 7 et 28 patchs (B)
Neupro 3 mg/24 h: 7 et 28 patchs (B)
Neupro 4 mg/24 h: 7 et 28 patchs (B)
Neupro 6 mg/24 h: 7 et 28 patchs (B)
Neupro 8 mg/24 h: 28 patchs (B)
Titulaire de l’autorisation
UCB-Pharma SA, Bulle.
Mise à jour de l’information
Mars 2021.