Composition
Principe actif: Fosaprépitant sous forme de fosaprépitant diméglumine.
Le fosaprépitant pour administration intraveineuse est la prodrogue lyophilisée de l'aprépitant. L'aprépitant est disponible également sous forme de capsules (Emend) pour administration orale.
Excipients: Sodium EDTA, polysorbate 80 (produit à partir de maïs génétiquement modifié), lactose, hydroxyde de sodium et/ou acide chlorhydrique pour l'ajustement du pH.
Forme galénique et quantité de principe actif par unité
Lyophilisat pour la reconstitution d'une solution pour perfusion. Chaque flacon ponctionnable pour administration intraveineuse contient 150 mg de fosaprépitant acide libre, correspondant à 245,3 mg de fosaprépitant diméglumine.
Indications/Possibilités d’emploi
Ivemend associé à un antagoniste 5-HT3 et à la dexaméthasone est indiqué chez les adultes pour la prévention des nausées et des vomissements aigus et retardés induits par une chimiothérapie hautement émétisante, y compris le cisplatine à des doses élevées, et par une chimiothérapie modérément émétisante.
Posologie/Mode d’emploi
Ivemend est une prodrogue lyophilisée de l'aprépitant à administrer par voie intraveineuse.
Ivemend 150 mg est administré le jour 1, en tant que perfusion de 20 à 30 minutes, environ 30 minutes avant la chimiothérapie. Ivemend doit être administré en association avec un corticostéroïde et un antagoniste 5-HT3 comme indiqué dans les tableaux ci-dessous. Avant de commencer le traitement par Ivemend 150 mg, l'information professionnelle de l'ondansétron doit être consultée.
Schéma thérapeutique recommandé pour la prévention des nausées et des vomissements dus à une chimiothérapie hautement émétisante:
|
|
Jour 1 |
Jour 2 |
Jour 3 |
Jour 4 |
|
Ivemend |
150 mg i.v. |
- |
- |
- |
|
Dexaméthason** |
12 mg p.o. |
8 mg p.o. |
8 mg p.o. 2x/j |
8 mg p.o. 2x/j |
|
Ondansétron |
Voir l'information professionnelle de l'ondansétron concernant le dosage approprié |
- |
- |
- |
** La dexaméthasone doit être administrée 30 minutes avant le début de la chimiothérapie au jour 1 ainsi que le matin des jours 2 à 4. La dexaméthasone doit aussi être administrée le soir des jours 3 et 4. La dose de dexaméthasone est déterminée en fonction des interactions médicamenteuses.
Schéma thérapeutique recommandé pour la prévention des nausées et des vomissements dus à une chimiothérapie modérément émétisante:
|
|
Jour 1 |
|
Ivemend |
150 mg i.v. |
|
Dexaméthason** |
12 mg p.o. |
|
Ondansétron*** |
2× 8 mg p.o. |
** La dexaméthasone doit être administrée 30 minutes avant le début de la chimiothérapie au jour 1. La dose de dexaméthasone est déterminée en fonction des interactions médicamenteuses.
*** La première capsule de 8 mg d'ondansétron doit être administrée 30 à 60 minutes avant le début de la chimiothérapie et une deuxième capsule de 8 mg doit être administrée 8 heures après la première dose au jour 1.
Pour la reconstitution de la solution injectable, voir la section «Remarques particulières»/«Remarques concernant la manipulation».
Informations générales
Pour plus d'informations concernant l'administration simultanée d'Ivemend et de corticostéroïdes, voir sous «Interactions».
Lors d'une association avec d'autres antiémétiques, il convient de consulter l'information professionnelle de l'antiémétique en question.
Instructions spéciales pour la posologie
Aucun ajustement posologique n'est nécessaire chez les patients souffrant d'insuffisance rénale sévère (clairance de la créatinine <30 ml/min) ni même chez les patients insuffisants rénaux au stade terminal et dialysés.
Aucun ajustement posologique n'est nécessaire chez les patients présentant une insuffisance hépatique légère ou modérée (score de Child-Pugh 5-9). On ne dispose d'aucune donnée clinique chez des patients souffrant d'insuffisance hépatique sévère (score de Child-Pugh >9).
La dose ne doit pas être adaptée en fonction de l'âge, du sexe, de l'appartenance ethnique ou de l'indice de masse corporelle (IMC).
Enfants et adolescents: la sécurité et l'efficacité d'Ivemend n'ont pas été étudiées chez l'enfant et l'adolescent.
Contre-indications
Ivemend est contre-indiqué chez les patients présentant une hypersensibilité au fosaprépitant, à l'aprépitant, au polysorbate 80 ou à un autre excipient de la composition.
Ivemend ne doit pas être administré en même temps que le pimozide, la terfénadine, l'astémizole ou le cisapride. L'inhibition en fonction de la dose de l'isoenzyme 3A4 du système du cytochrome P450 (CYP3A4) par l'aprépitant pourrait entraîner une élévation des concentrations plasmatiques de ces médicaments, susceptibles de provoquer des réactions sévères ou d'engager le pronostic vital (voir sous «Interactions»).
Mises en garde et précautions
Le fosaprépitant étant rapidement transformé en aprépitant, qui inhibe le CYP3A4 en fonction de la dose, Ivemend doit être utilisé avec précaution chez des patients prenant en même temps des médicaments métabolisés principalement par le CYP3A4; certains chimiothérapeutiques sont métabolisés par le CYP3A4 (voir sous «Interactions»). Les agents chimiothérapeutiques avec métabolisme connu par le CYP3A4 sont le docétaxel, le paclitaxel, l'étoposide, l'irinotécan, l'ifosfamide, l'imatinib, la vinorelbine, la vinblastine et la vincristine. L'inhibition faible du CYP3A4 par le fosaprépitant pourrait entraîner des concentrations plasmatiques plus élevées des médicaments utilisés en même temps (voir sous «Interactions»). Par conséquent, les agents chimiothérapeutiques métabolisés par le CYP3A4 doivent être utilisés avec précaution.
On a rapporté des cas de réactions d'hypersensibilité de type immédiat, y compris flushing, érythème, dyspnée et anaphylaxie/choc anaphylactique pendant ou peu après la perfusion de fosaprépitant. Ces réactions d'hypersensibilité ont généralement été traitées par un arrêt de la perfusion et un traitement approprié. Une reprise de la perfusion n'est pas recommandée chez les patients présentant une telle réaction d'hypersensibilité.
Des réactions au site de perfusion (ISRs) ont été rapportées lors de l'utilisation d'Ivemend (voir «Effets indésirables»). Dans la majorité des ISRs graves, y compris la thrombophlébite et la vasculite, on a rapporté une utilisation simultanée d'une chimiothérapie vésicante (p.ex. à base d'anthracycline), en particulier en conjonction avec une extravasation. Une nécrose a également été rapportée chez certains patients ayant reçu une chimiothérapie vésicante concomitante.
L'administration simultanée de fosaprépitant et de warfarine peut entraîner une diminution cliniquement significative du temps de prothrombine (INR). Le temps de prothrombine (INR) des patients traités à long terme à la warfarine doit être étroitement surveillé au cours des deux premières semaines et particulièrement aux jours 7 à 10 après le début du traitement au fosaprépitant associé à chacun des cycles de la chimiothérapie (voir sous «Interactions»). Aucune étude d'interaction n'a été réalisée avec l'acénocoumarol et la phenprocoumone. Chez les patients sous traitement à long terme à l'acénocoumarol ou à la phenprocoumone, le temps de prothrombine (INR) devrait être surveillé étroitement pendant 2 semaines après le début du traitement au fosaprépitant (voir sous «Interactions»).
L'efficacité des contraceptifs hormonaux peut être réduite pendant l'administration de fosaprépitant et pendant les 28 jours qui suivent. C'est pourquoi des méthodes contraceptives alternatives ou complémentaires doivent être utilisées durant le traitement par le fosaprépitant et 2 mois après l'administration de fosaprépitant (voir sous «Interactions»).
L'administration simultanée de fosaprépitant et de dérivés de l'ergotamine, qui sont des substrats du CYP3A4, peut provoquer des élévations des taux plasmatiques de ces substances. En raison du risque potentiel d'une toxicité due à l'ergotamine, la prudence est donc de mise.
Ivemend ne doit jamais être injecté en bolus. Il doit toujours être administré dilué, sous forme de perfusion intraveineuse lente. Ivemend ne doit jamais être administré par voie intramusculaire ou sous-cutanée. Une légère thrombose au site de ponction a été observée lors de l'administration de doses élevées. Si des signes ou symptômes d'une irritation locale se manifestent, l'injection/perfusion doit être arrêtée et poursuivie dans une autre veine.
Interactions
Le fosaprépitant administré par voie intraveineuse est rapidement transformé en aprépitant. On peut donc supposer que les médicaments interagissant avec l'aprépitant oral le feront également avec le fosaprépitant. Les informations suivantes proviennent des données obtenues avec l'aprépitant oral ainsi qu'avec le fosaprépitant en association avec la dexaméthasone orale, le midazolam oral ou le diltiazem oral.
L'aprépitant est un substrat, un inhibiteur faible à modéré (en fonction de la dose) et un inducteur du CYP3A4. L'aprépitant est également un inducteur du CYP2C9.
Ivemend 150 mg à une dose unique est un faible inhibiteur du CYP3A4.
Influence du fosaprépitant/aprépitant sur la pharmacocinétique d'autres médicaments
L'aprépitant en tant qu'inhibiteur faible à modéré du CYP3A4 et Ivemend 150 mg en tant que faible inhibiteur du CYP3A4 peuvent provoquer une élévation des concentrations plasmatiques d'autres médicaments oraux utilisés en même temps et métabolisés par le CYP3A4.
Le fosaprépitant ne doit pas être administré en même temps que le pimozide, la terfénadine, l'astémizole ou le cisapride. L'inhibition en fonction de la dose du CYP3A4 par l'aprépitant pourrait entraîner une élévation des concentrations plasmatiques de ces médicaments, susceptibles de provoquer des réactions sévères ou d'engager le pronostic vital (voir sous «Contre-indications»).
Il a été montré que l'aprépitant induit le métabolisme de la warfarine S(-) et du tolbutamide, tous deux métabolisés par le CYP2C9. L'administration simultanée de fosaprépitant et de ces médicaments ou d'autres substances connues pour être métabolisées par le CYP2C9, telles que la phénytoïne, peut entraîner une baisse des concentrations plasmatiques de ces produits.
Le fosaprépitant ne semble pas interagir avec des médicaments qui sont des substrats pour le transport réalisé par la P-glycoprotéine, comme une étude clinique sur les interactions médicamenteuses entre l'aprépitant oral et la digoxine l'a démontré.
Antagonistes 5-HT3: Lors d'études cliniques sur les interactions médicamenteuses, l'aprépitant administré selon un schéma thérapeutique avec 125 mg le jour 1 et 80 mg/jour les jours 2 et 3 n'a eu aucun effet cliniquement significatif sur la pharmacocinétique de l'ondansétron, du granisétron ou l'hydrodolasétron (le métabolite actif du dolasétron). Aucune étude n'a été réalisée avec le tropisétron.
Corticostéroïdes:
Dexaméthasone: En association avec une dose orale unique de 8 mg de dexaméthasone administrée les jours 1, 2 et 3, le fosaprépitant à dose unique de 150 mg administré par voie intraveineuse le jour 1 a entraîné, aux jours 1 et 2, une augmentation d'un facteur ~2 de l'AUC0-24h de la dexaméthasone, un substrat du CYP3A4. En association avec une administration de fosaprépitant 150 mg i.v. le jour 1, la dose orale de dexaméthasone doit être réduite d'environ 50% les jours 1 et 2 afin que les expositions de dexaméthasone correspondent à celles sans traitement complémentaire au fosaprépitant. Le schéma posologique recommandé inclut déjà cette réduction. (Voir sous «Posologie/mode d'emploi»).
Méthylprednisolone: L'aprépitant oral administré dans le cadre d'un schéma thérapeutique (une dose de 125 mg au jour 1 et 80 mg/jour aux jours 2 et 3) a augmenté l'AUC de la méthylprednisolone, un substrat du CYP3A4, d'un facteur 1,3 au jour 1 et 2,5 au jour 3 (dose de méthylprednisolone 125 mg i.v. au jour 1 et 40 mg par voie orale aux jours 2 et 3).
Cytostatiques: Au cours d'essais cliniques, l'aprépitant oral a été administré en même temps que les cytostatiques ci-après, métabolisés principalement ou en partie par le CYP3A4: l'étoposide, la vinorelbine, le docétaxel, l'ifosfamide, le cyclophosphamide, l'irinotécan et le paclitaxel. Les doses de ces cytostatiques n'ont pas été adaptées en fonction des interactions médicamenteuses potentielles. Néanmoins, chez des patients recevant de telles substances ou des autres cytostatiques métabolisés principalement par le CYP3A4, la prudence est de mise et un suivi minutieux supplémentaire est recommandé. Après l'introduction sur le marché, des événements concernant la neurotoxicité, un effet indésirable potentiel d'ifosfamide, ont été rapportés sous l'administration concomitante d'aprépitant avec l'ifosfamide (voir sous «Mises en garde et précautions»). En raison du petit nombre de patients inclus dans les études cliniques et ayant reçu les substrats du CYP3A4 le docétaxel, la vinblastine, la vincristine ou l'ifosfamide, une prudence particulière et un suivi attentif s'imposent chez les patients recevant ces agents chimiothérapeutiques ou d'autres agents chimiothérapeutiques métabolisés principalement par le CYP3A4 et n'ayant pas été examinés.
Docétaxel: La pharmacocinétique du docétaxel n'a pas été influencée par l'aprépitant oral (schéma thérapeutique pour la prévention des nausées et vomissements induits par une chimiothérapie) au cours d'une étude pharmacocinétique séparée.
Vinorelbine: La pharmacocinétique de la vinorelbine n'a pas été influencée par l'aprépitant oral (schéma thérapeutique pour la prévention des nausées et vomissements induits par une chimiothérapie) au cours d'une étude pharmacocinétique séparée.
Warfarine: Des volontaires sains, traités à long terme avec la warfarine à une dose stable, ont reçu une dose unique de 125 mg d'aprépitant oral au jour 1, suivie de 80 mg par jour aux jours 2 et 3. Bien qu'aucun effet de l'aprépitant oral sur l'AUC de la warfarine R(+) ou S(-) n'ait été constaté au jour 3, la concentration minimale de la warfarine S(-) (un substrat du CYP2C9) a diminué de 34% 5 jours après l'arrêt de l'emploi de l'aprépitant oral, et cette diminution était accompagnée d'une diminution de 14% du temps de prothrombine (INR). Cela laisse supposer une induction potentiellement significative sur le plan clinique du CYP2C9. Le temps de prothrombine (INR) des patients traités à long terme à la warfarine doit être étroitement surveillé au cours des deux premières semaines et particulièrement aux jours 7 à 10 après le début du traitement au fosaprépitant associé à chacun des cycles de la chimiothérapie. Aucune étude d'interaction avec l'acénocoumarol et la phenprocoumone n'a été réalisée. Chez les patients sous traitement à long terme à l'acénocoumarol ou à la phenprocoumone, le temps de prothrombine (INR) devrait être surveillé étroitement pendant 2 semaines après le début du traitement au fosaprépitant, et particulièrement aux jours 7 à 10.
Contraceptifs oraux: L'aprépitant, administré pendant 14 jours sous forme de capsule à la dose de 100 mg par jour en même temps qu'un contraceptif oral (35 µg d'éthinylestradiol et 1 mg de noréthistérone), a diminué l'AUC de l'éthinylestradiol de 43% et celle de la noréthistérone de 8%. Ces modifications pharmacocinétiques ont été associées à des saignements de privation anormaux.
Au cours d'une autre étude, un contraceptif oral (éthinylestradiol et noréthistérone) a été pris quotidiennement pendant 21 jours, et l'aprépitant oral a été administré selon le schéma thérapeutique suivant: 125 mg au jour 8 et 80 mg par jour aux jours 9 et 10; ondansétron 32 mg i.v. au jour 8 et dexaméthasone 12 mg par voie orale au jour 8 et 8 mg par jour aux jours 9, 10 et 11. Au cours de l'étude, l'AUC de l'éthinylestradiol avait baissé de 19% au jour 10 et les concentrations minimales de l'éthinylestradiol avaient baissé jusqu'à 64% aux jours 9 à 21. Bien que l'aprépitant oral n'ait pas provoqué de modification de l'AUC de la noréthistérone au jour 10, une diminution des concentrations minimales de la noréthistérone pouvant aller jusqu'à 60% a été notée aux jours 9 à 21.
L'efficacité des contraceptifs oraux peut diminuer durant et 28 jours après un traitement au fosaprépitant. Durant et 2 mois après le traitement de fosaprépitant, des mesures contraceptives alternatives ou supplémentaires doivent être prises.
Tolbutamide: Lorsque 500 mg de tolbutamide oral (un substrat du CYP2C9) sont administrés avant l'aprépitant oral et aux jours 4, 8 et 15 après l'aprépitant oral administré à la dose standard (125 mg au jour 1 et 80 mg par jour aux jours 2 et 3), l'AUC du tolbutamide baisse, par rapport à la valeur d'avant l'administration d'aprépitant oral, de 23% au jour 4, de 28% au jour 8 et de 15% au jour 15.
Midazolam: En association avec une dose orale unique de midazolam 2 mg les jours 1 et 4, le fosaprépitant 150 mg à dose unique intraveineuse administrée le jour 1 a provoqué une augmentation d'un facteur ~1,8 de l'AUC0-∞ du midazolam le jour 1 et n'a eu aucun effet (× 1,0) sur l'AUC le jour 4. Le fosaprépitant 150 mg i.v. administré à dose unique le jour 1 est un faible inhibiteur du CYP3A4, sans indices d'une inhibition ou induction significative du CYP3A4 le jour 4.
Les effets d'autres médicaments sur la pharmacocinétique de l'aprépitant
L'aprépitant est un substrat du CYP3A4; aussi l'association de fosaprépitant avec des médicaments inhibant l'activité du CYP3A4 peut-elle entraîner des concentrations plasmatiques plus élevées d'aprépitant. Par conséquent, l'association de fosaprépitant avec des inhibiteurs puissants du CYP3A4 (p.ex. le kétoconazole) ne doit être instaurée qu'avec prudence. Étant donné que des inhibiteurs modérés du CYP3A4 (p.ex. le diltiazem) entraînent un doublement des concentrations plasmatiques de l'aprépitant, une prudence particulière est de mise dans ce cas aussi.
L'aprépitant est un substrat du CYP3A4; aussi l'association de fosaprépitant avec des médicaments induisant fortement l'activité du CYP3A4 (tels que la rifampicine, la carbamazépine, la phénytoïne ou le millepertuis [Hypericum perforatum]) peut-elle entraîner des concentrations plasmatiques réduites d'aprépitant, avec pour résultat une diminution de son efficacité.
Kétoconazole: Lorsqu'une dose unique de 125 mg d'aprépitant oral a été administrée au jour 5 d'un schéma posologique de 10 jours à raison de 400 mg par jour de kétoconazole (un inhibiteur puissant du CYP3A4), l'AUC de l'aprépitant a été augmentée d'un facteur de 5 environ et la demi-vie terminale de l'aprépitant a été prolongée de 3 fois environ. L'association de fosaprépitant avec des inhibiteurs du CYP3A4 devrait être instaurée avec prudence.
Rifampicine: Lorsqu'une dose unique de 375 mg d'aprépitant oral a été administrée au jour 9 d'un schéma posologique de 14 jours à raison de 600 mg par jour de rifampicine (un inducteur puissant du CYP3A4), l'AUC de l'aprépitant a été divisée par 11 environ et la demi-vie terminale moyenne de l'aprépitant a été divisée par 3 fois environ. L'association de fosaprépitant avec des inducteurs puissants du CYP3A4 peut provoquer une baisse des concentrations plasmatiques de l'aprépitant et par conséquent une diminution de son efficacité.
Autres interactions
Diltiazem: Chez les patients souffrant d'hypertension artérielle légère à modérée, l'administration d'une perfusion de 100 mg de fosaprépitant associée à 120 mg de diltiazem trois fois par jour a entraîné une augmentation d'un facteur 1,5 de l'AUC de l'aprépitant et, simultanément, une augmentation d'un facteur 1,4 de l'AUC du diltiazem. Ces effets pharmacocinétiques ont entraîné une réduction de la pression artérielle diastolique (réduction de 16,8 mmHg sous fosaprépitant par rapport à une réduction de 10,5 mmHg sans fosaprépitant) et de la pression artérielle systolique (réduction de 24,4 mmHg sous fosaprépitant par rapport à une réduction de 18,8 mmHg sans fosaprépitant). Ces effets pharmacocinétiques n'ont cependant pas provoqué de modification cliniquement significative de la fréquence cardiaque ou de l'intervalle PR allant au-delà des effets provoqués par le diltiazem seul.
Paroxétine: L'association d'aprépitant sous forme de comprimé (correspondant à 85 mg ou 170 mg sous forme de capsule) 1× par jour et de 20 mg de paroxétine une fois par jour a provoqué, aussi bien pour l'aprépitant que pour la paroxétine, une diminution de l'AUC de 25% environ et une baisse de la Cmax de 20% environ.
Grossesse/Allaitement
Grossesse
Aucune étude clinique appropriée et contrôlée n'a été réalisée chez les femmes enceintes. Ivemend ne devrait être employé pendant la grossesse qu'en cas de nécessité absolue.
Allaitement
Le fosaprépitant administré par voie intraveineuse est rapidement transformé en aprépitant. L'aprépitant passe dans le lait de rates allaitantes. On ignore si la substance passe dans le lait de mères allaitant leur enfant. Étant donné que de nombreux médicaments passent dans le lait maternel et que des effets indésirables de l'aprépitant peuvent se manifester chez le nourrisson allaité, il faut décider, en tenant compte de l'importance du médicament pour la mère, si la mère doit arrêter l'allaitement ou le médicament.
Effet sur l’aptitude à la conduite et l’utilisation de machines
Aucune étude correspondante n'a été effectuée avec Ivemend. Néanmoins, il est possible que certains effets indésirables d'Ivemend affectent chez certains patients l'aptitude à la conduite de véhicules ou à l'utilisation de machines. Les réactions à Ivemend peuvent varier d'une personne à l'autre (voir sous «Effets indésirables»).
Effets indésirables
La sécurité globale du fosaprépitant a été évaluée chez environ 1600 sujets.
Prévention de nausées et vomissements induits par la chimiothérapie (NVIC)
Chimiothérapie modérément émétisante (CME)
Dans une étude clinique contrôlée avec comparateur actif auprès de patients sous CME, la sécurité a été examinée chez 504 patients ayant reçu une dose unique d'Ivemend associée à ondansétron et dexaméthasone (régime avec fosaprépitant) en comparaison avec 497 patients ayant reçu l'ondansétron et la dexaméthasone seule (régime de contrôle). Les effets indésirables médicamenteux cliniquement significatifs ci-après ont été rapportés chez des patients traités par le régime avec fosaprépitant et avec une incidence plus élevée que dans le groupe de contrôle.
[Fréquents (≥1/100, <1/10); occasionnels (≥1/1000, <1/100)]
Infections et infestations
Occasionnels: candidose orale.
Troubles du métabolisme et de la nutrition
Occasionnels: diminution de l'appétit.
Affections cardiaques
Occasionnels: palpitations.
Affections vasculaires
Occasionnels: bouffée de chaleur.
Affections respiratoires, thoraciques et médiastinales
Occasionnels: toux, douleurs oropharyngées, irritation de la gorge.
Affections gastro-intestinales
Fréquents: constipation.
Occasionnels: ballonnements abdominaux, douleur abdominale, douleur abdominale supérieure, dyspepsie.
Troubles généraux et anomalies au site d'administration
Fréquents: douleurs au site de ponction.
Occasionnels: asthénie.
Chimiothérapie hautement émétisante (CHE)
Dans une étude clinique contrôlée avec comparateur actif auprès de patients sous CHE, la sécurité a été examinée chez 1143 patients ayant reçu une dose unique d'Ivemend 150 mg, en comparaison avec 1169 patients ayant reçu Emend (aprépitant) selon le schéma d'administration de 3 jours. Le profil de sécurité a globalement été similaire à celui dans l' étude MEC avec fosaprépitant.
Les effets indésirables médicamenteux supplémentaires cliniquement significatifs ci-après ont été rapportés sous fosaprépitant 150 mg et n'ont pas été signalés dans les études cliniques antérieures avec l'aprépitant par voie orale ou dans les études MEC avec fosaprépitant.
[Occasionnels (≥1/1000, <1/100), rares (≥1/10000, <1/1000)]
Affections vasculaires
Occasionnels: flushing, thrombophlébite (généralement au site de ponction).
Affections de la peau et du tissu sous-cutané
Occasionnels: érythème.
Troubles généraux et anomalies au site d'administration
Occasionnels: érythème au site de ponction, prurit au site de ponction.
Rares: induration au site de ponction.
Investigations
Occasionnels: tension artérielle élevée.
Étant donné que le fosaprépitant est transformé en aprépitant, les effets indésirables associés à l'aprépitant peuvent également être attendus sous Ivemend. Pour toute information concernant la sécurité sur les études qui ont été menées avec l'aprépitant par voie orale, l'information professionnelle de celui-ci doit être consultée.
Effets rapportés après l'introduction sur le marché
Les effets indésirables suivants ont été signalés après l'introduction sur le marché. Vu que ces données reposent sur des rapports de manière spontanée issus d'une population de taille inconnue, il est généralement impossible de déterminer clairement leur incidence ou le rapport causal avec le médicament.
Affections du système immunitaire
Réactions d'hypersensibilité, y compris réactions anaphylactiques/choc anaphylactique.
Des réactions d'hypersensibilité de type immédiat - avec flushing, érythème et dyspnée - sont survenues pendant la perfusion de fosaprépitant (voir «Mises en garde et précautions»).
Affections de la peau et du tissu sous-cutané
Prurit, éruption, urticaire, dans de rares cas syndrome de Stevens-Johnson, nécrolyse épidermique toxique.
Surdosage
On ne dispose d'aucune donnée spécifique concernant le traitement d'un surdosage. Des doses uniques pouvant aller jusqu'à 200 mg de fosaprépitant i.v. ou jusqu'à 600 mg d'aprépitant ont généralement été bien tolérées par des volontaires sains. Sur 33 sujets sains ayant reçu 200 mg de fosaprépitant, 3 ont développé une légère thrombophlébite au site d'injection. Dans le cadre d'études sur d'autres indications que les nausées et les vomissements induits par des cytostatiques, l'aprépitant à des doses de 375 mg par jour a été bien toléré durant une période pouvant aller jusqu'à 42 jours. Chez 33 patients cancéreux, des doses de 375 mg d'aprépitant au jour 1 et de 250 mg une fois par jour aux jours 2 à 5 ont normalement été bien tolérées.
Un état d'obnubilation et des céphalées ont été rapportés chez un patient ayant absorbé 1440 mg d'aprépitant.
En cas de surdosage, Ivemend doit être arrêté et des mesures générales de soutien ainsi qu'une surveillance doivent être mises en œuvre. En raison de l'activité antiémétique de l'aprépitant, les médicaments provoquant des vomissements peuvent éventuellement ne pas être efficaces.
L'aprépitant ne peut pas être éliminé par hémodialyse.
Propriétés/Effets
Code ATC: A04AD12
Mécanisme d'action
Le fosaprépitant est une prodrogue (un précurseur) de l'aprépitant. Par conséquent, ses effets antiémétiques sont attribuables à l'aprépitant.
L'aprépitant est un antagoniste sélectif à affinité élevée pour les récepteurs de la substance P neurokinine 1 (NK1). L'aprépitant ne présente qu'une très faible affinité ou aucune affinité pour d'autres sites sur des enzymes, des transporteurs, des canaux ioniques et des récepteurs (y compris les récepteurs de la dopamine et de la sérotonine) visés par les thérapies employées jusqu'ici pour les nausées et les vomissements induits par des cytostatiques (NVIC).
Des études précliniques ont montré que les antagonistes du récepteur NK1 inhibent, par des mécanismes centraux, les vomissements induits par les effets cytotoxiques d'une chimiothérapie (p.ex. du cisplatine). Des études utilisant la tomographie par émission de positrons (TEP) chez l'animal et l'être humain ont montré que l'aprépitant traverse la barrière hémato-encéphalique et qu'il occupe les récepteurs NK1 dans le cerveau. En outre, il a été constaté, lors d'études précliniques, que l'aprépitant possède un effet central prolongé, qu'il inhibe les phases aiguës et retardées des vomissements dus au cisplatine et qu'il renforce l'activité antiémétique de l'antagoniste du récepteur 5-HT3 ondansétron et du corticostéroïde dexaméthasone sur les vomissements induits par le cisplatine.
Évaluation du degré d'occupation des récepteurs NK1 à l'aide de la tomographie par émission de positrons
Dans une étude effectuée à l'aide de la tomographie par émission de positrons auprès de d'hommes jeunes en bonne santé (n=8), avec administration d'une dose intraveineuse unique de 150 mg de fosaprépitant, on a constaté une occupation des récepteurs NK1 de ≥100% au moment de Tmax et au bout de 24 h ainsi que de ≥97% au bout de 48 h et de 41% à 75% 120 heures après l'administration. Dans cette étude, l'occupation des récepteurs NK1 dans le cerveau a présenté une bonne corrélation avec les concentrations plasmatiques d'aprépitant. Le rapport entre l'occupation des récepteurs NK1 et l'efficacité clinique de l'aprépitant n'a cependant pas été déterminé.
Efficacité clinique
Prévention des nausées et des vomissements dus à une chimiothérapie (NVIC)
Chimiothérapie hautement émétisante (CHE)
Une étude randomisée, réalisée en double aveugle par groupes parallèles avec contrôle actif, a comparé le fosaprépitant 150 mg (N=1147) en perfusion intraveineuse unique avec le schéma thérapeutique de 3 jours d'aprépitant (N=1175) chez des patients recevant une chimiothérapie hautement émétisante au cisplatine (≥70 mg/m2). D'autres agents chimiothérapeutiques associés et fréquemment utilisés étaient: fluorouracile, gemcitabine, paclitaxel et étoposide. Tous les patients des deux groupes ont reçu de la dexaméthasone et de l'ondansétron (voir le Tableau 1).
Tableau 1
Régime de traitement de l'étude HEC*
|
|
Jour 1 |
Jour 2 |
Jour 3 |
Jour 4 |
|
Régime NVIC avec fosaprépitant | ||||
|
Fosaprépitant |
150 mg par voie intraveineuse en 20 à 30 minutes, environ 30 minutes avant la chimiothérapie |
non |
non |
non |
|
Dexaméthasone orale† |
12 mg |
8 mg |
8 mg deux fois par jour |
8 mg deux fois par jour |
|
Ondansétron |
Ondansétron‡ |
non |
non |
non |
|
Régime NVIC avec aprépitant oral | ||||
|
Aprépitant |
125 mg |
80 mg |
80 mg |
non |
|
Dexaméthasone orale§ |
12 mg |
8 mg |
8 mg |
8 mg |
|
Ondansétron |
Ondansétron‡ |
non |
non |
non |
* Des placebos ont été substitués au fosaprépitant, aux capsules d'aprépitant et à la dexaméthasone (le soir des jours 3 et 4) afin de conserver la mise en aveugle.
† La dexaméthasone a été administrée 30 minutes avant la chimiothérapie le premier jour et le matin des jours 2 à 4, ainsi que le soir des jours 3 et 4. La dose de 12 mg de dexaméthasone de jour 1 et la dose quotidienne unique de 8 mg de jour 2 reflètent une adaptation de la dose en fonction d'une interaction médicamenteuse avec le régime avec fosaprépitant (voir «Interactions»).
‡ L'ondansétron 32 mg par voie intraveineuse a été utilisé dans les études cliniques avec fosaprépitant et aprépitant. Bien que cette dose ait été utilisée dans les études cliniques, elle n'est plus la dose actuellement recommandée. Consulter l'information professionnelle de l'ondansétron pour connaître la dose actuellement recommandée.
§ La dexaméthasone a été administrée 30 minutes avant la chimiothérapie le premier jour et le matin des jours 2 à 4. La dose de 12 mg de dexaméthasone de jour 1 et la dose quotidienne unique de 8 mg des jours 2 à 4 reflètent une adaptation de la dose en fonction d'une interaction médicamenteuse avec le régime avec aprépitant oral (voir «Interactions»).
Ivemend 150 mg a démontré sa non-infériorité par rapport au schéma thérapeutique de 3 jours d'aprépitant. Le Tableau 2 présente un résumé des critères primaires et secondaires de l'étude.
Tableau 2
Taux de réponse des patients sous chimiothérapie modérément émétisante par groupe et phase de traitement – cycle 1
|
Critères* |
Régime avec fosaprépitant |
Régime avec aprépitant |
Différence† |
|
Réponse complète‡ | |||
|
Total§ |
71,9 |
72,3 |
-0,4 (-4,1, 3,3) |
|
Phase retardée§§ |
74,3 |
74,2 |
0,1 (-3,5, 3,7) |
|
Aucun vomissement | |||
|
Total§ |
72,9 |
74,6 |
-1,7 (-5,3, 2,0) |
* Critère primaire en caractères italiques.
** n: nombre des patients inclus dans l'analyse primaire d'une réponse complète.
† La différence et l'intervalle de confiance (IC) ont été calculés selon la méthode proposée par Miettinen et Nurminen, avec ajustement en fonction du sexe.
‡ Réponse complète = aucun vomissement, aucune médication de secours.
§ Total = 0 à 120 h après le début de la chimiothérapie au cisplatine.
§§ Phase retardée = 25 à 120 heures après le début de la chimiothérapie au cisplatine.
Chimiothérapie modérément émétisante (CME)
Au cours d'une étude parallèle randomisée en double aveugle avec médicament actif de contrôle, du fosaprépitant 150 mg en perfusion intraveineuse unique (N=502) associé à l'ondansétron et à la dexaméthasone (régime avec fosaprépitant) a été comparé à l'ondansétron et la dexaméthasone seuls (régime de contrôle, N=498) (voir le Tableau 3) chez des patients suivant un régime de chimiothérapie modérément émétisante. Les agents de chimiothérapie CME les plus fréquemment employés étaient le carboplatine, l'oxaliplatine et le cyclophosphamide.
Tableau 3
Régime de traitement de l'étude CME*
|
|
Jour 1 |
Jour 2 |
Jour 3 |
|
Régime NVIC avec fosaprépitant | |||
|
Fosaprépitant |
150 mg par voie intraveineuse en 20 à 30 minutes, environ 30 minutes avant la chimiothérapie |
non |
non |
|
Dexaméthasone orale† |
12 mg |
non |
non |
|
Ondansétron oral‡ |
8 mg pour 2 doses |
non |
non |
|
Régime NVIC de contrôle | |||
|
Dexaméthasone orale |
20 mg |
non |
non |
|
Ondansétron oral‡ |
8 mg pour 2 doses |
8 mg deux fois par jour |
8 mg deux fois par jour |
* Des placebos ont été substitués au fosaprépitant et à la dexaméthasone (jour 1) afin de conserver la mise en aveugle.
†La dexaméthasone a été administrée 30 minutes avant la chimiothérapie le premier jour. La dose de 12 mg reflète une adaptation de la dose en fonction d'une interaction médicamenteuse avec le régime avec fosaprépitant (voir «Interactions»).
‡La première dose d'ondansétron a été administrée entre 30 et 60 minutes avant la chimiothérapie le premier jour et la deuxième dose 8 heures après la première dose d'ondansétron.
L'efficacité du fosaprépitant a été évaluée sur la base des critères d'évaluation principaux et secondaires du Tableau 4. Elle s'est avérée supérieure à celle du régime de contrôle en ce qui concerne la réponse complète, aussi bien dans l'ensemble que dans la phase retardée.
Tableau 4
Pourcentage de patients sous chimiothérapie modérément émétisante
Taux de réponse par groupe de traitement et par phase
|
Critères d'évaluation |
Régime avec fosaprépitant |
Régime de contrôle |
Valeur de P |
|
Critère d'évaluation principal | |||
|
Réponse complète† | |||
|
Phase retardée‡ |
78,9 |
68,5 |
<0,001 |
|
Critères d'évaluation secondaires importants | |||
|
Réponse complète† | |||
|
Total§ |
77,1 |
66,9 |
<0,001 |
|
Phase aiguë¶ |
93,2 |
91 |
0,184 |
* N: nombre de patients inclus dans la population en intention de traiter.
† Réponse complète = aucun vomissement, aucune médication de secours.
‡ Phase retardée = 25 à 120 heures après le début de la chimiothérapie.
§ Total = 0 à 120 h après le début de la chimiothérapie.
¶ Phase aiguë = 0 à 24 heures après le début de la chimiothérapie.
Les courbes de Kaplan-Meier de la Figure 1 montrent que le délai avant le premier vomissement était plus long avec le régime avec fosaprépitant qu'avec le régime de contrôle (valeur nominale de p <0,001 avec le test de log-rank).
Figure 1: Pourcentage de patients sous chimiothérapie modérément émétisante sans vomissements au cours du temps
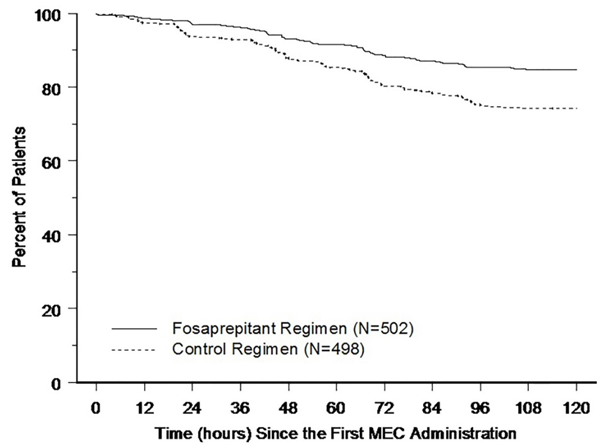
Régime avec fosaprépitant: fosaprépitant 150 mg le premier jour, associé à 16 mg d'ondansétron par voie orale le premier jour et 12 mg de dexaméthasone le premier jour. Des jours 2 à 3: placebo pour l'ondansétron toutes les 12 heures.
Régime de contrôle: 16 mg d'ondansétron par voie orale le premier jour, associé à 20 mg de dexaméthasone par voie orale le premier jour. Jours 2 à 3: 8 mg d'ondansétron par voie orale deux fois par jour.
Pharmacocinétique
Absorption
Après administration intraveineuse d'une dose unique de 150 mg de fosaprépitant sous forme de perfusion de 20 minutes, l'AUC0-∞ moyenne de l'aprépitant chez les sujets était de 35,0 µg•h/ml et la Cmax moyenne d'aprépitant était de 4,01 µg/ml.
Distribution
Le fosaprépitant est rapidement transformé en aprépitant.
L'aprépitant se lie à plus de 95% aux protéines plasmatiques. La moyenne géométrique du volume apparent de distribution à l'état stationnaire (Vdss) est d'environ 66 l chez l'homme.
L'aprépitant traverse le placenta chez les rats et traverse la barrière hémato-encéphalique chez les rats et les furets. Des essais par tomographie par émission de positrons chez l'homme ont montré que l'aprépitant traverse la barrière hémato-encéphalique (voir sous «Effets/Propriétés»).
Métabolisme
Dans des essais in vitro sur des préparations de foie de rat, de chien et de l'homme, le fosaprépitant a été transformé rapidement en aprépitant. En outre, le fosaprépitant subit une transformation rapide et presque complète en aprépitant dans des préparations S9 de différents autres tissus humains tels que le rein, le poumon et l'iléon. Il en ressort que la transformation du fosaprépitant en aprépitant s'effectue dans le foie et dans différents tissus extra-hépatiques. Chez l'homme, le fosaprépitant administré par voie intraveineuse est rapidement transformé en aprépitant, c'est-à-dire dans les 30 minutes suivant la fin de la perfusion.
L'aprépitant subit un métabolisme important. Chez le jeune adulte sain, l'aprépitant est responsable d'environ 24% de la radioactivité plasmatique mesurée 72 heures après une dose orale unique de 300 mg d'aprépitant marqué au [14C], ce qui indique une présence substantielle de métabolites dans le plasma. Sept métabolites seulement faiblement actifs de l'aprépitant ont été identifiés dans le plasma humain. Le métabolisme de l'aprépitant intervient largement via l'oxydation au niveau du cycle de la morpholine et de ses chaînes latérales. Des études réalisées in vitro sur des microsomes hépatiques humains indiquent que l'aprépitant est principalement métabolisé par le CYP3A4 et dans une faible mesure par les CYP1A2 et CYP2C19, mais non par les CYP2D6, CYP2C9 ou CYP2E1.
Après une dose intraveineuse de 100 mg de fosaprépitant radiomarqué au [14C], les mêmes métabolites qu'après une dose orale d'aprépitant radiomarqué au [14C] ont été retrouvés dans l'urine, les selles et le plasma. Après transformation de 245,3 mg de fosaprépitant diméglumine (correspondant à 150 mg de fosaprépitant acide libre) en aprépitant, 23,9 mg de phosphate et 95,3 mg de méglumine sont libérés.
Élimination
Après administration intraveineuse d'une dose unique de 100 mg de fosaprépitant radiomarqué au [14C] chez des sujets sains, 57% de la radioactivité ont été retrouvés dans l'urine et 45% dans les selles.
L'aprépitant est éliminé par métabolisme; il n'est pas éliminé par voie rénale. Après administration à des sujets sains d'une dose intraveineuse unique de 300 mg d'aprépitant marqué au [14C], 5% de la radioactivité ont été récupérés dans les urines sous forme de métabolites et 86% dans les fèces.
La clairance plasmatique apparente de l'aprépitant varie entre 60 et 84 ml/min; la demi-vie terminale apparente varie entre 9 et 13 heures environ.
Cinétique pour certains groupes de patients
Patients âgés: Après administration orale d'une dose unique de 125 mg d'aprépitant au jour 1 et de doses de 80 mg aux jours 2 à 5, l'AUC0-24h de l'aprépitant a été de 21% supérieure au jour 1 et de 36% supérieure au jour 5 chez le sujet âgé (dès 65 ans) par rapport à l'AUC0-24h du jeune adulte. La Cmax a été supérieure de 10% au jour 1 et de 24% au jour 5 chez le sujet âgé par rapport à celle mesurée chez le jeune adulte. Ces différences ne sont pas considérées comme étant cliniquement significatives. Aucun ajustement de la dose d'aprépitant n'est donc nécessaire chez les patients âgés.
Enfants et adolescents: Le fosaprépitant n'a pas été étudié chez les patients de moins de 18 ans.
Sexe: Après administration orale d'une dose unique d'aprépitant, l'AUC0-24h et la Cmax de l'aprépitant chez les femmes sont supérieures de 9% et 17%, respectivement, à celle mesurée chez les hommes. La demi-vie de l'aprépitant chez les femmes a été inférieure de 25% à celle mesurée chez des hommes, alors que le Tmax survient approximativement au même moment. Ces différences ne sont pas considérées comme étant cliniquement significatives. Aucun ajustement de la dose n'est donc nécessaire en fonction du sexe.
Indice de masse corporelle (IMC): L'exposition systémique (AUC) baisse de façon statistiquement significative avec un IMC croissant. Malgré la signification statistique, la réduction n'est que légère et une influence cliniquement significative est peu probable. Un ajustement de la dose en fonction de l'IMC n'est donc pas nécessaire.
Insuffisance hépatique: Le fosaprépitant est métabolisé par différents tissus extra-hépatiques; il est donc improbable qu'une insuffisance hépatique affecte la transformation du fosaprépitant en aprépitant.
L'aprépitant oral a été bien toléré par les patients souffrant d'insuffisance hépatique légère à modérée. Après la prise d'une dose unique de 125 mg d'aprépitant au jour 1 et de 80 mg une fois par jour aux jours 2 et 3, l'AUC0-24h de l'aprépitant chez les patients souffrant d'insuffisance hépatique légère (score Child-Pugh 5-6) était inférieure au jour 1 de 11% et au jour 3 de 36% à celle mesurée chez les volontaires sains sous la même dose. Chez les patients souffrant d'insuffisance hépatique modérée (score Child-Pugh 7-9), l'AUC0-24h de l'aprépitant était supérieure de 10% au jour 1 et de 18% au jour 3 par rapport à celle mesurée chez les volontaires sains sous la même dose. Aucune signification clinique n'est attribuée à ces différences au niveau de l'AUC0-24h, c'est pourquoi aucune adaptation de la dose n'est nécessaire chez les patients souffrant d'insuffisance hépatique légère à modérée.
On ne dispose d'aucune donnée clinique ni pharmacocinétique chez les patients souffrant d'insuffisance hépatique sévère (score de Child-Pugh >9).
Insuffisance rénale: Une dose unique de 240 mg d'aprépitant oral a été administrée à des patients souffrant d'insuffisance rénale sévère (clairance de la créatinine <30 ml/min) et à des patients dialysés souffrant d'une affection rénale au stade terminal.
Chez les patients souffrant d'insuffisance rénale sévère, l'AUC0-∞ de l'aprépitant total (lié ou non aux protéines) a diminué de 21% et la Cmax a diminué de 32% par rapport à celle des sujets sains. Chez les patients dialysés souffrant d'une néphropathie à un stade terminal sous dialyse, l'AUC0-∞ de l'aprépitant total a diminué de 42% et la Cmax a diminué de 32%. En raison d'une baisse modérée de la liaison aux protéines de l'aprépitant chez les patients atteints de néphropathie, l'AUC de la molécule non liée et pharmacologiquement active n'est pas affectée de façon significative chez les patients insuffisants rénaux par rapport à l'AUC chez les sujets sains. Une hémodialyse réalisée 4 et 48 heures après la prise n'a eu aucun effet significatif sur la pharmacocinétique de l'aprépitant; moins de 0,2% de la dose a été récupérée dans le dialysat.
Par conséquent, aucun ajustement posologique n'est nécessaire chez les patients souffrant d'insuffisance rénale sévère ni chez les patients atteints d'une néphropathie à un stade terminal sous dialyse.
Données précliniques
Les données précliniques ne révèlent aucun risque particulier pour l'homme sur la base des études conventionnelles sur la toxicité sous des doses uniques et sur la base d'études sur la toxicité chronique, la génotoxicité, le potentiel cancérigène et la toxicité sur la reproduction.
Remarques particulières
Incompatibilités
Ivemend est incompatible avec toutes les solutions contenant des cations bivalents (par exemple Ca2+, Mg2+), y compris la solution de Hartmann et la solution de Ringer. Ivemend ne doit pas être reconstitué ou mélangé avec une solution dont la compatibilité physico-chimique n'a pas été déterminée.
Remarques concernant le stockage
Les flacons ponctionnables doivent être conservés au réfrigérateur (entre 2 et 8 °C).
Remarques concernant la manipulation
Reconstitution de la solution perfusable d'Ivemend (150 mg)
1.Injectez 5 ml de solution de NaCl à 0,9% dans le flacon ponctionnable, en dirigeant le jet vers la paroi du flacon pour éviter de faire mousser la solution. Agiter doucement le flacon. Eviter les fortes secousses et les projections violentes de la solution de NaCl à 0,9% sous pression.
2.Préparez une poche de perfusion contenant 145 ml de solution de NaCl à 0,9%.
3.Aspirez la totalité du volume du flacon et transférez le liquide aspiré dans la poche de perfusion préparée contenant 145 ml de solution de NaCl à 0,9%, de façon à obtenir un volume total de 150 ml. Retournez doucement la poche 2 à 3 fois sur elle-même.
Stabilité
La solution de perfusion reconstituée reste chimiquement et physiquement stable pendant 24 heures à température ambiante (15 à 25 °C).
Les médicaments destinés à une administration parentérale doivent être contrôlés visuellement avant l'administration pour exclure la présence de particules et de colorations dans la mesure où la solution et le contenant permettent un tel contrôle.
Numéro d’autorisation
57913 (Swissmedic).
Présentation
Ivemend 150 mg est disponible en emballages de 1 flacon ponctionnable [B].
Titulaire de l’autorisation
MSD MERCK SHARP & DOHME AG, Lucerne.
Mise à jour de l’information
Août 2018.
S-WPC-MK0517-IV-052018/CHE-2018-017870