Composition
Principes actifs
Ticagrelorum.
Excipients
Comprimés pelliculés 60 mg
Mannitol (E421), hydrogénophosphate de calcium dihydraté, carboxyméthylamidon sodique (type A), hydroxypropylcellulose (E463), stéarate de magnésium (E470b), hypromellose (E464), dioxyde de titane (E171), macrogol 400, oxyde de fer noir (E172), oxyde de fer rouge (E172).
1 comprimé pelliculé de 60 mg contient 0,25 mg de sodium.
Comprimés pelliculés 90 mg
Mannitol (E421), hydrogénophosphate de calcium dihydraté, carboxyméthylamidon sodique (type A), hydroxypropylcellulose (E463), stéarate de magnésium (E470b), hypromellose (E464), dioxyde de titane (E171), talc (E553b), macrogol 400, oxyde de fer jaune (E172).
1 comprimé pelliculé de 90 mg contient 0,38 mg de sodium.
Comprimés orodispersibles
Mannitol (E421), cellulose microcristalline (E460), crospovidone (E1202), xylitol (E967), hydrogénophosphate de calcium anhydre (E341), fumarate de stéaryle sodique, hydroxypropylcellulose (E463), silice colloïdale anhydre.
1 comprimé orodispersible contient 0,53 mg de sodium.
Indications/Possibilités d’emploi
BRILIQUE 90 mg
BRILIQUE 90 mg, en association avec l'acide acétylsalicylique (AAS), est indiqué dans la prévention des événements athérothrombotiques (décès d'origine cardio-vasculaire, infarctus du myocarde, accident vasculaire cérébral) chez les patients ayant un syndrome coronarien aigu [SCA] (angor instable [AI], infarctus du myocarde sans sus-décalage du segment ST [NSTEMI] ou infarctus du myocarde avec sus-décalage du segment ST [STEMI]), incluant les patients traités médicalement et ceux traités par une intervention coronaire percutanée (ICP) ou un pontage aorto-coronarien (PAC).
BRILIQUE 60 mg
BRILIQUE 60 mg, en association avec l'acide acétylsalicylique (AAS), est indiqué dans la prévention des événements athérothrombotiques (décès d'origine cardiovasculaire, infarctus du myocarde, accident vasculaire cérébral) chez les patients ayant des antécédents d'infarctus du myocarde datant d'au moins 12 mois. De plus, ces patients doivent présenter au moins un autre facteur de risque cardiovasculaire (voir «Efficacité clinique»).
Posologie/Mode d’emploi
Posologie usuelle
BRILIQUE 90 mg
Chez les patients ayant un syndrome coronarien aigu, le traitement par BRILIQUE doit être instauré avec une dose initiale de 180 mg (2 comprimés à 90 mg) puis poursuivi avec une dose de 90 mg deux fois par jour. Le traitement est recommandé pendant au moins 12 mois, à moins que l'arrêt de BRILIQUE soit cliniquement indiqué (voir «Propriétés/Effets»).
Les patients sous BRILIQUE doivent également prendre une faible dose quotidienne d'AAS (75 à 150 mg) en traitement d'entretien, sauf contre-indication spécifique. Chez les patients ayant un syndrome coronarien aigu, une dose de charge initiale d'AAS est recommandée (voir «Propriétés/Effets»).
Les patients qui, après 12 mois de traitement par BRILIQUE 90 mg deux fois par jour suite à un événement athérothrombotique aigu, restent exposés à un risque élevé de développement d'un événement athérothrombotique peuvent poursuivre le traitement sans interruption par BRILIQUE 60 mg deux fois par jour. Un risque élevé est défini par la présence d'au moins un autre facteur de risque cardiovasculaire (voir «Efficacité clinique»).
BRILIQUE 60 mg
Une dose de charge initiale de BRILIQUE n'est pas nécessaire chez les patients ayant des antécédents d'infarctus du myocarde datant d'au moins 12 mois; la dose recommandée est de 60 mg deux fois par jour. Les données sur l'efficacité et la sécurité d'emploi de BRILIQUE au-delà d'une prolongation du traitement de 3 ans sont limitées. Il convient d'évaluer régulièrement et individuellement lors des examens de routine le bénéfice et le risque liés au traitement par BRILIQUE, en particulier si le traitement se poursuit au-delà de 3 ans.
Les patients sous BRILIQUE 60 mg doivent également prendre une faible dose quotidienne d'AAS (75 à 150 mg) en traitement d'entretien, sauf contre-indication spécifique.
Les patients peuvent initier le traitement par BRILIQUE 60 mg indépendamment de leur traitement antiagrégant plaquettaire antérieur et indépendamment de l'interruption ou non du traitement. Les patients doivent arrêter leur traitement antiagrégant actuel avant d'initier le traitement par BRILIQUE 60 mg deux fois par jour en association avec l'AAS à faible dose au moment de prise prévu.
Mode d'administration
Comprimés pelliculés:
Pour les patients qui ne sont pas capables d'avaler le(s) comprimé(s) en entier, les comprimés de BRILIQUE peuvent être écrasés en une poudre fine et mélangés dans un demi-verre d'eau et bus immédiatement. Le verre doit être rincé avec un autre demi-verre d'eau et le contenu doit être bu immédiatement. Le mélange peut également être administré via une sonde naso-gastrique (CH8 ou plus). Il est important de nettoyer la sonde naso-gastrique en y faisant passer de l'eau après administration du mélange.
Comprimés orodispersibles:
Les comprimés orodispersibles de 90 mg peuvent être utilisés comme alternative à BRILIQUE 90 mg, comprimés pelliculés chez les patients ayant des difficultés à avaler les comprimés pelliculés en entier. Le comprimé orodispersible doit être placé sur la langue, où il sera rapidement dispersé dans la salive. Il peut ensuite être avalé avec ou sans eau (voir «Pharmacocinétique»). Le comprimé orodispersible peut aussi être dispersé dans de l'eau et administré via une sonde naso-gastrique (CH8 ou plus). Il est important de rincer la sonde naso-gastrique en y faisant passer de l'eau après administration du mélange. Le comprimé orodispersible de 60 mg n'est pas disponible.
Les comprimés pelliculés ou les comprimés orodispersibles BRILIQUE peuvent être pris indépendamment des repas.
Omission d'une dose
Il est recommandé de ne pas interrompre le traitement. En cas d'oubli de prise de BRILIQUE, le patient ne prendra que la prochaine dose à l'heure de sa prise habituelle suivante.
Passage d'un autre traitement antiagrégant plaquettaire à BRILIQUE
Lors du passage d'un autre traitement antiagrégant plaquettaire à BRILIQUE après un SCA antérieur, les patients doivent prendre la première dose de BRILIQUE 24 heures après l'administration de la dernière dose de l'antiagrégant plaquettaire utilisé jusque-là (voir «Propriétés/Effets»).
L'arrêt prématuré de tout traitement antiagrégant plaquettaire, y compris de BRILIQUE, pourrait augmenter le risque de décès d'origine cardiovasculaire, d'infarctus du myocarde ou d'accident vasculaire cérébral lié à la pathologie sous-jacente du patient (voir «Mises en garde et précautions»).
Chez les patients présentant un syndrome coronarien aigu, la dose de charge de 180 mg doit être administrée aussi rapidement que possible, quel que soit le traitement antiagrégant plaquettaire que le patient a reçu auparavant.
Instructions posologiques particulières
Patients présentant des troubles de la fonction hépatique
Aucun ajustement de la posologie n'est nécessaire chez les patients présentant de troubles légers de la fonction hépatique. BRILIQUE n'a pas été étudié chez les patients présentant des troubles sévères de la fonction hépatique et seules des informations limitées sont disponibles concernant les patients présentant des troubles modérés de la fonction hépatique (voir «Contre-indications», «Mises en garde et précautions» ainsi que «Pharmacocinétique»).
Patients présentant des troubles de la fonction rénale
Aucun ajustement posologique n'est nécessaire chez les patients présentant des troubles de la fonction rénale (voir «Pharmacocinétique»).
Patients âgés
Aucune adaptation de la posologie n'est nécessaire chez les patients âgés (voir «Pharmacocinétique»).
Enfants et adolescents
La sécurité d'emploi et l'efficacité de BRILIQUE chez les enfants âgés de moins de 18 ans n'ont pas été établies pour l'indication approuvée chez l'adulte (voir «Propriétés/Effets»).
Contre-indications
Hypersensibilité au principe actif ou à l'un des excipients.
Saignement pathologique en cours.
Antécédent d'hémorragie intracrânienne (voir «Effets indésirables»).
Insuffisance hépatique sévère (voir «Posologie/Mode d'emploi», «Mises en garde et précautions» ainsi que «Pharmacocinétique».
Traitement prolongé chez les patients avec un saignement gastro-intestinal sévère au cours des 6 derniers mois.
Administration concomitante de BRILIQUE avec de puissants inhibiteurs du CYP3A4 (par exemple kétoconazole, clarithromycine, néfazodone, ritonavir et atazanavir).
Mises en garde et précautions
Risque de saignement
Comme pour d'autres antiagrégants plaquettaires, le traitement par BRILIQUE comprend un risque hémorragique accru, saignement intracrânien y compris. Chez les patients ayant un risque hémorragique accru connu, l'utilisation de BRILIQUE doit être évaluée au vu du rapport entre ces risques et les bénéfices en termes de prévention d'événements athérothrombotiques. L'utilisation de BRILIQUE est contre-indiquée chez les patients ayant un antécédent d'hémorragie intracrânienne. BRILIQUE doit être utilisé avec prudence chez les types de patients suivants:
·Patients à risque accru de saignement (en raison, par exemple, d'un traumatisme récent, d'une intervention chirurgicale importante au cours des 30 derniers jours, d'une intervention chirurgicale intracrânienne ou de la moelle épinière au cours des 5 dernières années, d'un trouble de la fonction hépatique modéré, d'un saignement gastro-intestinal actuel ou récent) ou patients présentant un risque accru d'hémorragie traumatique, y compris les hémorragies intracrâniennes traumatiques. L'utilisation de BRILIQUE est contre-indiquée chez les patients ayant un saignement pathologique en cours, les patients ayant un antécédent d'hémorragie intracrânienne et les patients ayant une insuffisance hépatique sévère (voir «Contre-indications»).
·Patients recevant de manière concomitante des médicaments susceptibles d'augmenter le risque de saignement, par exemple, d'autres antiagrégants plaquettaires (antagonistes du récepteur P2Y12 de l'ADP, inhibiteurs de la GPIIb/IIIa), anti-inflammatoires non stéroïdiens (AINS), traitement chronique par de l'héparine de bas poids moléculaire, anticoagulants oraux (antagonistes de la vitamine K, nouveaux anticoagulants oraux) et/ou fibrinolytiques dans les 24 heures autour de l'administration de BRILIQUE.
Dans deux études (TICO et TWILIGHT) contrôlées et randomisées, menées chez des patients avec un SCA ayant subi une ICP avec pose d'un stent libérant un médicament, il a été mis en évidence que le risque d'hémorragie diminue lors de l'arrêt de l'AAS au bout de 3 mois de double traitement antithrombotique (DAPT avec ticagrélor et AAS) et de traitement ultérieur par ticagrélor comme simple thérapie antiplaquettaire (SAPT) pendant 9 ou 12 mois. Ce faisant, par comparaison avec une DAPT continue, une augmentation significative du risque d'événements cardiovasculaires indésirables graves (MACE) n'est pas survenue. La décision d'arrêter l'AAS au bout de 3 mois de DAPT chez les patients présentant un risque d'hémorragie accru doit reposer sur l'évaluation individuelle du rapport bénéfice/risque (c.-à-d. l'estimation du risque d'hémorragie par rapport au risque d'événements thrombotiques).
Patients ayant des antécédents d'accident vasculaire cérébral ischémique
Les patients ayant présenté un SCA et ayant des antécédents d'accident vasculaire cérébral ischémique peuvent être traités par BRILIQUE 90 mg pendant une durée allant jusqu'à 12 mois (étude PLATO). Dans l'étude PEGASUS, aucun patient ayant des antécédents d'infarctus du myocarde avec accident vasculaire cérébral ischémique antérieur n'a été inclus. Par conséquent, en l'absence de données, la prolongation du traitement au-delà de 12 mois n'est pas recommandée chez ces patients.
La transfusion plaquettaire n'a pas permis la réversion de l'effet antithrombotique de BRILIQUE chez les volontaires sains. Il est peu probable qu'une transfusion plaquettaire présente un bénéfice clinique chez les patients qui présentent un saignement.
L'administration concomitante de BRILIQUE et de desmopressine ne diminuant pas le temps de saignement («template-bleeding time»), il est peu probable que la desmopressine soit efficace dans la prise en charge thérapeutique des événements hémorragiques.
Un traitement antifibrinolytique (acide aminocaproïque ou acide tranexamique) et/ou un traitement par le facteur VIIa recombinant peuvent améliorer l'hémostase. Le traitement par BRILIQUE peut être repris après l'identification de la cause des saignements et leur contrôle.
Chirurgie
·Si une opération est nécessaire chez un patient, le médecin doit tenir compte du profil clinique individuel du patient et peser les avantages et les risques d'une anticoagulation poursuivie avant de décider quand le traitement par BRILIQUE doit être arrêté.
·En raison de la liaison réversible de ticagrelor, le rétablissement de l'agrégation plaquettaire est plus rapide chez les patients traités par BRILIQUE que chez ceux traités au clopidogrel. Dans l'étude OFFSET, l'inhibition de l'agrégation plaquettaire (IAP) atteinte en moyenne sous BRILIQUE 72 heures après l'administration était comparable à l'IAP moyenne sous clopidogrel 120 heures après l'administration. La régression plus rapide des effets pourrait indiquer un plus faible risque de complications hémorragiques, par exemple dans les situations exigeant une suspension du traitement antithrombotique à cause d'une opération ou d'un traumatisme (voir «Propriétés/Effets»).
·Dans l'étude PLATO, chez les patients ayant eu un pontage aorto-coronarien (PAC), la fréquence de saignements majeurs sous BRILIQUE a été comparable à celle sous clopidogrel pour tous les jours après la fin du traitement, sauf pour le jour 1, pour lequel BRILIQUE a été associé à une fréquence supérieure de saignements majeurs (voir «Effets indésirables»).
·Si un patient doit avoir une intervention chirurgicale planifiée et que l'effet antiplaquettaire n'est pas souhaité, BRILIQUE doit être arrêté 7 jours avant la chirurgie.
Patients ayant des troubles modérés de la fonction hépatique
L'expérience avec BRILIQUE est limitée chez les patients avec des troubles modérés de la fonction hépatique. La prudence est donc de rigueur chez ces patients. L'utilisation de BRILIQUE est contre-indiquée chez les patients présentant une insuffisance hépatique sévère (voir «Posologie/Mode d'emploi», «Contre-indications» ainsi que «Pharmacocinétique»).
Bradyarythmie
Comparé au clopidogrel, la surveillance par Holter-ECG lors du traitement par le ticagrelor a mis en évidence une fréquence élevée de pauses ventriculaires essentiellement asymptomatiques. Les patients avec un risque accru d'événements bradycardiques (par exemple, patients sans stimulateur cardiaque ayant un syndrome de dysfonctionnement sinusal, un bloc auriculo-ventriculaire (AV) du 2e ou du 3e degré ou une syncope liée à une bradycardie) ont été exclus des études principales évaluant la tolérance et l'efficacité et du ticagrelor. Par conséquent, la prudence s'impose chez ces patients en raison de l'expérience clinique limitée (voir «Propriétés/Effets»).
Les données collectées après la mise sur le marché indiquent une accumulation d'événements bradyarythmiques, y compris de blocs AV, chez les patients avec un SCA traités par BRILIQUE (voir «Effets indésirables»), favorisée par une ischémie cardiaque et des traitements concomitants ayant un effet chronotrope/dromotrope négatif. Cela doit être pris en compte lors de l'ajustement du traitement chez ces patients.
De plus, BRILIQUE doit être administré avec précaution en cas d'association à des médicaments connus pour induire des bradycardies. Cependant, aucune manifestation d'un effet indésirable cliniquement significatif n'a été observée dans l'étude PLATO après l'administration concomitante de médicaments connus comme pouvant induire une bradycardie (par exemple 96 % de patients traités aux bêtabloquants, 33 % traités aux inhibiteurs calciques diltiazem et vérapamil, 4 % traités à la digoxine) (voir «Interactions»).
Une sous-étude Holter a été réalisée dans le cadre de l'étude PLATO. Dans cette sous-étude, un plus grand nombre de patients présentait des pauses ventriculaires ≥3 secondes avec BRILIQUE qu'avec le clopidogrel pendant la phase aiguë du syndrome coronarien aigu (SCA). L'augmentation du nombre de pauses ventriculaires détectées dans Holter sous BRILIQUE était plus importante chez les patients ayant une insuffisance cardiaque chronique que dans la population générale de l'étude durant la phase aiguë du SCA. Au bout d'un mois de traitement par BRILIQUE, il n'y avait plus de différence par rapport à la population générale ou les patients sous clopidogrel. Il n'y a pas eu d'effets indésirables cliniques associés à ce trouble (incluant des syncopes ou la pose d'un stimulateur cardiaque) dans cette population de patients (voir «Propriétés/Effets»).
Dyspnée
Une dyspnée a été observée chez des patients traités par BRILIQUE. La dyspnée est généralement légère à modérée et disparaît souvent sans qu'il soit nécessaire d'arrêter le traitement. Les patients présentant un asthme/une BPCO peuvent avoir une augmentation du risque absolu de présenter une dyspnée sous BRILIQUE (voir «Effets indésirables»). BRILIQUE doit être utilisé avec précaution chez les patients avec un antécédent d'asthme et/ou de BPCO. Le mécanisme n'a pas été élucidé (voir aussi «Propriétés/Effets» – Mécanisme lié à l'adénosine). Si un patient développe une dyspnée nouvelle, prolongée ou aggravée, une exploration complète est nécessaire, et si elle n'est pas tolérée, le traitement par BRILIQUE doit être interrompu.
Apnée centrale du sommeil
Des cas d'apnée centrale du sommeil incluant la respiration de Cheyne-Stokes ont été rapportés depuis la commercialisation chez des patients prenant BRILIQUE. Si une apnée centrale du sommeil est suspectée, une évaluation clinique supplémentaire peut être envisagée.
Purpura thrombotique thrombocytopénique (PTT)
Un purpura thrombotique thrombocytopénique a été observé très rarement en lien avec l'utilisation de BRILIQUE. Le PTT est une maladie grave et nécessite un traitement immédiat.
Interférence avec les tests de laboratoire
Tests de la fonction plaquettaire visant au diagnostic de la thrombocytopénie induite par l'héparine (TIH)
Des résultats faux négatifs au test de la fonction plaquettaire à la recherche d'une thrombocytopénie induite par l'héparine (TIH) ont été rapportés chez des patients qui avaient reçu du ticagrelor. Ceci est dû au fait qu'au cours du test, le ticagrelor induit dans le sérum/plasma du patient une inhibition du récepteur P2Y12 sur les thrombocytes de donneurs sains. Des informations concernant le traitement concomitant avec le ticagrelor sont nécessaires afin de pouvoir interpréter les tests de la fonction plaquettaire à la recherche d'une TIH.
Le rapport bénéfice-risque de la poursuite du traitement doit être évalué avant d'envisager un arrêt du traitement par le ticagrelor en tenant compte de l'état prothrombotique en présence d'une TIH, mais aussi du risque hémorragique accru de l'utilisation concomitante d'anticoagulants et de ticagrelor.
Autres
En se basant sur la relation observée dans PLATO entre la dose d'entretien d'AAS et l'efficacité relative de BRILIQUE comparé au clopidogrel, l'administration concomitante de BRILIQUE et d'une forte dose d'entretien d'AAS (>300 mg) n'est pas recommandée (voir «Propriétés/Effets»).
L'administration concomitante de BRILIQUE avec des inhibiteurs puissants du CYP3A4 (par exemple, kétoconazole, clarithromycine, néfazodone, ritonavir et atazanavir) est contre-indiquée, car elle peut conduire à une augmentation importante de l'exposition au ticagrelor (voir «Interactions»).
L'administration concomitante de ticagrelor avec des inducteurs puissants du CYP3A4 (par exemple rifampicine, phénytoïne, carbamazépine, phénobarbital et millepertuis) doit être évitée, car l'administration concomitante peut entraîner une diminution de la concentration et de l'efficacité du ticagrelor (voir «Interactions»).
L'administration concomitante de BRILIQUE et de substrats du CYP3A4 à marge thérapeutique étroite (par exemple cisapride et des alcaloïdes de l'ergot de seigle) n'est pas recommandée, étant donné que BRILIQUE peut augmenter l'exposition à ces médicaments. L'administration concomitante de BRILIQUE et de simvastatine ou de lovastatine à plus de 40 mg n'est pas recommandée.
Une étroite surveillance clinique et des analyses de laboratoire régulières sont recommandées quand la digoxine ou d'autres médicaments susceptibles d'influencer l'hémostase sont administrés de manière concomitante avec BRILIQUE.
Les inhibiteurs puissants de la glycoprotéine P (PGP) (par exemple vérapamil, quinidine, ciclosporine) augmentent l'exposition au ticagrelor. Si l'association ne peut pas être évitée, elle doit être utilisée avec prudence.
Arrêt du traitement
Les patients chez lesquels le traitement par BRILIQUE doit être arrêté sont exposés à un risque accru d'événements cardiaques ou d'accident vasculaire cérébral. Il est recommandé de ne pas interrompre le traitement prématurément. Si l'administration de BRILIQUE doit être suspendue en raison d'un effet indésirable, le traitement doit être repris dès que possible si les avantages du traitement l'emportent sur le risque de subir l'effet indésirable ou si ce dernier a régressé (voir «Posologie/Mode d'emploi»).
Sodium
Ce médicament contient moins de 1 mmol (23 mg) de sodium par comprimé pelliculé ou par comprimé orodispersible, c.-à-d. qu'il est essentiellement «sans sodium».
Interactions
Le ticagrelor est essentiellement un substrat du cytochrome CYP3A4 et un inhibiteur faible du CYP3A4. Le ticagrelor est aussi un substrat et un inhibiteur faible de la PGP et il peut augmenter l'exposition aux substrats de la PGP.
Effets d'autres médicaments sur le ticagrelor
Médicaments métabolisés par le CYP3A4
Inhibiteurs puissants du CYP3A4: L'administration concomitante de kétoconazole et de ticagrelor a multiplié par 2,4 la Cmax du ticagrelor et par 7,3 son aire sous la courbe (AUC). La Cmax et l'AUC du métabolite actif ont été diminuées de respectivement 89 % et 56 %. Les autres inhibiteurs puissants du CYP3A4 (clarithromycine, néfazodone, ritonavir et atazanavir) ont probablement des effets similaires, et leur administration concomitante avec BRILIQUE est contre-indiquée (voir «Contre-indications»).
Inhibiteurs modérés du CYP3A4: L'administration concomitante de diltiazem avec le ticagrelor a augmenté de 69 % la Cmax du ticagrelor et de 174 % son AUC, et a diminué la Cmax du métabolite actif de 38 %, alors que son AUC est restée inchangée. Une dose unique de 90 mg de ticagrelor n'a eu aucun effet sur les concentrations plasmatiques du diltiazem. Les autres inhibiteurs modérés du CYP3A4 (par exemple amprénavir, aprépitant, érythromycine, fluconazole, vérapamil et jus de pamplemousse) auraient probablement des effets similaires et peuvent donc également être co-administrés avec BRILIQUE.
Ciclosporine (inhibiteur de la PGP et du CYP3A)
L'administration concomitante de ciclosporine (600 mg) avec le ticagrelor a augmenté de 130 % la Cmax du ticagrelor et de 183 % son AUC. L'AUC du métabolite actif était augmentée de 33 % en présence de ciclosporine et la Cmax diminuée de 15 %. Le ticagrelor n'avait aucune influence sur la concentration plasmatique de la ciclosporine.
Inducteurs du CYP3A4: L'administration concomitante de rifampicine avec le ticagrelor a diminué de 73 % la Cmax du ticagrelor et de 86 % son AUC. La Cmax du métabolite actif est restée inchangée et son AUC a diminué de 46 %. Les autres inducteurs du CYP3A4 (par exemple phénytoïne, carbamazépine, phénobarbital et millepertuis) pourraient également diminuer l'exposition au ticagrelor et limiter ainsi son efficacité.
Autres
Les études d'interactions pharmacologiques ont montré que l'administration concomitante du ticagrelor avec l'héparine, l'énoxaparine et l'AAS n'a aucun effet sur les taux plasmatiques du ticagrelor ou de son métabolite actif. Si cliniquement indiqué, les médicaments altérant l'hémostase doivent être administrés avec prudence en association avec BRILIQUE (voir «Mises en garde et précautions»).
Aucune donnée n'est disponible concernant l'administration concomitante de BRILIQUE avec des inhibiteurs puissants de la PGP (par exemple vérapamil, quinidine, ciclosporine) qui pourraient augmenter l'exposition au ticagrelor. Si cliniquement indiquée, leur administration concomitante doit être réalisée avec prudence (voir «Mises en garde et précautions»).
Une exposition retardée ou réduite aux inhibiteurs du P2Y12 par voie orale, y compris le ticagrelor et son métabolite actif, a été rapportée chez des patients traités par la morphine (environ 35 % de réduction avec le ticagrelor). Cette interaction peut être liée à une diminution de la motilité gastro-intestinale et donc s'appliquer à d'autres opioïdes. La pertinence clinique de cette interaction est inconnue.
Effets du ticagrelor sur d'autres médicaments
Médicaments métabolisés par le CYP3A4
Le ticagrelor est un inhibiteur faible du CYP3A4. Une administration concomitante de BRILIQUE et de substrats du CYP3A4 à marge thérapeutique étroite (par exemple cisapride ou alcaloïdes de l'ergot de seigle) n'est pas recommandée, puisque le ticagrelor peut augmenter l'exposition à ces médicaments (voir «Mises en garde et précautions»).
Simvastatine: l'administration concomitante du ticagrelor avec la simvastatine a augmenté de 81 % la Cmax de la simvastatine et de 56 % son AUC, et elle a augmenté de 64 % la Cmax de la simvastatine acide et de 52 % son AUC, les valeurs individuelles étant multipliées par 2 à 3 dans certains cas.
L'administration concomitante de ticagrelor avec des doses de simvastatine dépassant 40 mg par jour pourrait causer des effets indésirables de la simvastatine qu'il convient d'évaluer par rapport aux bénéfices potentiels. La simvastatine en dose unique de 80 mg n'a pas eu d'effet sur les concentrations plasmatiques du ticagrelor. Il est possible que le ticagrelor ait des effets similaires sur la lovastatine. L'administration concomitante de BRILIQUE avec des doses de simvastatine ou de lovastatine supérieures à 40 mg n'est pas recommandée (voir «Mises en garde et précautions»).
Atorvastatine: l'administration concomitante d'atorvastatine et de ticagrelor a augmenté de 23 % la Cmax de l'atorvastatine acide et de 36 % son AUC. Des augmentations comparables de l'AUC et de la Cmax ont été observées pour tous les métabolites de l'atorvastatine acide. Ces augmentations ne sont pas considérées comme cliniquement significatives.
Un effet similaire sur les autres statines métabolisées par CYP3A4 ne peut pas être exclu. 93 % des patients traités au ticagrelor dans le cadre de l'étude PLATO prenaient différentes statines sans que cette association ait donné lieu à des réserves de sécurité concernant la prise de statines chez cette cohorte.
Médicaments métabolisés par le CYP2C9
Tolbutamide
L'administration concomitante de ticagrelor avec le tolbutamide n'a pas modifié les concentrations plasmatiques respectives de ces médicaments. Une dose unique de 500 mg de tolbutamide n'a pas eu d'influence sur le taux plasmatique de ticagrelor, ce qui suggère que le ticagrelor n'est pas un inhibiteur du CYP2C9 et qu'une interférence avec les médicaments métabolisés par le CYP2C9 comme la warfarine et le tolbutamide est peu probable.
Des interactions pharmacodynamiques avec des antagonistes de la vitamine K ne peuvent cependant pas être exclues (voir «Mises en garde et précautions»).
Contraceptifs oraux
L'administration concomitante de BRILIQUE, de lévonorgestrel et d'éthinylestradiol a augmenté l'exposition à l'éthinylestradiol d'environ 20 %, mais n'a pas modifié la pharmacocinétique du lévonorgestrel. Aucun effet cliniquement significatif n'est attendu sur l'efficacité contraceptive du lévonorgestrel et de l'éthinylestradiol lors d'une administration concomitante de BRILIQUE.
Médicaments métabolisés par le CYP2D6 – venlafaxine
L'administration concomitante de ticagrelor et de venlafaxine n'a influencé ni l'AUC ni la Cmax de l'O-desméthylvenlafaxine ni l'AUC de la venlafaxine. La Cmax de la venlafaxine était augmentée de 22 % après l'administration d'une dose unique de ticagrelor. La venlafaxine n'avait aucune influence sur la concentration plasmatique du ticagrelor. Ces résultats indiquent que le ticagrelor n'est pas un inhibiteur du CYP2D6 et qu'il est improbable que le ticagrelor influence le métabolisme de médicaments métabolisés par le CYP2D6 comme la venlafaxine ou le métoprolol.
Substrats de la glycoprotéine P (PGP)
L'administration concomitante de BRILIQUE a augmenté de 75 % la Cmax de la digoxine et de 28 % son AUC. La Cmin moyenne de la digoxine a été augmentée d'environ 30 % – et chez certains patients multipliée par deux – lors de l'administration concomitante de ticagrelor. En présence de digoxine, la Cmax et l'AUC du ticagrelor et de son métabolite actif n'ont pas été modifiées. Une surveillance clinique et/ou biologique appropriée est donc recommandée lors de l'administration concomitante de BRILIQUE avec des médicaments substrats de la PGP à marge thérapeutique étroite, comme la digoxine ou la ciclosporine.
Rosuvastatine (substrat de la BCRP)
Il a été démontré que le ticagrélor multiplie la Cmax de la rosuvastatine par 2,4 à 2,6 et son ASC par 2,3, ce qui peut accroître le risque de myopathie. Il convient de peser les bénéfices de la prévention des événements cardiovasculaires indésirables majeurs par l'administration de rosuvastatine, d'une part, et les risques liés à l'élévation de la concentration plasmatique de la rosuvastatine, d'autre part.
Autres traitements concomitants
En raison d'observations de pauses ventriculaires et de bradycardies le plus souvent asymptomatiques, BRILIQUE doit être administré avec précaution en cas d'association avec des médicaments connus pour induire des bradycardies (voir «Mises en garde et précautions»). Cependant, aucune manifestation d'un effet indésirable cliniquement significatif n'a été observée dans l'étude PLATO après l'administration concomitante de médicaments connus comme pouvant induire une bradycardie (par exemple 96 % de patients traités aux bêtabloquants, 33 % traités aux inhibiteurs calciques diltiazem et vérapamil, 4 % traités à la digoxine).
Dans des études cliniques, BRILIQUE a été fréquemment administré à long terme en même temps que les médicaments suivants selon les besoins et les maladies concomitantes: AAS, inhibiteurs de la pompe à protons, statines, bêtabloquants, inhibiteurs de l'enzyme de conversion de l'angiotensine et antagonistes des récepteurs de l'angiotensine. BRILIQUE a été administré pour de courtes périodes en association avec de l'héparine, de l'héparine de bas poids moléculaire et des inhibiteurs de la GpIIb/IIIa. On n'a pas observé de signes d'effets indésirables cliniquement significatifs avec ces divers médicaments.
En raison d'interactions pharmacodynamiques potentielles, toute administration concomitante de BRILIQUE avec des médicaments connus pour altérer l'hémostase doit être réalisée avec prudence (voir «Mises en garde et précautions»).
En raison de rapports de saignements cutanés anormaux sous ISRS (par exemple paroxétine, sertraline et citalopram), BRILIQUE doit être administré avec précaution en cas d'association avec les ISRS car cela peut augmenter le risque de saignement.
Grossesse, allaitement
Aucune étude clinique n'a été effectuée avec des femmes enceintes ou allaitant.
Grossesse
Les données sur l'utilisation du ticagrelor chez la femme enceinte sont absentes ou limitées. Les études chez l'animal ont montré une toxicité sur la reproduction (voir «Données précliniques»). BRILIQUE ne doit pas être utilisé pendant la grossesse, sauf nécessité absolue.
Allaitement
On ignore si le ticagrelor passe dans le lait maternel. Des études sur des rats ont montré que le ticagrelor et son métabolite actif passent dans le lait. BRILIQUE ne doit pas être utilisé pendant la période d'allaitement.
Effet sur l’aptitude à la conduite et l’utilisation de machines
Aucune étude n'a été réalisée sur les effets de BRILIQUE sur l'aptitude à conduire des véhicules et à utiliser des machines. Il est attendu que BRILIQUE n'a aucun ou qu'un effet négligeable sur l'aptitude à conduire des véhicules et à utiliser des machines. Des états de vertige et de confusion ont été rapportés pendant le traitement par BRILIQUE. Les patients chez lesquels ces symptômes se manifestent doivent donc être prudents pour la conduite d'un véhicule ou la manipulation de machines.
Effets indésirables
Résumé du profil de sécurité d'emploi
La sécurité d'emploi de BRILIQUE a été évaluée dans deux grandes études de phase III (PLATO et PEGASUS) ayant inclus plus de 39 000 patients (voir «Propriétés/Effets»). Les effets indésirables médicamenteux importants observés lors de ces études sont discutés ci-dessous.
La sécurité d'emploi de BRILIQUE 90 mg chez les patients présentant des syndromes coronariens aigus (angor instable, NSTEMI et STEMI) a été évaluée dans l'étude PLATO. Dans cette étude les patients traités par BRILIQUE 90 mg deux fois par jour ont été comparés à des patients traités par clopidogrel 75 mg une fois par jour, les deux groupes recevant en association de l'AAS ou d'autres traitements standard.
Dans l'étude PLATO, la durée médiane de traitement par BRILIQUE 90 mg était de 277 jours. L'incidence des arrêts dus à des événements indésirables a été plus élevée chez les patients traités par BRILIQUE 90 mg que sous clopidogrel (7,4 % vs 5,4 %).
Dans l'étude PEGASUS, la sécurité de BRILIQUE a été évaluée chez des patients ayant des antécédents connus d'infarctus du myocarde datant d'au moins 12 mois et un risque élevé de développer des événements athérothrombotiques; l'étude a comparé des patients traités par BRILIQUE 60 mg deux fois par jour ou 90 mg deux fois par jour en association avec l'AAS à des patients sous AAS en monothérapie et suivant d'autres traitements standard. La durée médiane de traitement par BRILIQUE 60 mg était de 29,4 mois. Dans l'étude PEGASUS, l'incidence des arrêts dus à des événements indésirables était plus élevée chez les patients sous BRILIQUE que chez ceux sous AAS en monothérapie (16,1 % pour BRILIQUE 60 mg avec AAS vs 8,5 % pour AAS en monothérapie). Les effets indésirables les plus fréquemment rapportés sous ticagrelor étaient les saignements et la dyspnée.
Les effets indésirables suivants ont été identifiés dans les études cliniques de phase III PEGASUS et PLATO.
Ces effets indésirables sont classés en fonction de leur classe de systèmes d'organes MedDRA et de leur fréquence. Les catégories de fréquence sont définies selon les conventions suivantes:
«très fréquents» (≥1/10),
«fréquents» (≥1/100 à <1/10),
«occasionnels» (≥1/1000 à <1/100),
«rares» (≥1/10 000 à <1/1000),
«très rares» (<1/10 000).
Tumeurs bénignes, malignes et non précisées (incl. kystes et polypes)
Occasionnel: saignements d'une tumeur (par exemple saignement dû à un cancer de la vessie, de l'estomac ou du côlon).
Affections hématologiques et du système lymphatique
Très fréquent: saignements dus à des troubles hématologiques (par exemple augmentation de la tendance aux ecchymoses, hématomes spontanés, diathèse hémorragique) (10,3 %).
Troubles du métabolisme et de la nutrition
Très fréquent: hyperuricémiea (22,1 %).
Fréquent: goutte.
Affections psychiatriques
Occasionnel: état confusionnel.
Affections du système nerveux
Fréquent: vertiges, syncope.
Occasionnel: hémorragie intracrânienne (c'est-à-dire une hémorragie intracrânienne spontanée, causée par une intervention ou par un traumatisme).
Affections oculaires
Occasionnel: hémorragie oculaire (par exemple intra-oculaire, conjonctivale, rétinienne).
Affections de l'oreille et du labyrinthe
Fréquent: vertiges.
Occasionnel: hémorragie de l'oreille.
Affections vasculaires
Fréquent: hypotension.
Affections respiratoires, thoraciques et médiastinales
Très fréquent: dyspnée (15.7 %).
Occasionnel: saignement des voies respiratoires (par exemple épistaxis, hémoptysie).
Affections gastro-intestinales
Fréquent: hémorragie gastro-intestinale.
(par exemple saignement gingival, saignement rectal, hémorragie d'un ulcère gastrique), diarrhée, nausée.
Occasionnel: hémorragie rétropéritonéale.
Affections de la peau et des tissus sous-cutanés
Fréquent: saignement sous-cutané ou dermique (par exemple ecchymoses, hémorragie cutanée, pétéchies), prurit.
Affections musculosquelettiques et du tissu conjonctif
Occasionnel: saignement musculaire (par exemple hémarthrose, hémorragie musculaire).
Affections du rein et des voies urinaires
Fréquent: saignement des voies urinaires (par exemple hématurie, cystite hémorragique).
Affections des organes de reproduction et du sein
Occasionnel: saignement des organes de reproduction (par exemple saignement vaginal, hémospermie, hémorragie post-ménopausique).
Investigations
Fréquent: augmentation du taux sanguin de créatininea.
Lésions, intoxications et complications d'interventions
Fréquent: hémorragies après une intervention, saignement traumatique (par exemple contusion, hématome traumatique, saignement hémorragique traumatique).
afréquences provenant de résultats d'analyses (augmentation de l'acide urique >limite supérieure de la normale à partir d'une valeur initiale inférieure à l'intervalle de référence ou dans celui-ci. Augmentation de la créatinine >50 % de la valeur initiale) et non pas fréquence brute des notifications de l'événement indésirable
Effets indésirables après commercialisation
Les effets indésirables suivants ont été observés après la mise sur le marché de BRILIQUE. Etant donné que ces données proviennent d'annonces spontanées issues d'une population de taille inconnue, il n'est pas toujours possible d'en estimer la fréquence de manière fiable.
Affections du système immunitaire
Réactions d'hypersensibilité, y compris angio-œdème (voir «Contre-indications»).
Affections du système nerveux
Apnée centrale du sommeil, y compris la respiration de Cheyne-Stokes (voir «Mises en garde et précautions»).
Affections de la peau et du tissu sous-cutané
Eruption cutanée.
Affections hématologiques et du système lymphatique
Purpura thrombotique thrombocytopénique (voir «Mises en garde et précautions»).
Affections cardiaques
Bradyarythmie, bloc AV (voir «Mises en garde et précautions»).
Description de certains effets indésirables
Résultats concernant les événements hémorragiques dans l'étude PLATO.
Les principaux critères d'exclusion étaient un risque accru de saignements, une thrombopénie cliniquement importante ou une anémie, des antécédents de saignements intracrâniens, un saignement gastro-intestinal au cours des 6 derniers mois ou une intervention chirurgicale importante au cours des 30 derniers jours.
Le tableau 1 présente les résultats généraux concernant les événements hémorragiques dans l'étude PLATO.
Tableau 1: Analyse des événements hémorragiques généraux, estimation de Kaplan-Meier des taux d'hémorragies après traitement à 12 mois (PLATO)
|
Critères de sécurité |
BRILIQUE 90 mg deux fois par jour |
Clopidogrel 75 mg une fois par jour |
Valeur p | |
|
|
KM % |
Hazard Ration |
|
|
|
Définition des catégories d'hémorragies dans l'étude PLATO | ||||
|
Hémorragies majeures au total |
11,6 |
1,04 |
11,2 |
0,4336 |
|
Hémorragies fatales / engageant le pronostic vital |
5,8 |
1.03 |
5,8 |
0,6988 |
|
Toutes les hémorragies majeures et mineures confondues |
16,1 |
1,11 |
14,6 |
0,0084 |
|
Hémorragies majeures non associées à un PAC |
4,5 |
1,19 |
3,8 |
0,0264 |
|
Hémorragies majeures non dues à une intervention |
3,1 |
1,31 |
2,3 |
0,0058 |
|
Hémorragies majeures et mineures non dues à une intervention |
5,9 |
1,39 |
4,3 |
<0,0001 |
|
Catégorie d'hémorragies selon l'échelle TIMI | ||||
|
Hémorragies majeures |
7,9 |
1,03 |
7,7 |
0,5669 |
|
Hémorragies majeures et mineures |
11,4 |
1,05 |
10,9 |
0,3272 |
L'étude PLATO a utilisé les définitions suivantes pour les hémorragies:
«Hémorragies mortelles/engageant le pronostic vital»: hémorragies mortelles, toute hémorragie intracrânienne ou intrapéricardique avec tamponnade cardiaque; ou avec hypovolémique choc ou hypotension sévère, nécessitant des vasopresseurs/médicaments à effet inotrope ou une intervention chirurgicale; ou hémorragie cliniquement évidente avec une perte d'hémoglobine de plus de ≥5 g/dl; ou transfusion de 4 unités ou davantage.
«Autres hémorragies majeures»: hémorragie associée à une invalidation notable ou cliniquement évidente avec une perte d'hémoglobine de 3 à 5 g/dl; ou transfusion de 2 à 3 unités (sang entier ou concentré d'érythrocytes) à cause d'une hémorragie.
«Hémorragies mineures»: exigeant une intervention médicale pour stopper ou traiter l'hémorragie
«Hémorragie TIMI majeure»: hémorragie engageant le pronostic vital ou toute hémorragie intracrânienne, ou signes d'hémorragie cliniquement manifeste associée à une réduction du taux d'hémoglobine supérieure à 5 g/dl, ou, si Hb non disponible, diminution de l'hématocrite de 15 %.
«Hémorragie TIMI mineure»: hémorragie cliniquement manifeste associée à une réduction du taux d'hémoglobine de 3 à 5 g/dl.
Dans l'étude PLATO, l'intervalle jusqu'au premier événement du groupe «hémorragies majeures au total» n'a pas été significativement différent entre BRILIQUE et le clopidogrel. Les hémorragies mortelles ont été rares dans cette étude: 20 (0,2 %) sous BRILIQUE 90 mg deux fois par jour et 23 (0,3 %) sous clopidogrel 75 mg une fois par jour. En incluant les hémorragies mineures, l'incidence des hémorragies majeures + mineures a été significativement supérieure sous BRILIQUE versus clopidogrel dans l'étude PLATO. Les incidences d'hémorragies selon la définition TIMI n'étaient pas significativement différentes entre BRILIQUE et le clopidogrel.
Hémorragies liées à un pontage aorto-coronarien: dans l'étude PLATO, 1584 patients (12 % de la cohorte) ont eu un PAC et 42 % d'entre eux ont eu un saignement majeur fatal ou engageant le pronostic vital selon le critère PLATO, sans différence entre les groupes de traitement. Des saignements fatals liés à un PAC sont survenus chez 6 patients dans chaque groupe de traitement (voir «Mises en garde et précautions»).
Saignements non liés à un pontage aorto-coronarien: BRILIQUE et clopidogrel ne se distinguent pas l'un de l'autre en ce qui concerne les hémorragies majeures fatales / engageant le pronostic vital (définition PLATO) qui n'étaient pas dues à un PAC. Cependant les hémorragies définies comme «hémorragies majeures au total» (définition PLATO), TIMI majeures et TIMI majeures + mineures étaient plus fréquentes avec BRILIQUE. De même, lorsqu'on exclut tous les saignements liés à une intervention, davantage de saignements sont survenus sous BRILIQUE que sous clopidogrel (tableau 1). Les arrêts de traitement liés à des saignements non liés à une intervention ont été plus fréquents sous BRILIQUE (2,9 %) que sous clopidogrel (1,2 %; p < 0,001).
Aucun des facteurs suivants, âge, sexe, poids, origine ethnique, origine géographique, maladies associées, traitements associés, antécédents médicaux incluant les accidents vasculaires cérébraux et les accidents ischémiques transitoires, n'a permis de prédire les saignements globaux ou les saignements majeurs (définition PLATO) non liés à une intervention. Il n'y a donc pas de sous-groupe identifié comme à risque de quelque forme de saignement que ce soit.
Saignement intracrânien: il y a eu plus de saignements intracrâniens non liés à une intervention dans le groupe BRILIQUE (n=27 saignements chez 26 patients, 0,3 %) que dans le groupe clopidogrel (n=14 saignements, 0,2 %), avec 11 saignements fatals sous BRILIQUE et un sous clopidogrel. Il n'y a pas eu de différence sur la totalité des hémorragies fatales. Considérant les comorbidités significatives et les facteurs de risque cardiovasculaire dans la population de patients examinée, l'incidence des saignements intracrâniens était basse dans les deux groupes de traitement.
Résultats concernant les événements hémorragiques dans l'étude PEGASUS
Les principaux critères d'exclusion étaient l'utilisation prévue de médicaments pouvant entraîner un risque accru de saignements, des troubles hémorragiques ou des antécédents d'AVC ou de saignement intracrânien, une tumeur du système nerveux central, une anomalie vasculaire intracrânienne, un saignement gastro-intestinal au cours des 6 derniers mois, une intervention chirurgicale intracrânienne ou de la moelle épinière au cours des 5 derniers mois ou une intervention chirurgicale importante au cours des 30 derniers jours.
Le tableau 2 présente les résultats généraux concernant les événements hémorragiques dans l'étude PEGASUS.
Tableau 2 – Analyse des événements hémorragiques généraux, estimation de Kaplan-Meier des taux d'hémorragies après traitement à 36 mois (PEGASUS)
|
|
BRILIQUE 60 mg deux fois par jour + AAS |
AAS en monothérapie |
| |
|
Critères de sécurité |
KM % |
Hazard Ratio |
KM % |
Valeur de p |
|
Catégories de saignements, définitions TIMI | ||||
|
Majeurs |
2,3 |
2,32 |
1,1 |
<0,0001 |
|
Fatals |
0,3 |
1,00 |
0,3 |
1,0000 |
|
Saignements intracrâniens |
0,6 |
1,33 |
0,5 |
0,3130 |
|
Autres majeurs |
1,6 |
3,61 |
0,5 |
<0,0001 |
|
Majeurs ou mineurs |
3,4 |
2,54 |
1,4 |
<0,0001 |
|
Majeurs ou mineurs ou nécessitant un traitement médical |
16,6 |
2,64 |
7,0 |
<0,0001 |
|
Catégories de saignements, définitions PLATO | ||||
|
Majeurs |
3,5 |
2,57 |
1,4 |
<0,0001 |
|
Fatals/engageant le pronostic vital |
2,4 |
2,38 |
1,1 |
<0,0001 |
|
Autres majeurs |
1,1 |
3,37 |
0,3 |
<0,0001 |
|
Majeurs ou mineurs |
15,2 |
2,71 |
6,2 |
<0,0001 |
Définitions des catégories de saignements:
Majeurs TIMI: saignement fatal OU tout saignement intracrânien, OU signes cliniquement évidents d'hémorragie avec une diminution de l'hémoglobinémie (Hb) ≥5 g/dl, ou, si Hb non disponible, diminution de l'hématocrite (Hct) > 15 %.
Fatal: événement hémorragique aboutissant directement au décès dans les 7 jours.
Autres majeurs, définition TIMI: saignements majeurs non fatals, non intracrâniens, définition TIMI.
Mineurs, définition TIMI: cliniquement évidents avec une diminution de 3 à 5 g/dl de l'hémoglobine.
Nécessitant un traitement médical, définition TIMI: nécessitant une intervention OU entraînant une hospitalisation OU nécessitant une évaluation.
Majeurs fatals/engageant le pronostic vital, définition PLATO: saignements fatals OU tout saignement intracrânien OU saignement intrapéricardique avec tamponnade cardiaque OU avec choc hypovolémique ou hypotension sévère nécessitant le recours à des vasopresseurs/inotropes ou une intervention chirurgicale OU cliniquement évidents avec une diminution >5 g/dl de l'hémoglobinémie OU la transfusion de ≥4 culots globulaires.
Autres majeurs, définition PLATO: entraînant un handicap significatif OU cliniquement évidents avec une diminution de 3 à 5 g/dl de l'hémoglobine OU la transfusion de 2 à 3 culots globulaires.
Mineurs, définition PLATO: nécessitant une intervention médicale pour arrêter ou traiter le saignement.
Dans l'étude PEGASUS, les saignements majeurs (définition TIMI) ont été plus fréquents sous BRILIQUE 60 mg administré deux fois par jour que sous AAS en monothérapie. Aucune augmentation du risque hémorragique n'a été observée pour les saignements fatals, et seule une augmentation mineure a été observée pour les hémorragies intracrâniennes comparativement à l'AAS en monothérapie. Quelques événements hémorragiques fatals sont survenus au cours de l'étude, 11 (0,3 %) pour BRILIQUE 60 mg et 12 (0,3 %) pour l'AAS en monothérapie. L'augmentation observée du risque de saignements majeurs TIMI sous BRILIQUE 60 mg a été principalement due à une fréquence plus élevée des autres saignements majeurs TIMI, liés à des événements gastro-intestinaux.
Des augmentations des profils de saignements similaires aux saignements majeurs TIMI ont été observées pour les catégories de saignements majeurs ou mineurs TIMI, majeurs PLATO et majeurs ou mineurs PLATO (voir tableau 2). L'arrêt du traitement en raison de saignements a été plus fréquent sous BRILIQUE 60 mg qu'avec l'AAS en monothérapie (respectivement 6,2 % et 1,5 %). La majorité de ces saignements a été de moindre sévérité (nécessitant un traitement médical selon la définition TIMI), par exemple épistaxis, ecchymose et hématomes.
Le profil des saignements sous BRILIQUE 60 mg a été uniforme dans plusieurs sous-groupes prédéfinis (par exemple par âge, sexe, poids, origine ethnique, région géographique, pathologies concomitantes, traitement concomitant et antécédents médicaux) pour les saignements majeurs TIMI, majeurs ou mineurs TIMI et majeurs PLATO.
Saignements intracrâniens: des saignements intracrâniens spontanés ont été rapportés à des taux similaires sous BRILIQUE 60 mg et sous AAS en monothérapie (n=13, 0,2 % dans les deux groupes de traitement). La fréquence des saignements intracrâniens d'origine traumatique ou dus à une procédure a été légèrement plus élevée sous BRILIQUE 60 mg (n=15, 0,2 %) que sous AAS en monothérapie (n=10, 0,1 %). Six saignements intracrâniens fatals sont survenus sous BRILIQUE 60 mg et 5 sous AAS en monothérapie. L'incidence des saignements intracrâniens a été faible dans les deux groupes de traitement, étant donné les comorbidités significatives et les facteurs de risque cardiovasculaire dans la population étudiée.
Dyspnée
Dans l'étude PLATO, des effets secondaires de type dyspnée ont été rapportés chez 13,8 % des patients traités par BRILIQUE 90 mg deux fois par jour et chez 7,8 % des patients traités par 75 mg de clopidogrel. La plupart des événements indésirables de type dyspnée étaient d'intensité légère à modérée et s'amélioraient souvent sans qu'il soit nécessaire d'arrêter le traitement. La dyspnée se présentait la plupart du temps sous forme d'épisodes uniques (chez 87 % des patients) survenant peu de temps après le début du traitement.
La dyspnée en tant qu'effet secondaire sévère a été observée chez 0,7 % des patients traités par BRILIQUE et chez 0,4 % des patients traités par clopidogrel. Les patients ayant rapporté une dyspnée avaient tendance à être plus âgés et avaient plus fréquemment présenté une dyspnée, une insuffisance cardiaque, une BPCO ou un asthme avant le début du traitement. Les résultats de PLATO ne permettent pas de conclure que la fréquence accrue sous BRILIQUE soit due à une affection cardiaque ou pulmonaire nouvelle ou s'aggravant (voir «Propriétés/Effets» - Mécanisme de l'adénosine). BRILIQUE ne modifie pas les explorations fonctionnelles respiratoires (voir «Mises en garde et précautions»).
Dans l'étude PEGASUS, des dyspnées ont été rapportées chez 14,2 % des patients traités par BRILIQUE 60 mg deux fois par jour et chez 5,5 % de patients traités par AAS en monothérapie. Comme dans l'étude PLATO, les cas de dyspnée rapportés ont été le plus souvent d'intensité légère à modérée (voir «Mises en garde et précautions»).
L'annonce d'effets secondaires présumés après l'autorisation est d'une grande importance. Elle permet un suivi continu du rapport bénéfice-risque du médicament. Les professionnels de santé sont tenus de déclarer toute suspicion d'effet secondaire nouveau ou grave via le portail d'annonce en ligne ElViS (Electronic Vigilance System). Vous trouverez des informations à ce sujet sur www.swissmedic.ch.
Surdosage
Il n'existe actuellement aucun antidote connu pour neutraliser les effets de ticagrelor, et il n'est pas attendu que le ticagrelor soit dialysable (voir «Mises en garde et précautions»). Le traitement du surdosage doit suivre la pratique médicale standard locale. L'effet pharmacologique attendu en cas de surdosage de ticagrelor est une prolongation de la durée du risque de saignements liés à l'inhibition de l'agrégation plaquettaire. Des mesures de soutien appropriées devront être prises si des saignements se produisent. Le ticagrelor est bien toléré jusqu'à des doses uniques de 900 mg. Une toxicité gastro-intestinale a été l'événement limitant dans une étude d'escalade de doses uniques. Les autres effets indésirables significatifs au plan clinique pouvant survenir en cas de surdosage sont la dyspnée et les pauses ventriculaires (voir «Effets indésirables»).
En cas de surdosage, il faudra rechercher ces effets indésirables potentiels et considérer une surveillance électrocardiographique continue.
Propriétés/Effets
Code ATC
B01AC24
Mécanisme d'action
BRILIQUE contient le principe actif ticagrelor, une substance appartenant à la classe chimique des cyclopentyltriazolopyrimidines (CPTP). Il s'agit d'un antagoniste oral, à action directe, sélectif et réversible du récepteur P2Y12 qui empêche l'activation et l'agrégation plaquettaires déclenchées par l'adénosine diphosphate (ADP) et dépendantes du P2Y12. Le ticagrelor n'empêche pas la liaison entre le récepteur et l'ADP, mais la transduction du signal médiée par l'ADP par sa liaison avec le récepteur P2Y12.
Comme les plaquettes sont impliquées dans le déclenchement et/ou le développement des complications thrombotiques observées dans l'athérosclérose, une inhibition de la fonction plaquettaire est en mesure d'atténuer le risque d'événements cardiovasculaires tels que décès par arrêt cardiaque, infarctus du myocarde ou accident vasculaire cérébral.
Le ticagrelor dispose d'un autre mécanisme d'action qui consiste en une augmentation du niveau endogène local d'adénosine par une inhibition du transporteur équilibrant type 1 de nucléosides (ENT-1). En présence d'hypoxie et de lésion tissulaire, l'adénosine est synthétisée localement par la dégradation de l'adénosine triphosphate et de l'adénosine diphosphate (ATP et ADP) libérées. Comme la dégradation de l'adénosine a lieu essentiellement dans l'espace intracellulaire, l'inhibition de l'ENT-1 par le ticagrelor entraîne un allongement de la demi-vie de l'adénosine. Le ticagrelor ne déploie pas d'action directe cliniquement importante sur les récepteurs de l'adénosine (A1, A2A, A2B, A3) et n'est pas métabolisé en adénosine. De nombreux effets sont documentés pour l'adénosine, parmi lesquels: vasodilatation, inhibition de l'agrégation plaquettaire, modulation de la réaction inflammatoire et induction d'une dyspnée, ces effets pouvant contribuer au profil clinique du ticagrelor.
Pharmacodynamie
Apparition de l'effet
L'inhibition de l'agrégation plaquettaire (IPA = inhibition of platelet aggregation) par le ticagrelor et le clopidogrel a été examinée dans le cadre d'une étude de 6 semaines au cours de laquelle l'effet antiagrégant aigu et chronique a été évalué en réaction à 20 µM d'ADP agissant comme agoniste de l'agrégation thrombocytaire chez des patients ayant une cardiopathie coronarienne stable et traités par de l'AAS. L'apparition de l'effet a été évaluée après une dose de charge de 180 mg de ticagrelor ou 600 mg de clopidogrel. Le ticagrelor démontre une rapidité de son effet pharmacologique, comme le montre l'inhibition moyenne de l'agrégation plaquettaire, qui est d'environ 41 %, 0,5 heure après une dose de charge de 180 mg de ticagrelor. L'inhibition maximale de l'agrégation plaquettaire, atteinte 2 à 4 heures après l'administration, est de 89 %. Elle se maintient pendant 2 à 8 heures. L'inhibition finale de l'agrégation plaquettaire est supérieure à 70 % 2 heures après l'administration du traitement chez 90 % des patients.
Disparition de l'effet
Si un pontage aorto-coronarien est prévu, le risque de saignement avec le ticagrelor est augmenté comparativement au clopidogrel quand il est arrêté moins de 96 heures avant l'intervention.
Données concernant le remplacement du traitement
Le remplacement du clopidogrel 75 mg une fois par jour par BRILIQUE 90 mg deux fois par jour conduit à une augmentation absolue de l'inhibition de l'agrégation plaquettaire de 26,4 %, et le remplacement de BRILIQUE par le clopidogrel entraîne une diminution absolue de l'IAP de 24,5 %. Les patients peuvent passer du clopidogrel au BRILIQUE sans interruption de l'effet antiagrégant plaquettaire (voir «Posologie/Mode d'emploi»).
Mécanisme de l'adénosine (ENT-1)
Le ticagrelor a entraîné une augmentation de la concentration plasmatique d'adénosine chez les patients ayant un SCA et a renforcé diverses réactions physiologiques à l'adénosine. L'adénosine est un vasodilatateur; il a été établi que le ticagrelor accroît l'augmentation induite par l'adénosine du débit sanguin coronaire chez des volontaires sains comme chez des patients ayant un SCA. L'adénosine est un antiagrégant plaquettaire endogène; il a été démontré que le ticagrelor renforce l'inhibition de l'agrégation plaquettaire liée à l'adénosine, en plus de son action anti-plaquettaire par antagonisme au niveau du récepteur P2Y12. De même, l'adénosine induit une dyspnée; il a été prouvé que le ticagrelor renforce la dyspnée induite par l'adénosine chez des volontaires sains. Il n'est donc pas exclu que la dyspnée observée chez certains patients sous ticagrelor soit partiellement médiée par l'adénosine (voir «Effets indésirables»).
Efficacité clinique
Les preuves cliniques de l'efficacité de BRILIQUE proviennent de deux études de phase III:
·L'étude PLATO [PLATO = PLATelet Inhibition and Patient Outcomes] qui a comparé BRILIQUE au clopidogrel, tous deux administrés en association avec l'AAS et d'autres traitements standard (syndrome coronarien aigu).
·L'étude PEGASUS TIMI-54 [PEGASUS = PrEvention with TicaGrelor of SecondAry Thrombotic Events in High-RiSk AcUte Coronary Syndrome Patients] qui a comparé BRILIQUE en association avec l'AAS et l'AAS en monothérapie (traitement prolongé avec antécédents d'infarctus du myocarde et la présence d'autres facteurs de risque cardiovasculaire).
Etude PLATO (syndrome coronarien aigu)
L'étude PLATO a inclus 18'624 patients ayant un syndrome coronarien aigu (angor instable [AI], infarctus du myocarde sans sus-décalage du segment ST [NSTEMI] ou infarctus du myocarde avec sus-décalage du segment ST [STEMI]) avec apparition des symptômes depuis moins de 24 heures et traités initialement soit médicalement, soit par intervention coronaire percutanée (ICP) ou par pontage aorto-coronarien (PAC) (voir «Indications/Possibilités d'emploi»).
Sur la base d'une administration quotidienne d'AAS, BRILIQUE à la dose de 90 mg administré 2 fois par jour s'est montré supérieur au clopidogrel à la dose de 75 mg administré une fois par jour, quant à la prévention du critère composite primaire de décès cardiovasculaire (CV), d'infarctus du myocarde (IM) ou d'accident vasculaire cérébral (AVC), avec une différence due essentiellement à une réduction des décès CV et des IM. Les patients ont reçu une dose de charge de 300 mg de clopidogrel (possibilité d'administrer 600 mg en cas d'ICP) ou 180 mg de BRILIQUE.
Ce résultat est apparu rapidement (avec une réduction du risque absolu [RRA] de 0,6 % et une réduction du risque relatif [RRR] de 12 % à trente jours), avec un effet constant du traitement pendant toute la période de 12 mois de l'étude, aboutissant à un RRA de 1,9 % et un RRR de 16 % à un an. Cela suggère qu'il est approprié de traiter les patients par BRILIQUE jusqu'à 12 mois (voir «Posologie/Mode d'emploi»). Traiter 54 patients avec syndrome coronarien aigu par BRILIQUE à la place du clopidogrel évitera 1 événement athérothrombotique. Traiter 91 patients évitera 1 décès cardiovasculaire (voir Figure 1 et Tableau 3).
L'effet du traitement par BRILIQUE versus clopidogrel apparaît de façon cohérente dans de nombreux sous-groupes, incluant poids; sexe; antécédents médicaux de diabète, d'accident ischémique transitoire, d'accident vasculaire cérébral non hémorragique ou de revascularisation; traitements concomitants incluant les héparines, les anti-GPIIb/IIIa et les inhibiteurs de la pompe à proton (voir «Interactions»); diagnostic final (STEMI, NSTEMI ou angor instable); et stratégie thérapeutique initialement envisagée lors de la randomisation (interventionnelle ou pharmacologique).
Une interaction faiblement significative a été observée en fonction des régions; le Hazard Ratio du critère principal d'évaluation est en faveur du clopidogrel en Amérique du Nord, région ou près de 10 % des patients de l'étude avaient été inclus, tandis qu'il est en faveur de BRILIQUE dans le reste monde (valeur p pour l'interaction = 0,045). Des analyses exploratoires suggèrent une association possible avec la dose d'AAS: une diminution de l'efficacité a été observée avec BRILIQUE en augmentant les doses d'AAS. Les doses d'AAS à long terme administrées parallèlement à BRILIQUE devraient être de 75 à 150 mg (voir «Posologie/Mode d'emploi» et «Mises en garde et précautions»).
La Figure 1 présente l'estimation du risque de première apparition d'un des événements du critère primaire composite.
Figure 1: Temps écoulé avant la première survenue d'un décès d'origine CV, d'un IM ou d'un AVC (PLATO)
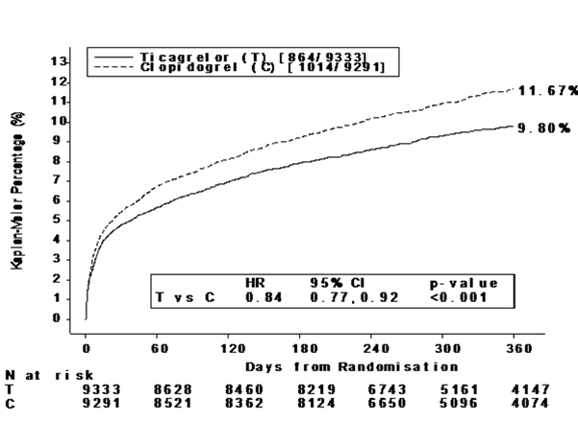
Aussi bien dans la population atteinte d'angor instable/NSTEMI que dans la population avec STEMI, BRILIQUE a diminué la survenue du critère principal composite d'évaluation comparativement au clopidogrel.
Tableau 3: Analyse des critères primaires et secondaires d'efficacité de l'étude PLATO
|
Critère primaire (CP) |
Patients avec événements |
|
|
| |
|
BRILIQUE 90 mg deux fois par jour |
Clopidogrel 75 mg une fois par jour (%) |
Réduction du risque relatif a (%) |
Hazard ratio |
Valeur p | |
|
Critère composite: décès CV/IM (sauf IM silencieux)/AVC |
9,3 |
10,9 |
16 |
0,84 |
p = 0,0003 |
|
Décès CV |
3,8 |
4,8 |
21 |
0,79 |
p = 0,0013 |
|
IM (hors IM silencieux) |
5,4 |
6,4 |
16 |
0,84 |
p = 0,0045 |
|
Accident vasculaire cérébral (AVC) |
1,3 |
1,1 |
-17 |
1,17 |
p= 0,2249 |
|
Critères secondaires | |||||
|
Critère composite: décès CV/IM (sauf IM silencieux)/AVC et traitement invasif prévu |
8,5 |
10,0 |
16 |
0,84 |
p = 0,0025 |
|
Critère composite: mortalité totale/IM (hors IM silencieux)/AVC |
9,7 |
11,5 |
16 |
0,84 |
p = 0,0001 |
|
Critère composite: décès CV/tous les IM/AVC/IRGb/ IRc/AITd/autres EATe |
13,8 |
15,7 |
12 |
0,88 |
p = 0,0006 |
|
Mortalité totale |
4,3 |
5,4 |
22 |
0,78 |
p = 0,0003** |
a RRR= réduction du risque relatif = (1-Hazard Ratio) x 100 %. Les valeurs avec une diminution relative négative du risque indiquent une augmentation relative du risque.
** Valeur nominale de p; toutes les autres valeurs sont formellement statistiquement significatives selon une analyse hiérarchisée prédéfinie.
b IRG = ischémie récurrente grave
c IR = ischémie récurrente
d AIT = accident ischémique transitoire
e EAT = événement athérothrombotique
Sous-étude Holter
Pour étudier la survenue de pauses ventriculaires et d'autres épisodes arythmiques pendant l'étude PLATO, les investigateurs ont pratiqué un enregistrement Holter chez une sous-population de près de 3'000 patients, dont environ 2'000 ont bénéficié d'enregistrements en phase aiguë de syndrome coronarien aigu et à un mois. La variable principale d'intérêt était la survenue de pauses ventriculaires ≥3 secondes. Le nombre de patients présentant des pauses ventriculaires était plus important sous BRILIQUE (6,0 %) que sous clopidogrel (3,5 %) pendant la phase aiguë, et atteignait respectivement 2,2 % et 1,6 % à un mois (voir «Mises en garde et précautions»). L'augmentation du nombre de pauses ventriculaires pendant la phase aiguë du syndrome coronarien aigu était plus prononcée chez les patients sous BRILIQUE ayant des antécédents d'insuffisance cardiaque chronique (9,2 % versus 5,4 % des patients sans antécédent d'insuffisance cardiaque chronique; pour les patients sous clopidogrel: 4,0 % de ceux ayant des antécédents d'insuffisance cardiaque chronique versus 3,6 % de ceux n'ayant pas d'antécédent d'ICC). Ce déséquilibre ne s'est pas produit à un mois: 2,0 % versus 2,1 % pour les patients sous BRILIQUE avec et sans antécédents d'insuffisance cardiaque chronique et 3,8 % versus 1,4 % sous clopidogrel. Il n'y a eu aucune conséquence clinique associée à ce déséquilibre (y compris la pose de stimulateurs cardiaques) dans cette population de patients.
Sous-étude génétique de PLATO
Le génotypage de 10'285 patients de l'étude PLATO pour les polymorphismes du CYP2C19 et de ABCB1 a permis d'obtenir des corrélations entre ces polymorphismes et les résultats de l'étude PLATO. La supériorité de BRILIQUE par rapport au clopidogrel en termes de réduction des événements cardiovasculaires majeurs n'est pas significativement modifiée par le génotype des patients pour CYP2C19 et ABCB1. Comme dans la totalité de l'étude PLATO, la fréquence des saignements majeurs suivant la définition PLATO n'est pas différente entre BRILIQUE et le clopidogrel, indépendamment du génotype pour CYP2C19 ou ABCB1. Les saignements majeurs suivant la définition PLATO survenant chez des patients n'ayant pas eu de PAC est plus élevée dans le groupe BRILIQUE que dans le groupe clopidogrel chez les patients ayant un ou deux allèles de perte de fonction du CYP2C19, mais similaire à celle du clopidogrel pour les patients n'ayant pas d'allèle de perte de fonction.
Critère composite d'efficacité et de sécurité
Un critère composite d'efficacité et de sécurité (décès CV, IM, AVC, ou événement du type «hémorragies majeures au total» selon la définition PLATO) confirme le bénéfice clinique de BRILIQUE versus clopidogrel (RRR 8 %, RAR 1,4 %, HR 0,92; p = 0,0257) pendant une période de 12 mois après un syndrome coronarien aigu.
Etude PEGASUS (traitement prolongé avec antécédents d'infarctus du myocarde et la présence d'autres facteurs de risque cardiovasculaire)
L'étude PEGASUS TIMI-54 est une étude événementielle, randomisée, en double aveugle, contrôlée versus placebo, en groupes parallèles, internationale et multicentrique menée chez 21'162 patients dans le but d'évaluer la prévention des événements athérothrombotiques par BRILIQUE administré en deux doses (90 mg deux fois par jour ou 60 mg deux fois par jour) en association avec de faibles doses d'AAS (75 à 150 mg) comparativement à l'AAS en monothérapie chez des patients ayant des antécédents d'infarctus du myocarde et présentant des facteurs additionnels de risque d'athérothrombose.
Facteurs de risque
Les patients étaient éligibles pour participer s'ils étaient âgés de 50 ans ou plus, avaient un antécédent d'infarctus du myocarde (1 à 3 ans avant la randomisation) et présentaient au moins un des facteurs de risque cardiovasculaire suivants:
·un second infarctus du myocarde dans les antécédents
·une coronaropathie multitronculaire documentée
·un diabète nécessitant un traitement
·âge ≥65 ans
·insuffisance rénale chronique non au stade terminal.
Figure 2 – Courbe de Kaplan-Meier et analyse du critère clinique composite principal de décès d'origine CV, d'infarctus du myocarde ou d'AVC dans PEGASUS
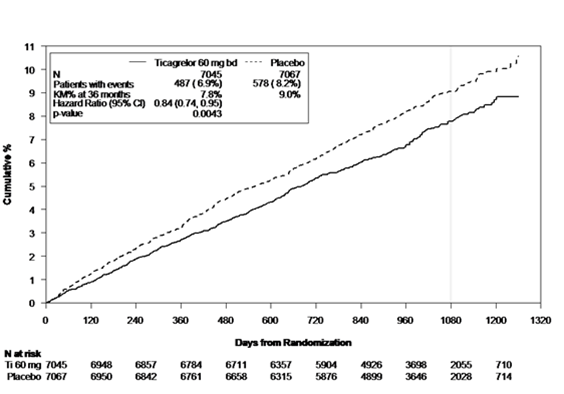
Tableau 4 – Analyse du critère principal et des critères secondaires d'efficacité dans PEGASUS
|
|
BRILIQUE 60 mg deux fois par jour + AAS |
AAS en monothérapie |
Valeur de p | |||||
|
Caractéristique |
Patients avec événements |
KM % |
HR (IC à 95 %) |
Patients avec événements |
KM % | |||
|
Critère principal | ||||||||
|
Critère composite décès CV/IM/AVC |
487 (6,9 %) |
7.8 % |
0,84 |
578 (8.2 %) |
9,0 % |
0,0043 (s) | ||
|
Décès CV |
174 (2,5 %) |
2.9 % |
0,83 |
210 (3.0 %) |
3,4 % |
0,0676 | ||
|
IM |
285 (4,0 %) |
4.5% |
0,84 |
338 (4.8 %) |
5,2 % |
0,0314 | ||
|
AVC |
91 (1,3 %) |
1,5 % |
0,75 |
122 (1,7 %) |
1,9 % |
0,0337 | ||
|
Critère secondaire | ||||||||
|
Décès CV |
174 (2,5 %) |
2,9 % |
0,83 |
210 (3.0 %) |
3,4 % |
- | ||
|
Mortalité toutes causes |
289 (4,1 %) |
4,7 % |
0,89 |
326 (4,6 %) |
5,2 % |
- | ||
Hazard ratio et valeurs de p pour BRILIQUE vs AAS en monothérapie calculés séparément à partir d'un modèle à risques proportionnels de Cox avec le groupe de traitement à titre de seule variable explicative.
Pourcentage selon Kaplan-Meier calculé à 36 mois.
Remarque: le nombre des premiers événements pour les composantes décès CV, infarctus du myocarde et AVC est le nombre réel des premiers événements pour chaque composante et ne s'ajoute pas au nombre d'événements dans le critère composite.
(s) Indique la significativité statistique.
IC = intervalle de confiance; HR = hazard ratio; KM = Kaplan-Meier; IM = infarctus du myocarde; N = nombre de patients.
Aux deux posologies de 60 mg deux fois par jour et de 90 mg deux fois par jour, BRILIQUE en association avec l'AAS a été supérieur à l'AAS en monothérapie dans la prévention des événements athérothrombotiques (critère composite: décès d'origine cardiovasculaire, infarctus du myocarde et AVC), avec un effet constant du traitement sur la totalité de la période d'étude, donnant une RRR de 16 % et une RRA de 1,27 % pour BRILIQUE 60 mg, et une RRR de 15 % et une RRA de 1,19 % pour BRILIQUE 90 mg.
Bien que les profils d'efficacité de BRILIQUE 90 mg et 60 mg soient similaires, des données indiquent que le profil de tolérance et de sécurité de la plus faible dose est meilleur au regard du risque de saignements et de dyspnée. Par conséquent, BRILIQUE 60 mg administré deux fois par jour en association avec l'AAS est recommandé pour la prévention des événements athérothrombotiques (décès d'origine cardiovasculaire, infarctus du myocarde et AVC) chez les patients ayant des antécédents d'infarctus du myocarde datant d'au moins 12 mois et à haut risque de développer un événement athérothrombotique.
Comparativement à l'AAS en monothérapie, l'administration de BRILIQUE 60 mg deux fois par jour a significativement réduit le critère composite principal de décès d'origine cardiovasculaire, d'infarctus du myocarde et d'AVC. Chacune des composantes a contribué à la réduction du critère composite principal (décès d'origine cardiovasculaire RRR 17 %, infarctus du myocarde RRR 16 % et AVC RRR 25 %).
Pour empêcher un événement du critère composite, il faut traiter 79 patients pendant 36 mois par BRILIQUE 60 mg deux fois par jour en association avec de l'AAS à la place de l'AAS en monothérapie.
L'utilité de BRILIQUE au niveau du critère composite principal est aussi apparue pour les deux critères d'évaluation secondaires, en l'occurrence sous la forme d'un recul du nombre autant des décès d'origine cardiovasculaire que de la mortalité globale sous BRILIQUE 60 mg en association avec l'AAS par rapport à l'AAS en monothérapie, la significativité statistique n'ayant toutefois pas été atteinte (voir Tableau 4).
Les RRR pour le critère composite de 1 à 360 jours (RRR 17 %) et à partir de 361 jours (RRR 16 %) ont été similaires avec un effet constant du traitement pendant toute l'étude qui a duré jusqu'à 48 mois (médiane 33 mois). La constance du RRR sur toute la durée suggère que la prolongation du traitement par BRILIQUE est indiquée aussi longtemps que le patient présente un risque élevé de développer un événement athérothrombotique (voir «Posologie/Mode d'emploi»).
L'effet du traitement par BRILIQUE 60 mg deux fois par jour par rapport à l'AAS en monothérapie est apparu de façon cohérente dans de nombreux sous-groupes de patients formés selon des données démographiques telles que poids, sexe, antécédents médicaux et région géographique.
Les bénéfices liés au traitement par BRILIQUE étaient par ailleurs indépendants de l'utilisation d'autres traitements cardiovasculaires, parmi lesquels hypolipémiants, bêtabloquants, inhibiteurs de l'ECA, antagonistes des récepteurs de l'angiotensine, inhibiteurs calciques, dérivés nitrés et inhibiteurs de la pompe à protons (voir «Interactions»).
Population pédiatrique
Dans une étude de phase III, randomisée, en double aveugle, contrôlée contre placebo, l'objectif principal portant sur la réduction du taux de crises vaso-occlusives chez des patients pédiatriques âgés de 2 ans à moins de 18 ans atteints de drépanocytose n'a pas été atteint.
Pharmacocinétique
Absorption
L'absorption du ticagrelor est rapide, avec un tmax médian d'environ 1,5 heure. La formation du métabolite principal, l'AR-C124910XX (également actif), à partir du ticagrelor est rapide, avec un tmax médian d'environ 2,5 heures. Après administration orale du ticagrelor 90 mg à jeun, la Cmax est de 529 ng/ml et l'AUC de 3451 ng*h/ml. Les rapports métabolite / substance mère sont de 0,28 pour Cmax et de 0,42 pour l'AUC.
La biodisponibilité absolue moyenne du ticagrelor a été estimée à 36 % (spectre de 25,4 % à 64,0 %). L'ingestion d'un repas riche en lipides a conduit à une augmentation de 21 % de l'AUC du ticagrelor et à une diminution de 22 % de la Cmax du métabolite actif, mais n'a eu d'effet ni sur la Cmax du ticagrelor, ni sur l'AUC du métabolite actif. Ces faibles modifications sont considérées comme ayant une signification clinique minime. Ainsi, le ticagrelor peut être administré avec ou sans aliments. Le ticagrelor et son métabolite actif sont des substrats de la PGP.
Comprimés pelliculés:
Les comprimés de ticagrelor, lorsqu'ils sont écrasés et mélangés dans de l'eau, administrés par voie orale ou par une sonde naso-gastrique dans l'estomac, présentent une biodisponibilité comparable à celle des comprimés entiers (AUC et Cmax entre 80 et 125 % pour le ticagrelor et le métabolite actif). L'exposition initiale (0,5 et 1 heure après la prise) de comprimés de ticagrelor écrasés et mélangés dans de l'eau est augmentée par rapport aux comprimés entiers. Cependant en général, après 2 à 48 heures, le profil général de concentration est identique.
Comprimés orodispersibles:
Les comprimés orodispersibles de ticagrelor, dispersés dans la salive et avalés sans eau ou mis en suspension dans de l'eau et administrés par une sonde naso-gastrique dans l'estomac, sont bioéquivalents aux comprimés pelliculés avalés (AUC et Cmax compris entre 80 et 125 % pour le ticagrelor et son métabolite actif). Lorsque le comprimé orodispersible est dispersé dans la salive et avalé avec de l'eau, l'AUC du ticagrelor est similaire, alors que la Cmax est diminuée d'environ 15 % par rapport au comprimé pelliculé. Il est peu probable que la différence observée pour la Cmax soit cliniquement pertinente.
Distribution
Le volume de distribution du ticagrelor à l'état l'équilibre est de 87,5 l. Le ticagrelor et son métabolite actif se lient fortement aux protéines plasmatiques humaines (> 99 %).
Métabolisme
Le CYP3A4 est la principale isoenzyme responsable du métabolisme du ticagrelor et de la formation du métabolite actif. Les interactions du ticagrelor et de son métabolite actif avec les autres substrats du CYP3A vont de l'activation à l'inhibition. Le ticagrelor et son métabolite actif sont de faibles inhibiteurs de la glycoprotéine P. Le ticagrelor est un inhibiteur de la BCRP.
Le métabolite principal du ticagrelor est l'AR-C124910XX. Il est également actif, comme le montre la fixation in vitro sur le P2Y12, récepteur plaquettaire à l'ADP. L'exposition systémique au métabolite actif atteint environ 30 à 40 % de celle du ticagrelor.
Élimination
La voie d'élimination principale du ticagrelor est le métabolisme hépatique. Après administration de ticagrelor radiomarqué, la récupération moyenne de la radioactivité est d'environ 84 % (57,8 % dans les fèces, 26,5 % dans l'urine). Les quantités de ticagrelor et de métabolite actif retrouvées dans l'urine ont été inférieures à 1 % de la dose administrée. La voie d'élimination principale du métabolite actif est probablement la sécrétion biliaire. La t1/2 moyenne a été d'environ 6,9 heures (fourchette de 4,5 à 12,8 heures) pour le ticagrelor et de 8,5 heures (fourchette de 6,5 à 12,8 heures) pour le métabolite actif.
Linéarité/non-linéarité
Le ticagrelor a une pharmacocinétique linéaire et l'exposition au ticagrelor et à son métabolite actif (AR-C124910XX) est approximativement proportionnelle à la dose dans le domaine des doses thérapeutiques.
Cinétique pour certains groupes de patients
Troubles de la fonction hépatique
La Cmax et l'AUC du ticagrelor ont été supérieures de respectivement 12 % et 23 % chez les patients présentant des troubles légers de la fonction hépatique comparativement aux sujets sains correspondants. Aucun ajustement de la posologie n'est nécessaire chez les patients présentant des troubles légers de la fonction hépatique (voir «Posologie/Mode d'emploi»). Le ticagrelor n'a pas été étudié chez les patients présentant des troubles sévères de la fonction hépatique et il n'y a pas d'informations pharmacocinétiques disponibles sur les patients présentant des troubles modérés de la fonction hépatique. (voir «Posologie/Mode d'emploi », «Contre-indications» et «Mises en garde et précautions»).
Troubles de la fonction rénale
L'exposition au ticagrelor a été inférieure d'environ 20 % et à son métabolite actif supérieure d'environ 17 % chez les patients présentant des troubles sévères de la fonction rénale comparativement aux sujets ayant une fonction rénale normale. Un ajustement de la posologie n'est pas nécessaire chez les patients présentant des troubles de la fonction rénale (voir «Posologie/Mode d'emploi»).
Chez les patients sous hémodialyse présentant une insuffisance rénale terminale, l'AUC et la Cmax après administration de 90 mg de BRILIQUE sur une journée sans dialyse étaient respectivement de 38 % et 51 % plus élevées par rapport aux patients ayant une fonction rénale normale. Une augmentation similaire de l'exposition était observée lorsque BRILIQUE était administré immédiatement avant la dialyse (respectivement 49 % et 61 %), ce qui montre que BRILIQUE n'est pas dialysable. L'exposition du métabolite actif a augmenté dans une moindre mesure (AUC 13 – 14 % et Cmax 17 – 36 %). L'effet IPA de BRILIQUE était indépendant de la dialyse chez les patients présentant une insuffisance rénale terminale et était similaire à celui des patients présentant une fonction rénale normale.
Patients âgés
Une exposition plus élevée au ticagrelor (environ 63 % pour la Cmax et 52 % pour l'AUC) et au métabolite actif (environ 60 % pour la Cmax et 48 % pour l'AUC) a été observée chez les patients âgés (≥65 ans) en comparaison avec aux patients plus jeunes. Ces différences ne sont pas considérées comme cliniquement significatives (voir «Posologie/Mode d'emploi»).
Enfants et adolescents
Les données cliniques dans la population pédiatrique sont limitées. BRILIQUE n'est pas indiqué dans la population pédiatrique (voir «Posologie/Mode d'emploi» et «Propriétés/Effets»).
Origine ethnique
Les patients d'origine asiatique ont une biodisponibilité supérieure de 39 % par rapport aux patients caucasiens. Les patients auto-identifiés comme Noirs ont une biodisponibilité de BRILIQUE de 18 % plus faible lorsque comparé aux patients caucasiens. Dans les études de pharmacologie clinique, l'exposition (Cmax et AUC) à BRILIQUE chez les sujets japonais a été supérieure d'environ 40 % (20 % après ajustement en fonction du poids corporel) à celle des Caucasiens. L'exposition a été similaire entre des patients auto-identifiés comme hispaniques ou latinos et des patients caucasiens.
Sexe
Une exposition plus élevée au ticagrelor et au métabolite actif a été observée chez la femme par rapport à l'homme. Les différences ne sont pas considérées comme cliniquement significatives.
Données précliniques
Pharmacologie de sécurité et toxicité en cas d'administration répétée
D'après les études conventionnelles sur la pharmacologie de sécurité, sur la toxicité après une administration unique et répétée et sur la génotoxicité potentielle, les données précliniques sur le ticagrelor et son métabolite principal (AR-C124910XX) n'ont pas démontré de risque inacceptable d'effets indésirables chez l'homme.
Les réactions indésirables suivantes n'ont pas été observées dans des études cliniques, mais dans des expérimentations animales avec des expositions similaires ou supérieures à celles examinées dans les études cliniques. Elles pourraient donc être significatives pour l'utilisation clinique: toxicité gastro-intestinale et irritations gastro-intestinales.
Carcinogénicité
Dans une étude de 2 ans sur des souris, avec administration de doses orales allant jusqu'à 250 mg/kg/jour (>18 fois l'exposition thérapeutique maximale chez l'homme), aucune tumeur induite par le médicament n'a été observée. Chez des rats mâles, des doses orales allant jusqu'à 120 mg/kg/jour (>15 fois l'exposition thérapeutique maximale chez l'homme) n'ont pas provoqué une augmentation des tumeurs. Chez des rats femelles, seules de fortes doses (>25 fois l'exposition thérapeutique maximale chez l'homme) ont été associées à une augmentation des adénocarcinomes utérins, des adénocarcinomes et adénomes hépatocellulaires, ainsi qu'à une réduction des adénomes hypophysaires et des fibro-adénomes mammaires. L'incidence de tumeurs est restée inchangée après l'administration de doses de 60 mg/kg/jour (8 fois l'exposition thérapeutique maximale chez l'homme). Il est apparu que les tumeurs utérines observées chez les rates sont dues à des effets endocriniens non génotoxiques, provenant d'un dérèglement hormonal après l'administration de ticagrelor fortement dosé. Les tumeurs bénignes du foie sont considérées comme une conséquence de la réaction du foie aux grandes exigences métaboliques dues aux doses élevées de ticagrelor. La signification de ces résultats pour l'homme n'est pas encore définitivement éclaircie à ce jour.
Génotoxicité
Le ticagrelor a été examiné dans une série de tests in vitro et in vivo. Aucune génotoxicité n'a été mise en évidence.
Toxicité sur la reproduction
Le ticagrelor provoque des cycles irréguliers (surtout allongés) chez les rats femelles mais n'affecte pas la fertilité globale des rats mâles et femelles.
Il a été montré que le ticagrelor administré à des rates à des doses orales allant jusqu'à 200 mg/kg/jour (environ 20 fois l'exposition thérapeutique maximale chez l'homme) et administré à des rats mâles à des doses allant jusqu'à 180 mg/kg/jour (15,7 fois l'exposition thérapeutique maximale chez l'homme) n'influence pas la fertilité.
Les études chez le rat et le lapin ont montré une toxicité sur la reproduction, avec une prise de poids maternelle légère, une viabilité néonatale retardée et un faible poids de naissance avec un retard de croissance.
Aux doses orales inférieures à 100 mg/kg/jour chez le rat (5,1 fois l'exposition thérapeutique maximale chez l'homme) et à des doses allant jusqu'à 42 mg/kg/jour chez le lapin (correspond à l'exposition thérapeutique maximale chez l'homme), le ticagrelor n'a eu aucun effet sur le développement fœtal. À part cela, le ticagrelor administré à des rats à des doses allant jusqu'à 60 mg/kg/jour (4,6 fois l'exposition thérapeutique maximale chez l'homme) n'a eu aucun effet sur la mise bas ou le développement postnatal des petits. Aux doses supérieures présentant une toxicité pour la mère, des effets non spécifiques ont été observés chez la progéniture.
Remarques particulières
Incompatibilités
Non applicable.
Stabilité
Le médicament ne doit pas être utilisé au-delà de la date figurant après la mention «EXP» sur l'emballage.
Remarques particulières concernant le stockage
Ne pas conserver au-dessus de 30°C. Conserver dans l'emballage d'origine.
Conserver hors de portée des enfants.
Numéro d’autorisation
61389 (Swissmedic)
66822 (Swissmedic)
Présentation
Comprimés pelliculés de 60 et 90 mg sous plaquettes thermoformées calendaires, boîtes de 56 et de 168 comprimés pelliculés. [B]
Comprimés pelliculés de 90 mg sous plaquettes thermoformées perforées, boîtes de 100 (10x10) comprimés pelliculés. [B]
Comprimés orodispersibles de 90 mg sous plaquettes thermoformées perforées, boîtes de 10 comprimés orodispersibles. [B]
Titulaire de l’autorisation
AstraZeneca AG, 6340 Baar
Mise à jour de l’information
Novembre 2025