Composition
Principes actifs
Bélimumab.
Excipients
Chlorhydrate de L-arginine, L-histidine, monochlorhydrate de L-histidine, polysorbate 80, chlorure de sodium, eau pour préparation injectable q.s. ad solutionem pro 1 mL.
Indications/Possibilités d’emploi
Benlysta est indiqué:
·pour réduire l'activité de la maladie chez les patients adultes atteints de lupus érythémateux disséminé (LED) actif avec présence d'auto-anticorps sous traitement de fond.
·pour traiter la néphrite lupique chez des patients adultes qui reçoivent un traitement standard.
Benlysta n'a pas été étudié chez des patients atteints de lupus sévère et actif avec atteinte du système nerveux central.
Posologie/Mode d’emploi
Remarques générales
On ne dispose pas de données ou de données suffisantes sur les effets du bélimumab chez les patients atteints de lupus sévère et actif avec atteinte du système nerveux central. Benlysta ne peut donc pas être recommandé pour le traitement de ces maladies.
L'efficacité et la sécurité de l'administration sous-cutanée ont été examinées dans le cadre de l'étude BEL112341 (voir «Propriétés/Effets»). Les données disponibles portent sur 1 année.
Les auto-injecteurs ne doivent PAS être utilisés pour des injections intraveineuses.
Le traitement avec Benlysta doit être instauré et surveillé par un médecin expérimenté dans le diagnostic et le traitement du LED. Il est recommandé de procéder au moins à la première injection sous-cutanée de Benlysta sous supervision médicale, dans un environnement suffisamment équipé pour la gestion d'éventuelles réactions d'hypersensibilité. Le médecin doit instruire le patient de façon adéquate sur la technique de l'injection sous-cutanée et l'informer des signes et symptômes des réactions d'hypersensibilité (voir «Mises en garde et précautions»). Après une information et une formation adéquates du patient, le médecin peut décider d'autoriser le patient à effectuer l'injection lui-même ou demander à un membre du personnel soignant de s'en charger (voir «Remarques concernant la manipulation»).
Benlysta peut être administré en injection sous-cutanée dans la région abdominale ou dans la cuisse. Si l'injection se fait à chaque fois dans la même région, on indiquera au patient de choisir pour chaque injection un nouveau site d'injection. Les injections ne doivent, en aucun cas, être effectuées dans des zones cutanées sensibles, lésées, rougies ou durcies. Lors de l'administration d'une dose de 400 mg dans la même région, il est recommandé d'injecter les deux doses de 200 mg en observant une distance de 5 cm au moins entre les deux sites d'injection.
Les doses oubliées doivent être rattrapées le plus rapidement possible une fois l'oubli constaté. Les patients peuvent ensuite poursuivre les administrations hebdomadaires au jour de la semaine habituel ou au jour de la semaine au cours duquel la dose oubliée a été rattrapée.
Posologie usuelle
Adultes
LED
La posologie recommandée est de 200 mg une fois par semaine, sous forme d'injection sous-cutanée dans l'abdomen ou dans la cuisse, administrée à chaque fois le même jour de la semaine.
En l'absence d'amélioration du contrôle de la maladie après un traitement de six mois par Benlysta, un arrêt du traitement devrait être considéré.
Néphrite lupique
Chez les patients débutant pour la première fois un traitement par Benlysta pour une néphrite lupique, le schéma posologique recommandé est de 400 mg (deux injections de 200 mg) une fois par semaine pour 4 doses, puis par la suite de 200 mg une fois par semaine.
Chez les patients qui poursuivent un traitement par Benlysta en raison d'une néphrite lupique, la posologie recommandée est de 200 mg une fois par semaine sous forme d'injection sous-cutanée dans l'abdomen ou dans la cuisse, administrée à chaque fois le même jour de la semaine.
Afin d'assurer la traçabilité des médicaments biotechnologiques, il convient de documenter, pour chaque traitement, le nom commercial et le numéro de lot.
Passage de la voie intraveineuse à la voie sous-cutanée
LED
Si un patient atteint de LED passe d'un traitement de Benlysta administré par voie intraveineuse à une administration sous-cutanée, la première injection sous-cutanée de 200 mg est administrée environ 2 semaines après la dernière administration intraveineuse (voir «Pharmacocinétique»).
Néphrite lupique
Si un patient atteint de néphrite lupique passe d'un traitement de Benlysta administré par voie intraveineuse de Benlysta à une administration sous-cutanée, la première dose sous-cutanée de 200 mg est administrée une à deux semaines après la dernière administration intraveineuse. Ce passage peut se faire à tout moment après l'administration des deux premières doses intraveineuses (voir «Pharmacocinétique»).
Instructions posologiques particulières
Patients présentant des troubles de la fonction hépatique
Aucune étude formelle n'a été effectuée sur l'utilisation du bélimumab chez les patients insuffisants hépatiques. Cependant, aucun ajustement de la dose n'est probablement nécessaire chez les patients insuffisants hépatiques (voir «Cinétique pour certains groupes de patients»).
Patients présentant des troubles de la fonction rénale
Aucune étude formelle n'a été effectuée sur l'utilisation du bélimumab chez les patients insuffisants rénaux.
Le bélimumab a été étudié chez un nombre limité de patients insuffisants rénaux atteints de LED. Aucun ajustement de la dose n'est requis chez les patients insuffisants rénaux (voir «Cinétique pour certains groupes de patients»).
Patients âgés
Bien que les données disponibles soient limitées, aucun ajustement de la dose n'est recommandé (voir «Cinétique pour certains groupes de patients»).
Enfants et adolescents
On ne dispose pas d'une expérience suffisante sur le bélimumab administré par voie sous-cutanée chez les enfants et les adolescents de moins de 18 ans.
Poids corporel
L'influence du poids corporel et de l'IMC sur l'exposition au bélimumab après administration sous-cutanée a été considérée comme non significative sur le plan clinique. Aucun ajustement de la dose n'est recommandé en fonction du poids corporel (voir «Cinétique pour certains groupes de patients»).
Contre-indications
Hypersensibilité au bélimumab ou à l'un des excipients.
Mises en garde et précautions
Les données disponibles ne confirment pas la sécurité et l'efficacité du rituximab lors de l'administration concomitante de bélimumab chez des patients atteints de LED (voir «Propriétés/Effets – Efficacité clinique»).
La prudence est de rigueur lors d'une utilisation de Benlysta en association avec d'autres traitements ciblant les lymphocytes B.
Réactions liées à l'injection et hypersensibilité
Des réactions systémiques liées à l'injection et des réactions d'hypersensibilité – éventuellement sévères ou même mortelles – peuvent se produire lors de l'administration de Benlysta. Ces réactions peuvent aussi survenir avec un effet retardé durant les jours qui suivent l'administration.
Dans le cas d'une réaction sévère, l'administration de Benlysta doit être interrompue et un traitement médical approprié doit être instauré. Un arrêt définitif du traitement a été rapporté chez 0,2% des patients traités par voie sous-cutanée. Les patients qui ont des allergies médicamenteuses multiples ou des antécédents d'hypersensibilité significative peuvent présenter un risque accru.
Une apparition tardive de réactions d'hypersensibilité a été observée ainsi qu'une réapparition de réactions cliniquement significatives après une régression initiale des symptômes suite à un traitement.
Les patients sous Benlysta doivent être avertis que des réactions d'hypersensibilité peuvent survenir le jour de l'administration ou encore plusieurs jours après celle-ci et ils doivent être informés des signes et symptômes possibles de telles réactions, ainsi que de la possibilité d'une réapparition de ces réactions. Les patients doivent être informés au sujet des risques potentiels de ce type de réactions et ils doivent être instruits de l'importance de contacter immédiatement le médecin dans un tel cas.
Les symptômes correspondants sont, par exemple, une réaction anaphylactique, une bradycardie, une hypotension, un angio-œdème et une dyspnée. Des réactions d'hypersensibilité tardives ou retardées peuvent survenir et se présenter par des symptômes tels qu'éruption cutanée respectivement urticaire, nausées, fièvre, fatigue, myalgie, céphalées, douleurs des membres et œdème du visage. Les patients doivent être instruits de signaler immédiatement tous les symptômes de telles réactions.
Dans les études cliniques avec la solution pour perfusion administrée par voie intraveineuse, on a rapporté des réactions liées à la perfusion chez 17% des patients sous bélimumab et 15% des patients sous placebo (dont des cas graves d'anaphylaxie, d'hypotension, d'angio-œdème, d'urticaire ou d'éruption cutanée, de prurit et de dyspnée, de bradycardie, de myalgie, de céphalées, de fièvre, d'hypertension, de sensation de vertige et d'arthralgie). Moins de 1% des patients ont souffert d'une réaction grave liée à la perfusion ou d'hypersensibilité.
Infections
Comme d'autres agents immunomodulateurs, le bélimumab présente un mécanisme d'action susceptible d'accroître le risque d'infections, y compris d'infections opportunistes.
Dans une étude de sécurité de phase IV, contrôlée contre placebo, portant sur l'administration intraveineuse du bélimumab, des infections mortelles sont apparues au cours d'une année plus fréquemment chez des patients traités par le bélimumab que chez ceux du groupe sous placebo (0,45% sous bélimumab contre 0,15% sous placebo).
Les patients qui avaient souffert jusqu'à 60 jours avant le début de l'étude d'une infection aiguë active ou chronique ayant nécessité un traitement spécifique ont été exclus de l'étude. Les infections mortelles sous bélimumab se sont produites essentiellement durant les 20 premières semaines de traitement et ont souvent concerné des patients avec une activité de la maladie élevée (SELENA-SLEDAI ≥10) et des patients traités simultanément par des corticostéroïdes (dose d'équivalent prednisone >7,5 mg/jour). Au bout d'un an, la mortalité globale des patients traités par le bélimumab était comparable à celle des patients sous placebo. Au total, l'incidence des infections graves était similaire dans les groupes sous bélimumab et sous placebo.
Les patients qui développent une infection sous Benlysta doivent être surveillés étroitement et un arrêt du traitement immunosuppresseur est à considérer. Les médecins doivent conseiller aux patients de consulter un médecin s'ils développent des symptômes d'une infection. La prudence est de rigueur lorsqu'un traitement par Benlysta est envisagé chez des patients ayant souffert d'infections graves ou chroniques auparavant. Le traitement par Benlysta ne doit pas être instauré chez les patients présentant une infection active cliniquement significative.
Leucoencéphalopathie multifocale progressive (LEMP)
Des cas de leucoencéphalopathie multifocale progressive (LEMP) ayant entraîné des déficits neurologiques, parfois avec une issue fatale, ont été rapportés chez des patients atteints de LED qui avaient reçu une pharmacothérapie immunosuppressive, entre autres par Benlysta. En principe, il faut toujours songer à la possibilité d'une LEMP chez les patients développant pour la première fois des signes et symptômes neurologiques ou subissant une aggravation de troubles neurologiques préexistants. En cas d'indication clinique, les patients concernés doivent être adressés à un neurologue ou à un autre spécialiste qualifié pour une évaluation approfondie. En cas de suspicion de LEMP, le traitement immunosuppresseur, y compris par Benlysta, doit être interrompu jusqu'à ce qu'une LEMP puisse être exclue. Si la LEMP est confirmée, le traitement immunosuppresseur – y compris Benlysta – doit être arrêté.
Risque de maladies cancéreuses
Comme d'autres agents immunomodulateurs, le bélimumab présente un mécanisme d'action susceptible d'accroître éventuellement le risque de maladies cancéreuses. Dans les études cliniques, aucune différence du risque de maladies cancéreuses n'a été constatée entre les patients sous bélimumab et les patients sous placebo.
Vaccinations
Aucun vaccin vivant ne doit être administré dans les 30 jours précédant et pendant le traitement par Benlysta, étant donné que la sécurité clinique n'est pas établie. On ne dispose pas de données concernant la transmission secondaire d'une infection à des patients sous bélimumab par des personnes ayant été vaccinées avec un vaccin vivant. Vu son mécanisme d'action, le bélimumab pourrait influencer la réaction d'immunisation. Il convient donc d'envisager de respecter un délai approprié entre la vaccination de rappel et le début du traitement par Benlysta.
Dans une étude qui portait sur la réponse à un vaccin pneumococcique 23-valent, les réponses immunitaires, après 4 semaines, aux différents sérotypes étaient globalement similaires chez les patients LED sous bélimumab et chez les patients LED n'ayant reçu aucun traitement au moment de la vaccination. Cette étude ne portait pas sur le maintien de la réponse au vaccin sur une période plus longue. Des données limitées suggèrent que le bélimumab n'a pas d'influence significative sur l'obtention d'une réaction immunitaire protectrice chez les patients ayant été immunisés avant de recevoir du bélimumab.
Maladies psychiatriques, dépression et tendances suicidaires
Dans les études cliniques contrôlées avec administration intraveineuse et sous-cutanée, des maladies psychiatriques (dépression, pensées suicidaires et comportement suicidaire), ont été plus fréquemment rapportées chez les patients traités par le bélimumab, dont un suicide chez un patient ayant reçu 10 mg/kg et un suicide chez un patient ayant reçu 1 mg/kg (voir «Effets indésirables»).
Avant de prescrire un traitement par le bélimumab, le médecin doit évaluer avec soin le risque dépressif et suicidaire du patient à l'aide de ses antécédents et de son état psychiatrique actuel et doit surveiller le patient en conséquence pendant le traitement. Le médecin doit indiquer au patient (et éventuellement aux aidants) de prendre contact avec lui si des nouveaux symptômes psychiatriques surviennent ou s'aggravent. Le rapport bénéfice-risque de la poursuite du traitement par le bélimumab doit être évalué avec soin chez les patients présentant de tels symptômes.
Interactions
Aucune étude d'interaction n'a été réalisée pour évaluer les interactions entre le bélimumab et d'autres médicaments.
Dans les études cliniques auprès de patients atteints de LED, l'administration concomitante de mycophénolate mofétil, de cyclophosphamide, d'azathioprine ou d'hydroxychloroquine n'a pas eu d'influence significative sur la pharmacocinétique du bélimumab (administré par voie intraveineuse ou sous-cutanée). De même, un large spectre d'autres médications concomitantes (AINS, aspirine et inhibiteurs de l'HMG-CoA réductase) n'a pas eu d'influence significative sur la pharmacocinétique du bélimumab. L'analyse pharmacocinétique de population a indiqué que la co-administration de corticostéroïdes, d'IEC et de bélimumab administré par voie intraveineuse entraîne une augmentation significative de la clairance systémique du bélimumab (6,0% et 8,5% respectivement). Ce n'est pas le cas lorsque le bélimumab est administré par voie sous-cutanée.
Grossesse, allaitement
Femmes en âge de procréer/Contraception chez la femme
Lorsque, au vu d'une évaluation du rapport bénéfice/risque (voir ci-dessous), la prévention d'une grossesse est indiquée, les femmes en âge de procréer doivent appliquer des mesures préventives de contraception appropriées pendant le traitement par Benlysta et au moins quatre mois au-delà de la dernière administration de Benlysta.
Grossesse
On ne dispose que de données limitées sur l'utilisation du bélimumab chez la femme enceinte. Des données post-commercialisation issues d'un registre prospectif des grossesses ont permis de rassembler des informations sur les grossesses de femmes ayant reçu du bélimumab. En raison de la petite taille de l'échantillon, il n'a pas été possible, à partir de ce registre, de tirer des conclusions définitives sur le risque éventuel d'anomalies congénitales après une exposition au bélimumab.
Les anticorps de type immunoglobulines G (IgG) – y compris le bélimumab – peuvent traverser le placenta. Benlysta ne doit être administré pendant la grossesse que si les avantages potentiels pour la mère justifient le risque potentiel pour le fœtus.Des malformations congénitales ont été observées chez des femmes sous bélimumab. Toutefois, les données limitées dont on dispose ne permettent pas de conclure à un lien de causalité.
Les expérimentations animales n'ont révélé aucun effet nuisible direct ou indirect dans le sens d'une toxicité maternelle, d'une perturbation sur le déroulement de la grossesse ou d'anomalies du développement embryonnaire et fœtal. Les constats pathologiques attribuables au traitement étaient limités à des réductions réversibles du taux de cellules B chez de jeunes singes (voir «Données précliniques»). Une régression du taux de lymphocytes B chez les nourrissons peut altérer la réponse aux vaccins (voir «Mises en garde et précautions»).
Allaitement
La sécurité d'utilisation de Benlysta pendant l'allaitement n'est pas établie. On ne dispose pas de données sur l'excrétion du bélimumab dans le lait maternel ou sur l'absorption systémique du bélimumab après une prise orale. Chez des macaques de Java, le bélimumab est toutefois passé dans le lait maternel.
Chez les mères allaitantes, il est recommandé au cours du traitement par Benlysta d'envisager l'arrêt de l'allaitement en tenant compte à la fois des avantages de l'allaitement maternel pour l'enfant et des avantages du traitement pour la femme, ainsi que d'éventuels effets indésirables pour l'enfant allaité qui sont liés à Benlysta ou à la pathologie sous-jacente de la mère. Il convient éventuellement de consulter un pédiatre concernant l'évaluation du risque pour l'enfant allaité.
Fertilité
On ne dispose pas de données concernant les effets du bélimumab sur la fertilité chez l'être humain. Aucune expérimentation animale n'a été conduite sur la fertilité masculine et féminine (voir «Données précliniques»).
Effet sur l’aptitude à la conduite et l’utilisation de machines
Aucune étude n'a été effectuée pour évaluer les effets du bélimumab sur l'aptitude à la conduite et à l'utilisation de machines. Les propriétés pharmacologiques du bélimumab ne suggèrent aucun effet négatif sur ce type d'activités.
Lors de l'appréciation de l'aptitude du patient à l'exécution de tâches exigeant une capacité de jugement et des capacités motrices et cognitives, on tiendra compte de l'état clinique du patient et du profil de sécurité du bélimumab.
Effets indésirables
La sécurité du bélimumab chez les patients atteints de LED a été évaluée dans 3 études contrôlées contre placebo portant sur l'administration intraveineuse, réalisées avant l'autorisation, dans une étude régionale subséquente contrôlée contre placebo portant sur l'administration intraveineuse et dans une étude contrôlée contre placebo portant sur l'administration sous-cutanée ainsi que dans deux études contrôlées contre placebo portant sur l'administration intraveineuse, réalisées après la commercialisation. La sécurité d'emploi chez des patients atteints de néphrite lupique a été évaluée dans une étude contrôlée contre placebo avec administration intraveineuse.
Les données présentées ci-dessous reposent sur le traitement intraveineux de 674 patients atteints de LED des trois études cliniques menées avant l'autorisation et de 470 patients de l'étude subséquente contrôlée contre placebo (10 mg/kg administrés sur une période de 1 heure aux jours 0, 14, 28 et ensuite tous les 28 jours, sur une période de 52 semaines), et sur le traitement sous-cutané de 556 patients atteints de LED (200 mg une fois par semaine pour une durée allant jusqu'à 52 semaines). Les données de sécurité présentées pour le traitement intraveineux englobent aussi des données qui s'étendent au-delà de la période de 52 semaines pour certains patients atteints de LED. En outre, il y a aussi des données de 224 patients atteints de néphrite lupique qui ont reçu Benlysta par voie intraveineuse (10 mg/kg pour une durée allant jusqu'à 104 semaines). Les données provenant d'annonces reçues après la mise en circulation ont également été prises en compte. La plupart des patients ont reçu également une ou plusieurs des médications concomitantes suivantes pour traiter le LED: corticostéroïdes, immunomodulateurs, antipaludiques et AINS.
Les effets indésirables sont décrits ci-dessous par classes de systèmes d'organes selon la terminologie MedDRA ainsi que par fréquence. Les catégories de fréquence suivantes ont été utilisées:
Très fréquents: ≥1/10
Fréquents: ≥1/100 à <1/10
Occasionnels: ≥1/1000 à <1/100
Infections et infestations
Très fréquents: Infections (70%).
Fréquents: Rhinopharyngite, bronchite, pharyngite, cystite, gastroentérite virale.
Affections hématologiques et du système lymphatique
Fréquents: Leucopénie.
Affections cardiaques
Occasionnels: Bradycardie.
Affections du système immunitaire
Fréquents: Réaction d'hypersensibilité.
Occasionnels: Réaction anaphylactique, angio-œdème, hypogammaglobulinémie.
Des réactions d'hypersensibilité tardives ou retardées peuvent survenir et s'annoncer par des symptômes tels qu'éruption, urticaire, nausées, fièvre, fatigue, myalgie, céphalées, douleurs des membres et œdème du visage.
Affections psychiatriques
Fréquents: Dépression.
Occasionnels: Pensées suicidaires, comportement suicidaire.
Affections du système nerveux
Fréquents: Migraine.
Affections gastro-intestinales
Très fréquents: Nausées (15%), diarrhée (12%).
Affections de la peau et du tissu sous-cutané
Fréquents: Réactions au site d'injection, éruption cutanée, urticaire.
Affections musculosquelettiques et du tissu conjonctif
Fréquents: Douleurs des membres.
Troubles généraux et anomalies au site d'administration
Fréquents: Pyrexie, réactions liées à la perfusion ou à l'injection, insomnie.
Description de certains effets indésirables
Immunogénicité
Dans les deux études de phase III avec administration intraveineuse dans le LED, 4 patients sur 563 (0,7%) du groupe recevant 10 mg/kg et 27 patients sur 559 (4,8%) du groupe recevant 1 mg/kg ont développé des anticorps anti-bélimumab persistants. La fréquence rapportée pour le groupe recevant 10 mg/kg est probablement inférieure à la fréquence réelle, car la sensibilité du test est réduite en présence de concentrations élevées du médicament.
Des anticorps neutralisants ont été constatés chez 3 patients atteints de LED sous bélimumab 1 mg/kg (intraveineux).
Dans l'étude de phase III avec administration sous-cutanée, il n'a pas été observé de formation d'anticorps anti-bélimumab chez les 556 patients atteints de LED qui ont reçu du bélimumab (200 mg une fois par semaine) pendant les 52 semaines de la période contrôlée contre placebo.
Au vu du faible nombre de patients qui présentent des anticorps anti-bélimumab, il n'est pas possible de tirer des conclusions définitives sur les répercussions d'une immunogénicité sur la pharmacocinétique du bélimumab.
Dans l'étude sur la néphrite lupique, au cours de laquelle 224 patients ont reçu Benlysta 10 mg/kg par voie intraveineuse, aucun anticorps anti-bélimumab n'a été détecté.
Cas de décès
Dans les études cliniques contrôlées avec administration intraveineuse, 14** cas de décès ont été enregistrés parmi les patients atteints de LED traités jusqu'à 52 semaines: 6 patients sur 673 traités par bélimumab 1 mg/kg, 0 patient sur 111 traités par bélimumab 4 mg/kg, 6 patients sur 674 traités par bélimumab 10 mg/kg et 3 patients sur 675 traités par placebo. Les causes de décès les plus fréquentes ont correspondu à ce qu'on pouvait attendre chez une population atteinte de LED: entre autres infection, poussée due au lupus, complications cardio-vasculaires et suicide. Aucun cas de décès supplémentaire ne s'est produit parmi les patients traités jusqu'à 76 semaines.
** En plus de ce nombre, un patient du groupe traité par 1 mg/kg est décédé après la fin de l'étude.
Dans l'étude de phase III avec administration sous-cutanée dans le LED, 5 cas de décès sont survenus au total dans la phase en double aveugle, dont 2 (0,7%) dans le groupe sous placebo et 3 (0,5%) dans le groupe sous bélimumab 200 mg par voie sous-cutanée. Les trois cas de décès survenus dans le groupe sous bélimumab étaient tous imputables à une infection, les 2 cas de décès du groupe sous placebo étaient dus à un arrêt cardiaque et à une thrombocytopénie.
Infections
Dans l'étude clinique avec administration sous-cutanée, l'incidence globale d'infections a été de 55% dans le groupe sous bélimumab et de 57% dans le groupe sous placebo. Des infections bactériennes des voies urinaires sont survenues chez 7,6% des patients sous bélimumab et chez 6,4% des patients sous placebo. Des infections graves sont survenues chez 4% des patients sous bélimumab et chez 5% des patients sous placebo, 0,2% respectivement 0% de ces patients ayant subi des infections graves dues à des germes opportunistes.
Dans des études cliniques sur 18 mois (données mises en commun pour l'administration intraveineuse et sous-cutanée), les infections opportunistes graves observées ont été numériquement plus fréquentes sous bélimumab (10/2014, 0,5% contre 0/955 sous placebo). Dans une étude de sécurité randomisée (1:1), en double aveugle, contrôlée contre placebo, d'une durée de 52 semaines (BEL115467), réalisée après la commercialisation, menée auprès de 4003 patients atteints de LED et qui a évalué la mortalité et les événements indésirables spécifiques chez l'adulte, 3,7% des patients ayant reçu 10 mg/kg de bélimumab par voie intraveineuse et 4,1% des patients sous placebo ont développé des infections sévères. Des infections mortelles se sont produites chez 0,45% (9/2002) des patients sous bélimumab et chez 0,15% (3/2001) des patients sous placebo. L'incidence de la mortalité globale était de 0,50% (10/2002) dans le groupe sous bélimumab et de 0,40% (8/2001) dans le groupe sous placebo.
Dans l'étude sur la néphrite lupique, les patients ont reçu un traitement de base standard (voir «Études cliniques»). 13,8% des patients qui ont reçu le bélimumab et 17,0% des patients qui ont reçu le placebo ont développé des infections sévères. Des infections d'issue fatale ont été observées chez 0,9% (2/224) des patients sous bélimumab et chez 0,9% (2/224) des patients sous placebo.
Affections psychiatriques
Dans les trois études cliniques avec administration intraveineuse dans le LED (n=2133), réalisées avant l'autorisation, des événements psychiatriques ont été rapportés plus fréquemment sous Benlysta (16%) que sous placebo (12%), ceux-ci ayant été essentiellement des événements liés à une dépression (6,3% sous Benlysta et 4,7% sous placebo), de l'insomnie (6,0% sous Benlysta et 5,3% sous placebo) et une anxiété (3,9% sous Benlysta et 2,8% sous placebo). Des événements psychiatriques sévères ont été rapportés chez 0,8% des patients sous Benlysta (0,6% avec 1 mg/kg, 1,2% avec 10 mg/kg) et chez 0,4% des patients sous placebo. Une dépression sévère est survenue chez 0,4% (6/1458) des patients sous Benlysta et chez 0,1% (1/675) des patients sous placebo. Deux suicides (0,1%) ont été recensés chez les patients sous Benlysta.
Dans une étude sur le LED randomisée, en double aveugle, contrôlée contre placebo avec administration intraveineuse de 10 mg/kg de bélimumab, réalisée après la commercialisation, des événements psychiatriques sévères ont été rapportés chez 1,0% (20/2002) des patients sous bélimumab et chez 0,3% (6/2001) des patients sous placebo. Des dépressions sévères ont été observées chez 0,3% (7/2002) des patients sous bélimumab et chez <0,1% (1/2001) des patients sous placebo. L'incidence globale des pensées suicidaires sévères, du comportement suicidaire sévère ou des automutilations sévères sans intention suicidaire a été de 0,7% (15/2002) dans le groupe bélimumab et de 0,2% (5/2001) dans le groupe placebo.
Sur l'échelle Columbia-Suicide-Severity-Rating-Scale (C-SSRS), 2,4% (48/1974) des patients sous bélimumab ont indiqué avoir des pensées suicidaires ou un comportement suicidaire, contre 2,0% (39/1988) des patients sous placebo. Aucun suicide n'a été rapporté dans aucun groupe.
Dans l'étude sur le LED susmentionnée avec administration intraveineuse, les patients ayant des antécédents de maladies psychiatriques n'ont pas été exclus.
En revanche, dans l'étude clinique sur le LED avec administration sous-cutanée, les patients ayant des antécédents connus de maladies psychiatriques ont été exclus et des événements psychiatriques sévères ont été rapportés chez 0,2% (1/556) des patients sous bélimumab et chez aucun des patients sous placebo. Des événements sévères liés à une dépression ou des suicides ne sont survenus dans aucun groupe. Sur l'échelle C-SSRS, 1,3% (7/554) des patients sous bélimumab et 0,7% (2/277) des patients sous placebo ont indiqué avoir des pensées suicidaires ou un comportement suicidaire.
Réactions au site d'injection
Dans l'étude clinique sur le LED avec administration sous-cutanée, des réactions au site d'injection sont survenues chez 6,1% (34/556) des patients sous bélimumab et chez 2,5% (7/280) des patients sous placebo. Ces réactions au site d'injection (généralement des douleurs, un érythème, un hématome, un prurit et une induration) avaient un degré de gravité léger à modéré. Deux participants (0,4%, 2/556) du groupe sous bélimumab ont arrêté l'étude en raison d'une réaction locale au site d'injection.
L'annonce d'effets secondaires présumés après l'autorisation est d'une grande importance. Elle permet un suivi continu du rapport bénéfice-risque du médicament. Les professionnels de santé sont tenus de déclarer toute suspicion d'effet secondaire nouveau ou grave via le portail d'annonce en ligne ElViS (Electronic Vigilance System). Vous trouverez des informations à ce sujet sur www.swissmedic.ch.
Surdosage
On dispose d'une expérience limitée en ce qui concerne un surdosage de bélimumab. Les effets indésirables rapportés en lien avec des cas de surdosage reflétaient le profil de sécurité connu du bélimumab.
Propriétés/Effets
Code ATC
L04AG04
Mécanisme d'action
Le stimulateur de lymphocytes B (BLyS, également appelé BAFF et TNFSF13) appartient à la famille des ligands du facteur de nécrose tumorale (TNF). C'est la molécule cible du bélimumab. Le BLyS inhibe l'apoptose des lymphocytes B et stimule la prolifération et la différenciation des lymphocytes B en plasmocytes producteurs d'immunoglobulines.
Le BLyS est surexprimé chez les patients atteints de LED. Il existe une forte corrélation entre l'activité du LED (évaluée à l'aide du «Safety of Estrogen in Lupus Erythematosus National Assessment-Systemic Lupus Erythematosus Disease Activity Index» [SELENA-SLEDAI]) et les taux plasmatiques de BLyS.
Le bélimumab est un anticorps monoclonal humain de type IgG1λ d'environ 147 kDa. Il se fixe spécifiquement au BLyS soluble humain et inhibe son activité biologique. Le bélimumab ne se fixe pas directement aux lymphocytes B, mais inhibe la survie des lymphocytes B – y compris des lymphocytes B auto-réactifs – et réduit la différenciation des lymphocytes B en plasmocytes producteurs d'immunoglobulines par une liaison et une neutralisation du BLyS.
Pharmacodynamique
Dans des études avec administration intraveineuse ou sous-cutanée, les concentrations médianes d'IgG ont diminué de 11 à 15% jusqu'à la 52e semaine chez les patients atteints de LED sous bélimumab et de 2,5 à 0,7% chez les patients sous placebo.
Parmi les patients atteints de LED ayant présenté des anticorps anti-ADN double brin au début de l'étude, une réduction a été observée sous bélimumab dès la semaine 4. À 52 semaines, les anticorps anti-ADN double brin étaient indétectables chez 16 à 18% des patients sous bélimumab, par rapport à 7 à 15% des patients sous placebo.
Chez les patients atteints de LED ayant présenté de faibles taux de complément au début de l'étude, le traitement par bélimumab a provoqué une augmentation des taux de complément (C3 et C4). Cette augmentation a été observée dès la semaine 4 et s'est maintenue par la suite. À 52 semaines, les taux de C3 et de C4 étaient normalisés chez 38 à 42% et 44 à 53% des patients sous bélimumab, par rapport à 17 à 21% et 19 à 20% des patients sous placebo.
Sous bélimumab, une diminution des lymphocytes B circulants, qu'il s'agisse des lymphocytes B transitionnels, naïfs et activés, des plasmocytes et des lymphocytes B auto-réactifs du LED, a été observée à la semaine 52. Les réductions de lymphocytes naïfs, de plasmocytes, de plasmocytes à courte durée de vie et de lymphocytes B auto-réactifs du LED étaient constatables dès la semaine 8. Le taux de lymphocytes B à mémoire a augmenté au début, puis baissé lentement aux valeurs initiales jusqu'à la semaine 52.
Dans une étude d'extension à long terme non contrôlée avec administration intraveineuse, menée chez des patients atteints de LED, des lymphocytes B (entre autres des lymphocytes naïfs, activés, des plasmocytes et le sous-groupe de lymphocytes B auto-réactifs du LED) ainsi que les concentrations d'IgG ont été observés sur plus de 7 ans sous traitement. Une régression marquée, durable et progressive de différents sous-groupes de lymphocytes B a été constatée, qui a entraîné une diminution médiane des lymphocytes B naïfs de 87%, des lymphocytes B à mémoire de 67%, des lymphocytes B activés de 99% et des plasmocytes de 92% après un traitement de plus de 7 ans. Après environ 7 ans, une diminution médiane des concentrations d'IgG de 28% a été observée, 1,6% des patients ayant montré une régression des concentrations d'IgG au-dessous de 400 mg/dl. L'incidence des effets indésirables signalés est restée globalement stable ou a légèrement reculé au cours de l'étude.
Après un traitement par Benlysta (10 mg/kg par voie intraveineuse) ou par placebo chez des patients atteints de néphrite lupique, une élévation des taux sériques d'IgG, associée à une diminution de la protéinurie, a été observée. Par rapport au placebo, on a observé une plus faible élévation du taux sérique d'IgG dans le groupe sous Benlysta, comme l'on pouvait s'y attendre au vu du mécanisme connu du bélimumab. À la semaine 104, le pourcentage médian d'élévation des taux d'IgG par rapport à la valeur initiale était de 17% pour Benlysta et de 37% pour le placebo. Les réductions observées des auto-anticorps, les élévations du complément et les réductions des lymphocytes B circulants totaux et des sous-types de lymphocytes B étaient conformes aux résultats des études sur le LED.
Efficacité clinique
Injection sous-cutanée dans le LED
L'efficacité du bélimumab administré par voie sous-cutanée a été examinée dans le cadre d'une étude de phase III randomisée, en double aveugle, contrôlée par placebo, d'une durée de 52 semaines (HGS1006-C1115; BEL112341), menée auprès de 836 patients atteints de LED diagnostiqué cliniquement selon les critères de classification de l'American College of Rheumatology. Les patients remplissant les critères de l'étude avaient un LED actif, défini par un score SELENA-SLEDAI de 8 au minimum, avec des résultats positifs de tests pour les anticorps antinucléaires (AAN ou anti-ADNdb; titre d'AAN de 1:80 au minimum et/ou anticorps anti-ADNdb de 30 unités/mL au minimum) lors de la sélection. Les patients étaient sous un régime thérapeutique stable pour leur LED (traitement standard) incluant les composants suivants (seuls ou en association): corticostéroïdes, antipaludiques, AINS ou autres immunosuppresseurs. Étaient exclus les patients souffrant d'un lupus sévère et actif avec atteinte du système nerveux central ou de néphrite lupique active sévère, les patients qui avaient reçu un autre médicament biologique encore en phase expérimentale au cours des 3 mois précédents, les patients qui avaient été testés positifs pour des anticorps anti-VIH, les antigènes de surface de l'hépatite B ou les anticorps dirigés contre l'hépatite C, ainsi que les patients transplantés et les patients souffrant d'hypogammaglobulinémie ou de déficit en IgA.
Cette étude a été menée aux États-Unis, en Amérique du Sud, en Europe et en Asie, Les patients avaient un âge médian de 37 ans (fourchette: de 18 à 77 ans) et étaient en majorité (94%) de sexe féminin. Les patients ont été randomisés dans un rapport de 2:1 pour recevoir en sous-cutané, pendant 52 semaines, soit le bélimumab 200 mg soit le placebo une fois par semaine.
Le critère primaire d'efficacité était un critère composite (SLE-Responder-Index, SRI), à l'aide duquel ont été définis comme répondeurs les patients qui, au bout de 52 semaines, satisfaisaient à tous les critères suivants par rapport au début de l'étude:
·régression du score SELENA-SLEDAI de 4 points au minimum et
·aucune nouvelle implication de systèmes d'organes BILAG-A ou absence de 2 nouvelles implications de systèmes d'organes BILAG-B (BILAG=British Isles Lupus Assessment Group), ainsi que
·aucune aggravation (élévation de <0,30 point) du score PGA (PGA=Physician's Global Assessment).
L'index SLE-Responder se base sur le score SELENA-SLEDAI comme mesure objective de la diminution de l'activité globale de la maladie, sur l'index BILAG pour exclure une aggravation significative dans un système d'organes spécifique et sur le score PGA pour vérifier que des améliorations de l'activité de la maladie ne s'obtiennent pas aux dépens de l'état général du patient.
Les critères secondaires d'efficacité étaient notamment le temps écoulé jusqu'à la première poussée sévère (obtenu à l'aide de l'index SELENA-SLEDAI-SLE-Flare modifié) et le pourcentage de patients avec une réduction de la dose moyenne de prednisone de ≥25% à ≤7,5 mg/jour pendant la période de la semaine 40 à 52, par rapport au début de l'étude. Un critère pour le succès du traitement portait sur la variation moyenne du score sur l'échelle FACIT-Fatigue (FACIT=Functional Assessment of Chronic Illness Therapy) au bout de 52 semaines.
Le bélimumab a entraîné des améliorations significatives de l'index SLE-Responder et du score SELENA-SLEDAI ainsi que de ses composantes individuelles (voir Tableau 1).
Tableau 1: Taux de réponse à la semaine 52
|
Réponse1 |
Placebo |
Bélimumab |
|
Index SLE-Responder |
48,4% |
61,4% |
|
Composantes de l'index SLE-Responder | ||
|
Pourcentage de patients présentant une régression de SELENA-SLEDAI ≥4 (%) |
49,1% |
62,3% |
|
Pourcentage de patients sans aggravation selon l'index BILAG (%) |
74,2% |
80,9% |
|
Pourcentage de patients sans aggravation selon PGA (%) |
72,8% |
81,2% |
¹ Dans les analyses, tous les sujets pour lesquels une valeur initiale de l'un des composants manquait ont été exclus (1 dans le groupe placebo; 2 dans le groupe bélimumab).
Les différences entre les groupes de traitement étaient statistiquement significatives à partir de la semaine 16 et le sont restées jusqu'à la semaine 52 (Figure 1).
Figure 1: Pourcentage de SRI-Responder après visite

Les poussées sévères de LED ont été déterminées au moyen de l'index SELENA-SLEDAI-SLE-Flare modifié, la modification permettant d'exclure la saisie de poussées sévères exclusivement sur la base d'une valeur supérieure à 12 du score SELENA-SLEDAI. Le risque de poussées sévères a chuté de 49% (Hazard-Ratio=0,51; p=0,0004) dans le groupe sous bélimumab pendant l'observation de 52 semaines, par rapport au groupe sous placebo.
Au début de l'étude, 60% des patients recevaient de la prednisone (ou un équivalent de la prednisone) à des dosages de >7,5 mg/jour. Parmi ces patients, 18,2% ont réduit sous bélimumab leur dose moyenne de prednisone de 25% au moins à ≤7,5 mg/jour pendant les semaines 40 à 52, par rapport à 11,9% des patients sous placebo. Cette différence n'était cependant pas statistiquement significative (p=0,0732).
Le bélimumab, administré aussi bien par voie intraveineuse que par voie sous-cutanée a amélioré la fatigue par rapport au placebo, selon le score FACIT-Fatigue.
L'analyse par sous-groupes du critère primaire des études pivots sur l'administration intraveineuse et sous-cutanée a montré que ce sont les patients qui présentent une activité plus élevée de la maladie au début qui profitent le plus du traitement, notamment aussi les patients qui ont des scores SELENA-SLEDAI de 10 ou davantage, les patients qui ont des taux de complément abaissés, les patients qui ont besoin de stéroïdes pour contrôler la maladie ainsi que les patients qui ont à la fois des anticorps anti-ADNdb positifs et des taux de complément abaissés.
L'efficacité et la sécurité de Benlysta en association avec un seul cycle de rituximab ont été évaluées dans une étude de phase III randomisée, en double aveugle, contrôlée contre placebo, de 104 semaines, réalisée chez 292 patients (BLISS-BELIEVE). Le critère d'évaluation principal était la proportion de patients présentant le statut d'une maladie contrôlée, défini par un score SLEDAI-2K ≤2, qui était atteint à la semaine 52 sans immunosuppresseurs et avec des corticostéroïdes à une dose équivalente à ≤5 mg de prednisone par jour. Ceci a été atteint chez 19,4% (n = 28/144) des patients traités par Benlysta en association avec le rituximab et chez 16,7% (n = 12/72) des patients traités par Benlysta en association avec le placebo (Odds-Ratio 1,27; IC à 95%: 0,60; 2,71; p = 0,5342). Chez les patients traités par Benlysta en association avec le rituximab, des effets indésirables (91,7% vs 87,5%), des effets indésirables sévères (22,2% vs 13,9%) et des infections sévères (9,0% vs 2,8%) ont été observés plus fréquemment que chez les patients traités par Benlysta en association avec le placebo.
Perfusion intraveineuse dans la néphrite lupique
L'efficacité et la sécurité d'emploi du bélimumab 10 mg/kg, administré par voie intraveineuse sur une période d'une heure aux jours 0, 14 et 28 et ensuite tous les 28 jours, ont été évaluées dans une étude de phase III (BEL114054) randomisée (1:1), contrôlée contre placebo, en double aveugle, d'une durée de 104 semaines et réalisée chez 448 patients atteints de néphrite lupique active.
Au moment de la sélection, les patients présentaient un diagnostic clinique de LED posé selon les critères de classification de l'ACR, une néphrite lupique de classe III, IV et/ou V confirmée par une biopsie ainsi qu'une maladie rénale active qui rendait nécessaire un traitement standard (corticostéroïdes avec mycophénolate-mofétil pour l'induction et l'entretien ou cyclophosphamide pour le traitement d'induction, suivi d'azathioprine pour le traitement d'entretien). L'étude a été menée en Asie, en Amérique du Nord, en Amérique du Sud et en Europe. L'âge médian des patients était de 31 ans (fourchette: de 18 à 77 ans). Les participants étaient majoritairement de sexe féminin (88%).
Le critère principal d'efficacité était la réponse rénale primaire (Primary Efficacy Renal Response, PERR) à la semaine 104, définie comme une réponse à la semaine 100 confirmée par la répétition de la mesure des paramètres suivants à la semaine 104: rapport protéines urinaires/créatinine (uPCR) ≤0,7 et débit de filtration glomérulaire estimé (DFGe) ≥60 ml/min/1,73 m2 ou absence d'une chute du DFGe de >20% par rapport à la valeur mesurée avant la poussée.
Les principaux critères secondaires comprenaient:
·une réponse rénale complète (Complete Renal Response, CRR), définie comme une réponse à la semaine 100 confirmée par la répétition de la mesure des paramètres suivants à la semaine 104: uPCR <0,5 et DFGe ≥90 ml/min/1,73 m2 ou absence d'une chute du DFGe de >10% par rapport à la valeur mesurée avant la poussée
·une PERR à la semaine 52
·le temps écoulé jusqu'à un événement rénal ou jusqu'au décès (événement rénal défini comme le premier événement d'une insuffisance rénale terminale, doublement de la valeur de la créatinine sérique, détérioration de la fonction rénale [définie comme une augmentation de la protéinurie et/ou une altération de la fonction rénale] ou administration d'une thérapie non autorisée pour le traitement de maladies rénales)
En ce qui concerne les critères de PERR et CRR, les patients qui avaient quitté prématurément l'étude ou avaient reçu des médicaments non autorisés ont été considérés comme des non-répondeurs. Pour que le patient soit considéré comme répondeur en ce qui concerne les critères pertinents, le traitement par stéroïdes devait être réduit à ≤10 mg/jour à partir de la semaine 24.
Le pourcentage de patients qui ont atteint une PERR à la semaine 104 était significativement plus élevé dans le groupe sous bélimumab que dans le groupe sous placebo. De même, en ce qui concerne les principaux critères secondaires, le bélimumab a permis d'obtenir des résultats significativement meilleurs par rapport au placebo (Tableau 2).
Tableau 2: Résultats d'efficacité chez des patients adultes atteints de néphrite lupique
|
Critère d'efficacité |
Placebo |
Bélimumab |
Différence observée vs placebo |
Odds/Hazard Ratio vs placebo |
Valeur P |
|
PERR à la semaine 1041 |
32,3% |
43,0% |
10,8% |
OR 1,55 |
0,0311 |
|
Composantes de la PERR | |||||
|
Rapport protéines urinaires/créatinine |
33,6% |
44,4% |
10,8% |
OR 1,54 |
0,0320 |
|
DGFe ≥60 ml/min/1,73 m2 |
50,2% |
57,4% |
7,2% |
OR 1,32 |
0,1599 |
|
Pas d'échec du traitement |
74,4% |
83,0% |
8,5% |
OR 1,65 |
0,0364 |
|
CRR à la semaine 1041 |
19,7% |
30,0% |
10,3% |
OR 1,74 |
0,0167 |
|
Composantes de la CRR | |||||
|
Rapport protéines urinaires/créatinine |
28,7% |
39,5% |
10,8% |
OR 1,58 |
0,0268 |
|
DFGe ≥90 ml/min/1,73 m2 |
39,9% |
46,6% |
6,7% |
OR 1,33 |
0,1539 |
|
Pas d'échec du traitement |
74,4% |
83,0% |
8,5% |
OR 1,65 |
0,0364 |
|
PERR à la semaine 521 |
35,4% |
46,6% |
11,2% |
OR 1,59 |
0,0245 |
|
Temps écoulé jusqu'à un événement rénal ou jusqu'au décès.1 |
28,3% |
15,7% |
- |
|
|
|
Temps écoulé jusqu'à l'événement [Hazard Ratio (IC à 95%)] |
|
|
- |
0,51 |
0,0014 |
|
1
La PERR à la semaine 104 était l'analyse d'efficacité primaire; la CRR à la semaine 104, la PERR à la semaine 52 et le temps écoulé jusqu'à l'événement rénal ou le décès faisaient partie de la hiérarchie des tests préalablement définis. | |||||
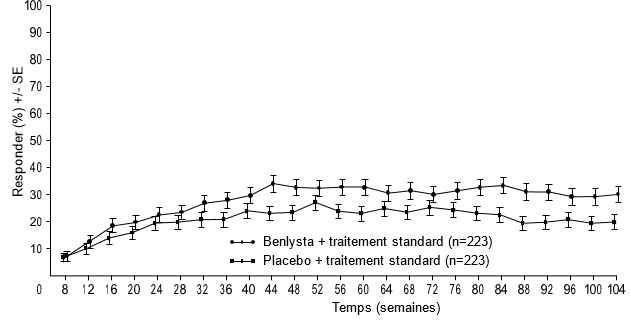
À partir de la semaine 24, un pourcentage numériquement plus élevé de patients sous bélimumab a atteint la PERR par rapport aux patients sous placebo. Cette différence entre traitements s'est maintenue jusqu'à la semaine 104. À partir de la semaine 12, un pourcentage numériquement plus élevé de patients sous bélimumab a atteint la CRR par rapport aux patients sous placebo. Cette différence numérique s'est maintenue jusqu'à la semaine 104 (Figure 2).
Figure 2: Taux de réponse des adultes atteints de néphrite lupique par visite
Réponse rénale primaire (Primary Efficacy Renal Response, PERR)
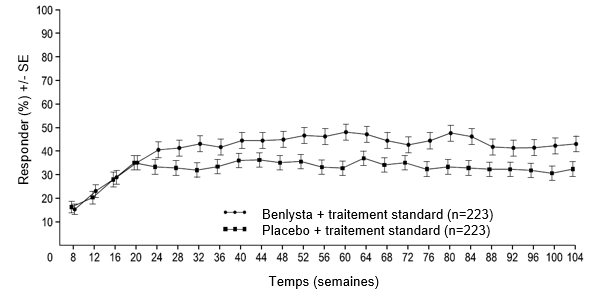
Réponse rénale complète (Complete Renal Response, CRR)
Dans des analyses descriptives de sous-groupes, les taux de PERR et de CRR ont été examinés par régime d'induction (mycophénolate ou cyclophosphamide), par classe de biopsie (classe III ou IV, classe III + V ou classe IV + V ou classe V) et selon les valeurs initiales de l'uPCR (<3 g/g ou ≥3 g/g; analyse post hoc) ont été examinés (Figure 3).
Figure 3: Odds Ratio pour la PERR et la CRR à la semaine 104 dans les divers sous-groupes
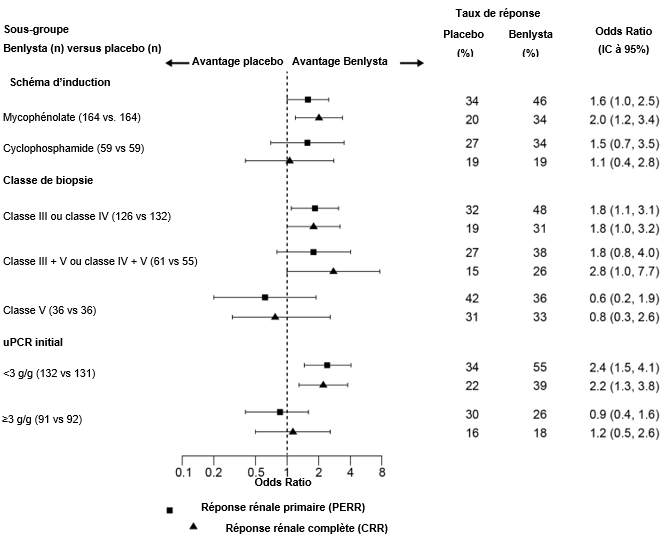
¹ Classe III=néphrite lupique focale proliférative; classe IV=néphrite lupique diffuse proliférative; classe V=néphrite lupique membraneuse; classe III + V=néphrite lupique mixte membraneuse – focale proliférative; classe IV + V=néphrite lupique mixte membraneuse – diffuse proliférative.
² Le rapport protéines urinaires initiales:créatinine (uPCR) était une analyse post hoc.
Pharmacocinétique
Études sur le LED
Les paramètres pharmacocinétiques intraveineux décrits ci-dessous reposent sur des estimations des paramètres de la population chez les 563 patients atteints de LED et ayant reçu du bélimumab intraveineux dosé à 10 mg/kg dans les deux études de phase III (aux jours 0, 14, 28 et ensuite tous les 28 jours sur une durée allant jusqu'à 52 semaines).
Les paramètres pharmacocinétiques de l'administration sous-cutanée traités ci-après se basent sur des estimations pharmacocinétiques de population tirées des données de 661 participants, dont 554 patients atteints de LED et 107 personnes en bonne santé, qui ont reçu le bélimumab par voie sous-cutanée.
Absorption
Après administration sous-cutanée, la concentration sérique maximale (Cmax) du bélimumab s'est élevée à 108 µg/mL, le temps requis pour atteindre la Cmax à l'équilibre d'accumulation (Tmax) a été de 2,6 jours. La biodisponibilité du bélimumab était d'environ 74%.
Distribution
Le bélimumab s'est réparti dans les tissus avec un volume total de distribution de 5 L environ.
Métabolisme
Le bélimumab est une protéine. Il est dégradé en petits peptides et en acides aminés individuels.
Élimination
Après administration sous-cutanée, la demi-vie terminale du bélimumab a été de 18,3 jours. La demi-vie de distribution a été de 1,1 jour. La diminution biphasique observée lors d'une administration intraveineuse était masquée par la lente phase d'absorption. La clairance systémique a été de 204 mL/jour.
Étude sur la néphrite lupique
Une analyse pharmacocinétique de population a été effectuée chez 224 patients adultes atteints de néphrite lupique qui recevaient Benlysta 10 mg/kg par voie intraveineuse (aux jours 0, 14 et 28 et ensuite tous les 28 jours jusqu'à 104 semaines). Chez les patients atteints de néphrite lupique, la clairance du bélimumab était initialement plus élevée que celle observée dans les études sur le LED, en raison de l'activité de la maladie rénale. Cependant, après 24 semaines de traitement et pendant tout le reste de la durée de l'étude, la clairance et l'exposition du bélimumab étaient similaires à celles observées chez des patients adultes atteints de LED ayant reçu 10 mg/kg de bélimumab par voie intraveineuse.
Sur la base de la modélisation et de la simulation pharmacocinétique de population, les concentrations moyennes à l'état d'équilibre lors de l'administration sous-cutanée une fois par semaine de 200 mg de bélimumab à des adultes atteints de néphrite lupique sont supposées être similaires à celles observées chez des patients adultes atteints de néphrite lupique qui reçoivent 10 mg/kg de bélimumab par voie intraveineuse toutes les 4 semaines.
Cinétique pour certains groupes de patients
Troubles de la fonction hépatique
Aucune étude formelle n'a été effectuée pour évaluer l'influence d'une insuffisance hépatique sur la pharmacocinétique du bélimumab. Les immunoglobulines comme le bélimumab sont dégradées par le système réticulo-endothélial. Il est donc peu probable que les altérations de la fonction hépatique aient une influence sur l'élimination du bélimumab.
Troubles de la fonction rénale
Aucune étude formelle n'a été effectuée pour évaluer l'influence d'une insuffisance rénale sur la pharmacocinétique du bélimumab. Au cours de son développement clinique, le bélimumab (intraveineux et sous-cutané) a été évalué chez un nombre restreint de patients insuffisants rénaux (clairance de la créatinine <60 mL/min, dont certains patients avec une clairance de la créatinine <30 mL/min) atteints de LED. Une protéinurie (≥2 g par jour) et une clairance réduite de la créatinine n'ont pas entraîné des modifications cliniquement significatives de la clairance du bélimumab. Un ajustement de la dose n'est donc pas recommandé chez les patients insuffisants rénaux.
Patients âgés
Le bélimumab n'a été évalué que chez un nombre restreint de patients âgés. Selon l'analyse pharmacocinétique de population, l'âge n'avait pas d'effet sur l'exposition au bélimumab. Toutefois, vu le faible nombre de personnes âgées de ≥65 ans dans la population évaluée, une influence de l'âge ne peut pas être définitivement exclue.
Enfants et adolescents
On ne dispose pas de données pharmacocinétiques concernant les enfants et les adolescents.
Autres caractéristiques de patients
Le sexe, la race ou l'origine ethnique n'ont pas eu d'influence significative sur la pharmacocinétique du bélimumab. L'effet du poids corporel et de l'IMC sur l'exposition au bélimumab après administration sous-cutanée a été considéré comme cliniquement non significatif. Il n'a pas été constaté d'incidences liées au poids sur l'efficacité ou la sécurité.
Passage de la voie intraveineuse à la voie sous-cutanée
LED
Les patients atteints de LED qui sont passés, dans un intervalle de 1 à 4 semaines, d'une dose de 10 mg/kg administrée par voie intraveineuse toutes les 4 semaines à une dose de 200 mg administrée par voie sous-cutanée une fois par semaine présentaient, avant leur première injection sous-cutanée, des concentrations sériques de bélimumab qui se rapprochaient étroitement de leur future concentration minimale à l'état d'équilibre obtenue après administration par voie sous-cutanée (voir «Posologie/Mode d'emploi»). Sur la base de simulations avec des paramètres pharmacocinétiques de population, les concentrations moyennes à l'état d'équilibre de bélimumab administré à raison de 200 mg par voie sous-cutanée une fois par semaine étaient similaires aux concentrations de bélimumab administré à raison de 10 mg/kg par voie intraveineuse toutes les 4 semaines.
Néphrite lupique
D'après des simulations de pharmacocinétique de population, les patients atteints de néphrite lupique qui passent de 10 mg/kg par voie intraveineuse une à deux semaines après la fin des deux premières doses intraveineuses à 200 mg par voie sous-cutanée chaque semaine devraient avoir des concentrations sériques moyennes de bélimumab similaires à celles des patients ayant reçu 10 mg/kg par voie intraveineuse toutes les 4 semaines (voir «Posologie/Mode d'emploi»).
Données précliniques
Dans les études sur la toxicité de doses répétées et les études sur la toxicité de reproduction, les données précliniques ne révèlent aucun risque particulier pour l'être humain.
Toxicité en cas d'administration répétée
L'administration intraveineuse et sous-cutanée a conduit chez le singe à la réduction attendue du taux de lymphocytes B périphériques et des tissus lymphatiques, sans que cet effet ait été associé à des constats toxicologiques.
Génotoxicité
Le bélimumab étant un anticorps monoclonal, aucune étude de génotoxicité n'a été effectuée.
Carcinogénicité
Aucune étude de carcinogénicité n'a été effectuée.
Toxicité sur la reproduction
Aucune étude n'a été effectuée sur l'effet du bélimumab sur la fertilité (masculine ou féminine).
Dans les études de reproduction, des femelles gravides de macaques de Java ont reçu du bélimumab 150 mg/kg en perfusion intraveineuse toutes les 2 semaines pendant des périodes atteignant jusqu'à 21 semaines (cette dose correspond à une exposition environ 9 fois supérieure à l'exposition maximale attendue chez l'homme). Le traitement par le bélimumab n'a pas été associé à des effets nuisibles directs ou indirects dans le sens d'une toxicité maternelle, d'une toxicité développementale ou d'une tératogénicité. Les constats pathologiques attribuables au traitement étaient limités aux réductions réversibles attendues du taux de lymphocytes B chez la mère et chez les petits ainsi qu'à une réduction réversible des taux d'IgM chez les petits. Après l'arrêt d'administration du bélimumab, le taux de lymphocytes B est revenu à la normale chez les guenons adultes en l'espace d'un an post-partum et chez les petits en l'espace de 3 mois après la naissance. Les taux d'IgM des petits exposés in utero se sont normalisés en l'espace de 6 mois après la naissance.
Remarques particulières
Stabilité
Le médicament ne doit pas être utilisé au-delà de la date figurant après la mention «EXP» sur l'emballage.
Remarques particulières concernant le stockage
Conserver entre 2°C et 8°C. Ne pas congeler.
Protéger de la lumière. Conserver dans l'emballage d'origine jusqu'à l'utilisation. Conserver hors de portée des enfants.
Remarques concernant la manipulation
La solution pour injection est limpide à opalescente, incolore à jaune pâle.
Des instructions complètes illustrées pour l'administration se trouvent dans la notice d'emballage correspondante.
Numéro d’autorisation
66585 (Swissmedic).
Présentation
Emballage avec 1 auto-injecteur contenant 200 mg (B).
Emballage avec 4 auto-injecteurs contenant 200 mg (B).
Titulaire de l’autorisation
GlaxoSmithKline AG, 6340 Baar.
Mise à jour de l’information
Juillet 2025