Composition
Principes actifs
Pitolisantum (ut Pitolisanti hydrochloridum)
Excipients
Cellulosum microcristallinum, Crospovidonum, Talcum, Poly(alcool vinylicus), Magnesii stearas, Macrogolum 3350, Silica colloidalis anhydrica, Color E 171
Indications/Possibilités d’emploi
Wakix est indiqué chez l’adulte, l’adolescent et l’enfant de plus de 6 ans pour le traitement de la narcolepsie avec ou sans cataplexie (voir aussi « Propriétés/Effets »).
Posologie/Mode d’emploi
Le traitement doit être instauré par un médecin expérimenté dans la prise en charge des troubles du sommeil.
Posologie
Adultes
Wakix doit être utilisé à la dose efficace la plus faible, en fonction de la réponse et de la tolérance du patient selon le schéma posologique ci-dessous. La dose thérapeutique optimale doit être atteinte par palier, sans dépasser la dose de 36 mg/jour :
-1ère semaine : Une posologie initiale de 9 mg (2 comprimés à 4,5 mg) par jour.
-2ème semaine : la posologie peut être augmentée à 18 mg (1 comprimé à 18 mg) par jour ou diminuée à 4,5 mg (1 comprimé à 4,5 mg) par jour.
-3ème semaine : la posologie peut être augmentée à 36 mg (2 comprimés à 18 mg) par jour.
À tout moment, la dose peut être diminuée (jusqu’à 4,5 mg par jour) ou augmentée (jusqu’à 36 mg par jour) selon l’évaluation du médecin et la réponse du patient.
La dose quotidienne totale devrait être administrée en une seule prise le matin au cours du petit-déjeuner.
Maintien de l’efficacité
Les données d’efficacité à long terme étant limitées (voir « Propriétés/Effets »), le maintien de l’efficacité du traitement doit être évalué régulièrement par le médecin.
Populations particulières
Patients âgés
Les données disponibles chez les patients âgés sont limitées. Par conséquent, des adaptations de la dose peuvent être nécessaires en fonction du statut de la fonction rénale et hépatique de ces patients.
Insuffisance rénale
Chez les patients présentant une insuffisance rénale, la dose maximale quotidienne ne doit pas dépasser 18 mg.
Insuffisance hépatique
Chez les patients présentant une insuffisance hépatique modérée (Child-Pugh B), deux semaines après l’instauration du traitement, la dose quotidienne peut être augmentée. Une dose maximale de 18 mg ne doit pas être dépassée (voir « Pharmacocinétique »).
Pitolisant est contre-indiqué chez les patients ayant une insuffisance hépatique sévère (Child-Pugh C) (voir « Contre-indications »).
Aucun ajustement de dose n’est nécessaire chez les patients ayant une insuffisance hépatique légère.
Enfants et adolescents
Wakix doit être utilisé à la dose optimale, en fonction de la réponse individuelle du patient et de sa tolérance, selon un schéma d'augmentation de la dose par palier, sans dépasser la dose de 36 mg/jour (18 mg/jour chez l'enfant de moins de 40 kg).
-Semaine 1 : posologie initiale de 4,5 mg (un comprimé à 4,5 mg) par jour.
-Semaine 2 : la posologie peut être augmentée à 9 mg (deux comprimés à 4,5 mg) par jour.
-Semaine 3 : la posologie peut être augmentée à 18 mg (un comprimé à 18 mg) par jour.
-Semaine 4 : chez les enfants pesant 40 kg et plus, la posologie peut être augmentée à 36 mg (deux comprimés à 18 mg) par jour.
A tout moment, la posologie peut être diminuée (jusqu'à 4,5 mg par jour) ou augmentée (jusqu'à 36 mg par jour chez l'enfant de 40 kg et plus ou 18 mg par jour chez l'enfant de moins de 40 kg) selon l'évaluation du médecin et la réponse du patient.
La dose quotidienne totale devrait être administrée en une seule prise le matin au cours du petit-déjeuner.
Métaboliseurs lents
Par rapport aux métaboliseurs normaux de CYP2D6, on observe chez les métaboliseurs lents de CYP2D6 une plus grande exposition systémique (jusqu'à 3 fois). Le schéma de titration par palier devra tenir compte de cette exposition plus élevée.
Mode d’administration
Le comprimé doit être avalé entier avec suffisamment de liquide.
Contre-indications
·Hypersensibilité à la substance active ou à l'un des excipients selon sa composition.
·Insuffisance hépatique sévère (Child-Pugh C).
·Allaitement (voir « Grossesse, Allaitement »).
Mises en garde et précautions
Affections psychiatriques
Le pitolisant ne devrait être utilisé chez les patients souffrant de troubles psychiatriques, tels que des troubles anxieux graves ou une dépression grave avec tendance au suicide, qu’après une évaluation individuelle avantages-risques en raison d'une aggravation possible de ces tableaux cliniques. Des cas d’idées suicidaires ont été rapportés chez des patients ayant des antécédents psychiatriques traités par le pitolisant.
Insuffisance rénale ou hépatique
Le pitolisant doit être administré avec prudence chez les patients présentant une insuffisance rénale ou une insuffisance hépatique modérée (Child-Pugh B). La posologie doit être adaptée conformément à la rubrique « Posologie/Mode d’emploi ».
Affections gastro-intestinales
Des cas de troubles gastriques ont été rapportés avec le pitolisant, par conséquent, il doit être administré avec prudence chez les patients présentant des troubles gastriques liés à l'hyperacidité (voir « Effets indésirables ») ou en cas de co-administration avec des médicaments nocifs pour la muqueuse de l'estomac tels que les corticostéroïdes ou AINS.
Troubles de la nutrition
Le pitolisant doit être administré avec prudence chez les patients atteints d'obésité sévère ou d'anorexie sévère (voir « Effets indésirables »). En cas de changement de poids significatif, le traitement doit être réévalué par le médecin.
Troubles cardiaques
Dans deux études spécifiques, des doses de pitolisant supra-thérapeutiques (3 à 6 fois la dose thérapeutique, c’est-à-dire de 108 mg à 216 mg) ont produit une prolongation légère à modérée de l'intervalle QTc (10-13 ms) Dans les essais cliniques, aucun signal particulier de sécurité cardiaque n’a été identifié aux doses thérapeutiques de pitolisant. Néanmoins, les patients souffrant de maladie cardiaque, les patients traités avec d'autres médicaments allongeant l’intervalle QT ou pouvant augmenter le risque de troubles de la repolarisation, ou traités avec des médicaments augmentant de manière significative la Cmax et l’ASC (aire sous la courbe) du pitolisant (voir rubrique « Interactions ») ou les patients atteints d'insuffisance rénale sévère ou d’insuffisance hépatique modérée (voir « Avertissements et mesures de précaution ») doivent être étroitement surveillés, en particulier avec contrôle d'électrolytes et ECG (voir « Interactions »).
Épilepsie
Des convulsions ont été rapportées à des doses élevées dans des modèles animaux (voir « Données précliniques »). Dans les essais cliniques, un cas d’aggravation d'épilepsie a été rapporté chez un patient épileptique. Il convient d’être prudent chez les patients souffrant d'épilepsie sévère.
Femmes en âge de procréer
Les femmes en âge de procréer doivent utiliser une contraception efficace pendant le traitement et au moins jusqu'à 21 jours après l'arrêt du traitement (sur la base de la demi-vie du pitolisant et de ses métabolites). Le pitolisant peut réduire l'efficacité des contraceptifs hormonaux. Par conséquent, une autre méthode de contraception efficace doit être utilisée si la patiente utilise des contraceptifs hormonaux (voir rubriques « Interactions » et « Grossesse, Allaitement »).
Interaction avec d’autres médicaments
L’association du pitolisant avec les substrats du CYP3A4 et ayant une marge thérapeutique étroite doit être évitée (voir « Interactions »).
Effet rebond
Aucun effet rebond n’a été observé au cours des études cliniques. Cependant, l’arrêt du traitement doit être surveillé.
Abus
Les données cliniques (étude spécifique du potentiel d’abus chez l’homme à des doses de 36 à 216 mg chez les patients adultes, et effets indésirables liés à l’abus observés dans les études de phase 3) ont montré un potentiel d’abus faible ou nul pour le pitolisant.
Interactions
Antidépresseurs
Les antidépresseurs tricycliques ou tétracycliques (par exemple imipramine, clomipramine ou mirtazapine) peuvent modifier l'efficacité du pitolisant car ils possèdent des propriétés antagonistes du récepteur de l’histamine H1 et peuvent annuler l'effet de l'histamine endogène libérée dans le cerveau sous l’effet du traitement par le pitolisant.
Antihistaminiques
Les antihistaminiques (antagonistes du récepteur H1) qui traversent la barrière hémato-encéphalique (par exemple maléate de phéniramine, chlorphéniramine, diphenhydramine, prométhazine, mépyramine et doxylamine) peuvent altérer l’efficacité du pitolisant.
Médicaments allongeant l’intervalle QT ou connus pour augmenter le risque de troubles de la repolarisation
L’association avec le pitolisant doit être faite sous une surveillance étroite (voir « Avertissements et précautions »).
Interactions pharmacocinétiques
Médicaments affectant le métabolisme du pitolisant
Inducteurs d’enzyme
L'administration concomitante de doses répétées de rifampicine diminue de manière significative la Cmax moyenne et l’ASC (aire sous la courbe) du pitolisant d'environ 39 % et 50 %, respectivement. Par conséquent, l'administration concomitante de pitolisant avec des inducteurs puissants du CYP3A4 (par exemple rifampicine, phénobarbital, carbamazépine, phénytoïne) doit être faite avec prudence. Le millepertuis (Hypericum perforatum) ayant un effet inducteur puissant du CYP3A4, il convient d’être prudent lorsqu'il est associé au pitolisant. Une surveillance clinique doit être mise en place lorsque les deux substances actives sont associées avec éventuellement un ajustement de la posologie pendant l'association et une semaine après le traitement par l'inducteur.
Dans une étude clinique à doses répétées, l’association du pitolisant avec le probenecid a diminué l’AUC du pitolisant d’environ 34 %.
Inhibiteurs du cytochrome CYP2D6
La co-administration du pitolisant avec la paroxétine augmente de manière significative la Cmax moyenne et l’ASC0—72h du pitolisant d’environ 47 % et 105 %, respectivement. Compte tenu du doublement de l'exposition au pitolisant, son administration concomitante avec des inhibiteurs du CYP2D6 (par exemple paroxétine, fluoxétine, venlafaxine, duloxétine, bupropion, quinidine, terbinafine, cinacalcet) doit être faite avec prudence. Un ajustement de la posologie pendant l'association pourra éventuellement être envisagé.
Médicaments dont le métabolisme peut être affecté par le pitolisant
Substrats des CYP3A4 et CYP2B6
Selon les données in vitro, le pitolisant et ses principaux métabolites peuvent induire les cytochromes CYP3A4 et CYP2B6 aux concentrations thérapeutiques, et par extrapolation les CYP2C, UGTs et glycoprotéines P. Aucune donnée clinique sur l'ampleur de cette interaction n’est disponible. Par conséquent, l’association du pitolisant avec des médicaments substrats du CYP3A4 et ayant une marge thérapeutique étroite (par exemple, immunosuppresseurs, docétaxel, inhibiteurs de kinases, cisapride, pimozide, halofantrine) doit être évitée (voir « Avertissements et précautions »). La prudence est de rigueur lors de l’association avec d'autres substrats du CYP3A4, du CYP2B6 (par exemple éfavirenz, bupropion), CYP2C (par exemple répaglinide, phénytoine, warfarine), glycoprotéines P (par exemple dabigatran, digoxine) et UGT (par exemple morphine, paracétamol, irinotécan) et une surveillance clinique de leur efficacité sera mise en place.
L’association du pitolisant avec les contraceptifs oraux devra être évitée et une autre méthode de contraception fiable sera utilisée.
Substrats de l’OCT1
Le pitolisant a montré une inhibition supérieure à 50 % envers l’OCT1 (transporteurs de cations organiques 1) à 1,33 µM, la concentration inhibitrice CI50 extrapolée du pitolisant est de 0,795 µM.
Même si la pertinence clinique de cet effet n’est pas démontrée, la prudence est recommandée lorsque le pitolisant est administré avec un substrat de l’OCT1 (par exemple, metformine (biguanides)) (voir « Pharmacocinétique »).
L’association du pitolisant avec le modafinil et l’oxybate de sodium, les traitements usuels de la narcolepsie, a été évaluée chez des volontaires sains, aux doses thérapeutiques. Aucune interaction médicamenteuse pharmacocinétique cliniquement significative n'a été mise en évidence soit avec le modafinil ou avec l’oxybate de sodium.
Enfants et adolescents
Les études d'interaction ont été réalisées uniquement chez l'adulte.
Grossesse, Allaitement
Femmes en âge de procréer
Les femmes en âge de procréer doivent utiliser une contraception efficace pendant le traitement et au moins jusqu'à 21 jours après l'arrêt du traitement (sur la base de la demi-vie du pitolisant et de ses métabolites). Le pitolisant et ses métabolites peuvent réduire l'efficacité des contraceptifs hormonaux. Par conséquent, une autre méthode de contraception efficace doit être utilisée si la patiente utilise des contraceptifs hormonaux (voir « Interactions »).
Grossesse
Il n'y a pas ou peu de données sur l'utilisation du pitolisant chez la femme enceinte. Les études chez l'animal ont montré une toxicité sur la reproduction, y compris une tératogénicité. Chez le rat, le pitolisant et/ou ses métabolites traversent la barrière placentaire (voir « Données précliniques »).
Le pitolisant ne doit pas être utilisé pendant la grossesse si cela n’est pas absolument nécessaire.
Allaitement
Une étude animale a montré que le pitolisant et/ou ses métabolites sont excrétés dans le lait. Par conséquent, l'allaitement est contre-indiqué pendant le traitement avec le pitolisant (voir « Contre-indications »).
Fertilité
Les études sur animaux ont montré des effets sur les paramètres du sperme, sans impact significatif sur la reproduction chez les mâles. Chez les femelles traitées, les études ont montré une diminution du pourcentage de fœtus vivants (voir « Données précliniques »).
Effet sur l’aptitude à la conduite et l’utilisation de machines
Le pitolisant a une légère influence sur l’aptitude à la conduite ou l’utilisation de machines.
Les patients présentant une somnolence anormale recevant du pitolisant doivent être avertis que leur niveau de veille peut ne pas redevenir normal. Chez les patients présentant une somnolence excessive, y compris les patients prenant du pitolisant, il convient de réévaluer périodiquement le niveau de somnolence et le cas échéant, de recommander aux patients d'éviter de conduire ou d'effectuer d'autres activités potentiellement dangereuses.
Effets indésirables
Résumé du profil de tolérance
Les effets indésirables les plus fréquents rapportés avec le pitolisant chez les patients adultes sont les suivants : insomnie (8,4 %), céphalées (7,7 %), nausées (4,8 %), anxiété (2,1 %), irritabilité (1,8 %), sensations vertigineuses (1,4 %), dépression (1,3 %), tremblements (1,2 %), troubles du sommeil (1,1 %), fatigue (1,1 %), vomissements (1,0 %), vertiges (1,0 %) dyspepsie (1,0 %), augmentation du poids (0,9 %), douleur abdominale haute (0,9 %). Les effets indésirables les plus graves rapportés avec le pitolisant sont une diminution anormale du poids (0,09 %) et un avortement spontané (0,09 %).
Les effets indésirables suivants ont été reportés lors des essais cliniques conduits avec le pitolisant dans la narcolepsie et autres indications. Ils sont listés selon les termes préférentiels MedDRA par classe de systèmes d’organes. Les fréquences sont définies comme : très fréquent (≥ 1/10), fréquent (≥ 1/100 à < 1/10), peu fréquent (≥ 1/1 000 à < 1/100), rare (≥ 1/10 000 , < 1 000), très rare(<1/10 000). Dans chaque groupe de fréquence, les effets indésirables sont présentés par ordre décroissant de gravité.
Troubles du métabolisme et de la nutrition
Peu fréquent: diminution de l’appétit, augmentation de l’appétit, rétention d’eau, augmentation du poids corporel, diminution du poids corporel.
Rare: anorexie, hyperphagie, troubles de l’appétit.
Affections psychiatriques
Fréquent: insomnie, anxiété, irritabilité, dépression, troubles du sommeil.
Peu fréquent: agitation, hallucinations visuelles / auditives, labilité émotionnelle, rêves anormaux, dyssomnie, insomnie de maintien, insomnie initiale, insomnie terminale, nervosité, tension, apathie, cauchemars, impatience, crise d'angoisse, idées suicidaires, diminution de la libido, augmentation de la libido.
Rare: comportement anormal, état confusionnel, humeur dépressive, excitabilité, pensées obsessionnelles, dysphorie, hallucinations hypnopompiques, symptôme dépressif, hallucinations hypnagogiques, déficience mentale.
Affections du système nerveux
Fréquent: céphalées, vertiges, tremblements.
Peu fréquent: dyskinésie, trouble de l'équilibre, cataplexie, troubles de l'attention, dystonie, phénomène on/off, hypersomnie, migraine, hyperactivité psychomotrice, syndrome des jambes sans repos, somnolence, épilepsie bradykinésie, paresthésie.
Rare: perte de conscience, céphalée de tension, troubles de la mémoire, mauvaise qualité du sommeil.
Affections oculaires
Peu fréquent: diminution de l’acuité visuelle, blépharospasme.
Affections de l’oreille et du labyrinthe
Fréquent: vertiges.
Peu fréquent: acouphènes.
Affections cardiaques
Peu fréquent: extrasystoles, bradycardie, prolongation de l’intervalle QT sur l’électrocardiogramme, augmentation de la fréquence cardiaque.
Rare: ECG : Troubles de la repolarisation sur l’électrocardiogramme, ECG : Inversion de l'onde T sur l’électrocardiogramme.
Affections vasculaires
Peu fréquent: hypertension, hypotension, bouffées de chaleur.
Affections respiratoires, thoraciques et médiastinales
Peu fréquent: bâillements.
Affections gastro-intestinales
Fréquent: nausées, vomissements, dyspepsie
Peu fréquent: bouche sèche, douleur abdominale, diarrhée, gêne abdominale, douleur abdominale haute, constipation, reflux gastro-œsophagien, gastrite, douleurs gastro-intestinales, hyperacidité, paresthésie orale, douleurs stomacales.
Rare: distension abdominale, dysphagie, flatulence, odynophagie, entérocolite.
Affections hépatobiliaires
Peu fréquent: Augmentation des enzymes hépatiques, augmentation des gamma GT.
Affections de la peau et du tissu sous-cutané
Peu fréquent: érythème, prurit, éruption, hyperhydrose, transpiration.
Rare: éruption cutanée toxique, photosensibilité.
Affections musculosquelettiques et du tissu conjonctif
Peu fréquent: arthralgies, dorsalgies, rigidité musculaire, faiblesse musculaire, douleurs musculo-squelettiques, myalgies, douleurs aux extrémités.
Rare: douleur dans le cou, douleur thoracique musculo-squelettique, augmentation de la créatine phosphokinase.
Affections du rein et des voies urinaires
Peu fréquent: pollakiurie.
Affections gravidiques, puerpérales et périnatales
Rare: avortement spontané.
Affections des organes de reproduction et du sein
Peu fréquent: métrorrhagie.
Troubles généraux et anomalies au site d'administration
Fréquent: fatigue.
Peu fréquent: asthénie, douleur dans la poitrine, sensation anormale, malaise, œdème, œdème périphérique.
Rare: douleur, sueurs nocturnes, sensation d’oppression, anomalie de l’état général
Description de certains effets indésirables
Céphalée et insomnie
Au cours des études cliniques, des épisodes de maux de tête et d’insomnie ont été rapportés (7,7 % et 8,4 %). La plupart de ces effets indésirables étaient d’intensité légère à modérée. Si les symptômes persistent, une diminution de la dose quotidienne ou l’arrêt du traitement doivent être envisagés.
Troubles gastriques
Des troubles gastriques, causés par l’hyperacidité, ont été rapportés au cours des études cliniques chez 3,5 % des patients recevant du pitolisant. Ces effets étaient généralement d’intensité légère à modérée. Si ces effets persistent, un traitement correctif avec un inhibiteur de la pompe à protons peut être initié.
Population pédiatrique (6 à 17 ans)
La population pédiatrique a été étudiée dans un essai multicentrique randomisé en double aveugle contre placebo ; un total de 73 enfants et adolescents atteints de narcolepsie avec ou sans cataplexie ont été traités par pitolisant pendant 8 semaines.
La fréquence, le type et la sévérité des effets indésirables chez les enfants et les adolescents étaient similaires à ceux observés chez les adultes. Les effets indésirables liés au médicament les plus fréquemment rapportés dans cette population étaient céphalées (11 %), insomnie (5,5 %), hypertension (2,7 %). Aucune donnée n’est disponible à ce jour concernant les éventuels risques liés à une prise prolongée.
L’annonce d’effets secondaires présumés après l’autorisation est d’une grande importance. Elle permet un suivi continu du rapport bénéfice-risque du médicament. Les professionnels de santé sont tenus de déclarer toute suspicion d’effet secondaire nouveau ou grave via le portail d’annonce en ligne ElViS (Electronic Vigilance System). Vous trouverez des informations à ce sujet sur www.swissmedic.ch.
Surdosage
Symptômes
Les symptômes d’un surdosage en Wakix peuvent comporter des céphalées, une insomnie, une irritabilité, des nausées et des douleurs abdominales.
Traitement
En cas de surdosage, il est recommandé d’hospitaliser le patient et de surveiller ses fonctions vitales. Aucun antidote spécifique n’a été identifié.
Propriétés/Effets
Code ATC
N07XX11
Mécanisme d’action
Pitolisant est un antagoniste des récepteurs de l'histamine H3 ou agoniste inversé du récepteur de l'histamine H3 puissant et actif par voie orale qui améliore l'activité des neurones histaminergiques dans le cerveau, un système d'excitation important avec des projections étendues dans tout le cerveau, en bloquant les autorécepteurs de l'histamine. De plus, Pitolisant module divers systèmes de neurotransmetteurs, augmentant ainsi la libération d'acétylcholine, de norépinéphrine et de dopamine dans le cerveau. Cependant, une libération accrue de dopamine dans le striatum, y compris dans le noyau accumbens, n'a pas été démontrée en relation avec le pitolisant.
Pharmacodynamique
Chez des patients narcoleptiques avec ou sans cataplexie, les mesures objectives de la capacité à maintenir l’état de veille (par exemple Test de Maintien d’Eveil (MWT= Maintenance of Wakefulness Test)) et tâche d’attention soutenue (SART)) ont montré que le pitolisant améliore le niveau et la durée de l’état d’éveil et de la vigilance diurne.
Efficacité clinique
Population adulte
La narcolepsie (avec ou sans cataplexie) est une maladie chronique. L’efficacité du pitolisant jusqu’à 36 mg par jour dans le traitement de la narcolepsie avec ou sans cataplexie a été démontrée dans 2 études principales, multicentriques, randomisées en double aveugle, contrôlées contre placebo et en groupes parallèles, d’une durée de 8 semaines (Harmony I et Harmony CTP). Harmony Ibis, étude avec un schéma similaire, a été conduite à une dose allant jusqu’à 18 mg par jour. Des données de sécurité à long terme du pitolisant dans cette indication sont disponibles dans l'étude à long terme en ouvert HARMONY III.
La première étude pivot (Harmony I) versus placebo et modafinil (400 mg/jour), en groupes parallèles avec adaptation flexible de la dose, a été menée chez 94 patients (31 traités par pitolisant, 30 par placebo et 33 par modafinil). Le traitement a été instauré à 9 mg une fois par jour et a été augmenté, selon l’efficacité et la tolérance, à 18 mg ou 36 mg une fois par jour par intervalles d’une semaine. La plupart des patients (60 %) ont atteint la dose de 36 mg une fois par jour. Pour évaluer l'efficacité du Pitolisant sur la somnolence diurne excessive, le score sur l'échelle de somnolence d'Epworth (Epworth Sleepiness Scale, ESS) a été utilisé comme critère principal d'efficacité. Les résultats ont été significativement supérieurs dans le groupe pitolisant que dans le groupe placebo (différence moyenne : -3,33 ; IC95 % [-5,83 à -0,83] ; p<0,05), mais n’étaient pas significativement différents de ceux observés dans le groupe traité par le modafinil (différence moyenne : 0,12 ; IC95 % [-2,5 à 2,7]). L’effet éveillant a été établi avec des valeurs similaires pour les deux médicaments actifs (Figure 1).
Figure 1 : Modification du score moyen sur l’échelle de somnolence d’Epworth (ESS : Epworth Sleepiness Scale) de l’inclusion dans l’étude à la semaine 8 (jour 56) lors de l’étude Harmony I
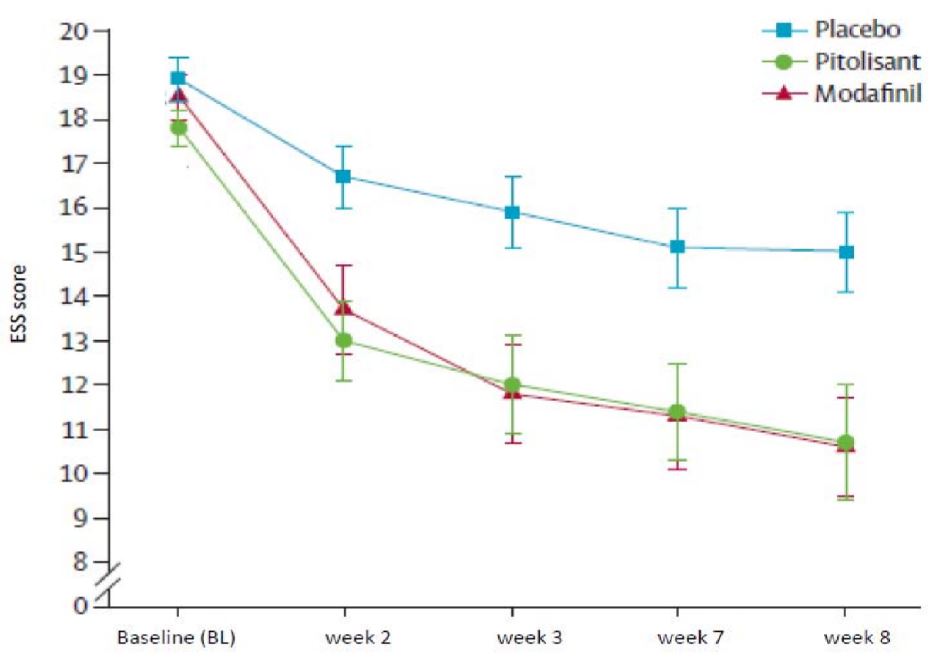
Ces résultats sur l’ESS ont été confirmés par deux tests de laboratoire de maintien de l’éveil et de l’attention (Test de maintien de l’éveil (MWT : Maintenance of Wakefulness Test) (p=0,044)) et Tâche de maintien de l’attention à une réponse (SART : Sustained Attention to Response Task) (p=0,053)).
La fréquence des attaques de cataplexie chez les patients présentant ce symptôme a été diminuée de manière significative (p = 0,034) avec le pitolisant (-65 %) par rapport au placebo (-10 %). Le taux journalier de cataplexies (moyenne géométrique) était de 0,52 au début et de 0,18 à la dernière visite dans le groupe pitolisant et de 0,43 au début et de 0,39 à la dernière visite dans le groupe placebo, avec un risque relatif rR = 0,38 [0,16 ; 0,93] (p = 0,034).
La deuxième étude pivot (Harmony Ibis) a inclus 165 patients (67 traités par pitolisant, 33 par placebo et 65 par modafinil). La conception de l'étude était semblable à celle de l'étude Harmony I ; la seule différence était que la dose maximale de pitolisant atteinte par 75 % des patients était de 18 mg une fois par jour au lieu de 36 mg une fois par jour (comme dans l'étude Harmony I). Étant donné qu'un déséquilibre significatif a conduit à la comparaison des résultats avec ou sans regroupement des sites, l'approche la plus conservatrice pour le pitolisant a montré une réduction insignifiante du score ESS par rapport au placebo (placebo pitolisant = -1,94 ; p=0,065). Les résultats des taux de cataplexie à 18 mg une fois par jour ne sont pas cohérents avec ceux de la première étude pivot (36 mg une fois par jour).
L’amélioration des deux tests objectifs d'éveil et d'attention, MWT et SART, avec le pitolisant était significative par rapport au placebo (p = 0,009 et p = 0,002, respectivement) et non significative par rapport au modafinil (p = 0,713 et p = 0,294, respectivement).
Harmony CTP, la troisième étude clé en double aveugle, randomisée, en groupes parallèles, pitolisant versus placebo, a été conçue pour établir l'efficacité du pitolisant chez les patients narcoleptiques avec une haute fréquence de cataplexies. Le critère principal d'efficacité était le changement du nombre moyen d'attaques de cataplexie par semaine entre les 2 semaines d’inclusion et les 4 semaines de la période de traitement stable à la fin de l'étude. 105 patients narcoleptiques avec une fréquence initiale élevée de cataplexies hebdomadaires ont été inclus (54 patients traités avec pitolisant et 51 avec placebo). Le traitement a été instauré à 4,5 mg une fois par jour et a été augmenté, selon l’efficacité et la tolérance,à 9 mg, 18 mg ou 36 mg une fois par jour par intervalles d’une semaine. La plupart des patients (65 %) ont atteint la dose de 36 mg une fois par jour.
D'après le critère principal d'efficacité, taux hebdomadaire des épisodes de cataplexie, les résultats avec pitolisant étaient significativement supérieurs à ceux du groupe placebo (p <0,0001), avec une diminution progressive de 64 % entre le début et la fin du traitement (Figure 2). À l’inclusion, la moyenne géométrique des taux hebdomadaires des épisodes de cataplexie était de 7,31 (médiane=6,5 [4,5; 12]) et 9,15 (médiane=8,5 [5,5; 15.5]) dans les groupes placebo et pitolisant respectivement. Au cours de la période stable (jusqu'à la fin du traitement), la moyenne géométrique a diminué pour atteindre 6,79 (médiane=6 [3 ; 15]) et 3,28 (médiane=3 [1,3 ; 6]) dans les groupes placebo et pitolisant respectivement, chez les patients ayant eu au moins un épisode de cataplexie. Le taux hebdomadaire des épisodes de cataplexie observé dans le groupe pitolisant était environ la moitié de celui observé dans le groupe placebo : l'effet de taille du pitolisant par rapport au placebo est résumé par le rapport rR (Pt/Pb), rR = 0,512 ; IC95 % [0,435 à 0,603] ; p <0,0001.
Figure 2 : Modification du taux hebdomadaire des épisodes de cataplexie (moyenne géométrique) entre l’inclusion et la semaine 7 lors de l’étude Harmony CTP
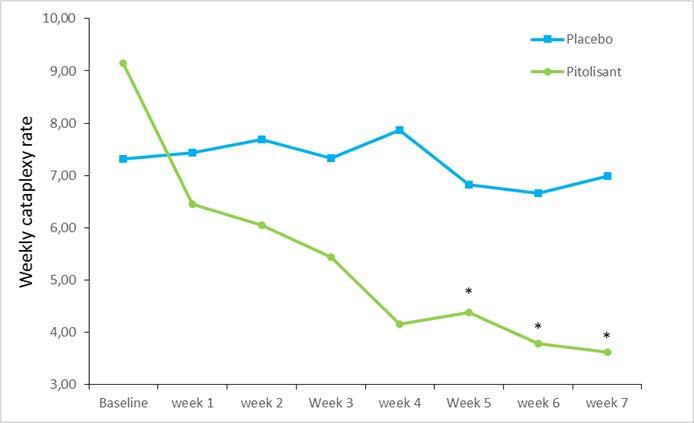
*p<0.0001 vs placebo
L'effet du pitolisant sur la somnolence diurne excessive a également été évalué dans cette population en utilisant le score ESS. Dans le groupe pitolisant, l’ESS a diminué de façon significative entre le début et la fin du traitement par rapport au placebo avec une variation moyenne observée de -1,9 ± 4,3 et -5,4 ± 4,3 (moyenne ± écart-type) pour le placebo et le pitolisant respectivement (p <0,0001). Cet effet sur la somnolence diurne excessive a été confirmé par les résultats du test de maintien de l'éveil (MWT). La moyenne géométrique des ratios (MWTFinal/MWT Baseline) était de 1,8 (IC95 % 1,19 - 2,71, p = 0,005). La valeur du MWT dans le groupe pitolisant était 80 % plus élevée que dans le groupe placebo.
L’étude de phase III à long terme en ouvert (HARMONY III) a évalué la sécurité à long terme du pitolisant chez des patients souffrant de narcolepsie (avec ou sans cataplexie) sur 12 mois et avec une période d’extension jusqu'à 5 ans. 102 patients narcoleptiques avec ou sans cataplexie ont été inclus dans la période de suivi de 12 mois. 68 patients ont terminé la première période de 12 mois. 45, 38, 34 et 14 patients ont terminé les périodes de suivi de 2, 3, 4 et 5 ans, respectivement.
La dose maximale reçue pendant l'étude était de 36 mg / jour chez 85% des patients. Après 12 mois de traitement, les améliorations de la somnolence diurne excessive évaluées par le score ESS des patients restants sont de la même ampleur que celles observées dans les autres essais menés chez les patients narcoleptiques. La diminution du score ESS moyen (DS) était de -3,62 (4,63) après 1 an.
Après 12 mois de traitement avec le pitolisant, la fréquence des symptômes tels que attaques de sommeil, paralysies du sommeil, cataplexies et hallucinations a été améliorée.
Aucun problème majeur de sécurité n'a été identifié. Les résultats de sécurité observés étaient similaires à ceux rapportés dans les essais précédents où le pitolisant était administré à 36 mg une fois par jour pendant 3 mois seulement.
Population pédiatrique
L'efficacité du pitolisant à des posologies allant jusqu'à 36 mg une fois par jour pour le traitement de la narcolepsie avec ou sans cataplexie chez les enfants âgés de 6 à moins de 18 ans a été étudiée dans une étude de 8 semaines, multicentrique, randomisée, en double aveugle, versus placebo, en groupes parallèles. 110 patients ont été randomisés (72 patients dans le groupe pitolisant, 38 dans le groupe placebo). Le traitement a été initié à 4,5 mg une fois par jour et a été augmenté, selon l’efficacité et la tolérance, à 18 mg ou 36 mg une fois par jour par intervalle d'une semaine. Les patients pesant moins de 40 kg sont restés à une posologie maximale de 18 mg. La majorité des patients (60 %) ont atteint la posologie de 36 mg une fois par jour. 35 patients (31,8 %) étaient âgés de 6 à 11 ans et 75 patients (68,2 %) étaient âgés de 12 à moins de 18 ans. Pour évaluer l'efficacité du pitolisant sur la somnolence diurne excessive et la cataplexie , le score total de l'échelle de narcolepsie d'Ullanlinna (UNS) a été utilisé comme critère d'efficacité principal, évalué par le changement entre l’inclusion et la fin de la période en double aveugle. L'estimation de la différence des moyennes des MC (moindres carrés) (ES) [IC à 95 %] de l'UNS entre les groupes de traitement (pitolisant moins placebo) était de -3,69 (1,37) [-6,38 ; -0,99], p=0,0073. Les critères d'évaluation secondaires comprenaient l'échelle pédiatrique de somnolence diurne (PDSS), le sous-score UNS de cataplexie et le taux hebdomadaire de cataplexie (WRC). L'estimation de la différence des moyennes des MC (SE) [IC à 95 %] du score total PDSS entre les groupes de traitement (pitolisant moins placebo) était de -3,41 (1,07) [-5,52 ; -1,31], p=0,0015. Dans le sous-groupe de patients narcoleptiques de type 1, qui n'avaient pas de niveau minimum de cataplexie requis à l'inclusion (N=61 dans le groupe pitolisant ; N=29 dans le groupe placebo), l'estimation de la différence des moyennes des MC (SE) [IC à 95 %] du sous-score de cataplexie entre les groupes de traitement (pitolisant moins placebo) était de -1,77 (0,78) [-3,29 ; -0,24], p=0,0229 et le ratio entre le WRC dans le groupe pitolisant et le WRC dans le groupe placebo, ajusté par rapport à l'inclusion, était en faveur du pitolisant (0,42 [IC à 95 % : 0,18 ; 1,01], p=0,0540).
Tableau 1: Résumé des résultats d’efficacité après 8 semaines dans l’étude pédiatrique de phase 3
|
|
Placebo (n= 38) |
Pitolisant (n= 72) |
|
Echelle Ullanlinna de Narcolepsie (UNS) | ||
|
Score total | ||
|
Moyenne à l’inclusion (DS) |
23.68 (9.08) |
24.63 (7.80) |
|
Score pédiatrique de somnolence diurne (PDSS) | ||
|
Moyenne à l’inclusion (DS) |
20.00 (3.49) |
20.16 (3.64) |
|
|
Placebo (n= 29) |
Pitolisant (n= 61) |
|
UNS-Sous-score de cataplexie* | ||
|
Moyenne à l’inclusion (DS) |
9.03 (4.33) |
8.93 (3.96) |
|
Taux hebdomadaire de cataplexie* (WRC) | ||
|
Moyenne à l’inclusion (DS) |
13.44 (26.92) |
8.63 (17.73) |
*mesuré uniquement chez les patients avec narcolepsie de type IDS : déviation standard ; MC : moindres carrés ; ES : erreur standard ; IC : intervalle de confiance
Figure 3 : Evolution du score total moyen de l'échelle de narcolepsie Ullanlinna (moyenne ± SEM) entre le début et la fin du traitement (analyse sur l’ensemble des données)
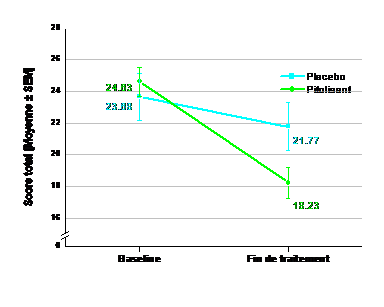
Baseline = [score V1 (J-14) + score V2 (J0)]/2
Fin de traitement = [score V6 (J49) + score V7 (J56)]/2
SEM = erreur standard de la moyenne
Pharmacocinétique
L’exposition au pitolisant a été évaluée chez des volontaires sains lors d’études menées chez plus de 200 sujets qui ont reçu des doses de pitolisant par administration unique allant jusqu’à 216 mg et pendant des durées allant jusqu’à 28 jours.
Absorption
Le pitolisant est rapidement absorbé, et sa concentration plasmatique atteint sa valeur maximale environ trois heures après l’administration.
Distribution
Le pitolisant est fortement lié aux protéines sériques (> 90 %) et montre une distribution pratiquement égale entre les globules rouges et le plasma.
Métabolisme
Le métabolisme du pitolisant chez l’homme est entièrement caractérisé. Les principaux métabolites non conjugués sont des dérivés hydroxylés dans plusieurs positions et des formes clivées de pitolisant conduisant à un métabolite acide carboxylique inactif majeur, présent dans l’urine et le sérum. Ils sont formés sous l’action des cytochromes CYP3A4 et CYP2D6. Plusieurs métabolites conjugués ont été identifiés, les principaux (inactifs) étant deux conjugués de glycine de métabolites acides du pitolisant et un conjugué glucuronide d’un métabolite cétone du pitolisant mono hydroxylé.
Sur des microsomes hépatiques, le pitolisant et ses principaux métabolites n’ont pas significativement inhibé l’activité des cytochromes CYP1A2, CYP2C9, CYP2C19, CYP2C8, CYP2B6, CYP2E1 et CYP3A4 et des isoformes des uridine diphospho-glucuronosyl transférases UGT1A1, UGT1A4, UGT1A6, UGT1A9 et UGT2B7 jusqu’à la concentration de 13,3 µM, qui est considérablement plus élevée que celle atteinte avec une dose thérapeutique. Le pitolisant inhibe le CYP2D6 avec une puissance modérée (CI50 = 2,6 µM).
Le pitolisant induit les cytochromes CYP3A4, CYP1A2 et CYP2B6 in vitro. Des interactions cliniquement pertinentes sont possibles avec les substrats des CYP3A4 et CYP2B6, et par extrapolation les substrats des UGT, CYP2C et glycoprotéine P (voir « Interactions »).
Des études in vitro montrent que le pitolisant n’est ni un substrat ni un inhibiteur de la P-gp (glycoprotéine P) humaine et de la BCRP (protéine de résistance du cancer du sein). Le pitolisant n’est pas un substrat des OATP1B1 et OATP1B3. Le pitolisant n’est pas un inhibiteur important des OAT1, OAT3, OCT2, OATP1B1, OATP1B3, MATE1, ou MATE2K à la concentration testée. Le pitolisant a montré une inhibition supérieure à 50 % envers l’OCT1 (transporteurs de cations organiques 1) à 1,33 µM, la concentration inhibitrice CI50 extrapolée du pitolisant est de 0,795 µM (voir « Interactions »).
Élimination
La demi-vie plasmatique du pitolisant est de 10 à 12 heures. Lors d’administrations répétées, l’état d’équilibre est atteint entre cinq et six jours, avec un doublement de la concentration sérique. La variabilité interindividuelle est relativement élevée, certains volontaires ont montré un profil d’exposition largement supérieur à la moyenne, sans problème de tolérance.
L’élimination est principalement urinaire (environ 63 %) sous la forme d’un métabolite inactif non conjugué (BP2.951) et d’un métabolite conjugué de glycine. 25 % de la dose sont excrétés dans l’air expiré et une petite fraction (< 3 %) est retrouvée dans les fèces, où la quantité de pitolisant ou de BP2.951 est négligeable.
Linéarité/non-linéarité
Lorsque la dose de pitolisant est doublée de 27 à 54 mg, l’ASC0-∞ augmente d’un facteur d’environ 2,3.
Cinétique pour certains groupes de patients
Patients âgés
Les paramètres pharmacocinétiques du pitolisant ne sont pas modifiés entre des patients âgés de 68 à 80 ans et des patients plus jeunes (18 à 45 ans). Une légère variation de la cinétique sans pertinence clinique a été observée au-delà de 80 ans. Les données disponibles chez les patients âgés sont limitées. Par conséquent, la dose doit être ajustée en fonction de la fonction rénale et hépatique du patient (voir « Posologie/Mode d’emploi » et « Mises en garde et précautions »).
Insuffisance rénale
Chez les patients atteints d'insuffisance rénale (stades 2 à 4 selon la classification internationale des maladies rénales chroniques, c'est-à-dire clairance de la créatinine entre 15 ml/min et 89 ml/min), la Cmax et l'ASC avaient tendance à augmenter d'un facteur 2,5, sans effet sur la demi-vie (voir « Posologie/Mode d’emploi »).
Insuffisance hépatique
Les paramètres pharmacocinétiques ne sont pas significativement différents entre des patients présentant une insuffisance hépatique légère (Child-Pugh A) et des volontaires sains. L’ASC a augmenté d’un facteur d’environ 2,4 et la demi-vie a doublé chez des patients présentant une insuffisance hépatique modérée (Child-Pugh B) (voir « Posologie/Mode d’emploi »). La pharmacocinétique du pitolisant après administration répétée chez des patients souffrant d’insuffisance hépatique n’a pas encore été évaluée.
Métaboliseurs lents du CYP2D6
L'exposition au pitolisant était plus élevée chez les métaboliseurs lents du CYP2D6 après une dose unique et à l'état d'équilibre. La Cmax et l’AUC(0-tau) étaient environ 2,7 fois et 3,2 fois plus grand au jour 1 et 2,1 fois et 2,4 fois au jour 7. La demi-vie sérique du pitolisant était plus longue chez les métaboliseurs lents du CYP2D6 par rapport aux métaboliseurs normaux.
Origine ethnique
L’effet de l’origine ethnique sur le métabolisme du pitolisant n’a pas été évalué.
Population pédiatrique
La pharmacocinétique du pitolisant à la dose de 18 mg chez les enfants âgés de 6 à moins de 18 ans atteints de narcolepsie a été étudiée dans un essai multicentrique à dose unique. Par comparaison à l’exposition chez les patients adultes, dans une analyse pharmacocinétique de population avec un modèle dépendant du poids corporel, l'exposition systémique au pitolisant à la dose de 18 mg estimée par la Cmax et l'ASC0-10h est environ 3 fois plus élevée chez les enfants de poids inférieur à 40 kg et 2 fois plus élevée chez les adolescents pesant plus de 40 kg, par rapport aux patients adultes. En conséquence, la titration de dose doit être initiée à la dose la plus faible de 4,5 mg et limitée à 18 mg chez les enfants pesant moins de 40 kg (voir « Posologie/Mode d’emploi »).
Données précliniques
Toxicité en cas d’administration répétée
Au bout d’un mois chez la souris, six mois chez le rat et neuf mois chez le singe, la dose sans effet indésirable observé (NOAEL no observed adverse effect level) a été de respectivement 75, 30 et 12 mg/kg/jour p.o, procurant des marges de sécurité de respectivement 9, 1 et 0,4 comparativement à l’exposition au médicament à la dose thérapeutique chez l’homme. Chez le rat, des épisodes de convulsions transitoires réversibles sont survenus à Tmax et pourraient être imputables à un métabolite abondant chez cette espèce mais non chez l’homme. Chez le singe et aux plus fortes doses, des signes cliniques transitoires liés au système nerveux central, dont des vomissements, des tremblements et des convulsions ont été rapportés. Aux plus fortes doses, aucune modification histopathologique n’a été rapportée chez le singe, et les rats ont présenté des modifications histopathologiques limitées dans certains organes (foie, duodénum, thymus, glandes surrénales et poumons).
Mutagénicité et carcinogénicité
Le pitolisant ne présente aucun effet génotoxique ni cancérogène.
Toxicité sur la reproduction
Un effet tératogène du pitolisant a été observé à des doses toxiques maternelles (marge de sécurité pour la tératogenèse < 1 chez le rat et le lapin). À fortes doses, le pitolisant a entrainé des anomalies morphologiques des spermatozoïdes et une diminution de la motilité, sans effet significatif sur les indices de fertilité chez les rats mâles ; le pitolisant a également entraîné une diminution du pourcentage d’embryons vivants et une augmentation des pertes post-implantatoires chez les rats femelles (marge de sécurité de 1). Le pitolisant a entraîné un retard de développement post-natal (marge de sécurité de 1).
Le pitolisant et/ou ses métabolites traversent la barrière placentaire chez l’animal.
Autres données
Les études de toxicité juvénile chez le rat ont indiqué que l’administration de fortes doses de pitolisant avait induit une mortalité liée à la dose et des épisodes de convulsions qui pourraient être attribuables à un métabolite abondant chez le rat mais non chez l’homme.
Le pitolisant bloque le canal hERG avec une CI50 dépassant les concentrations thérapeutiques et induit un faible allongement du QTc chez le chien.
Des études précliniques réalisées chez la souris, le singe et le rat ont évalué la dépendance et le potentiel addictogène. Cependant, aucune conclusion définitive ne peut être tirée des études de tolérance, dépendance et d'auto-administration.
Remarques particulières
Incompatibilités
Non pertinent.
Stabilité
Le médicament ne doit pas être utilisé au-delà de la date figurant après la mention «EXP» sur l’emballage.
Remarques particulières concernant le stockage
Conserver hors de portée des enfants.
Ne pas conserver au-dessus de 30°C.
Numéro d’autorisation
67007 (Swissmedic)
Présentation
Flacon en polyéthylène haute densité (PEHD) muni d’un bouchon inviolable à fermeture de sécurité enfant en polypropylène muni d’un dessicant (gel de silice).
4,5 comprimés pelliculés : flacon de 30 comprimés [B]
18 comprimés pelliculés : flacon de 30 comprimés [B]
Titulaire de l’autorisation
Future Health Pharma GmbH, 8620 Wetzikon
Mise à jour de l’information
Mai 2024