Composition
Principes actifs
Le brolucizumab est un fragment d'anticorps monoclonal humanisé à chaîne unique Fv(scFv) de poids moléculaire d'environ 26 kDa produit dans des cellules d'Escherichia coli par la technologie de l'ADN recombinant.
Excipients
2,58 mg/ml de citrate de sodium, 58 mg/ml de saccharose, 0,2 mg/ml de polysorbate 80, hydroxyde de sodium (pour ajuster le pH à environ 7,2) et eau pour injection.
Indications/Possibilités d’emploi
Beovu est indiqué dans le traitement de la dégénérescence maculaire liée à l'âge (DMLA) néovasculaire (humide).
Posologie/Mode d’emploi
Flacon à usage unique ou seringue préremplie à usage unique. Pour administration intravitréenne exclusivement. Chaque flacon ou seringue ne peut être utilisé(e) que pour le traitement d'un seul œil.
Beovu doit être administré par un médecin qualifié.
Posologie usuelle
La dose recommandée pour Beovu est de 6 mg (0,05 ml), administrée par injection intravitréenne, les trois premières injections étant effectuées toutes les 4 semaines (mensuellement). Beovu est ensuite administré toutes les 12 semaines (3 mois). L'intervalle de traitement peut être ajusté à toutes les 8 semaines (2 mois) (voir «Propriétés/Effets»). Le médecin peut déterminer les intervalles de traitement sur une base individuelle en fonction de l'activité de la maladie, mesurée par l'acuité visuelle et des paramètres anatomiques.
Afin d'assurer la traçabilité des médicaments biotechnologiques, il convient de documenter pour chaque traitement le nom commercial et le numéro de lot.
Instructions posologiques particulières
Patients présentant des troubles de la fonction hépatique
Aucune étude n'a été menée chez des patients atteints d'insuffisance hépatique.
Patients présentant des troubles de la fonction rénale
Aucun ajustement posologique n'est recommandé chez les patients atteints d'insuffisance rénale. Il n'existe que des données limitées concernant les patients ayant une insuffisance rénale modérée et aucune donnée concernant les patients ayant une forte insuffisance rénale (voir «Propriétés/Effets»).
Patients âgés
Aucun ajustement posologique n'est nécessaire chez les patients âgés de 65 ans et plus.
Enfants et adolescents
La sécurité et l'efficacité de Beovu chez les enfants et les adolescents n'ont pas été établies.
Mode d'administration
Comme tous les médicaments à usage intravitréen, Beovu doit être inspecté visuellement avant son administration (voir «Remarques concernant la manipulation»).
L'injection intravitréenne doit être réalisée dans des conditions aseptiques. Cela comprend la désinfection chirurgicale des mains, des gants chirurgicaux stériles, un champ stérile et un spéculum de paupière stérile (ou un instrument comparable). Des instruments pour la paracentèse stérile doivent être disponibles par mesure de précaution. Avant l'injection intravitréenne, il faut procéder à une anamnèse approfondie des réactions d'hypersensibilité possibles (voir «Contre-indications»). Une anesthésie et une désinfection adéquates de la peau périoculaire, de la paupière et de la surface oculaire au moyen d'un antiseptique topique à large spectre doivent être effectuées avant l'injection.
Pour plus d'informations sur la préparation de Beovu, voir Remarques concernant la manipulation (voir «Remarques particulières»).
L'aiguille d'injection doit être insérée dans le corps vitré de 3,5 à 4,0 mm en arrière du limbe, en évitant le méridien horizontal et en visant le milieu du globe oculaire. Le volume d'injection de 0,05 ml peut alors être injecté lentement. Les injections subséquentes doivent être effectuées à différents endroits de la sclérotique.
La sécurité et l'efficacité du traitement par Beovu dans les deux yeux simultanément n'ont pas été étudiées.
Contre-indications
·Hypersensibilité au principe actif ou à l'un des excipients.
·Infection oculaire ou périoculaire existante ou suspectée.
·Inflammation intraoculaire existante.
Mises en garde et précautions
Réactions associées à l'injection intravitréenne
Les injections intravitréennes, y compris celles de Beovu, sont associées à une endophtalmie, inflammation intraoculaire et décollement de la rétine (voir «Effets indésirables»). Beovu doit toujours être administré dans des conditions d'injection aseptiques. Les patients doivent être informés de la nécessité de signaler immédiatement tout symptôme potentiellement en lien avec l'un ou l'autre des événements énumérés ci-dessus.
Une augmentation transitoire de la pression intraoculaire a été observée dans les 30 premières minutes suivant l'injection, comme c'est le cas à l'administration intravitréenne d'autres inhibiteurs du VEGF (voir «Effets indésirables»). Une pression intraoculaire élevée et soutenue a également été rapportée. La pression intraoculaire ainsi que la perfusion du point de passage du nerf optique doivent être contrôlées et traitées si nécessaire.
Événements thromboemboliques artériels
Il existe un risque potentiel d'événements thromboemboliques artériels dans le cas de l'application intravitréenne des inhibiteurs du VEGF. Le risque peut être augmenté chez les patients présentant un risque connu d'AVC ou d'infarctus du myocarde.
Interactions
Aucune étude officielle n'a été effectuée pour consigner les interactions.
Grossesse/Allaitement
Femmes en âge de procréer
Les femmes en âge de procréer doivent utiliser une méthode contraceptive fiable pendant le traitement par Beovu et pendant au moins un mois après la fin du traitement par Beovu.
Grossesse
Il n'existe pas d'études suffisantes et bien contrôlées sur l'administration de Beovu à des femmes enceintes. Aucune étude sur la reproduction n'a été effectuée chez l'animal. Le risque potentiel lié à l'utilisation de Beovu pendant la grossesse est inconnu. Toutefois, en raison du mécanisme d'action anti-VEGF, le brolucizumab doit être considéré comme potentiellement tératogène et embryotoxique/fœtotoxique. En conséquence, Beovu ne doit pas être administré pendant la grossesse, sauf en cas d'absolue nécessité.
Allaitement
On ne sait pas si le brolucizumab passe dans le lait maternel après l'administration de Beovu. Il n'existe pas de données concernant les effets de Beovu sur les enfants allaités ou sur la production de lait. En raison du risque d'effets indésirables chez les enfants allaités, l'allaitement est déconseillé pendant le traitement et pendant au moins un mois après le traitement.
Fertilité
Il n'existe aucune donnée correspondante.
Effet sur l’aptitude à la conduite et l’utilisation de machines
Après l'injection intravitréenne de Beovu et l'examen oculaire associé, la vision du patient peut être temporairement altérée. Il convient donc d'indiquer aux patients de ne pas reprendre la route et de ne pas utiliser de machines tant que leur fonction visuelle n'est pas suffisamment rétablie.
Effets indésirables
Un total de1'088 patients traités par Beovu ont formé la population de patients dans les deux études de phase III HAWK et HARRIER. L'exposition cumulative à Beovu était de 96 semaines et 730 patients ont reçu la dose recommandée de 6 mg.
Les effets indésirables les plus fréquemment signalés (chez plus de 5% des patients traités par Beovu 6 mg) ont été une diminution de l'acuité visuelle (7,3%), une cataracte (7,0%), une hémorragie conjonctivale (6,3%) et des mouches volantes (5,1%).
Les effets indésirables graves apparus rarement, chez moins de 1% des patients traités par Beovu 6 mg, étaient: endophtalmie, cécité, occlusion de l'artère rétinienne et décollement de la rétine.
Les effets indésirables des médicaments lors d'études cliniques sont énumérés selon la fréquence, les effets indésirables les plus fréquents étant mentionnés en premier. En outre, la catégorie de fréquence respective de chaque effet indésirable est basée sur la convention suivante (CIOMS III): très fréquents (≥1/10), fréquents (≥1/100 à <1/10), occasionnels (≥1/1'000 à <1/100), rares (≥1/10'000 à <1/1'000) et très rares (<1/10'000).
Affections du système immunitaire
Fréquents: Hypersensibilitéa
Affections oculaires
Fréquents: Baisse de l'acuité visuelle, cataracte, hémorragie conjonctivale, mouches volantes, douleur oculaire, hémorragie rétinienne, décollement du vitré, augmentation de la pression intraoculaire, conjonctivite, déchirure du pigment épithélial rétinien, vision trouble, uvéite, érosions cornéennes, kératite ponctuée, iritis, déchirure rétinienne
Occasionnels: Hyperémie conjonctivale, augmentation de la sécrétion lacrymale, cécité, occlusion d'une artère rétinienne, anomalies sensorielles de l'œil, endophtalmie, décollement de la rétine, décollement de l'épithélium pigmentaire de la rétine, inflammation du corps vitré, inflammation de la chambre antérieure, iridocyclite, Phénomène de Tyndall de la chambre antérieure, œdème de la cornée, hémorragie vitrée
a Y compris urticaire, rash, prurit, érythème.
Immunogénicité
Comme pour toutes les protéines thérapeutiques, les patients traités par Beovu sont potentiellement à risque pour le déclenchement d'une réponse immunitaire. L'immunogénicité de Beovu a été étudiée dans des échantillons de sérum. Les données sur l'immunogénicité reflètent le pourcentage de patients dont les résultats aux tests immunologiques ont été jugés positifs pour les anticorps contre Beovu. La détection d'une réponse immunitaire dépend fortement de la sensibilité et de la spécificité des tests utilisés, de la manipulation des échantillons, du moment du prélèvement, des médicaments concomitants et de la maladie sous-jacente. Pour ces raisons, la comparaison de l'incidence des anticorps dirigés contre Beovu avec l'incidence des anticorps dirigés contre d'autres produits peut être trompeuse. Chez les patients naïfs de tout traitement, des anticorps, y compris des anticorps à chaîne unique, dirigés contre un grand nombre de protéines thérapeutiques produites par biotechnologie ont été détectés avant même le début du traitement.
L'incidence des anticorps dirigés contre le brolucizumab avant traitement était de 35 à 52%. Après l'administration de Beovu, des anticorps liés au traitement dirigés contre le brolucizumab ont été détectés chez 23 à 25% des patients pendant 88 semaines.
L'apparition d'anticorps dirigés contre le brolucizumab n'a été associée à aucune influence sur l'efficacité clinique. Un nombre plus élevé d'événements inflammatoires intraoculaires a été observé chez les patients présentant des anticorps liés au traitement. La signification clinique des anticorps dirigés contre le brolucizumab en termes de sécurité reste encore à définir à l'heure actuelle.
L'annonce d'effets secondaires présumés après l'autorisation est d'une grande importance. Elle permet un suivi continu du rapport bénéfice-risque du médicament. Les professionnels de santé sont tenus de déclarer toute suspicion d'effet secondaire nouveau ou grave via le portail d'annonce en ligne ElViS (Electronic Vigilance System). Vous trouverez des informations à ce sujet sur www.swissmedic.ch.
Surdosage
Une surdose supérieure au volume d'injection recommandé peut entraîner une augmentation de la pression intraoculaire. Par conséquent, en cas de surdosage, la pression intraoculaire doit être surveillée et, selon l'évaluation du médecin traitant, traitée si nécessaire.
Propriétés/Effets
Code ATC
S01LA06
Mécanisme d'action
Une transmission accrue du signal par la voie VEGF-A (facteur de croissance vasculaire endothélial A) est associée à une angiogenèse oculaire pathologique et à un œdème rétinien. Le brolucizumab se lie avec une grande affinité aux isoformes VEGF-A (p.ex. VEGF110, VEGF121 et VEGF165) et empêche ainsi le VEGF-A de se fixer à ses récepteurs VEGFR-1 et VEGFR-2. En inhibant la liaison au VEGF-A, le brolucizumab supprime la prolifération des cellules endothéliales, réduisant ainsi la néovascularisation pathologique et la perméabilité vasculaire.
Pharmacodynamie
La dégénérescence maculaire liée à l'âge (DMLA) néovasculaire (humide) est caractérisée par une néovascularisation choroïdienne (NVC) pathologique. La perte de sang et de liquide due à la NVC peut entraîner un épaississement ou un œdème de la rétine ou un saignement sous/intrarétinien, entraînant une perte d'acuité visuelle.
Dans les études HAWK et HARRIER, les paramètres anatomiques connexes faisaient partie de l'évaluation de l'activité de la maladie, qui a servi de base aux décisions thérapeutiques. Une réduction de l'épaisseur centrale de la rétine (ECR) et la présence de liquide intrarétinien/sous-rétinien (LIR/LSR) ou de liquide épithélial pigmentaire sous-rétinien (sous-EPR) ont été observées chez des patients traités par Beovu dès les 4 semaines suivant le début du traitement et jusqu'aux semaines 48 et 96.
Dans ces études, une réduction de la taille des lésions de NVC a été observée chez les patients traités avec Beovu dès 12 semaines après le début du traitement et aux semaines 48 et 96 après le début du traitement.
Efficacité clinique
L'innocuité et l'efficacité de Beovu ont été évaluées dans deux études de phase III (HAWK et HARRIER) randomisées, multicentriques, en double aveugle avec contrôle actif, menées auprès de patients atteints de DMLA néovasculaire. Au total, 1'817 patients ont été traités dans ces essais pendant deux ans (1'088 par Beovu et 729 par aflibercept). Les patients étaient âgés de 50 à 97 ans, avec une valeur moyenne de 76 ans.
Dans l'étude HAWK, les patients ont été randomisés selon un rapport 1:1:1 et affectés à l'un des schémas posologiques suivants:
1.Beovu 3 mg administré toutes les 12 ou 8 semaines («q12w/q8w») après les 3 premières doses mensuelles.
2.Beovu 6 mg, administré toutes les 12 ou 8 semaines («q12w/q8w») après les 3 premières doses mensuelles.
3.Aflibercept 2 mg, administré toutes les 8 semaines («q8w») après les 3 premières doses mensuelles.
Dans l'étude HARRIER, les patients ont été randomisés selon un rapport 1:1 et affectés à l'un des schémas posologiques suivants:
1.Beovu 6 mg, administré toutes les 12 ou 8 semaines («q12w/q8w») après les 3 premières doses mensuelles.
2.Aflibercept 2 mg, administré toutes les 8 semaines («q8w») après les 3 premières doses mensuelles.
Dans les deux études, les patients traités par le brolucizumab ont été traités toutes les 12 semaines après les 3 premières doses mensuelles (semaines 0, 4 et 8) avec la possibilité de passer à un intervalle de traitement de 8 semaines en fonction de l'activité de la maladie. L'activité de la maladie a été évaluée par un médecin au cours des 12 premières semaines (semaines 16 et 20) et à chacune des 12 semaines de traitement prévues. Les patients qui présentaient une activité pathologique au cours de l'une de ces visites (p.ex. diminution de l'acuité visuelle, augmentation de l'épaisseur centrale de la rétine (ECR) ou présence de liquides rétiniens (LIR/LSR, sous-EPR) ont reçu un traitement toutes les huit semaines.
Résultats
Le critère d'efficacité primaire des essais était la variation de la meilleure acuité visuelle corrigée (MAVC) par rapport à l'acuité visuelle initiale à la semaine 48, telle que mesurée par les tableaux des lettres ETDRS, dans le but principal de démontrer la non-infériorité de Beovu par rapport à l'aflibercept. Les deux études ont démontré que Beovu (administré selon un schéma thérapeutique de 12/8 semaines) n'était pas inférieur à l'aflibercept à la dose de 2 mg (administré toutes les 8 semaines) en termes d'efficacité.
Dans l'étude HAWK, les patients des groupes Beovu 6 mg et aflibercept ont atteint, à la semaine 48, une variation moyenne de respectivement +6,6 lettres et +6,8 lettres (p <0,0001), par rapport à la situation initiale. La variation moyenne par rapport à la situation initiale dans le groupe recevant 3 mg de Beovu était de +6,1 lettres (p = 0,0003). La proportion de patients ayant gagné au moins 15 lettres d'acuité visuelle par rapport à la situation initiale était de 33,6% dans le groupe brolucizumab contre 25,4% dans le groupe aflibercept. La proportion de patients ayant perdu 15 lettres ou plus d'acuité visuelle par rapport à la situation initiale était de 6,4% dans le groupe recevant 6 mg de brolucizumab contre 5,5% dans le groupe recevant l'aflibercept.
Dans l'étude HARRIER, les patients des groupes Beovu et aflibercept ont atteint, à la semaine 48, une variation moyenne de respectivement +6,9 lettres et +7,6 lettres (p <0,0001) par rapport à la situation initiale. La proportion de patients ayant gagné au moins 15 lettres d'acuité visuelle par rapport à la situation initiale était de 29,3% dans le groupe brolucizumab contre 29,9% dans le groupe aflibercept. La proportion de patients ayant perdu 15 lettres ou plus d'acuité visuelle par rapport à la situation initiale était de 3,8% dans le groupe recevant 6 mg de brolucizumab contre 4,8% dans le groupe recevant l'aflibercept.
Le gain d'acuité visuelle observée la première année a été maintenu la deuxième année.
Figure 0-1: Variation moyenne de l'acuité visuelle par rapport à la situation initiale jusqu'à la semaine 96 dans les études HAWK et HARRIER
Étude HAWK
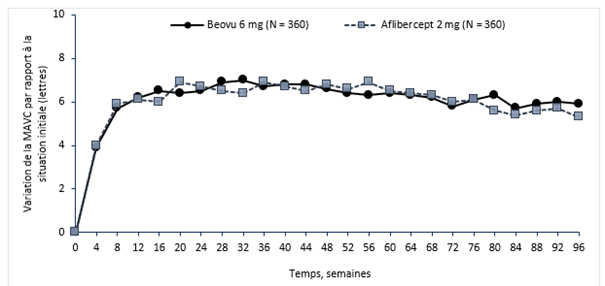
Étude HARRIER
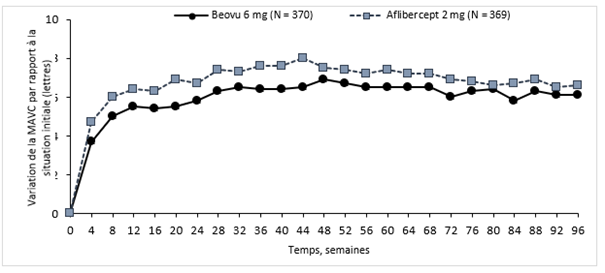
Dans les études HAWK et HARRIER, respectivement 56% et 51% des patients traités par Beovu 6 mg à 12 semaines d'intervalle ont atteint ce gain d'acuité visuelle à la semaine 48 (variation moyenne par rapport à la situation initiale), et respectivement 45% et 39% des patients à la semaine 96.
Parmi les patients considérés comme adaptés à cet intervalle de traitement au cours du premier intervalle de 12 semaines de traitement, respectivement 85% et 82% ont maintenu l'intervalle de 12 semaines jusqu'à la semaine 48. Chez respectivement 82% et 75% des patients traités à la semaine 48 avec l'intervalle de traitement de 12 semaines, l'intervalle de traitement de 12 semaines a été maintenu de la semaine 48 à la semaine 96.
Les effets du traitement dans les sous-groupes évaluables (p.ex. âge, sexe, origine ethnique, acuité visuelle en situation initiale, épaisseur de la rétine en situation initiale, type de lésion, taille de la lésion, état hydrique) dans les deux études concordaient en grande partie avec les résultats dans la population totale.
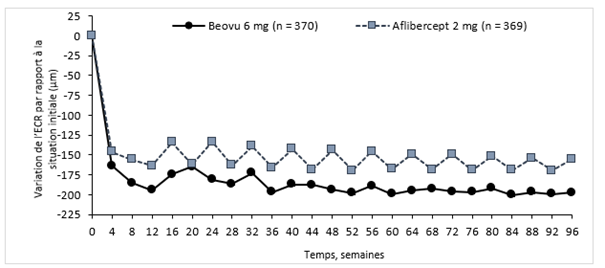
L'activité de la maladie a été évaluée sur la base des variations de l'acuité visuelle ou de critères morphologiques, notamment l'épaisseur centrale de la rétine (ECR) et la présence de liquides rétiniens (LIR/LSR, sous-EPR). À la semaine 16, lorsque l'activité de la maladie a été évaluée pour la première fois pour déterminer l'intervalle de traitement, le nombre de patients ayant montré une activité de la maladie était statistiquement moins élevé dans le groupe sous Beovu à la dose de 6 mg que dans le groupe sous aflibercept à la dose de 2 mg (24% contre 35% dans HAWK, p = 0,0013; 23% contre 32% dans HARRIER, p = 0,0021). L'activité de la maladie a été évaluée tout au long des études. Les critères morphologiques de l'activité de la maladie étaient plus faibles au cours des semaines 48 et 96 dans le groupe Beovu comparativement au groupe aflibercept (Tableau 0-2).
Tableau 0-1: Évaluation de l'activité de la maladie dans les études HAWK et HARRIER jusqu'à la semaine 96
|
|
|
Étude HAWK |
|
Étude HARRIER |
| ||
|
Résultats d'efficacité (critères d'évaluation secondaires préétablis) |
À la semaine |
Beovu 6 mg |
Aflibercept |
Différence (IC à 95%) brolucizumab et aflibercept |
Beovu 6 mg |
Aflibercept |
Différence (IC à 95%) brolucizumab et aflibercept |
|
Variation moyenne de l'ECR par rapport à la situation initiale (µm) |
16 c) |
-161,4 |
-133,6 |
-27,8 |
-174,4 |
-134,2 |
-40,2 |
|
48 |
-172.8 |
-143,7 |
-29,0 |
-193,8 |
-143,9 |
-49,9 | |
|
96 |
-174.8 |
-148,7 |
-26,0 |
-197,7 |
-155,1 |
-42,6 | |
ECR: épaisseur centrale de la rétine, LIR/LSR: liquide intrarétinien/liquide sous-rétinien, EPR: épithélium pigmentaire rétinien
a) Critère d'évaluation secondaire dans l'étude HARRIER, analyse de confirmation dans l'étude HAWK. Valeurs p unilatérales pour la supériorité du brolucizumab
b) Critères d'évaluation secondaire dans les études HAWK et HARRIER; valeurs de p bilatéral
c) Jusqu'à la semaine 16, l'exposition au traitement était identique, permettant une comparaison coordonnée de Beovu avec l'aflibercept.
Figure 0-2: Variation de l'épaisseur centrale de la rétine, en situation initiale jusqu'à la semaine 96 dans les études HAWK et HARRIER
Étude HAWK
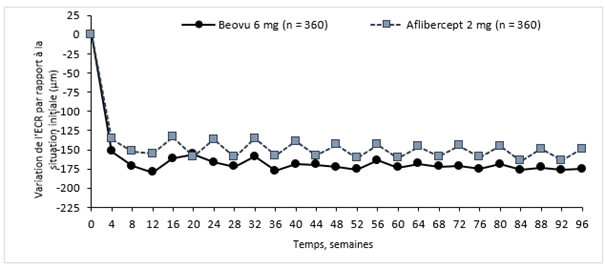
Étude HARRIER
Dans les deux études, le traitement par Beovu a entraîné des modifications cliniquement significatives par rapport à la situation initiale en ce qui concerne le critère d'évaluation secondaire de l'efficacité préspécifié des «résultats rapportés par les patients» enregistrés au moyen du questionnaire NEI VFQ-25 sur la santé oculaire de l'Institut national des yeux des États-Unis. L'ampleur de ces modifications était comparable à celle des études publiées et correspondait à une augmentation de 15 lettres dans la meilleure acuité visuelle corrigée (MACV). Le bénéfice des résultats rapportés par les patients a été maintenu au cours de la deuxième année.
Aucune différence cliniquement significative n'a été observée entre Beovu et l'aflibercept en ce qui concerne les variations du score total NEI VFQ-25 et des sous-échelles entre la situation initiale et la semaine 48 (vision générale, douleur oculaire, vision de près, vision de loin, activité sociale, bien-être mental, difficultés à exercer des rôles sociaux, dépendance envers autrui, conduite automobile, vision des couleurs et vision périphérique).
Pharmacocinétique
Absorption
Beovu est administré directement dans le corps vitré pour obtenir des effets locaux dans l'œil.
Distribution
Après administration intravitréenne de 6 mg de brolucizumab par œil à des patients atteints de DMLAn, la Cmax moyenne du brolucizumab libre dans le sérum était de 49,0 ng/ml (intervalle: 8,97 à 548 ng/ml) et a été atteinte en un jour. L'ASC moyenne était de 6000 h*ng/ml (intervalle: 1420 à 60 400 h*ng/ml)
Métabolisme
Le brolucizumab est un fragment d'anticorps monoclonal et aucune étude n'a été menée sur le métabolisme du principe actif. Le brolucizumab étant un fragment d'anticorps à chaîne unique, le brolucizumab libre devrait être excrété à la fois par élimination ciblée par liaison au VEGF endogène libre et par élimination rénale passive et métabolisation par protéolyse.
Élimination
Après injection intravitréenne, le brolucizumab était éliminé avec une demi-vie systémique apparente de 4,4 jours. Il n'y avait pas d'accumulation de Beovu dans le sérum lorsqu'il était administré par voie intravitréenne toutes les 4 semaines.
Cinétique pour certains groupes de patients
Troubles de la fonction hépatique
La pharmacocinétique chez les patients atteints d'insuffisance hépatique n'a pas été étudiée.
Troubles de la fonction rénale
La pharmacocinétique systémique du brolucizumab a été évaluée chez des patients atteints de DMAn dont les données pharmacocinétiques sur le brolucizumab sérique et la clairance de la créatinine du brolucizumab étaient disponibles. Le rapport de la moyenne géométrique (IC à 90%) chez les patients présentant une insuffisance rénale légère (60 à <90 ml/min (n = 22)) et modérée (30 à <60 ml/min (n = 3)) par rapport aux patients avec une fonction rénale normale est Cmax 1,4 (0,7; 2,9) et. 1,7 (1,0; 2,8) pour le brolucizumab et le rapport pour l'ASCinf 1,4 (0,7; 2,9) et 1.0 (0.5, 2.0). Aucun patient présentant une insuffisance rénale sévère (<30 ml/min) n'a été examiné.
Patients âgés
Les données limitées concernant la pharmacocinétique du brolucizumab chez les patients âgés ne permettent pas de tirer des conclusions quant à un effet de l'âge sur la pharmacocinétique du brolucizumab.
Polymorphisme génétique
Groupes ethniques
Dans une étude portant sur 24 patients caucasiens et 26 patients japonais, aucune différence ethnique en ce qui concerne les propriétés pharmacocinétiques systémiques n'a été observée suite à l'injection intravitréenne.
Données précliniques
Toxicité à long terme (ou toxicité en cas d'administration répétée)
L'injection intravitréenne de brolucizumab à des doses allant jusqu'à 6 mg/œil toutes les 4 semaines pendant 26 semaines n'a produit aucun effet oculaire ou systémique chez le singe cynomolgus et a été bien tolérée.
Mutagénicité/carcinogénicité
Aucune étude n'a été réalisée pour clarifier le potentiel mutagène ou carcinogène de Beovu.
Toxicité sur la reproduction
Aucune étude sur la reproduction ou la fertilité n'a été effectuée. Il a été démontré que l'inhibition du VEGF influence la maturation folliculaire, la fonction du corps jaune et la fertilité. Sur la base du mécanisme d'action des inhibiteurs du VEGF, il existe un risque potentiel pour la reproduction féminine et le développement embryofœtal.
Remarques particulières
Incompatibilités
Aucune étude de tolérance n'ayant été effectuée, ce médicament ne doit pas être mélangé à d'autres médicaments.
Conservation
Le médicament ne doit pas être utilisé au-delà de la date figurant après la mention «EXP» sur le flacon ou la seringue.
Remarques particulières concernant le stockage
Tenir hors de portée des enfants.
Flacons: Conserver dans l'emballage d'origine, à l'abri de la lumière. Conserver au réfrigérateur (2 à 8 °C). Ne pas congeler.
Le flacon non ouvert peut être conservé à température ambiante (25 °C) jusqu'à 24 heures avant utilisation.
Seringue préremplie: Conserver dans la plaquette thermoformée scellée dans l'emballage d'origine, à l'abri de la lumière. Conserver au réfrigérateur (2 à 8 °C). Ne pas congeler.
La plaquette thermoformée non ouverte peut être conservée à température ambiante (25 °C) jusqu'à 24 heures avant utilisation.
Pour des informations complémentaires, veuillez consulter «Remarques concernant la manipulation».
Numéro d’autorisation
67245, 67244 (Swissmedic)
Présentation
1 flacon de 0,23 ml comprenant 1 aiguille filtre. [B]
1 seringue préremplie de 0,165 ml. [B]
Titulaire de l’autorisation
Novartis Pharma Schweiz AG, Risch; domiciliée 6343 Rotkreuz
Mise à jour de l’information
Janvier 2020
Remarques concernant la manipulation
Pour préparer Beovu à l'usage intravitréen, veuillez procéder comme suit:
|
1 |
|
Retirer le papier aluminium de la plaquette thermoformée et extraire la seringue dans des conditions aseptiques. | |
|
2 |
|
|
Rompre le capuchon de la seringue (ne pas tourner ni dévisser). |
|
3 |
|
Fixer une aiguille d'injection 30 G x ½″ dans des conditions aseptiques sur la seringue. | |
|
4 |
|
|
Pour vérifier l'absence de bulles d'air, tenir la seringue avec l'aiguille dirigée vers le haut. Si des bulles d'air sont présentes, tapoter avec précaution la seringue du doigt jusqu'à ce que les bulles d'air remontent vers le haut. |
|
5 |
|
|
Tenir la seringue à hauteur des yeux et appuyer doucement sur le piston jusqu'à ce que le bord situé sous le dôme du bouchon en caoutchouc se trouve aligné avec la marque de dosage 0,05 ml. La seringue est maintenant prête pour l'injection. |
|
6 |
|
Injecter la solution lentement jusqu'à ce que le bouchon en caoutchouc atteigne l'extrémité du cylindre de la seringue de sorte que le volume total de 0,05 ml soit administré. Vérifier que toute la dose a été injectée en contrôlant que le bouchon en caoutchouc a atteint l'extrémité du cylindre de la seringue. |