ZusammensetzungWirkstoffe
Tenecteplase (mittels rekombinanter DNA-Technologie in Ovarialzellen des chinesischen Hamsters (CHO) hergestellt).
Hilfsstoffe
·Pulver: L-Arginin, Phosphorsäure, Polysorbat 20
·Lösungsmittel: Wasser für Injektionszwecke
·Spurenrückstände: Gentamicin aus dem Herstellungsprozess
Indikationen/AnwendungsmöglichkeitenBelegte Indikationen
Thrombolytische Therapie bei akutem Myokardinfarkt.
Dosierung/AnwendungMetalyse sollte so früh wie möglich nach Symptombeginn verabreicht werden.
Metalyse wird körpergewichtsbezogen verabreicht, mit einer maximalen Dosis von 10'000 U. Das Injektionsvolumen zur Verabreichung der richtigen Dosis kann mittels des folgenden Schemas ermittelt werden:
|
Körpergewicht des Patienten (kg)
|
Tenecteplase
(U)
|
Tenecteplase
(mg)
|
Volumen der gebrauchsfertigen Lösung (ml)
| |
< 60
|
6'000
|
30
|
6
| |
≥ 60 bis < 70
|
7'000
|
35
|
7
| |
≥ 70 bis < 80
|
8'000
|
40
|
8
| |
≥ 80 bis < 90
|
9'000
|
45
|
9
| |
≥ 90
|
10'000
|
50
|
10
|
Herstellung der Injektionslösung: siehe «Korrekte Art der Anwendung».
Begleittherapie
Es wird empfohlen, die antithrombotische Begleittherapie entsprechend den derzeitigen internationalen Richtlinien zur Behandlung von Myokardinfarkt mit ST-Hebung durchzuführen.
Für eine primäre perkutane koronare Intervention (PCI) siehe «Warnhinweise und Vorsichtsmassnahmen».
Zur Kombination von Metalyse mit Enoxaparin bzw. mit Abciximab siehe ASSENT-3-Studie in Kapitel «Eigenschaften/Wirkungen».
Spezielle Dosierungsanweisungen
Kinder und Jugendliche
Die Anwendung und Sicherheit von Metalyse, Injektionspräparat bei Kindern und Jugendlichen unter 18 Jahren ist bisher nicht geprüft worden.
Korrekte Art der Anwendung
Die rekonstituierte Lösung sollte intravenös verabreicht werden und ist zum sofortigen Gebrauch bestimmt.
Die erforderliche Dosis ist als intravenöser Einfach-Bolus innerhalb von 5-10 Sekunden zu verabreichen.
Zubereitung der Injektionslösung:
Die Herstellung der Metalyse-Lösung erfolgt durch Zugabe der gesamten Menge Wasser für Injektionszwecke aus der Fertigspritze in die Durchstechflasche mit dem Pulver.
1.Prüfen Sie die Unversehrtheit der Verschlusskappe der Durchstechflasche und entfernen Sie die Verschlusskappe (Abbildung 1).
2.Entfernen Sie ebenfalls die Verschlusskappe der Fertigspritze (Abbildung 1).
3.Schrauben Sie unverzüglich die Fertigspritze auf den Adapter (Abbildung 2) und durchstechen Sie den Verschluss der Durchstechflasche mit Hilfe der Adapterspitze (Abbildung 3).
4.Injizieren Sie das Wasser in die Durchstechflasche durch langsames Niederdrücken des Spritzenstempels, um Schäumen zu vermeiden (Abbildung 4).
5.Lassen Sie die Fertigspritze am Adapter befestigt und lösen Sie das Pulver durch leichtes Schwenken (Abbildung 5).
6.Die rekonstituierte Lösung ist farblos bis schwach gelblich und klar. Nur klare, partikelfreie Lösungen verwenden.
7.Drehen Sie die Durchstechflasche samt Spritze unmittelbar vor Gabe der Lösung um, so dass sich nun die Spritze unten befindet (Abbildung 6).
8.Ziehen Sie das erforderliche Volumen Metalyse-Lösung in die Spritze auf, entsprechend dem Körpergewicht des Patienten (Abbildung 6) (siehe auch die entsprechende Tabelle unter «Dosierung/Anwendung»).
9.Schrauben Sie die Spritze vom Adapter ab (Abbildung 7).
10.Ein bereits vorhandener intravenöser Zugang, der ausschliesslich der Gabe von 0,9%iger Natriumchloridlösung diente, kann für die Verabreichung von Metalyse verwendet werden. Metalyse darf nicht mit anderen Arzneimitteln gemischt werden, weder in derselben Durchstechflasche noch über denselben intravenösen Zugang (auch nicht mit Heparin). Die Lösung nicht über eine Infusionsleitung geben, die Glukose enthält, da Metalyse mit Glukoselösung inkompatibel ist.
11.Metalyse wird intravenös innerhalb von 5-10 Sekunden verabreicht.
12.Der Zugang sollte nach der Metalyse-Injektion mit 0.9%iger Natriumchloridlösung durchgespült werden, um eine einwandfreie Abgabe zu gewährleisten.
13.Nicht verbrauchte Lösung sollte verworfen werden.
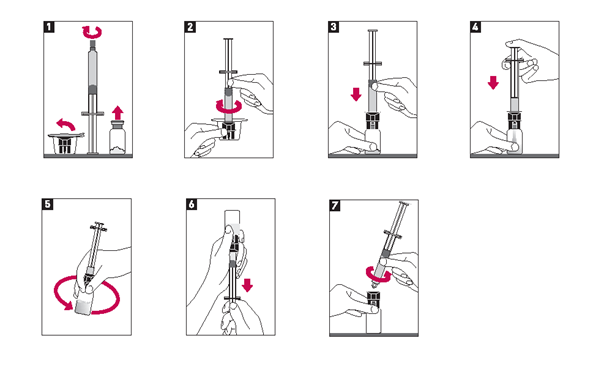
Alternativ kann die Zubereitung der Injektionslösung mit einer Injektionsnadel anstelle des mitgelieferten Adapters erfolgen.
Rückverfolgbarkeit
Um die Rückverfolgbarkeit von biotechnologisch hergestellten Arzneimitteln sicherzustellen, wird empfohlen Handelsname und Chargennummer bei jeder Behandlung zu dokumentieren.
KontraindikationenMetalyse ist kontraindiziert:
·bei Patienten mit bekannter Überempfindlichkeit gegenüber dem Wirkstoff Tenecteplase, Gentamicin (Spurenrückstände aus dem Herstellungsprozess) oder einem der Bestandteile
·in Situationen, welche mit einem Blutungsrisiko verbunden sind, wie:
·Schwerwiegender Blutung (akut oder innerhalb der vergangenen 6 Monate)
·Patienten, die eine wirksame orale Antikoagulanzientherapie erhalten (z.B. Vitamin K-Antagonisten) (INR >1,3) (siehe «Warnhinweise und Vorsichtsmassnahmen»).
·Jede Erkrankung des zentralen Nervensystems (z. B. Neoplasma, Aneurysma, intrakranielle oder intraspinale Operation) in der Anamnese
·Bekannte hämorrhagische Diathese
·Schwere, nicht kontrollierte Hypertonie (siehe «Warnhinweise und Vorsichtsmassnahmen»)
·Grosse Operation, Biopsie eines parenchymatösen Organs oder schweres Trauma in den letzten zwei Monaten (einschliesslich jeglicher mit dem akuten Herzinfarkt zusammenhängender Traumen)
·Kürzlich erlittene Kopf- oder Schädelverletzungen
·Schwere Leberfunktionsstörung einschliesslich Leberversagen, Zirrhose, Pfortaderhochdruck (Ösophagusvarizen) und aktiver Hepatitis
·Aktive ulzerative Magendarmerkrankung
·Bekannte Arterielle Aneurysmen und/oder arteriovenöse Missbildungen
·Neoplasma mit erhöhtem Blutungsrisiko
·Bakterielle Endokarditis, Perikarditis
·Akute Pankreatitis
·Entbindung
·Kürzlich zurückliegende Punktion eines nicht abdrückbaren Blutgefässes z.B. der V. subclavia oder jugularis
·Hämorrhagischer Hirnschlag oder Hirnschlag unbekannten Ursprungs zu irgendeinem vorausgegangenen Zeitpunkt
·Ischämischer Hirnschlag oder transitorische ischämische Attacke (TIA) in den letzten 6 Monaten. Zur thrombolytischen Behandlung bei akutem ischämischem Hirnschlag innerhalb von 3 Stunden sind andere Präparate zugelassen.
Warnhinweise und VorsichtsmassnahmenAllgemein
Bei Patienten mit starkem ST-Hebungsinfarkt sollten die Ärzte entweder eine Thrombolyse oder eine PCI als primäre Behandlungsstrategie für die Reperfusion wählen. Rescue-PCI oder anschliessende elektive PCI können nach Verabreichung der Thrombolyse-Therapien durchgeführt werden, sofern dies medizinisch angebracht ist; die optimale Anwendung begleitender antithrombotischer Medikamente und Thrombozytenaggregationshemmer in dieser Situation ist jedoch nicht bekannt.
Bei der Entscheidung, einen Patienten mit akutem Herzinfarkt mit Metalyse zu behandeln, sollte ein mit der Thrombolyse erfahrener Arzt beratend zugezogen werden. Es wird empfohlen, bei der Gabe von Metalyse, wie bei anderen Thrombolytika auch, Standard-Wiederbelebungs-Geräte und -Medikamente in allen Fällen bereitzustellen.
Nach Reanimation sind vor der Applikation die Grösse des Traumas und die Gefahr von Rippenfrakturen zu berücksichtigen.
Blutungen
Die am häufigsten beobachtete Nebenwirkung unter Metalyse sind Blutungen. Die Begleittherapie mit Heparin kann hierbei mitverantwortlich sein. Da die Therapie mit Metalyse zu einer Auflösung von Fibrin führt, kann es zu Blutungen aus frischen Punktionsstellen kommen. Während der thrombolytischen Therapie müssen deshalb mögliche Blutungsquellen sorgfältig beobachtet werden (dies schliesst Zugänge für Katheter, arterielle und venöse Punktionsstellen, Abbindungsstellen und Einstichstellen ein). Die Anwendung starrer Katheter, intramuskuläre Injektionen und nicht unbedingt erforderliche Massnahmen am Patienten sollten während der Therapie mit Metalyse unterbleiben.
Am häufigsten werden Blutungen an der Injektionsstelle, gelegentlich Blutungen im Urogenitaltrakt und Zahnfleischbluten beobachtet.
Bei schweren Blutungen, besonders bei zerebralen Blutungen, sollte eine Begleittherapie mit Heparin sofort beendet werden. Sofern Heparin innerhalb von 4 Stunden vor Beginn der Blutung gegeben wurde, sollte die Gabe von Protamin erwogen werden.
Bei den wenigen Patienten, bei denen diese konservativen Massnahmen nicht helfen, ist der vernünftige Gebrauch von Tranfusionsprodukten angezeigt. Die Transfusion von Kryopräzipitat, frisch gefrorenem Plasma, und Plättchen mit einer jeweiligen Neubeurteilung der Klinik und der Laborwerte nach jeder Administration sollte erwogen werden. Der Zielwert von 1 g/l Fibrinogen ist bei der Kryopräzipitat-Infusion wünschenswert. Als letzte Alternative werden antifibrinolytische Substanzen empfohlen.
In den folgenden Fällen ist die Anwendung von Metalyse mit einem erhöhten Risiko verbunden und sollte hinsichtlich dessen Nutzens abgewogen werden:
·Diabetische hämorrhagische Retinopathie oder andere Blutungsereignisse im Auge
·Systolischer Blutdruck >160 mm Hg
·Kurz zurückliegende gastrointestinale oder urogenitale Blutung (in den vergangenen 10 Tagen)
·Verdacht auf Hämostasestörungen, auch sekundär wegen schwerer Lebererkrankung (siehe auch unter «Kontraindikationen»)
·kürzlich erfolgte intramuskuläre Injektion oder kurz zurückliegende kleine Traumata, wie z.B. Biopsien, Punktionen grosser Gefässe
·Fortgeschrittenes Alter (d.h. 75 Jahre oder älter)
In einer explorativen Studie wurde unter der Standarddosierung mit Metalyse gefolgt von einer PCI bei Bedarf, bei Patienten mit einem Alter >75 Jahren eine erhöhte Inzidenz von intrakraniellen Blutungen festgestellt (siehe unter «Eigenschaften/Wirkungen»).
·Körpergewicht <50 kg
·Zur Kombination mit Enoxaparin oder Abciximab, siehe ASSENT-3-Studie in Kapitel «Eigenschaften/Wirkungen». Der gleichzeitige Gebrauch von GPIIb/IIIa Antagonisten erhöht das Blutungsrisiko.
·Patienten, die eine orale Antikoagulantien-Therapie erhalten. Die Anwendung von Metalyse kann erst in Erwägung gezogen werden, wenn durch geeignete Tests gezeigt werden kann, dass die antikoagulatorische Aktivität keine klinisch relevante Aktivität mehr zeigt (INR < 1.3, siehe unter Kontraindikationen).
Hypersensitivität
Nach Behandlung mit Tenecteplase wurde keine permanente Bildung von Antikörpern beobachtet.
Es liegen jedoch keine systematischen Erfahrungen mit einer wiederholten Anwendung von Metalyse vor.
Anaphylaktoide Reaktionen in Verbindung mit der Gabe von Metalyse sind selten und können durch Überempfindlichkeit gegenüber dem Wirkstoff Tenecteplase, Gentamicin (Spurenrückstände aus dem Herstellungsprozess) oder einem der Hilfsstoffe verursacht werden.
Im Fall einer anaphylaktischen Reaktion sollte die Injektion beendet und eine geeignete Behandlung eingeleitet werden.
Thromboembolie
Der Gebrauch von Metalyse kann das Risiko thromboembolistischer Ereignisse bei Patienten mit bestehenden Thromben, z.B. Linksherzthrombus (Mitralstenose, Vorhofflimmern, etc) erhöhen.
Koronare Intervention
Überweisung in eine für koronare Interventionen eingerichtete Klinik für eine PCI (perkutane koronare Intervention):
Patienten, die Metalyse für eine primäre koronare Rekanalisation erhalten, sollten unverzüglich in eine Einrichtung überwiesen werden, welche für eine Angiographie und eine zeitnahe Koronarintervention innerhalb von 6-24 Stunden oder früher, ausgerüstet ist, wenn medizinisch indiziert (siehe auch «Klinische Wirksamkeit»).
Perkutane koronare Intervention (PCI)
Das für Metalyse in der ASSENT-4-Studie angewendete Behandlungsschema darf nicht angewendet werden (siehe «Eigenschaften/Wirkungen»), wenn eine primäre PCI entsprechend den Behandlungsrichtlinien beabsichtigt wird.
Arrhythmien
Koronare Thrombolyse kann Arrhythmien assoziiert mit Reperfusion auslösen. Reperfusionsarrhythmien können zum Herzstillstand führen, lebensbedrohlich sein und eine konventionelle antiarrhythmische Therapie erfordern.
Kinder und Jugendliche
Metalyse wird bei Kindern und Jugendlichen unter 18 Jahren aufgrund des Fehlens von Daten zur Sicherheit und Wirksamkeit nicht empfohlen.
Hilfsstoffe
Metalyse 10'000 U (50 mg) enthält 4,0 mg Polysorbat 20 in jeder 50 mg Durchstechflasche. Polysorbate können allergische Reaktionen hervorrufen.
InteraktionenArzneimittel, welche die Blutgerinnung beeinflussen oder die Thrombozytenfunktion (Thrombozytenaggregationshemmer, Cumarinderivate, Heparin und andere) verändern, können die Blutungsgefahr vor, während oder nach einer Behandlung mit Metalyse erhöhen.
Schwangerschaft, StillzeitSchwangerschaft
Zur Anwendung von Metalyse bei Schwangeren liegt eine begrenzte Anzahl Daten vor. Präklinische Studien mit Tenecteplase führten beim Muttertier zu Blutungen und aufgrund der bekannten pharmakologischen Aktivität des Präparats als Sekundärfolge zu Todesfällen. In einigen Fällen kam es zu Aborten und fetaler Resorption (wurde nur bei wiederholter Gabe beobachtet). Tenecteplase wird als nicht teratogen betrachtet (siehe «Präklinische Daten»).
Erfahrungen zur Anwendung von Metalyse bei Schwangeren liegen nicht vor. Metalyse soll in der Schwangerschaft und Stillzeit nicht angewendet werden, es sei denn, dies ist eindeutig notwendig.
Stillzeit
Es ist nicht bekannt, ob Tenecteplase beim Menschen in die Milch übergeht. Vorsicht ist geboten, wenn Metalyse einer stillenden Frau verabreicht wird. Muttermilch sollte innerhalb der ersten 24 Stunden nach der thrombolytischen Therapie verworfen werden.
Wirkung auf die Fahrtüchtigkeit und auf das Bedienen von MaschinenDie Wirkung auf die Fahrtüchtigkeit und auf das Bedienen von Maschinen wurde nicht untersucht.
Unerwünschte WirkungenWie bei anderen Thrombolytika waren Blutungen die am häufigsten beobachtete unerwünschte Wirkung unter Metalyse. Es können Blutungen an jeglichen Körperstellen oder -kavitäten, inklusive intrakranieller Blutungen, auftreten und zu lebensbedrohlichen Situationen, bleibender Behinderung oder zum Tod führen. Die Blutungsereignisse können in zwei Kategorien eingeteilt werden:
·Oberflächliche Blutungen, in der Regel an der Injektionsstelle
·Innere Blutungen an jeglichen Körperstellen oder kavitäten.
Neurologische Symptome wie Somnolenz, Aphasie, Hemiparese, Konvulsion können mit intrakraniellen Blutungen assoziiert sein.
In der doppelblinden ASSENT-2-Studie wurde die Sicherheit und Wirksamkeit von Metalyse (30-50 mg körpergewichtsbezogen) mit der beschleunigten Infusion von Alteplase (≤100 mg) bei 16'949 Patienten mit akutem Myokardinfarkt verglichen (Lyse innerhalb 6 Stunden nach Symptombeginn). Es zeigte sich, dass die Inzidenz schwerer Blutungen und CABGs (Coronary Artery Bypass Graft) signifikant geringer als bei Alteplase ist und eine therapeutische Äquivalenz bezüglich 30-Tage-Mortalität besteht. Schwere nicht-zerebrale Blutungen unter Metalyse wurden bei 4,7% der Patienten beobachtet (bei Alteplase 5,9%). Die Inzidenz intrakranieller Blutungen lag bei 0,93% für Metalyse (Alteplase 0,94%) in der ASSENT-2-Studie.
Im Rahmen der ASSENT-3-Studie wurden für die verschiedenen Behandlungsgruppen (Gruppe A: Metalyse und unfraktioniertes Heparin; Gruppe B: Metalyse und Enoxaparin; Gruppe C: Metalyse in halber Dosierung und Abciximab und unfraktioniertes Heparin – genaue Dosisschemata siehe Kapitel «Eigenschaften/Wirkungen») die folgenden Nebenwirkungen ermittelt:
|
Unerwünschtes Ereignis während des stationären Aufenthaltes
|
Gruppe A
(N = 2038) (%)
|
Gruppe B
(N = 2040) (%)
|
Gruppe C
(N = 2017) (%)
|
p-Wert (insgesamt)
| |
Intrakranielle Blutung
|
0,93
|
0,88
|
0,94
|
0,98
| |
Ischämischer Insult
|
1,52
|
1,62
|
1,49
|
0,94
| |
Transfusionspflichtige Blutungen oder Blutungen mit Kreislaufinsuffizienz (schwerwiegende Blutungen)
|
2,2
|
3,0
|
4,4
|
0,0005
| |
Sonstige Blutungen
|
18,7
|
22,6
|
35,3
|
< 0,0001
| |
Bluttransfusionen
|
2,3
|
3,4
|
4,2
|
0,0032
| |
Schwere behandlungsbedürftige Thrombopenie
|
0,20
|
0,10
|
0,59
|
0,0101
| |
Alle Thrombopenien
|
1,3
|
1,2
|
3,2
|
< 0,0001
|
Hirnschlag und intrakranielle Blutungen
Insgesamt erlitten 94 Patienten während des stationären Aufenthaltes einen Schlaganfall, von welchen 56 als intrakranielle Blutung eingestuft wurden. Weitere 16 Patienten erlitten zwischen der Entlassung und dem Tag 30 nach Studieneinschluss einen Schlaganfall. Davon befanden sich 3 in Gruppe A, 6 in Gruppe B und 7 in Gruppe C. Einer dieser Fälle aus Gruppe A wurde als intrakranielle Blutung gewertet.
Klinisch relevante Blutungen und Bluttransfusionen
Transfusionen während des stationären Aufenthaltes waren in den Gruppen B und C signifikant (p=0,0327 bzw. 0,0010) häufiger als in der Gruppe A indiziert.
Thrombopenie
Thrombopenien traten in Gruppe C signifikant (p=0,0001) häufiger als in den Gruppen A und B auf. In keinem der 19 vorliegenden schweren Fälle wurde Tenecteplase alleine als möglicher Auslöser eingestuft. Die Rate von Thrombopenien in Gruppe C glich derjenigen, die für Abciximab in Kombination mit unfraktioniertem Heparin bekannt ist.
Die verwendeten Häufigkeitskategorien sind wie folgt definiert: sehr häufig (≥1/10), häufig (≥1/100, <1/10), gelegentlich (≥1/1000, <1/100), selten (≥1/10'000, <1/1000), sehr selten (<1/10'000), Häufigkeit nicht bekannt (kann aus den verfügbaren Daten nicht abgeschätzt werden).
Erkrankungen des Immunsystems
Selten: anaphylaktoide Reaktionen (einschliesslich Hautausschlag, Urtikaria, Bronchospasmen, Larynxödem)
Erkrankungen des Nervensystems
Gelegentlich: Intrakranielle Blutungen (wie Hirnblutung, zerebrale Hämatome, hämorrhagischer Hirnschlag, hämorrhagische Transformation eines Hirnschlags, intrakranielles Hämatom, subarachnoidale Blutung), einschliesslich Begleitsymptome wie Schläfrigkeit, Sprachstörungen, Hemiparese und Krampfanfälle
Augenerkrankungen
Gelegentlich: Blutungen im Auge, insbesondere bei Patienten mit einer diabetischen Retinopathie
Herzerkrankungen
Gelegentlich: Reperfusionsarrhythmien (wie Arrhythmie, akzelerierte idioventrikuläre Arrhythmie, Asystolie, Extrasystolen, Vorhofflimmern, AV-Block ersten Grades bis zum kompletten AV-Block, Bradykardie, Tachykardie, ventrikuläre Arrhythmie, Kammerflimmern, ventrikuläre Tachykardie) kommen im engen zeitlichen Zusammenhang mit einer Tenecteplase-Behandlung vor.
Selten: Blutungen am Perikard
Gefässerkrankungen
Sehr häufig: Blutungen (21%)
Selten: Embolie (Thromboembolie)
Erkrankungen der Atemwege, des Brustraums und Mediastinums
Häufig: Nasenbluten
Selten: pulmonale Blutungen
Erkrankungen des Gastrointestinaltrakts
Häufig: gastrointestinale Blutungen (wie Magenblutungen, Magenulkusblutungen, rektale Blutungen, Hämatemesis, Melaena, Blutungen im Mund)
Gelegentlich: retroperitoneale Blutungen (wie retroperitoneales Hämatom)
Häufigkeit nicht bekannt: Übelkeit, Erbrechen
Erkrankungen der Haut und des Unterhautgewebes
Häufig: Ekchymosen
Erkrankungen der Nieren und Harnwege
Häufig: Urogenitale Blutungen (wie Hämaturie, Blutungen der Harnwege)
Allgemeine Erkrankungen und Beschwerden am Verabreichungsort
Häufig: Blutungen an der Injektionsstelle bzw. Punktionsstelle
Untersuchungen
Selten: Blutdrucksenkung
Häufigkeit nicht bekannt: erhöhte Körpertemperatur
Verletzung, Vergiftungen und durch Eingriffe bedingte Komplikationen
Häufigkeit nicht bekannt: Fettembolien (Embolien durch Cholesterinkristalle), welche zu entsprechenden Folgen in den betroffenen Organen führen kann
Chirurgische und medizinische Eingriffe
Häufigkeit nicht bekannt: Bluttransfusionen erforderlich
Wie bei anderen Thrombolytika wurden folgende Ereignisse als Folge des Herzinfarkts und/oder der thrombolytischen Therapie berichtet:
·Sehr häufig (>10%): Niedriger Blutdruck, Herzfrequenz- und Herzrhythmusstörungen, Angina pectoris
·Häufig (<10%-1%): Erneute Ischämie, Herzinsuffizienz, Reinfarkt, kardiogener Schock, Perikarditis, Lungenödem
·Gelegentlich (<1%-0,1%): Herzstillstand, Mitralklappeninsuffizienz, Perikarderguss, venöse Thrombosen, Herztamponade, Myokardruptur
·Selten (<0,1%-0,01%): Lungenembolie.
Diese kardiovaskulären Ereignisse können lebensbedrohlich sein und zum Tod führen.
Die Meldung des Verdachts auf Nebenwirkungen nach der Zulassung ist von grosser Wichtigkeit. Sie ermöglicht eine kontinuierliche Überwachung des Nutzen-Risiko-Verhältnisses des Arzneimittels. Angehörige von Gesundheitsberufen sind aufgefordert, jeden Verdacht einer neuen oder schwerwiegenden Nebenwirkung über das Online-Portal ElViS (Electronic Vigilance System) anzuzeigen. Informationen dazu finden Sie unter www.swissmedic.ch.
ÜberdosierungAnzeichen und Symptome
Im Falle einer Überdosierung könnte eine erhöhte Blutungsgefahr bestehen.
Behandlung
Bei schweren, lang anhaltenden Blutungen kann eine Substitutionstherapie (Plasma, Plättchen) erwogen werden. Siehe auch «Warnhinweise und Vorsichtsmassnahmen».
Eigenschaften/WirkungenATC-Code
B01AD11
Wirkungsmechanismus
Tenecteplase ist ein rekombinanter fibrinspezifischer Plasminogen-Aktivator (t-PA), der durch Modifizierung von natürlichem t-PA an drei Stellen des Moleküls entsteht. Er bindet an das Fibrin eines Thrombus (Blutgerinnsel) und wandelt an den Thrombus gebundenes Plasminogen in Plasmin um und führt so zur Auflösung des Thrombus. Tenecteplase weist eine höhere Fibrinspezifität als natürliches t-PA auf und wird weniger durch den endogenen Inhibitor (PAI-1) inaktiviert.
Pharmakodynamik
Nach Gabe von Tenecteplase wurde ein dosisabhängiger Verbrauch von α2-Antiplasmin (dem plasmatischen Inhibitor des Plasmins) mit gleichzeitiger Zunahme einer systemischen Plasminbildung beobachtet. Diese Beobachtung stimmt mit einer erwarteten Plasminogenaktivierung überein. In Vergleichsstudien (Tenecteplase versus Alteplase) wurde bei Patienten, die mit der Maximaldosis von Tenecteplase (10'000 U, entsprechend 50 mg) behandelt wurden, ein Abfall des Fibrinogens um weniger als 15% und des Plasminogens um weniger als 25% beobachtet. Demgegenüber kam es unter Alteplase zu einem Abfall der Fibrinogen- und Plasminogenspiegel um ca. 50%. Eine klinisch relevante Antikörperbildung wurde bis 30 Tage nicht beobachtet. Daten zur wiederholten Gabe liegen allerdings nicht vor.
Klinische Wirksamkeit
Daten zur Wiedereröffnungsrate der Koronararterien in angiographisch kontrollierten Phase I und II Studien zeigen, dass Tenecteplase als intravenöser Einfach-Bolus dosisabhängig Thromben in der Infarktarterie von Patienten mit akutem Herzinfarkt auflöst.
ASSENT 2 Studie
Eine grosse doppelblinde Vergleichsstudie (ASSENT-2) mit etwa 17'000 Patienten zeigte, dass Tenecteplase im Vergleich zu Alteplase hinsichtlich der Sterblichkeitssenkung therapeutisch äquivalent ist (6,2% in beiden Therapiegruppen nach 30 Tagen) und dass es unter Tenecteplase zu signifikant weniger nichtzerebralen Blutungen kam (26,4% versus 28,9%, p=0,0003).
Die Senkung des Blutungsrisikos hängt wahrscheinlich mit der erhöhten Fibrinspezifität von Tenecteplase und der körpergewichtsadaptierten Dosierung zusammen.
Dies bedingte eine signifikant geringere Bluttransfusionsrate (4,3% versus 5,5%, p=0,0002). Die zerebrale Blutungsrate betrug 0,93% für Tenecteplase bzw. 0,94% für Alteplase.
ASSENT 3 Studie
Ziel der ASSENT 3 Studie war es, die antithrombotische Begleittherapie mit Tenecteplase so zu optimieren, dass die frühen Offenheitsraten verbessert werden und die Perfusion aufrechterhalten bleibt. Dazu sollte vor allem der paradoxe prokoagulatorische Effekt beseitigt werden, der durch die lysebedingte Freisetzung von gebundenem Thrombin entsteht. Drei unterschiedliche antithrombotische Begleittherapien wurden an 6'095 Patienten verglichen:
Gruppe A: Volle Dosis Tenecteplase + unfraktioniertes Heparin (UFH) versus Gruppe B: volle Dosis Tenecteplase + niedermolekulares Heparin (Enoxaparin) versus Gruppe C: halbe Dosis Tenecteplase + unfraktioniertes Heparin + volle Dosis Abciximab.
Unfraktioniertes Heparin wurde entsprechend den Empfehlungen der Richtlinien der American College of Cardiology und der American Heart Association (AHA/ACC-Richtlinien) nach dem folgenden, vollständig gewichtadaptierten Niedrigdosis-Schema gegeben: Zuerst ein intravenöser Einfach-Bolus von 60 U/kg (höchstens 4'000 U), unmittelbar anschliessend eine intravenöse Infusion von 12 U/kg/Stunde (höchstens 1'000 U/Stunde) für die ersten 3 Stunden, dann Einstellung der Infusion unter aPTT-Kontrolle so, dass eine aPTT von 50-70 Sekunden erreicht wird, für bis zu 48 Stunden.
Die Raten für die 30-Tage-Mortalität betragen 6,0%, 5,4% bzw. 6,6%, für grössere Blutungsereignisse (ausser intrakranielle Blutungen) intrastationär 2,16%, 3,04% bzw. 4,32% und für intrakranielle Blutungen 0,93%, 0,88% bzw. 0,94%.
Das von der ACC/AHA empfohlene Therapieschema, eine vollständig gewichtadaptierte Gabe von unfraktioniertem Heparin in niedriger Dosierung, das in ASSENT 3 gleichzeitig mit Tenecteplase angewendet wurde, führt zu einer Abnahme systemischer Blutungen, verglichen mit dem aggressiveren Dosierungsschema mit unfraktioniertem Heparin, das in ASSENT 2 geprüft wurde, dies bei vergleichbaren Raten intrakranieller Blutungen, jedoch ohne Wirksamkeit einzubüssen.
ASSENT 3 PLUS Studie
ASSENT 3 PLUS, eine Ergänzungsstudie zu ASSENT 3, sollte den Einsatz in der prästationären Situation untersuchen. Wirksamkeit und Sicherheit einer vollen Dosis Tenecteplase + unfraktioniertes Heparin (UFH) versus einer vollen Dosis Tenecteplase + niedermolekulares Heparin (Enoxaparin) wurden an 1'639 Patienten untersucht.
Studiendesign und Arzneimitteldosierung sind identisch mit denjenigen in ASSENT 3. Eine prästationäre Reperfusionstherapie mit Tenecteplase und UFH oder Enoxaparin ermöglichte bei >50 % der Patienten mit STEMI eine Behandlung innerhalb von 2 Stunden nach Einsetzen der Symptome.
Nach den Ergebnissen der Studien ASSENT 3 und 3 PLUS verringerte sowohl eine prästationäre als auch eine stationäre Zusatztherapie mit Enoxaparin die Inzidenz ischämischer Komplikationen, verglichen mit einer Zusatztherapie mit UFH: Die Inzidenz des zusammengesetzten Endpunkts für Wirksamkeit nach 30 Tagen (Tod, Reinfarkt, refraktäre Ischämie) betrug 11,4% versus 15,4% in ASSENT 3 bzw. 14,2% versus 17,4 % in ASSENT 3 PLUS. In der prästationären Situation war jedoch Tenecteplase plus Enoxaparin in der eingesetzten Dosierung mit einem erhöhten Risiko für grössere Blutungsereignisse und intrakranielle Blutungen bei Patienten >75 Jahre verbunden.
Daten zur Offenheit der Koronargefässe und begrenzte Daten zum klinischen Ergebnis zeigten, dass Patienten mit akutem Myokardinfarkt auch noch mehr als 6 Stunden nach Symptombeginn erfolgreich behandelt wurden.
ASSENT 4 PCI Studie
Die ASSENT-4 PCI-Studie sollte an 4'000 teilnehmenden Patienten mit ausgedehntem Myokardinfarkt zeigen, ob bessere Ergebnisse als mit einer primären perkutanen koronare Intervention (PCI) allein erzielt werden, wenn innerhalb von 60 bis 180 Minuten vor der PCI mit einer vollen Dosis Tenecteplase und der gleichzeitigen Gabe eines Einfach-Bolus von bis zu 4'000 U unfraktionierten Heparins vorbehandelt wird. Die Studie wurde nach 1'667 randomisierten Patienten vorzeitig abgebrochen, weil die Mortalität in der PCI plus Tenecteplase-Gruppe erhöht war.
Der primäre Endpunkt, zusammengesetzt aus Tod, kardiogenem Schock und Stauungsinsuffizienz innerhalb von 90 Tagen, trat signifikant häufiger auf in der Gruppe, die die explorative Therapie – Tenecteplase, sofort gefolgt von der routinemässigen PCI – erhielt: 18,6% (151/810) gegenüber 13,4% (110/819) in der Gruppe mit PCI allein, p=0,0045.
Dieser signifikante Unterschied zwischen den Gruppen in Bezug auf den primären Endpunkt nach 90 Tagen zeigte sich bereits während des stationären Aufenthalts und nach 30 Tagen.
Numerisch sprachen alle Komponenten des zusammengesetzten klinischen Endpunkts für die Behandlung mit der PCI allein: Tod: 6,7% versus 4,9%, p=0,14; kardiogener Schock: 6,3% versus 4,8%, p=0,19; Stauungsinsuffizienz: 12,0% versus 9,2%, p=0,06. Die sekundären Endpunkte Reinfarkt und erneute Revaskularisierung des betreffenden Gefässes waren in der mit Tenecteplase vorbehandelten Gruppe signifikant erhöht: Reinfarkt: 6,1% versus 3,7%, p=0,0279; erneute Revaskularisierung des Gefässes: 6,6% versus 3,4%, p=0,0041.
Die folgenden unerwünschten Ereignisse traten in der mit Tenecteplase vorbehandelten PCI-Gruppe häufiger auf: Intrakranielle Blutung: 1% versus 0%, p=0,0037; Schlaganfall: 1,8% versus 0%, p<0,0001; stärkere Blutungen: 5,6% versus 4,4%, p=0,3118; leichtere Blutungen: 25,3% versus 19,0%, p=0,0021; Bluttransfusionen: 6,2% versus 4,2%, p=0,0873; plötzlicher Gefässverschluss: 1,9% versus 0,1%, p=0,0001.
STREAM-Studie
In der explorativen STREAM-Studie wurde die Sicherheit von Metalyse gefolgt von einer fakultativen PCI verglichen mit derjenigen einer primären PCI. Eingeschlossen wurden Patienten mit einem akuten Myokardinfarkt, mit ST-Hebung innerhalb von 3 Stunden nach dem Einsetzen der Symptome, bei denen innerhalb von 1 Stunde nach dem ersten medizinischen Kontakt keine primäre PCI durchgeführt werden konnte. Die Kombinationstherapie bestand aus einer frühzeitigen Fibrinolyse mit Tenecteplase als Bolusinjektion und einer zusätzlichen Antiplättchen- und Antikoagulanzien-Therapie, gefolgt von einer Angiographie innerhalb von 6 bis 24 Stunden, mit einer notfallmässigen Koronarintervention (Rescue-PCI) bei Bedarf.
Die Studienpopulation umfasste 1892 Patienten.
Die beobachtete Inzidenz von schweren und leichten nicht-intrakraniellen Blutungen war wie folgt:
|
|
Pharmako-invasiv (n=944)
|
Primäre PCI (n=948)
|
P
| |
Schwere nicht-intrakranielle Blutung
|
61/939 (6,5%)
|
45/944 (4,8%)
|
0,11
| |
Leichte nicht-intrakranielle Blutung
|
205/939 (21,8%)
|
191/944 (20,2%)
|
0,40
|
Die Inzidenz der gesamten Schlaganfälle und intrakraniellen Blutungen betrug:
|
|
Pharmako-invasiv (n=944)
|
Primäre PCI (n=948)
|
P
| |
Schlaganfälle insgesamt (alle Typen)
|
15/939 (1,6%)
|
5/946 (0,5%)
|
0,03
| |
Intrakranielle Blutung
|
9/939 (0,96%)
|
2/946 (0,21%)
|
0,04
| |
Intrakranielle Blutung nach der Protokolländerung auf die halbe Dosis bei Patienten ≥75 Jahre
|
4/747 (0,5%)
|
2/758 (0,3%)
|
0,45
|
PharmakokinetikAbsorption / Distribution
Tenecteplase ist ein intravenös verabreichtes rekombinantes Protein, das Plasminogen aktiviert.
Nach einem intravenösen Bolus von 30 mg Tenecteplase bei Patienten mit akutem Herzinfarkt liegt die geschätzte Tenecteplase Plasmakonzentration bei 6.45 + 3.60 µg/ml (Mittel + SD). Die Distributionsphase liegt nach Verabreichung von Dosen von 5 bis 50 mg zwischen 31% + 22% und 69% + 15% (Mittel + SD) der Gesamt-AUC.
Die mittlere Verweilzeit im Körper beträgt ca. 1 Stunde und das mittlere (+ SD) Verteilungsvolumen im steady-state (Vss) liegt zwischen 6.3 + 2 l und 15 ± 7 l.
Metabolismus
Tenecteplase wird durch Bindung an spezifische Leberrezeptoren und nachfolgende Spaltung in kleine Peptide aus dem Blutkreislauf eliminiert. Im Vergleich zu natürlichem t-PA ist die Bindung an die Leberrezeptoren weniger stark ausgeprägt, was zu einer verlängerten Halbwertszeit führt.
Elimination
Nach einem intravenösen Einfach-Bolus von Tenecteplase bei Patienten mit akutem Herzinfarkt ergibt sich für Tenecteplase Antigen eine biphasische Elimination aus dem Plasma. Im therapeutischen Bereich findet sich keine Dosisabhängigkeit für die Elimination von Tenecteplase. Die α-Halbwertszeit beträgt 24 ± 5,5 Minuten eine fünffache Verlängerung im Vergleich zu natürlichem t-PA. Die β-Halbwertszeit beträgt 129 ± 87 Minuten und die Plasmaclearance 119 ± 49 ml/Min.
Mit steigendem Körpergewicht nimmt die Plasmaclearance von Tenecteplase etwas zu (pro Zunahme des Körpergewichts um 10 kg, steigt die Plasmaclearance durchschnittlich um 9,6 ml/Min.), höheres Alter führt zu einer etwas niedrigeren Clearance (bei älteren Patienten nimmt die Plasmaclearance pro 10 Jahre um durchschnittlich 16,1 ml/Min. ab). Frauen weisen im Allgemeinen eine niedrigere Clearance als Männer auf, was sich durch das allgemein niedrigere Körpergewicht erklären lässt.
Linearität/Nicht Linearität
Die Dosis Linearitätsanalyse basierend auf die AUC deutet darauf hin, dass Tenecteplase im Dosisbereich von 5 bis 50 mg eine nicht-lineare Pharmakokinetik aufweist.
Kinetik spezieller Patientengruppen
Leberfunktionsstörungen
Die Auswirkungen einer Leberinsuffizienz auf die Pharmakokinetik bei Menschen sind nicht bekannt.
Nierenfunktionsstörungen
Da Tenecteplase offensichtlich nicht über die Nieren ausgeschieden wird, ist nicht zu erwarten, dass die Pharmakokinetik durch eine Niereninsuffizienz beeinflusst wird.
Präklinische DatenDiese Klasse der rekombinanten Proteine wird weder als mutagen noch als karzinogen eingestuft und daher wurde auf Untersuchungen zur Genotoxizität und Karzinogenität verzichtet.
Im Hinblick auf die Indikation und die Einmalgabe beim Menschen wurde die Reproduktionstoxizität auf Kaninchen als sensitiver Tierspezies beschränkt. Tenecteplase führte zu keiner Teratogenizität. Wiederholte Gabe führte zu Blutungen mit Todesfällen als Sekundärfolge. In einigen Fällen kam es zu Abort und Resorption der Föten. Nach Einmalgabe von Tenecteplase wurden diese Effekte nicht beobachtet.
Weder nach intravenöser, noch nach intraarterieller oder nach paravenöser Gabe der endgültigen Formulierung von Tenecteplase kam es zu lokalen Reizungen der Blutgefässe.
Sonstige HinweiseInkompatibilitäten
Metalyse ist mit Glukoselösungen nicht kompatibel. Metalyse darf nicht mit anderen Arzneimitteln gemischt werden noch sollten diese über denselben intravenösen Zugang wie Metalyse appliziert werden (siehe auch Rubrik «Dosierung/Anwendung» unter «Korrekte Art der Anwendung»).
Haltbarkeit
Das Arzneimittel darf nur bis zu dem auf dem Behälter mit «EXP» bezeichneten Datum verwendet werden.
Haltbarkeit der gebrauchsfertigen Lösung
Es wurde eine chemische und physikalische Stabilität der rekonstituierten Lösung von 24 Stunden bei 2-8 ºC und von 8 Stunden bei 30 ºC nachgewiesen.
Aus mikrobiologischer Sicht sollte die gebrauchsfertige Lösung sofort nach ihrer Herstellung verwendet werden. Falls aseptisch hergestellt, sollten üblicherweise 24 Stunden bei 2-8 ºC nicht überschritten werden.
Besondere Lagerungshinweise
Ausser Reichweite von Kindern aufbewahren.
Nicht über 30 ºC lagern. Das Behältnis in der Faltschachtel aufbewahren, um den Inhalt vor Licht zu schützen.
Zulassungsnummer55'418 (Swissmedic)
PackungenPackung mit 1 Durchstechflasche mit 10'000 U (50 mg) Tenecteplase und 1 Fertigspritze mit 10 ml Wasser für Injektionszwecke.
Abgabekategorie: B
ZulassungsinhaberinBoehringer Ingelheim (Schweiz) GmbH, Basel
Stand der InformationFebruar 2025
|