Eigenschaften/WirkungenATC-Code
L04AJ01
Soliris ist ein rekombinanter humanisierter monoklonaler IgG2/4k-Antikörper, der an das humane Komplementprotein C5 bindet und die Aktivierung des terminalen Komplements hemmt. Der Soliris-Antikörper enthält humanisierte konstante Regionen und murine Komplementarität bestimmende Regionen, die auf die variablen Regionen der leichten und schweren Ketten des menschlichen Gerüsts aufgesetzt sind. Soliris besteht aus zwei schweren Ketten mit 448 Aminosäuren und zwei leichten Ketten mit 214 Aminosäuren und hat ein Molekulargewicht von ca. 148 kDa.
Soliris wird in einem murinen Myelom-(NS0-Zelllinien-) Expressionssystem hergestellt und wird durch Affinitäts- und Ionenaustauschchromatographie gereinigt. Der Herstellungsprozess des Arzneistoffs umfasst ebenfalls spezifische Virusinaktivierungs- und -suppressionsschritte.
Wirkungsmechanismus
Eculizumab, der Wirkstoff in Soliris, ist ein terminaler Komplementinhibitor, der spezifisch und mit hoher Affinität an das Komplementprotein C5 bindet und dadurch dessen Spaltung in die Fragmente C5a und C5b blockiert und somit die Bildung des terminalen Komplementkomplexes C5b-9 verhindert. Eculizumab erhält die frühen Komponenten der Komplementaktivierung, die von wesentlicher Bedeutung für die Opsonisierung von Mikroorganismen und die Elimination (Clearance) von Immunkomplexen sind.
Bei PNH-Patienten werden die unkontrollierte terminale Komplementaktivierung und die daraus resultierende komplementvermittelte intravaskuläre Hämolyse durch die Behandlung mit Soliris blockiert.
Bei den meisten PNH-Patienten reichen Eculizumab-Serumkonzentrationen von etwa 35 µg/ml für eine praktisch vollständige Hemmung der terminalen komplementvermittelten intravaskulären Hämolyse aus.
Die dauerhafte Verabreichung von Soliris bei PNH führte zu einer raschen und nachhaltigen Verringerung der komplementvermittelten hämolytischen Aktivität. Bei aHUS-Patienten werden die unkontrollierte terminale Komplementaktivierung und die daraus resultierende komplementvermittelte thrombotische Mikroangiopathie durch die Behandlung mit Soliris blockiert.
Alle aHUS-Patienten, die nach dem empfohlenen Dosierungsschema mit Soliris behandelt wurden, erreichten eine rasche und anhaltende Abnahme der terminalen Komplementaktivität. Bei allen Patienten mit aHUS reichen Eculizumab-Serumkonzentrationen von etwa 50-100 µg/ml für eine praktisch vollständige Hemmung der terminalen Komplementaktivität aus.
Die dauerhafte Verabreichung von Soliris bei aHUS führte zu einer raschen und nachhaltigen Verringerung der komplementvermittelten thrombotischen Mikroangiopathie.
Bei Patienten mit refraktärer gMG verursacht die unkontrollierte terminale Komplementaktivierung eine vom Membranangriffskomplex (MAC) abhängige Lyse und eine C5a-abhängige Entzündung an der neuromuskulären Endplatte, was zum Ausfall der neuromuskulären Übertragung führt. Die dauerhafte Verabreichung von Soliris führt zu einer sofortigen, vollständigen und nachhaltigen Hemmung der terminalen Komplementaktivität (Eculizumab-Serumkonzentrationen ≥116 µg/ml).
Bei Patienten mit NMOSD führt eine durch Autoantikörper gegen AQP4 verursachte unkontrollierte Aktivierung des terminalen Komplements zur Entstehung der MAC- und C5a-abhängigen Entzündung, die Astrozytennekrose und eine erhöhte Durchlässigkeit der Blut-Hirn-Schranke sowie das Absterben der umgebenden Oligodendrozyten und Neuronen zur Folge hat. Die dauerhafte Anwendung von Soliris führt zu einer sofortigen, vollständigen und anhaltenden Hemmung der terminalen Komplementaktivität (Eculizumab-Serumkonzentrationen ≥116 µg/ml).
Pharmakodynamik
siehe «Wirkungsmechanismus»
Klinische Wirksamkeit
Paroxysmale nächtliche Hämoglobinurie
Die Sicherheit und Wirksamkeit von Soliris bei PNH-Patienten mit Hämolyse wurden in einer 26-wöchigen randomisierten, doppelblinden, placebokontrollierten Studie (C04-001) untersucht. PNH-Patienten wurden ebenfalls in einer 52-wöchigen einarmigen Studie (C04-002) sowie in einer Langzeit-Fortsetzungsstudie (E05-001) mit Soliris behandelt. Die Patienten erhielten vor Verabreichung von Soliris eine Meningokokkenimpfung. In allen Studien betrug die Eculizumab-Dosis 600 mg alle 7 ± 2 Tage über 4 Wochen, gefolgt von 900 mg 7 ± 2 Tage später und anschliessend 900 mg alle 14 ± 2 Tage für die Dauer der Studie. Soliris wurde als intravenöse Infusion über 25 bis 45 Minuten (35 ± 10 Minuten) verabreicht. Zusätzlich wurde ein nicht-interventionelles Beobachtungsregister (M07-001) bei PNH Patienten initiiert, um den natürlichen Verlauf der PNH bei unbehandelten Patienten, sowie die klinischen Ergebnisse unter Soliris-Therapie zu charakterisieren.
In die Studie C04-001 (TRIUMPH) wurden PNH-Patienten mit mindestens 4 Transfusionen in den vorangegangenen 12 Monaten, einem mittels Durchflusszytometrie bestätigten Anteil von mindestens 10 % PNH-Zellen und einer Thrombozytenzahl von mindestens 100.000/Mikroliter randomisiert entweder der Soliris- (n = 43) oder der Placebo-Behandlung (n = 44) zugeordnet. Vor der Randomisierung durchliefen alle Patienten eine anfängliche Beobachtungsphase, um den Bedarf an Erythrozytentransfusion zu bestätigen und die Hämoglobinkonzentration (den „Sollwert“) zu ermitteln, welche die Hämoglobinstabilisierung und die Transfusionsergebnisse jedes Patienten bestimmen würde. Der Hämoglobin-Sollwert war bei Patienten mit Symptomen ≤ 9 g/dl und bei Patienten ohne Symptome ≤ 7 g/dl. Primäre Endpunkte waren die Hämoglobin-Stabilisierung (Patienten, die eine über dem Hämoglobinsollwert liegende Hämoglobinkonzentration aufrechterhielten und ohne weitere Erythrozytentransfusion während des gesamten 26-wöchigen Zeitraums auskamen) und der Bedarf an Bluttransfusion. Fatigue und Quality of Life (krankheitsbedingte Lebensqualität) waren relevante sekundäre Endpunkte. Die Hämolyse wurde hauptsächlich durch Messung der LDH-Spiegel im Serum überwacht, und der Anteil der PNH-Erythrozyten wurde mittels Durchflusszytometrie kontrolliert. Patienten, die Antikoagulantien und systemische Kortikosteroide zu Beginn erhielten, setzten die Einnahme dieser Medikamente fort. Die wichtigsten Ausgangsmerkmale waren in beiden Behandlungsarmen vergleichbar (siehe Tabelle 2).
In der nicht-kontrollierten Studie C04-002 (SHEPHERD) erhielten PNH-Patienten mit mindestens einer Transfusion in den vorausgegangenen 24 Monaten und mindestens 30.000 Thrombozyten/Mikroliter Soliris über einen 52-wöchigen Zeitraum. Zu den Begleittherapien gehörten Antithrombotika bei 63 % der Patienten und systemische Kortikosteroide bei 40 % der Patienten. Die Ausgangsparameter sind in Tabelle 2 dargestellt.
Tabelle 2: Demografische Patientendaten und parameter in den Studien C04-001 und C04-002
|
|
C04-001
|
C04-002
| |
Parameter
|
Placebo
N = 44
|
Soliris
N = 43
|
Soliris
N = 97
| |
Mittleres Alter (SD)
|
38,4 (13,4)
|
42,1 (15,5)
|
41,1 (14,4)
| |
Geschlecht - weiblich (%)
|
29 (65,9)
|
23 (53,5)
|
49 (50,5)
| |
Aplastische Anämie oder MDS in der Anamnese (%)
|
12 (27,3)
|
8 (18,7)
|
29 (29,9)
| |
Begleitmedikation Antikoagulantien (%)
|
20 (45,5)
|
24 (55,8)
|
59 (61)
| |
Begleitmedikation Steroiden/Immunsuppressiva (%)
|
16 (36,4)
|
14 (32,6)
|
46 (47,4)
| |
Behandlungsabbruch
|
10
|
2
|
1
| |
Erythrozytenkonzentrate in den vorangegangenen 12 Monaten (Median (Q1, Q3))
|
17,0 (13,5/25,0)
|
18,0 (12,0/24,0)
|
8,0 (4,0/24,0)
| |
Mittlerer Hb-Spiegel (g/dl) am Sollwert (SD)
|
7,7 (0,75)
|
7,8 (0,79)
|
n.a.
| |
Prätherapeutische LDH-Spiegel (Median, U/l)
|
2234,5
|
2032,0
|
2051,0
| |
Freies Hämoglobin bei Studienbeginn (Median, mg/dl)
|
46,2
|
40,5
|
34,9
|
In der TRIUMPH Studie zeigten die mit Soliris behandelten Patienten eine signifikante Verminderung der Hämolyse (p<0,001), was Verbesserungen der Anämie bewirkte, was sich durch eine erhöhte Hämoglobin-Stabilisierung und einen verminderten Bedarf an Erythrozyten-Transfusionen im Vergleich zu den mit Placebo behandelten Patienten äusserte (siehe Tabelle 3). Die Verbesserung konnte in allen 3 Erythrozyten-Transfusion-Untergruppen (4–14 Einheiten; 15–25 Einheiten; > 25 Einheiten) nachgewiesen werden. Nach 3 Wochen Soliris-Therapie zeigten die Patienten eine Verminderung der Fatigue und eine verbesserte Quality of Life (gesundheitsbezogene Lebensqualität). Aufgrund des Stichprobenumfangs und der Dauer der Studie konnten die Wirkungen von Soliris auf thrombotische Ereignisse nicht ermittelt werden. In der SHEPHERD-Studie beendeten 96 der 97 eingeschlossenen Patienten die Studie (ein Patient starb nach einem thrombotischen Ereignis). Die Reduktion intravaskulärer Hämolyse, die anhand der LDH-Spiegel im Serum gemessen wurde, hielt während des Behandlungszeitraums an und führte zu einer erhöhten Transfusionsvermeidung, einem verringerten Bedarf an Erythrozyten-Transfusion und zu geringerer Fatigue (siehe Tabelle 3).
Tabelle 3: Wirksamkeitsergebnisse in den Studien C04-001 und C04-002
|
|
C04-001
|
C04-002*
| |
|
Placebo
N = 44
|
SOLIRIS
N = 43
|
p-Wert
|
SOLIRIS
N = 97
|
p-Wert
| |
Prozentualer Anteil der Patienten mit stabilisierten Hämoglobinspiegeln am Ende der Studie
|
0
|
49
|
< 0,001
|
n.a.
| |
Transfundierte Einheiten Erythrozytenkonzentrat während der Behandlung (Median)
|
10
|
0
|
< 0,001
|
0
|
< 0,001
| |
Verminderter Bedarf an Transfusionen während der Behandlung (%)
|
0
|
51
|
< 0,001
|
51
|
< 0,001
| |
LDH-Spiegel am Ende der Studie (Median, U/l)
|
2167
|
239
|
< 0,001
|
269
|
< 0,001
| |
LDH-AUC am Ende der Studie (Median, U/l x Tag)
|
411822
|
58587
|
< 0,001
|
-632264
|
< 0,001
| |
Freies Hämoglobin am Ende der Studie (Median, mg/dl)
|
62
|
5
|
< 0,001
|
5
|
< 0,001
| |
FACIT-Fatigue (Effektgrösse)
|
|
1,12
|
< 0,001
|
1,14
|
< 0,001
|
* Die Ergebnisse aus der Studie C04-002 beziehen sich auf Vergleiche vor und nach der Behandlung.
195 mit Soliris behandelte Patienten aus den Studien C04-001, C04-002 in anderen Anfangsstudien wurden Langzeit-Fortsetzungs-Studie (E05-001) eingeschlossen. Bei allen Patienten konnte eine Reduktion der intravaskulären Hämolyse während der gesamten Dauer der Soliris-Behandlung von 10 bis 54 Monaten nachgewiesen werden. Im Vergleich zur gleichen Zeitdauer vor der Behandlung traten unter der Behandlung mit Soliris weniger thrombotische Ereignisse auf. Dieses Ergebnis wurde jedoch in einer nicht-kontrollierten klinischen Studie festgestellt.
Das PNH Register (M07-001) wurde genutzt, um die Wirksamkeit von Soliris bei PNH-Patienten ohne Transfusionen mit Erythrozytenkonzentraten in der Historie zu beurteilen. Diese Patienten hatten eine hohe Krankheitsaktivität, die durch eine erhöhte Hämolyse (LDH ≥1,5xONW) und das Vorhandensein eines oder mehrerer der damit verbundenen klinischen Symptome definiert ist: Fatigue, Hämoglobinurie, abdominelle Schmerzen, Kurzatmigkeit (Dyspnoe), Anämie (Hämoglobin <100 g/l), schwere unerwünschte vaskuläre Ereignisse (einschliesslich Thrombosen), Dysphagie oder erektile Dysfunktion.
Im PNH Register wurde, bei mit Soliris behandelten Patienten, eine Reduktion der Hämolyse und der damit verbundene Symptome beobachtet. Nach 6 Monaten hatten die mit Soliris behandelten Patienten ohne Transfusionen mit Erythrozytenkonzentraten in der Historie signifikant (p<0,001) reduzierte LDH-Spiegel (medianer LDH-Spiegel von 305 U/l; Tabelle 4). Weiterhin erfuhren 74% der mit Soliris behandelten Patienten ohne Transfusionshistorie klinisch relevante Verbesserungen im FACIT-Fatigue Score (d.h. Erhöhung um 4 oder mehr Punkte) und 84% im EORTC Fatigue Score (d.h. Abnahme um 10 oder mehr Punkte).
Tabelle 4: Ergebnisse zur Wirksamkeit (LDH-Spiegel und FACIT-Fatigue) bei PNH-Patienten ohne Transfusionshistorie im M07-001
|
|
M07-001
| |
Parameter
|
Soliris
Keine Transfusion
| |
LDH Spiegel zu Beginn der Studie
(Median, U/l)
|
N=43
1447
| |
LDH Spiegel nach 6 Monaten
(Median, U/l)
|
N=36
305
| |
FACIT-Fatigue Score zu Beginn der Studie
(Median)
|
N=25
32
| |
FACIT-Fatigue Score der letzten verfügbaren Auswertung (Median)
|
N=31
44
|
FACIT-Fatigue wird auf einer Skala von 0-52 ermittelt, wobei höhere Werte auf weniger Fatigue hinweisen
Atypisches Hämolytisch-Urämisches Syndrom
Die Wirksamkeit von Soliris in der Behandlung des aHUS wurde in vier prospektiven kontrollierten klinischen Studien mit 100 Patienten (drei Studien bei Erwachsenen und Jugendlichen (C08-002A/B, C08-003A/B, C10-004), einer Studie bei Kindern und Jugendlichen (C10-003) und einer retrospektiven Studie (C09-001r) mit 30 Patienten untersucht.
Bei Studie C08-002A/B handelte es sich um eine prospektive, kontrollierte, offene Studie, in die Patienten in der Frühphase eines aHUS mit Anzeichen einer klinisch manifestierten thrombotischen Mikroangiopathie (Thrombozytenzahl von ≤ 150 x 109/l trotz Plasmaaustausch/Plasmainfusion und LDH und Serum-Kreatinin oberhalb der oberen Grenze des Normalbereichs) eingeschlossen wurden. Bei Studie C08-003A/B handelte es sich um eine prospektive, kontrollierte, offene Studie, in die Patienten mit länger bestehendem aHUS ohne offensichtliche Hinweise auf eine klinisch manifestierte thrombotische Mikroangiopathie eingeschlossen wurden. Diese Patienten hatten über längere Zeit Plasmaaustausch/Plasmainfusionen (PA/PI) erhalten (≥ 1 PA/PI-Sitzung alle zwei Wochen und nicht mehr als 3 PA/PI-Sitzungen/Woche über mindestens 8 Wochen vor der ersten Dosis). In beiden prospektiven Studien wurden die Patienten über 26 Wochen mit Soliris behandelt. Die meisten dieser Patienten wurden danach in eine offene Verlängerungsstudie aufgenommen. Alle Patienten, die in die beiden prospektiven Studien aufgenommen wurden, hatten einen ADAMTS-13-Wert über 5 %.
Vor Therapieeinleitung mit Soliris wurden die Patienten gegen Meningokokken geimpft oder erhielten eine geeignete Antibiotikaprophylaxe bis 2 Wochen nach Impfung. In allen Studien betrug die Soliris-Dosis bei Erwachsenen und Jugendlichen mit aHUS 900 mg alle 7 ± 2 Tage über 4 Wochen, gefolgt von 1200 mg 7 ± 2 Tage später, dann 1200 mg alle 14 ± 2 Tage über die gesamte Studiendauer. Soliris wurde als intravenöse Infusion über 35 Minuten verabreicht. Das Dosierungsschema bei Kindern und Jugendlichen mit einem Körpergewicht unter 40 kg wurde mithilfe einer pharmakokinetischen Simulation festgelegt, mit der die empfohlene Dosis und das Dosierungsschema auf Basis des Körpergewichts ermittelt wurde (siehe «Dosierung / Anwendung»).
Zu den primären Endpunkten gehörte in Studie C08-002A/B die Änderung der Thrombozytenzahl gegenüber dem Ausgangswert und in Studie C08-003A/B die Abwesenheit von Ereignissen einer thrombotischen Mikroangiopathie (TMA). Weitere Endpunkte waren TMA-Interventionsrate, Normalisierung hämatologischer Parameter, vollständiges Ansprechen der TMA, Änderungen der LDH, Nierenfunktion und gesundheitsbezogenen Lebensqualität. Die Abwesenheit von TMA-Ereignissen war definiert als Abwesenheit folgender Ereignisse über mindestens 12 Wochen: Abnahme der Thrombozytenzahl von > 25% gegenüber dem Ausgangswert, Plasmaaustausch/Plasmainfusion und neu eingeleitete Dialyse. TMA-Interventionen waren definiert als Plasmaaustausch/Plasmainfusion oder neu eingeleitete Dialyse. Normalisierung hämatologischer Parameter war definiert als Normalisierung der Thrombozytenzahl und der LDH-Spiegel während ≥ 2 aufeinanderfolgender Messungen während ≥ 4 Wochen. Vollständiges Ansprechen der TMA war definiert als Normalisierung hämatologischer Parameter und anhaltende Abnahme des Serumkreatinins um ≥ 25% in ≥ 2 aufeinanderfolgenden Messungen über ≥ 4 Wochen.
Die Ausgangsparameter für die jeweiligen Studien sind in Tabelle 5 dargestellt.
Tabelle 5: Demografische Patientendaten und -parameter in den Studien C08-002A/B und C08-003A/B
|
Parameter
|
C08-002A/B
|
C08-003A/B
| |
Soliris
N = 17
|
Soliris
N = 20
| |
Zeit von der Erstdiagnose bis zum Screening, Median in Monaten (min/max)
|
10 (0,26/236)
|
48 (0,66/286)
| |
Zeit von der Manifestierung der bestehenden TMA bis zum Screening, Median in Monaten (min/max)
|
< 1 (< 1/4)
|
9 (1/45)
| |
Anzahl der Sitzungen für Plasmaaustausch/Plasmainfusion zur Behandlung einer bestehenden TMA, Median (min/max)
|
17 (2/37)
|
62 (20/230)
| |
Anzahl der Sitzungen für Plasmaaustausch/Plasmainfusion innerhalb 7 Tagen vor der ersten Eculizumab-Verabreichung, Median (min/max)
|
6 (0/7)
|
2 (1/3)
| |
Thrombozytenzahl, Ausgangswert (× 109/l), Mittelwert (SD)
|
109 (32)
|
228 (78)
| |
LDH Ausgangswert (U/l),
Mittelwert (SD)
|
323 (138)
|
223 (70)
| |
Patienten ohne identifizierte Mutation,
n (%)
|
4 (24)
|
6 (30)
|
Patienten in der aHUS-Studie C08-002A/B wurden mindestens 26 Wochen mit Soliris behandelt. Nach Abschluss der initialen 26-wöchigen Behandlungsphase führten die meisten Patienten die Behandlung im Rahmen einer Verlängerungsstudie fort. In der aHUS-Studie C08-002A/B betrug die mediane Behandlungsdauer mit Soliris 100 Wochen (Spanne: 2 - 145 Wochen).
Nach Beginn der Soliris-Behandlung waren eine Abnahme der terminalen Komplementaktivität sowie ein Anstieg der Thrombozytenzahlen im Vergleich zu den Ausgangswerten zu beobachten. Die Abnahme der terminalen Komplementaktivität war bei allen Patienten nach Beginn der Soliris-Behandlung zu beobachten. Daten zur Wirksamkeit von Soliris in Studie C08-002A/B sind in Tabelle 6 zusammengefasst. Sämtliche Wirksamkeitsparameter haben sich während der 2 Behandlungsjahre verbessert oder erhalten. Das vollständige Ansprechen der TMA blieb bei allen Respondern aufrechterhalten. Von den Patienten, bei denen die Behandlung länger als 26 Wochen durchgeführt wurde, haben 2 weitere ein vollständiges Ansprechen der TMA erreicht und aufrechterhalten, was mit der Normalisierung des LDH-Werts (1 Patient) und der Senkung des Serum-Kreatinins (2 Patienten) zusammenhängt.
Unter Behandlung mit Soliris verbesserte und hielt sich die Nierenfunktion, gemessen als geschätzte glomeruläre Filtrationsrate (eGFR). Vier der 5 Patienten, die zu Studienbeginn dialysepflichtig waren, konnten die Dialyse für die Dauer der Soliris-Behandlung unterbrechen. Ein Patient benötigte eine neue Dialyse. Die Patienten berichteten eine Verbesserung ihrer gesundheitsbezogenen Lebensqualität (QoL).
In der aHUS-Studie C08-002A/B zeigten Patienten mit und ohne identifizierte Mutationen in Genen, die Proteine für Komplement-regulierende Faktoren kodieren, ein vergleichbares Ansprechen auf Soliris.
Patienten in der aHUS-Studie C08-003A/B wurden mindestens 26 Wochen mit Soliris behandelt. Nach Abschluss der initialen 26-wöchigen Behandlungsphase führten die meisten Patienten die Behandlung im Rahmen einer Verlängerungsstudie fort. In der aHUS-Studie C08-003A/B betrug die mediane Behandlungsdauer mit Soliris 114 Wochen (Spanne: 26-129 Wochen). Daten zur Wirksamkeit von Soliris in Studie C08-003A/B sind in Tabelle 6 zusammengefasst.
In der aHUS-Studie C08-003A/B zeigten Patienten mit und ohne identifizierte Mutationen in Genen, die Proteine für Komplement-regulierende Faktoren kodieren, ein vergleichbares Ansprechen auf Soliris. Bei allen Patienten wurde nach Beginn der Soliris Behandlung eine Abnahme der terminalen Komplementaktivität beobachtet. Sämtliche Wirksamkeitsparameter verbesserten oder hielten sich während der 2 Behandlungsjahre. Ein vollständiges Ansprechen der TMA blieb bei allen Respondern erhalten. Von den Patienten, bei denen die Behandlung länger als 26 Wochen durchgeführt wurde, erreichten 6 weitere ein vollständiges Ansprechen der TMA und konnten es erhalten, was mit der Senkung des Serum-Kreatinins zusammenhängt. Kein Patient benötigte eine neue Dialyse. Die Nierenfunktion, gemessen als geschätzte glomeruläre Filtrationsrate (eGFR), verbesserte sich unter Soliris-Behandlung.
Tabelle 6: Ergebnisse zur Wirksamkeit in den prospektiven aHUS-Studien C08-002A/B und C08-003A/B
|
|
C08-002A/B
N = 17
|
C08-003A/B
N = 20
| |
|
Nach 26 Wochen
|
Nach 2 Jahren1
|
Nach 26 Wochen
|
Nach 2 Jahren1
| |
Normalisierung der Thrombozytenzahl
Alle Patienten, n (%) (95 % KI)
Patienten mit abnormem Ausgangswert, n/n (%)
|
14 (82)
(57-96)
13/15 (87)
|
15 (88)
(64-99)
13/15 (87)
|
18 (90)
(68-99)
3/20 (15)
|
18 (90)
(68-99)
1/3 (33)
| |
Abwesenheit von TMA-Ereignissen,
n (%) (95 % KI)
|
15 (88)
(64-99)
|
15 (88)
(64-99)
|
16 (80)
(56-94)
|
19 (95)
(75-99)
| |
TMA Interventionsrate, Median pro Tag (min/max)
- prä-Eculizumab
- unter Eculizumab
p-Wert
|
0,88
(0,04/1,59)
0 (0/0,31)
p < 0,0001
|
0,88
(0,04/1,59)
0 (0/0,31)
p <0,0001
|
0,23
(0,05/1,09)
0
p < 0,0001
|
0,23
(0,05/1,09)
0
P<0,0001
| |
CKD Verbesserung um ≥ 1 Stadium
n (%) (95 % KI)
|
10 (59)
(33-82)
|
12 (71)
(44-90)
|
7 (35)
(15-59)
|
12 (60)
(36-81)
| |
eGFR Veränderung ml/min/1,73 m2: Median (Spanne)
|
20 (-1;98)
|
28 (3;82)
|
5 (-1;20)
|
11(-42;30)
| |
eGFR Veränderung ≥ 15 ml/min/1,73m2; n (%) (95 % KI)
|
8 (47)
(23-72)
|
10 (59)
(33-82)
|
1 (5)
(0-25)
|
8 (40)
(19-64)
| |
Veränderung des Hb > 20g/l,
n (%) (95 % KI)
|
11 (65)
(38-86)2
|
13 (76)
(50-93)
|
9 (45)
(23-68)3
|
13 (65)
(41-85)
| |
Normalisierung der hämatologischen Parameter, n (%) (95 % KI)
|
13 (76)
(50-93)
|
15 (88)
(64-99)
|
18 (90)
(68-99)
|
18 (90)
(68-99)
| |
Vollständiges Ansprechen der TMA,
n (%) (95 % KI)
|
11 (65)
(38-86)
|
13 (76)
(50-93)
|
5 (25)
(9-49)
|
11 (55)
(32-77)
|
1 bei cut-off (20.April 2012)
2 Studie C008-002: 3 Patienten erhielten Erythropoiese stimulierende Substanzen, die nach Beginn der Soliris-Behandlung abgesetzt wurden.
3 Studie C008-003: 8 Patienten erhielten Erythropoiese stimulierende Substanzen, die bei 3 Patienten während der Soliris-Behandlung abgesetzt wurden.
Die Studie C10-004 untersuchte 41 Patienten mit Anzeichen von thrombotischer Mikroangiopathie (TMA). Einschlusskriterien waren: Thrombozytenzahl niedriger als die untere Grenze des Normalbereichs, Anzeichen für eine Hämolyse wie erhöhter Serum-LDH-Spiegel und Serum-Kreatinin-Spiegel oberhalb der oberen Grenze des Normalbereichs, ohne chronischen Dialysebedarf. Das mediane Alter der Patienten betrug 35 Jahre (zwischen 18 und 80 Jahre). Alle in die Studie C10-004 aufgenommenen Patienten hatten einen ADAMTS-13-Wert über 5 %. 51 % der Patienten hatten eine festgestellte Mutation eines Komplement-regulierenden Faktors oder Autoantikörper. Insgesamt erhielten 35 Patienten eine Plasmainfusion oder einen Plasmaaustausch, oder eine Soliris-Gabe vor Beginn der Behandlung mit Eculizumab. Tabelle 7 fasst die klinischen Eigenschaften und die Eigenschaften, die in Zusammenhang mit der Erkrankung der Patienten bei Beginn der Studie C10-004 stehen, zusammen.
Tabelle 7: Eigenschaften der Patienten bei Beginn der klinischen Studie zu aHUS C10-004
|
Parameter
|
Studie aHUS C10-004
n = 41
| |
Zeit von der Erstdiagnose der aHUS bis zur ersten Studienmedikation (Monat), Median (min, max)
|
0,79 (0,03 – 311)
| |
Zeit von der Manifestierung der bestehenden TMA und erster verabreichter Dosis innerhalb der Studie (Monat), Median (min, max)
|
0,52 (0,03 – 19)
| |
Thrombozytenzahl bei Screening (× 109/l), Median (min, max)
|
125 (16 – 332)
| |
LDH Ausgangswert (U/l), Median (min, max)
|
375 (131 – 3318)
| |
eGFR, Ausgangswert (ml/min/1- 73m3)
Median (min; max)
|
10 (6; 53)
|
Die Patienten der Studie C10-004 wurden mindestens 26 Wochen mit Soliris behandelt. Nach Abschluss der initialen 26-wöchigen Behandlungsphase führten die meisten Patienten die Behandlung dauerhaft fort. Bei cut-off betrug die mediane Behandlungsdauer mit Soliris ungefähr 50 Wochen (Spanne: 13-86 Wochen).
Nach dem Beginn der Behandlung mit Soliris konnten eine Abnahme der terminalen Komplementaktivität und ein Anstieg der Thrombozytenzahl gegenüber dem Studieneinschluss beobachtet werden. Soliris reduzierte die Anzeichen einer Komplement-vermittelten TMA, was der Anstieg der medianen Thrombozytenzahl zwischen Studieneinschluss und Woche 26 zeigt. In der Studie stieg die mediane Thrombozytenzahl von 119 ± 66 x109/l bei Studieneinschluss auf 200 ± 84 x109/l nach Woche 1; diese Entwicklung konnte 26 Wochen lang aufrechterhalten werden (mediane Thrombozytenzahl in Woche 26: 252 ± 70 x109/l). Die Nierenfunktion, die anhand der medianen eGFR bewertet wurde, konnte unter Soliris verbessert werden. 20 der 24 Patienten, die vor Studienbeginn auf eine Dialyse angewiesen waren, konnten während der Dauer der Solirisbehandlung die Dialysebehandlung unterbrechen. Tabelle 8 fasst die Ergebnisse aus Studie C10-004 hinsichtlich der Wirksamkeit zusammen.
Tabelle 8: Ergebnisse zur Wirksamkeit in der prospektiven aHUS Studie C10-004
|
Wirksamkeitsparameter
|
Studie aHUS C10-004
(n = 41)
Bis 26 Wochen
| |
Veränderung der Thrombozytenzahl zwischen Screening und Woche 26 (109/l)
|
111 (-122; 362)
| |
Normalisierung der Blutwerte, n (%)
Dauer bis zur Normalisierung der Blutwerte, Median in Wochen (min, max)
|
36 (88)
46 (10; 74)
| |
Vollständiges Ansprechen der TMA, n (%)
Dauer bis zum vollständigen Ansprechen der TMA, Median in Wochen (min, max)
|
23 (56)
42 (6; 74)
| |
Fehlende Anzeichen von TMA, n (%)
KI 95 %
|
37 (90)
77; 97
| |
TMA Interventionsrate, Median pro Tag (min, max):
prä-Eculizumab
unter Eculizumab
|
0,63 (0; 1,38)
0 (0; 0,58)
|
1 Bei cut-off (4. September 2012), bei medianer Dauer der Soliris-Therapie von 50 Wochen (Spanne: 13 bis 86 Wochen)
Eine Langzeitbehandlung mit Soliris (Median 52 Wochen, im Bereich von 15 bis 126 Wochen) war mit einem erhöhten Anteil von klinisch bedeutsamen Verbesserungen bei erwachsenen aHUS-Patienten verbunden. Als die Soliris-Therapie länger als 26 Wochen beibehalten wurde, erreichten 3 weitere Patienten (63 % der Patienten insgesamt) ein vollständiges MAT-Ansprechen. Weitere 4 Patienten (98 % der Patienten insgesamt) erreichten eine Normalisierung der hämatologischen Parameter. Bei der letzten Auswertung erreichten 25 der 41 Patienten (61 %) eine Verbesserung der eGFR ≥ 15 ml/min / 1,73 m2 im Vergleich zum Ausgangswert.
Refraktäre generalisierte Myasthenia gravis
Die Wirksamkeit von Soliris bei der Behandlung von Patienten mit refraktärer gMG wurde aus den Daten von 139 Patienten in zwei prospektiven kontrollierten klinischen Studien (C08-001 und ECU-MG-301) und einer offenen Verlängerungsstudie (ECU-MG-302) ermittelt.
Bei der Studie ECU-MG-301 (REGAIN) handelte es sich um eine 26-wöchige doppelblinde, randomisierte, placebo-kontrollierte, multizentrische Phase-3-Studie mit Soliris bei Patienten, die auf vorangegangene Therapien nicht angesprochen hatten und weiterhin symptomatisch waren. Einhundertachtzehn (118) der 125 (94 %) Patienten schlossen die 26-wöchige Behandlungsphase ab und 117 (94 %) Patienten wurden anschliessend in die Studie ECU-MG-302 aufgenommen, eine offene, multizentrische Verlängerungsstudie zur Untersuchung der langfristigen Wirksamkeit und Sicherheit, bei der alle Patienten eine Behandlung mit Soliris erhielten.
In der Studie ECU-MG-301 wurden gMG-Patienten mit einem positiven serologischen Test auf Anti-AChR-Antikörper, mit klinischer MGFA-Klassifizierung (Myasthenia Gravis Foundation of America) der Klassen II bis IV und einem MG-ADL-Gesamtscore von mindestens 6 randomisiert entweder Soliris (n = 62) oder Placebo (n = 63) zugeordnet. Alle in die Studie eingeschlossenen Patienten hatten refraktäre gMG und erfüllten die folgenden im Vorfeld festgelegten Kriterien:
1) Seit mindestens einem Jahr erfolglose Behandlung mit 2 oder mehr immunsuppressiven Therapien (entweder in Kombination oder als Monotherapie), d.h. Patienten, deren Alltagsaktivitäten trotz immunsuppressiver Therapien weiterhin eingeschränkt waren
ODER
2) Erfolglose Behandlung mit mindestens einer immunsuppressiven Therapie und Notwendigkeit eines dauerhaften Plasmaaustauschs oder von intravenösen Immunglobulinen (IVIg) zur Kontrolle der Symptome, d.h. die Patienten benötigten mindestens alle 3 Monate über die letzten 12 Monate regelmässig Plasmaaustausch oder IVIg zur Behandlung von Muskelschwäche.
Vor Beginn der Behandlung mit Soliris wurden die Patienten gegen Meningokokken geimpft oder erhielten eine geeignete Antibiotikaprophylaxe bis 2 Wochen nach der Impfung. In den Studien ECU-MG-301 und ECU-MG-302 betrug die Soliris-Dosis bei erwachsenen Patienten mit refraktärer gMG 900 mg alle 7 ± 2 Tage über 4 Wochen, gefolgt von 1.200 mg in Woche 5 ± 2 Tage und anschliessend 1.200 mg alle 14 ± 2 Tage für die Dauer der Studie. Soliris wurde als intravenöse Infusion über 35 Minuten verabreicht. Die Tabelle 9 zeigt die Ausgangsparameter der in die Studie ECU-MG-301 aufgenommenen Patienten mit refraktärer gMG.
Tabelle 9: Demografische Patientendaten und -parameter in der Studie ECU-MG-301
|
|
Soliris (n=62)
|
Placebo (n=63)
| |
Alter zum Zeitpunkt der MG-Diagnose (in Jahren),
Mittelwert (Min, Max)
|
38,0 (5,9; 70,8)
|
38,1 (7,7; 78,0)
| |
Weiblich, n (%)
|
41 (66,1)
|
41 (65,1)
| |
Dauer der MG (Jahre),
Mittelwert (Min, Max)
|
9,9 (1,3; 29,7)
|
9,2 (1,0; 33,8)
| |
Ausgangswert MG-ADL-Score
|
|
| |
Mittelwert (SD)
|
10,5 (3,06)
|
9,9 (2,58)
| |
Median
|
10,0
|
9,0
| |
Ausgangswert QMG-Score
|
|
| |
Mittelwert (SD)
|
17,3 (5,10)
|
16,9 (5,56)
| |
Median
|
17,0
|
16,0
| |
≥3 vorangegangene immunsuppressive Therapien* seit der Diagnose, n (%)
|
31 (50,0)
|
34 (54,0)
| |
Anzahl Patienten mit vorangegangenen Exazerbationen seit der Diagnose, n (%)
|
46 (74,2)
|
52 (82,5)
| |
Anzahl Patienten mit vorangegangener MG-Krise seit der Diagnose, n (%)
|
13 (21,0)
|
10 (15,9)
| |
Vorangegangene künstliche Beatmung seit der Diagnose, n (%)
|
15 (24,2)
|
14 (22,2)
| |
Vorangegangene Intubation seit der Diagnose (MGFA-Klasse V), n (%)
|
11 (17,7)
|
9 (14,3)
|
* Immunsuppressiva waren unter anderem Kortikosteroide, Azathioprin, Mycophenolat, Methotrexat, Ciclosporin, Tacrolimus und Cyclophosphamid.
Der primäre Endpunkt der Studie ECU-MG-301 war die Veränderung des MG-ADL-Gesamtscores (ADL, Activities of Daily Living Profile – eine vom Patienten gemeldete Outcome-Variable, die bei gMG bewertet wird) in Woche 26 gegenüber dem Ausgangswert. Die primäre Analyse des MG-ADL war eine Worst-Rank-ANCOVA mit einem durchschnittlichen Rang von 56,6 für Soliris und 68,3 für Placebo, basierend auf 125 Patienten in der Studie (p = 0,0698).
Der wichtigste sekundäre Endpunkt war die Veränderung des Gesamtscores des Quantitative MG Scoring System (QMG – eine vom Arzt gemeldete Outcome-Variable, die bei gMG bewertet wird) in Woche 26 gegenüber dem Ausgangswert. Die primäre Analyse des QMG war eine Worst-Rank-ANCOVA mit einem durchschnittlichen Rang von 54,7 für Soliris und 70,7 für Placebo, basierend auf 125 Patienten der Studie (p = 0,0129).
Die Ergebnisse zur Wirksamkeit für die zuvor festgelegten Analysen der primären und sekundären Endpunkte mit wiederholten Messungen sind in Tabelle 10 aufgeführt.
Tabelle 10: ECU-MG-301 Ergebnisse zur Wirksamkeit: Veränderung in Woche 26 gegenüber dem Ausgangswert
|
Wirksamkeitsendpunkte: Veränderung des Gesamtscores in Woche 26 gegenüber dem Ausgangswert
|
Soliris
(n=62)
(SEM)
|
Placebo
(n=63)
(SEM)
|
Veränderung unter Soliris gegenüber Placebo – Differenz der Kleinste-Quadrate-Mittelwerte (95% KI)
|
p-Wert (mittels Analyse mit wiederholten Messungen)
| |
MG-ADL
|
-4,2 (0,49)
|
-2,3 (0,48)
|
-1,9
(-3,3; -0,6)
|
0,0058
| |
QMG
|
-4,6 (0,60)
|
-1,6 (0,59)
|
-3,0
(-4,6; -1,3)
|
0,0006
| |
MGC
|
-8,1 (0,96)
|
-4,8 (0,94)
|
-3,4
(-6,0; -0,7)
|
0,0134
| |
MG-QoL-15
|
-12,6 (1,52)
|
-5,4 (1,49)
|
-7,2
(-11,5; -3,0)
|
0,0010
|
SEM = Standardfehler des Mittelwertes; KI = Konfidenzintervall; MGC = Myasthenia Gravis Composite; MG-QoL15 = Myasthenia Gravis Quality of Life 15
In der Studie ECU-MG-301 wurde für einen klinischen Responder beim MG-ADL-Gesamtscore eine Verbesserung um mindestens 3 Punkte definiert. Der Anteil der klinischen Responder, die bis einschliesslich Woche 26 keine Notfallbehandlung erhielten, betrug unter Soliris 59,7 %, verglichen mit 39,7 % unter Placebo (p = 0,0229). In Studie ECU-MG-301 wurde für einen klinischen Responder beim QMG-Gesamtscore eine Verbesserung um mindestens 5 Punkte definiert. Der Anteil der klinischen Responder, die in Woche 26 keine Notfallbehandlung erhielten, betrug bei Soliris 45,2 % verglichen mit 19 % unter Placebo (p = 0,0018).
Tabelle 11 zeigt eine Übersicht der Patienten, die in den 26 Wochen über eine klinische Verschlechterung berichteten, und der Patienten, die in diesem Zeitraum eine Notfallbehandlung benötigten.
Tabelle 11: Klinische Verschlechterung und Notfallbehandlung in ECU-MG-301
|
Variable
|
Statistik
|
Placebo
(N=63)
|
Soliris
(N=62)
| |
Gesamtzahl Patienten, die über eine klinische Verschlechterung berichteten
|
n (%)
|
15 (23,8)
|
6 (9,7)
| |
Gesamtzahl Patienten, die eine Notfallbehandlung benötigten
|
n (%)
|
12 (19,0)
|
6 (9,7)
|
Von den 125 Patienten der Studie ECU-MG-301 wurden 117 Patienten anschliessend in die Langzeit-Verlängerungsstudie (Studie ECU-MG-302) aufgenommen, in der alle Patienten Soliris erhielten. Patienten, die zuvor in der Studie ECU-MG-301 mit Soliris behandelt worden waren, zeigten bei allen Zielgrössen (MG-ADL, QMG, MGC und MG-QoL15) über einen zusätzlichen Eculizumab-Behandlungszeitraum von 130 Wochen weiterhin eine nachhaltige Wirkung von Soliris. Bei Patienten, die in der Studie ECU-MG-301 (Placebo/Eculizumab-Arm der Studie ECU-MG-302) Placebo erhielten, trat nach Beginn der Behandlung mit Eculizumab eine Verbesserung ein, die in der Studie ECU-MG-302 über mehr als 130 Wochen aufrechterhalten wurde. Abbildung 1 zeigt die Veränderung gegenüber dem Ausgangswert bei MG-ADL (A) und QMG (B) nach 26wöchiger Behandlung in Studie ECU-MG-301 und nach 130-wöchiger Behandlung (n = 80 Patienten) in Studie ECU-MG-302.

Abbildung 1: Durchschnittliche Veränderungen gegenüber dem Ausgangswert bei MG-ADL (1A) und QMG (1B) in den Studien ECU-MG-301 und ECU-MG-302
In der Studie ECU-MG-302 hatten Ärzte die Möglichkeit, bestehende Hintergrundtherapien mit Immunsuppressiva anzupassen. In diesem Rahmen verringerten 65,0% der Patienten ihre tägliche Dosis von mindestens 1 immunsuppressiven Therapie (IST); 43,6% der Patienten brachen eine bestehende IST ab. Der häufigste Grund für eine Änderung der IST-Therapie war die Verbesserung der MG-Symptome.
Neuromyelitis-optica-Spektrumerkrankungen
Daten von 143 Patienten in einer kontrollierten Studie (ECU-NMO-301) und von 119 Patienten, die in einer offenen Verlängerungsstudie (Studie ECU-NMO-302) weiterbehandelt wurden, wurden verwendet, um die Wirksamkeit und Sicherheit von Soliris bei der Behandlung von Patienten mit NMOSD zu beurteilen.
Studie ECU-NMO-301 war eine doppelblinde, randomisierte, placebokontrollierte, multizentrische Phase-III-Studie mit Soliris bei Patienten mit NMOSD.
In Studie ECU-NMO-301 wurden NMOSD-Patienten mit positivem Serumtest auf Anti-AQP4-Antikörper, einer Anamnese mit mindestens 2 Schüben in den letzten 12 Monaten oder 3 Schüben in den letzten 24 Monaten und mindestens 1 Schub in den 12 Monaten vor dem Screening sowie einem Expanded Disability Status Scale (EDSS) Score von ≤ 7 im Verhältnis 2:1 auf Soliris (n = 96) oder Placebo (n = 47) randomisiert.
Den Patienten war es gestattet, während der Studie eine Hintergrundbehandlung mit Immunsuppressiva in stabiler Dosis, mit Ausnahme von Rituximab und Mitoxantron anzuwenden.
Die Patienten erhielten entweder mindestens 2 Wochen vor Beginn der Behandlung mit Soliris eine Meningokokken-Impfung oder bis 2 Wochen nach der Impfung eine prophylaktische Behandlung mit geeigneten Antibiotika. In dem klinischen Entwicklungsprogramm für Eculizumab bei NMOSD betrug die Soliris-Dosis bei erwachsenen NMOSD-Patienten 900 mg alle 7 ± 2 Tage über 4 Wochen, gefolgt von 1200 mg in Woche 5 ± 2 Tage und anschliessend 1200 mg alle 14 ± 2 Tage für die Dauer der Studie. Soliris wurde als intravenöse Infusion über 35 Minuten gegeben.
Die Mehrheit (90,9 %) der Patienten war weiblich. Ungefähr die Hälfte der Patienten (49,0 %) war weiss. Das Durchschnittsalter bei der ersten Gabe des Studienmedikaments betrug 45 Jahre.
Tabelle 12: Krankheitsanamnese und Ausgangsparameter der Patienten in Studie ECU-NMO-301
|
Variable
|
Statistik
|
Placebo
(N = 47)
|
Eculizumab
(N = 96)
|
Insgesamt
(N = 143)
| |
NMOSD-Anamnese
| |
Alter bei der ersten klinischen Manifestation der NMOSD (Jahre)
|
Mittelwert (SD)
|
38,5 (14,98)
|
35,8 (14,03)
|
36,6 (14,35)
| |
Median
|
38,0
|
35,5
|
36,0
| |
Min, Max
|
12; 73
|
5; 66
|
5; 73
| |
Zeitraum von der ersten klinischen Manifestation der NMOSD bis zur Anwendung der ersten Dosis des Studienmedikaments (Jahre)
|
Mittelwert (SD)
|
6,601 (6,5863)
|
8,156 (8,5792)
|
7,645 (7,9894)
| |
Median
|
3,760
|
5,030
|
4,800
| |
Min, Max
|
0,51; 29,10
|
0.41, 44.85
|
0,41; 44,85
| |
Anamnestische annualisierte Schubrate innerhalb von 24 Monaten vor dem Screening
|
Mittelwert (SD)
|
2,07 (1,037)
|
1,94 (0,896)
|
1,99 (0,.943)
| |
Median
|
1,92
|
1,85
|
1,92
| |
Min, Max
|
1,0; 6,4
|
1.0, 5.7
|
1,0; 6,4
| |
Ausgangsparameter
| |
EDSS-Ausgangsscore
|
Mittelwert (SD)
|
4,26 (1,510)
|
4,15 (1,646)
|
4,18 (1,598)
| |
Median
|
4,00
|
4,00
|
4,00
| |
Min, Max
|
1,0; 6,5
|
1,0; 7,0
|
1,0; 7,0
| |
Keine IST-Anwendung zu Studienbeginn
|
n (%)
|
13 (27,7)
|
21 (21,9)
|
34 (23,8)
|
Abkürzungen: EDSS = Expanded Disability Status Scale; IST = Immunsupressiva -Therapie; Max = Maximum; Min = Minimum; NMOSD = Neuromyelitis-optica-Spektrum-Erkrankungen; SD = Standardabweichung (standard deviation).
Der primäre Endpunkt von Studie ECU-NMO-301 war die Dauer bis zum ersten Schub während der Studie gemäss der Bestätigung durch ein unabhängiges und für die Behandlung verblindetes Komitee. Während der Studie wurde ein signifikanter Effekt von Eculizumab im Vergleich zu Placebo bezüglich der Dauer bis zum Auftreten des ersten bestätigten Schubes beobachtet (relative Risikoreduktion 94 %; Hazard Ratio 0,058; p<0,0001) (Abbildung 2). Die mit Soliris behandelten Patienten zeigten eine ähnliche Verbesserung der Dauer bis zum ersten bestätigten Schub während der Studie mit oder ohne begleitende IST-Therapie.
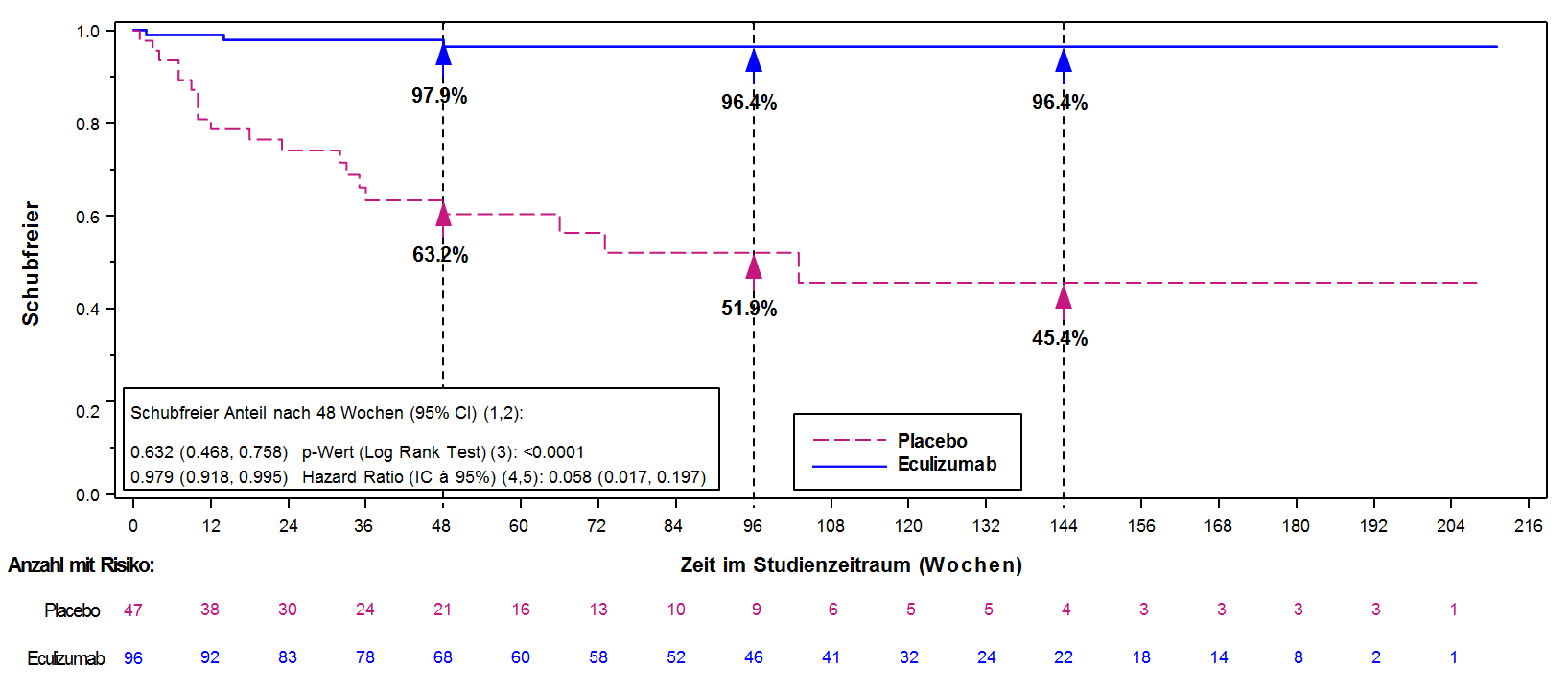
Abbildung 2: Kaplan-Meier-Kurve für die Zeit bis zum ersten bestätigten Schub während der Studie ECU-NMO-301 – vollständiges Analyseset
Hinweis: Patienten, bei denen kein bestätigter Schub während der Studie auftrat, wurden am Ende des Studienzeitraums zensiert.
Stratifizierte Analysen basieren auf vier Randomisierungsstraten:
(i) niedriger EDSS bei Randomisierung (<= 2,0), (ii) hoher EDSS (> = 2,5 bis <= 7) und nicht vorbehandelt bei Randomisierung, (iii) hoher EDSS (> = 2,5 bis <= 7) und Fortsetzung derselben IST(s) seit dem letzten Schub bei der Randomisierung, (iv) hoher EDSS (> = 2,5 bis <= 7) und Änderungen bei den IST(s) seit dem letzten Schub bei der Randomisierung.
1 Basierend auf der Kaplan-Meier-Methode.
2 Basierend auf der komplementären Log-Log-Transformation.
3 Basierend auf einem stratifizierten Log-Rank-Test.
4 Basierend auf einem stratifizierten Cox-Proportional-Hazards-Modell.
5 Wald-Konfidenzintervall.
Abkürzungen: KI = Konfidenzintervall (confidence interval); EDSS = Expanded Disability Status Scale; IST = Immunsuppressiva-Therapie
Das Verhältnis der bestätigten jährlichen Schubrate (annualized relapse rate, ARR) während der Studie (95% KI) für Eculizumab im Vergleich zu Placebo betrug 0,045 (0,013; 0,151). Dies entspricht einer relativen Reduktion der bestätigten ARR während der Studie von 95,5% bei Patienten, die mit Eculizumab behandelt wurden, im Vergleich zu Placebo (p <0,0001) (Tabelle 13).
Tabelle 13: Bestätigte annualisierte Schubrate während der Studie – vollständiges Analyseset
|
Variable
|
Statistik
|
Placebo
(N = 47)
|
Eculizumab
(N = 96)
| |
Gesamtzahl der Schübe
|
Summe
|
21
|
3
| |
Gesamtzahl der Patientenjahre im Studienzeitraum
|
n
|
52,41
|
171,32
| |
Adjustierte bestätigte ARRa
|
Rate
|
0,350
|
0,016
| |
95% KI
|
0,199; 0,616
|
0,005; 0,050
| |
Behandlungseffekta
|
Rate Ratio (Eculizumab/Placebo)
|
…
|
0,045
| |
95% KI
|
…
|
0,013; 0,151
| |
p-Wert
|
…
|
<0,0001
| |
a
Basierend auf einer Poisson-Regression, angepasst um Randomisierungsstraten und die historische ARR in 24 Monaten vor dem Screening.
Abkürzungen: ARR = annualisierte Schubrate; KI = Konfidenzintervall.
|
Verglichen mit Patienten, die Placebo erhielten, wiesen die mit Soliris behandelten Patienten niedrigere annualisierte Raten auf für stationäre Behandlungen (0,04 für Soliris versus 0,31 für Placebo), intravenöse Kortikosteroid-Anwendungen zur Behandlung von akuten Schüben (0,07 für Soliris versus 0,42 für Placebo) und Plasmaaustausch-Behandlungen (0,02 für Soliris versus 0,19 für Placebo).
Die Messung der Veränderungen von Studienbeginn bis Studienende bei den sekundären Endpunkten neurologische Behinderungen (EDSS-Score [p-Wert = 0,0597] und mRS [nominaler p-Wert = 0,0154]), Funktionseinschränkung (HAI [nominaler p-Wert = 0,0002]) und Lebensqualität (EQ-5D VAS [nominaler p-Wert = 0,0309] und EQ-5D Index [nominaler p-Wert = 0,0077]) fiel zu Gunsten von Eculizumab gegenüber Placebo aus.
Die finale Analyse der Studie ECU-NMO-302 (mediane Behandlungsdauer 20.00 Wochen [0.1;198.4 Wochen]) zeigte eine statistisch signifikante und klinisch bedeutsame Abnahme der ARR während der Studie (gemäss Entscheidung des behandelnden Arztes) unter der Eculizumab-Behandlung, basierend auf der medianen (Min, Max) Veränderung (-1,825 [-6,38; 1,02], p <0,0001) gegenüber der anamnestischen ARR (24 Monate vor dem Screening in der Studie ECU-NMO-301). Insgesamt liegen für die Behandlung mit Soliris über 20 Wochen nur limitierte Daten zu Wirksamkeit und Sicherheit vor.
In der Studie ECU-NMO-302 hatten Ärzte die Möglichkeit, bestehende Hintergrundtherapien mit Immunsuppressiva anzupassen. In diesem Rahmen war die häufigste Änderung der immunsuppressiven Therapie eine Verringerung der Immunsuppressiva-Dosis, die bei 21,0 % der Patienten erfolgte. Zudem brachen 15,1 % der Patienten eine bestehende IST-Therapie ab.
Soliris (Eculizumab) wurde nicht im Hinblick auf die Behandlung von akuten Schüben bei NMOSD-Patienten untersucht.
Sicherheit und Wirksamkeit bei älteren Patienten
Refraktäre gMG
Zweiundzwanzig (22) (17,6 %) der älteren Patienten mit refraktärer gMG (> 65 Jahre) wurden im Rahmen der klinischen Studien mit Soliris behandelt. Dabei wurden keine wesentlichen Unterschiede in Bezug auf Sicherheit und Wirksamkeit in Zusammenhang mit dem Lebensalter festgestellt.
Neuromyelitis-optica-Spektrumerkrankungen
Soliris wurde nur in insgesamt 9 älteren Patienten mit NMOSD (3 unter Placebo, 6 unter Eculizumab untersucht, so dass keine verlässlichen Aussagen über Sicherheit und Wirksamkeit in dieser Population getroffen werden können.
Sicherheit und Wirksamkeit bei pädiatrischen Patienten
Paroxysmale Nächtliche Hämoglobinurie
In der Studie M07-005 wurden insgesamt 7 pädiatrische Patienten mit PNH mit einem medianen Körpergewicht von 57,2 kg (Spanne 48,6 bis 69,8 kg) und im Alter von 11 bis 17 Jahren (medianes Alter: 15,6 Jahre) mit Soliris behandelt.
Die Behandlung mit Eculizumab entsprechend dem Dosierungsschema für Kinder und Jugendliche führte zu einer Verminderung der intravaskulären Hämolyse, gemessen anhand des Serum-LDH Spiegels. Die Behandlung führte auch zu einer deutlichen Reduktion oder Elimination des Bedarfes an Bluttransfusionen und tendenziell zu einer generellen Verbesserung des Allgemeinzustandes. Die Wirksamkeit einer Behandlung mit Eculizumab bei pädiatrischen Patienten mit PNH scheint mit der Wirksamkeit bei Erwachsenen mit PNH übereinzustimmen, die in den Zulassungsstudien (C04-001 und C04-002) behandelt wurden (Tabelle 3 und 14).
Tabelle 14: Ergebnisse zur Wirksamkeit bei pädiatrischen Patienten mit PNH in Studie M07-005
|
|
|
P – Wert
| |
|
Mittelwert (SD)
|
Wilcoxon-Vorzeichen-Rang-Test
| |
Veränderung LDH-Wert nach 12 Wochen gegenüber dem Ausgangswert (U/l)
|
-771 (914)
|
0,0156
| |
LDH-AUC
(U/l x Tag)
|
-60.634 (72.916)
|
0,0156
|
Atypisches Hämolytisch-Urämisches Syndrom
In Studie C09-001r wurden insgesamt 15 pädiatrische Patienten (Alter 2 Monate bis <12 Jahre) mit Soliris behandelt. Siebenundvierzig Prozent dieser Patienten hatten eine nachgewiesene Mutation eines Komplementregulierenden Faktors oder Autoantikörper. Die mediane Zeit von der aHUS-Diagnose bis zur ersten Verabreichung von Soliris betrug 14 Monate (Spanne <1 bis 110 Monate). Die mediane Zeit von der Manifestation der bestehenden thrombotischen Mikroangiopathie bis zur ersten Verabreichung von Soliris betrug 1 Monat (Spanne <1 bis 16 Monate). Die mediane Dauer der Soliris-Behandlung betrug in der Gruppe der Patienten unter 2 Jahren 16 Wochen (Spanne 4 bis 70 Wochen; n = 5), in der Gruppe der 2 bis < 12-jährigen Patienten 31 Wochen (Spanne 19-63 Wochen; n = 10).
Insgesamt erscheinen die Ergebnisse zur Wirksamkeit bei diesen pädiatrischen Patienten konsistent mit den Ergebnissen der Patienten in den aHUS Pivot-Studien C008-002 und C008-003 (Tabelle 6). Keiner der pädiatrischen Patienten benötigte eine neue Dialyse.
Tabelle 15: Ergebnisse zur Wirksamkeit bei pädiatrischen Patienten in Studie C09-001r
|
Wirksamkeits-Parameter
|
< 2 Jahre
(n = 5)
|
2 bis < 12 Jahre
(n = 10)
|
< 12 Jahre
(n = 15)
| |
Patienten mit Normalisierung der Thrombozytenzahl, n (%)
|
4 (80)
|
10 (100)
|
14 (93)
| |
Vollständiges Ansprechen der TMA, n (%)
|
2 (40)
|
5 (50)
|
7 (50)
| |
TMA Interventionsrate, Median pro Tag (Spanne)
prä-Eculizumab
unter Eculizumab
|
1 (0/2)
<1 (0/<1)
|
<1 (0,07/1.46)
0 (0/<1)
|
<1 (0/2)
0 (0/<1)
| |
Patienten mit eGFR Veränderung ≥15 ml/min/1,73 m2, n (%)
|
2 (40)
|
6 (60)
|
8 (53)
|
Bei pädiatrischen Patienten mit einer kürzeren Dauer einer bestehenden schweren klinischen Manifestation der thrombotischen Mikroangiopathie (TMA) vor Eculizumab-Behandlung wurden eine Kontrolle der TMA und eine Verbesserung der Nierenfunktion unter Eculizumab-Behandlung erreicht (Tabelle 15).
Bei pädiatrischen Patienten mit einer längeren Dauer einer bestehenden schweren klinischen Manifestation der thrombotischen Mikroangiopathie (TMA) vor Eculizumab-Behandlung wurde eine Kontrolle der TMA unter Eculizumab-Behandlung erreicht. Die Nierenfunktion verbesserte sich jedoch auf Grund der bereits bestehenden irreversiblen Nierenschädigung nicht (Tabelle 16).
Tabelle 16: Ergebnisse zur Wirksamkeit bei pädiatrischen Patienten in Studie C09-001r in Bezug auf die Dauer der aktuellen schweren klinischen Manifestation einer thrombotischen Mikroangiopathie (TMA)
|
|
Dauer der aktuellen schweren klinischen Manifestation einer TMA
| |
< 2 Monate
(N = 10) %
|
> 2 Monate
(N= 5) %
| |
Patienten mit Normalisierung der Thrombozytenzahl, n (%)
|
9 (90)
|
5 (100)
| |
Abwesenheit von TMA-Ereignissen, n (%)
|
8 (80)
|
3 (60)
| |
Vollständiges Ansprechen der TMA, n (%)
|
7 (70)
|
0
| |
Patienten mit eGFR Veränderung
≥15 ml/min/1,73 m2, n (%)
|
7 (70)
|
0*
|
*Ein Patient erreichte eine Verbesserung der eGFR nach Nierentransplantation
Insgesamt erhielten 22 Kinder oder Jugendliche (im Alter zwischen 5 Monaten und 17 Jahren) eine Behandlung mit Soliris im Rahmen der Studie C10-003.
Um in die Studie aufgenommen werden zu können, mussten die Patienten eine Thrombozytenzahl aufweisen, die unterhalb der unteren Grenze des Normalbereichs lag, zudem Anzeichen für eine Hämolyse wie einen erhöhten Serum-LDH-Spiegel oberhalb des oberen Normalbereichs und einen Serum-Kreatinin-Spiegel ≥ 97. Perzentile im Verhältnis zum Alter ohne chronischen Dialysebedarf. Das mediane Alter der Patienten lag bei 6,5 Jahren (Spanne: 5 Monate bis 17 Jahre). Die Patienten, die in die Studie zu aHUS C10-003 aufgenommen wurden, hatten einen ADAMTS-13-Wert über 5 %. 50 % der Patienten hatten eine festgestellte Mutation eines Komplement-regulierenden Faktors oder Autoantikörper. Insgesamt erhielten 10 Patienten eine Plasmainfusion, einen Plasmaaustausch oder eine Soliris-Gabe vor Beginn der Behandlung mit Soliris. Tabelle 17 fasst die klinischen Eigenschaften und die Eigenschaften, die in Zusammenhang mit der Erkrankung der Patienten bei Beginn der Studie C10-003 stehen, zusammen.
Tabelle 17: Eigenschaften der pädiatrischen Patienten bei Beginn der Studie zu aHUS C10-003
|
Parameter
|
1 Monat bis < 12 Jahre
(n=18)
|
Alle Patienten
(n=22)
| |
Zeit von der Erstdiagnose der aHUS bis zur ersten Studienmedikation (Monat) Median (min, max)
|
0,51 (0,03 – 58)
|
0,56 (0,03-191)
| |
Zeit von der Manifestierung der bestehenden TMA und erster verabreichter Dosis innerhalb der Studie, (Monat) Median (min, max)
|
0,23 (0,03 – 4)
|
0,2 (0,03-4)
| |
Thrombozytenzahl bei Screening (× 109/l), Median (min, max)
|
110 (19-146)
|
91 (19-146)
| |
LDH-Ausgangswert (U/l), Median (min, max)
|
1510 (282-7164)
|
1244 (282-7164)
| |
eGFR, Ausgangswert (ml/min/1.73m2)
Median (min, max)
|
22 (10,105)
|
22 (10, 105)
|
Patienten der Studie C10-003 wurden mindestens 26 Wochen mit Soliris behandelt. Nach Abschluss der initialen 26-wöchigen Behandlungsphase führten die meisten Patienten die Behandlung dauerhaft fort.
Nach dem Beginn der Behandlung mit Soliris konnte eine Abnahme der terminalen Komplementaktivität beobachtet werden. Soliris reduzierte die Anzeichen einer Komplement-vermittelten TMA, was der Anstieg der medianen Thrombozytenzahl zwischen Studieneinschluss und Woche 26 zeigt. In der Studie stieg die mediane Thrombozytenzahl von 88±42x109/l bei Studieneinschluss auf 281±123x109/l nach Woche 1; diese Entwicklung konnte 26 Wochen lang aufrechterhalten werden (mediane Thrombozytenzahl in Woche 26: 293±106 x109/l).
Die Nierenfunktion, die anhand der eGFR bewertet wurde, konnte unter Soliris verbessert werden. 9 der 11 Patienten, die vor Studienbeginn dialysepflichtig waren, waren nach Tag 15 der Behandlung mit Soliris nicht mehr auf eine Dialyse angewiesen. Das Ansprechen war bei allen Altersstufen zwischen 5 Monaten und 17 Jahren vergleichbar. In der Studie C10-003 zeigten Patienten mit und ohne identifizierte Mutationen in Genen, die Proteine für Komplement-regulierende Faktoren oder gegen den Faktor H gerichtete Autoantikörper kodieren, ein vergleichbares Ansprechen auf Soliris.
Tabelle 18 fasst die Ergebnisse aus der Studie zu aHUS C10-003 hinsichtlich der Wirksamkeit zusammen.
Tabelle 18: Ergebnisse zur Wirksamkeit der prospektiven aHUS Studie C10-003
|
Wirksamkeitsparameter
|
1 Monat bis < 12 Jahre
(n = 18)
Bis 26 Wochen
|
Alle Patienten
(n = 22)
Bis 26 Wochen
| |
Vollständige Normalisierung der hämatologischen Parameter, n (%)
Dauer bis zur vollständigen Normalisierung der hämatologischen Parameter, Median in Wochen (min, max)
|
14 (78)
35 (13; 78)
|
18 (82)
35 (13; 78)
| |
Vollständiges Ansprechen der TMA, n (%)
Dauer bis zum vollständigen Ansprechen der TMA, Median in Wochen (min, max)1
|
11 (61)
40 (13; 78)
|
14 (64)
37 (13; 78)
| |
Abwesenheit von TMA-Ereignissen, n (%)
95 % KI
|
17 (94)
n.a.
|
21 (96)
77; 99
| |
TMA Interventionsrate, Median pro Tag (min, max):
prä-Eculizumab,
unter Eculizumab
|
n.a.
n.a.
|
0,4 (0; 1,7)
0 (0; 1.01)
| |
Verbesserung der eGFR ≥ 15 ml/min/ 1,73•m2, n (%)
|
16 (89)
|
19 (86)
| |
Veränderung der eGFR (≥ 15 ml/min/1.73 m2) in Woche 26, Median (min, max)
|
64 (0; 146)
|
58 (0; 146)
| |
Verbesserung der CKD ≥ 1 Stadium, n (%)
|
14/16 (88)
|
17/20 (85)
| |
Keine Plasmainfusion, Plasmaaustausch oder Gabe von Soliris, in (%)
Keine neue Dialyse, n (%)
KI 95 %
|
16 (89)
18 (100)
n.a.
|
20 (91)
22 (100)
85; 100
|
1Bei cut-off (12. Oktober 2012) mit einer medianen Dauer der Soliris-Therapie von 44 Wochen (Spanne: 1 Dosis bis 88 Wochen).
Eine Langzeitbehandlung mit Soliris (Median 55 Wochen im Bereich von 1 Tag bis 107 Wochen) war mit einem erhöhten Anteil von klinisch bedeutsamen Verbesserungen bei aHUS-Patienten im Kindes- und Jugendalter verbunden. Wenn die Soliris-Therapie länger als 26 Wochen beibehalten wurde, erreichte 1 weiterer Patient (68 % der Patienten insgesamt) ein vollständiges TMA-Ansprechen. Weitere 2 Patienten (91 % der Patienten insgesamt) erreichten eine Normalisierung der hämatologischen Parameter. Bei der letzten Auswertung erreichten 19 der 22 Patienten (86 %) eine Verbesserung der eGFR ≥ 15 ml / min / 1,73 m2, im Vergleich zum Ausgangswert. Kein Patient benötigte unter Soliris eine erneute Dialyse.
Refraktäre generalisierte Myasthenia gravis
Insgesamt 11 pädiatrische Patienten mit refraktärer gMG in der Studie ECU-MG-303 erhielten Soliris. Das mittlere (Spanne) Körpergewicht der behandelten Patienten zu Studienbeginn betrug 59,7 kg (37,2 bis 91,2 kg) und das mittlere (Spanne) Alter beim Screening war 15 Jahre (12 bis 17 Jahre). Bei allen in die Studie einbezogenen Patienten handelte es sich um Patienten mit refraktärer gMG, die eine oder mehrere der folgenden Eigenschaften aufwiesen:
1. Fehlgeschlagene Behandlung ≥ 1 Jahr mit mindestens 1 immunsuppressiven Therapie, definiert als: (i) anhaltende Schwäche mit Beeinträchtigung der Aktivitäten des täglichen Lebens oder (ii) Verschlimmerung und/oder myasthene Krise während der Behandlung oder (iii) Unverträglichkeit gegenüber IST aufgrund von Nebenwirkungen oder Begleiterkrankungen.
2. Benötigen eine Erhaltungstherapie mit PE oder IVIg zur Symptomkontrolle (d. h. Patienten, die regelmässig PE oder IVIg zur Behandlung von Muskelschwäche benötigen, mindestens alle 3 Monate in den letzten 12 Monaten vor dem Screening).
Die Ausgangsparameter der pädiatrischen Patienten mit refraktärer gMG, die in die Studie ECU-MG-303 aufgenommen wurden, sind in Tabelle 19 aufgeführt.
|
Tabelle 19: Demografische Patientendaten und -parameter in der Studie ECU-MG-303
| |
|
Eculizumab (n = 11)
| |
Weiblich
|
n (%)
|
9 (81,8 %)
| |
Dauer der MG (Zeit von der MG-Diagnosestellung bis zur ersten Studienmedikation [Jahre])
|
Mittelwert (SD)
Median (min, max)
|
3,99 (2,909)
2,90 (0,1; 8,8)
| |
Ausgangswert MG-ADL-Gesamtscore
|
Mittelwert (SD)
Median (min, max)
|
5,0 (5,25)
4,0 (0; 19)
| |
Ausgangswert QMG-Gesamtscore
|
Mittelwert (SD)
Median (min, max)
|
16,7 (5,64)
15,0 (10; 28)
| |
MGFA-Klassifizierung beim Screening
IIa
IIb
IIIa
IIIb
IVa
IVb
|
n (%)
|
2 (18,2)
3 (27,3)
3 (27,3)
0
3 (27,3)
0
| |
Anzahl Patienten mit vorangegangenen Exazerbationen der MG, einschliesslich MG-Krise, seit der Diagnose
Nein
Ja
Exazerbation
MG-Krise
|
n (%)
|
4 (36,4)
7 (63,6)
6 (54,5)
3 (27,3)
| |
IVIg-Langzeittherapie bei Studienbeginn
Ja
Nein
|
n (%)
|
6 (54,5)
5 (45,5)
| |
Anzahl der Immunsupressiva-Therapien bei Studienbeginn
0
1
2
|
n (%)
|
2 (18,2)
4 (36,4)
5 (45,5)
| |
Patienten mit Immunsupressiva-Therapiena zu Studienbeginn n (%)
Kortikosteroide
Azathioprin
Mycophenolat-Mofetil
Tacrolimus
|
n (%)
|
8 (72,7)
1 (9,1)
2 (18,2)
3 (27,3)
|
aZu den Immunsuppressiva-Therapien gehörten Kortikosteroide, Azathioprin, Cyclophosphamid, Ciclosporin, Methotrexat, Mycophenolat-Mofetil oder Tacrolimus. Zu Studienbeginn erhielt keiner der Patienten Cyclosporin, Cyclophosphamid oder Methotrexat.
Abkürzungen: IVIg = intravenöses Immunglobulin; max = Maxiumum; MG = Myasthenia gravis; MG ADL = Myasthenia Gravis Activities of Daily Living (Myasthenia-Gravis-Profil für Aktivitäten des täglichen Lebens); MGFA = Myasthenia Gravis Foundation of America; min = Minimum; QMG = Quantitative Myasthenia Gravis score for disease severity (Quantitativer Myasthenia Gravis-Score für die Schwere der Erkrankung); SD = Standardabweichung
Der primäre Endpunkt der Studie ECU-MG-303 war die Veränderung des QMG-Gesamtscores gegenüber dem Ausgangswert im Laufe der Zeit, unabhängig von der Notfallbehandlung. Mit Soliris behandelte pädiatrische Patienten zeigten im QMG-Gesamtscore über den gesamten 26wöchigen primären Behandlungszeitraum eine statistisch signifikante Verbesserung gegenüber dem Ausgangswert. Die Ergebnisse für die primären und sekundären Endpunkte der Studie ECU-MG-303 sind in Tabelle 20 aufgeführt.
Die Wirksamkeit der Soliris-Behandlung bei pädiatrischen Patienten mit refraktärer gMG entsprach derjenigen, die bei erwachsenen Patienten mit refraktärer gMG im Rahmen der Zulassungsstudie ECU MG 301 beobachtet wurde (Tabelle 10).
Tabelle 20: Wirksamkeitsergebnisse der Studie ECU-MG-303
|
Wirksamkeitsendpunkte: Veränderung des Gesamtscores in Woche 26 gegenüber dem Ausgangswert
|
Differenz der Kleinste-Quadrate-Mittelwerte (SEM)
95% KI
| |
QMG
|
-5,8 (1,2)
(-8,40; -3,13)
na = 10
| |
MG-ADL-Gesamtscore
|
-2,3 (0,6)
(-3,63; -1,03)
na = 10
| |
MGC-Gesamtscore
|
-8,8 (1,9)
(-12,93; -4,69)
na = 9
|
na ist die Anzahl der Patienten in Woche 26
Abkürzungen: KI = Konfidenzintervall; LS = kleinste Quadrate; MG-ADL = Myasthenia Gravis Activities of Daily Living (Myasthenia-Gravis-Profil für Aktivitäten des täglichen Lebens); MGC = Myasthenia-Gravis-Komposit; QMG = Quantitative Myasthenia Gravis score for disease severity (Quantitativer Myasthenia Gravis-Score für die Schwere der Erkrankung); SEM = Standardfehler des Mittelwerts;
In der Studie ECU-MG-303 wurde ein klinisches Ansprechen bei den QMG- und MG-ADL-Gesamtscores als eine Verbesserung von mindestens 5 bzw. 3 Punkten gegenüber dem Ausgangswert definiert. Der Anteil der klinischen Responder bei den QMG- und MG-ADL-Gesamtscores betrug in Woche 26, unabhängig von der Notfallbehandlung, 70 % bzw. 50 %. Die 10 Patienten, die den Besuch nach 26 Wochen wahrnahmen, erreichten zu diesem Zeitpunkt einen verbesserten MGFA-postinterventionellen Status (MGFA PIS). Sieben (70 %) Patienten erreichten in Woche 26 eine minimale Manifestation der refraktären gMG.
Bei einem Patienten (9,1 %) wurde während der primären Auswertung des Behandlungszeitraums eine klinische Verschlechterung (myasthene Krise) beobachtet, die eine Notfalltherapie (PE) erforderte, welche zwischen den Studienbesuchen in Woche 22 und Woche 24 verabreicht wurde. Infolgedessen und aufgrund der Entscheidung des Arztes wurden bei diesem Patienten nach Woche 20 keine QMG-, MG-ADL- oder andere Wirksamkeitsbeurteilungen durchgeführt und er wurde nicht in den Verlängerungszeitraum aufgenommen.
Während der primären Auswertung des Behandlungszeitraums bei pädiatrischen Patienten mit refraktärer gMG (Studie ECU-MG-303) reduzierte 1 von 11 Patienten (9,1 %) die tägliche Anticholinesterase-Dosis und 3 von 11 Patienten (27,3 %) reduzierten ihre tägliche Kortikosteroid-Dosis aufgrund einer Verbesserung der MG-Symptome.
Neuromyelitis-optica-Spektrumerkrankungen
Soliris wurde nicht bei Kindern und Jugendlichen mit NMOSD untersucht.
|