Eigenschaften/WirkungenATC-Code
R07AX30
Wirkungsmechanismus
Pharmakotherapeutische Gruppe: Andere Mittel für den Respirationstrakt
Das CFTR-Protein ist ein Chloridkanal an der Oberfläche von Epithelzellen in verschiedenen Organen. Die F508del-Mutation wirkt sich in verschiedener Weise auf das CFTR-Protein aus und verursacht in erster Linie einen Defekt in der zellulären Verarbeitung und Transportsteuerung, der zu einer Verringerung der CFTR-Menge an der Zelloberfläche führt. Die kleine Menge an F508del-CFTR, die die Zelloberfläche erreicht, besitzt eine geringe Öffnungswahrscheinlichkeit des Kanals (defektes Gating; Kanalschaltverhalten). Lumacaftor ist ein CFTR-Korrektor, der direkt auf das F508del-CFTR einwirkt, um dessen zelluläre Verarbeitung und Transportsteuerung zu verbessern und dadurch die Menge an funktionellem CFTR an der Zelloberfläche zu erhöhen. Ivacaftor ist ein CFTR-Potentiator, der einen erhöhten Chloridtransport ermöglicht, indem er die Öffnungswahrscheinlichkeit (Gating) des CFTR-Kanals an der Zelloberfläche erhöht. Das Zusammenwirken von Lumacaftor und Ivacaftor führt zu einer erhöhten Menge und Funktion von F508del-CFTR an der Zelloberfläche, was einen erhöhten Chloridionentransport zur Folge hat. Die genauen Mechanismen, durch welche Lumacaftor die zelluläre Verarbeitung und Transportsteuerung von F508del-CFTR verbessert und Ivacaftor F508del-CFTR verstärkt, sind nicht bekannt.
Pharmakodynamik
Wirkungen auf die Schweisschloridkonzentration
Die Veränderungen der Schweisschloridkonzentration als Reaktion auf Lumacaftor allein oder in Kombination mit Ivacaftor wurden in einer doppelblinden, placebokontrollierten klinischen Phase-2-Studie bei CF-Patienten ab 18 Jahren bewertet. In dieser Studie schlossen 10 Patienten (homozygot für die F508del-CFTR-Mutation) die Behandlung mit 400 mg Lumacaftor allein alle 12 Stunden (q12h) für 28 Tage und anschliessender Zugabe von 250 mg Ivacaftor q12h für weitere 28 Tage ab, 25 Patienten (homozygot oder heterozygot für F508del) schlossen die Behandlung mit einem Placebo ab. Der Behandlungsunterschied zwischen 400 mg Lumacaftor q12h allein und Placebo, bewertet als mittlere Veränderung der Schweisschloridkonzentration von Baseline bis Tag 28, war mit -8,2 mmol/l (95 %-KI: -14, -2) statistisch signifikant. Der Behandlungsunterschied zwischen der Kombination 400 mg Lumacaftor/250 mg Ivacaftor q12h und Placebo, bewertet als mittlere Veränderung der Schweisschloridkonzentration von Baseline bis Tag 56, war mit -11 mmol/l (95 %-KI: -18, -4) ebenfalls statistisch signifikant.
Veränderungen der Schweisschloridkonzentration als Reaktion auf Lumacaftor/Ivacaftor wurden im Rahmen einer 24-wöchigen, placebokontrollierten klinischen Phase-3-Studie (Studie VX14-809-109) an 204 Patienten mit CF im Alter von 6 bis unter 12 Jahren untersucht, die homozygot für die F508del-Mutation im CFTR-Gen waren (103 erhielten Lumacaftor 200 mg/Ivacaftor 250 mg alle 12 Stunden und 101 erhielten Placebo). Bei der Behandlung mit Lumacaftor/Ivacaftor zeigte sich eine statistisch signifikante Abnahme der Schweisschloridkonzentration im Vergleich zu Placebo, die über die 24-wöchige Behandlung hinweg anhielt. Der Behandlungsunterschied (LS-Mittelwert) bei der Schweisschloridkonzentration für die durchschnittliche absolute Veränderung an Tag 15 und in Woche 4 im Vergleich zu Placebo betrug -20,8 mmol/l (95%-KI: -23,4; -18,2; p<0,0001). Der Behandlungsunterschied (LS-Mittelwert) bei der absoluten Veränderung der Schweisschloridkonzentration in Woche 24 im Vergleich zu Placebo betrug -24,9 mmol/l (p<0,0001).
In Studie VX15-809-115 bei Patienten im Alter von 2 bis 5 Jahren, die homozygot für die F508del-CFTR-Mutation waren, betrug die mittlere absolute Veränderung der Schweisschloridkonzentration innerhalb der Gruppe ab Baseline bis Woche 24 -31,7 mmol/l (95% KI: -35,7, -27,6). Ausserdem war die mittlere absolute Veränderung der Schweisschloridkonzentration von Woche 24 in Woche 26 nach der 2-wöchigen Auswaschphase (zur Bewertung der Reaktion ohne das Arzneimittel) ein Anstieg von 33,0 mmol/l (95% KI: 28,9; 37,1; nominal p <0,0001), was auf eine Rückkehr zu den Ausgangswerten nach Auswaschen der Behandlung hinweist. Bis Woche 24 zeigten 16 % der Kinder eine Abnahme der Schweisschloridkonzentration auf unter 60 mmol/l, während es bei keinem Kind zu einer Abnahme unter 30 mmol/l kam.
In Studie VX16-809-122 bei Patienten im Alter von 1 Jahr bis unter 2 Jahren, die homozygot für die F508del-CFTR-Mutation waren, zeigte sich unter der Behandlung mit Lumacaftor/Ivacaftor in Woche 4 eine Abnahme der Schweisschloridkonzentration, die bis einschliesslich Woche 24 erhalten blieb. Die mittlere absolute Veränderung der Schweisschloridkonzentration gegenüber Baseline in Woche 24 betrug -29,1 (13,5) mmol/l (95%-KI: -34,8; -23,4). Darüber hinaus betrug die mittlere (SD) absolute Veränderung der Schweisschloridkonzentration von Woche 24 bis Woche 26 nach der 2-wöchigen Auswaschphase 27,3 (11,1) mmol/l (95%-KI: 22,3; 32,3). Diese Veränderung zeigt eine Rückkehr zur Baseline nach der Auswaschphase der Behandlung.
Veränderungen des FEV1
Die Veränderungen des ppFEV1 mit Lumacaftor allein oder in Kombination mit Ivacaftor wurden in der doppelblinden, placebokontrollierten Phase-2-Studie an Patienten mit CF ab 18 Jahren ebenfalls untersucht. Der Behandlungsunterschied zwischen Lumacaftor 400 mg q12h allein und Placebo, bewertet als mittlere absolute Veränderung des ppFEV1, betrug -4,6 Prozentpunkte (95 %-KI: -9,6; 0,4) von Baseline bis Tag 28, 4,2 Prozentpunkte (95 %-KI: –1,3; 9,7) von Baseline bis Tag 56 und 7,7 Prozentpunkte (95 %-KI: 2,6; 12,8;) von Tag 28 bis Tag 56 (nach Zugabe von Ivacaftor zur Lumacaftor-Monotherapie).
Abnahme der Herzfrequenz
Während der 24-wöchigen, placebokontrollierten Phase-3-Studien wurde an Tag 1 und Tag 15 etwa 4 bis 6 Stunden nach der Einnahme eine maximale Abnahme der mittleren Herzfrequenz um 6 Schläge pro Minute gegenüber Baseline beobachtet. Nach Tag 15 wurde die Herzfrequenz in diesen Studien im Zeitraum nach der Einnahme nicht mehr kontrolliert. Ab Woche 4 lag die Veränderung der vor der Einnahme gemessenen mittleren Herzfrequenz bei den mit Lumacaftor/Ivacaftor behandelten Patienten zwischen 1 und 2 Schlägen pro Minute unter dem Baseline-Wert. Der prozentuale Anteil von Patienten mit Herzfrequenzwerten <50 Schlägen pro Minute unter der Behandlung betrug 11 % bei den Patienten, die Lumacaftor/Ivacaftor erhielten, im Vergleich zu 4,9 % bei den Patienten, die Placebo erhielten.
Kardiale Elektrophysiologie
Im Rahmen einer eingehenden klinischen QT-Studie zur Beurteilung von Lumacaftor 600 mg einmal täglich/Ivacaftor 250 mg q12h und Lumacaftor 1000 mg einmal täglich/Ivacaftor 450 mg q12h wurden keine bedeutsamen Veränderungen des QTc-Intervalls oder des Blutdrucks beobachtet.
Klinische Wirksamkeit
Klinische Wirksamkeit und Sicherheit
Studien bei CF-Patienten ab dem Alter von 12 Jahren, die homozygot für die F508del-Mutation im CFTR-Gen sind
Die Wirksamkeit von Lumacaftor/Ivacaftor bei CF-Patienten, die homozygot für die F508del-Mutation im CFTR-Gen sind, wurde in zwei randomisierten, doppelblinden, placebokontrollierten klinischen Studien bei 1108 klinisch stabilen CF-Patienten untersucht, wobei 737 Patienten randomisiert mit Lumacaftor/Ivacaftor behandelt wurden. In beiden Studien wurden die Patienten im Verhältnis 1:1:1 randomisiert und erhielten 600 mg Lumacaftor einmal täglich/250 mg Ivacaftor q12h, 400 mg Lumacaftor q12h/250 mg Ivacaftor q12h oder Placebo. Die Patienten nahmen zusätzlich zu ihren verordneten CF-Therapien (z.B. Bronchodilatatoren, inhalative Antibiotika, Dornase alfa und hypertone Kochsalzlösung) 24 Wochen lang das Prüfpräparat zusammen mit einer fetthaltigen Mahlzeit ein. Patienten aus diesen Studien waren für die Übernahme in eine verblindete Verlängerungsstudie qualifiziert.
Es gibt keine klinische Studie, die einen direkten Vergleich der Überlegenheit der Kombinationstherapie mit den Komponenten der Monotherapie durchgeführt hätte.
Studie VX12-809-103 wertete die Daten von 549 CF-Patienten ab 12 Jahren (Durchschnittsalter 25,1 Jahre) mit einem FEV1 in Prozent des Sollwerts (ppFEV1) zwischen 40 und 90 beim Screening (mittleres ppFEV1 60,7 zu Baseline [Bereich: 31,1 bis 94,0]) aus. Studie VX12-809-104 wertete die Daten von 559 Patienten ab 12 Jahren (Durchschnittsalter 25,0 Jahre) mit einem ppFEV1 zwischen 40 und 90 beim Screening (mittleres ppFEV1 60,5 zu Baseline [Bereich: 31,3 bis 99,8]) aus. Patienten mit einer Vorgeschichte von Kolonisierung mit Organismen wie Burkholderia cenocepacia, Burkholderia dolosa oder Mycobacterium abscessus sowie Patienten, bei denen drei oder mehr Leberfunktionstests auffällige Werte ergaben (ALT, AST, AP, GGT ≥3 x ULN oder Gesamtbilirubin ≥2 x ULN), wurden ausgeschlossen.
Primärer Wirksamkeitsendpunkt in beiden Studien war die absolute Veränderung des ppFEV1 von Baseline bis Behandlungswoche 24. Weitere Wirksamkeitsvariablen waren die relative Veränderung des ppFEV1 von Baseline, die absolute Veränderung des BMI von Baseline, die absolute Veränderung des CFQ-R-Scores für die Atemwegssymptomatik von Baseline, der Anteil Patienten, die eine relative Veränderung des ppFEV1 ≥5 % von Baseline bis Behandlungswoche 24 erreichten, und die Anzahl an Fällen mit pulmonalen Exazerbationen (einschliesslich solcher, die einen Spitalaufenthalt oder eine intravenöse Antibiotikatherapie erforderlich machten) bis Behandlungswoche 24.
In beiden Studien führte die Behandlung mit Lumacaftor/Ivacaftor zu einer statistisch signifikanten Verbesserung des ppFEV1 (siehe Tabelle 7). Die durchschnittliche Verbesserung des ppFEV1 setzte schnell ein (Tag 15) und hielt über den gesamten 24wöchigen Behandlungszeitraum an. Der Behandlungsunterschied zwischen 400 mg Lumacaftor/250 mg Ivacaftor q12h und Placebo bei der mittleren absoluten Veränderung (95 %-KI) des ppFEV11 von Baseline bis Tag 15 betrug 2,51 Prozentpunkte in den gepoolten Studien VX12-809-103 und VX12-809-104 (P < 0,0001). Verbesserungen beim ppFEV1 wurden unabhängig von Alter, Schwere der Erkrankung, Geschlecht und geographischer Region beobachtet. Die Phase-3-Studien zu Lumacaftor/Ivacaftor umfassten 81 Patienten mit ppFEV1 < 40 zu Baseline. Der Behandlungsunterschied in dieser Untergruppe war mit dem bei Patienten mit ppFEV1 ≥40 beobachteten vergleichbar. Der Behandlungsunterschied zwischen 400 mg Lumacaftor/250 mg Ivacaftor q12h und Placebo bei der mittleren absoluten Veränderung (95 %-KI) des ppFEV1 von Baseline bis Woche 24 in den gepoolten Studien VX12-809-103 und VX12-809-104 betrug 3,39 Prozentpunkte (P = 0,0382) bei Patienten mit einem ppFEV1 < 40 und 2,47 Prozentpunkte (P < 0,0001) bei Patienten mit einem ppFEV1 ≥40.
|
Tabelle 7: Zusammenfassung der primären Endpunkte und wichtigsten sekundären Endpunkte in Studie VX12-809-103 und Studie VX12-809-104*
| |
|
Studie VX12-809-103
|
Studie VX12-809-104
|
Gepoolt
(Studie VX12-809-103 und Studie VX12- 809-104)
| |
|
Placebo
(n = 184)
|
400 mg LUM q12h/250 mg IVA q12h
(n = 182)
|
Placebo
(n = 187)
|
400 mg LUM q12h/250 mg IVA q12h
(n = 187)
|
Placebo
(n = 371)
|
400 mg LUM q12h/250 mg IVA q12h
(n = 369)
| |
Absolute Veränderung des ppFEV1 bis Woche 24 (Prozentpunkte)
|
Behandlungsunterschied
|
–
|
2,41
(P = 0,0003) †
|
–
|
2,65
(P = 0,0011) †
|
–
|
2,55
(P < 0,0001)
| |
|
Veränderung innerhalb der Gruppe
|
-0,73
(P = 0,2168)
|
1,68
(P = 0,0051)
|
-0,02
(P = 0,9730)
|
2,63
(P < 0,0001)
|
-0,39
(P < 0,3494)
|
2,16
(P < 0,0001)
| |
Relative Veränderung des ppFEV1 bis Woche 24 (%)
|
Behandlungsunterschied
|
–
|
4,15
(P = 0,0028)†
|
–
|
4,69
(P = 0,0009)†
|
–
|
4,4
(P < 0,0001)
| |
|
Veränderung innerhalb der Gruppe
|
-0,85
(P = 0,3934)
|
3,3
(P = 0,0011)
|
0,16
(P = 0,8793)
|
4,85
(P < 0,0001)
|
-0,34
(P = 0,6375)
|
4,1
(P < 0,0001)
| |
Absolute Veränderung des BMI bis Woche 24 (kg/m2)
|
Behandlungsunterschied
|
–
|
0,13
(P = 0,1938)
|
–
|
0,36
(P < 0,0001)†
|
–
|
0,24
(P = 0,0004)
| |
|
Veränderung innerhalb der Gruppe
|
0,19
(P = 0,0065)
|
0,32
(P < 0,0001)
|
0,07
(P = 0,2892)
|
0,43
(P < 0,0001)
|
0,13
(P = 0,0066)
|
0,37
(P < 0,0001)
| |
Absolute Veränderung des CFQ-R-Scores für die Atemwegs-symptomatik bis Woche 24 (Punkte)
|
Behandlungsunterschied
|
–
|
1,5
(P = 0,3569)
|
–
|
2,9
(P = 0,0736)
|
–
|
2,2
(P = 0,0512)
| |
|
Veränderung innerhalb der Gruppe
|
1,1
(P = 0,3423)
|
2,6
(P = 0,0295)
|
2,8
(P = 0,0152)
|
5,7
(P < 0,0001)
|
1,9
(P = 0,0213)
|
4,1
(P < 0,0001)
| |
Anteil Patienten mit ≥5 % relativer Veränderung des ppFEV1 bis Woche 24
|
%
|
25%
|
32%
|
26%
|
41%
|
26%
|
37%
| |
|
Quotenverhältnis (Odds Ratio)
|
–
|
1,43
(P = 0,1208)
|
–
|
1,90
(P = 0,0032)
|
–
|
1,66
(P = 0,0013)
| |
Anzahl der pulmonalen Exazerbationen bis Woche 24
|
Anzahl Ereignisse (Rate pro 48 Wochen)
|
112 (1,07)
|
73 (0,71)
|
139 (1,18)
|
79 (0,67)
|
251 (1,14)
|
152 (0,70)
| |
|
Rate Ratio
|
–
|
0,66
(P = 0,0169)
|
–
|
0,57
(P = 0,0002)
|
–
|
0,61
(P < 0,0001)
| |
* In jeder Studie wurde innerhalb jeder Verumgruppe für primäre und sekundäre Endpunkte vs. Placebo ein hierarchisches Testverfahren durchgeführt; bei jedem Schritt war statistische Signifikanz nur dann gegeben, wenn der Unterschied zum Niveau P ≤0,0250 signifikant war und dieses Niveau auch in allen vorangegangenen Tests erreicht wurde.
† Zeigt an, dass die statistische Signifikanz im hierarchischen Testverfahren bestätigt wurde.
|
Zu Woche 24 war der Anteil Patienten ohne pulmonale Exazerbationen bei den mit Lumacaftor/Ivacaftor behandelten Patienten signifikant höher als bei den Placebo-Patienten. In der gepoolten Analyse lag die Rate Ratio (Ratenverhältnis) für Exazerbationen bis Woche 24 bei mit Lumacaftor/Ivacaftor behandelten Patienten (400 mg Lumacaftor/250 mg Ivacaftor q12h; n = 369) bei 0,61 (P < 0,0001), was einer Reduktion um 39 % gegenüber Placebo entspricht. Die Ereignisrate pro Jahr, berechnet auf 48 Wochen, lag bei 0,70 in der Lumacaftor/Ivacaftor-Gruppe und bei 1,14 in der Placebo-Gruppe. Die Behandlung mit Lumacaftor/Ivacaftor verringerte signifikant das Risiko von Exazerbationen, die einen Spitalaufenthalt notwendig machten, gegenüber Placebo um 61 % (Rate Ratio = 0,39, P < 0,0001; Ereignisrate pro 48 Wochen 0,17 bei Lumacaftor/Ivacaftor und 0,45 bei Placebo) und reduzierte die Fälle von Exazerbationen, die eine Behandlung mit intravenösen Antibiotika erforderten, um 56 % (Rate Ratio = 0,44, P < 0,0001; Ereignisrate pro 48 Wochen 0,25 bei Lumacaftor/Ivacaftor und 0,58 bei Placebo). Diese Ergebnisse wurden im Rahmen der Testhierarchie für die Einzelstudien nicht als statistisch signifikant eingestuft.
Langzeit-Rollover-Studie zur Sicherheit und Wirksamkeit
Studie VX12-809-105 war eine multizentrische Parallelgruppen-Phase-3-Rollover-Verlängerungsstudie, die CF-Patienten ab 12 Jahren aus Studie VX12-809-103 und Studie VX12-809-104 einschloss. Diese Verlängerungsstudie war dazu angelegt, die Sicherheit und Wirksamkeit einer Langzeitbehandlung mit Lumacaftor/Ivacaftor zu untersuchen. Von den 1108 Patienten, die in Studie VX12-809-103 oder Studie VX12-809-104 eine Behandlung erhielten, wurden 1029 (93 %) in Studie VX12-809-105 bis zu weitere 96 Wochen (d.h. bis zu insgesamt 120 Wochen) mit Verum behandelt (600 mg Lumacaftor einmal täglich/ 250 mg Ivacaftor q12h oder 400 mg Lumacaftor q12h / 250 mg Ivacaftor q12h). Die primäre Wirksamkeitsanalyse dieser Verlängerungsstudie umfasste Daten bis zu Woche 72 von Studie VX12-809-105, wobei zusätzlich eine Sensitivitätsanalyse erfolgte, die Daten bis zu Woche 96 von Studie VX12-809-105 einschloss.
Die in Studie VX12-809-103 und Studie VX12-809-104 mit Lumacaftor/Ivacaftor behandelten Patienten zeigten eine Wirkung, die nach weiteren 96 Wochen in Studie VX12-809-105 im Verhältnis zum Ausgangswert erhalten blieb. Bei Patienten, die von Placebo auf die Verumbehandlung umgestellt wurden, waren ähnliche Veränderungen festzustellen wie bei Patienten, die in Studie VX12-809-103 und Studie VX12-809-104 mit Lumacaftor/Ivacaftor behandelt wurden (siehe Tabelle 7). Die Ergebnisse von Studie VX12-809-105 sind in Abbildung 1 und Tabelle 8 dargestellt.
Abbildung 1. Absolute Veränderung des ppFEV1 gegenüber dem Ausgangswert bei jedem Untersuchungszeitpunkt†
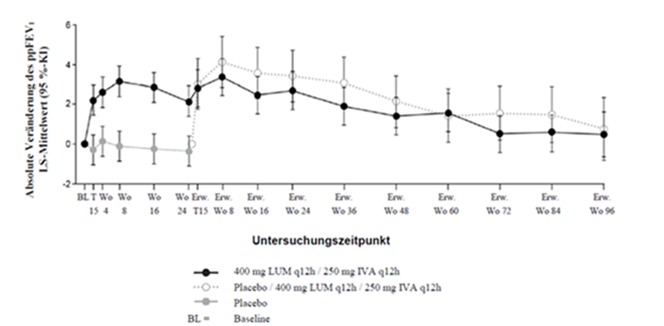
† Aus den Studien VX12-809-103, VX12-809-104 und VX12-809-105.
|
Tabelle 8: Langzeitwirkung von Lumacaftor/Ivacaftor in Studie VX12-809-105*
| |
|
Umstellung von Placebo auf
400 mg Lumacaftor q12h/
250 mg Ivacaftor q12h
(n=176)**
|
400 mg Lumacaftor q12h/
250 mg Ivacaftor q12h
(n=369)†
| |
Ausgangswert und Endpunkt
|
Mittelwert (SD)
|
LS Mittelwert (95 %-KI)
|
P-Wert
|
Mittelwert (SD)
|
LS Mittelwert (95 %-KI)
|
P-Wert
| |
Absolute Veränderung vs. Ausgangswert des ppFEV1 (Prozentpunkte)
| |
Ausgangswert des ppFEV1‡
|
60,2 (14,7)
|
|
|
60,5 (14,1)
|
|
| |
Verlängerungsstudie Woche 72
|
|
(n=134)
1,5
(0,2; 2,9)
|
0,0254
|
|
(n=273)
0,5
(-0,4; 1,5)
|
0,2806
| |
Verlängerungsstudie Woche 96
|
|
(n=75)
0,8
(-0,8; 2,3)
|
0,3495
|
|
(n=147)
0,5
(-0,7; 1,6)
|
0,4231
| |
Relative Veränderung vs. Ausgangswert des ppFEV1 (%)
| |
Verlängerungsstudie Woche 72
|
|
(n=134)
2,6
(0,2; 5,0)
|
0,0332
|
|
(n=273)
1,4
(-0,3; 3,2)
|
0,1074
| |
Verlängerungsstudie Woche 96
|
|
(n=75)
1,1
(-1,7; 3,9)
|
0,4415
|
|
(n=147)
1,2
(-0,8; 3,3)
|
0,2372
| |
Ausgangswert des BMI (kg/m2)‡
|
20,9 (2,8)
|
|
|
21,5 (3,0)
|
|
| |
Absolute Veränderung vs. Ausgangswert des BMI (kg/m2)
| |
Verlängerungsstudie Woche 72
|
|
(n=145)
0,62
(0,45; 0,79)
|
<0,0001
|
|
(n=289)
0,69
(0,56; 0,81)
|
<0,0001
| |
Verlängerungsstudie Woche 96
|
|
(n=80)
0,76
(0,56; 0,97)
|
<0,0001
|
|
(n=155)
0,96
(0,81; 1,11)
|
<0,0001
| |
Ausgangswert des Scores der respiratorischen Domäne des CFQ-R (Punkte)‡
|
70,4 (18,5)
|
|
|
68,3 (18,0)
|
|
| |
Absolute Veränderung des Scores der respiratorischen Domäne des CFQ-R (Punkte)
| |
Verlängerungsstudie Woche 72
|
|
(n=135)
3,3
(0,7; 5,9)
|
0,0124
|
|
(n=269)
5,7
(3,8; 7,5)
|
<0,0001
| |
Verlängerungsstudie Woche 96
|
|
(n=81)
0,5
(-2,7; 3,6)
|
0,7665
|
|
(n=165)
3,5
(1,3; 5,8)
|
0,0018
| |
Anzahl der pulmonalen Exazerbationen (Ereignisse) ** † ***
| |
Anzahl der Ereignisse pro Patientenjahr (95 %-KI) (Rate pro 48 Wochen)
|
|
0,69
(0,56; 0,85)
|
|
|
0,65
(0,56; 0,75)
|
| |
Anzahl der Ereignisse mit Hospitalisierungsbedarf pro Patientenjahr (95 %-KI) (Rate pro 48 Wochen)
|
|
0,30
(0,22; 0,40)
|
|
|
0,24
(0,19; 0,29)
|
| |
Anzahl der Ereignisse mit Bedarf für intravenöse Antibiotika pro Patientenjahr (95 %-KI) (Rate pro 48 Wochen)
|
|
0,37
(0,29; 0,49)
|
|
|
0,32
(0,26; 0,38)
|
| |
* Insgesamt 82 % (421 von 516 geeigneten Patienten) schlossen 72 Wochen dieser Studie ab; 42 % schlossen 96 Wochen ab. Die meisten Patienten, die die Studie abbrachen, taten dies aus anderen Gründen als der Sicherheit.
** Bei den aus Studie VX12-809-103 und VX12-809-104 übernommenen Patienten (Gruppe mit Umstellung von Placebo auf Lumacaftor/Ivacaftor) betrug die Gesamtexposition bis zu 96 Wochen. Die dargestellte Dosisgruppe mit 400 mg Lumacaftor q12h / 250 mg Ivacaftor q12h entspricht der empfohlenen Dosierung.
*** Die Ereignisrate pro Patientenjahr wurde auf 48 Wochen umgerechnet.
† Bei den aus Studie VX12-809-103 und VX12-809-104 übernommenen Patienten (Gruppe, die durchwegs mit Lumacaftor/Ivacaftor behandelt wurde) betrug die Gesamtexposition bis zu 120 Wochen. Die dargestellte Dosisgruppe mit 400 mg Lumacaftor q12h / 250 mg Ivacaftor q12h entspricht der empfohlenen Dosierung.
‡ Die Ausgangswerte der Gruppe mit Umstellung von Placebo auf 400 mg Lumacaftor q12h / 250 mg Ivacaftor q12h waren die Ausgangswerte von Studie VX12-809-105. Die Ausgangswerte der Gruppe mit 400 mg Lumacaftor q12h / 250 mg Ivacaftor q12h waren die Ausgangswerte von Studie VX12-809-103 und VX12-809-104.
|
Studie bei CF-Patienten, die heterozygot für die F508del-Mutation im CFTR-Gen sind
Studie VX09-809-102 war eine multizentrische, doppelblinde, randomisierte, placebokontrollierte Phase-2-Studie mit 125 CF-Patienten ab 18 Jahren, die ein ppFEV1 von 40 bis einschliesslich 90 hatten und die F508del-Mutation auf einem Allel plus ein zweites Allel mit einer Mutation, die voraussichtlich zu einer fehlenden CFTR-Produktion oder einem CFTR-Protein führt, das in vitro auf Ivacaftor nicht anspricht, aufweisen.
Die Patienten erhielten entweder Lumacaftor/Ivacaftor (n = 62) oder Placebo (n = 63) zusätzlich zu ihren verordneten CF-Therapien. Primärer Endpunkt war die Verbesserung der Lungenfunktion, ermittelt anhand der mittleren absoluten Veränderung des ppFEV1 von Baseline bis Tag 56. Die Behandlung mit Lumacaftor/Ivacaftor führte bei CF-Patienten, die heterozygot für die F508del-Mutation im CFTR-Gen sind, zu keiner signifikanten Verbesserung des ppFEV1 gegenüber Placebo (Behandlungsunterschied 0,60 [P = 0,5978]) sowie zu keiner wesentlichen Verbesserung des BMI oder Gewichts (siehe «Warnhinweise und Vorsichtsmassnahmen»).
Kinder und Jugendliche
Klinische Studien an CF-Patienten im Alter von 6 bis unter 12 Jahren, die homozygot für die F508del-Mutation im CFTR-Gen sind
Studie VX14-809-109 war eine 24-wöchige, placebokontrollierte klinische Phase 3-Studie an 204 CF-Patienten im Alter von 6 bis unter 12 Jahren (mittleres Alter 8,8 Jahre). In dieser Studie wurden Patienten mit einem Lungenclearance-Index (LCI2,5) ≥7,5 beim ersten Screening-Besuch (mittlerer LCI2,5 10,28 bei Ausgangswerterhebung [Bereich: 6,55 bis 16,38]) und einem ppFEV1 ≥70 beim Screening (mittlerer ppFEV1 89,8 bei Ausgangswerterhebung [Bereich: 48,6 bis 119,6]) untersucht. Die Patienten erhielten entweder Lumacaftor 200 mg/Ivacaftor 250 mg alle 12 Stunden (n=103) oder Placebo (n=101) zusätzlich zu den ihnen verschriebenen CF-Therapien. Patienten mit 2 oder mehr abnormalen Leberfunktionstests (ALT, AST, AP, GGT ≥3 x ULN) oder ALT oder AST > 5 x ULN oder Gesamtbilirubin > 2 x ULN wurden ausgeschlossen.
Der primäre Wirksamkeitsendpunkt war die absolute Veränderung des LCI2,5 gegenüber dem Ausgangswert bis Woche 24. Die wichtigsten sekundären Endpunkte waren die durchschnittliche absolute Veränderung der Schweisschloridkonzentration gegenüber dem Ausgangswert an Tag 15 und in Woche 4 sowie in Woche 24 (siehe «Pharmakodynamik»), die absolute Veränderung des BMI gegenüber dem Ausgangswert in Woche 24, sowie die absolute Veränderung des Scores der respiratorischen Domäne des CFQ-R bis Woche 24. Die Ergebnisse sind in Tabelle 9 unten gezeigt:
|
Tabelle 9: Zusammenfassung der primären und massgeblichen sekundären Zielkriterien von Studie VX14-809-109
| |
|
Placebo
(n=101)
|
LUM 200 mg/IVA 250 mg alle 12 Std.
(n=103)
| |
Primärer Endpunkt
| |
Absolute Veränderung des Lungenclearance-Index (LCI2,5) gegenüber dem Ausgangswert bis Woche 24
|
Behandlungsunterschied
|
–
|
-1,09
(p<0,0001)
| |
|
Veränderung innerhalb der Gruppe
|
0,08
(p=0,5390)
|
-1,01 (p<0,0001)
| |
Massgebliche sekundäre Endpunkte*
| |
Absolute Veränderung des BMI in Woche 24 (kg/m2)
|
Behandlungsunterschied
|
–
|
0,11
(p=0,2522)
| |
|
Veränderung innerhalb der Gruppe
|
0,27
(p=0,0002)
|
0,38
(p<0,0001)
| |
Absolute Veränderung des Scores der respiratorischen Domäne des CFQ-R bis Woche 24 (Punkte)
|
Behandlungsunterschied
|
–
|
2,5
(P=0,0628)
| |
|
Veränderung innerhalb der Gruppe
|
3,0
(p=0,0035)
|
5,5
(p<0,0001)
| |
* Die Studie schloss massgebliche sekundäre und weitere sekundäre Endpunkte ein.
|
Das FEV1 in Prozent des Sollwerts wurde ebenfalls als ein klinisch bedeutsamer weiterer sekundärer Endpunkt ausgewertet. Bei den Patienten unter Lumacaftor/Ivacaftor lag der Behandlungsunterschied bei der absoluten Veränderung des ppFEV1 gegenüber dem Ausgangswert bis Woche 24 bei 2,4 (p=0,0182).
Studie VX15-809-115: Studie zur Sicherheit und Verträglichkeit bei Kindern mit CF im Alter von 2 bis 5 Jahren, die homozygot für die F508del-Mutation im CFTR-Gen sind
Studie VX15-809-115 untersuchte 60 Patienten, die beim Screening 2 bis 5 Jahre alt waren (mittleres Alter bei Baseline 3,7 Jahre). Entsprechend ihrem Körpergewicht beim Screening erhielten die Patienten alle 12 Stunden Granulat vermischt mit einer Speise in einer Dosis von 100 mg Lumacaftor/125 mg Ivacaftor Granulat bei Patienten unter 14 kg (n = 19) bzw. 150 mg Lumacaftor/188 mg Ivacaftor bei Patienten von 14 kg oder mehr (n = 41) über 24 Wochen zusätzlich zu ihren verordneten CF-Therapien. Zur Bewertung der Wirkungen ohne das Arzneimittel wurde bei den Patienten nach einer 2-wöchigen Auswaschphase eine Follow-up-Visite zur Sicherheit durchgeführt.
Sekundäre Endpunkte waren die absolute Veränderung der Schweisschloridkonzentration von Baseline in Woche 24 sowie die absolute Veränderung der Schweisschloridkonzentration von Woche 24 in Woche 26 (siehe «Pharmakodynamik»). Darüber hinaus galten die in Tabelle 10 genannten Endpunkte. Die klinische Relevanz des Ausmasses dieser Veränderungen bei Kindern im Alter von 2 bis 5 Jahren mit zystischer Fibrose bei Langzeitbehandlung konnte bisher noch nicht eindeutig geklärt werden.
|
Tabelle 10: Zusammenfassung der sekundären Endpunkte von Studie VX15-809-115
| |
Sekundäre Endpunkte*
|
LUM/IVA
| |
Absolute Veränderung des Body Mass Index (BMI) gegenüber Baseline
|
n = 57
0,27
95% KI: 0,07; 0,47; p = 0,0091
| |
LCI2,5
|
n = 17
-0,58
95% KI: -1,17; 0,02; p = 0,0559
|
Hinweis: Die p-Werte in der Tabelle sind nominale Werte.
* Die absolute Veränderung gegenüber Baseline für die aufgeführten Endpunkte ist die absolute mittlere Veränderung gegenüber Baseline in Woche 24.
Studie VX16-809-122: Sicherheits- und Verträglichkeitsstudie bei Kindern mit CF im Alter von 1 Jahr bis unter 2 Jahren, die homozygot für die F508del-Mutation im CFTR-Gen sind
In Studie VX16-809-122 Teil B wurde der primäre Endpunkt Sicherheit und Verträglichkeit bei 46 Patienten über 24 Wochen ausgewertet (mittleres Alter bei Baseline 18,1 Monate). Die ausgewerteten sekundären Endpunkte waren die Pharmakokinetik und die absolute Veränderung der Schweisschloridkonzentration von Baseline in Woche 24 (siehe «Pharmakodynamik»). Entsprechend ihrem Körpergewicht beim Screening erhielten die Patienten mit einer Mahlzeit vermischtes Granulat alle 12 Stunden für 24 Wochen in einer Dosis von Lumacaftor 75 mg/Ivacaftor 94 mg Granulat (Patienten mit einem Körpergewicht von 7 kg bis <9 kg) oder Lumacaftor 100 mg/Ivacaftor 125 mg Granulat (Patienten mit einem Körpergewicht von 9 kg bis <14 kg) oder Lumacaftor 150 mg/Ivacaftor 188 mg Granulat (Patienten mit einem Körpergewicht ≥14 kg) zusätzlich zu ihren verordneten CF-Therapien. Zur Untersuchung von Auswirkungen nach dem Absetzen des Arzneimittels (off-drug effects) nahmen die Patienten im Anschluss an die 2-wöchige Auswaschphase an einer Sicherheitsnachuntersuchung teil.
Studie VX15-809-110: 96-wöchige Verlängerungsstudie
Patienten mit CF ab 6 Jahren aus Studie VX13-809-011B und Studie VX14-809-109 wurden in eine multizentrische Rollover-Verlängerungsstudie der Phase 3 (Studie VX15-809-110) übernommen. Diese nicht kontrollierte Verlängerungsstudie war dazu ausgelegt, die Sicherheit und deskriptive Wirksamkeit einer Langzeitbehandlung mit Lumacaftor/Ivacaftor zu bewerten. Von den 262 Patienten, die in Studie VX13-809-011B oder Studie VX14-809-109 eine der Behandlungen erhielten, wurden in der Verlängerungsstudie 239 (91 %) behandelt und erhielten alle die Behandlung mit Lumacaftor/Ivacaftor (Patienten im Alter von 6 bis <12 Jahren erhielten Lumacaftor 200 mg q12h/Ivacaftor 250 mg q12h; Patienten ≥12 Jahre erhielten Lumacaftor 400 mg q12h/Ivacaftor 250 mg q12h) für bis zu zusätzlich 96 Wochen (d.h. für bis zu insgesamt 120 Wochen) (siehe «Unerwünschte Wirkungen»). Die sekundären deskriptiven Wirksamkeitsergebnisse und die Anzahl der pulmonalen Exazerbationen pro Patientenjahr sind in Tabelle 11 zusammengestellt.
|
Tabelle 11: Langzeitwirkung von Lumacaftor/Ivacaftor in Studie VX15-809-110
| |
|
Umstellung von Placebo auf
Lumacaftor / Ivacaftor
(P-L/I)
(n = 96)*
|
Lumacaftor / Ivacaftor –
Lumacaftor / Ivacaftor
(L/I-L/I)
(n = 143)*
| |
Baseline und Endpunkt
|
Mittelwert (SD)
|
LS-Mittelwert (95 % KI)
|
Mittelwert (SD)
|
LS-Mittelwert (95 % KI)
| |
Absolute Veränderung gegenüber Baseline bei LCI2,5
| |
|
n = 101
|
|
n = 128
|
| |
Baseline LCI2,5‡**
|
10,26
(2,24)
|
|
10,24
(2,42)
|
| |
Verlängerungsstudie Woche 96
|
|
(n = 69)
-0,86
(-1,33; -0,38)
|
|
(n = 88)
-0,85
(-1,25; -0,45)
| |
|
n = 101
|
|
n = 159
|
| |
Baseline ppFEV11 (Prozentpunkte)‡
|
90,7 (10.8)
|
|
89,7 (13.8)
|
| |
Absolute Veränderung gegenüber Baseline des ppFEV1 (Prozentpunkte)
| |
Verlängerungsstudie Woche 96
|
|
n = 79
0,0
(-2,7, 2,7)
|
|
n = 123
3,1
(1,0, 5,1)
| |
|
n = 96
|
|
n = 143
|
| |
Baseline BMI für das Alter, Z-Score (kg/m2)‡
|
-0,16
(0,90)
|
|
-0,09
(0,88)
|
| |
Absolute Veränderung gegenüber Baseline beim BMI für das Alter, Z-Score (kg/m2)
| |
Verlängerungsstudie Woche 96
|
|
(n = 83)
0,31
(0,19; 0,42)
|
|
(n =130)
0,17
(0,08; 0,26)
| |
|
n = 78
|
|
n = 135
|
| |
Baseline CFQ-R‡ Respiratorische-Domäne-Score (Punkte)
|
77,1
(15,5)
|
|
78,5
(14,3)
|
| |
Absolute Veränderung des Respiratorischen-Domäne-Scores des CFQ-R (Punkte)
| |
Verlängerungsstudie Woche 96
|
|
(n = 65)
6,6
(3,1; 10,0)
|
|
(n = 108)
7,4
(4,8; 10,0)
| |
Anzahl der pulmonalen Exazerbationen (Ereignisse) (Studie VX14-809-109 FAS und ROS)†
| |
Anzahl der Ereignisse pro Patientenjahr (95 % KI)
|
|
n = 96
0,30
(0,21; 0,43)
|
|
n = 103
0,45
(0,33; 0,61)
| |
* Studienteilnehmer, die in Studie VX14-809-109 mit Placebo behandelt wurden (n=96) und in der Verlängerungsstudie auf eine Verumbehandlung mit LUM/IVA umgestellt wurden (P-L/I).
Studienteilnehmer, die in einer der beiden Hauptstudien [Studie VX13-809-011B (n=49) oder Studie VX14-809-109 (n=94)] mit LUM/IVA behandelt wurden und in der Verlängerung weiterhin eine Verumbehandlung mit LUM/IVA erhielten (L/I-L/I).
‡ Die Baseline für beide Gruppen (P-L/I und L/I-L/I) war die Studie-VX13-809-011B- und Studie-VX14-809-109- (Hauptstudie) Baseline und die entsprechende Zahl «n» bezieht sich auf das Analyseset der Hauptstudie.
** Die LCI-Substudie schloss 117 Teilnehmer in der L/I-L/I-Gruppe und 96 Teilnehmer in der P-L/I-Gruppe ein.
† FAS = vollständiges Analyseset (n=103), umfasst Teilnehmer, die in Studie VX13-809-109 und Studie VX15-809-110 L/I erhielten und über den kumulativen Studienzeitraum für L/I ausgewertet wurden; ROS = Rollover-Set (n=96), umfasst Teilnehmer, die in Studie VX14-809-109 Placebo erhielten und in Studie VX15-809-110 L/I und über den laufenden Studienzeitraum für Studie VX15-809-110 bewertet wurden.
|
Studie VX16-809-116: 96–wöchige Verlängerungsstudie
Patienten mit CF ab 2 Jahren von Studie VX15-809-115 wurden in eine multizentrische Rollover-Verlängerungsstudie der Phase 3 übernommen (Studie VX16-809-116). Diese nicht kontrollierte Verlängerungsstudie war dazu ausgelegt, die Sicherheit und deskriptive Wirksamkeit einer Langzeitbehandlung mit Lumacaftor/Ivacaftor zu bewerten. Von den 60 Patienten, die in Studie VX15-809-115 eine der Behandlungen erhielten, wurden in der Verlängerungsstudie 57 (95 %) weiterbehandelt. 33,3 % der Patienten waren < 3 Jahre alt und 66,7 % der Patienten waren ≥3 Jahre alt. In den unkontrollierten, deskriptiven Wirksamkeitsuntersuchungen war die Behandlung mit Lumacaftor/Ivacaftor mit einer Verbesserung der Parameter Gewicht und Körpergrösse (BMI, Gewicht, Grösse) einschliesslich z-Scores verbunden. Bei Patienten, die Lumacaftor/Ivacaftor in der Hauptstudie VX15-809-115 erhalten hatten, bevor sie in die Verlängerungsstudie VX16-809-116 aufgenommen wurden, konnte nach einer zusätzlichen 96-wöchigen Behandlung keine klinisch bedeutsame Veränderung in Bezug auf den LCI2,5 (N=17: -0,20, (95% KI -0,99; 0,60)) oder den ppFEV1 (N=13: 0,4, (95% KI -7,5, 8,4)) festgestellt werden.
|