ZusammensetzungWirkstoffe
Acalabrutinib (als Acalabrutinibmaleat-Monohydrat)
Hilfsstoffe
Tablettenkern:
Mannitol (E421)
Mikrokristalline Cellulose
Niedrig substituierte Hydroxypropylcellulose
Natriumstearylfumarat
Filmüberzug:
Hypromellose
Copovidon
Titandioxid (E171)
Macrogol 3350
Mittelkettige Triglyceride
Gelbes Eisenoxid (E172)
Rotes Eisenoxid (E172)
1 Filmtablette enthält 0.59 mg Natrium.
Indikationen/AnwendungsmöglichkeitenBefristet zugelassene Indikation
Mantelzell-Lymphom (MCL)
CALQUENCE in Kombination mit Bendamustin und Rituximab (BR) ist indiziert zur Behandlung von erwachsenen Patienten mit bisher unbehandeltem MCL, die für eine autologe Stammzelltransplantation nicht geeignet sind (siehe «Warnhinweise und Vorsichtsmassnahmen» sowie «Eigenschaften /Wirkungen»).
Aufgrund einer zum Zeitpunkt der Begutachtung des Gesuches unvollständigen Dokumentation, wurde diese Indikation befristet zugelassen (Art. 9a Heilmittelgesetz). Die befristete Zulassung ist zwingend an die zeitgerechte Erfüllung von Auflagen gebunden. Nach deren Erfüllung kann die befristete Zulassung in eine Zulassung ohne besondere Auflagen überführt werden.
Nicht befristet zugelassene Indikationen
Mantelzell-Lymphom (MCL)
CALQUENCE als Monotherapie ist indiziert zur Behandlung von erwachsenen Patienten mit MCL, bei denen kein partielles Ansprechen erreicht wurde mit vorheriger Therapie oder die eine Progression nach der vorherigen Therapie gezeigt haben (siehe «Eigenschaften/Wirkungen»).
Chronische lymphatische Leukämie (CLL)
CALQUENCE als Monotherapie oder in Kombination mit Obinutuzumab ist indiziert zur Behandlung von erwachsenen Patienten mit bisher unbehandelter (CLL), die 65 Jahre und älter sind oder Begleiterkrankungen haben (siehe «Eigenschaften/Wirkungen»).
CALQUENCE als Monotherapie ist indiziert zur Behandlung von erwachsenen Patienten mit CLL, die mindestens eine Vortherapie erhalten haben (siehe «Eigenschaften/Wirkungen»).
Dosierung/AnwendungDie Behandlung mit CALQUENCE sollte von einem in der Krebstherapie erfahrenen Arzt eingeleitet und überwacht werden.
Übliche Dosierung
MCL
Die empfohlene Dosierung von CALQUENCE als Monotherapie beträgt 100 mg (1 Tablette) zweimal täglich. Für die Kombinationstherapie entnehmen Sie bitte die empfohlene Dosierung der Fachinformation des jeweiligen Arzneimittels. Weitere Informationen zur Kombinationstherapie siehe «Eigenschaften/Wirkungen». Die Behandlung mit CALQUENCE sollte bis zum Fortschreiten der Krankheit oder Auftreten einer nicht akzeptablen Toxizität fortgesetzt werden.
CLL
Die empfohlene Dosierung von CALQUENCE beträgt bei der Behandlung der CLL zweimal täglich 100 mg (1 Tablette), entweder als Monotherapie oder in Kombination mit Obinutuzumab. Bitte entnehmen Sie die empfohlene Dosierung von Obinutuzumab der Fachinformation von Obinutuzumab. (Weitere Informationen zur Kombinationstherapie siehe «Eigenschaften/Wirkungen»).
Die Dosen sollten in einem ungefähren Abstand von 12 Stunden eingenommen werden.
Die Behandlung mit CALQUENCE sollte bis zum Fortschreiten der Krankheit oder Auftreten einer nicht akzeptablen Toxizität fortgesetzt werden.
Art der Anwendung
CALQUENCE sollte jeden Tag morgens und abends ungefähr zur gleichen Zeit mit Wasser als Ganzes geschluckt werden. CALQUENCE kann mit oder ohne Nahrung eingenommen werden. Die Tablette darf nicht zerkaut, zerdrückt, aufgelöst oder geteilt werden.
Versäumte Dosis
Hat ein Patient eine Dosis von CALQUENCE um mehr als 3 Stunden versäumt, ist die nächste Dosis zum geplanten Zeitpunkt einzunehmen. Es dürfen keine zusätzlichen Tabletten als Ausgleich für versäumte Dosen eingenommen werden.
Dosisanpassungen aufgrund unerwünschter Wirkungen/Interaktionen
Empfohlene Dosisanpassungen von CALQUENCE für unerwünschte Wirkungen Grad ≥3 bei Patienten, die CALQUENCE als Monotherapie und CALQUENCE in Kombination mit Obinutuzumab erhalten, sind in Tabelle 1 angegeben.
Empfohlene Dosisanpassungen für unerwünschte Wirkungen Grad ≥3 bei Patienten, die CALQUENCE in Kombination mit Bendamustin und Rituximab erhalten, sind in Tabelle 2 angegeben.
Tabelle 1: Empfohlene Dosisanpassungen für unerwünschte Wirkungen bei Patienten, die CALQUENCE als Monotherapie und CALQUENCE in Kombination mit Obinutuzumab erhalten*
|
Unerwünschte Wirkung
|
Auftreten der unerwünschten Wirkung
|
Dosisanpassung
(Anfangsdosis = 100 mg ca. alle 12 Stunden)
| |
Thrombozytopenie Grad 3 mit Blutung,
Thrombozytopenie Grad 4
Oder
Länger als 7 Tage anhaltende Neutropenie Grad 4
Jede andere nicht behandelbare Grad 3 Toxizität, oder jede andere Grad 4 Toxizität
|
Erstes und zweites Mal
|
Aussetzen von CALQUENCE
Nach Abklingen der Toxizität auf Grad 1 oder Wiederherstellung des Ausgangszustands kann die Behandlung mit CALQUENCE 100 mg ca. alle 12 Stunden wiederaufgenommen werden
| |
Drittes Mal
|
Aussetzen von CALQUENCE
Nach Abklingen der Toxizität auf Grad 1 oder Wiederherstellung des Ausgangszustands kann die Behandlung mit CALQUENCE mit reduzierter Verabreichungsfrequenz von 100 mg einmal täglich wiederaufgenommen werden
| |
Viertes Mal
|
Absetzen von CALQUENCE
|
*Der Schweregrad der unerwünschten Wirkungen wird gemäss Version 4.03 der CTCAE-Kriterien (Common Terminology Criteria for Adverse Events) des National Cancer Institute (NCI) eingestuft.
Tabelle 2. Empfohlene Dosisanpassungen für unerwünschte Wirkungen* Grad ≥3 bei Patienten, die CALQUENCE in Kombination mit Bendamustin und Rituximab erhalten
|
Unerwünschte Wirkung
|
Dosisanpassung Bendamustin†
|
Dosisanpassung CALQUENCE
| |
Neutropenie
|
Bei Neutropenie Grad 3 oder Grad 4:
Aussetzen von Bendamustin.
Nach Abklingen der Toxizität auf Grad ≤2 oder Wiederherstellung des Ausgangszustands kann die Behandlung mit Bendamustin 70 mg/m2 wiederaufgenommen werden.
Absetzen von Bendamustin, wenn eine weitere Dosisreduktion erforderlich ist.
|
Bei länger als 7 Tage anhaltender Neutropenie Grad 4 Aussetzen von CALQUENCE.
Nach Abklingen der Toxizität auf Grad ≤2 oder Wiederherstellung des Ausgangszustands kann die Behandlung mit CALQUENCE mit der Anfangsdosis (1. Auftreten der unerwünschten Wirkung) oder mit reduzierter Verabreichungsfrequenz von 100 mg einmal täglich (2. und 3. Auftreten der unerwünschten Wirkung) wiederaufgenommen werden.
Absetzen von CALQUENCE, wenn die unerwünschte Wirkung zum 4. Mal auftritt.
| |
Thrombozytopenie
|
Bei Thrombozytopenie Grad 3 oder Grad 4:
Aussetzen von Bendamustin.
Nach Abklingen der Toxizität auf Grad 2 oder Wiederherstellung des Ausgangszustands kann die Behandlung mit Bendamustin 70 mg/m2 wiederaufgenommen werden.
Absetzen von Bendamustin, wenn eine weitere Dosisreduktion erforderlich ist.
|
Bei Thrombozytopenie Grad 3 mit relevanter Blutung oder Grad 4 Aussetzen von CALQUENCE.
Nach Abklingen der Toxizität auf Grad ≤2 oder Wiederherstellung des Ausgangszustands kann die Behandlung mit CALQUENCE mit der Anfangsdosis (1. Auftreten der unerwünschten Wirkung) oder mit reduzierter Verabreichungsfrequenz von 100 mg einmal täglich (2. und 3. Auftreten) wiederaufgenommen werden.
Absetzen von CALQUENCE, wenn die unerwünschte Wirkung einer Thrombozytopenie mit relevanter Blutung zum 3. Mal auftritt.
Absetzen von CALQUENCE, wenn die unerwünschte Wirkung zum 4. Mal auftritt.
| |
Andere hämatologische Toxizität Grad 4‡ oder unbehandelbare Toxizität Grad 3
|
Aussetzen von Bendamustin.
Nach Abklingen der Toxizität auf Grad ≤2 oder Wiederherstellung des Ausgangszustands kann die Behandlung mit Bendamustin 70 mg/m2 wiederaufgenommen werden.
Absetzen von Bendamustin, wenn eine weitere Dosisreduktion erforderlich ist.
|
Aussetzen von CALQUENCE.
Nach Abklingen der Toxizität auf Grad ≤2 oder Wiederherstellung des Ausgangszustands kann die Behandlung mit CALQUENCE mit der Anfangsdosis (1. Auftreten der unerwünschten Wirkung) oder mit reduzierter Verabreichungsfrequenz von 100 mg einmal täglich (2. und 3. Auftreten der unerwünschten Wirkung) wiederaufgenommen werden.
Absetzen von CALQUENCE, wenn die unerwünschte Wirkung zum 4. Mal auftritt.
| |
Nicht hämatologische Toxizitäten Grad 3 oder höher
|
Aussetzen von Bendamustin.
Nach Abklingen der Toxizität auf Grad 1 oder Wiederherstellung des Ausgangszustands kann die Behandlung mit Bendamustin 70 mg/m2 wiederaufgenommen werden.
Absetzen von Bendamustin, wenn eine weitere Dosisreduktion erforderlich ist.
|
Aussetzen von CALQUENCE.
Nach Abklingen der Toxizität auf Grad 2 oder Wiederherstellung des Ausgangszustands kann die Behandlung mit CALQUENCE mit der Anfangsdosis (1. Auftreten der unerwünschten Wirkung) oder mit reduzierter Verabreichungsfrequenz von 100 mg einmal täglich (2. Auftreten der unerwünschten Wirkung) wiederaufgenommen werden.
Absetzen von CALQUENCE, wenn die unerwünschte Wirkung zum 3. Mal auftritt.
|
*Der Schweregrad der unerwünschten Wirkungen wird gemäss Version 4.03 der CTCAE-Kriterien (Common Terminology Criteria for Adverse Events) des National Cancer Institute (NCI) eingestuft.
†Toxizitäten, die nicht in dieser Tabelle aufgeführt sind, sind der lokalen Fachinformation von Bendamustin zu entnehmen.
‡ Lymphopenie Grad 4 ist ein zu erwartendes Ergebnis der Behandlung mit Bendamustin und Rituximab. Eine Dosisanpassung aufgrund einer Lymphopenie wird nur erwartet, wenn der Prüfarzt dies als klinisch wichtig erachtet, z.B. bei wiederkehrenden Infektionen.
Weitere Informationen zur Behandlung von Toxizitäten sind der Fachinformation des jeweiligen in Kombination mit CALQUENCE angewendeten Arzneimittels zu entnehmen.
Tabelle 3 enthält Empfehlungen zur Anwendung von Calquence mit CYP3A-Inhibitoren oder -Induktoren.
Tabelle 3: Anwendung mit CYP3A4 - Inhibitoren oder - Induktoren
|
|
Gleichzeitig verabreichtes Arzneimittel
|
Empfohlene Anwendung von CALQUENCE
| |
CYP3A-Inhibitoren
|
Starke CYP3A-Inhibitoren
|
Gleichzeitige Anwendung vermeiden.
Wenn diese Inhibitoren kurzfristig angewendet werden (z.B. Antiinfektiva bis zu 7 Tage), Aussetzen von CALQUENCE.
| |
Moderate CYP3A-Inhibitoren
|
Keine Dosisanpassung. Patienten sollten engmaschig in Bezug auf Nebenwirkungen überwacht werden, wenn sie gleichzeitig moderate CYP3A4-Inhibitoren einnehmen.
| |
CYP3A-Induktoren
|
Starke CYP3A-Induktoren
|
Gleichzeitige Anwendung vermeiden; alternative Wirkstoffe mit geringerer CYP3A-Induktion erwägen.
|
Spezielle Dosierungsanweisungen
Patienten mit Nierenfunktionsstörungen
Bei Patienten mit leichter bis mässiger Nierenfunktionsstörung (eGFR ≥30 ml/min/1,73 m2) wird gemäss MDRD (Modification of Diet in Renal Disease Equation - Formel zur Anpassung der Ernährung bei Nierenerkrankungen) keine Dosisanpassung empfohlen. Die Pharmakokinetik und Sicherheit von CALQUENCE bei Patienten mit schwerer Nierenfunktionsstörung (eGFR <29 ml/min/1,73 m2) oder terminaler Niereninsuffizienz wurden nicht untersucht (siehe «Pharmakokinetik»).
Patienten mit Leberfunktionsstörungen
Bei Patienten mit leichter oder mässiger Einschränkung der Leberfunktion (Child-Pugh A, Child-Pugh B oder Gesamt-Bilirubin zwischen der 1,5- und 3-fachen Obergrenze des Normbereichs (Upper Limit of Normal, ULN) und jeglicher AST (Aspartat-Aminotransferase)) wird keine Dosisanpassung empfohlen. Patienten mit schwerer Einschränkung der Leberfunktion (Child-Pugh C oder Gesamtbilirubin in Höhe des >3-fachen der ULN und beliebige AST) sollten CALQUENCE nicht einnehmen (siehe «Pharmakokinetik»).
Ältere Patienten (≥65 Jahre)
Es sind keine altersbedingten Dosisanpassungen nötig (siehe «Pharmakokinetik»).
Kinder und Jugendliche
Die Sicherheit und Wirksamkeit von CALQUENCE bei Kindern und Jugendlichen unter 18 Jahren ist nicht erwiesen.
KontraindikationenBekannte Überempfindlichkeit gegenüber dem Wirkstoff oder einem der Hilfsstoffe.
Warnhinweise und VorsichtsmassnahmenZweite primäre maligne Erkrankungen
Bei 17,6% der Patienten mit hämatologischen Malignom, die CALQUENCE als Monotherapie erhielten (n = 1478) und bei 20,4% der mit CALQUENCE in Kombination mit anderen Arzneimitteln behandelten Patienten wurde ein zweiter Primärtumor beschrieben. Es handelte sich dabei um Hautneoplasmen und andere Neoplasmen. Dabei waren nicht-melanomatöse Hautneoplasmen der häufigste zweite Primärtumor und traten bei 9,9% (alle Grade) der Patienten, welche mit CALQUENCE Monotherapie und bei 12,5% der Patienten, welche mit CALQUENCE in Kombination mit anderen Arzneimitteln behandelt wurden, auf.
Patienten sind bezüglich des Auftretens von Zweittumoren zu überwachen und sollten Sonnenexposition vermeiden.
Infektionen
Infektionen traten bei Patienten mit hämatologischem Malignom unter CALQUENCE als Monotherapie (74,3%, n=1478) und unter CALQUENCE als Kombinationstherapie mit anderen Arzneimitteln (81,3%, n=520) auf. Am häufigsten waren Infektionen der oberen Atemwege (25,8% bzw. 26,7%), Pneumonie (15,8% bzw. 15,4%) und Sinusitis (11,4% bzw. 11,2%). Schwere Infektionen (Bakterien-, Viren- oder Pilzinfektionen), darunter auch Ereignisse mit tödlichem Ausgang, traten bei Patienten mit hämatologischem Malignom auf, die CALQUENCE als Monotherapie (25,3%) und CALQUENCE als Kombinationstherapie mit anderen Arzneimitteln (37,1%) erhielten. Diese Infektionen traten vorwiegend ohne Vorliegen einer Neutropenie Grad 3 oder 4 auf. Infektionen aufgrund einer Reaktivierung des Hepatitis-B-Virus (HBV) und Herpes-zoster-Virus (HSV), Aspergillose und progressive multifokale Leukenzephalopathie (PML) sind aufgetreten (siehe Rubrik «Unerwünschte Wirkungen»).
Bei mit CALQUENCE behandelten Patienten wurde über die Reaktivierung einer Hepatitis B berichtet. Vor Einleitung einer Behandlung mit CALQUENCE ist der Hepatitis-B-Virus (HBV)-Status zu erheben. Wenn Patienten eine positive Hepatitis-B-Serologie aufweisen, wird empfohlen, vor Therapiebeginn einen Experten für Lebererkrankungen zu konsultieren. Die Patienten sind bezüglich der Prävention einer Hepatitis-B-Reaktivierung zu überwachen und zu behandeln.
Nach Anwendung von CALQUENCE bei früherer oder gleichzeitiger immunsuppressiver Therapie wurde über das Auftreten progressiver multifokaler Leukenzephalopathie (PML) einschliesslich Fälle mit tödlichem Ausgang berichtet. Bei Patienten mit neu auftretenden oder sich verschlechternden neurologischen, kognitiven oder verhaltensbezogenen Anzeichen oder Symptomen sollte der Arzt eine PML bei der Differentialdiagnose in Erwägung ziehen. Bei Verdacht auf PML sind angemessene diagnostische Beurteilungen durchzuführen und die Behandlung mit CALQUENCE sollte bis zum Ausschluss einer PML ausgesetzt werden. Wenn irgendwelche Zweifel bestehen, sind die Überweisung an einen Neurologen und angemessene Diagnoseverfahren für PML einschliesslich einer MRT-Untersuchung, vorzugsweise mit Kontrastmittel, Liquortests auf DNA des JC-Virus und wiederholte neurologische Kontrolluntersuchungen in Erwägung zu ziehen.
Bei Patienten mit erhöhtem Risiko für opportunistische Infektionen ist eine Prophylaxe gemäss Therapiestandard zu erwägen. Die Patienten sind auf Anzeichen und Symptome von Infektionen zu überwachen und gegebenenfalls entsprechend zu behandeln.
Blutungen
Bei Patienten mit hämatologischem Malignom, die CALQUENCE als Monotherapie und CALQUENCE als Kombinationstherapie mit anderen Arzneimitteln erhielten, wurden schwerwiegende hämorrhagische Ereignisse berichtet. Darunter waren auch Ereignisse mit tödlichem Ausgang. Schwere Blutungen (Blutungsereignisse Grad 3 oder höher, schwerwiegende oder das Zentralnervensystem betreffende Ereignisse) wurden bei 4,5% bzw. 4,0% der Patienten beobachtet und verliefen in 0,1% bzw. 0,2% der Fälle tödlich. Die häufigsten schweren Blutungen waren gastrointestinale Blutung (0,5 bzw. 0,4%), Hämaturie (0,4% bzw. 0,8%), Hämatom (0,5% bzw. 0,2%), Epistaxis (0,3% bzw. 0%), intrakranielle Blutung (0,3% bzw. 0%), Netzhautblutung (0,3% bzw. 0,2%) und Magenblutung (0,2% bzw. 0,2%). Insgesamt traten bei 46,1% bzw. 41,3% der Patienten mit Blutungsereignisse (alle Grade) auf, darunter Hämatome und Petechien.
Der diesen Blutungsereignissen zugrunde liegende Mechanismus ist noch unklar.
Warfarin oder andere Vitamin K-Antagonisten sollten nicht gleichzeitig mit CALQUENCE verabreicht werden.
Patienten, die eine antithrombotische Therapie erhalten, können von einem erhöhten Blutungsrisiko betroffen sein. Bei Anwendung zusammen mit antithrombotischen Substanzen ist Vorsicht geboten. Wenn die gemeinsame Anwendung medizinisch erforderlich ist, ist eine zusätzliche Überwachung der Patienten auf Anzeichen und Symptome von Blutungen erforderlich.
Aufgrund einer Nutzen-Risiko-Abschätzung sollte CALQUENCE im Zeitraum von mindestens 3 Tagen vor und 3 Tagen nach einer Operation nicht angewendet werden.
Zytopenien
Zytopenien traten bei Patienten mit hämatologischem Malignom auf, die mit CALQUENCE als Monotherapie und mit CALQUENCE als Kombinationstherapie mit anderen Arzneimitteln behandelt wurden. Die Gesamthäufigkeit für Neutropenie betrug 15,5% bzw. 34,6%, für Anämie 16,6% bzw. 18,8% und für Thrombozytopenie 8,7% bzw. 12,3%. Bei Patienten mit hämatologischem Malignom, die CALQUENCE als Monotherapie (n = 1478) und CALQUENCE als Kombinationstherapie mit anderen Arzneimitteln (n = 520) erhielten, traten unter der Behandlung labormedizinisch nachweisbare Zytopenien Grad ≥3, einschliesslich Neutropenie (17,5% bzw. 42,1%), Anämie (9,5% bzw. 8,5%) und Thrombozytopenie (6,2% bzw. 9,4%) auf.
Eine Überwachung des grossen Blutbildes wird daher empfohlen sofern medizinisch angemessen.
Vorhofflimmern
Bei 2,3% der Patienten mit hämatologischem Malignom, die CALQUENCE als Monotherapie erhielten (n=1478), wurde Vorhofflimmern/-flattern Grad 3 oder höher beschrieben, während insgesamt bei 7,4% Vorhofflimmern/-flattern jeglichen Grades auftrat. Die Patienten sind auf Symptome (z.B. Palpitationen, Schwindelgefühl, Synkope, Thoraxschmerz, Dyspnoe) von Vorhofflimmern oder -flattern zu überwachen, wenn nötig sollte ein EKG aufgezeichnet werden.
Tumorlysesyndrom (TLS)
In Zusammenhang mit der Anwendung von Acalabrutinib wurde über Tumorlysesyndrom berichtet. Bei Patienten mit hoher Tumorlast vor der Behandlung besteht die Gefahr eines Tumorlysesyndroms. Die Patienten sind engmaschig zu überwachen und es müssen entsprechende Vorsichtsmassnahmen getroffen werden.
Hepatotoxizität, einschliesslich arzneimittelinduzierte Leberschädigung (drug-induced liver injury, DILI)
Hepatotoxizität, einschliesslich schwerer, lebensbedrohlicher und potenziell tödlicher Fälle von arzneimittelinduzierter Leberschädigung (drug-induced liver injury, DILI), ist bei Patienten aufgetreten, die mit Bruton-Tyrosinkinase-Inhibitoren, einschliesslich CALQUENCE, behandelt wurden. Vor Beginn und während der Behandlung mit CALQUENCE sind die Bilirubin- und Transaminasenwerte zu überwachen. Patienten, die nach Anwendung von CALQUENCE abnormale Leberfunktionswerte entwickeln, sollten häufiger auf Abnormalitäten in Leberfunktionstests und klinische Anzeichen einer Hepatotoxizität überwacht werden. Bei Verdacht auf eine DILI ist die Therapie mit CALQUENCE auszusetzen. Nach Bestätigung einer DILI muss CALQUENCE abgesetzt werden.
Potentielle Risikopopulationen, die nicht untersucht wurden
Patienten mit Lymphom des Zentralnervensystems (ZNS) oder Leukämie, bekannter Prolymphozytenleukämie oder anamnestisch bekanntem bzw. aktuell vermutetem Richter-Transformation, relevanter kardiovaskulärer Erkrankung, unkontrollierter aktiver systemischer Bakterien-, Viren- oder Pilzinfektion oder einer anderen Infektion, einschliesslich aktiver Hepatitis B oder C, anamnestisch bekannter HIV-Infektion, arzneimittelinduzierter Pneumonitis, anamnestisch bekanntem Schlaganfall oder intrakranieller Blutung innerhalb der letzten 6 Monate vor Verabreichung der ersten Dosis des Prüfpräparats, anamnestisch bekannter Blutungsdiathese, Antikoagulation mit Warfarin oder äquivalenten Vitamin-K-Antagonisten; Behandlung mit Protonenpumpeninhibitoren oder Bedarf an Steroiden mit einer täglichen systemischen Exposition von >20 mg eines Prednison-Äquivalents waren in den klinischen Studien ausgeschlossen.
Dieses Arzneimittel enthält weniger als 1 mmol Natrium (23 mg) pro Tablette, d.h. es ist nahezu «natriumfrei».
InteraktionenWirkstoffe, die die Plasmakonzentration von Acalabrutinib erhöhen können
CYP3A-Inhibitoren
Die gleichzeitige Verabreichung eines starken CYP3A-Inhibitors (200 mg Itraconazol einmal täglich für 5 Tage) hatte bei gesunden Probanden (n=17) einen Anstieg der Cmax von Acalabrutinib auf das 3,9-fache und einen Anstieg der AUC auf das 5,1-fache zur Folge.
Eine gleichzeitige Anwendung von starken CYP3A-/P-gp-Inhibitoren sollte vermieden werden. Falls die starken CYP3A-/P-gp-Inhibitoren (z.B. Ketoconazol, Conivaptan, Clarithromycin, Indinavir, Itraconazol, Ritonavir, Telaprevir, Posaconazol, Voriconazol) kurzzeitig angewendet werden, sollte die Behandlung mit Calquence unterbrochen werden (siehe «Dosierung/Anwendung»).
Die gleichzeitige Gabe moderater CYP3A4-Inhibitoren (400 mg Fluconazol als Einzeldosis oder 200 mg Isavuconazol in wiederholter Dosis für 5 Tage) erhöhte bei gesunden Probanden die Cmax und die AUC von Acalabrutinib um das 1,4-Fache bis 2-Fache, wohingegen die Cmax und die AUC des aktiven Metaboliten ACP-5862 um das 0,65-Fache bis 0,88-Fache bezogen auf die alleinige Gabe von Acalabrutinib abnahmen. In Kombination mit moderaten CYP3A4-Inhibitoren ist keine Dosisanpassung erforderlich. Patienten sollten engmaschig auf Nebenwirkungen überwacht werden (siehe «Dosierung/Anwendung»).
Wirkstoffe, die die Plasmakonzentration von Acalabrutinib senken können
CYP3A-Induktoren
Die gleichzeitige Verabreichung eines starken CYP3A-Induktors (600 mg Rifampicin einmal täglich für 9 Tage) hatte bei gesunden Probanden (n=24) eine Abnahme der Cmax von Acalabrutinib um 68% und eine Abnahme der AUC um 77% zur Folge.
Magensäurereduzierende Medikamente
Es wurden keine klinisch signifikanten Unterschiede in der Pharmakokinetik von Acalabrutinib festgestellt, wenn das Arzneimittel gleichzeitig mit dem Protonenpumpenhemmer Rabeprazol angewendet wurde. Acalabrutinib Tabletten können gleichzeitig mit magensäurereduzierenden Wirkstoffen (Protonenpumpenhemmer, H2-Rezeptorantagonisten, Antazida) verabreicht werden.
Wirkung von Acalabrutinib und seinem aktiven Metaboliten ACP-5862 auf den Metabolismus anderer Substanzen
In vitro ist Acalabrutinib ein schwacher Inhibitor von CYP3A4/5, CYP2C8 und CYP2C9, hat jedoch keine hemmende Wirkung auf CYP1A2, CYP2B6, CYP2C19, CYP2D6, UGT1A1 und UGT2B7.
ACP-5862 ist in vitro ein schwacher Inhibitor von CYP2C8, CYP2C9 und CYP2C19, während es CYP1A2, CYP2B6, CYP2D6, CYP3A4/5, UGT1A1 und UGT2B7 nicht hemmt.
Acalabrutinib ist ein schwacher Induktor von CYP1A2-, CYP2B6- und CYP3A4-mRNA; ACP-5862 bewirkt eine schwache Induktion von CYP3A4.
CYP3A-Substrate
Basierend auf invitro-Daten, klinischen Daten und PBPK-Modellierung wird in den klinisch relevanten Konzentrationen keine Wechselwirkung mit CYP3A4-Substraten erwartet (siehe «Eigenschaften/Wirkungen»).
Wirkung von Acalabrutinib und seinem aktiven Metaboliten, ACP-5862, auf Wirkstofftransportsysteme
Durch Hemmung von BCRP (Breast Cancer Resistance Protein) im Darm kann Acalabrutinib die Exposition gegenüber gleichzeitig verabreichten Substraten von BCRP (z.B. Methotrexat) erhöhen.
Durch Hemmung von MATE1 kann ACP-5862 die Exposition gegenüber gleichzeitig verabreichten Substraten von MATE1 (z.B. Metformin) erhöhen.
Wechselwirkung mit Transportproteinen
In vitro sind Acalabrutinib und sein aktiver Metabolit, ACP-5862, Substrate von P-Glycoprotein (P-gp) und Breast Cancer Resistance Protein (BCRP). Acalabrutinib ist in vitro kein Substrat der renalen Aufnahmetransporter OAT1, OAT3 und OCT2 oder der hepatischen Transporter OATP1B1 und OATP1B3. ACP-5862 ist kein Substrat von OATP1B1 oder OATP1B3.
Acalabrutinib und ACP-5862 haben in klinisch relevanten Konzentrationen keinen hemmenden Einfluss auf P-gp, OAT1, OAT3, OCT2, OATP1B1, OATP1B3 und MATE2-K.
Schwangerschaft, StillzeitSchwangerschaft
Es liegen keine ausreichenden klinischen Daten zur Anwendung von CALQUENCE bei Schwangeren vor. Aufgrund von Befunden aus tierexperimentellen Studien kann bei Exposition gegenüber Acalabrutinib während der Schwangerschaft ein Risiko für den Fetus und einen gestörten Geburtsverlauf (Dystokie) bestehen (siehe «Präklinische Daten»).
Während der Schwangerschaft darf das CALQUENCE nicht verabreicht werden, es sei denn, dies ist eindeutig erforderlich.
Frauen im gebärfähigen Alter respektive Patienten mit einer Partnerin im gebärfähigen Alter sollen während der Behandlung mit CALQUENCE und für mindestens 1 Woche nach Erhalt der letzten Dosis eine sehr zuverlässige Verhütungsmethode anwenden. Bei Anwendung einer hormonalen Methode zur Empfängnisverhütung sollte zusätzlich eine Barrieremethode angewendet werden.
Wenn die Patientin während der Einnahme von CALQUENCE schwanger wird, muss sie über die mögliche Gefährdung des Föten informiert werden.
Stillzeit
Es ist nicht bekannt, ob Acalabrutinib bzw. seine Metaboliten in die Muttermilch übergehen. Daten zu den Auswirkungen von Acalabrutinib auf einen gestillten Säugling oder die Milchproduktion liegen nicht vor. Acalabrutinib und sein aktiver Metabolit wurden über die Milch von Ratten ausgeschieden (siehe «Präklinische Daten»). Ein Risiko für den gestillten Säugling kann nicht ausgeschlossen werden. Stillenden Müttern wird empfohlen, während der Behandlung mit CALQUENCE und für mindestens 2 Wochen nach Erhalt der letzten Dosis nicht zu stillen.
Fertilität
Es liegen keine Daten zu den Auswirkungen von CALQUENCE auf die Fertilität beim Menschen vor. In einer präklinischen Studie zu Acalabrutinib wurden bei männlichen und weiblichen Ratten keine unerwünschten Effekte auf Fertilitätsparameter beobachtet (siehe «Präklinische Daten»).
Wirkung auf die Fahrtüchtigkeit und auf das Bedienen von MaschinenDie Wirkung von CALQUENCE auf die Fahrtüchtigkeit und das Bedienen von Maschinen ist nicht untersucht worden. Da während der Behandlung mit CALQUENCE jedoch von Kopfschmerzen, Fatigue, Schwindel, Stürzen und Synkopen berichtet wurde, sollten Patienten, bei denen diese Symptome auftreten, angewiesen werden, bis zum Abklingen dieser Symptome nicht Auto zu fahren oder Maschinen zu bedienen. Patienten sollten auf das mögliche Auftreten dieser Wirkungen hingewiesen werden (siehe «Unerwünschte Wirkungen»).
Unerwünschte WirkungenDas Gesamtsicherheitsprofil von Acalabrutinib basiert auf den gepoolten Daten von 1478 Patienten mit hämatologischen Malignom, die Acalabrutinib als Monotherapie erhielten und den gepoolten Daten von 520 Patienten, die mit der Kombination Acalabrutinib und entweder Obinutuzumab (n=223 Patienten) oder Bendamustin und Rituximab (n=297 Patienten) behandelt wurden. Die mediane Dauer der Behandlung mit CALQUENCE betrug 28.2 Monate für die Monotherapie und 49.7 Monate für die Kombinationstherapie.
CALQUENCE als Monotherapie
Bei Patienten unter Behandlung mit CALQUENCE als Monotherapie waren die häufigsten (≥10%) gemeldeten unerwünschten Wirkungen jeden Grades Hämoglobin erniedrigt (47,4%), absolute Neutrophilenzahl erniedrigt (43,9%), Thrombozyten erniedrigt (36,9%), Diarrhoe (36,7%), Kopfschmerzen (36,5%), Schmerzen des Muskel- und Skelettsystems (31,9%), Bluterguss (30,9%), Infektion der oberen Atemwege (25,8%), Husten (25,2%), Arthralgie (24,0%), Ermüdung (23,6%), Übelkeit (21,8%), Leukopenie (20,8%), Ausschlag (20,3%), Kontusion (20,2%), Neutropenie (19,4%), zweiter Primärtumor (17,6%), Anämie (17,1%), Schwindelgefühl/Schwindel (16,5%), Blutung / Hämatom (16,3%), Pneumonie (15,8%), Obstipation (15,2%), Abdominalschmerz (14,5%), Erbrechen (14,0%), Thrombozytopenie (11,5%), Sinusitis (11,4%) und Bluthochdruck (11,2%).
Die am häufigsten (≥5%) gemeldeten unerwünschten Wirkungen Grad ≥3 waren absolute Neutrophilenzahl erniedrigt (24%), Leukopenie (18,2%), Neutropenie (17,5%), Hämoglobin erniedrigt (10,8%), Thrombozyten erniedrigt (9,5%), Anämie (9,5%), Pneumonie (8,7%), zweiter Primärtumor (6,7%), Thrombozytopenie (6,2%) und zweiter Primärtumor ohne Nicht-Melanom-Hautkrebs (5,5%).
Die häufigsten schwerwiegenden unerwünschten Wirkungen (≥1%), die auch letale Ereignisse umfassten, waren Infektionen, darunter Pneumonie (8,3%) und Sepsis (2,8%) zweite Primärtumoren (7,1%) sowie Blutungen / Hämatome (2,8%), Leukopenie (2,2%), Neutropenie (2,2%) und Anämie (2,7%). Diese Infektionen traten vorwiegend ohne Vorliegen einer Neutropenie Grad 3 oder 4 auf (siehe Abschnitt «Warnhinweise und Vorsichtsmassnahmen»).
Bei 5,9% der Patienten wurden Dosisreduktionen aufgrund von unerwünschten Ereignissen berichtet. Bei 15,8% der Patienten wurde die Behandlung aufgrund von unerwünschten Ereignissen abgebrochen, wobei Pneumonie (0,8%), COVID-19 (0,6%), COVID-19-Pneumonie (0,5%) und Thrombozytopenie (0,4%) die häufigsten Ereignisse waren, die zum Abbruch führten.
CALQUENCE als Kombinationstherapie
Bei den 520 Patienten unter Behandlung mit CALQUENCE als Kombinationstherapie waren die häufigsten (≥10%) gemeldeten unerwünschten Wirkungen jeden Grades absolute Neutrophilenzahl erniedrigt (69,8%), Hämoglobin erniedrigt (66,7%), Thrombozyten erniedrigt (60,2%), Leukopenie (48,7%), Neutropenie (46,0%), Diarrhoe (42,5%), Schmerzen des Muskel- und Skelettsystems (42,1%), Übelkeit (37,7%), Ausschlag (36,3%), Kopfschmerzen (36,0%), Ermüdung (31,2%), Husten (30,8%), Arthralgie (27,9%), Bluterguss (26,9%), Infektion der oberen Atemwege (26,7%), Erbrechen (24,8%), Obstipation (24,4%), Schwindelgefühl/Schwindel (23,7%), Kontusion (20,6%), zweiter Primärtumor (20,4%), Anämie (20,2%), Thrombozytopenie (19,8%), Blutung / Hämatom (19,0%), Abdominalschmerz (16,0%), Pneumonie (15,4%), Bluthochdruck (14,4%), Harnwegsinfektion (14,0%), Nicht-Melanom-Hautkrebs (12,5%), zweiter Primärtumor ohne Nicht-Melanom-Hautkrebs (11,3%), Herpes-Virusinfektionen (11,3%), Sinusitis (11,2%), Nasopharyngitis (10,4%), und Asthenie (10,0%).
Die am häufigsten (≥5%) gemeldeten unerwünschten Wirkungen Grad ≥3 waren absolute Neutrophilenzahl erniedrigt (48,5%), Leukopenie (44,0%), Neutropenie (42,1%), Thrombozyten erniedrigt (15,6%), Hämoglobin erniedrigt (10,6%), Thrombozytopenie (9,4%), Anämie (8,5%), zweiter Primärtumor (8,1%), Pneumonie (8,1%), Ausschlag (6,3%), zweiter Primärtumor ohne Nicht-Melanom-Hautkrebs (6,0%), and Bluthochdruck (5,4%).
In gepoolten Analysen der Patienten, die mit der Kombination Acalabrutinib plus Obinutuzumab behandelt wurden (n=223), wurde gegenüber den Patienten unter Monotherapie mit Acalabrutinib (n=1040) eine höhere Gesamthäufigkeit folgender unerwünschter Wirkungen beobachtet: Infektionen (74 vs. 66,7%) einschliesslich Infektionen Grad ≥3 (21,5 vs. 17,6%), Infektionen der oberen Atemwege (31,4 vs. 22%) und anderer sehr häufiger Infektionen, Skelettmuskulatur-, Bindegewebs- und Knochenerkrankungen (58,3 vs. 51,6%), vorwiegend Arthralgie (26,9 vs. 19,1%) und Schmerz in einer Extremität (13,9 vs. 8,9%), Ermüdung (30,5 vs. 21,3%), Kontusion (27,4 vs. 21,7%), Schwindelgefühl (23,8 vs. 13,4%) und Stürze (14,8 vs. 7,9%). Die Gesamthäufigkeit von unerwünschten Ereignissen ≥Grad 3 (70,4 vs. 54,1%) war im Kombinationspool gegenüber dem Monotherapiepool ebenfalls erhöht und vorwiegend durch eine höhere Inzidenz von Neutropenie Grad ≥3 (23,8 vs. 11,2%) bedingt. Zusätzlich wurden höhere Raten mit ≥10% PT-Differenz für Neutropenie (25,1 vs. 12,3%), Reaktionen im Zusammenhang mit einer Infusion (19,3 vs. 0,8%) und makulo-papulöser Ausschlag (17 vs. 4,9%) beobachtet.
Die unerwünschten Wirkungen in klinischen Studien mit Patienten unter Acalabrutinib-Monotherapie vs. Kombinationstherapie mit Acalabrutinib und anderen Arzneimitteln (Obinutuzumab oder Acalabrutinib mit Bendamustin und Rituximab) sind in Tabelle 4 angegeben.
Unerwünschte Wirkungen sind nach den Systemorganklassen (SOC) gemäss MedDRA aufgeführt. Innerhalb jeder Systemorganklasse sind die unerwünschten Wirkungen nach Häufigkeit dargestellt, wobei die häufigste Wirkung zuerst genannt wird. Zusätzlich basiert die jeweilige Häufigkeitskategorie jeder unerwünschten Wirkung auf der CIOMS-III-Konvention und sind wie folgt definiert: sehr häufig (≥1/10), häufig (≥1/100, <1/10); gelegentlich (≥1/1.000, <1/100); selten (≥1/10.000, <1/1000); sehr selten (<1/10.000); nicht bekannt (Häufigkeit auf Grundlage der verfügbaren Daten nicht abschätzbar).
Tabelle 4. Unerwünschte Wirkungen* bei Patienten mit hämatologischem Malignom, die Acalabrutinib als Monotherapie erhielten (n=1478) oder als Kombinationstherapie mit anderen Arzneimitteln (n=520)
|
MedDRA-SOC
|
MedDRA-Term
|
CIOMS-Deskriptor/ Gesamthäufigkeit
aller CTCAE-Grade [Häufigkeit CTCAE-Grad ≥3]†
| |
Monotherapie
|
Kombinationstherapie
| |
Infektionen und parasitäre Erkrankungen
|
Infektion der oberen Atemwege
|
Sehr häufig (25,8%) [1,2%]
|
Sehr häufig (26,7%) [1,2%]
| |
Sinusitis
|
Sehr häufig (11,4%) [0,4%]
|
Sehr häufig (11,2%) [0,4%]
| |
Pneumonie
|
Sehr häufig (15,8%) [8,7%]
|
Sehr häufig (15,4%) [8,1%]
| |
Harnwegsinfektion
|
Häufig (9,9%) [1,8%]
|
Sehr häufig (14,0%) [1,5%]
| |
Nasopharyngitis
|
Häufig (8,3%) [0%]
|
Sehr häufig (10,4%) [0,2%]
| |
Bronchitis
|
Häufig (9,7%) [0,6%]
|
Häufig (9,8%) [0,6%]
| |
Herpes-Virusinfektionen1
|
Häufig (8,9%) [0,9%]
|
Sehr häufig (11,3%) [1,3%]
| |
Sepsis1
|
Häufig (3,2%) [3,0%]
|
Häufig (4,8%) [4,8%]
| |
Aspergillus-Infektionen1
|
Gelegentlich (0,3%) [0,2%]
|
Sehr selten (0%) [0%]
| |
Reaktivierung einer Hepatitis B
|
Gelegentlich (0,4%) [0,3%]
|
Häufig (1,2%) [0,2%]
| |
Gutartige, bösartige und unspezifische Neubildungen6
|
Zweiter Primärtumor2
|
Sehr häufig (17,6%) [6,7%]
|
Sehr häufig (20,4%) [8,1%]
| |
Zweiter Primärtumor ohne Nicht-Melanom-Hautkrebs3
|
Häufig (9,7%) [5,5%]
|
Sehr häufig (11,3%) [6,0%]
| |
Nicht-Melanom-Hautkrebs
|
Häufig (9,9%) [1,4%]
|
Sehr häufig (12,5%) [2,3%]
| |
Erkrankungen des Blutes und des Lymphsystems
|
Neutropenie1
|
Sehr häufig (19,4%) [17,5%]
|
Sehr häufig (46,0%) [42,1%]
| |
Anämie1
|
Sehr häufig (17,1%) [9,5%]
|
Sehr häufig (20,2%) [8,5%]
| |
Thrombozytopenie1
|
Sehr häufig (11,5%) [6,2%]
|
Sehr häufig (19,8%) [9,4%]
| |
Leukopenie1
|
Sehr häufig (20,8%) [18,2%]
|
Sehr häufig (48,7%) [44,0%]
| |
Lymphozytose
|
Gelegentlich (0,5%) [0,3%]
|
Gelegentlich (0,6%) [0,2%]
| |
Absolute Neutrophilenzahl erniedrigt7
|
Sehr häufig (43,9%) [24,0%]
|
Sehr häufig (69,8%) [48,5%]
| |
Hämoglobin erniedrigt7
|
Sehr häufig (47,4%) [10,8%]
|
Sehr häufig (66,7%) [10,6%]
| |
Thrombozyten erniedrigt7
|
Sehr häufig (36,9%) [9,5%]
|
Sehr häufig (60,2%) [15,6%]
| |
Stoffwechsel- und Ernährungsstörungen
|
Tumorlysesyndrom
|
Gelegentlich (0,5%) [0,4%]
|
Häufig (1,5%) [1,5%]
| |
Erkrankungen des Nervensystems
|
Kopfschmerzen
|
Sehr häufig (36,5%) [1,2%]
|
Sehr häufig (36,0%) [1,3%]
| |
Schwindelgefühl / Schwindel1
|
Sehr häufig (16,5%) [0,3%]
|
Sehr häufig (23,7%) [1,0%]
| |
Herzerkrankungen
|
Vorhofflimmern/-flattern4
|
Häufig (7,4%) [2,3%]
|
Häufig (7,1%) [3,1%]
| |
Gefässerkrankungen
|
Bluterguss1
|
Sehr häufig (30,9%) [0%]
|
Sehr häufig (26,9%) [0,2%]
| |
Kontusion
|
Sehr häufig (20,2%) [0%]
|
Sehr häufig (20,6%) [0%]
| |
Petechien
|
Häufig (8,9%) [0%]
|
Häufig (6,2%) [0%]
| |
Ekchymosen
|
Häufig (5,7%) [0%]
|
Häufig (3,1%) [0,2%]
| |
Blutung / Hämatom1
|
Sehr häufig (16,3%) [3,2%]
|
Sehr häufig (19,0%) [2,5%]
| |
Gastrointestinale Blutung
|
Gelegentlich (0,6%) [0,5%]
|
Gelegentlich (0,4%) [0,4%]
| |
Intrakranielle Blutung
|
Gelegentlich (0,3%) [0,2%]
|
Sehr selten (0%) [0%]
| |
Epistaxis
|
Häufig (8,0%) [0,3%]
|
Häufig (6,3%) [0%]
| |
Bluthochdruck
|
Sehr häufig (11,2%) [4,7%]
|
Sehr häufig (14,4%) [5,4%]
| |
Erkrankungen der Atemwege, des Brustraums und Mediastinums
|
Husten
|
Sehr häufig (25,2%) [0,4%]
|
Sehr häufig (30,8%) [0,4%]
| |
Erkrankungen des Gastrointestinaltrakts
|
Diarrhoe
|
Sehr häufig (36,7%) [2,6%]
|
Sehr häufig (42,5%) [4,6%]
| |
Übelkeit
|
Sehr häufig (21,8%) [0,8%]
|
Sehr häufig (37,7%) [0,8%]
| |
Obstipation
|
Sehr häufig (15,2%) [0,1%]
|
Sehr häufig (24,4%) [0,6%]
| |
Abdominalschmerz1
|
Sehr häufig (14,5%) [1,2%]
|
Sehr häufig (16,0%) [2,1%]
| |
Erbrechen
|
Sehr häufig (14,0%) [0,7%]
|
Sehr häufig (24,8%) [1,0%]
| |
Erkrankungen der Haut und des Unterhautzellgewebes
|
Ausschlag1
|
Sehr häufig (20,3%) [0,9%]
|
Sehr häufig (36,3%) [6,3%]
| |
Skelettmuskulatur-, Bindegewebs- und Knochenerkrankungen
|
Schmerzen des Muskel- und Skelettsystems5
|
Sehr häufig (31,9%) [1,8%]
|
Sehr häufig (42,1%) [3,3%]
| |
Arthralgie
|
Sehr häufig (24,0%) [0,9%]
|
Sehr häufig (27,9%) [1,3%]
| |
Allgemeine Erkrankungen und Beschwerden am Verabreichungsort
|
Ermüdung
|
Sehr häufig (23,6%) [2,0%]
|
Sehr häufig (31,2%) [2,5%]
| |
Asthenie
|
Häufig (7,0%) [0,9%]
|
Sehr häufig (10,0%) [0,8%]
| |
*Gemäss Version 4.03 der NCI-CTCAE-Kriterien (Common Terminology Criteria for Adverse Events des National Cancer Institute).
1 Umfasst mehrere UAW-Begriffe.
2 Zweite Primärtumoren waren definiert als SMQ Malignome (einschliesslich hämatologischer Malignome SMQ und nicht-hämatologischer Malignome SMQ), SMQ Maligne Lymphome [narrow] und SMQ Myelodysplastisches Syndrom [narrow].
3 Zweite Primärtumoren (ausgenommen Nicht-Melanom-Hautkrebs) waren durch die Kriterien für zweite Primärtumoren ausgenommen Preferred Terms (PT) unter dem High Level Term «Neubildungen der Haut bösartig und nicht spezifiziert (ausschl. Melanome)» definiert.
4 Umfasst alle PT mit Vorhofflimmern oder Vorhofflattern.
5 Umfasst Rückenschmerzen, Knochenschmerzen, Schmerzen des Muskel- und Skelettsystems am Brustkorb, Schmerzen des Muskel- und Skelettsystems, muskuloskelettale Beschwerden, myofasziales Schmerz-Syndrom, Nackenschmerzen, Schmerz in einer Extremität, Myalgie, Wirbelsäulenschmerz.
6 Umfasst Ereignisse nach Ende der Meldephase im Rahmen der Studien
7 Behandlungsbedingte hämatologische Laborwertabweichungen
|
Beschreibung spezifischer unerwünschter Wirkungen und Zusatzinformationen
Hepatotoxizität
Über Hepatotoxizität, vorwiegend in Form einer Erhöhung der Transaminasen, wurde bei Patienten, die Calquence erhielten, berichtet. Es wurden auch schwere Fälle von Hepatotoxizität beobachtet. Ein kausaler Zusammenhang zwischen Calquence und Hepatotoxizität/Transaminasenerhöhungen ist nicht nachgewiesen worden.
Synkopen und Stürze
In klinischen Studien und in der Postmarketing-Phase wurden Synkopen und Stürze bei mit Calquence behandelten Patienten beobachtet (siehe «Wirkung auf die Fahrtüchtigkeit und auf das Bedienen von Maschinen»).
Ältere Patienten
Von den 1478 Studienteilnehmern der klinischen Studien zu CALQUENCE als Monotherapie gehörten 42,2% zur Altersgruppe ≥65 Jahre und <75 Jahre sowie 20,6% zur Altersgruppe ≥75 Jahre. Bei Patienten ab 75 Jahren waren unerwünschte Ereignisse vom Grad ≥3 häufiger (77,4%) als bei Patienten der Altersgruppe ≥65 Jahre und <75 Jahre (68,4%) oder Patienten unter 65 Jahren (59,3%). Höhere Raten wurden bei Patienten ab 75 Jahren im Vergleich zu den anderen beiden Altersgruppen beobachtet für Pneumonie jeden Grades (19,7%, 15,9% bzw. 13,5%) einschliesslich Pneumonie Grad ≥3 (14,4%, 8,2% bzw. 6,0%).
Die Meldung des Verdachts auf Nebenwirkungen nach der Zulassung ist von grosser Wichtigkeit. Sie ermöglicht eine kontinuierliche Überwachung des Nutzen-Risiko-Verhältnisses des Arzneimittels. Angehörige von Gesundheitsberufen sind aufgefordert, jeden Verdacht einer neuen oder schwerwiegenden Nebenwirkung über das Online-Portal ElViS (Electronic Vigilance System) anzuzeigen. Informationen dazu finden Sie unter www.swissmedic.ch.
ÜberdosierungEs gibt keine spezifische Behandlung für eine CALQUENCE-Überdosierung und die Symptome einer Überdosierung sind nicht bekannt. Im Falle einer Überdosierung müssen die Patienten engmaschig auf Befunde und Symptome von unerwünschten Wirkungen überwacht werden und es muss eine geeignete symptomatische Behandlung eingeleitet werden.
Eigenschaften/WirkungenATC-Code
L01EL02
Wirkungsmechanismus
Acalabrutinib ist ein selektiver niedermolekularer Bruton-Tyrosinkinase (BTK)-Inhibitor. BTK ist ein Signalmolekül der B-Zell-Antigen-Rezeptor- (BCR) und Zytokin-Rezeptor-Signalwege. Die Signalgebung über BTK führt in B-Zellen zum Überleben und zur Proliferation der Zellen und ist für zelluläre Adhäsion, Trafficking und Chemotaxis erforderlich.
Acalabrutinib und sein aktiver Metabolit (ACP-5862) bilden eine kovalente Bindung mit einem Cysteinrest im aktiven Zentrum von BTK, was zur irreversiblen und selektiven Inaktivierung von BTK (IC50 ≤5nM) mit minimalen off-target Interaktionen führt. In einem Screening von 380 Säugetier-Wildtyp-Kinasen waren die einzigen zusätzlichen Kinasen, die durch klinisch relevante Konzentrationen von Acalabrutinib und ACP-5862 beeinflusst wurden, BMX und ERBB4, und diese Beeinflussung war von 3- bis 4-fach geringerer Potenz als bei BTK.
In nicht-klinischen Studien inhibierte Acalabrutinib die BTK-vermittelte Aktivierung der nachgeschalteten Signalproteine CD86 und CD69, hemmte die Proliferation und das Überleben maligner B-Zellen und wies eine minimale Aktivität auf andere Immunzellen (T-Zellen und NK-Zellen) auf.
Pharmakodynamik
Bei Patienten mit malignen B-Zell-Erkrankungen, die zweimal täglich 100 mg Acalabrutinib einnahmen, betrug die mediane BTK-Belegung im peripheren Blut im Steady-State ≥95% und blieb über 12 Stunden aufrechterhalten. Dies führte zur Inaktivierung von BTK über das gesamte empfohlene Verabreichungsintervall.
Kardiale Elektrophysiologie
In einer spezifischen QT-Studie wurde durch Acalabrutinib nach Gabe von 100 mg und 400 mg Einzeldosen, keine klinisch relevante Verlängerung des QT/QTc-Intervalls beobachtet (z.B. nicht länger als oder gleich 10 ms).
Klinische Wirksamkeit
Patienten mit nicht vorbehandelter CLL
Die Sicherheit und Wirksamkeit von CALQUENCE bei Patienten mit nicht vorbehandelter CLL wurde in einer multizentrischen, randomisierten, unverblindeten Phase-III-Studie (ELEVATE-TN) untersucht, an der 535 Patienten teilnahmen. Es musste sich um eine CD20+, gemäss IWCLL 2008 Kriterien diagnostizierte und aktive/behandlungsbedürftige CLL handeln. Darüber hinaus mussten absolute Neutrophilenzahl und Thrombozyten unabhängig von Wachstumsfaktor- respektive Transfusions-Unterstützung sein und über 750 respektive 50.000 Zellen/μl liegen (im Falle von Knochenmarksbeteiligung über 500 respektive 30.000 Zellen/μl). Patienten mit ZNS-Beteiligung, prolymphozytischer Leukämie oder Richter-Transformation waren von der Teilnahme ausgeschlossen. Die Patienten erhielten CALQUENCE plus Obinutuzumab, CALQUENCE als Monotherapie oder Obinutuzumab plus Chlorambucil. Die ELEVATE-TN-Studie schloss Patienten im Alter von 65 Jahren oder darüber und solche zwischen 18 und 65 Jahren mit Begleiterkrankungen (Kreatinin-Clearance 30-69 ml/min und/oder CIRS-G-Score >6) ein. Die Patienten durften begleitend antithrombotische Wirkstoffe erhalten, mit Ausnahme von Warfarin und äquivalenten Vitamin-K-Antagonisten.
Die Patienten wurden im Verhältnis 1:1:1 randomisiert und einem der folgenden 3 Behandlungsarme zugeteilt:
·CALQUENCE plus Obinutuzumab (CALQUENCE+G): CALQUENCE wurde, beginnend an Tag 1 von Zyklus 1, bis zu einem Fortschreiten der Krankheit oder bis zum Auftreten einer nicht vertretbaren Toxizität in einer Dosis von 100 mg zweimal täglich verabreicht. Obinutuzumab wurde, beginnend an Tag 1 von Zyklus 2, für maximal 6 Behandlungszyklen verabreicht. Obinutuzumab wurde in einer Dosis von 1'000 mg an den Tagen 1 und 2 (100 mg an Tag 1 und 900 mg an Tag 2), 8 und 15 von Zyklus 2 verabreicht, gefolgt von 1'000 mg an Tag 1 der Zyklen 3 bis 7. Ein Behandlungszyklus dauerte 28 Tage.
·CALQUENCE-Monotherapie: CALQUENCE wurde bis zu einem Fortschreiten der Krankheit oder bis zum Auftreten einer nicht vertretbaren Toxizität in einer Dosis von 100 mg zweimal täglich verabreicht.
·Obinutuzumab plus Chlorambucil (GClb): Obinutuzumab und Chlorambucil wurden für maximal 6 Behandlungszyklen verabreicht. Obinutuzumab wurde in einer Dosis von 1.000 mg an den Tagen 1 und 2 (100 mg an Tag 1 und 900 mg an Tag 2), 8 und 15 von Zyklus 1 verabreicht, gefolgt von 1.000 mg an Tag 1 der Zyklen 2 bis 6. Chlorambucil wurde in einer Dosis von 0,5 mg/kg an den Tagen 1 und 15 der Zyklen 1 bis 6 verabreicht. Ein Behandlungszyklus dauerte 28 Tage.
Die Patienten wurden gemäss 17p-Deletions-Mutationsstatus (Ja/Nein), ECOG-Performance-Status (Status gemäss Eastern Cooperative Oncology Group, 0 oder 1 versus 2) und geographische Region (Nordamerika und Westeuropa versus Andere) stratifiziert. Nach bestätigtem Fortschreiten der Krankheit erhielten 45 in den GClb-Arm randomisierte Patienten im Rahmen eines Crossovers die CALQUENCE-Monotherapie.
Die Ausgangsmerkmale waren in den drei Armen (Calquence plus Obinutuzumab [n=179], Calquence-Monotherapie [n=179] und Obinutuzumab plus Chlorambucil [n=177]) generell ausgewogen: medianes Alter 70, 70 bzw. 71 Jahre; 62%, 62% bzw. 59,9% waren männlich; 94,4%, 92,2% bzw. 94,4% hatten einen ECOG-PS von 0-1; die mediane Zeit seit Diagnosestellung betrug 30,5, 24,4 bzw. 30,7 Monate; die zytogenetischen Faktoren (del17p, del11q, TP53-Mutation, nicht-mutiertes IGHV, komplexer Karyotyp) sowie das Rai-Stadium waren generell ausgewogen.
Der primäre Endpunkt war das progressionsfreie Überleben (PFS) gemäss Beurteilung durch ein unabhängiges Gremium (IRC) auf Grundlage der IWCLL (International Workshop on Chronic Lymphocytic Leukemia)-Kriterien aus dem Jahr 2008 unter Berücksichtigung der Abklärung betreffend behandlungsbedingter Lymphozytose (Cheson 2012). Nach einer medianen Nachbeobachtungsdauer von 28,3 Monaten zeigte das durch das IRC ermittelte PFS bei den Patienten mit nicht vorbehandelter CLL im CALQUENCE+G-Behandlungsarm im Vergleich zum GClb-Behandlungsarm eine statistisch signifikante Reduktion des Risikos für Fortschreiten der Krankheit oder Tod von 90%. Zum Zeitpunkt der Analyse war das mediane Gesamtüberleben mit insgesamt 37 Todesfällen noch in keinem Behandlungsarm erreicht worden: 9 (5%) im CALQUENCE+G-Behandlungsarm, 11 (6,1%) im CALQUENCE-Monotherapie-Behandlungsarm und 17 (9,6%) im GClb-Arm. Die Wirksamkeitsdaten sind in Tabelle 5 aufgeführt.
Tabelle 5: Wirksamkeitsergebnisse bei Patienten mit CLL (ELEVATE-TN)
|
Merkmal
|
CALQUENCE plus Obinutuzumab
N=179
|
CALQUENCE-Monotherapie
n=179
|
Obinutuzumab plus Chlorambucil
n=177
| |
Progressionsfreies Überleben*
| |
Anzahl der Ereignisse (%)
|
14 (7,8)
|
26 (14,5)
|
93 (52,5)
| |
Median (95%-KI), Monate
|
n.e.
|
n.e. (34,2; n.e.)
|
22.6 (20,2; 27,6)
| |
HR† (95%-KI)
|
0,10 (0,06; 0,17)
|
0,20 (0,13; 0,30)
|
-
| |
Gesamtansprechrate*
| |
ORR, n (%)
(95%-KI)
|
168 (93,9)
(89,3; 96,5)
|
153 (85.5)
(79,6; 89,9)
|
139 (78.5)
(71,9; 83,9)
| |
CR, n (%)
|
23 (12,8)
|
1 (0,6)
|
8 (4,5)
|
KI=Konfidenzintervall; HR=Hazard Ratio; n.e.=nicht erreicht; CR=Vollremission (Complete Response); CRi=Vollremission mit unvollständiger Erholung der Blutzellzahlen; nPR=noduläre Teilremission; PR=Teilremission (Partial Response);
*Gemäss IRC-Beurteilung
†Basierend auf einem stratifizierten Cox-Proportional-Hazards-Modell
Die PFS-Ergebnisse für die Behandlung mit CALQUENCE mit oder ohne Obinutuzumab fielen in den verschiedenen Untergruppen, darunter solche mit Hochrisiko-Merkmalen (17p-Deletion, 11q-Deletion, TP53-Mutation und nicht-mutiertes IGHV), vergleichbar aus.
Eine Richter-Transformation trat bei 6 Patienten (3,4%) im Acalabrutinib-Monotherapie-Arm (und bei keinem Patienten im Kombinationstherapie-Arm) während der randomisierten Phase und bei einem Patienten (2,2%) im Chlorambucil/Obinutuzumab-Arm während der Crossover-Phase auf.
Patienten mit CLL, die mindestens eine Vorbehandlung erhalten haben
Die Sicherheit und Wirksamkeit von CALQUENCE bei Patienten mit rezidivierter oder refraktärer CLL wurden in einer multizentrischen, randomisierten, unverblindeten Phase-III-Studie (ASCEND) untersucht, an der 310 Patienten teilnahmen, die mindestens eine vorherige Behandlung erhalten hatten. Eine vorgängige Behandlung mit einem B-Zell Lymphom (BCL)-2-Inhibitor (z.B. Venetoclax), einem B-Zell Rezeptor (BCR) Inhibitor (z.B. BTK- oder PI3K-Inhibitoren) oder eine Radio- oder Toxin-konjugierte Antikörper-Therapie war nicht gestattet. Es musste sich um eine CD20+, gemäss IWCLL 2008 Kriterien diagnostizierte und aktive/behandlungsbedürftige CLL handeln. Darüber hinaus mussten absolute Neutrophilenzahl und Thrombozyten unabhängig von Wachstumsfaktor- respektive Transfusions-Unterstützung sein und über 750 respektive 50.000 Zellen/μl liegen (im Falle von Knochenmarksbeteiligung über 500 respektive 30.000 Zellen/μl). Patienten mit ZNS-Beteiligung, prolymphozytischer Leukämie oder Richter-Transformation waren von der Teilnahme ausgeschlossen. Die Patienten erhielten eine CALQUENCE-Monotherapie oder eine Behandlung nach Wahl des Prüfarztes, die entweder in Idelalisib plus Rituximab oder in Bendamustin plus Rituximab bestehen konnte. Die Anwendung antithrombotischer Wirkstoffe war erlaubt, mit Ausnahme von Warfarin und äquivalenten Vitamin-K-Antagonisten.
Die Patienten wurden im Verhältnis 1:1 randomisiert und einer der folgenden Behandlungsgruppen zugeteilt:
·CALQUENCE 100 mg zweimal täglich bis zu einem Fortschreiten der Krankheit oder bis zum Auftreten einer nicht vertretbaren Toxizität, oder
·nach Wahl des Prüfarztes:
·Idelalisib 150 mg zweimal täglich bis zu einem Fortschreiten der Krankheit oder bis zum Auftreten einer nicht vertretbaren Toxizität, in Kombination mit ≤8 Infusionen Rituximab (375 mg/m2/500 mg/m2) an Tag 1 jedes 28-tägigen Zyklus für bis zu 6 Zyklen
·Bendamustin 70 mg/m2 (Tag 1 und 2 jedes 28-tägigen Zyklus) in Kombination mit Rituximab (375 mg/m2/500 mg/m2) an Tag 1 jedes 28-tägigen Zyklus für bis zu 6 Zyklen
Die Patienten wurden gemäss Faktoren 17p-Deletions-Mutationsstatus (Ja/Nein), ECOG-Performance-Status (0 oder 1 versus 2) und Anzahl der Vorbehandlungen (1 bis 3 versus ≥4) stratifiziert. Nach bestätigtem Fortschreiten der Krankheit erhielten 35 Patienten, die zunächst nach Wahl des Prüfarztes entweder mit Idelalisib plus Rituximab oder mit Bendamustin plus Rituximab behandelt worden waren, im Rahmen eines Crossovers CALQUENCE.
Die Ausgangsmerkmale waren in den zwei Armen (CALQUENCE-Monotherapie [n=155] bzw. vom Prüfarzt gewählte Behandlung mit Idelalisib + Rituximab oder Bendamustin + Rituximab [n=155]) generell ausgewogen: medianes Alter 68 bzw. 67 Jahre; 69,7% bzw. 64,5% waren männlich; 87,7% bzw. 86,5% hatten einen ECOG-PS von 0-1; die mediane Zeit seit Diagnosestellung betrug 85,3 bzw. 79 Monate; die mediane Zeit zwischen der letzten vorherigen CLL-Therapie und der ersten Dosis betrug 26,4 bzw. 22,7 Monate; die zytogenetischen Faktoren (del17p, del11q, TP53-Mutation, nicht-mutiertes IGHV, komplexer Karyotyp) sowie das Rai-Stadium waren generell ausgewogen.
Primärer Endpunkt war das durch ein IRC beurteilte PFS auf Grundlage der IWCLL-Kriterien aus dem Jahr 2008 unter Berücksichtigung der Abklärung betreffend behandlungsbedingter Lymphozytose (Cheson 2012). Zum Zeitpunkt der ersten präspezifizierten Analyse mit einer medianen Nachbeobachtungsdauer von 16,1 Monaten zeigte CALQUENCE eine klinisch relevante und statistisch signifikante Verbesserung des IRC-beurteilten PFS im Vergleich zum IR/BR-Arm (Hazard Ratio 0,31 [95%-KI 0,20 bis 0,49] p<0,0001). Zum Zeitpunkt dieser Analyse war das mediane Gesamtüberleben mit insgesamt 33 Todesfällen noch in keinem Behandlungsarm erreicht worden: 15 (9,7%) im CALQUENCE-Monotherapie-Arm und 18 (11,6%) im Behandlungsarm mit der vom Prüfarzt gewählten Behandlung (Idelalisib plus Rituximab oder Bendamustin plus Rituximab).In einer weiteren nicht präspezifizierten Analyse nach einer medianen Nachbeobachtungsdauer von 22 Monaten wurde das mediane PFS, welches im Gegensatz zum primären Endpunkt vom Prüfarzt beurteilt wurde, im CALQUENCE-Arm nicht erreicht und betrug im IR/BR-Arm 16,8 Monate (Hazard Ratio 0,27 [95%-KI 0,18 bis 0,40]. Die Daten zum Gesamtüberleben waren weiterhin unreif mit 21 (13,5%) und 26 (16,8%) Ereignissen im CALQUENCE- respektive Vergleichsarm. Die Wirksamkeitsergebnisse der präspezifizierten Analyse sind in Tabelle 6 dargestellt.
Tabelle 6: Wirksamkeitsergebnisse bei Patienten mit CLL (ASCEND)
|
|
CALQUENCE
Monotherapie
n=155
|
Vom Prüfarzt gewählte Behandlung mit Idelalisib + Rituximab (n=119) oder Bendamustin + Rituximab (n=36)
n=155
| |
Progressionsfreies Überleben*
| |
Anzahl der Ereignisse (%)
|
27 (17,4)
|
68 (43,9)
| |
Median (95%-KI), Monate
|
n.e.
|
16.5 (14,0; 17,1)
| |
HR† (95%-KI)
|
0.31 (0,20; 0,49)
| |
Gesamtansprechrate*
| |
ORR, n (%)
(95%-KI)
|
126 (81,3)
(74,4; 86,6)
|
117 (75,5)
(68,1; 81,6)
| |
CR, n (%)
|
0
|
2 (1,3)
| |
Dauer des Ansprechens* (DoR)
| |
Median (95%-KI), Monate
|
n.e.
|
13,6 (11,9; n.e.)
|
KI=Konfidenzintervall; HR=Hazard Ratio; n.e.=nicht erreicht; CR=Vollremission (Complete Response); PR=Teilremission (Partial Response)
*Gemäss IRC-Beurteilung
†Basierend auf einem stratifizierten Cox-Proportional-Hazards-Modell
Die PFS-Ergebnisse für die Behandlung mit CALQUENCE fielen in den verschiedenen Untergruppen, darunter solche mit Hochrisiko-Merkmalen (17p-Deletion, 11q-Deletion, TP53-Mutation und nicht-mutiertes IGHV), vergleichbar aus.
Eine Richter-Transformation trat bei 4 Patienten (2,6%) im Acalabrutinib-Monotherapie-Arm und 3 Patienten (2,0%) im Idelalisib plus Rituximab- / Bendamustin plus Rituximab-Arm während der randomisierten Phase und bei 2 Patienten (5,7%) im Idelalisib plus Rituximab- / Bendamustin plus Rituximab-Arm während der Crossover-Phase auf.
Patienten mit bisher unbehandeltem MCL
Die Wirksamkeit von CALQUENCE bei Patienten mit bisher unbehandeltem MCL wurden in ECHO, einer randomisierten, doppelblinden, placebokontrollierten, multizentrischen Phase-III-Studie, untersucht. An ECHO nahmen 598 Patienten im Alter von 65 Jahren und älter mit bestätigtem, bisher unbehandeltem MCL teil. Patienten mit dem Behandlungsziel einer Tumorreduktion vor autologer Stammzelltransplantation waren aus der Studie ausgeschlossen. Die Randomisierung der Patienten erfolgte stratifiziert nach geographischer Region (Nordamerika versus Westeuropa versus Andere) und vereinfachtem MIPI (Mantle Cell Lymphoma International Prognostic Index)-Score (0–3 versus 4–5 versus 6–11). In die Studie wurden Patienten während der COVID-19-Pandemie aufgenommen.
Die Patienten wurden im Verhältnis 1:1 auf 2 Arme randomisiert und erhielten:
·CALQUENCE plus Bendamustin und Rituximab (CALQUENCE + BR)-Arm – CALQUENCE 100 mg wurde ab Tag 1 von Zyklus 1 zweimal täglich kontinuierlich verabreicht. Bendamustin, 90 mg/m2, wurde an den Tagen 1 und 2 jedes der sechs 28-tägigen Zyklen intravenös über 30 Minuten verabreicht und Rituximab, 375 mg/m2, wurde an Tag 1 jedes der sechs 28-tägigen Zyklen intravenös verabreicht. CALQUENCE + BR wurden für maximal 6 Behandlungszyklen verabreicht (Induktionsbehandlung).
·Placebo plus Bendamustin und Rituximab (Placebo + BR)-Arm – Placebo wurde ab Tag 1 von Zyklus 1 zweimal täglich kontinuierlich verabreicht. Bendamustin, 90 mg/m2, wurde an den Tagen 1 und 2 jedes der sechs 28-tägigen Zyklen intravenös über 30 Minuten verabreicht und Rituximab, 375 mg/m2, wurde an Tag 1 jedes der sechs 28-tägigen Zyklen intravenös verabreicht. Placebo + BR wurden für maximal 6 Behandlungszyklen verabreicht (Induktionsbehandlung).
CALQUENCE oder Placebo wurden kontinuierlich bis zu einem Fortschreiten der Krankheit oder bis zum Auftreten einer nicht akzeptablen Toxizität verabreicht. Nach der Induktionsbehandlung erhielten die Patienten im Placebo + BR-Arm, die ein Ansprechen (Teilremission oder Vollremission) erreicht hatten, eine Rituximab-Erhaltungsdosis von 375 mg/m2 an Tag 1 jedes zweiten Zyklus für maximal 12 zusätzliche Dosen bis Zyklus 30. Auf den Placebo + BR-Arm randomisierte Patienten, bei denen ein Fortschreiten der Krankheit bestätigt wurde, konnten im Rahmen eines Crossovers bis zu einem zweiten Fortschreiten der Krankheit oder dem zweiten Auftreten einer nicht vertretbaren Toxizität auf die CALQUENCE-Monotherapie mit einer Dosis von zweimal täglich 100 mg umgestellt werden.
Das mediane Alter betrug 71 Jahre (65–86); 70,7% waren männlich; 78,3% waren Weisse; 93,1% hatten einen ECOG-Performance-Status von 0–1. Der vereinfachte MIPI-Score war bei 33,1% der Patienten niedrig (0–3), bei 42,8% intermediär (4–5) und bei 24,1% hoch (6–11). Insgesamt 37,7% der Patienten hatten ein Tumorvolumen von ≥5 cm und 86% wiesen das Ann-Arbor-Stadium IV auf. Aggressive Varianten des MCL wie blastoide und pleomorphe Formen traten bei 7,7% bzw. 5,5% der Patienten auf. Insgesamt 47,8% der Patienten hatten einen Ki-67-Score von ≥30%. Die Ausgangsmerkmale waren in beiden Behandlungsarmen ähnlich.
Der primäre Endpunkt war das progressionsfreie Überleben (PFS), das von einem unabhängigen Gremium (IRC, Independent Review Committee) gemäss der Lugano-Klassifikation für NHL bei Patienten mit bisher unbehandeltem MCL beurteilt wurde. Weitere multiplizitätskontrollierte Wirksamkeitsendpunkte waren die vom IRC beurteilte Gesamtansprechrate (ORR) und das Gesamtüberleben (OS). Das Studiendesign ohne eine 2. Randomisierung für die Post-Induktionstherapie lässt keine Aussage über den Vorteil der Dauertherapie mit Calquence bis zum Fortschreiten der Erkrankung oder inakzeptabler Toxizität zu.
Bei einer medianen Nachbeobachtungsdauer von 46,1 Monaten im CALQUENCE + BR-Arm und 44,4 Monaten im Placebo + BR-Arm ergab die Beurteilung des PFS durch das IRC eine statistisch signifikante Reduktion des Risikos für ein Fortschreiten der Krankheit oder den Tod bei den mit CALQUENCE + BR behandelten Patienten um 27% im Vergleich zu Placebo + BR. Es fielen allerdings Inkonsistenzen in gewissen Subgruppen auf, diese zeigten sich sowohl für das PFS als auch für das OS. Besonders erwähnenswert ist die Inkonsistenz in Bezug auf das Geschlecht. Die HR für PFS bei den 423 untersuchten Männern betrug 0.91 (0.68, 1.21) im Vergleich zu 0.34 (0.19,0.58) bei den 175 Frauen, die entsprechende HR für OS war 1.01 (0.74, 1.38) für die Männer und 0.52 (0.28, 0.94) für die Frauen.Die vom IRC beurteilte ORR zeigte keinen statistisch signifikanten Unterschied Calquence plus BR und Placebo plus BR.
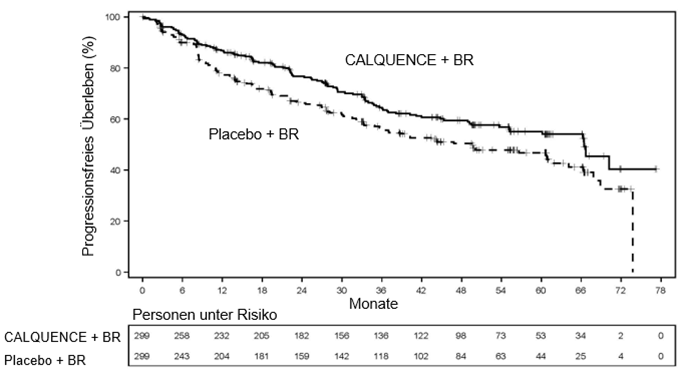
Zum Zeitpunkt der PFS-Analyse war das mediane OS bei insgesamt 203 Todesfällen in keinem der beiden Arme erreicht worden: 97 (32,4%) im CALQUENCE + BR-Arm, 106 (35,5%) im Placebo + BR-Arm, und das OS zeigte keinen statistisch signifikanten Unterschied zwischen den Behandlungsarmen: HR (95%-KI) (stratifiziert) 0.86 (0.65, 1.13). Die Wirksamkeitsergebnisse sind in Tabelle 7 aufgeführt. Die Kaplan-Meier-Kurven des PFS sind in Abbildung 1 dargestellt.
Tabelle 7: Wirksamkeitsergebnisse bei Patienten mit bisher unbehandeltem MCL in ECHO
|
|
CALQUENCE + BR
N = 299
|
Placebo + BR
N = 299
| |
Vom IRC beurteiltes PFS
| |
Median (95%-KI)
|
66,4 (55,1; n.a.)
|
49,6 (36,0; 64,1)
| |
HR (95%-KI) (stratifiziert)*
|
0,73 (0,57; 0,94)
| |
p-Wert‡
|
0,0160
| |
Vom IRC beurteiltes ORR
| |
CR + PR n (%)
|
272 (91,0)
|
263 (88,0)
| |
95%-KI
|
87,3; 93,8
|
83,9; 91,3
| |
CR n (%)
|
199 (66,6)
|
160 (53,5)
| |
PR n (%)
|
73 (24,4)
|
103 (34,4)
| |
ORR-Differenz (vs. PBR-Arm)
|
3,0%
|
-
| |
p-Wert
|
0,2196
|
-
|
HR = Hazard Ratio, CR = Vollremission (Complete Response), PR = Teilremission (Partial Response), n.a. – nicht auswertbar
* Stratifiziert nach Randomisierungsstratifizierungsfaktoren: Geographische Regionen (Nordamerika, Westeuropa, Andere) und vereinfachter MIPI-Score (niedriges Risiko [0 bis 3], intermediäres Risiko [4 bis 5], hohes Risiko [6 bis 11]), wie über IXRS erhoben. Geschätzt auf der Grundlage eines stratifizierten Cox-Proportional-Hazards-Modells für die Hazard Ratio (95%-KI).
‡ Geschätzt auf der Grundlage eines stratifizierten Log-Rank-Tests für den p-Wert
Abbildung 1. Kaplan-Meier-Kurve des PFS gemäss Beurteilung durch das IRC bei Patienten mit bisher unbehandeltem MCL (ECHO)
Patienten mit MCL, die mindestens eine Vortherapie erhalten haben
Die Sicherheit und Wirksamkeit von CALQUENCE bei MCL wurden in einer offenen, multizentrischen, einarmigen Phase-II-Studie (ACE-LY-004) untersucht, an der 124 vorbehandelte Patienten teilnahmen, bei denen mit vorheriger Therapie kein partielles Ansprechen erreicht wurde oder die nach der vorherigen Therapie eine Progression gezeigt hatten. Alle Patienten erhielten CALQUENCE 100 mg zweimal täglich oral bis zu einem Fortschreiten der Erkrankung oder bis zum Auftreten einer nicht vertretbaren Toxizität. An der Studie nahmen keine Patienten teil, die zuvor mit BTK-Inhibitoren, anderen Inhibitoren des B-Zell-Rezeptor-Signalwegs (Phosphoinositid-3 Kinase [PI3K] oder SYK) oder einem BCL-2 Inhibitor behandelt worden waren. Der primäre Endpunkt war die vom Prüfarzt beurteilte Gesamtansprechrate (ORR) gemäss der Lugano-Klassifikation für Non-Hodgkin-Lymphome (NHL). Die Dauer des Ansprechens (DoR) war ein zusätzliches Ergebnismass. Die Wirksamkeitsergebnisse der primären (12 Monate) und der finalen (54 Monate) Analyse sind in Tabelle 8 dargestellt.
Bei der primären Analyse betrug das mediane Alter 68 (Bereich 42 bis 90) Jahre, 79,8% waren männlich und 74,2% waren Weisse. Zu Studienbeginn hatten 92,8% der Patienten einen ECOG-Performance-Status von 0 oder 1. Die mediane Zeit seit Diagnosestellung betrug 46,3 Monate und die mediane Anzahl der Vorbehandlungen lag bei 2 (Bereich 1 bis 5), darunter 17,7% mit vorangegangener Stammzelltransplantation. Die häufigsten vorherigen Therapieschemata waren CHOP-basiert (51,6%) und ARA-C (33,9%). Zu Studienbeginn hatten 37,1% der Patienten mindestens einen Tumor mit einem längsten Durchmesser von ≥5 cm; 72,6% wiesen einen zusätzlichen Knotenbefall auf, davon 50,8% einen Knochenmarkbefall. Der vereinfachte MIPI-Score (der das Alter, den ECOG-Score sowie den Ausgangswert für die Laktatdehydrogenase und die Leukozytenzahl berücksichtigt) war bei 43,5% der Patienten intermediär und bei 16,9% hoch.
Tabelle 8. Gesamtansprechrate und Dauer des Ansprechens bei (ACE-LY-004) Patienten mit MCL bei der 12- und der 54-Monats-Analyse
|
|
Beurteilung des Prüfarztes nach 12 Monaten
n = 124
n (%) (95%-KI*)
|
Beurteilung des Prüfarztes nach 54 Monaten
n = 124
n (%) (95%-KI*)
| |
Gesamtansprechrate (ORR)
| |
Gesamtansprechrate
|
100 (80,6%) (72,6; 87,2)
|
101 (81,5%) (73,5; 87,9)
| |
Vollremission
|
49 (39,5%) (30,9; 48,7)
|
59 (47,6%) (38,5; 56,7)
| |
Teilremission
|
51 (41,1%) (32,4; 50,3)
|
42 (33,9%) (25,6; 42,9)
| |
Stabile Erkrankung
|
11 (8,9%) (4,5; 15,3)
|
10 (8,1%) (3,9; 14,3)
| |
Krankheitsprogression
|
10 (8,1%) (3,9; 14,3)
|
10 (8,1%) (3,9; 14,3)
| |
Nicht auswertbar†
|
3 (2,4%) (0,5; 6,9)
|
3 (2,4%) (0,5; 6,9)
| |
Dauer des Ansprechens (DoR)
| |
Median (Monate)
|
n. e. (13,5; n.e.)
|
28,6 (17,5; 39,1)
| |
KI = Konfidenzintervall; n. e. = nicht erreicht
*Exaktes binominales Konfidenzintervall von 95%.
†Umfasst Probanden ohne hinreichende Krankheitsbeurteilung nach Studienbeginn.
|
PharmakokinetikDie Pharmakokinetik von Acalabrutinib und seinem aktiven Metaboliten, ACP-5862, wurde bei gesunden Probanden und bei Patienten mit B-Zell-Malignom untersucht. Acalabrutinib zeigt Dosisproportionalität und sowohl Acalabrutinib als auch ACP-5862 zeigen über eine Dosisspanne von 75 bis 250 mg eine fast lineare Pharmakokinetik. Eine populationspharmakokinetische Modellierung weist darauf hin, dass die Pharmakokinetik von Acalabrutinib und ACP-5862 bei Patienten mit verschiedenen B-Zell-Malignomen vergleichbar ausfällt. In der empfohlenen Dosis von 100 mg zweimal täglich betrugen der geometrische Mittelwert der Fläche unter der Plasmakonzentrations-Zeit-Kurve über 24 Stunden (AUC24h) und die maximalen Plasmakonzentrationen (Cmax) von Acalabrutinib bei Patienten mit B-Zell-Malignom (einschliesslich Mantelzell-Lymphom (MCL) und CLL) 1679 ng•h/ml bzw. 438 ng/ml. Die entsprechenden Werte für ACP-5862 betrugen 4166 ng•h/ml und 446 ng/ml.
CALQUENCE Filmtabletten und CALQUENCE Kapseln sind nachweislich bioäquivalent.
Absorption
Die mediane Zeit bis zum Erreichen der maximalen Plasmakonzentrationen (tmax) betrug 0,5 Stunden (Spannweite: 0,2 bis 3,0) Stunden für CALQUENCE bzw. 0,75 (0,5 bis 4,0) Stunden für ACP-5862. Die absolute Bioverfügbarkeit von CALQUENCE betrug 25%.
Wirkung von Nahrung auf Acalabrutinib
Bei gesunden Probanden hatte die Einnahme einer Einzeldosis von 100 mg Acalabrutinib mit einer fettreichen, kalorienreichen Mahlzeit (etwa 918 Kalorien, 59 Gramm Kohlenhydrate, 59 Gramm Fett und 39 Gramm Protein) im Vergleich zur Einnahme im nüchternen Zustand keinen Einfluss auf die mittlere AUC. Der resultierende Cmax-Wert sank um 54% und tmax war um 1-2 Stunden verzögert.
Distribution
Acalabrutinib bindet zu 97,5% und ACP-5862 zu 98,6% reversibel an Plasmaproteine des Menschen. Das mittlere Blut-zu-Plasma-Verhältnis betrug in vitro für Acalabrutinib 0,8. Für ACP-5862 betrug es 0,7. Das mittlere Distributionsvolumen im Steady-State (Vss) betrug für Acalabrutinib etwa 34 Liter.
Metabolismus
In vitro wird Acalabrutinib hauptsächlich durch CYP3A-Enzyme und zu einem geringen Teil durch Glutathionkonjugation und Amidhydrolyse metabolisiert. ACP-5862 wurde als Hauptmetabolit im Plasma identifiziert, wobei die mittlere geometrische Exposition (AUC) das etwa 2- bis 3-fache der Exposition gegenüber Acalabrutinib beträgt. ACP-5862 erzielt eine um etwa 50% schwächere BTK-Inhibition als Acalabrutinib.
Elimination
Nach einmaliger oraler Einnahme von 100 mg Acalabrutinib betrug der arithmetische Mittelwert der terminalen Halbwertszeit (t1/2) von Acalabrutinib 1,6 Stunden (Spannweite: 0,8 bis 9,0 h). Der arithmetische Mittelwert der t1/2 des aktiven Metaboliten, ACP-5862, betrug 6.9 Stunden (Spannweite: 2,5 bis 10,1 h).
Die mittlere orale Clearance (CL/F) betrug für Acalabrutinib 134 l/h. Für ACP-5862 betrug sie 22 l/h.
Nach Verabreichung einer einzelnen radioaktiv markierten [14C]-Acalabrutinib-Dosis von 100 mg an gesunde Probanden wurden 84% der Dosis im Stuhl und 12% der Dosis im Urin wiedergefunden, wobei weniger als 2% der Dosis als unverändertes Acalabrutinib im Stuhl oder Urin ausgeschieden wurden.
Kinetik spezieller Patientengruppen
Basierend auf populationspharmakokinetischen Analysen hatten weder Alter, Geschlecht, Ethnie (Kaukasier, Afroamerikaner) noch das Körpergewicht klinisch signifikante Auswirkungen auf die Pharmakokinetik von Acalabrutinib und seinem aktiven Metaboliten, ACP-5862.
Nierenfunktionsstörungen
Acalabrutinib unterliegt einer minimalen renalen Elimination. Eine pharmakokinetische Studie an Patienten mit eingeschränkter Nierenfunktion wurde nicht durchgeführt.
In einer pharmakokinetischen Populationsanalyse unterschied sich die Pharmakokinetik bei 408 Probanden mit leichter Nierenfunktionsstörung (eGFR zwischen 60 und 89 ml/min/1,73 m2, geschätzt anhand der MDRD-Formel) und 109 Probanden mit mässiger Nierenfunktionsstörung (eGFR zwischen 30 und 59 ml/min/1,73 m2) nicht in klinisch relevanter Weise von der bei 192 Probanden mit normaler Nierenfunktion (eGFR grösser oder gleich 90 ml/min/1,73 m2). Die Pharmakokinetik von Acalabrutinib bei Patienten mit schwerer Nierenfunktionsstörung (eGFR <29 ml/min/1,73 m2) oder dialysepflichtiger Niereninsuffizienz wurde nicht untersucht. Patienten mit einem Kreatininspiegel von mehr als dem 2,5-fachen der ULN wurden nicht in die klinischen Studien eingeschlossen (siehe «Dosierung/Anwendung»).
Leberfunktionsstörungen
Acalabrutinib wird in der Leber metabolisiert. In speziellen Untersuchungen zu Leberfunktionsstörungen war die Acalabrutinib-Exposition (AUC) bei Patienten mit leichter (n=6) (Child-Pugh A), mässiger (n=6) (Child-Pugh B) und schwerer (n=8) (Child-Pugh C) Leberfunktionsstörung im Vergleich zu Teilnehmern mit normaler Leberfunktion (n=6) 1,9-fach, 1,5-fach bzw. 5,3-fach erhöht. Populationspharmakokinetische Analysen ergaben keinen klinisch relevanten Unterschied zwischen Probanden mit leichter (n=79) oder mässiger (n=6) Leberfunktionsstörung (Gesamtbilirubin in Höhe des 1,5- bis 3-fachen der ULN und beliebige AST) im Vergleich zu Probanden mit normaler (n=613) Leberfunktion (Gesamtbilirubin und AST im Normbereich).
Präklinische DatenToxizität bei wiederholter Verabreichung
In Toxizitätsstudien wurden Ratten bis zu 6 Monate und Hunden bis zu 9 Monate orale Tagesdosen von Acalabrutinib verabreicht. Es wurden dabei Expositionsmengen toleriert, welche die therapeutische Exposition im Menschen in der empfohlenen Dosis überstiegen (1,1 fach bei Ratten, 8,2-fach bei Hunden, basierend auf der AUC).
Bei Ratten wurden bei einer AUC, die dem ≥7-fachen der empfohlenen Humandosis entsprach, renale Effekte einschliesslich tubulärer Degeneration beobachtet. Bei Ratten, die dem 4,2-fachen der empfohlenen Humandosis ausgesetzt waren, waren die renalen Effekte reversibel mit vollständiger Erholung. Bei Ratten, die höheren Expositionen (6,8-fach oder höher) ausgesetzt waren, waren die Effekte nur teilweise reversibel.
Bei Ratten wurden dosisabhängige, reversible Leberbefunde einschliesslich Hepatozytennekrose nach Expositionen in Höhe des ≥4,2-fachen der empfohlenen Humandosis beobachtet.
Bei Ratten, die Dosen exponiert waren, welche dem ≥6,8-fachen der empfohlenen humanen Dosis entsprachen, wurde eine kardiale Toxizität (myokardiale Blutung, Entzündung, Nekrose) beobachtet. Diese Ratten starben während der Studie. Die Reversibilität der Herzbefunde konnte nicht untersucht werden, da diese nur bei Dosen oberhalb der maximalen verträglichen Dosis (MTD) beobachtet wurden. Bei Expositionen, die dem 4,2-fachen der empfohlenen humanen Dosis entsprachen, wurden keine kardialen Toxizitäten beobachtet.
Genotoxizität/Mutagenität
Acalabrutinib zeigte weder im Rückmutationstest an Bakterien noch in vitro im Chromosomen-Aberrationstest oder in vivo im Maus-Knochenmark-Mikronukleustest Mutagenität.
Karzinogenität
Es wurden keine Karzinogenitätsstudien mit Acalabrutinib durchgeführt.
Reproduktionstoxizität
Bei Expositionen in Höhe des 10- bzw. 9-Fachen der beim Menschen unter der empfohlenen Dosis erzielten Exposition (AUC) konnten keine Auswirkungen auf die Fertilität von männlichen oder weiblichen Ratten festgestellt werden.
In einer kombinierten Studie zur Fertilität und embryofetalen Entwicklung wurde Acalabrutinib ab 14 Tagen vor der Paarung bis zum Gestationstag (GD) 17 in oralen Tagesdosen von bis zu 200 mg/kg an weibliche Ratten verabreicht. Es wurden keine Auswirkungen auf die embryofetale Entwicklung und das Überleben beobachtet. Die AUC bei oralen Tagesdosen von 200 mg/kg betrug etwa das 9-fache der AUC bei Patienten in der empfohlenen Dosis von 100 mg zweimal täglich. Acalabrutinib und sein aktiver Metabolit wurden im Plasma von Ratten-Feten nachgewiesen.
In einer Studie zur embryofetalen Entwicklung wurde Acalabrutinib trächtigen Kaninchen während der Organogenese (GD 6 – GD 18) in oralen Tagesdosen von bis zu 200 mg/kg verabreicht. Unter Expositionen, die beim Muttertier eine Toxizität verursachten (Tagesdosen von ≥100 mg/kg) und etwa dem 2,4-fachen der beim Menschen durch die empfohlene Dosis erzielten Exposition entsprachen, wurden verringertes fetales Körpergewicht und eine verzögerte Ossifikation beobachtet. In einer Reproduktionsstudie wurde bei Ratten unter Expositionen in Höhe des >2,3-fachen der durch 100 mg zweimal täglich erzielten klinischen Exposition Dystokie (verlängerte/schwierige Geburt) beobachtet.
Acalabrutinib und sein aktiver Metabolit wurden in der Milch säugender Ratten nachgewiesen.
In einer Studie zur prä- und postnatalen Entwicklung bei Ratten wurde Acalabrutinib trächtigen Tieren während der Organogenese, Geburt und Laktation in Dosen von 50, 100 und 150 mg/kg/Tag oral verabreicht. Bei Dosen ≥100 mg/kg/Tag wurden Dystokie (verlängerte oder starke Wehen) und Mortalität der Jungen beobachtet. Die AUC bei 100 mg/kg/Tag betrug bei trächtigen Ratten etwa das 2-fache der AUC bei Patienten, die etwa alle 12 Stunden 100 mg erhielten. In der F1-Generation wurden bei 150 mg/kg/Tag auch unterentwickelte Nierenpapillen beobachtet, die AUC betrug etwa das 5-fache der AUC bei Patienten, die etwa alle 12 Stunden 100 mg erhielten.
Sonstige HinweiseHaltbarkeit
Das Arzneimittel darf nur bis zu dem auf dem Behälter mit «EXP» bezeichneten Datum verwendet werden.
Besondere Lagerungshinweise
In der Originalverpackung aufbewahren.
Nicht über 30°C lagern.
Ausser Reichweite von Kindern aufbewahren.
Zulassungsnummer68817 (Swissmedic).
PackungenCALQUENCE 100 mg Filmtabletten:
Aluminium/Aluminium-Blisterpackungen. Schachteln mit 6 x 10 Filmtabletten [A].
ZulassungsinhaberinAstraZeneca AG, 6340 Baar.
Stand der InformationJanuar 2025.
|