ZusammensetzungWirkstoffe
Somapacitanum*
*Hergestellt durch rekombinante DNA-Technologie in Escherichia coli mit anschliessender Anbringung einer albuminbindenden Einheit.
Hilfsstoffe
Histidinum
Mannitolum (E 421)
Poloxamerum 188
Phenolum
Aqua ad iniectabile
Acidum hydrochloridum (zur Einstellung des pH-Werts)
Natrii hydroxidum (zur Einstellung des pH-Werts);
Die maximale Menge Natrium beträgt 0.05 mg pro ml Injektionslösung.
Indikationen/AnwendungsmöglichkeitenSubstitution des endogenen Wachstumshormons (Growth Hormone, GH) bei pädiatrischen Patienten ab dem Alter von 3 Jahren mit Wachstumsstörungen aufgrund eines nachgewiesenen Wachstumshormonmangels (Growth Hormone Deficiency, GHD).
Dosierung/AnwendungDie Therapie mit Somapacitan sollte von Ärzten eingeleitet und überwacht werden, die in der Diagnostik und Behandlung des GH-Mangels angemessen qualifiziert sind und über entsprechende Erfahrung verfügen (z.B. Endokrinologen).
Dokumentation der Chargennummer
Um die Rückverfolgbarkeit von biotechnologisch hergestellten Arzneimitteln sicherzustellen, wird empfohlen, Handelsname und Chargennummer bei jeder Behandlung zu dokumentieren.
Art der Anwendung
Sogroya ist einmal wöchentlich zu einer beliebigen Tageszeit zu verabreichen. Sogroya wird subkutan in die Bauchdecke, den Oberschenkel, das Gesäss oder den Oberarm injiziert. Die Injektionsstelle sollte jede Woche gewechselt werden. Für weitere Informationen siehe «Hinweise für die Handhabung».
Die Injektion sollte jeweils am selben Wochentag erfolgen. Falls die Verabreichung am geplanten Wochentag nicht möglich ist, kann die Dosis bis zu 2 Tage vor oder 3 Tage nach dem geplanten Anwendungstag appliziert werden. Zwischen zwei Dosen muss jedoch jeweils ein Mindestabstand von 4 Tagen (96 Stunden) eingehalten werden (siehe auch «Versäumte Anwendung» unten). Danach soll am nächsten planmässigen Verabreichungstag wieder mit dem üblichen Dosierungsschema fortgefahren werden.
Zu den verfügbaren Pens und deren Dosisschritten siehe «Darreichungsform und Wirkstoffmenge pro Einheit».
Übliche Dosierung
Die empfohlene Dosis beträgt 0.08-0.16 mg/kg/Woche.
Bei therapienaiven Patienten sollte die Behandlung mit einer Dosis von 0.12 mg/kg/Woche eingeleitet und bei Bedarf wie im Abschnitt «Dosisanpassung» beschrieben angepasst werden.
Bei bereits mit GH vorbehandelten Patienten sollte sich die Somapacitan-Startdosis an der zuvor benötigten Dosis orientieren.
Dosen >0.16 mg/kg/Woche wurden nicht untersucht.
Dosisanpassung
Die Überwachung der Therapie kann anhand der Wachstumsgeschwindigkeit und der Konzentrationen des insulinähnlichen Wachstumsfaktors 1 (Insulin-like Growth Factor 1, IGF-1) erfolgen. Zu diesem Zweck sollten die Proben jeweils 4 Tage nach Applikation der letzten Dosis entnommen werden. Der Zielwert für den IGF-1 SDS (Standard Deviation Score) sollte dabei im oberen Normbereich liegen und 2 SDS nicht überschreiten.
Bei unzureichender Wirksamkeit (ungenügender Anstieg der IGF-1-Konzentrationen, ungenügende Steigerung der Wachstumsgeschwindigkeit) kann die Dosis in Intervallen von 4-6 Wochen in Schritten von 0.02 mg/kg/Woche gesteigert werden. Eine Maximaldosis von 0.16 mg/kg/Woche darf jedoch nicht überschritten werden.
Bei erhöhten IGF-1-Konzentrationen sowie bei Auftreten schwerer unerwünschter Wirkungen kann die Sogroya-Dosis wie folgt angepasst werden:
Im Falle eines IGF-1 SDS >2 sollte zunächst 4-6 Wochen später eine erneute Bestimmung erfolgen. Falls der Wert weiterhin bei >2 liegt, soll die Dosis um 0.02 mg/kg/Woche reduziert werden. Die nächste Kontrolle des IGF-1 sollte 4-6 Wochen nach der Dosisreduktion erfolgen. Falls der Wert immer noch bei >2 liegt, soll die Dosis in Schritten von 0.02 mg/kg/Woche weiter reduziert werden. Falls der IGF-1 SDS auch unter einer Dosis von 0.08 mg/kg noch bei >2 liegt, muss das Nutzen-Risiko-Verhältnis einer Fortführung der Therapie individuell abgewogen werden.
Während der Langzeittherapie mit Sogroya sollten Wirksamkeit und Sicherheit in 6-12monatigen Intervallen überprüft werden. Hierzu sollten insbesondere Wachstumsgeschwindigkeit und IGF-1 sowie die Glucosespiegel bestimmt werden.
Therapiedauer
Die Behandlung mit Sogroya sollte beendet werden, wenn die Endgrösse (annähernd) erreicht ist, d.h. bei einer Wachstumsgeschwindigkeit von <2 cm/Jahr, sowie bei Erreichen eines Knochenalters >14 Jahre bei Mädchen bzw. >16 Jahre bei Knaben (was dem Verschluss der Epiphysenfugen entspricht).
Versäumte Anwendung
Wird innerhalb von drei Tagen bemerkt, dass eine Dosis versäumt wurde, so soll die Injektion so bald wie möglich nachgeholt werden. In diesem Fall kann anschliessend das gewohnte wöchentliche Dosierungsschema fortgesetzt, d.h. der übliche Wochentag der Injektion beibehalten werden.
Sind seit dem vorgesehenen Anwendungstag bereits mehr als drei Tage vergangen oder wurde mehr als eine Dosis vergessen, soll(en) die versäumte Dosis / die versäumten Dosen ausgelassen und die nächste Dosis am geplanten Verabreichungstag appliziert werden. Danach kann das ursprüngliche Dosierungsschema fortgeführt werden.
Änderung des Injektionstages
Der Wochentag der Injektion kann gewechselt werden, sofern zwischen zwei Dosen mindestens vier Tage (96 Stunden) liegen. Nach der Wahl eines neuen Verabreichungstags soll das gewohnte wöchentliche Dosierungsschema wieder aufgenommen werden.
Umstellung von anderen Wachstumshormon-Präparaten
Eine Umstellung von einem anderen GH-Präparat sollte nur durch einen Arzt mit Erfahrung in der Behandlung von Wachstumshormonmangel vorgenommen werden. Beim Wechsel von einem täglich verabreichten Wachstumshormon zu Sogroya einmal wöchentlich soll zunächst ein Wochentag für die Anwendung festgelegt werden. Die letzte Dosis der täglichen Behandlung erfolgt dann einen Tag (bzw. mindestens acht Stunden) vor der Anwendung der ersten Dosis von Sogroya.
Patienten, die von einem einmal wöchentlich verabreichten GH-Präparat zu Sogroya wechseln, wird empfohlen, ihr gewohntes wöchentliches Dosierungsschema (d.h. den gewählten Wochentag für die Injektion) beizubehalten.
Spezielle Dosierungsanweisungen
Kinder unter 3 Jahren
Zur Sicherheit und Wirksamkeit von Somapacitan bei Kindern <3 Jahren liegen nur limitierte Daten vor; bei Patienten <2.5 Jahre wurde Somapacitan nicht untersucht. In dieser Altersgruppe wird eine Anwendung daher nicht empfohlen.
Ältere Patienten
Sogroya ist nur für die Anwendung bei pädiatrischen Patienten zugelassen.
Patienten mit Leberfunktionsstörungen
Somapacitan wurde bei pädiatrischen Patienten mit Leberinsuffizienz nicht untersucht. Zu den Befunden bei Erwachsenen siehe «Pharmakokinetik», Abschnitt «Kinetik spezieller Patientengruppen».
Bei leichter Leberinsuffizienz (Child Pugh A) ist keine Dosisanpassung erforderlich. Die Anwendung sollte jedoch unter besonderer Vorsicht und regelmässiger Überwachung des IGF-1 erfolgen.
Bei Patienten mit moderater oder schwerer Leberinsuffizienz (Child Pugh B und C) wird eine Anwendung von Sogroya nicht empfohlen.
Patienten mit Nierenfunktionsstörungen
Somapacitan wurde bei pädiatrischen Patienten mit Niereninsuffizienz nicht untersucht; zu den Befunden bei Erwachsenen siehe «Pharmakokinetik», Abschnitt «Kinetik spezieller Patientengruppen». Eine Anpassung der Anfangsdosis ist bei eingeschränkter Nierenfunktion nicht erforderlich, die Anwendung von Sogroya sollte aber unter besonderer Vorsicht (sowie ggf. Überwachung des IGF-1) erfolgen.
Kontraindikationen·Hinweise auf eine Tumoraktivität (siehe «Warnhinweise und Vorsichtsmassnahmen»)
·Kritisch kranke Patienten mit akuter respiratorischer Insuffizienz oder mit Komplikationen nach grösseren abdominal- oder herzchirurgischen Eingriffen oder nach Polytrauma
·Bloom-Syndrom
·Fanconi-Anämie
·Überempfindlichkeit gegenüber dem Wirkstoff oder einem der Hilfsstoffe
Warnhinweise und VorsichtsmassnahmenNach Verschluss der Epiphysenfugen ist Somapacitan nicht mehr zur Wachstumsförderung wirksam und soll nicht weiter eingesetzt werden.
Ein intrakranieller Tumor muss inaktiv und eine Antitumor-Therapie vor Beginn der Behandlung mit Somapacitan abgeschlossen sein. Falls Hinweise auf ein Tumorwachstum auftreten, muss die Behandlung abgebrochen werden.
Unter Anwendung von Somapacitan wurden jeweils zu Beginn des Dosierungsintervalles höhere IGF-1-Konzentrationen beobachtet als unter einer konventionellen GH-Therapie mit täglich applizierten GH-Präparaten. Insbesondere kam es häufiger als bei der konventionellen Therapie zu einer Überschreitung der oberen Normgrenze von 2 SDS. Es ist nicht bekannt, ob dies mit erhöhten Langzeitrisiken (wie Tumoren oder Diabetes mellitus Typ II) verbunden ist.
Die nachfolgend beschriebenen Risiken wurden unter Anwendung von rekombinantem Wachstumshormon beobachtet. Es ist zu erwarten, dass sie in ähnlicher Weise auch auf Somapacitan zutreffen. Für Somapacitan selbst liegen bisher nur limitierte Erfahrungen vor.
Neoplasien
Patienten unter einer Behandlung mit Somapacitan sollten grundsätzlich sorgfältig auf eine mögliche Entwicklung von Tumoren hin überwacht werden.
Patienten mit Tumorerkrankungen in der Anamnese (einschliesslich intrakranieller Tumoren) waren aus den klinischen Studien ausgeschlossen. Sogroya sollte daher aus Vorsichtsgründen bei diesen Patienten nicht angewendet werden. Falls doch eine Behandlung erfolgen soll, muss der Patient unter der Therapie mit Somapacitan engmaschig auf mögliche Tumorrezidive hin überwacht werden.
Bei einer kleinen Anzahl von Kindern, die mit GH behandelt wurden, wurde über das Auftreten einer Leukämie berichtet. Es gibt jedoch keine Hinweise, dass eine GH-Therapie bei Patienten ohne prädisponierende Faktoren die Inzidenz von Leukämien erhöht.
Bei Patienten nach in der Kindheit überstandener maligner Erkrankung (sogenannte «childhood cancer survivors»), die mit Wachstumshormon behandelt wurden, wurde insgesamt ein leichter Anstieg von Zweitneoplasien beobachtet. Am häufigsten handelte es sich dabei um intrakranielle Tumore, insbesondere Meningeome. Diese Tumoren wurden v.a. bei Patienten beobachtet, welche zur Therapie ihrer ersten Neoplasie eine Strahlentherapie des Kopfes erhalten hatten.
Benigne intrakranielle Hypertonie
Bei schweren oder rezidivierenden Kopfschmerzen, Sehstörungen, Übelkeit und/oder Erbrechen wird eine Fundoskopie zur Erkennung eines möglichen Papillenödems empfohlen. Wird eine Stauungspapille bestätigt, sollte die Diagnose einer benignen intrakraniellen Hypertonie (Pseudotumor cerebri) in Betracht gezogen und gegebenenfalls die Wachstumshormontherapie abgebrochen werden.
Über die weitere Behandlung von Patienten nach intrakranieller Hypertonie liegen nur ungenügende Erfahrungen vor. Falls die Wachstumshormontherapie wieder aufgenommen wird, ist eine sorgfältige Überwachung auf mögliche Symptome einer intrakraniellen Drucksteigerung erforderlich.
Störungen des Glukosestoffwechsels
Eine Behandlung mit Wachstumshormon kann die Insulinempfindlichkeit reduzieren, insbesondere bei prädisponierten Patienten unter höheren Dosen. Daher kann bei Patienten mit unzureichender insulinsekretorischer Kapazität eine Hyperglykämie auftreten. Infolgedessen können sich während der Wachstumshormontherapie eine zuvor nicht diagnostizierte eingeschränkte Glukosetoleranz oder ein manifester Diabetes mellitus manifestieren. Aus diesem Grund sollte der Blutzuckerspiegel bei allen Patienten, die mit Wachstumshormon behandelt werden, in regelmässigen Abständen kontrolliert werden, insbesondere bei solchen mit Risikofaktoren für Diabetes mellitus, wie z.B. Adipositas, positive Familienanamnese für Diabetes oder Komedikation mit Kortikosteroiden.
Bei Patienten mit bereits bestehendem Diabetes mellitus oder eingeschränkter Glukosetoleranz soll Somapacitan nur mit Vorsicht und unter engmaschiger Überwachung des Glukosestoffwechsels angewendet werden. Zu Beginn einer Wachstumshormontherapie kann eine Anpassung der antidiabetischen Therapie erforderlich sein (siehe auch «Interaktionen»).
Schilddrüsenfunktion
Wachstumshormon erhöht die extrathyreoidale Umwandlung von T4 zu T3 und kann somit eine latente Hypothyreose aufdecken. Vor allem bei Patienten mit einer fortschreitenden Hypophysenerkrankung kann sich eine Hypothyreose entwickeln. Da eine Hypothyreose das Ansprechen auf eine Wachstumshormontherapie beeinträchtigt, sollte die Schilddrüsenfunktion regelmässig überprüft und ggf. eine Substitutionstherapie mit Schilddrüsenhormon eingeleitet werden.
Pankreatitis
Es liegen Berichte über das Auftreten einer Pankreatitis unter einer GH-Behandlung vor, insbesondere bei Kindern. Treten bei einem Patienten unter Behandlung mit Sogroya akute Oberbauchbeschwerden auf, sollte die Möglichkeit einer Pankreatitis in Betracht gezogen werden.
Nebennierenrinden (NNR)-Insuffizienz
Unter einer GH-Therapie kann es zu einer Reduktion der Aktivität des an der Cortisolsynthese beteiligten Enzyms 11β-Hydroxysteroid-Dehydrogenase Typ 1 kommen. Dadurch kann eine latente sekundäre NNR-Insuffizienz demaskiert werden, wodurch eine Glukokortikoid-Substitutionstherapie erforderlich werden kann. Darüber hinaus kann bei Patienten, die bereits eine Glukokortikoid-Substitutionstherapie aufgrund einer bekannten NNR-Insuffizienz erhalten, nach Beginn der GH-Therapie eine Erhöhung der Erhaltungs- und/oder der Stressdosis erforderlich sein (siehe «Interaktionen»). Die Patienten sollten daher auf erniedrigte Kortisolspiegel überwacht werden. Bei Patienten mit bekannter NNR-Insuffizienz ist insbesondere auf die Notwendigkeit einer Steigerung der Glukokortikoid-Dosis zu achten.
Skelettveränderungen
Eine Epiphyseolysis capitis femoris kann bei Patienten mit endokrinen Störungen (wie z.B. GH-Mangel) gehäuft vorkommen. Auch ein Morbus Legg-Calvé-Perthes kann bei kleinwüchsigen Patienten gehäuft vorkommen. Dies kann sich durch Hinken oder Hüft- bzw. Knieschmerzen äussern. Die Eltern sollten informiert werden, auf solche Symptome zu achten und sie ggf. unverzüglich dem behandelnden Arzt mitzuteilen.
Eine Skoliose kann sich bei jedem Kind während Phasen eines schnellen Wachstums manifestieren oder verschlechtern. Da Somapacitan die Wachstumsrate steigert, sollte während der Behandlung auf mögliche Anzeichen einer Skoliose geachtet werden. Es gibt jedoch bisher keine Hinweise, dass eine Behandlung mit Wachstumshormon Inzidenz oder Schweregrad einer Skoliose erhöht.
Reaktionen an der Applikationsstelle
In den klinischen Studien wurden bei ca. 5% der Patienten Reaktionen an der Injektionsstelle beobachtet. Diese waren meist leichtgradig und bildeten sich in der Mehrzahl nach kurzer Zeit zurück. Am häufigsten handelte es sich dabei um Hämatome (3%), Schmerzen (1.5%) oder Schwellung (0.8%).
Immunogenität
Unter einer Therapie mit Wachstumshormon kann es zur Entwicklung von Antikörpern gegen das jeweilige Präparat kommen. In der pivotalen Studie unterschied sich die Inzidenz von Antikörpern nicht in relevanter Weise zwischen Somapacitan und dem als Komparator eingesetzten konventionellen GH-Präparat. Die Bindungskapazität der gefundenen Antikörper war gering, und ein Einfluss auf Wirksamkeit oder Sicherheit war nicht erkennbar. Neutralisierende Antikörper wurden nicht nachgewiesen. Trotzdem sollten bei jedem Patienten, der nicht auf die Therapie anspricht, neben der Abklärung anderer möglicher Ursachen auch die Antikörper gegen Somapacitan bestimmt werden.
Überempfindlichkeitsreaktionen
Unter der Anwendung anderer GH-Präparate wurde über schwerwiegende systemische Überempfindlichkeitsreaktionen (z.B. Anaphylaxie, Angioödem) berichtet. Falls es zu einer solchen Reaktion kommt, muss die Anwendung von Sogroya umgehend beendet, eine entsprechende Behandlung eingeleitet und der Patient bis zum Abklingen der Symptome überwacht werden.
Lipohypertrophie
Wenn Sogroya über einen längeren Zeitraum hinweg an der gleichen Stelle verabreicht wird, kann eine Lipohypertrophie auftreten. Um das Risiko zu reduzieren, sollte die Injektionsstelle regelmässig gewechselt werden (siehe «Dosierung/Anwendung»).
Interaktion mit exogenen Östrogenen
Exogene Östrogene (z.B. in kombinierten hormonalen Kontrazeptiva) können, insbesondere bei oraler Gabe, die IGF-1-Konzentration im Serum reduzieren und dadurch die Wirksamkeit einer Wachstumshormon-Behandlung abschwächen. Zur Kontrazeption sollten daher ggf. nicht-hormonale Methoden eingesetzt und die Patientinnen entsprechend beraten werden.
Akute kritische Erkrankungen
Die Anwendung von GH in pharmakologischen Dosen wurde bei Patienten mit akuten kritischen Erkrankungen nach operativen Eingriffen am offenen Herzen bzw. im Abdominalbereich, nach Polytrauma oder bei akuter respiratorischer Insuffizienz mit einer erhöhten Mortalität in Verbindung gebracht (siehe «Kontraindikationen»). Für eine Substitutionstherapie bei Patienten mit GH-Mangel liegen keine entsprechenden Daten vor. Bei Patienten, die mit Sogroya behandelt werden und akut kritisch erkranken, sollte der erwartete Nutzen einer Weiterbehandlung sorgfältig gegen das mögliche Risiko abgewogen werden.
Weitere Vorsichtsmassnahmen
Bei eingeschränkter Leberfunktion ist die Somapacitan-Exposition erhöht (siehe «Pharmakokinetik», Abschnitt «Kinetik spezieller Patientengruppen»). Andererseits ist bei Leberfunktionsstörungen die Fähigkeit der Leber zur vermehrten Sekretion von IGF-1 nach Stimulation durch GH reduziert, sodass das Ansprechen auf die Therapie vermindert sein kann. Bei Patienten mit moderater oder schwerer Leberinsuffizienz (Child Pugh B und C) wird eine Anwendung von Sogroya daher nicht empfohlen.
In einer Placebo-kontrollierten Studie bei Erwachsenen wurde ein Shift von normalen zu erhöhten Phosphatkonzentrationen im Serum unter Somapacitan häufiger beobachtet als unter Placebo. Auch in einer pädiatrischen Studie (mit limitierter Fallzahl) war ein solcher Shift unter Somapacitan häufiger als in der Vergleichsgruppe unter einem täglich applizierten Somatropin-Präparat.
Bei erwachsenen Patienten mit GH-Mangel ist bekannt, dass Frauen einen höheren GH-Bedarf aufweisen als Männer. Zu möglichen geschlechtsabhängigen Unterschieden bei Patientinnen mit kindlichem GH-Mangel nach Erreichen der Pubertät liegen bisher keine ausreichenden Daten vor. Es kann jedoch nicht ausgeschlossen werden, dass bei gleicher Dosierung Mädchen nach der Pubertät mit einem geringeren Therapieerfolg rechnen müssen als Knaben. Es wird daher empfohlen, bei Mädchen das Ansprechen auf die GH-Therapie nach Eintritt der Pubertät besonders sorgfältig zu überwachen.
In der pädiatrischen Population wurde Somapacitan ausschliesslich bei pädiatrischen Patienten mit GH-Mangel (sogenanntem hypophysärem Minderwuchs) untersucht. Für andere Formen einer Wachstumsstörung wie Minderwuchs bei Turner-Syndrom, Wachstumsstörungen bei chronischer Niereninsuffizienz, Prader-Willi-Syndrom oder Wachstumsstörungen bei Patienten mit intrauterinem Kleinwuchs (SGA) liegen keine Daten vor. Sogroya soll daher in diesen Patientengruppen nicht angewendet werden.
Dieses Arzneimittel enthält weniger als 1 mmol Natrium (23 mg) pro ml, d.h. es ist nahezu «natriumfrei».
InteraktionenMit Somapacitan wurden keine Interaktionsstudien durchgeführt. Die nachfolgenden Angaben beruhen auf den entsprechenden Erfahrungen mit täglich appliziertem rekombinantem GH. Es ist zu erwarten, dass diese auch für Somapacitan gelten.
Pharmakokinetische Interaktionen
Einfluss von Wachstumshormon auf die Pharmakokinetik anderer Substanzen
Cytochrom P450-Substrate
Die Elimination von Arzneimitteln, die unter Beteiligung von Cytochrom P450-Isoenzymen metabolisiert werden, kann bei gleichzeitiger Anwendung von Wachstumshormon beschleunigt sein, was zu einer Erniedrigung der Plasmaspiegel dieser Wirkstoffe führen kann. Dies gilt z.B. für Kortikosteroide, Sexualsteroide, Antikonvulsiva oder Ciclosporin. Die klinische Relevanz dieser Interaktion ist unbekannt.
Pharmakodynamische Interaktionen
Einfluss anderer Arzneimittel auf die Wirksamkeit von Wachstumshormon
Die Wirksamkeit von Wachstumshormon kann durch begleitende Therapien mit anderen Hormonen, z.B. Testosteron oder Schilddrüsenhormone, beeinflusst werden.
Einfluss von Wachstumshormon auf die Wirksamkeit anderer Arzneimittel
Glukokortikoide
Wachstumshormon reduziert die Umwandlung von Kortison in Kortisol und könnte eine latente sekundäre NNR-Insuffizienz aufdecken oder eine niedrigdosierte Substitutionstherapie mit Glukokortikoiden unwirksam machen (s. «Warnhinweise und Vorsichtsmassnahmen»).
Antidiabetika
Bei Diabetikern kann bei gleichzeitiger Gabe von Somapacitan eine Dosisanpassung der antidiabetischen Therapie erforderlich sein, da Somapacitan die Insulinempfindlichkeit reduzieren kann.
Schwangerschaft, StillzeitSchwangerschaft
Es liegen keine Daten zur Anwendung von Somapacitan bei schwangeren Frauen vor. Tierexperimentelle Studien haben eine Reproduktionstoxizität gezeigt (siehe «Präklinische Daten»). Die Anwendung von Sogroya während der Schwangerschaft wird daher nicht empfohlen.
Bei Patientinnen im gebärfähigen Alter sollte Sogroya nicht ohne zuverlässige Kontrazeption angewendet werden, wobei nicht-hormonale Methoden zu wählen sind (siehe «Warnhinweise und Vorsichtsmassnahmen»). Wird bei Mädchen die Behandlung mit Sogroya nach der Menarche fortgeführt, muss daher eine Beratung bezüglich nicht-hormonaler Kontrazeptionsmethoden erfolgen.
Stillzeit
Es ist nicht bekannt, ob Somapacitan und/oder seine Metaboliten in die Muttermilch übergehen. Verfügbare pharmakodynamische/toxikologische Daten aus Tierstudien deuten auf einen Übertritt von Somapacitan in die Muttermilch hin.
Ein Risiko für gestillte Säuglinge kann daher nicht ausgeschlossen werden.
Es muss eine Entscheidung darüber getroffen werden, ob das Stillen unterlassen oder die Behandlung mit Sogroya abgebrochen/ausgesetzt werden soll. Dabei ist sowohl der Nutzen des Stillens für das Kind als auch der Nutzen der Therapie für die Frau zu berücksichtigen.
Fertilität
Es liegen keine klinischen Erfahrungen zu potenziellen Auswirkungen von Somapacitan auf die Fertilität vor. In tierexperimentellen Studien wurden keine unerwünschten Wirkungen auf die Fertilität beobachtet (siehe «Präklinische Daten»).
Wirkung auf die Fahrtüchtigkeit und auf das Bedienen von MaschinenEntsprechende Studien wurden nicht durchgeführt. Ein Einfluss von Somapacitan auf die Fahrtüchtigkeit oder die Fähigkeit, Maschinen zu bedienen, wird jedoch nicht erwartet.
Unerwünschte WirkungenZu den schwerwiegenden Risiken einer Therapie mit Wachstumshormon wird auch auf die Rubrik «Warnhinweise und Vorsichtsmassnahmen» verwiesen.
Die nachstehend aufgeführten unerwünschten Wirkungen beruhen auf den Sicherheitsdaten aus einer 52-wöchigen Phase 3-Studie, in welcher n=132 pädiatrische Patienten mit Wachstumshormonmangel (GHD) mit Somapacitan behandelt wurden.
Die in den klinischen Studien mit Somapacitan am häufigsten beobachteten unerwünschten Wirkungen waren Kopfschmerzen (12%), Hypothyreose (5%), Reaktionen an der Injektionsstelle (5%) und periphere Ödeme (3%).
In der Regel waren die unerwünschten Wirkungen vorübergehend.
Die unerwünschten Arzneimittelwirkungen sind nachfolgend nach MedDRA-Systemorganklassen und Häufigkeit gemäss folgender Konvention geordnet: Sehr häufig (≥1/10); häufig (≥1/100, <1/10); gelegentlich (≥1/1'000, <1/100); selten (≥1/10'000, <1/1'000); sehr selten (<1/10'000).
Erkrankungen des Immunsystems
Häufig: Überempfindlichkeitsreaktionen (wie Hautausschlag, Pruritus, Urtikaria)
Endokrine Erkrankungen
Häufig: Hypothyreose, NNR-Insuffizienz
Stoffwechsel- und Ernährungsstörungen
Häufig: Hyperglykämie
Erkrankungen des Nervensystems
Sehr häufig: Kopfschmerzen (12%)
Skelettmuskulatur-, Bindegewebs- und Knochenerkrankungen
Häufig: Arthralgien, Myalgien
Allgemeine Erkrankungen und Beschwerden am Verabreichungsort
Häufig: periphere Ödeme, Reaktionen an der Injektionsstelle (z.B. Hämatom, Schmerzen, Schwellung; siehe auch «Warnhinweise und Vorsichtsmassnahmen»), Müdigkeit
Gelegentlich: Lipodystrophie
Meldung des Verdachts auf Nebenwirkungen
Die Meldung des Verdachts auf Nebenwirkungen nach der Zulassung ist von grosser Wichtigkeit. Sie ermöglicht eine kontinuierliche Überwachung des Nutzen-Risiko-Verhältnisses des Arzneimittels. Angehörige von Gesundheitsberufen sind aufgefordert, jeden Verdacht einer neuen oder schwerwiegenden Nebenwirkung über das Online-Portal ElViS (Electronic Vigilance System) anzuzeigen. Informationen dazu finden Sie unter www.swissmedic.ch.
ÜberdosierungEs liegen nur limitierte klinische Erfahrungen mit einer Überdosierung von Somapacitan vor. Eine kurzfristige Überdosierung kann zunächst zu hypoglykämischen Reaktionen und anschliessend zu Hyperglykämien führen. Die niedrigen Blutzuckerwerte konnten nur biochemisch festgestellt werden und verliefen klinisch asymptomatisch. Bei chronischer Überdosierung kann es zu den typischen Symptomen eines Überschusses an humanem Wachstumshormon kommen.
Eigenschaften/WirkungenATC-Code
H01AC07
Wirkungsmechanismus
Somapacitan ist ein langwirksames, rekombinantes Derivat des humanen Wachstumshormons. Es besteht aus der Aminosäuresequenz des endogenen humanen Wachstumshormons mit einer einzelnen Substitution (L101C), an welche eine albuminbindende Einheit angebracht wurde. Die reversible Bindung an endogenes Albumin verzögert die Elimination von Somapacitan und verlängert dadurch Halbwertszeit und Wirkungsdauer in vivo.
Die albuminbindende Einheit (Seitenkette) besteht aus einer Fettsäureeinheit und einem an die Proteinposition 101 gebundenen hydrophilen Spacer. Der Wirkungsmechanismus von Somapacitan setzt entweder direkt über den Wachstumshormonrezeptor an und/oder indirekt über IGF-1, das ubiquitär im Organismus, vorwiegend aber in der Leber produziert wird.
Somapacitan verteilt sich in die Epiphysen. Es stimuliert das Längenwachstum infolge seiner Wirkung auf die Epiphysenfugen der Knochen.
Pharmakodynamik
Somapacitan stimuliert bei pädiatrischen Patienten mit Wachstumshormonmangel das Längenwachstum und steigert die Wachstumsgeschwindigkeit.
Durch die Gabe von Somapacitan wird ein dosisabhängiger Anstieg des IGF-1, eines anerkannten Biomarkers für die Wirksamkeit einer GH-Therapie, induziert. Das Steady State der IGF-1-Konzentrationen wurde nach 1–2 Dosen erreicht.
Bei pädiatrischen Patienten mit Wachstumshormonmangel induzierte Somapacitan eine dosislineare IGF-1-Antwort, wobei eine Veränderung von 0.02 mg/kg durchschnittlich zu einer Veränderung des IGF-1-Standardabweichungs-Scores (SDS) von 0.32 führte.
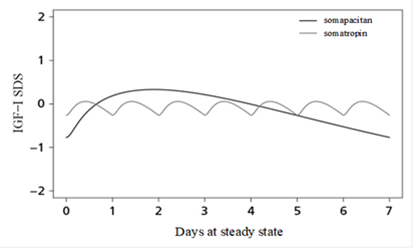
Abbildung 1: Modellierte IGF-1-Profile im Steady-State von Somapacitan und Somatropin (basierend auf Daten von AGHD = Adult Growth Hormone Deficiency (Wachstumshormonmangel bei Erwachsenen))
QTc-Dauer
Es wurde keine tQT-Studie durchgeführt. Die potenzielle Wirkung von Somapacitan auf die kardiale Repolarisation bei Erwachsenen wurde in einer Phase 3-Studie anhand von EKGs beurteilt, die etwa zum Zeitpunkt der Cmax von Somapacitan in therapeutischen Dosen aufgezeichnet wurden. Es fand sich kein Zusammenhang zwischen der Veränderung der QTcF gegenüber Baseline und der Konzentration von Somapacitan.
Klinische Wirksamkeit
Wirksamkeit und Sicherheit von Somapacitan einmal wöchentlich wurden in einer 52-wöchigen multizentrischen, offenen, randomisierten, aktiv kontrollierten Phase-3-Parallelgruppenstudie (REAL 4) an n=200 therapienaiven, präpubertären pädiatrischen Patienten mit Wachstumshormonmangel untersucht. Die Patienten erhielten 0.16 mg/kg/Woche Somapacitan einmal wöchentlich (n=132) oder 0.034 mg/kg/Tag Somatropin einmal täglich (n=68).
Eingeschlossen wurden Kinder mit nachgewiesenem GH-Mangel (d.h. hypophysärem Minderwuchs) im Alter von 2.5 Jahren bis <10 Jahre (Mädchen) bzw. <11 Jahre (Knaben). Patienten mit anderen Ursachen einer Wachstumsstörung waren von der Studienteilnahme ausgeschlossen. Einschlusskriterium waren eine Wachstumsgeschwindigkeit unterhalb der 25. Perzentile sowie ein IGF-1 SDS <-1.
Primärer Wirksamkeitsendpunkt war die Wachstumsgeschwindigkeit (HV) nach 12-monatiger Behandlung. Wesentlicher Sekundärendpunkt war z.B. die Veränderung des Körpergrössen-SDS gegenüber dem Ausgangswert.
Das mittlere Alter bei Studieneintritt betrug 6.4 Jahre (Spanne: 2.5 bis 11). 25.5% der Patienten waren weiblich, 74.5% männlich. 57% der Patienten waren Weisse, 37% Asiaten. Die mittlere Körpergrösse lag in der Somapacitan-Gruppe bei -2.99 SDS, in der Komparatorgruppe bei -3.47 SDS. Der Anteil der Patienten mit einem IGF-1 SDS <-2 (d.h. höherem Schweregrad des GH-Mangels) lag in der Somapacitan-Gruppe bei 42%, in der Komparatorgruppe bei 57%.
Die 52-wöchige Behandlung mit Somapacitan einmal wöchentlich führte zu einem annualisierten Längenwachstum von 11.2 cm/Jahr. Patienten, die täglich mit Somatropin behandelt wurden, erreichten nach einer 52-wöchigen Behandlung ein annualisiertes Längenwachstum von 11.7 cm/Jahr. Die Therapiedifferenz (-0.5; 95%-KI -1.1; 0.2) erfüllte dabei das vordefinierte Kriterium der Nichtunterlegenheit von Somapacitan gegenüber einmal täglich appliziertem Somatropin.
Die Befunde für die wesentlichen Sekundärendpunkte waren hierzu konsistent und zeigten ebenfalls eine vergleichbare Wirksamkeit von Somapacitan und einmal täglich appliziertem Somatropin.
Darüber hinaus liegen aus der Verlängerung einer Phase-II-Studie an initial n=59 ebenfalls präpubertären, therapienaiven Patienten mit GH-Mangel limitierte Langzeitdaten über eine Behandlungsdauer von bis zu 4 Jahren vor. Diese limitierten Daten deuten darauf hin, dass die Wirksamkeit von Somapacitan auch bei längerfristiger Anwendung aufrechterhalten bleibt. Auch bei Patienten, welche nach 3 Jahren von einem einmal täglich applizierten Somatropin-Präparat auf Somapacitan umgestellt wurden, konnte die Wirksamkeit der Vorbehandlung aufrechterhalten werden. Nach 4-jähriger Therapie lag der mittlere Körpergrössen-SDS bei -1.06.
PharmakokinetikAbsorption
Bei pädiatrischen Patienten mit Wachstumshormonmangel lag die mediane tmax unter Dosen von 0.02 mg/kg/Woche bis 0.16 mg/kg/Woche zwischen 8 und 25.5 Stunden. Die Steady-State-Exposition wurde nach 1–2 wöchentlichen Gaben erreicht.
Die absolute Bioverfügbarkeit von Somapacitan beim Menschen wurde nicht untersucht.
Distribution
Somapacitan ist weitgehend an Plasmaproteine gebunden (> 99%) und wird erwartungsgemäss wie Albumin verteilt. Basierend auf populationspharmakokinetischen Analysen lag das geschätzte Verteilungsvolumen (V/F) bei pädiatrischen Patienten mit Wachstumshormonmangel bei 1.7 l.
Metabolismus
Somapacitan unterliegt einer ausgeprägten Metabolisierung durch Proteolyse und Spaltung der Linkersequenz zwischen dem Peptid und der albuminbindenden Einheit.
Elimination
Nach einer Einzeldosis von 0.16 mg/kg/Woche lag die terminale Halbwertszeit bei pädiatrischen Patienten mit Wachstumshormonmangel bei 34 Stunden.
Es wurde wenig bis keine Akkumulation (mittlere Akkumulationsrate: 1–2) von Somapacitan nach Mehrfachdosen beobachtet.
Daten zur Elimination von Somapacitan liegen nur von erwachsenen Patienten mit GH-Mangel vor. Dabei wurde Somapacitan in Form seiner Metaboliten überwiegend mit dem Urin (81%) und nur zu einem kleineren Anteil (13%) über die Faeces ausgeschieden. Unverändertes Somapacitan wurde weder im Urin noch in den Faeces nachgewiesen.
Linearität/Nicht-Linearität
Bei pädiatrischen Patienten wurde die Pharmakokinetik von Somapacitan nach subkutaner Verabreichung bei Dosierungen von 0.02 bis 0.16 mg/kg/Woche untersucht. Dabei zeigte Somapacitan eine nicht-lineare Pharmakokinetik.
Kinetik spezieller Patientengruppen
Basierend auf populationspharmakokinetischen Analysen haben Geschlecht, ethnische Herkunft und Körpergewicht bei pädiatrischen Patienten mit Wachstumshormonmangel bei einer gewichtsbasierten Dosierung keinen klinisch relevanten Einfluss auf die Pharmakokinetik von Somapacitan. Somapacitan wurde bei pädiatrischen Patienten mit einem Körpergewicht von bis zu 62 kg verabreicht.
Patienten mit Leberfunktionsstörungen
Zur Pharmakokinetik von Somapacitan bei pädiatrischen Patienten mit eingeschränkter Leberfunktion liegen keine Daten vor.
Bei Erwachsenen führte eine Somapacitan-Dosis von 0.08 mg/kg im Steady State bei Patienten mit moderater Leberfunktionsstörung zu höheren Expositionen; das Verhältnis zur normalen Leberfunktion betrug 4.69 für die AUC0-168h und 3.52 für die Cmax. Eine leichtgradige Leberfunktionsstörung hatte keinen Einfluss auf die Somapacitan-Exposition.
Patienten mit Nierenfunktionsstörungen
Zur Pharmakokinetik von Somapacitan bei pädiatrischen Patienten mit eingeschränkter Nierenfunktion liegen keine Daten vor.
Bei Erwachsenen war bei Patienten mit eingeschränkter Nierenfunktion die Exposition im Steady State unter einer Somapacitan-Dosis von 0.08 mg/kg erhöht. Dies zeigte sich insbesondere bei Patienten mit schwerer Nierenfunktionsstörung und bei dialysepflichtigen Patienten; hier lag das Verhältnis für die AUC0-168h im Vergleich zur normalen Nierenfunktion bei 1.75 bzw. 1.63. Im Allgemeinen stieg die Somapacitan-Exposition mit abnehmender GFR an.
Präklinische DatenBasierend auf konventionellen Studien zur Sicherheitspharmakologie, Toxizität bei wiederholter Gabe, Genotoxizität oder prä- und postnatalen Entwicklung lassen die präklinischen Daten keine besonderen Gefahren für den Menschen erkennen.
Kanzerogenität
Es wurden keine Kanzerogenitätsstudien mit Somapacitan durchgeführt.
Reproduktionstoxizität
Bei männlichen und weiblichen Ratten wurden bei einer Dosis, die zu einer mindestens 5-mal höheren Exposition als die erwartete maximale klinische Exposition bei 0.16 mg/kg/Woche (in pädiatrischen Patienten) führte, keine unerwünschten Wirkungen auf die Fertilität beobachtet. Jedoch wurde bei allen verabreichten Dosen ein unregelmässiger weiblicher Östruszyklus beobachtet.
Bei subkutaner Verabreichung von Somapacitan-Dosen an trächtige Ratten und Kaninchen während der Organogenese, die zu Expositionen 4- bis 6-fach über den erwarteten Expositionen bei der klinischen Maximaldosis in pädiatrischen Patienten von 0.16 mg/kg/Woche führten, wurden keine Hinweise auf fetale Schäden identifiziert. Bei hohen Dosen, die zu einer mindestens 50-mal höheren Exposition als die erwartete klinische Exposition bei 0.16 mg/kg/Woche führten, wurden bei den Jungtieren von weiblichen Ratten, denen Somapacitan verabreicht wurde, kurze/gekrümmte/verdickte Röhrenknochen beobachtet. Derartige Befunde bei Ratten haben sich als nach der Geburt reversibel erwiesen und werden eher als geringfügige Missbildungen denn als dauerhafte Anomalien betrachtet.
In der Studie mit trächtigen Kaninchen führte die subkutane Verabreichung von Somapacitan bei allen Dosen zu einem verminderten Fetuswachstum. Bei der niedrigen Dosis (Exposition ca. 4-fach über der erwarteten klinischen Exposition bei 0.16 mg/kg/Woche) waren die Effekte leicht und wurden nicht als advers eingestuft.
Toxizitätsprüfungen mit juvenilen Tieren
Es wurde keine Toxizitätsprüfungen mit juvenilen Tieren durchgeführt, da in den Toxizitätsstudien kein konkret zu untersuchendes Zielgewebe für pädiatrische Patienten identifiziert wurde.
Sonstige HinweiseInkompatibilitäten
Da keine Kompatibilitätsstudien durchgeführt wurden, darf das Arzneimittel nicht mit anderen Arzneimitteln gemischt werden.
Haltbarkeit
Das Arzneimittel darf nur bis zu dem auf der Packung mit «EXP» bezeichneten Datum verwendet werden.
Haltbarkeit nach Anbruch
Nach der ersten Anwendung
Haltbarkeit des angebrochenen Pens: 6 Wochen
Im Kühlschrank lagern (2°C - 8°C). Siehe zudem «Besondere Lagerungshinweise».
Besondere Lagerungshinweise
Ausser Reichweite von Kindern aufbewahren.
Vor und nach der ersten Anwendung
Mit aufgesetzter Penkappe im Kühlschrank bei 2–8°C lagern. In der Originalverpackung aufbewahren, um den Inhalt vor Licht zu schützen. Nicht einfrieren und nicht direkt neben dem Kühlelement des Kühlschranks lagern. Sogroya nicht verwenden, wenn es einmal gefroren war. Den Fertigpen entsorgen, wenn er über 30°C gelagert wurde. Direkte oder übermässige Hitze vermeiden. Sonnenlicht vermeiden.
Wenn eine Kühlung nicht möglich ist (z.B. auf Reisen), kann Sogroya über einen Zeitraum von insgesamt 72 Stunden (3 Tage) bei einer Temperatur bis zu 30°C aufbewahrt werden. Sogroya muss nach Aufbewahrung bei dieser Temperatur wieder in den Kühlschrank gelegt werden. Falls das Produkt mehrmals ausserhalb des Kühlschranks gelagert und dann wieder in den Kühlschrank gelegt wird, darf die Gesamtdauer der Lagerung ausserhalb des Kühlschranks 3 Tage nicht überschreiten. Dies muss sorgfältig überwacht werden. Der Sogroya Pen muss entsorgt werden, wenn er länger als 72 Stunden (3 Tage) bei bis zu 30°C oder für eine beliebige Zeitspanne bei über 30°C aufbewahrt wurde.
Hinweise für die Handhabung
Für detaillierte Angaben siehe Gebrauchsanweisung in der Patienteninformation (Packungsbeilage).
Das Datum der ersten Verwendung muss im dafür vorgesehenen Leerfeld auf der Originalverpackung vermerkt werden.
Bitte raten Sie dem Patienten resp. seiner Betreuungsperson, die Hinweise zur Handhabung in der Packungsbeilage sehr sorgfältig zu lesen, bevor Sogroya angewendet wird.
Der Pen darf nur von einer Person verwendet werden.
Sogroya darf nur verwendet werden, wenn die Lösung klar bis leicht schimmernd, farblos oder leicht gelblich und frei von sichtbaren Partikeln ist.
Sogroya kann mit Injektionsnadeln von bis zu 8 mm Länge verabreicht werden. Der Pen ist für die Anwendung mit NovoFine® oder NovoTwist® Einwegnadeln vorgesehen. Die Injektionsnadeln sind nicht in der Packung enthalten.
Die Injektionsnadel ist nach jeder Injektion zu entfernen und sicher zu entsorgen. Der Sogroya Fertigpen ist ohne befestigte Injektionsnadel zu lagern. Für jede Injektion ist eine neue Kanüle zu verwenden, um Verunreinigungen zu vermeiden.
Die Patrone darf nicht aus dem Fertigpen herausgenommen und wiederaufgefüllt werden.
Injektionsadeln und sonstiges Abfallmaterial sollten gemäss den nationalen Bestimmungen entsorgt werden.
Zulassungsnummer69063 (Swissmedic)
PackungenSogroya 5 mg/1.5 ml Somapacitan Fertigpen (blaugrün): Packungen zu 1 Stück. (A)
Sogroya 10 mg/1.5 ml Somapacitan Fertigpen (gelb): Packungen zu 1 Stück. (A)
Sogroya 15 mg/1.5 ml Somapacitan Fertigpen (rot): Packungen zu 1 Stück. (A)
Injektionsnadeln sind nicht im Lieferumfang enthalten.
ZulassungsinhaberinNovo Nordisk Pharma AG, Kloten
Domizil: Zürich
Stand der InformationDezember 2023
|