ZusammensetzungWirkstoffe
Rozanolixizumab ist ein rekombinanter, humanisierter monoklonaler Antikörper des Immunglobulins G 4P (IgG4P) gegen den neonatalen Fc-Rezeptor (FcRn), der in gentechnisch veränderten CHO (Chinese Hamster Ovary)-Zellen durch rekombinante DNA-Technologie hergestellt wird.
Ein ml enthält 140 mg Rozanolixizumab.
Hilfsstoffe
L-Histidin, L-Histidinhydrochlorid-Monohydrat, L-Prolin, Polysorbat 80, Wasser für Injektionszwecke.
Indikationen/AnwendungsmöglichkeitenRystiggo wird angewendet als Zusatzbehandlung zur Standardtherapie von generalisierter Myasthenia gravis (gMG) bei erwachsenen Patienten, die Antikörper-positiv bezüglich Anti-AChR(Acetylcholin-Rezeptor)- oder Anti-MuSK(Muskelspezifische Tyrosinkinase) sind (siehe Rubrik Klinische Wirksamkeit).
Dosierung/AnwendungDie Behandlung sollte von Ärzten eingeleitet und überwacht werden, die in der Behandlung von Patienten mit neuromuskulären oder neuroinflammatorischen Erkrankungen erfahren sind.
Dosierung
Um die Rückverfolgbarkeit von biotechnologischen Arzneimitteln sicherzustellen, wird empfohlen, Handelsnamen und die Chargennummer bei jeder Behandlung zu dokumentieren.
Ein Behandlungszyklus besteht aus einer subkutan verabreichten Dosis pro Woche über einen Zeitraum von sechs Wochen.
Die folgende Tabelle zeigt die empfohlene wöchentliche Gesamtdosis von Rozanolixizumab nach Körpergewicht des Patienten:
|
Körpergewicht
|
≥35 bis <50 kg
|
≥50 bis < 70 kg
|
≥ 70 to < 100 kg
|
≥100 kg
| |
Wöchentliche Dosis (mg)
|
280 mg
|
420 mg
|
560 mg
|
840 mg
| |
Wöchentliche Dosis (ml)
|
2 ml
|
3 ml
|
4 ml
|
6 ml
| |
Anzahl der zu verwendenden Durchstechflaschen*
|
1
|
2
|
2
|
3
|
*jede Durchstechflasche enthält ein Übervolumen zum Vorfüllen der Infusionsleitung, siehe «Art der Anwendung».
Es liegen keine Erfahrungen mit Patienten unter 35 kg und über 155 kg vor. Patienten mit Myasthenia Gravis Foundation of America (MGFA) Klasse IVb und V wurden im klinischen Studienprogramm von Rystiggo nicht untersucht.
Nachfolgende Behandlungszyklen werden entsprechend der klinischen Beurteilung verabreicht.
Die Häufigkeit der Behandlungszyklen kann je nach Patient variieren. Bei einer Verschlechterung der Symptome, die eine zusätzliche Behandlung erforderlich macht (definiert als ein Anstieg um mindestens 3 Punkte auf der Quantitative-Myasthenia Gravis (QMG) Skala und/oder um 2 Punkte auf der Myasthenia gravis-Activities of Daily Living (MG-ADL) Skala seit dem Ende des letzten Therapiezyklus), können die Patienten einen weiteren 6-wöchigen Behandlungszyklus erhalten. Im klinischen Entwicklungsprogramm hatten die meisten Teilnehmer behandlungsfreie Intervalle von 4-13 Wochen zwischen den Zyklen. Der Mindestabstand zwischen zwei Behandlungszyklen beträgt vier Wochen (d.h. Beginn des nachfolgenden Zyklus mindestens 28 Tage nach letzter Dosis des vorausgegangenen Zyklus).
Der Nutzen der Behandlung sollte regelmässig überprüft werden. Wenn ein Patient auf zwei aufeinanderfolgende sechswöchige Behandlungszyklen nicht anspricht (definiert als eine Verbesserung um weniger als 3 Punkte auf der QMG-Skala und/oder um weniger als 2 Punkte auf der MG-ADL-Skala gegenüber dem Beginn des jeweiligen Zyklus), sollte die Behandlung mit Rystiggo nicht fortgeführt werden.
Wenn eine geplante Infusion versäumt wird, kann Rystiggo bis zu 4 Tage nach dem geplanten Zeitpunkt verabreicht werden. Anschliessend sollte das ursprüngliche Dosierungsschema wieder aufgenommen werden, bis der Behandlungszyklus abgeschlossen ist.
Spezielle Dosierungsanweisungen
Patienten mit Leberfunktionsstörungen
Für Patienten mit Leberfunktionsstörung liegen keine Daten vor. Eine Dosisanpassung wird nicht als notwendig erachtet, da die Pharmakokinetik von Rozanolixizumab durch Leberfunktionsstörungen wahrscheinlich nicht beeinflusst wird (siehe Rubrik Pharmakokinetik).
Patienten mit Nierenfunktionsstörungen
Bei Patienten mit leichter bis mittelschwerer Nierenfunktionsstörung (eGFR > 45 ml/min/1,73 m2) liegen nur begrenzte Daten zur Sicherheit und Wirksamkeit vor. Für Patienten mit schwerer Nierenfunktionsstörung liegen keine Daten vor. Eine Dosisanpassung wird nicht als notwendig erachtet, da die Pharmakokinetik von Rozanolixizumab durch eine Nierenfunktionsstörungen wahrscheinlich nicht beeinflusst wird (siehe Rubrik Pharmakokinetik).
Ältere Patienten
Eine Dosisanpassung ist nicht erforderlich (siehe Rubrik Pharmakokinetik). Zur Sicherheit und Wirksamkeit bei Patienten >85 Jahren stehen bislang keine Daten zur Verfügung. Für die Altersgruppe ≥65 bis 85 Jahre ist die Datenlage limitiert (siehe Rubrik Warnhinweise und Vorsichtsmassnahmen sowie Unerwünschte Wirkungen).
Kinder und Jugendliche
Die Sicherheit und Wirksamkeit von Rozanolixizumab bei Kindern und Jugendlichen unter 18 Jahren sind nicht erwiesen. Es liegen keine Daten vor. Rystiggo ist für die Anwendung in der pädiatrischen Population nicht zugelassen.
Art der Anwendung
Zur subkutanen Infusion mit einer Pumpe.
Es müssen Infusionspumpen, Spritzen und Infusionssets verwednet werden, die für die subkutane Anwendung von Arzneimitteln geeignet sind. Es wird empfohlen, Pumpen zu verwenden, bei denen das verabreichte Volumen voreingestellt werden kann, da jede Durchstechflasche überschüssiges Volumen zum Füllen der Infusionsleitung enthält.
Es wird empfohlen, Rozanolixizumab subkutan zu verabreichen, vorzugsweise in den rechten oder linken unteren Teil des Bauches, unterhalb des Bauchnabels. Andere Injektionsstellen wurden im Rahmen des klinischen Entwicklungsprogramms nicht untersucht. Infusionen sollten nicht an Stellen verabreicht werden, an denen die Haut empfindlich, gerötet oder verhärtet ist.
Während der Anwendung aller Einzeldosen des gesamten ersten Behandlungszyklus und der Anwendung der ersten Dosis des zweiten Behandlungszyklus von Rozanolixizumab soll eine angemessene Behandlung für Injektions- und Überempfindlichkeitsreaktionen unmittelbar verfügbar sein (siehe Rubrik Warnhinweise und Vorsichtsmassnahmen).
Infusionsrate
Rozanolixizumab wird mit Hilfe einer Infusionspumpe mit einer konstanten Flussrate von bis zu 20 ml/Std. verabreicht.
Vor der Verabreichung von Rozanolixizumab muss die Gebrauchsanweisung sorgfältig gelesen werden.
KontraindikationenÜberempfindlichkeit gegen den Wirkstoff oder einen der im Abschnitt Zusammensetzung genannten sonstigen Bestandteile.
Warnhinweise und VorsichtsmassnahmenKlinisches Monitoring
Im klinischen Studienprogramm wurde die Wirksamkeit und Sicherheit der wiederholten zyklischen Behandlung mit der zugelassenen Dosierung von Rystiggo nur an einer begrenzten Anzahl von Patienten untersucht. Rystiggo wird als symptombedingte zyklische Behandlung verabreicht.
Angesichts individueller, nicht vorhersehbarer, Unterschiede im klinischen Ansprechen sollen Patienten engmaschig überwacht werden. Im klinischen Studienprogramm kam es trotz wiederholter zyklischer Behandlung mit Rystiggo in einer Dosierung von etwa (≈) 7mg/kg oder ≈ 10mg/kg bei 13,8% der Patienten zu einer Verschlechterung der Grunderkrankung bis zur myasthenen Krise (2,1%) (siehe Rubrik Klinische Wirksamkeit und Unerwünschte Wirkungen).
Zur Wirksamkeit und Sicherheit von Rystiggo zur Behandlung der myasthenen Krise liegen keine Daten vor.
Körpergewicht <50kg
Im klinischen Studienprogramm wurde nur eine sehr begrenzte Anzahl von Patienten mit einem Körpergewicht unter 50kg untersucht. Die Wirksamkeit der wiederholten zyklischen Behandlung mit Rozanolixizumab wurde in dieser Gruppe bislang nicht zuverlässig gezeigt (siehe Rubrik Klinische Wirksamkeit).
Ältere Patienten
Im klinischen Studienprogramm wurde nur eine begrenzte Anzahl von Patienten im Alter von ≥65 bis 85 Jahren untersucht Der Behandlungseffekt einer wiederholten zyklischen Behandlung war in dieser Altersgruppe geringer und weniger robust gegenüber Patienten <65 Jahren. Zugleich zeigten ältere Patienten ein ungünstigeres Nebenwirkungsprofil: So kam es in einer doppelt verblindeten Studie und einer offenen Erweiterungsstudie bei 39,6% der älteren Patienten (≥65 Jahre) zu schweren unerwünschten Wirkungen gegenüber 30,7% in der Gruppe <65 Jahren. Eine myasthene Krise trat bei 6,3% der Patienten ≥65 Jahren und bei 0,7% der Patienten <65 Jahren auf (siehe Rubrik Klinische Wirksamkeit).
Myasthene Krise
Die Behandlung von Patienten mit drohender oder manifester myasthener Krise mit Rozanolixizumab wurde nicht untersucht. Rozanolixizumab ist für die Behandlung der drohenden oder manifesten myasthenen Krise nicht zugelassen. Im Fall der drohenden oder manifesten myasthenen Krise muss die Interaktion zwischen Rozanolixizumab und etablierten Therapien der myasthenen Krise beachtet werden, da Rozanolixizumab die Wirksamkeit dieser Therapien beeinträchtigen kann (siehe Rubrik Interaktionen).
Aseptische Meningitis
Nach Behandlung mit Rozanolixizumab wurde über arzneimittelinduzierte aseptische Meningitis berichtet (siehe Abschnitt Unerwünschte Wirkungen). Treten Symptome auf, die auf eine arzneimittelinduzierte aseptische Meningitis hindeuten, müssen Diagnosestellung und Behandlung gemäss Versorgungsstandard erfolgen.
Hypogammaglobulinämie
Aufgrund des Wirkmechanmismus kann Rystiggo zu einem deutlichen Abfall der Immunglobulin G (IgG) Spiegel führen (siehe Abschnitt Unerwünschte Wirkungen). Im klinischen Entwicklungsprogramm wurde die Behandlung mit Rystiggo vorübergehend unterbrochen, wenn das Gesamt-IgG im Serum <1g/L fiel und zwar unabhängig davon, ob zugleich eine Infektion vorlag. Sobald der IgG-Spiegel wieder ≥2g/L lag, konnte die Behandlung mit Rystiggo wieder aufgenommen werden. Im Fall einer nicht- schwerwiegenden anhaltenden oder wiederkehrenden Infektion bei einem Gesamt-IgG im Serum ≥1 and <2g/L wurde die Therapie mit Rystiggo ebenfalls vorübergehend solange ausgesetzt, bis die Infektion abgeklungen und das IgG wieder auf ≥2g/L angestiegen war.
Infektionen
Da Rozanolixizumab eine vorübergehende Reduktion der IgG-Spiegel verursacht, kann das Infektionsrisiko ansteigen (siehe Abschnitt Eigenschaften/Wirkungen). Die Behandlung mit Rozanolixizumab sollte bei Patienten mit einer klinisch bedeutsamen aktiven Infektion nicht begonnen werden, bis die Infektion abgeklungen oder angemessen behandelt ist. Während der Behandlung mit Rozanolixizumab ist auf klinische Anzeichen und Symptome von Infektionen zu achten. Wenn eine klinisch bedeutsame aktive Infektion auftritt, sollte die Rozanolixizumab-Behandlung unterbrochen werden, bis die Infektion abgeklungen ist.
Impfung
Die Immunisierung mit Impfstoffen während der Rozanolixizumab-Therapie wurde nicht untersucht. Die Sicherheit der Immunisierung mit Lebendimpfstoffen oder attenuierten Lebendimpfstoffen und die Reaktion auf die Immunisierung mit Impfstoffen sind nicht bekannt. Alle Impfstoffe sollen gemäss den Impfrichtlinien und mindestens 4 Wochen vor Beginn der Behandlung mit Rozanolixizumab verabreicht werden. Während der Behandlung mit Rozanolixizumab wird eine Impfung der Patienten mit Lebendimpfstoffen oder attenuierten Lebendimpfstoffen nicht empfohlen. Bei allen anderen Impfstoffen soll die Impfung mindestens 2 Wochen nach der letzten Infusion eines Behandlungszyklus und 4 Wochen vor Beginn des nächsten Zyklus mit Rozanolixizumab erfolgen.
Überempfindlichkeit
Infusionsreaktionen wie Ausschlag oder Angioödem können auftreten (siehe Abschnitt Unerwünschte Wirkungen). Im klinischen Studienprogramm waren diese leicht bis mittelschwer. Patienten müssen während der Behandlung mit Rozanolixizumab und noch 15 Minuten nach Abschluss der Anwendung von Rozanolixizumab auf klinische Anzeichen und Symptome von Überempfindlichkeitsreaktionen überwacht werden. Wenn eine Überempfindlichkeitsreaktion während der Anwendung des Arzneimittels auftritt (siehe Abschnitt Unerwünschte Wirkungen), muss die Rozanolixizumab-Infusion abgebrochen werden und es sind bei Bedarf geeignete Massnahmen einzuleiten. Nach dem Abklingen kann die Anwendung wieder aufgenommen werden.
Immunogenität
In den gepoolten zyklischen Behandlungsdaten aus dem Phase-III-Programm entwickelten nach 1 Behandlungszyklus mit 6 wöchentlichen Rozanolixizumab-Dosen 26,9 % (42/156) der Patienten Antikörper gegen den Wirkstoff (Anti-Wirkstoff-Antikörper, antidrug antibodies) und 10,3 % (16/156) Antikörper, die als neutralisierend klassifiziert wurden. Bei Wiederaufnahme der Therapie stieg der Anteil der Patienten, die Antikörper gegen den Wirkstoff bzw. neutralisierende Antikörper entwickelten, nach 5 Behandlungszyklen auf 61,4 % (35/57) bzw. 43,9% (25/57) an. Die Entwicklung neutralisierender Antikörper war mit einer Abnahme der Gesamtplasmaexposition von Rozanolixizumab um 24 % verbunden. Die Immunogenität hatte keinen deutlichen Einfluss auf die Wirksamkeit (siehe Abschnitt Eigenschaften/Wirkungen). Studienteilnehmer mit ADAs berichteten aber mehr als zweimal häufiger über bestimmte Nebenwirkungen im Vergleich zu Studienteilnehmern ohne ADAs (siehe Abschnitt Unerwünschte Wirkungen).
Hilfsstoff Prolin
Dieses Arzneimittel enthält 29 mg Prolin pro ml.
Die Anwendung bei Patienten mit Hyperprolinämie sollte auf Fälle beschränkt bleiben, in denen keine alternative Behandlung verfügbar ist.
InteraktionenEs wurden keine Interaktionsstudien durchgeführt. Da Rozanolixizumab in den Wiederaufnahmemechanismus des neonatalen Fc-Rezeptors (FcRn) von Immunglobulin G (IgG) eingreift, wird erwartet, dass die Serumkonzentrationen von IgG-basierten Arzneimitteln (z.B. monoklonale Antikörper und intravenöses Immunglobulin [IVIg]) und Fc-Peptid-Fusionsproteinen bei gleichzeitiger Verabreichung mit Rozanolixizumab verringert werden. Zwei Wochen nach einer Rozanolixizumab-Infusion ist es unwahrscheinlich, dass eine klinisch relevante Wirkung von Rozanolixizumab auf die PK oder Wirksamkeit dieser Arzneimittel auftritt. Es wird empfohlen, diese Medikamente 2 Wochen nach einer Rozanolixizumab-Infusion zu verabreichen und auf eine abgeschwächte Wirksamkeit dieser Medikamente bei gleichzeitiger Verabreichung mit Rozanolixizumab zu achten.
Wechselwirkungen mit stark proteingebundenen Medikamenten oder Medikamenten, die Substrate, Induktoren oder Inhibitoren von Cytochrom-P450-Enzymen oder Transportern sind, sind unwahrscheinlich.
Die Behandlung mit i.v. (intravenös) oder s.c. (subkutan) verabreichten Immunglobulinen, PLEX/Plasmapherese oder Immunadsorption kann die zirkulierenden Konzentrationen von Rozanolixizumab senken.
Schwangerschaft, StillzeitSchwangerschaft
Begrenzte Daten lassen keine Rückschlüsse auf die Anwendung von Rozanolixizumab bei schwangeren Frauen zu. In Studien an Tieren wurden Auswirkungen auf Fehlgeburten beobachtet. Ausserdem, wie aufgrund der pharmakologischen Wirkungsweise von Rozanolixizumab zu erwarten, wiesen die Nachkommen behandelter Muttertiere bei der Geburt sehr niedrige IgG-Spiegel auf. Rozanolixizumab zeigte in Tieren keine Auswirkung auf die fötale Entwicklung, Geburt oder postnatale Entwicklung (siehe Rubrik Präklinische Daten). Die Behandlung von schwangeren Frauen mit Rozanolixizumab sollte nur dann in Betracht gezogen werden, wenn der klinische Nutzen die Risiken überwiegt.
Da erwartet wird, dass Rozanolixizumab die mütterlichen IgG-Spiegel senkt und auch die Übertragung von mütterlichen IgG auf den Fötus hemmt, ist mit einem reduzierten Nestschutz beim Neugeborenen zu rechnen. Daher sollen Risiken und Nutzen der Anwendung von Lebendimpfstoffen/attenuierten Lebendimpfstoffen bei Säuglingen von mit Rozanolixizumab behandelten Schwangeren in den ersten 2 Monaten nach der Geburt gegeneinander abgewogen werden (siehe Abschnitt Warnhinweise und Vorsichtsmassnahmen - Impfung).
Stillzeit
Es ist nicht bekannt, ob Rozanolixizumab in die Muttermilch übergeht. Maternales IgG geht bekanntermassen in den ersten Tagen nach der Geburt in geringen Mengen in die Muttermilch über, was aber bald darauf abnimmt. Auf der Basis dieser theoretischen Überlegungen kann ein Risiko für gestillte Säuglinge insbesondere in den ersten Tagen nach der Geburt nicht ausgeschlossen werden. Nach dieser frühen Phase sollten bei der Entscheidung, ob die Anwendung von Rozanolixizumab gestoppt oder auf das Stillen verzichtet werden soll, die potenziellen Vorteile des Stillens, der klinische Bedarf der Mutter für die Behandlung mit Rozanolixizumab und die möglichen schädlichen Auswirkungen von Rozanolixizumab auf den gestillten Säugling gegeneinander abgewogen werden.
Fertilität
Die Wirkungen von Rozanolixizumab auf die menschliche Fertilität sind nicht bekannt. Tierexperimentelle Studien weisen nicht auf schädliche Wirkungen in Bezug auf die Fruchtbarkeit hin (siehe Rubrik Präklinische Daten).
Wirkung auf die Fahrtüchtigkeit und auf das Bedienen von MaschinenEs wurden keine Untersuchungen zur Wirkung von Rozanolixizumab auf die Fahrtüchtigkeit und auf das Bedienen von Maschinen durchgeführt. Theoretische Erwägungen legen keinen bzw. einen vernachlässigbaren Einfluss auf die Verkehrstüchtigkeit und die Fähigkeit, Maschinen zu bedienen, nahe.
Unerwünschte WirkungenZusammenfassung des Sicherheitsprofils
In einer doppelblinden, placebokontrollierten klinischen Studie zur Myasthenia gravis wurden insgesamt 133 Patienten mit Rozanolixizumab behandelt. Die am häufigsten gemeldeten Nebenwirkungen, in der Doppelblindstudie und einer offenen Erweiterungsstudie (188 Patienten), waren Kopfschmerzen (51,6 %), Diarrhöe (33,5 %), Infektionen der oberen Atemwege (25,5%), Fieber (20,7 %), Übelkeit (17,6%), Infusions/Injektionsreaktionen (12,2%) und Arthralgie (12,2%).
Liste der unerwünschten Wirkungen
Die unerwünschten Wirkungen aus klinischen Studien mit gMG sind nach MedDRA-Systemorganklassen gemäss folgender Konvention geordnet. Innerhalb jeder Systemorganklasse sind die unerwünschten Wirkungen nach ihrer Häufigkeit geordnet, wobei die häufigsten Wirkungen an erster Stelle stehen.
Die Häufigkeitskategorien sind wie folgt definiert: Sehr häufig (≥1/10); häufig (≥1/100 bis < 1/10); gelegentlich (≥1/1000 bis < 1/100); selten (≥1/10'000 bis < 1/1000); sehr selten (< 1/10'000), nicht bekannt (kann aus den verfügbaren Daten nicht abgeschätzt werden).
Tabelle 1: Liste der unerwünschten Wirkungen
|
MedDRA-Systemorganklasse
|
Häufigkeits-kategorie
|
Unerwünschte Wirkungen
| |
Erkrankungen des Blutes und des Lymphsystems
|
Sehr häufig
|
Reduzierter IgG-Blutspiegel (11,2%)
| |
Infektionen und parasitäre Erkrankungen
|
Sehr häufig
|
Infektionen der oberen Atemwege (25,5%)1
| |
Häufig
|
Virale Infektionen2
Infektionen der unteren Atemwege3
Herpes-Virus-Infektionen4
| |
Erkrankungen des Nervensystems
|
Sehr häufig
|
Kopfschmerzen (51,6%)5
| |
Selten
|
Aseptische Meningitis
| |
Erkrankungen der Atemwege, des Brustraums und Mediastinums
|
Häufig
|
Oropharyngeale Schmerzen
| |
Erkrankungen des Gastrointestinaltrakts
|
Sehr häufig
|
Diarrhöe (33,5%),
abdominelle Beschwerden (16,5%)6
Übelkeit (17,6%)
| |
Häufig
|
Erbrechen
| |
Erkrankungen der Haut und des
Unterhautzellgewebes
|
Häufig
|
Ausschlag7,
| |
Gelegentlich
|
Angioödem8
| |
Skelettmuskulatur-, Bindegewebs- und Knochenerkrankungen
|
Sehr häufig
|
Arthralgie (12,2%)
| |
Häufig
|
Myalgie
Nackenschmerzen
Muskelkrämpfe
| |
Allgemeine Erkrankungen und
Beschwerden am Verabreichungsort
|
Sehr häufig
|
Fieber (20,7%)
Reaktionen an der Injektions/Infusionsstelle (12,2%)9
| |
Häufig
|
Influenza ähnliche Erkrankung
Brustschmerzen
|
1 Umfasst akute und chronische Sinusitis, Laryngitis, Nasopharyngitis, Pharyngitis, Rhinitis, Sinusitis, Tonsillitis, Infektion der oberen Atemwege
2 Umfasst virale Gastroenteritis, virale Atemwegsinfektion, virale Infektion, virale Infektion der oberen Atemwege, jedoch ohne COVID-19-Infektionen
3 Umfasst Bronchitis, Infektion der unteren Atemwege, Pneumonie
4 Umfasst Herpes simplex, Herpes Virus, Herpes zoster, ophthalmischer Herpes simplex, orale Herpes Infektionen
5 Einschliesslich Kopfschmerzen und Migräne
6 Umfasst abdomineller Schmerz, Oberbauch/Unterbauchbeschwerden, gastrointestinaler Schmerz, abdominelle Beschwerden
7 Umfasst Ausschlag, Ausschlag papulös und Hautausschlag erythematös
8 Umfasst geschwollene Zunge
9 Umfasst, aber nicht beschränkt auf, Bluterguss an der Injektionsstelle, Hautausschlag an der Injektionsstelle, Reaktion an der Injektionsstelle, Erythem an der Injektionsstelle, Pruritus an der Injektionsstelle, Erythem an der Infusionsstelle, Reaktion an der Infusionsstelle.
Beschreibung spezifischer unerwünschter Wirkungen und Zusatzinformationen
Kopfschmerzen
In MG0003 waren Kopfschmerzen das häufigste Ereignis, das bei 58 (43.6 %) und bei 13 (19,4 %) Patienten unter Placebo berichtet wurde. Kopfschmerzen traten am häufigsten nach der ersten Infusion von Rozanolixizumab und innerhalb von 1 bis 4 Tagen nach der Infusion auf. Die Kopfschmerzen waren nicht stark, meist leicht oder mittelschwer, und die Inzidenz stieg bei wiederholter zyklischer Behandlung nicht an.
Immunogenität
Die Nachweis von Anti-Drug-Antikörpern (ADAs) hängt in hohem Mass von der Empfindlichkeit und Spezifität des Tests ab. Darüber hinaus kann die beobachtete Inzidenz der Antikörperpositivität (einschliesslich neutralisierender Antikörper) in einem Test von mehreren Faktoren beeinflusst werden, beispielsweise von der Testmethodik, der Probenhandhabung, dem Zeitpunkt der Probengewinnung, der Begleitmedikation und der Grundkrankheit. Aus diesem Grund kann der Vergleich der Inzidenz von Antikörpern gegen Rozanolixizumab mit der Inzidenz von Antikörpern gegen andere Arzneimittel irreführend sein.
In den gepoolten zyklischen Behandlungsdaten aus dem Phase-III-Programm entwickelten nach einem Behandlungszyklus mit 6 wöchentlichen Rozanolixizumab-Dosen 26,9 % (42/156) der Patienten Anti-Drug-Antikörper und 10,3 % (16/156) Antikörper, die als neutralisierend klassifiziert wurden. Bei Wiederaufnahme der Therapie stieg der Anteil der Patienten, die Anti-Drug-Antikörper und neutralisierende Antikörper entwickelten, nach 5 Behandlungszyklen auf 61,4 % (35/57) bzw. 43,9 % (25/57) an. Die Entwicklung neutralisierender Antikörper war mit einer Abnahme der Gesamtplasmaexposition von Rozanolixizumab um 24 % verbunden.
Die Reduktion der Gesamt-IgG-Konzentration durch Rozanolixizumab bei Patienten mit neutralisierenden Antikörpern unterschied sich nicht von Patienten, bei denen keine Anti-Drug Antikörper nachgewiesen wurden.
Die Immunogenität hatte keinen deutlichen Einfluss auf die Wirksamkeit und Sicherheit insgesamt.
Die Häufigkeit bestimmter unerwünschter Wirkungen (Oberbauchbeschwerden, Infektionen der oberen Atemwege, erniedrigte Anzahl neutrophiler Granulozyten, Hypertonie, Dys/Parästhesie, Atemnot, Dyslipidämie) war bei Studienteilnehmern mit ADA allerdings mindestens doppelt so hoch wie bei Studienteilnehmern ohne ADAs. Ein direkter ursächlicher Zusammenhang zwischen dem Auftreten dieser Nebenwirkungen und ADAs wurde bislang nicht gezeigt. Patienten ohne ADAs wurden häufiger mit Immunsuppressiva, systemischen Steroiden und Chemotherapeutika behandelt.
Die Meldung des Verdachts auf Nebenwirkungen nach der Zulassung ist von grosser Wichtigkeit. Sie ermöglicht eine kontinuierliche Überwachung des Nutzen-Risiko-Verhältnisses des Arzneimittels. Angehörige von Gesundheitsberufen sind aufgefordert, jeden Verdacht einer neuen oder schwerwiegen den Nebenwirkung über das Online-Portal ElViS (Electronic Vigilance System) anzuzeigen. Informationen dazu finden Sie unter www.swissmedic.ch.
ÜberdosierungAnzeichen und Symptome
Es gibt keine Daten über Symptome im Zusammenhang mit einer Überdosierung. In klinischen Studien wurden subkutane Einzeldosen von bis zu 20 mg/kg (2162 mg) und wöchentliche subkutane Dosen von etwa 10 mg/kg (1120 mg) über einen Zeitraum von bis zu 52 Wochen ohne dosislimitierende Toxizität verabreicht.
Behandlung
Im Falle einer Überdosierung wird empfohlen, die Patienten engmaschig auf unerwünschte Wirkungen zu überwachen und sofort geeignete Unterstützungsmassnahmen zu ergreifen.
Eigenschaften/WirkungenATC-Code
L04AG16
Wirkungsmechanismus
Rozanolixizumab ist ein humanisierter monoklonaler Immunglobulin (Ig)-G4-Antikörper, der die IgG-Konzentration im Serum senkt, indem er die Bindung von IgG an den neonatalen Fc-Rezeptor (FcRn) hemmt, einen Rezeptor, der normalerweise IgG vor intrazellulärem Abbau schützt und IgG wieder an die Zelloberfläche zurückführt.
Über denselben Mechanismus senkt Rozanolixizumab die Konzentration der pathogenen IgG-Autoantikörper, die mit gMG assoziiert sind. Klinische Daten mit Rozanolixizumab haben keine klinisch relevanten Auswirkungen auf den Albuminspiegel ergeben, der an einer anderen Stelle des FcRn bindet.
Pharmakodynamik
Die wöchentliche subkutane Verabreichung von Rozanolixizumab führte zu einer raschen und anhaltenden Senkung der Gesamt-IgG-Serumkonzentrationen mit einer signifikanten Senkung des IgG um 45 % im Vergleich zum Ausgangswert innerhalb einer Woche und einem maximalen Rückgang um 73 % nach etwa drei Wochen. Nach Beendigung der Verabreichung gingen die IgG-Konzentrationen innerhalb von etwa 8 Wochen auf die Baseline zurück. Ähnliche Wirkungen wurden für alle IgG-Unterklassen beobachtet.
Klinische Wirksamkeit
Phase-III-Studien
Die Sicherheit und Wirksamkeit von Rozanolixizumab wurde bei Patienten mit generalisierter Myasthenia gravis in drei Phase-III-Studien untersucht... Die Patienten waren mindestens 18 Jahre alt, hatten ein Körpergewicht von ≥35 kg, die Diagnose einer generalisierten Myasthenia gravis,wiesen Autoantikörper gegen AChR oder MuSK auf, entsprachen einem Krankheitsstadium Klasse II bis IVa nach der Myasthenia Gravis Foundation of America (MGFA) Klassifikation, hatten einen MG-Activities of Daily Living (MG-ADL) Score von mindestens 3 (mit ≥3 Punkten für nicht-okuläre Symptome), und einen Quantitative Myasthenia Gravis (QMG) Score von mindestens 11 und erfüllten die Kriterien für eine zusätzliche Behandlung mit Plasmapherese (plasma exchange (PLEX)) oder intravenösen Immunglobulinen (IVIg).
Patienten wurden von der Teilnahme an der Studie ausgeschlossen, wenn sie Folgendes hatten:
·eine klinisch relevante aktive Infektion oder schwere Infektionen, mykobakterielle Infektionen, Hepatitis-B-, Hepatitis-C-, HIV-Infektionen
·Primäre Immundefizienz oder IgA-Mangel in der Anamnese, Splenektomie oder Transplantation (solide Organtransplantation oder hämatopoetische Stammzelltransplantation/ Knochenmarkstransplantation)
·Thymektomie in den letzten 6 Monaten vor Behandlungsbeginn oder Thymom zu einem beliebigen Zeitpunkt, das eine Chemotherapie und/oder Bestrahlung erforderte, und aktive neoplastische Erkrankung oder eine neoplastische Erkrankung in der Anamnese in den letzten 5 Jahren
·Therapie mit PLEX, IVIg 1 Monat vor Behandlungsbeginn und mit monoklonalen Antikörpern 3 bis 6 Monate vor Behandlungsbeginn
·einen IgG-Gesamtspiegel im Serum von ≤5,5 g/l oder eine absolute Neutrophilenzahl < 1 500 Zellen/mm3
Die Sicherheit und Wirksamkeit von Rozanolixizumab wurden bei Patienten mit gMG in der Phase-III-Pivotstudie MG0003 untersucht. Die langfristige Sicherheit, Verträglichkeit und Wirksamkeit von Rozanolixizumab wurden in 2 offenen Verlängerungsstudien (OLE [open-label extension]) der Phase III beurteilt, wobei Rozanolixizumab in der OLE MG0007 in 6-wöchigen Behandlungszyklen auf Basis der klinischen Bedürfnisse über einen Zeitraum von bis zu 2 Jahren (s.u.) verabreicht wurde.
Studie MG0003
In der Studie MG0003 wurden 200 Patienten über einen Zeitraum von bis zu 18 Wochen untersucht, wobei die Patienten nach dem Zufallsprinzip entweder gewichtsabgestufte Dosen von Rozanolixizumab, die etwa (≈) 7 mg/kg oder ≈10 mg/kg entsprachen, oder einen Placebo erhielten. Die Behandlung bestand aus einer Dosis pro Woche über einen Zeitraum von 6 Wochen, gefolgt von einem 8-wöchigen Beobachtungszeitraum.
Die Wirksamkeit von Rozanolixizumab wurde im Hinblick auf die Auswirkungen auf die Werte von MG-ADL, MG-C, QMG und eine Reihe anderer von Patienten berichteter Ergebnisinstrumente, einschliesslich des MG-Symptoms-PRO-Scores, bewertet. Der primäre Endpunkt war die Veränderung des MG-ADL-Scores von der Baseline bis zum Tag 43. Sekundäre Wirksamkeitsendpunkte waren die Veränderung von der Baseline bis zum Tag 43 des MG-C-Scores, und des QMG -Scores sowie der MG-ADL-Responder-Status am Tag 43.
Im Allgemeinen waren die demografischen Daten der Patienten und die Krankheitsmerkmale bei Baseline über die Behandlungsgruppen hinweg ausgewogen. Die Mehrheit der Patienten in der Gruppe mit Rozanolixizumab mit einer Dosis von ≈7 mg/kg war weiblich (59,1 %), unter 65 Jahre alt (74,2 %), kaukasischer (62,1 %) oder asiatischer Abstammung (13,5 %) und stellte sich mit gMG-MGFA-Klasse II oder III vor (95,4 %). Das mediane Alter bei MG-Diagnose war 44,0 Jahre und die mediane Zeit seit der Diagnose betrug 5,3 Jahre. Die Autoantikörperverteilung bei den Patienten war 7,6 % Anti-MuSK-positiv und 90,9 % Anti-AChR-positiv. Insgesamt wurden 95,5 % der Patienten bei Baseline mit mindestens einem MG-Medikament behandelt, das sie während der Studie weiter erhielten, wobei 83,3 % Acetylcholinesterase-Hemmer, 63,6 % Kortikosteroide, 47,0 % Immunsuppressiva in stabilen Dosen erhielten. In der Rozanolixizumab- und der Placebogruppe lag der mediane MG-ADL-Gesamt-Score bei 8,0 und der mediane QMG-Gesamt-Score bei 15,0 zu Beginn der Studie.
Die Ergebnisse für die primären und sekundären Wirksamkeitsendpunkte sind in der nachstehenden Tabelle 2 aufgeführt.
Eine statistisch signifikante und klinisch bedeutsame Verbesserung der MG-Symptome am Tag 43 wurde bei den MG-ADL, MG-C-, und QMG- Scores für beide Rozanolixizumab-Behandlungsgruppen im Vergleich zum Placebo beobachtet.
Tabelle 2: Veränderung der Wirksamkeitsergebnisse von der Baseline bis Tag 43
|
|
Placebo
(N=67)
|
Rozanolixizumab
≈7 mg/kg
(N=66)
| |
MG-ADL
| |
Baseline Mittelwert
|
8,4
|
8,4
| |
Veränderung gegenüber Baseline
LS Mittelwert (SE)
|
-0,784 (0,488)
|
-3,370 (0,486)
| |
Differenz zu Placebo
|
NA
|
-2,586
| |
95 % KI für die Differenz
|
NA
|
-4,091, -1,249
| |
P-Wert für die Differenz
|
NA
|
< 0,001
| |
MG-C
| |
Baseline Mittelwert
|
15,6
|
15,9
| |
Veränderung gegenüber Baseline
LS Mittelwert (SE)
|
-2,029 (0,917)
|
-5,930 (0,916)
| |
Differenz zu Placebo
|
NA
|
-3,901
| |
95 % KI für die Differenz
|
NA
|
-6,634, -1,245
| |
P-Wert für die Differenz
|
NA
|
< 0,001
| |
QMG
| |
Baseline Mittelwert
|
15,8
|
15,4
| |
Veränderung gegenüber Baseline
LS Mittelwert (SE)
|
-1,915 (0,682)
|
-5,398 (0,679)
| |
Differenz zu Placebo
|
NA
|
-3,483
| |
95 % KI für die Differenz
|
NA
|
-5,614, -1,584
| |
P-Wert für die Differenz
|
NA
|
< 0,001
|
≈=ungefähre Dosis; KI=Konfidenzintervall; N=Gesamtzahl der Patienten in der Behandlungsgruppe; n=Anzahl der Patienten; LS=kleinstes Quadrat; SE=Standardfehler.
Ein MG-ADL Responder musste zu jedem Zeitpunkt während des Behandlungs- und Beobachtungszeitraums der Studie eine Verbesserung von mindestens 2 Punkten gegenüber der Baseline aufweisen. Der Anteil der MG-ADL Responder an Tag 43 war in der ≈7 mg/kg- Rozanolixizumab Gruppe (46 [71,9 %]) mehr als doppelt so hoch wie in der Placebo-Gruppe (20 [31,3 %]). Der Anteil der QMG-Responder am Tag 43 lag in der Placebo-Gruppe bei 39,1% gegenüber 54,7% in der ≈7 mg/kg-Rozanolixizumab Gruppe.
Ein QMG Responder musste zu jedem Zeitpunkt während des Behandlungs- und Beobachtungszeitraums der Studie eine Verbesserung von mindestens 3 Punkten gegenüber der Baseline aufweisen.
Die Behandlung mit Rozanolixizumab war mit einer raschen Verbesserung der MG-ADL-, MG-C- und QMG-Scores verbunden, die bereits eine Woche nach der ersten Dosis bei den Patienten zu beobachten war. Die grössten Verbesserungen wurden zwischen Tag 36 und Tag 43 in der ≈7 mg/kg Rozanolixizumab-Behandlungsgruppen berichtet.
OLE Studien
Die OLE-Studie MG0007 untersuchte die Wirksamkeit von wiederholten 6-wöchigen Rozanolixizumab-Behandlungszyklen. Patienten, bei denen sich nach Ansicht des Prüfers eine Verschlechterung einstellte (Anstieg um mindestens 2 Punkte auf der MG-ADL-Skala oder 3 Punkte auf der QMG Skala), konnten einen weiteren Behandlungszyklus erhalten. Insgesamt erhielten 25 Patienten mehr als 3 Behandlungszyklen mit ≈7mg/kg Rozanolixizumab. Einzelne Patienten erhielten bis zu 15 Zyklen. Patienten, die auf die wiederholte Behandlung mit ≈7mg/kg Rozanolixizumab ansprachen, zeigten eine stabile Therapieantwort auf die Gabe weiterer Behandlungszyklen.
In der Gruppe der ausschliesslich mit der zugelassenen Dosierung von Rozanolixizumab behandelten Patienten lagen die Ansprechraten nach der MG-ADL-Skala in den Zyklen 1 bis 4 zwischen 55,3% und 87,5%, und nach der QMG-Skala zwischen 57,9% und 71,4%.
Wirksamkeit bei älteren Menschen
Die Studie MG0003 untersuchte auch die Wirksamkeit von Rozanolixizumab-Behandlungsgruppen im Vergleich zu Placebo bei älteren Patienten (≥65 Jahre). In der ausschliesslich mit ≈7mg/kg Rozanolixizumab-Gruppe waren 17 Patienten ≥65 Jahre alt. Die Differenz gegenüber Plazebo behandelten Patienten in der Verbesserung des MG-ADL Scores an Tag 43 gegenüber Baseline lag in der Gruppe ≥65 Jahren bei -2,287 (SE 1.257) gegenüber -2,577 (SE 0.524) in der Altersgruppe <65 Jahre.
In der OLE-Studie MG0007 wurden 14 ältere Patienten (≥65 Jahre) mit wiederholten Zyklen von Rozanolixizumab in der zugelassenen Dosierung behandelt (1 Zyklus erhielten 14 Patienten, 2 Zyklen 12 Patienten, 3 Zyklen 7 Patienten, 4 und mehr Zyklen: ≤5 Patienten). Dabei lag die mittlere Verbesserung des MG-ADL Scores an Tag 43 gegenüber Baseline des jeweiligen Zyklus niedriger als in der Gruppe <65 Jahren (Zyklus 1: -2,5 (SD 3,6) vs. -3,4 (SD 3,2), Zyklus 2: -1,6 (SD 3,8) vs. -2,7 (SD 2,5), Zyklus 3: -1,7 (SD 3,1) vs. -3,3 (SD 2,6) Punkte).
Wirksamkeit bei AChR- und MuSK-Autoantikörper-positiven Teilnehmern
Es wurde eine Untergruppenanalyse nach MG-spezifischen Autoantikörpern, MuSK+ und AChR+, durchgeführt.
Am Tag 43 wurden bei wiederholter zyklischer Behandlung für die Zyklen 1 bis 4 MG-ADL-, MG-C- und QMG-Ansprechraten von >40 % für Anti-AChR-positive Studienteilnehmer (28 Teilnehmer in Zyklus 1, 27 Teilnehmer in Zyklus 2, 21 Teilnehmer in Zyklus 3 and 15 Teilnehmer in Zyklus 4) und >50 % für Anti-MuSK-positive Studienteilnehmer (jeweils 4 Teilnehmer in Zyklus 1 bis 4) berichtet.
Diese Angaben beziehen sich auf Studienteilnehmer, die ausschliesslich mit der zugelassenen Dosierung von ≈7mg/kg Körpergewicht Rozanolixizumab behandelt wurden.
Wirksamkeit bei Patienten mit einem Körpergewicht <50kg
Die Anzahl der Studienteilnehmer mit einem Körpergewicht <50kg, die eine wiederholte zyklische Behandlung mit ausschliesslich ≈7mg/kg Rozanolixizumab in der OLE MG0007 erhielten, war klein (Zyklus 1-5: bis zu 4 Studienteilnehmer pro Zyklus). In dieser Gruppe variierten die MG-ADL Responder-Raten zwischen 0 und 100% über die Zyklen 1 bis 5 hinweg, wobei die mittlere Änderung des MG-ADL Scores in drei der fünf Zyklen die Grenze für die klinische Bedeutsamkeit (‚clinical meaningfulness', definiert als mindestens ≥2 Punkte am Tag 43 vs. Baseline des jeweiligen Zyklus) nicht erreichte.
Pädiatrie
Für Informationen zur pädiatrischen Anwendung siehe Rubrik Dosierung/Anwendung.
PharmakokinetikAbsorption
Nach subkutaner Verabreichung von Rozanolixizumab werden die maximalen Plasmaspiegel nach etwa 2 Tagen erreicht. Die absolute Bioverfügbarkeit von Rozanolixizumab nach subkutaner Verabreichung lag bei etwa 70 %, wie durch eine populationspharmakokinetische Analyse geschätzt wurde.
Distribution
Das scheinbare Verteilungsvolumen von Rozanolixizumab beträgt etwa 7 l, geschätzt durch populationspharmakokinetische Analysen.
Metabolismus
Es wird erwartet, dass Rozanolixizumab ähnlich wie endogenes IgG über katabolische Wege in kleine Peptide und Aminosäuren abgebaut wird.
Elimination
Die scheinbare lineare Clearance für den freien Wirkstoff beträgt etwa 0,9 l/Tag. Die Halbwertszeit von Rozanolixizumab ist konzentrationsabhängig und kann nicht berechnet werden. Die Rozanolixizumab-Plasmakonzentrationen sind innerhalb einer Woche nach der Dosisgabe nicht nachweisbar.
Linearität/Nicht Linearität
Rozanolixizumab zeigte eine nichtlineare Pharmakokinetik, die typisch für einen monoklonalen Antikörper ist, der eine zielvermittelte Medikamentendisposition durchläuft.
Im Steady-State wurden die maximalen Plasmakonzentrationen bzw. die Fläche unter der Konzentrations-Zeit-Kurve (area under the concentration time curve, AUC) bei gewichtsspezifischen Dosen von ≈ 10 mg/kg als 3-fach bzw. 4-fach höher vorhergesagt als bei ≈ 7 mg/kg.
Kinetik spezieller Patientengruppen
Leberfunktionsstörungen oder Nierenfunktionsstörungen
Es wurden keine speziellen Studien bei Patienten mit eingeschränkter Nieren- oder Leberfunktion durchgeführt. Es ist jedoch nicht zu erwarten, dass eine Beeinträchtigung der Nieren- oder Leberfunktion die Pharmakokinetik von Rozanolixizumab beeinflusst. Auf der Grundlage einer populationspharmakokinetischen Analyse hatten die Nierenfunktion (geschätzte glomeruläre Filtrationsrate [eGFR] 38-161 ml/min/1,73 m2) oder die Biochemie- und Funktionstests der Leber (ALT, AST, alkalische Phosphatase und Bilirubin) keinen klinisch signifikanten Einfluss auf die scheinbare lineare Clearance von Rozanolixizumab.
Alter, Geschlecht oder ethnische Zugehörigkeit
Eine populationspharmakokinetische Analyse ergab keine klinisch signifikanten Auswirkungen von Alter (zwischen 18 - 89 Jahren), Geschlecht oder ethnische Zugehörigkeit auf die Pharmakokinetik von Rozanolixizumab.
Präklinische DatenToxizität bei wiederholter Gabe
Nichtklinische Daten lassen auf der Grundlage von Studien zur Toxizität bei wiederholter Verabreichung keine besondere Gefahr für den Menschen erkennen. Die Verabreichung an Javaner- und Rhesusaffen führte zu der erwarteten Verringerung des IgG-Gehalts.
Die T-Zell-abhängige Antikörperantwort (TDAR) während der Behandlungsphase führte zu normalen IgM-Spiegeln und einer geringen IgG-Antwort aufgrund eines beschleunigten Abbaus. Eine weitere Immunisierung nach der Rozanolixizumab-Clearance führte jedoch zu einer normalen IgM- und IgG-Antwort.
Kanzerogenität
Es wurden keine Karzinogenitätsstudien durchgeführt.
Gentoxizität
Da Rozanolixizumab ein monoklonaler Antikörper ist, wurden keine Studien zur Genotoxizität durchgeführt.
Reproduktionstoxizität
In einer Studie mit Behandlung von Cynomolgus-Affen ab dem 20. Tag der Trächtigkeit war die Inzidenz von Fehlgeburten in den Rozanolixizumab-Gruppen im Vergleich zur Kontrollgruppe erhöht (vor allem zwischen dem 20. und 50. Tag der Trächtigkeit). Es wurden keine Auswirkungen der Rozanolixizumab-Behandlung auf die fötale Entwicklung, die Geburt und die postnatale Entwicklung beobachtet. Die Nachkommen der behandelten Muttertiere wiesen sehr niedrige IgG-Werte auf, wie aufgrund der Pharmakologie zu erwarten war. Der IgG-Spiegel erreichte innerhalb von 60 Tagen Kontrollwerte oder mehr. Es gab keine Auswirkungen auf die Immunfunktion der Jungtiere der behandelten Mütter, wie mit einem TDAR-Test festgestellt wurde.
Beeinträchtigung der Fertilität
Es wurden keine Fertilitätsstudien (männlich oder weiblich) durchgeführt.
In der 26-wöchigen Toxizitätsstudie mit wiederholter Verabreichung von Rozanolixizumab an Affen wurden keine behandlungsbedingten Veränderungen in den Fortpflanzungsorganen der geschlechtsreifen Tiere festgestellt. Eine Bewertung des Menstruationszyklus und der männlichen Reproduktionsendpunkte (Ejakulatgewicht, Spermienzahl, Spermienmotilität und -morphologie) zeigte keine behandlungsbedingten Veränderungen.
Sonstige HinweiseInkompatibilitäten
Da keine Kompatibilitätsstudien durchgeführt wurden, darf das Arzneimittel nicht mit anderen Arzneimitteln zur Infusion gemischt werden.
Haltbarkeit
Das Arzneimittel darf nur bis zu dem auf der Packung mit «EXP» bezeichneten Datum verwendet werden.
Besondere Lagerungshinweise
Im Kühlschrank (2-8 °C) lagern.
Nicht einfrieren.
Die Durchstechflasche im Umkarton aufbewahren, um den Inhalt vor Licht zu schützen.
Ausser Reichweite von Kindern aufbewahren.
Packungen2 ml Lösung in einer Durchstechflasche (Typ I Glas) mit einem Elastomerstopfen, der mit einem Bördelverschluss und einer Flip-Off-Kappe versiegelt ist.
Packungsgrösse: 1 Durchstechflasche [A].
ZulassungsinhaberinUCB-Pharma AG, Bulle
Stand der InformationOktober 2024
Hinweise für die Handhabung
Materielle Besonderheiten
Rozanolixizumab-Injektionslösung kann mit Polypropylen-Spritzen sowie Infusionssets verabreicht werden, die Polyethylen (PE), Polyethylen niedriger Dichte (low density polyethylene, LDPE), Polyester, Polyvinylchlorid (PVC ohne DEHP), Polycarbonat (PC), fluoriertes Ethylenpolypropylen (FEP), Urethan/Acrylat, Polyurethan, Methylmethacrylat-Acrylnitril-Butadien-Styrol (MABS), Silikon oder Cyclohexanon enthalten. Zur Anwendung dürfen keine Geräte verwendet werden, die laut Kennzeichnung Di(2-ethylhexyl)-Phthalat (DEHP) enthalten.
Um mögliche Unterbrechungen der Verabreichung von Rozanolixizumab zu vermeiden, sollten die folgenden Kriterien beachtet werden:
·Die Alarmgrenzen für die Verstopfung der Spritzenpumpe müssen auf den maximalen Wert eingestellt werden.
·Es wird ein Verabreichungsschlauch mit einer Länge von 61 cm oder kürzer empfohlen.
·Es sollte ein Infusionsset mit einer Nadel der Stärke 26 G oder grösser verwendet werden.
Vor der Verabreichung von Rozanolixizumab müssen die Anweisungen sorgfältig gelesen werden.
Rozanolixizumab enthält keine Konservierungsstoffe und jede Durchstechflasche ist nur zum einmaligen Gebrauch bestimmt. Daher darf in der Durchstechflasche verbliebenes, nicht verwendetes Arzneimittel oder Abfallmaterial nicht verwendet werden und ist entsprechend den nationalen Anforderungen zu beseitigen.
Die folgenden Informationen sind nur für medizinisches Fachpersonal bestimmt.
Gebrauchsanweisung für medizinisches Fachpersonal
Handhabung von Rystiggo mittels einer gerätegestützten Infusionstechnik z.B. eine Infusionspumpe
Nur zur subkutanen Anwendung.
Die Anzahl der zu verwendenden Durchstechflaschen (2 ml pro Durchstechflasche) hängt vom Körpergewicht des Patienten ab. Zur Verabreichung der 280-mg-Dosis an Patienten mit einem Gewicht von ≥35 bis < 50 kg sind 2 ml erforderlich. Zur Verabreichung der 420-mg-Dosis an Patienten mit einem Gewicht von ≥50 kg bis < 70 kg sind 3 ml erforderlich. Zur Verabreichung der 560-mg-Dosis an Patienten mit einem Gewicht von ≥70 bis < 100 kg sind 4 ml erforderlich. Zur Verabreichung der 840-mg-Dosis an Patienten mit einem Gewicht von ≥100 kg sind 6 ml erforderlich.
Lesen Sie ALLE nachstehenden Anweisungen, bevor Sie Rozanolixizumab-Lösung verabreichen.
1. Nehmen Sie Rystiggo aus der Faltschachtel:
·Lassen Sie die Durchstechflasche Raumtemperatur annehmen. Dies kann mindestens 30 Minuten und bis zu 120 Minuten dauern. Verwenden Sie keine Geräte zum Erwärmen.
·Überprüfen Sie jede Durchstechflasche vor der Verwendung:
·Verfallsdatum: Nach Ablauf des Verfallsdatums nicht mehr verwenden.
·Farbe: Die Lösung muss farblos bis schwach bräunlich-gelb, klar bis leicht opalisierend sein. Verwenden Sie die Durchstechflasche nicht, wenn die Flüssigkeit trüb aussieht, Fremdpartikel enthält oder sich die Farbe verändert hat.
·Kappe: Nicht verwenden, wenn die Schutzkappe der Durchstechflasche fehlt oder defekt ist.
2. Legen Sie alle Utensilien bereit:
·Legen Sie alle Utensilien für die Infusion bereit. Neben der/den Durchstechflasche(n) gehören dazu die folgenden Utensilien, die nicht mitgeliefert werden: Spritze, Spritzennadel(n), Alkoholtupfer, Infusionsset, Pflaster oder durchsichtiger Verband, Infusionspumpe und Behälter für scharfe Gegenstände.
3. Wenden Sie bei der Vorbereitung und Verabreichung dieses Produkts aseptische Techniken an
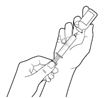
4. Rystiggo für die Infusion vorbereiten
·Füllen Sie die Spritze mit Transfernadeln.
·Verwenden Sie Mischkanülen zum Befüllen der Spritze.
·Nehmen Sie die Schutzkappe von der Durchstechflasche ab und reinigen Sie den Stopfen der Durchstechflasche mit einem Alkoholtupfer. Trocknen lassen.
·Übertragen Sie den gesamten Inhalt der Durchstechflasche in die Spritze. Eine kleine Menge verbleibt in der Durchstechflasche und muss entsorgt werden.
·Bei mehreren Durchstechflaschen muss eine frische Nadel verwendet und die vorherigen Schritte wiederholt werden.
·Entfernen Sie die Nadel von der Spritze und befestigen Sie das Infusionsset an der Spritze.
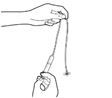
5. Die Infusion vorbereiten
·Die Infusionspumpe wird gemäss den ihr beiliegenden Anweisungen vorbereitet und die Infusionsleitung befüllt. Die Verabreichung erfolgt unmittelbar nach dem Vorfüllen des Infusionssets.
·Jede Durchstechflasche enthält ein Übervolumen (für das Vorfüllen der Infusionsleitung); daher muss die Pumpe so voreingestellt werden, dass das verschriebene Volumen verabreicht wird. Bei Pumpen, die nicht vorab eingestellt werden können, muss das zu verabreichende Volumen nach dem Vorfüllen der Infusionsleitung durch Ausstossen des Übervolumens angepasst werden.
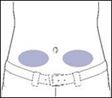
6. Die Infusionsstelle vorbereiten
·Wählen des Infusionsbereichss: rechter oder linker Unterbauch, unterhalb des Bauchnabels. In Bereiche, in denen die Haut Blutergüsse aufweist oder empfindlich, gerötet oder hart ist, darf niemals infundiert werden. Infusionen in Narben oder Dehnungsstreifen sind zu vermeiden.
·Reinigen Sie die Infusionsstelle mit einem Alkoholtupfer. Trocknen lassen.
7. Einführen der Infusionssetnadel
·Fassen Sie eine Bauchhautfalte mit zwei Fingern.
·Führen Sie die Nadel des Infusionssets in das subkutane Gewebe ein.

8. Fixieren Sie die Nadel an der Haut
·Falls erforderlich, wird die Nadel mit Klebeband oder einem transparenten Verband fixiert.

9. Infusion starten
·Befolgen Sie die Anweisungen des Herstellers für die Verwendung der Pumpe.
10. Infusion beenden
·Nach Abschluss der Infusion darf die Infusionsleitung nicht gespült werden, da das Infusionsvolumen unter Berücksichtigung der Verluste in der Leitung ermittellt wurde.
·Ziehen Sie die Nadel aus der Infusionsstelle.
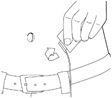
11. Aufräumen
·Entsorgen Sie alle Utensilien, die Produktreste enthalten, d.h. teilweise verwendete Durchstechflaschen, Infusionssets und jegliches Verabreichungszubehör, in einem Nadelabwurfbehälter.
|