Zusammensetzung
Wirkstoffe
Delgocitinib.
Hilfsstoffe
10 mg/g Benzylalkohol (E1519), 0,2 mg/g Butylhydroxyanisol (E320), 72 mg/g Cetostearylalkohol, Citronensäure-Monohydrat, Natriumedetat, Salzsäure, dickflüssiges Paraffin, Macrogol-22cetostearylether, gereinigtes Wasser.
Indikationen/Anwendungsmöglichkeiten
Anzupgo® ist für die Behandlung von mittelschwerem bis schwerem chronischem Handekzem (CHE) bei Erwachsenen indiziert, die auf eine Therapie mit potenten bis hochpotenten topischen Kortikosteroiden nur unzureichend angesprochen haben oder bei denen diese nicht empfohlen wird. Die Vermeidung von Kontakten mit der auslösenden Noxe, Hautschutz und Basispflege sind wichtige Bestandteile der Therapie.
Dosierung/Anwendung
Übliche Dosierung
2x täglich eine dünne Schicht Anzupgo® auf die betroffenen Hautstellen der Hände und Handgelenke auftragen, bis die Haut frei oder fast frei von Symptomen ist.
Bei einem erneuten Auftreten der Anzeichen und Symptome (Schübe) kann die 2x tägliche Behandlung der betroffenen Hautstellen nach Bedarf wieder aufgenommen werden.
Es soll nicht mehr als 1 Tube à 60 g pro Monat appliziert werden.
Spezielle Dosierungsanweisungen
Patienten mit Leberfunktionsstörungen
Eine Dosisanpassung wird aufgrund der geringen systemischen Exposition von topisch angewendetem Delgocitinib nicht empfohlen (siehe «Pharmakokinetik»).
Patienten mit Nierenfunktionsstörungen
Es wurden keine Studien mit Anzupgo® bei Patienten mit schweren Nierenfunktionsstörungen durchgeführt. Allerdings ist eine Dosisanpassung aufgrund der geringen systemischen Exposition von topisch angewendetem Delgocitinib nicht empfohlen (siehe «Pharmakokinetik»).
Ältere Patienten
Für ältere Patienten (≥ 65 Jahre) wird keine Dosisanpassung empfohlen.
Kinder und Jugendliche
Anzupgo® ist für die Anwendung in der pädiatrischen Population nicht zugelassen. Die Sicherheit und Wirksamkeit von Anzupgo® bei Kindern und Jugendlichen unter 18 Jahren ist bisher noch nicht geprüft worden (siehe «Eigenschaften/Wirkungen»). Es liegen keine Daten vor.
Verspätete Anwendung
Falls eine Anwendung vergessen wurde, soll Anzupgo® so schnell wie möglich aufgetragen werden. Danach die Anwendung zum regulär vorgesehenen Zeitpunkt fortsetzen.
Art der Anwendung
Anzupgo® ist zur topischen Anwendung bestimmt.
2x täglich eine dünne Schicht Anzupgo® auf die saubere und trockene Haut der betroffenen Stellen der Hände und Handgelenke auftragen. Unmittelbar vor und nach der Anwendung von Anzupgo® sollen keine anderen topischen Präparate angewendet werden. Die gleichzeitige Anwendung mit Emollientien innerhalb von 2 Stunden vor und nach der Anwendung von Delgocitinib wurde nicht untersucht.
Wenn eine andere Person Anzupgo® aufträgt, sollte diese Person darauf hingewiesen werden, sich danach die Hände zu waschen.
Der Kontakt mit gesunder Haut, Augen, Mund, Genitalien oder anderen Schleimhäuten ist zu vermeiden. Bei Kontakt gründlich mit Wasser abspülen.
Kontraindikationen
Überempfindlichkeit gegenüber dem Wirkstoff oder einem der Hilfsstoffe gemäss Zusammensetzung.
Warnhinweise und Vorsichtsmassnahmen
Unter der Behandlung mit Anzupgo® wurden bei den meisten Patienten geringe systemische Delgocitinib-Konzentrationen beobachtet und infolgedessen ist eine systemische pharmakologische Wirkung eher nicht zu erwarten. Bei einigen Patienten wurden vorübergehend erhöhte Delgocitinib-Konzentrationen beobachtet, so dass unerwünschte systemische pharmakologische Wirkungen, einschliesslich der unten aufgeführten Klasseneffekte von oralen JAK-Inhibitoren für chronisch-inflammatorische Erkrankungen nicht ausgeschlossen werden können. Wägen Sie daher die Risiken und den Nutzen ab vor einer Behandlung mit Anzupgo®.
Folgende unerwünschte Wirkungen wurden mit anderen systemischen JAK-Inhibitoren für chronisch-inflammatorische Erkrankungen beobachtet:
Schwerwiegende Infektionen
Bei Patienten, die orale JAK-Inhibitoren erhalten haben, wurden schwerwiegende und manchmal tödliche Infektionen durch Bakterien, Mykobakterien, invasive Pilze, Viren oder andere opportunistische Erreger beobachtet.
Die Anwendung von Anzupgo® ist bei folgenden Patienten mit einer aktiven, schweren Infektion, einschliesslich lokalisierter Infektionen ist zu vermeiden:
§Patienten mit chronischen oder rekurrenten Infektionen;
§Patienten mit einer schweren oder opportunistischen Infektion in der Vorgeschichte;
§Patienten die einer Tuberkulose exponiert waren;
§Patienten die sich in Gebieten mit endemischer Tuberkulose oder endemischen Mykosen aufgehalten haben oder dorthin gereist sind; oder
§Patienten mit Grunderkrankungen, die sie für eine Infektion prädisponieren könnten.
Überwachen Sie die Patienten während und nach der Behandlung mit Anzupgo® engmaschig auf Anzeichen und Symptome einer Infektion.
Unterbrechen Sie die Behandlung mit Anzupgo®, wenn ein Patient eine schwerwiegende Infektion, eine opportunistische Infektion oder eine Sepsis entwickelt.
Nehmen Sie die Behandlung mit Anzupgo® erst wieder auf, wenn die Infektion unter Kontrolle ist.
Tuberkulose (TB)
Fälle von aktiver Tuberkulose wurden in klinischen Studien mit oralen JAK-Inhibitoren zur Behandlung chronischer inflammatorischer Erkrankungen berichtet. Erwägen Sie, Patienten vor der Verabreichung von Anzupgo® auf latente und aktive TB-Infektionen zu untersuchen. Während der Anwendung von Anzupgo®, sollten die Patienten auf die Entwicklung von Anzeichen und Symptomen einer TB überwacht werden.
Virusreaktivierung
Virusreaktivierung einschliesslich Fälle einer Herpesvirus-Reaktivierung (z. B. Herpes zoster) wurden in klinischen Studien mit JAK-Inhibitoren berichtet. Wenn ein Patient einen Herpes zoster entwickelt, ist eine vorübergehende Unterbrechung von Anzupgo® bis zum Abklingen der Episode in Erwägung zu ziehen.
Die Auswirkungen von JAK-Inhibitoren zur Behandlung entzündlicher Erkrankungen auf die Reaktivierung einer chronischen Virushepatitis sind nicht bekannt. Patienten mit einer Hepatitis-B- oder -C-Infektion in der Anamnese waren von den klinischen Studien ausgeschlossen.
Erhöhungen der Hepatitis-B-Viruslast (HBV-DNA-Titer), mit oder ohne begleitende Erhöhungen der Alanin-Aminotransferase und Aspartat-Aminotransferase, wurden bei Patienten mit chronischer HBV-Infektion berichtet, die einen oralen JAK-Inhibitor einnahmen.
Maligne Tumorerkrankungen (ausser NMSC), schwerwiegende unerwünschte kardiovaskuläre Ereignisse (MACE), thromboembolische Ereignisse und Gesamtmortalität
In einer grossen, randomisierten Sicherheitsstudie nach Markteinführung eines oralen JAK-Inhibitors bei Patienten ab 50 Jahren mit rheumatoider Arthritis (RA) die mindestens einen kardiovaskulären Risikofaktor aufwiesen, wurde bei Patienten, die mit dem oralen JAK-Inhibitor behandelt wurden, folgende unerwünschte Wirkungen häufiger beobachtet im Vergleich zu Patienten, die mit TNF-Blockern behandelt wurden.
·Maligne Tumorerkrankungen (ausser NMSC), insbesondere Lungenkrebs und Lymphome
MACE, definiert als kardiovaskulärer Tod, nicht-tödlicher Myokardinfarkt (MI) und nicht-tödlicher Schlaganfall.
Die Patienten sollten über die Symptome von schwerwiegenden kardiovaskulären Ereignissen und die zu ergreifenden Massnahmen bei deren Auftreten informiert werden. Setzen Sie Anzupgo® bei Patienten ab die einen Myokardinfarkt oder Schlaganfall erlitten haben.
·Thromboembolische Ereignisse, einschliesslich -tiefer Venenthrombosen und Lungenembolien, sowie arterielle Thrombosen. Viele dieser unerwünschten Wirkungen waren schwerwiegend und einige führten zum Tod.
Vermeiden Sie Anzupgo® bei Patienten mit erhöhtem Thromboserisiko. Wenn Symptome einer Thrombose auftreten, setzen Sie Anzupgo® ab und untersuchen und behandeln Sie die Patienten entsprechend.
·Gesamtmortalität
Raucher und Ex-Raucher hatten ein zusätzlich erhöhtes Risiko für diese unerwünschten Wirkungen.
Nicht-melanozytärer Hautkrebs (NMSC)
Unter der Behandlung mit Anzupgo® wurden NMSC (Basalzellkarzinome) beobachtet. Mit einem anderen topischen JAK-Inhibitor traten auch Plattenepithelkarzinome auf. Regelmässige Hautuntersuchungen werden für alle Patienten empfohlen, insbesondere für Patienten mit Risikofaktoren für Hautkrebs. Die Exposition gegenüber Sonnenlicht und UV-Licht sollte durch das Tragen von Schutzkleidung und der Anwendung von Breitspektrum-Sonnenschutzmitteln eingeschränkt werden.
Thrombozytopenie, Anämie und Neutropenie
In den klinischen Studien mit JAK- Inhibitoren wurde über Thrombozytopenie, Anämie und Neutropenie berichtet. Führen Sie eine Überwachung des Blutbildes durch, wenn dies klinisch angezeigt ist. Wenn Anzeichen bzw. Symptome einer klinisch signifikanten Thrombozytopenie, Anämie oder Neutropenie auftreten, sollten die Behandlung mit Anzupgo® abgebrochen werden.
Versehentliche Exposition
Als Vorsichtsmassnahme sollte unmittelbar nach dem Auftragen der Creme auf die Hände und/oder Handgelenke der direkte Hautkontakt mit anderen Personen (insbesondere mit Neugeborenen und Säuglingen, siehe «Stillzeit») vermieden werden. Im Falle einer versehentlichen Übertragung der Creme kann diese abgewischt werden. Ein Risiko für lokale und systemische Effekte durch unbeabsichtigte Übertragung von Anzupgo® auf andere Personen (insbesondere Neugeborene oder Säuglinge) kann nicht ausgeschlossen werden (siehe auch «Stillzeit»).
Wenn eine andere Person Anzupgo® aufträgt, sollte diese Person darauf hingewiesen werden, sich danach die Hände zu waschen.
Der Kontakt mit gesunder Haut, Augen, Mund oder anderen Schleimhäuten ist zu vermeiden. Bei Kontakt gründlich mit Wasser abspülen.
Dieses Arzneimittel enthält 10 mg/g Benzylalkohol pro g Creme. Benzylalkohol kann allergische Reaktionen und leichte lokale Reizungen hervorrufen. Dieses Arzneimittel enthält ausserdem 0,2 mg/g Butylhydroxyanisol und 72 mg/g Cetylstearylalkohol, welche örtlich begrenzt Hautreizungen (z. B. Kontaktdermatitis) hervorrufen können. Butylhydroxyanisol kann auch Reizungen der Augen und der Schleimhäute hervorrufen.
Interaktionen
Der Metabolismus von Delgocitinib ist gering. Daher besteht ein geringes Risiko für potentielle Interaktionen mit systemischen Arzneimitteln (siehe «Pharmakokinetik»).
Basierend auf in vitro-Daten hemmt oder induziert Delgocitinib weder Cytochrom-P450-Enzyme noch hemmt es Transportersysteme wie organische Anionentransporter (Organic Anion Transporter, OAT), organische Anionentransportproteine (Organic Anion Transporting Protein, OATP), organische Kationentransporter (Organic Cation Transporter, OCT), Brustkrebsresistenzprotein (Breast Cancer Resistance Protein, BCRP) oder multimikrobielles Extrusionsprotein (Multi-Antimicrobial Extrusion Protein, MATE) in klinisch relevanten Konzentrationen.
Es wurden keine klinischen Arzneimittelinteraktionsstudien durchgeführt.Anzupgo® wurde nicht in Kombination mit anderen topischen Arzneimitteln untersucht und die gleichzeitige Anwendung auf derselben Hautstelle wird nicht empfohlen.
Schwangerschaft, Stillzeit
Schwangerschaft
Es liegen nur begrenzte Daten über die Anwendung von Delgocitinib bei Schwangeren vor.
Studien, in welchen Tieren Delgocitinib oral verabreicht wurde, zeigten eine Reproduktionstoxizität bei Expositionen, welche ausreichend weit über der entsprechenden Exposition nach topischer Anwendung beim Menschen lagen (siehe «Präklinische Daten»).
Als Vorsichtsmassnahme soll Anzupgo® während der Schwangerschaft nicht angewendet werden.
Stillzeit
Es ist nicht bekannt, ob Delgocitinib in die Muttermilch übergeht.
Nach oraler Verabreichung war Delgocitinib in der Milch säugender Ratten vorhanden (siehe «Präklinische Daten»).
Ein Risiko für den Säugling kann nicht ausgeschlossen werden. Als Vorsichtsmassnahme soll Anzupgo® während der Stillzeit nicht angewendet werden. Siehe «Warnhinweise und Vorsichtsmassnahmen»/Versehentliche Exposition.
Wenn Anzupgo® während der Stillzeit angewendet wird, sollte darauf geachtet werden, direkten Kontakt mit der Brustwarze oder dem umliegenden Bereich zu vermeiden, nachdem die Creme auf die Hände und/oder Handgelenke aufgetragen wurde.
Fertilität
Es liegen keine Daten zu Auswirkungen von Delgocitinib auf die Fertilität beim Menschen vor.
Ausgehend von Ergebnissen bei weiblichen Ratten führte die orale Verabreichung von Delgocitinib zu einer verminderten Fertilität bei Expositionen, die als ausreichend weit über die Exposition beim Menschen hinaus erachtet wurden (siehe «Präklinische Daten»).
Tierstudien zeigten keine Hinweise auf Auswirkungen in Bezug auf die Fertilität bei männlichen Tieren.
Wirkung auf die Fahrtüchtigkeit und auf das Bedienen von Maschinen
Anzupgo® hat einen vernachlässigbaren Einfluss auf die Fahrtüchtigkeit und die Fähigkeit zum Bedienen von Maschinen.
Unerwünschte Wirkungen
Zusammenfassung des Sicherheitsprofils
Die häufigsten unerwünschten Wirkungen waren Reaktionen am Verabreichungsort (1,0%).
Die nachfolgend beschriebenen Sicherheitsdaten basieren auf einem Pool von drei Vehikel-kontrollierten klinischen Studien an 1062 Patienten mit chronischem Handekzem, von denen 691 2x täglich für bis zu 16 Wochen mit Delgocitinib Creme (20 mg/g) behandelt wurden. In der offenen Langzeitverlängerungsstudie (siehe «Eigenschaften/Wirkungen») wurde die Sicherheit bis zu 52 Wochen beobachtet.
Liste der unerwünschten Wirkungen
Die folgenden unerwünschten Wirkungen wurden in klinischen Studien beobachtet und sind nach MedDRA-Systemorganklassen und den unten aufgeführten Häufigkeitskategorien aufgeführt:
«sehr häufig» (≥1/10)
«häufig» (≥1/100, <1/10)
«gelegentlich» (≥1/1000, <1/100)
«selten» (≥1/10’000, <1/1000)
«sehr selten» (<1/10’000)
Erkrankungen der Haut und des Unterhautgewebes
Häufig: Reaktionen am Verabreichungsort
Beschreibung spezifischer unerwünschter Wirkungen und Zusatzinformationen
Reaktionen am Verabreichungsort
Im Pool von drei Vehikel-kontrollierten klinischen Studien über einen Zeitraum von 16 Wochen wurden Reaktionen am Verabreichungsort, einschliesslich Schmerzen, Parästhesie, Pruritus und Erythem am Verabreichungsort, bei 1,0% der mit Delgocitinib Creme behandelten Patienten im Vergleich zu 2,5% der mit Vehikel-Creme behandelten Patienten berichtet. Die Mehrzahl dieser Reaktionen war mild. Es wurden keine schweren oder schwerwiegenden Ereignisse gemeldet. Von den Reaktionen, welche bei mit Delgocitinib behandelten Patienten beobachtet wurden, traten über 75% innerhalb der ersten Behandlungswoche auf, keine führte zu einer Unterbrechung der Behandlung und die mediane Zeit bis zum Abklingen betrug 3 Tage.
Die Ereignisrate von Reaktionen am Verabreichungsort in der Langzeitverlängerungsstudie war mit 0,56 Ereignissen pro 100 Patientenjahre Beobachtung niedriger als in den 16-wöchigen, Vehikel-kontrollierten klinischen Studien (4,11 Ereignisse pro 100 Patientenjahre Beobachtung).
Dermale Sicherheitsstudien
Die Ergebnisse klinischer Studien an gesunden Teilnehmern zeigten, dass Delgocitinib keine phototoxischen oder photoallergischen Hautreaktionen verursacht.
Kardiologie/Elektrophysiologie
In einer Konzentration QT-Analyse an gesunden Teilnehmern gab es keine Hinweise auf eine QTc-verlängernde Wirkung nach oral verabreichtem Delgocitinib in Einzeldosen von bis zu 12 mg (etwa das 200-fache der menschlichen Exposition nach topischer Anwendung, basierend auf Cmax). Es ist daher nicht zu erwarten, dass Anzupgo® unter klinischen Anwendungsbedingungen die kardiale Repolarisation beeinflusst.
Die Meldung des Verdachts auf Nebenwirkungen nach der Zulassung ist von grosser Wichtigkeit. Sie ermöglicht eine kontinuierliche Überwachung des Nutzen-Risiko-Verhältnisses des Arzneimittels. Angehörige von Gesundheitsberufen sind aufgefordert, jeden Verdacht einer neuen oder schwerwiegenden Nebenwirkung über das Online-Portal ElViS (Electronic Vigilance System) anzuzeigen. Informationen dazu finden Sie unter www.swissmedic.ch.
Überdosierung
Aufgrund der geringen systemischen Absorption von Delgocitinib kann bei einer Überdosierung nach topischer Anwendung von Anzupgo® ein Risiko für systemische unerwünschte Wirkungen nicht vollständig ausgeschlossen werden . Bei Anwendung einer zu grossen Menge sollte die überschüssige Creme abgewischt werden. (Siehe auch die Abschnitte «Art der Anwendung» und «Warnhinweise und Vorsichtsmassnahmen»/Versehentliche Exposition).
Eigenschaften/Wirkungen
ATC-Code
D11AH11
Wirkungsmechanismus
Delgocitinib ist ein Pan-Janus-Kinase (JAK)-Inhibitor, der konzentrationsabhängig auf die Aktivität aller vier Mitglieder der JAK-Enzymfamilie, bestehend aus JAK1, JAK2, JAK3 und Tyrosinkinase 2 (TYK2), abzielt. Janus-Kinasen sind intrazelluläre Enzyme, die mit Zytokinrezeptorketten assoziiert sind und Signale von Zytokinen übertragen, um ein breites Spektrum physiologischer und pathologischer Prozesse, einschliesslich Entzündungsreaktionen, zu regulieren. Innerhalb des Signalwegs werden JAKs bei der Interaktion zwischen Zytokin und Rezeptor aktiviert. Sie phosphorylieren und aktivieren anschliessend Signalwandler und Transkriptionsaktivatoren (Signal Transducers and Activators of Transcription, STATs). Aktivierte STATs wiederum aktivieren die Expression von Zytokin-responsiven Genen, um spezifische biologische Reaktionen in Zielzellen auszulösen. Die Hemmung der JAK-Aktivität mit Delgocitinib verhindert die Phosphorylierung und Aktivierung von STATs.
Die Ergebnisse zellulärer Studien am Menschen zeigten, dass die Hemmung des JAK-STAT-Signalwegs durch Delgocitinib die Signalübertragung mehrerer proinflammatorischer Zytokine (einschliesslich Interleukin(IL)-2, IL-4, IL-6, IL-13, IL-21, IL-23, Granulozyten-Makrophagen-Kolonie-stimulierender Faktor (Granulocyte-Macrophage-Colony-Stimulating Factor, GM-CSF) und Interferon(IFN)-α) abschwächt, wodurch die Immun- und Entzündungsreaktionen in Zellen, die für die Pathologie des chronischen Handekzems relevant sind, herunterreguliert werden.
Pharmakodynamik
Bei Patienten mit chronischem Handekzem führte die Behandlung mit Delgocitinib zu einer Verringerung der proinflammatorischen Marker des chronischen Handekzems, wie z. B. S100 Calcium-bindendes Protein A9/12 (S100A9/12), und Serpin-Familie B Mitglied 3 (SERPINB3).
Die Behandlung mit topisch aufgetragenem Delgocitinib führte auch zu einer erhöhten Expression von Genen, die an der Haut-Barrierefunktion (z. B. Filaggrin, Loricrin, Claudine) der läsionalen Haut beteiligt sind.
Die Besiedlung der Haut mit Staphylococcus aureus wurde unter Behandlung mit Delgocitinib im Vergleich zur Behandlung mit Vehikel-Creme stark reduziert.
Klinische Wirksamkeit
Die Sicherheit und Wirksamkeit von Anzupgo® wurde in zwei pivotalen, randomisierten, doppelblinden, Vehikel-kontrollierten Studien mit gleichem Design (DELTA 1, DELTA 2) geprüft. An den Studien nahmen 960 Patienten im Alter von mindestens 18 Jahren mit mittelschwerem bis schwerem chronischem Handekzem gemäss der Definition von IGA-CHE (Investigator's Global Assessment for Chronic Hand Eczema) mit einem Score von 3 oder 4 (mittelschwer oder schwer) teil (siehe Tabelle 1), die bei Baseline einen Juckreiz-Score von ≥ 4 Punkten im Handekzem-Symptomtagebuch (Hand Eczema Symptom Diary, HESD) aufweisen mussten. In Frage kamen Patienten, die zuvor unzureichend auf potente bis hochpotente topische Kortikosteroide angesprochen hatten oder bei denen potente bis hochpotente topische Kortikosteroide nicht empfohlen sind.
Tabelle 1: Investigator’s Global Assessment für chronisches Handekzem (IGA-CHE)
|
IGA-CHE- Schweregrad |
IGA-CHE Score |
Anzeichen und Intensität |
|
Erscheinungsfrei |
0 |
Keine Anzeichen von Erythem, Schuppung, Hyperkeratose/Lichenifikation, Vesikelbildung, Ödem oder Fissuren |
|
Fast erscheinungsfrei |
1 |
Kaum wahrnehmbares Erythem, keine Anzeichen von Schuppung, Hyperkeratose/Lichenifikation, Vesikelbildung, Ödem oder Fissuren |
|
Leicht |
2 |
Mindestens eines: |
|
Mittelschwer |
3 |
Mindestens eines: |
|
Schwer |
4 |
Mindestens eines: |
In DELTA 1 und DELTA 2 trugen die Patienten während 16 Wochen 2x täglich entweder Delgocitinib Creme (20 mg/g) oder eine Vehikel-Creme auf die betroffenen Stellen an Händen und Handgelenken auf. Alle Patienten, welche die beiden pivotalen Studien abgeschlossen hatten, konnten an der Langzeitverlängerungsstudie DELTA 3 teilnehmen.
Endpunkte
In DELTA 1 und DELTA 2 war der primäre Endpunkt, das Erreichen eines IGA-CHE-Behandlungserfolgs (IGA-CHE TS (Treatment Success)), definiert als ein IGA-CHE-Score von 0 (frei von Symptomen) oder 1 (fast frei von Symptomen: nur kaum wahrnehmbares Erythem) mit einer mindestens zweistufigen Verbesserung von Baseline bis Woche 16. Der IGA-CHE bewertet den Schweregrad der Erkrankung und basiert auf einer 5-Punkte-Skala, die von 0 (frei von Symptomen) bis 4 (schwer) reicht (siehe Tabelle 1).
Zu den weiteren Ergebnissen zur Wirksamkeit gehörten der Hand Eczema Severity Index (HECSI) und das HESD zu verschiedenen Zeitpunkten. Der HECSI bewertet den Schweregrad von sechs klinischen Anzeichen (Erythem, Infiltration/Papulation, Bläschen, Risse, Schuppung, Ödem) und das Ausmass der Läsionen an jeder der fünf Handregionen (Fingerspitzen, Finger, Handfläche, Handrücken, Handgelenke). Im HESD wurden die von den Patienten berichteten Ergebnisse (Patient Reported Outcome, PRO) täglich mit 6 Punkten erfasst. Es dient dazu, den schlimmsten Schweregrad der Anzeichen und Symptome des chronischen Handekzems (Juckreiz, Schmerzen, Rissbildung, Rötung, Trockenheit, Schuppenbildung) anhand einer 11-stufigen numerischen Bewertungsskala zu beurteilen.
Baseline-Merkmale
In allen Behandlungsgruppen in DELTA 1 und DELTA 2 betrug das Durchschnittsalter 44,1 Jahre. 7,6% der Patienten waren 65 Jahre alt oder älter, 64,4% waren weiblich, 3,5% waren asiatischer Herkunft, 90,4% waren Patienten mit weisser und 0,7% mit schwarzer Hautfarbe. Die Häufigkeit von CHE nach Hauptsubtypen betrug 35,9 % atopisches Handekzem, 21,5 % hyperkeratotisches Ekzem, 19,6 % irritatives Kontaktekzem, 13,9 % allergisches Kontaktekzem, 9,1 % vesikuläres Handekzem (Pompholyx) und 0,1 % Kontakturtikaria/Proteinkontaktdermatitis. In DELTA 1 und DELTA 2 hatten 28,4% der Patienten einen Baseline-IGA-CHE-Score von 4 (schweres chronisches Handekzem). Der mittlere Baseline-Score im Dermatology Life Quality Index (DLQI) lag bei 12,5, der HECSI-Score bei 71,6 und der HESD-Score bei 7,1. Die mittleren HESD-Scores für Juckreiz und Schmerzen betrugen 7,1 bzw. 6,7.
Klinisches Ansprechen
DELTA 1 und DELTA 2
In DELTA 1 und DELTA 2 erreichte ein signifikant grösserer Anteil der Patienten in der Delgocitinib-Gruppe den primären Endpunkt IGA-CHE TS in Woche 16 im Vergleich zur Vehikel-Gruppe. Die Ergebnisse für den primären und ausgewählte andere multiplizitätskontrollierte sekundäre Endpunkte sind in Tabelle 2 aufgeführt. Abbildung 1 zeigt den Anteil derjenigen Patienten, die im Laufe der Zeit in DELTA 1 und DELTA 2 eine Verbesserung bei HECSI-75, HESD-Juckreiz ≥ 4 Punkte und HESD-Schmerz ≥ 4 Punkte erreichten.
Tabelle 2: Ergebnisse zur Wirksamkeit von Delgocitinib in Woche 16 in DELTA 1 und DELTA 2
|
|
DELTA 1 |
DELTA 2 | ||
|
|
Delgocitinib |
Vehikel |
Delgocitinib |
Vehikel |
|
IGA-CHE TS, % der Respondera, b |
19,7# |
9,9 |
29,1§ |
6,9 |
|
HECSI-90, % der Respondera, c |
29,5§ |
12,3 |
31,0§ |
8,8 |
|
HECSI-75, % der Respondera, d, e |
49,2§ |
23,5 |
49,5§ |
18,2 |
|
HECSI, mittlere prozentuale (%) LS-Veränderung gegenüber Baseline (± SE)f |
-56,5§ |
-21,2 |
-58,9§ |
-13,4 |
|
HESD-Juckreiz-Score, mittlere LS-Veränderung gegenüber Baseline (± SE)f |
-3,6§ |
-1,9 |
-3,4§ |
-1,4 |
|
HESD-Schmerz-Score, mittlere LS-Veränderung gegenüber Baseline (± SE)f |
-3,4§ |
-1,8 |
-3,3§ |
-1,3 |
|
HESD-Score, mittlere LS-Veränderung gegenüber Baseline (± SE)f |
-3,4§ |
-1,7 |
-3,2§ |
-1,4 |
|
HESD-Juckreiz ≥ 4-Punkte-Verbesserung, % der Respondera, g, h |
47,1§ |
23,0 |
47,2§ |
19,9 |
|
HESD-Schmerz ≥ 4-Punkte-Verbesserung, % der Respondera, b, g |
49,1§ |
27,5 |
48,6§ |
22,7 |
|
HESD ≥ 4-Punkte-Verbesserung, % der Respondera, b, g |
47,2§ |
24,4 |
44,5§ |
20,9 |
#p<0,01, §p<0,001
Alle p-Werte waren im Vergleich zum Vehikel statistisch signifikant mit Anpassung für Multiplizität.
Abkürzungen: LS = kleinste Quadrate (least squares); N = Anzahl der Patienten im vollständigen Analysesatz (alle Patienten, die randomisiert und dosiert wurden); SE = Standardfehler (standard error)
a.Daten nach Einleitung einer Notfallbehandlung, nach dauerhaftem Abbruch der Behandlung oder deren Daten fehlten, wurden als Non-Response betrachtet.
b.Statistisch signifikant im Vergleich zum Vehikel mit Anpassung für Multiplizität in Woche 4 und 8 in DELTA 1 und DELTA 2.
c.HECSI-90-Responder waren Patienten mit einer Verbesserung des HECSI um ≥ 90 % gegenüber Baseline.
d.HECSI-75-Responder waren Patienten mit einer Verbesserung des HECSI um ≥ 75 % gegenüber Baseline.
e.Statistisch signifikant im Vergleich zum Vehikel mit Anpassung für Multiplizität in Woche 8 in DELTA 1 und DELTA 2.
f.Daten nach Einleitung einer Notfallbehandlung oder nach dauerhaftem Abbruch der Behandlung oder deren Daten wurden als Non-Response betrachtet, indem die schlechteste Beobachtung übertragen wurde.
g.Basierend auf der Anzahl der Patienten, deren Baseline-Score ≥ 4 war (Skala von 0 bis 10).
h.Statistisch signifikant im Vergleich zum Vehikel mit Anpassung für Multiplizität in Woche 2, 4 und 8 in DELTA 1 und DELTA 2.
Im Vergleich zur Behandlung mit Vehikel-Creme wurden 1 Tag bzw. 3 Tage nach Beginn der Delgocitinib-Behandlung stärkere Verbesserungen, gemessen an der mittleren Veränderung der HESD-Scores für Juckreiz und Schmerzen, beobachtet. In Woche 2 wurden bei allen HESD-Punkten (Juckreiz, Schmerzen, Rissbildung, Rötung, Trockenheit, Schuppenbildung) stärkere Verbesserungen gegenüber der Behandlung mit Vehikel-Creme beobachtet.
Abbildung 1: Anteil der Patienten, die im Laufe der Zeit in DELTA 1 und DELTA 2 eine Verbesserung bei HECSI-75, HESD-Juckreiz ≥ 4 Punkte und HESD-Schmerz ≥ 4 Punkte erreichten – gepoolte Daten aus DELTA 1 und DELTA 2
|
HECSI-75a |
HESD-Juckreiz ≥ 4-Punkte-Verbesserungb |
HESD-Schmerz ≥ 4-Punkte-Verbesserungb |
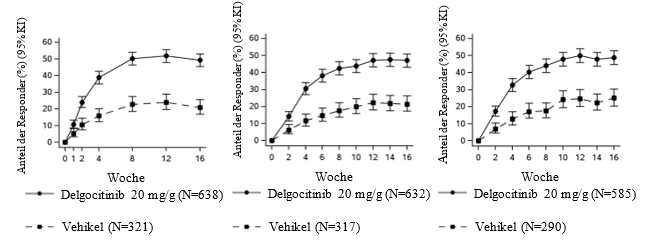
KI = Konfidenzintervall.
a.HECSI-75-Responder waren Patienten mit einer Verbesserung des HECSI um ≥ 75 % gegenüber Baseline.
b.Basierend auf der Anzahl der Patienten, deren Baseline-Score ≥ 4 war (Skala von 0 bis 10).
In DELTA 1 und DELTA 2 waren die Behandlungseffekte in den Untergruppen (Gewicht, Alter, Geschlecht, Schweregrad der Erkrankung, Dauer des chronischen Handekzems und vorherige Behandlung) mit den Ergebnissen in der gesamten Studienpopulation vergleichbar.
Die Punktschätzungen des Behandlungseffekts für den primären und die wichtigsten sekundären Endpunkte in Woche 16 fielen bei allen CHE-Subtypen durchweg zugunsten von Delgocitinib aus. Beim hyperkeratotischen Handekzem betrug der geschätzte Behandlungsunterschied für IGA-CHE TS jedoch 2,1%, 95% CI (-6,8:11,0). Bei allen CHE-Subtypen kam es in Woche 16 zu einer statistisch signifikant stärkeren Verbesserung des Juckreizes und der Schmerzen bei HESD (≥ 4 Punkte) im Vergleich zur Vehikel-Creme.
Zusätzliche Lebensqualität - Ergebnisberichte von Patienten
Sowohl in DELTA 1 als auch in DELTA 2 zeigten Patienten, die mit Delgocitinib-Creme behandelt wurden, eine signifikante Verbesserung gegenüber der Baseline bis zur 16. Woche im Vergleich zu mit Vehikel in der Hand Eczema Impact Scale (HEIS)(proximale Einschränkungen bei täglichen Aktivitäten, Scham, Frustration, Schlaf, Arbeit und körperliche Funktion [Fähigkeit, Gegenstände zu halten oder zu greifen]) (siehe Tabelle 3).
In DELTA 1 und DELTA 2 wurden bei mit Delgocitinib behandelten Patienten im Vergleich zu mit Vehikel-Creme behandelten Patienten in Woche 16 signifikant bedeutende Verbesserungen der gesundheitsbezogenen Lebensqualität, erfasst mittels DLQI, beobachtet (siehe Tabelle 3). Von den vier Bereichen «Arbeitsproduktivität und Aktivitätsbeeinträchtigung: Chronisches Handekzem» (Abwesenheit, Anwesenheit, Verlust der Arbeitsproduktivität, Beeinträchtigung der Aktivität) zeigten Patienten, die Delgocitinib erhielten, in Woche 16 bedeutendere Verbesserungen in allen Bereichen ausser bei der Abwesenheit, gegenüber der Behandlung mit Vehikel-Creme.
Tabelle 3: Zusätzliche Lebensqualität - Ergebnisberichte von Patienten unter Behandlung mit Delgocitinib in Woche 16 in DELTA 1 und DELTA 2
|
|
DELTA 1 |
DELTA 2 | ||
|
|
Delgocitinib |
Vehikel |
Delgocitinib |
Vehikel |
|
DLQI, mittlere LS-Veränderung gegenüber Baseline (± SE)a |
-7,6§ |
-3,9 |
-7,0§ |
-3,1 |
|
HEIS, mittlere LS-Veränderung gegenüber Baseline (± SE)a |
-1,46§ |
-0,82 |
-1,45§ |
-0,64 |
|
HEIS PDAL, mittlere LS-Veränderung gegenüber Baseline (± SE)a, b |
-1,46§ |
-0,86 |
-1,48§ |
-0,66 |
|
DLQI ≥ 4-Punkte-Verbesserung, |
74,4§ |
50,0 |
72,2§ |
45,8 |
§p<0,001
Alle p-Werte waren im Vergleich zum Vehikel statistisch signifikant mit Anpassung für Multiplizität.
Abkürzungen: LS = kleinste Quadrate (least squares); N = Anzahl der Patienten im vollständigen Analysesatz (alle Patienten, die randomisiert und dosiert wurden); PDAL = proximale tägliche Aktivitätseinschränkungen (proximal daily activity limitations); SE = Standardfehler (standard error)
a.Daten nach Einleitung einer Notfallbehandlung, nach dauerhaftem Abbruch der Behandlung, oder deren Daten wurden als Non-Response betrachtet, indem die schlechteste Beobachtung übertragen wurde.
b.HEIS PDAL beurteilt die Fähigkeit des Patienten, Seifen/Reinigungsmittel zu benutzen, den Haushalt zu führen und sich zu waschen.
c.Daten nach Einleitung einer Notfallbehandlung, nach dauerhaftem Abbruch der Behandlung, oder deren Daten fehlten, wurden als Non-Response betrachtet.
d.Basierend auf der Anzahl der Patienten, deren Baseline-Score ≥ 4 war.
Verlängerungsstudie (DELTA 3)
Patienten, die entweder die Studie DELTA 1 oder DELTA 2 abgeschlossen hatten, konnten an einer 36-wöchigen offenen Verlängerungsstudie (DELTA 3) teilnehmen. In DELTA 3 wurde die langfristige Sicherheit und Wirksamkeit einer bedarfsgerechten Delgocitinib-Behandlung bei 801 Patienten untersucht. Die Patienten begannen mit der Anwendung von Delgocitinib 2x täglich auf die betroffenen Stellen, wenn der IGA-CHE-Score ≥ 2 (leicht oder schlechter) war, und beendeten die Behandlung, wenn ein IGA-CHE-Score von 0 oder 1 (frei von Symptomen oder fast frei von Symptomen) erreicht wurde. Patienten, die mit einem IGA-CHE-Score von 0 oder 1 in DELTA 3 eintraten, wurden nicht mehr behandelt bis zum Verlust des Ansprechens (IGA-CHE-Score ≥ 2).
Patienten, die nach der anfänglichen 16-wöchigen Behandlung mit Delgocitinib eine Verbesserung beim IGA-CHE 0 oder 1, beim HECSI-75, beim HECSI-90, beim HESD-Juckreiz ≥ 4 Punkte und beim HESD-Schmerz ≥ 4 Punkte erreichten, wurden bis zur Woche 52 mit einer Behandlung nach Bedarf weiterbehandelt. Bei den 560 Patienten, die in den pivotalen Studien (DELTA 1 und DELTA 2) in die Delgocitinib-Gruppe randomisiert wurden und anschliessend an der DELTA 3 Studie teilnahmen, betrug die durchschnittliche Anzahl der Behandlungsperioden 1,5 (Spanne 0 bis 6), die durchschnittliche Dauer der Behandlungsperiode 123 Tage und die durchschnittliche kumulative Anzahl der Tage des Ansprechens (Tage mit einem IGA-CHE-Score von 0 oder 1 innerhalb des 36-wöchigen Behandlungszeitraums) 46. Bei denjenigen Patienten, die in den pivotalen Studien in Woche 16 einen IGA-CHE TS erreichten, lag die mittlere kumulative Anzahl der Tage des Ansprechens bei 111.
Von denjenigen Patienten, die in den pivotalen Studien in die Delgocitinib-Gruppe randomisiert wurden und in Woche 16 einen IGA-CHE TS erreichten, lag die mediane Dauer des Ansprechens während des Absetzens der Behandlung bei 4 Wochen, wobei 28% das Ansprechen für mindestens 8 Wochen beibehielten. Die mediane Dauer bis zum Wiedererreichen eines IGA-CHE-Scores von 0 oder 1 nach Wiederaufnahme der Behandlung betrug 8 Wochen. Von denjenigen Patienten, die in den pivotalen Studien in Woche 16 unter Delgocitinib-Behandlung keinen IGA-CHE TS erreicht hatten, erreichten 48,1% unter fortgesetzter Delgocitinib-Behandlung in DELTA 3 einen IGA-CHE von 0 oder 1.
Direkte Vergleichsstudie der Phase 3 mit Alitretinoin (DELTA Force)
Die Wirksamkeit und Sicherheit von Delgocitinib-Creme 20 mg/g, die 2x täglich aufgetragen wird, wurde in einer randomisierten, Gutachter-verblindeten Studie im Vergleich zu Alitretinoin-Kapseln 30 mg (mit der Option, während der Studie auf 10 mg zu reduzieren), die einmal täglich verabreicht werden, bei erwachsenen Patienten mit schwerem CHE untersucht. Die Behandlungsdauer betrug bis zu 24 Wochen.
Für Delgocitinib-Creme wurde im Vergleich zu Alitretinoin eine statistisch signifikant grössere Verbesserung für den primären Endpunkt, die Veränderung des HECSI-Scores vom Ausgangswert bis zur 12. Woche, erzielt. Statistisch signifikant grössere Verbesserungen wurden für Delgocitinib auch bei den wichtigsten sekundären Endpunkten erzielt, darunter HESD-Juckreiz und HESD-Schmerz. Die Ergebnisse für die primären und ausgewählten multiplizitätskontrollierten sekundären Endpunkte sind in Tabelle 4 dargestellt.
Tabelle 4: Ergebnisse zur Wirksamkeit in Woche 12 aus DELTA Force - Delgocitinib im Vergleich zu Alitretinoin
|
|
DELTA Force | |
|
|
Delgocitinib |
Alitretinoin |
|
HECSI, mittlere LS-Veränderung gegenüber Baseline (± SE)a |
-67.6§ |
-51,5 |
|
HECSI-90, % der Responderb |
38,6# |
26,0 |
|
IGA-CHE TS, % der Responderb |
27,2# |
16,6 |
|
HESD Juckreiz-Score, mittlere LS Veräderung gegenüber Baseline (± SE)a |
-3,0# |
-2,4 |
|
HESD Schmerz-Score, mittlere LS Veränderung gegenüber Baseline (± SE)a |
-2,9* |
-2,3 |
*p=0.018, #p<0,01, §p<0,001
Alle p-Werte waren im Vergleich zu Alitretinoin statistisch signifikant mit Anpassung für Multiplizität.
Abkürzungen: LS = kleinste Quadrate (least squares); N = Anzahl der Patienten im vollständigen Analysesatz; SE = Standardfehler (standard error)
a.Daten nach Einleitung einer NotfallBehandlung, nach dauerhaftem Abbruch der Behandlung, oder fehlende Daten wurden als Non-Response gewertet, indem die schlechteste Beobachtung übertragen wurde.
b.Daten nach Einleitung einer Notfallbehandlung, nach dauerhaftem Abbruch der Behandlung, oder denen Daten fehlten, galten als Non-Response
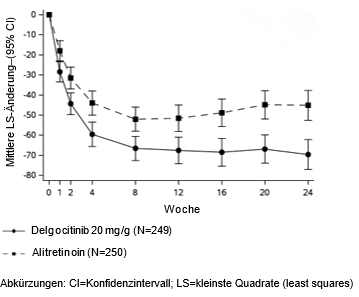
Grössere Verbesserungen bei Delgocitinib-Creme, gemessen an der mittleren Änderung des HECSI-Scores im Vergleich zu Alitretinoin-Kapseln, wurden bereits in Woche 1 beobachtet und verbesserten sich bis Woche 24 weiter. Die Veränderung der HECSI-Scores vom Ausgangswert bis Woche 24 für Delgocitinib und Alitretinoin sind in Abbildung 2 dargestellt.
Abbildung 2: Mittlere LS-Änderung des HECSI-Scores gegenüber dem Ausgangswert im Laufe der Zeit aus DELTA Force
Pharmakokinetik
Absorption
Die Pharmakokinetik der Delgocitinib-Creme (20mg/g) wurde in einer Studie mit 15 erwachsenen Patienten im Alter von 22 bis 69 Jahren mit mittelschwerem bis schwerem chronischem Handekzem untersucht. Die Patienten trugen Delgocitinib während 8 Tagen 2x täglich auf die betroffenen Stellen an den Händen und Handgelenken auf.
Die geometrische mittlere ± SD maximale Plasmakonzentration (Cmax) und der Fläche unter der Konzentrationskurve vom Zeitpunkt 0 bis 12 Stunden (AUC0-12) an Tag 8 betrug 0,46 ng/ml ± 0,28 bzw. 3,7 ng*h/ml ± 1,88. Es wurde keine Akkumulation beobachtet.Die systemische Exposition (AUC und Cmax) zwischen Tag 1 und Tag 8 war ähnlich.
Nach 2x täglicher Anwendung von Delgocitinib in DELTA 2 war die geometrische mittlere Plasmakonzentration (CV%), die 2 bis 6 Stunden nach der Anwendung an Tag 113 beobachtet wurde, um 48% niedriger als die an Tag 8 (0,11 ng/ml (460%) bzw. 0,21 ng/ml (218%)), wobei eine hohe Variabilität beobachtet wurde.
Die mittlere relative Bioverfügbarkeit von Delgocitinib nach topischer Anwendung von Anzupgo® beträgt etwa 0,6% im Vergleich nach oraler Verabreichung von Delgocitinib-Tabletten.
Distribution
Die Plasmaproteinbindung von Delgocitinib beträgt 22 bis 29%.
Metabolismus
Da Delgocitinib unbeträchtlich metabolisiert wird, ist die Hauptplasmakomponente unverändertes Delgocitinib. Nach oraler Verabreichung wurden vier Metaboliten (gebildet durch Oxidation und Glucuronid-Konjugation) mit < 2% der durchschnittlichen unveränderten Delgocitinib-Plasmakonzentrationen nachgewiesen. Der begrenzte Metabolismus von Delgocitinib erfolgt hauptsächlich über CYP3A4/5 und in geringerem Masse über CYP2C9, CYP2C19 und CYP2D6.
Elimination
Delgocitinib wird hauptsächlich über die Nieren ausgeschieden, da etwa 75% der Gesamtdosis nach oraler Verabreichung unverändert im Urin gefunden wurden.
Nach wiederholter topischer Anwendung wurde die durchschnittliche Halbwertszeit von Delgocitinib auf 20,3 Stunden geschätzt. Die beobachtete Halbwertszeit nach topischer Verabreichung ist aufgrund der langsamen Resorption nach topischer Anwendung länger als nach oraler Verabreichung.
Kinetik spezieller Patientengruppen
Leberfunktionsstörungen
Es wurden keine Studien mit Delgocitinib Creme bei Patienten mit Leberfunktionsstörungen durchgeführt.
Aufgrund der geringen systemischen Exposition von topisch angewendetem Delgocitinib und der begrenzten Metabolisierung von Delgocitinib ist es unwahrscheinlich, dass Veränderungen der Leberfunktion Auswirkungen auf die Elimination von Delgocitinib haben. Daher sind bei Patienten mit Leberfunktionsstörungen keine Dosisanpassungen erforderlich (siehe«Dosierung/Anwendung»).
Nierenfunktionsstörungen
Die pharmakokinetischen Parameter von Delgocitinib wurden bei 96 Patienten mit leichter oder mittelschwerer Nierenfunktionsstörung (eGFR 30 bis 89 ml/min/1,73 m2) in der Studie DELTA 2 analysiert. Die in Woche 1 gemessenen geometrisch mittleren Plasmakonzentrationen betrugen 0,18 ng/ml (n=201), 0,29 ng/ml (n=80) und 0.41 ng/ml (n=6) bei nicht, leicht und mässig eingeschränkter Nierenfunktion. Aufgrund der geringensystemischen Exposition von topisch angewendetem Delgocitinib sind Veränderungen der Plasmakonzentration bei Nierenfunktionsstörungen wahrscheinlich nicht von klinischer Bedeutung. Daher sind bei Patienten mit Nierenfunktionsstörung keine Dosisanpassungen erforderlich (siehe «Dosierung/Anwendung»).
Präklinische Daten
Basierend auf konventionellen Studien zur Sicherheitspharmakologie, Genotoxizität, Phototoxizität, lokalen Verträglichkeit, Hautsensibilisierung und juvenilen Toxizität lassen die präklinischen Daten keine besonderen Gefahren für den Menschen erkennen. Auswirkungen in Studien zur Toxizität bei Wiederholungsdosen wurden nur bei Expositionen beobachtet, die als ausreichend weit über der maximalen Exposition des Menschen nach topischer Anwendung liegend angesehen wurden.
Kanzerogenität
In einer 2-jährigen dermalen Kanzerogenitätsstudie an Mäusen wurden keine lokalen oder systemischen neoplastischen Befunde bei Stärken bis zu 50 mg/g Delgocitinib-Salbe beobachtet (Systemische Expositionen bis zum etwa 600fachen der menschlichen Exposition nach topischer Anwendung basierend auf der AUC).
Die Ergebnisse einer 2-jährigen oralen Kanzerogenitätsstudie an Ratten umfassten Adenome der Bauchspeicheldrüse und Lipome der Haut/Unterhaut (nur bei männlichen Ratten), Thymome (nur bei weiblichen Ratten) und Leydig Zelltumore bei Expositionen, die etwa dem 160-, 580- bzw. 1800fachen der menschlichen Exposition nach topischer Anwendung entsprachen. Die klinische Relevanz der Tumorbefunde bei Ratten ist angesichts der Tumorarten bei einer einzigen Spezies und einem einzigen Geschlecht, der Expositionen, bei denen die Tumore auftraten, und der negativen Befunde in der 2-jährigen Studie zur dermalen Karzinogenität bei Mäusen gering.
Fertilität und frühe embryonale Entwicklung
Oral verabreichtes Delgocitinib hatte bei keiner der untersuchten Dosen bei männlichen Ratten Auswirkungen auf die Fertilität (Expositionen, die etwa dem 1700fachen der menschlichen Exposition nach topischer Anwendung entsprachen). Bei weiblichen Ratten führte oral verabreichtes Delgocitinib zu Auswirkungen auf die weibliche Fertilität (niedrigerer Fertilitätsindex, weniger Corpora lutea und weniger Implantationen) bei Expositionen, die etwa dem 5800fachen der menschlichen Exposition nach topischer Anwendung entsprachen. Postimplantationsverluste und eine Abnahme der Anzahl lebender Embryonen wurden bei Expositionen beobachtet, die etwa dem 432- bzw. 1000fachen der menschlichen Exposition entsprachen.
Embryo-fötale Entwicklung
Oral verabreichtes Delgocitinib führte bei Ratten oder Kaninchen bei einer Exposition, die etwa dem 120- bzw. 194-fachen der menschlichen Exposition nach topischer Anwendung entsprach, nicht zu schädlichen Auswirkungen auf den Fötus.
Bei Ratten wurde eine Abnahme des Fötusgewichts und Skelettveränderungen (Variationen) bei einer Exposition, die dem 512fachen der menschlichen Exposition nach topischer Anwendung entsprach, beobachtet. Ein Anstieg der Postimplantationsverluste, verzögertes fötales Wachstum und verzögerte Ossifikation wurden bei einer Exposition beobachtet, die etwa dem 1400fachen der menschlichen Exposition nach topischer Anwendung entsprach. Bei Kaninchen wurden bei einer Exposition, die etwa dem 992fachen der menschlichen Exposition nach topischer Anwendung entsprach, ein Anstieg der Postimplantationsverluste, eine geringere Anzahl lebender Föten und eine Tendenz zu einem Rückgang des Fötusgewichts beobachtet.
Prä- und postnatale Entwicklung
Oral verabreichtes Delgocitinib führte bei Ratten zu einer verringerten Lebensfähigkeit des Fötus, verlängerter Tragzeit, längerer Dauer des Geburtsvorgangs und einem geringeren Gewicht der Jungtiere in der frühen postnatalen Periode bei einer Exposition, die etwa dem 1400fachen der menschlichen Exposition nach topischer Anwendung entsprach. Es gab bei keiner der untersuchten Dosen eine Auswirkung auf das Verhalten und die Lernfähigkeit, die sexuelle Reifung oder die Reproduktionsleistung der Nachkommen.
Nach oraler Verabreichung an säugenden Ratten wurde Delgocitinib in der Milch in Mengen ausgeschieden, die etwa dem 3fachen der Konzentrationen im Blutplasma (basierend auf der AUC) entsprachen.
Sonstige Hinweise
Haltbarkeit
Das Arzneimittel darf nur bis zu dem auf der Packung mit «EXP» bezeichneten Datum verwendet werden.
Haltbarkeit nach dem Öffnen
Nach dem Öffnen 12 Monate haltbar.
Besondere Lagerungshinweise
Nicht über 30°C lagern. Nicht einfrieren.
Ausser Reichweite von Kindern aufbewahren.
Zulassungsnummer
69330 (Swissmedic)
Packungen
Tube à 60 g [B]
Zulassungsinhaberin
LEO Pharmaceutical Products Sarath Ltd., Kloten
Domizil: Zürich
Stand der Information
Juni 2024