ZusammensetzungWirkstoffe
Ravulizumab, aus gentechnisch veränderten Ovarialzellen des chinesischen Hamsters hergestellt.
Hilfsstoffe
Natriumdihydrogenphosphat-Monohydrat
Natriummonohydrogenphosphat-Heptahydrat
Ultomiris 300 mg/3 ml und 1100 mg/11 ml: L-Arginin
Ultomiris 300 mg/3 ml und 1100 mg/11 ml: Saccharose
Ultomiris 300 mg/30 ml: Natriumchlorid
Ultomiris 300 mg/30 ml: 115 mg Natrium pro 30ml-Durchstechflasche
Ultomiris 300 mg/3 ml: 4,6 mg Natrium pro 3ml-Durchstechflasche
Ultomiris 1100 mg/11 ml: 16,8 mg Natrium pro 11ml-Durchstechflasche
Polysorbat 80
Wasser für Injektionszwecke
Darreichungsform und Wirkstoffmenge pro EinheitKonzentrat zur Herstellung einer Infusionslösung.
Ultomiris 300 mg/30 ml:
Klare bis durchscheinende, leicht weissliche Lösung, pH-Wert 7,0.
Eine Durchstechflasche mit 30 ml enthält 300 mg Ravulizumab (10 mg/ml) und 115 mg Natrium.
Nach Verdünnung beträgt die endgültige Konzentration der zu infundierenden Lösung 5 mg/ml.
Ultomiris 300 mg/3 ml:
Durchscheinende, klare bis gelbliche Lösung, pH-Wert 7,4.
Eine Durchstechflasche mit 3 ml enthält 300 mg Ravulizumab (100 mg/ml) und 4,6 mg Natrium.
Nach Verdünnung beträgt die endgültige Konzentration der zu infundierenden Lösung 50 mg/ml.
Ultomiris 1100 mg/11 ml:
Durchscheinende, klare bis gelbliche Lösung, pH-Wert 7,4.
Eine Durchstechflasche mit 11 ml enthält 1100 mg Ravulizumab (100 mg/ml) und 16,8 mg Natrium.
Nach Verdünnung beträgt die endgültige Konzentration der zu infundierenden Lösung 50 mg/ml.
Indikationen/AnwendungsmöglichkeitenParoxysmale nächtliche Hämoglobinurie (PNH)
Ultomiris wird angewendet zur Behandlung erwachsener und pädiatrischer Patienten ab einem Körpergewicht von 10 kg mit PNH:
- bei Patienten mit Hämolyse zusammen mit einem oder mehreren klinischen Symptomen als Hinweis auf eine hohe Krankheitsaktivität,
- bei Patienten, die klinisch stabil sind, nachdem sie mindestens während der vergangenen 6 Monate mit Eculizumab behandelt wurden.
Atypisches hämolytisch-urämisches Syndrom (aHUS)
Ultomiris wird angewendet zur Behandlung erwachsener und pädiatrischer Patienten ab einem Körpergewicht von 10 kg mit aHUS, die zuvor nicht mit Komplementinhibitoren behandelt wurden (Behandlungs-naive Patienten bezüglich Komplementinhibitoren) oder Eculizumab mindestens 3 Monate lang erhalten haben und nachweislich auf Eculizumab ansprachen.
Generalisierte Myasthenia gravis (gMG)
Ultomiris wird angewendet als Zusatztherapie zu einer Standardbehandlung bei erwachsenen Azetylcholinrezeptor (AChR)-Antikörper-positiven Patienten mit gMG.
Neuromyelitis-optica-Spektrum-Erkrankungen (NMOSD)
Ultomiris wird angewendet zur Behandlung erwachsener Patienten mit NMOSD, die positiv für Anti-Aquaporin-4 (AQP4)-Antikörper sind (siehe Abschnitt "Eigenschaften/Wirkungen" ).
Dosierung/AnwendungRavulizumab muss von medizinischem Fachpersonal und unter der Aufsicht eines in der Behandlung von Patienten mit hämatologischen Erkrankungen, Nierenerkrankungen, neuromuskulären oder neuroinflammatorischen Erkrankungen erfahrenen Arztes verabreicht werden.
Vor Beginn der Behandlung sollte sichergestellt sein, dass keine aktive Meningokokkeninfektion/-sepsis vorliegt und gemäss den offiziellen Impfempfehlungen ein ausreichender Impfschutz gegen Meningokokken besteht (siehe "Kontraindikationen" , "Warnhinweise und Vorsichtsmassnahmen" ).
Um die Rückverfolgbarkeit von biotechnologisch hergestellten Arzneimitteln sicherzustellen, wird empfohlen, Handelsname und Chargennummer bei jeder Behandlung zu dokumentieren.
Erwachsene Patienten mit PNH, aHUS, gMG oder NMOSD
Das empfohlene Dosierungsschema besteht aus einer Initialdosis gefolgt von Erhaltungsdosen, die als intravenöse Infusion verabreicht werden. Die zu verabreichenden Dosen basieren auf dem Körpergewicht des Patienten, wie in Tabelle 1 dargestellt. Bei erwachsenen Patienten (im Alter von ≥ 18 Jahren) müssen die Erhaltungsdosen jeweils im Abstand von 8 Wochen verabreicht werden, beginnend 2 Wochen nach Verabreichung der Initialdosis.
Das Dosierungsschema darf in Einzelfällen um ± 7 Tage vom planmässigen Infusionstag abweichen (ausser bei der ersten Erhaltungsdosis von Ravulizumab), die darauffolgende Dosis sollte jedoch gemäss dem ursprünglichen Schema verabreicht werden.
Bei Patienten, die von Eculizumab auf Ravulizumab umgestellt werden, sollte die Initialdosis 2 Wochen nach der letzten Eculizumab-Infusion verabreicht werden, anschliessend wird alle 8 Wochen eine Erhaltungsdosis verabreicht, beginnend 2 Wochen nach Verabreichung der Initialdosis, wie in Tabelle 1 gezeigt.
Tabelle 1: Körpergewichtsbasiertes Dosierungsschema für Ravulizumab bei erwachsenen Patienten mit einem Körpergewicht ≥ 40 kg.
|
Körpergewicht (kg)
|
Initialdosis (mg)
|
Erhaltungsdosis (mg)*
|
Dosierungsintervall
| |
≥ 40 bis < 60
|
2400
|
3000
|
Alle 8 Wochen
| |
≥ 60 bis < 100
|
2700
|
3300
|
Alle 8 Wochen
| |
≥ 100
|
3000
|
3600
|
Alle 8 Wochen
|
*Die erste Erhaltungsdosis wird 2 Wochen nach der Initialdosis gegeben.
Ergänzungsdosis nach Plasmaaustausch (PE), Plasmapherese (PP) oder intravenösem Immunglobulin (IVIg)
Plasmaaustausch (PE), Plasmapherese (PP) und intravenöses Immunglobulin (IVIg) senken nachweislich die Ravulizumab-Serumspiegel. Bei Behandlungen wie PE, PP oder IVIg ist eine zusätzliche Dosis Ravulizumab erforderlich (Tabelle 2).
Tabelle 2: Ergänzungsdosis Ravulizumab nach PP, PE oder IVIg
|
Körpergewicht (kg)
|
Zuletzt gegebene Ravulizumab-Dosis (mg)
|
Ergänzungsdosis (mg) nach jedem PE oder jeder PP
|
Ergänzungsdosis (mg) nach Abschluss eines IVIg-Behandlungszyklus
| |
≥ 40 bis < 60
|
2400
|
1200
|
600
| |
3000
|
1500
| |
≥ 60 bis < 100
|
2700
|
1500
|
600
| |
3300
|
1800
| |
≥ 100
|
3000
|
1500
|
600
| |
3600
|
1800
| |
Zeitpunkt der Ergänzungsdosis Ravulizumab
|
Innerhalb von 4 Stunden nach jedem PE oder jeder PP
|
Innerhalb von 4 Stunden nach Abschluss eines IVIg-Behandlungszyklus
|
Abkürzungen: IVIg = intravenöses Immunglobulin, kg = Kilogramm, PE = Plasmaaustausch, PP = Plasmapherese
PNH ist eine chronische Erkrankung. Es wird daher empfohlen, die Behandlung mit Ravulizumab über die gesamte Lebensdauer des Patienten fortzusetzen, sofern das Absetzen von Ravulizumab nicht klinisch angezeigt ist (siehe Abschnitt "Warnhinweise und Vorsichtsmassnahmen" ).
Bei aHUS sollte die Behandlung mit Ravulizumab zur Beseitigung der Manifestationen der thrombotischen Mikroangiopathie (TMA) über mindestens 6 Monate durchgeführt werden. Danach muss die Behandlungsdauer für jeden Patienten individuell festgesetzt werden. Bei Patienten, bei denen nach Feststellung durch den behandelnden Arzt (oder gemäß der klinischen Indikation) ein höheres Risiko für ein TMA-Rezidiv besteht, kann eine Langzeitbehandlung erforderlich sein (siehe Abschnitt "Warnhinweise und Vorsichtsmassnahmen" ).
Bei Patienten mit gMG oder NMOSD wurde die Behandlung mit Ravulizumab nur im Rahmen der chronischen Anwendung untersucht (siehe Abschnitt "Warnhinweise und Vorsichtsmassnahmen" ).
Ravulizumab wurde bei gMG-Patienten mit MGFA-Klasse V nicht untersucht.
Patienten mit Leberfunktionsstörungen
Die Sicherheit und Wirksamkeit von Ravulizumab wurden bei Patienten mit Leberfunktionsbeeinträchtigung nicht untersucht; allerdings legen pharmakokinetische Daten nahe, dass bei Patienten mit Leberfunktionsbeeinträchtigung keine Dosisanpassung erforderlich ist.
Patienten mit Nierenfunktionsstörungen
Bei Patienten mit einer Nierenfunktionsbeeinträchtigung ist keine Dosisanpassung erforderlich (siehe Abschnitt "Pharmakokinetik" ).
Ältere Patienten
Bei Patienten mit PNH, aHUS, gMG oder NMOSD im Alter von 65 Jahren oder älter ist keine Dosisanpassung erforderlich. Es liegen keine Hinweise vor, dass bei der Behandlung von geriatrischen Patienten besondere Vorsichtsmassnahmen notwendig sind. Es liegen jedoch nur begrenzte Erfahrungen zu Ravulizumab bei älteren Patienten mit PNH, aHUS oder NMOSD vor.
Kinder und Jugendliche
Kinder und Jugendliche mit PNH und aHUS und einem Körpergewicht ≥ 40 kg werden gemäss den Dosierungsempfehlungen für Erwachsene behandelt (Tabelle 1). Die nach dem Körpergewicht bemessenen Dosen und Dosierungsintervalle für Kinder und Jugendliche mit einem Körpergewicht von ≥ 10 kg bis < 40 kg sind in Tabelle 3 gezeigt.
Bei Patienten, die von Eculizumab auf Ravulizumab umgestellt werden, sollte die Initialdosis von Ravulizumab 2 Wochen nach der letzten Eculizumab-Infusion gegeben werden. Die anschliessenden Erhaltungsdosen sollten auf der Grundlage eines körpergewichtsbasierten Dosierungsschemas, wie es in Tabelle 3 gezeigt ist, ab 2 Wochen nach der Initialdosis gegeben werden.
Tabelle 3: Körpergewichtsbasiertes Dosierungsschema von Ravulizumab bei Kindern und Jugendlichen unter 40 kg mit PNH oder aHUS
|
Körpergewicht (kg)
|
Initialdosis
(mg)
|
Erhaltungsdosis
(mg)*
|
Dosierungsintervall
| |
≥ 10 bis < 20
|
600
|
600
|
Alle 4 Wochen
| |
≥ 20 bis < 30
|
900
|
2.100
|
Alle 8 Wochen
| |
≥ 30 bis < 40
|
1200
|
2.700
|
Alle 8 Wochen
|
*Die Erhaltungsdosis wird 2 Wochen nach der Initialdosis gegeben.
Die Sicherheit und Wirksamkeit von Ravulizumab bei Kindern mit PNH oder aHUS und einem Körpergewicht unter 10 kg wurde noch nicht untersucht. Die aktuell vorliegenden Daten sind in Abschnitt "Unerwünschte Wirkungen" beschrieben, aber es können keine Dosierungsempfehlungen gegeben werden.
Ravulizumab wurde bei Kindern und Jugendlichen mit PNH und einem Körpergewicht unter 30 kg nicht untersucht. Die Dosierung von Ravulizumab bei Kindern und Jugendlichen unter 30 kg stützt sich auf die bei Kindern und Jugendlichen mit aHUS angewendete Dosierung und basiert auf den pharmakokinetischen/pharmakodynamischen (PK/PD) Daten, die für mit Ravulizumab behandelte aHUS- und PNH-Patienten verfügbar sind.
Die Sicherheit und Wirksamkeit von Ravulizumab bei Kindern mit gMG oder NMOSD wurde noch nicht untersucht. Es liegen keine Daten vor.
Art der Anwendung
Nur zur intravenösen Infusion.
Dieses Arzneimittel muss durch einen 0,2-µm-Filter verabreicht werden und ist nicht als intravenöse Druck- oder Bolusinjektion zu verabreichen.
Ultomiris 300 mg/30 ml Konzentrat zur Herstellung einer Infusionslösung darf nicht mit Ultomiris 300 mg/3 ml oder 1100 mg/11 ml Konzentrat zur Herstellung einer Infusionslösung gemischt werden.
Ultomiris 300 mg/3 ml und 1100 mg/11 ml Konzentrat zur Herstellung einer Infusionslösung
Ultomiris Konzentrat zur Herstellung einer Infusionslösung wird in 3 ml- und 11 ml-Durchstechflaschen (100 mg/ml) angeboten und muss auf eine Endkonzentration von 50 mg/ml verdünnt werden. Nach der Verdünnung wird Ultomiris als intravenöse Infusion mittels Spritzenpumpe oder Infusionspumpe über einen Mindestzeitraum von 10 bis 75 Minuten (0,17 bis 1,3 Stunden), abhängig vom Körpergewicht, gegeben (siehe nachstehende Tabellen 4 und 5 unten).
Tabelle 4: Infusionsrate für Dosen von Ultomiris 300 mg/3 ml und 1100 mg/11 ml Konzentrat zur Herstellung einer Infusionslösung
|
Körpergewicht
(kg)a
|
Initialdosis (mg)
|
Mindestdauer der Infusion
Minuten (Stunden)
|
Erhaltungs-dosis (mg)
|
Mindestdauer der Infusion
Minuten (Stunden)
| |
≥ 10 bis < 20b
|
600
|
45 (0,8)
|
600
|
45 (0,8)
| |
≥ 20 bis < 30b
|
900
|
35 (0,6)
|
2100
|
75 (1,3)
| |
≥ 30 bis < 40b
|
1200
|
31 (0,5)
|
2700
|
65 (1,1)
| |
≥ 40 bis < 60
|
2400
|
45 (0,8)
|
3000
|
55 (0,9)
| |
≥ 60 bis < 100
|
2700
|
35 (0,6)
|
3300
|
40 (0,7)
| |
≥ 100
|
3000
|
25 (0,4)
|
3600
|
30 (0,5)
|
a Körpergewicht zum Behandlungszeitpunkt.
b Nur bei der Indikation PNH und aHUS
Tabelle 5: Infusionsrate für Ergänzungsdosen von Ultomiris 300 mg/3 ml und 1.100 mg/11 ml Konzentrat zur Herstellung einer Infusionslösung
|
Körpergewicht (kg)a
|
Ergänzungsdosisb (mg)
|
Mindestdauer der Infusion
Minuten (Stunden)
| |
≥ 40 bis < 60
|
600
|
15 (0,25)
| |
1200
|
25 (0,42)
| |
1500
|
30 (0,5)
| |
≥ 60 bis < 100
|
600
|
12 (0,20)
| |
1500
|
22 (0,36)
| |
1800
|
25 (0,42)
| |
≥ 100
|
600
|
10 (0,17)
| |
1500
|
15 (0,25)
| |
1800
|
17 (0,28)
|
a Körpergewicht zum Behandlungszeitpunkt.
b Siehe Tabelle 2 zur Auswahl der Ergänzungsdosis von Ravulizumab
Ultomiris 300 mg/30 ml Konzentrat zur Herstellung einer Infusionslösung
Ultomiris Konzentrat zur Herstellung einer Infusionslösung wird in 30 ml-Durchstechflaschen (10 mg/ml) angeboten und muss auf eine Endkonzentration von 5 mg/ml verdünnt werden. Nach der Verdünnung wird Ultomiris als intravenöse Infusion mittels Spritzenpumpe oder Infusionspumpe über einen Mindestzeitraum von 22 bis 194 Minuten (0,4 bis 3,3 Stunden), abhängig vom Körpergewicht, gegeben (siehe nachstehende Tabellen 6 und 7).
Tabelle 6: Infusionsrate für Dosen von Ultomiris 300 mg/30 ml Konzentrat zur Herstellung einer Infusionslösung
|
Körpergewicht a (kg)
|
Initialdosis (mg)
|
Mindestdauer der Infusion
Minuten (Stunden)
|
Erhaltungsdosis (mg)
|
Mindestdauer der Infusion
Minuten (Stunden)
| |
≥ 10 bis < 20b
|
600
|
113 (1,9)
|
600
|
113 (1,9)
| |
≥ 20 bis < 30b
|
900
|
86 (1,5)
|
2100
|
194 (3,3)
| |
≥ 30 bis < 40b
|
1200
|
77 (1,3)
|
2700
|
167 (2,8)
| |
≥ 40 bis < 60
|
2400
|
114 (1,9)
|
3000
|
140 (2,3)
| |
≥ 60 bis < 100
|
2700
|
102 (1,7)
|
3300
|
120 (2,0)
| |
≥ 100
|
3000
|
108 (1,8)
|
3600
|
132 (2,2)
|
a Körpergewicht zum Behandlungszeitpunkt.
b Nur bei der Indikation PNH und aHUS.
Tabelle 7: Infusionsrate für Ergänzungsdosen von Ultomiris 300 mg/30 ml Konzentrat zur Herstellung einer Infusionslösung
|
Körpergewicht (kg)a
|
Ergänzungsdosisb (mg)
|
Mindestdauer der Infusion
Minuten (Stunden)
| |
≥ 40 bis < 60
|
600
|
30 (0,5)
| |
1200
|
60 (1,0)
| |
1500
|
72 (1,2)
| |
≥ 60 bis < 100
|
600
|
23 (0,4)
| |
1500
|
60 (1,0)
| |
1800
|
65 (1,1)
| |
≥ 100
|
600
|
22 (0,4)
| |
1500
|
60 (1,0)
| |
1800
|
65 (1,1)
|
a Körpergewicht zum Behandlungszeitpunkt.
b Siehe Tabelle 2 zur Auswahl der Ergänzungsdosis von Ravulizumab
Hinweise zur Verdünnung des Arzneimittels vor der Anwendung, siehe Abschnitt "Hinweise für die Handhabung" .
Kontraindikationen- Überempfindlichkeit gegen den Wirkstoff oder einen der in Abschnitt "Hilfsstoffe" genannten sonstigen Bestandteile.
- Patienten mit nicht ausgeheilter Infektion mit Neisseria meningitidis bei Behandlungsbeginn (siehe Abschnitt "Warnhinweise und Vorsichtsmassnahmen" ).
- Patienten ohne aktuellen Impfschutz gegen Neisseria meningitidis, es sei denn, sie erhalten eine geeignete Antibiotikaprophylaxe bis zu zwei Wochen nach der Impfung (siehe Abschnitt "Warnhinweise und Vorsichtsmassnahmen" ).
- Patienten mit hereditären Komplementdefekten (siehe Abschnitt "Warnhinweise und Vorsichtsmassnahmen" ).
Warnhinweise und VorsichtsmassnahmenSchwere Meningokokkeninfektion
Aufgrund seines Wirkmechanismus erhöht Ravulizumab die Anfälligkeit des Patienten für eine Meningokokkeninfektion/-Sepsis (Neisseria meningitidis). Eine Meningokokkenerkrankung kann durch jedwede Serogruppe auftreten (siehe Abschnitt "Unerwünschte Wirkungen" ). Zur Verringerung dieses Infektionsrisikos müssen alle Patienten mindestens zwei Wochen vor Beginn der Behandlung mit Ravulizumab gegen Meningokokkeninfektionen geimpft werden, es sei denn, das Risiko eines Aufschubs der Behandlung mit Ravulizumab überwiegt das Risiko des Auftretens einer Meningokokkeninfektion. Patienten, bei denen eine Meningokokkenimpfung zu Beginn der Behandlung mit Ravulizumab weniger als 2 Wochen zurückliegt, müssen bis 2 Wochen nach der Impfung eine geeignete Antibiotikaprophylaxe erhalten. Zur Vorbeugung gegen die häufig pathogenen Meningokokken-Serogruppen werden, sofern verfügbar, Impfstoffe gegen die Serogruppen A, C, Y, W135 und B empfohlen. Die Patienten müssen gemäss den offiziellen Impfempfehlungen geimpft oder nachgeimpft werden. Wird der Patient von einer Eculizumab-Behandlung umgestellt, sollte der Arzt überprüfen, dass gemäss den offiziellen Impfempfehlungen ein ausreichender Impfschutz gegen Meningokokken besteht.
Eine Impfung ist unter Umständen nicht ausreichend, um eine Meningokokkeninfektion zu verhindern. Die offiziellen Empfehlungen zur indikationsgerechten Anwendung von Antibiotika sollten berücksichtigt werden. Bei Patienten, die mit Ravulizumab behandelt wurden, und bei Patienten, die mit anderen terminalen Komplementinhibitoren behandelt wurden, wurde über schwere oder tödliche Meningokokkeninfektionen/-Sepsen berichtet. Alle Patienten sollten auf Frühzeichen von Meningokokkeninfektion und -Sepsis überwacht, bei Infektionsverdacht sofort untersucht und mit geeigneten Antibiotika behandelt werden. Die Patienten sollten über diese Anzeichen und Symptome informiert werden und sich unverzüglich in ärztliche Behandlung begeben. Ärzte sollten den Patienten die Patienten-Informationsbroschüre und die Patientenkarte aushändigen.
Immunisierung
Vor dem Beginn der Therapie mit Ravulizumab wird empfohlen, dass Patienten mit Impfungen entsprechend den aktuellen Impfrichtlinien beginnen.
Eine Impfung kann das Komplement zusätzlich aktivieren. Folglich können sich bei Patienten mit komplementvermittelten Erkrankungen die Anzeichen und Symptome ihrer Grunderkrankung verstärken. Daher sollten die Patienten im Anschluss an die empfohlene Impfung engmaschig auf Krankheitssymptome überwacht werden.
Patienten unter 18 Jahren müssen gegen Haemophilus influenzae und Pneumokokkeninfektionen geimpft werden, wobei die nationalen Impfempfehlungen für jede Altersgruppe streng eingehalten werden müssen.
Sonstige systemische Infektionen
Die Therapie mit Ravulizumab sollte bei Patienten mit aktiven systemischen Infektionen mit Vorsicht durchgeführt werden. Ravulizumab hemmt die terminale Komplementaktivierung, daher kann es bei den Patienten zu einer erhöhten Anfälligkeit für durch Neisseria-Spezies und bekapselte Bakterien verursachte Infektionen kommen. Es wurden schwerwiegende Infektionen durch Neisseria-Spezies (ausser Neisseria meningitidis) beobachtet, einschliesslich disseminierte Gonokokken-Infektionen.
Die Patienten sollen über mögliche schwere Infektionen und deren Anzeichen und Symptome informiert werden. Ärzte sollten Patienten in Hinblick auf die Prävention von Gonorrhö beraten.
Infusionsbedingte Reaktionen
Die Verabreichung von Ravulizumab kann zu systemischen infusionsbedingten Reaktionen sowie zu allergischen Reaktionen oder Überempfindlichkeitsreaktionen einschliesslich Anaphylaxie führen (siehe Abschnitt "Unerwünschte Wirkungen).
In klinischen Prüfungen kam es häufig (1 %) zu Infusionsbedingten Reaktionen. Diese Reaktionen waren leicht bis mittelschwer und vorübergehend, (einschliesslich Schmerzen im unteren Rückenbereich, Bauchschmerzen, Muskelkrämpfe, Abfall des Blutdrucks, Blutdruckanstieg, Muskelstarre, Gliederbeschwerden, Arzneimittelüberempfindlichkeit (allergische Reaktion), Dysgeusie (Geschmacksstörung) und Benommenheit. Im Falle einer systemischen infusionsbedingten Reaktion, wenn Anzeichen einer kardiovaskulären Instabilität oder einer Beeinträchtigung der Atmung auftreten, sollte die Verabreichung von Ravulizumab unterbrochen und es sollten geeignete unterstützende Massnahmen ergriffen werden.
Behandlungsabbruch bei PNH
Wenn die Behandlung mit Ravulizumab bei Patienten mit PNH abgesetzt wird, sollten sie auf Anzeichen und Symptome einer schweren intravaskulären Hämolyse engmaschig überwacht werden. Eine schwere Hämolyse ist an erhöhten LDH (Lactatdehydrogenase)-Werten in Verbindung mit Folgendem erkennbar: plötzliche Verkleinerung des PNH-Klons oder plötzliche Abnahme des Hämoglobins oder erneutes Auftreten von Symptomen wie Ermüdung/Fatigue, Hämoglobinurie, Abdominalschmerz, Kurzatmigkeit (Dyspnoe), einem schwerwiegenden unerwünschten vaskulären Ereignis (einschliesslich Thrombose), Dysphagie oder Erektionsstörung. Patienten, bei denen die Therapie mit Ravulizumab abgesetzt wird, sollten mindestens 16 Wochen lang überwacht werden, damit Hämolysen und andere Reaktionen erkannt werden können. Wenn nach Absetzen Anzeichen oder Symptome einer Hämolyse auftreten, einschliesslich erhöhter LDH-Werte, sollte eine erneute Anwendung von Ravulizumab in Betracht gezogen werden.
Behandlungsabbruch bei aHUS
Es liegen keine spezifischen Daten zum Absetzen von Ravulizumab vor. In einer prospektiven Langzeit-Beobachtungsstudie führte das Absetzen der Behandlung mit dem Komplement-C5-Inhibitor (Eculizumab) zu einer 13,5-fach höheren Rate von TMA-Rezidiven und es bestand eine Tendenz zur Abnahme der Nierenfunktion im Vergleich zu Patienten, die die Behandlung fortsetzten.
Wenn Patienten die Behandlung mit Ravulizumab absetzen müssen, sollten sie fortlaufend engmaschig auf Anzeichen und Symptome einer TMA überwacht werden. Es ist jedoch möglich, dass eine Überwachung nicht ausreicht, um schwere TMA-Komplikationen vorherzusagen oder ihnen vorzubeugen.
Komplikationen durch eine TMA nach dem Absetzen der Behandlung lassen sich anhand einer der folgenden Beobachtungen identifizieren:
(i) Mindestens zwei der folgenden Laborbefunde liegen gleichzeitig vor: eine Abnahme der Thrombozytenzahl um mindestens 25 % im Vergleich zu entweder der Ausgangs- oder höchsten Thrombozytenzahl während der Ravulizumab-Behandlung; Anstieg des Serumkreatinins um mindestens 25 % im Vergleich zum Ausgangswert oder zum Tiefstwert während der Ravulizumab-Behandlung; oder Anstieg des Serum-LDH um mindestens 25 % im Vergleich zum Ausgangswert oder zum Tiefstwert während der Ravulizumab-Behandlung (die Ergebnisse sollten durch eine zweite Messung bestätigt werden) oder
(ii) eines der folgenden Symptome einer TMA: Veränderung des mentalen Zustandes oder Krampfanfälle oder andere extrarenale Manifestationen einer TMA, einschließlich kardiovaskulärer Anomalien, Perikarditis, gastrointestinale Symptome/Diarrhoe oder Thrombose.
Wenn nach dem Absetzen von Ravulizumab Komplikationen durch eine TMA auftreten, ist eine Wiederaufnahme der Ravulizumab-Behandlung mit der Initial- und Erhaltungsdosis in Erwägung zu ziehen (siehe Abschnitt "Dosierung/Anwendung" ).
Behandlungsabbruch bei gMG
Da es sich bei gMG um eine chronische Erkrankung handelt, sollten Patienten, die von einer Behandlung mit Ravulizumab profitieren und die Behandlung abbrechen, auf Symptome der Grunderkrankung überwacht werden. Wenn nach dem Absetzen gMG-Symptome auftreten, ist eine Wiederaufnahme der Behandlung mit Ravulizumab in Betracht zu ziehen.
Umstellung von Eculizumab zu Ravulizumab.
Bei Patienten mit gMG, die nicht auf das für Eculizumab zugelassene Dosierungsschema ansprechen, wird eine Behandlung mit Ravulizumab nicht empfohlen.
Behandlungsabbruch bei NMOSD
Die Anwendung von Ravulizumab zur Behandlung von NMOSD wurde nur im Rahmen einer dauerhaften Anwendung untersucht und die Wirkung des Absetzens von Ravulizumab wurde nicht beschrieben. Patienten, bei denen die Ultomiris-Behandlung abgesetzt wird, sollten sorgfältig auf Anzeichen eines möglichen NMOSD-Schubs überwacht werden.
Patienten mit Hämolytisch-Urämischem Syndrom im Zusammenhang mit E. coli Shiga-Toxin (STEC-HUS)
Es liegen keine Daten vor zur Anwendung von Ultomiris bei Patienten mit STEC-HUS.
Natriumgehalt
Ultomiris 300 mg/30 ml Konzentrat zur Herstellung einer Infusionslösung
Nach Verdünnung mit Natriumchlorid 9 mg/ml (0,9 %) Injektionslösung enthält die Höchstdosis dieses Arzneimittels 2,65 g Natrium pro 720 ml, entsprechend 133 % der von der WHO für einen Erwachsenen empfohlenen maximalen täglichen Natriumaufnahme mit der Nahrung von 2 g.
Ultomiris 300 mg/3 ml und 1100 mg/11 ml Konzentrat zur Herstellung einer Infusionslösung
Nach Verdünnung mit Natriumchlorid 9 mg/ml (0,9 %) Injektionslösung enthält die Höchstdosis dieses Arzneimittels 0,18 g Natrium pro 72 ml, entsprechend 9,1 % der von der WHO für einen Erwachsenen empfohlenen maximalen täglichen Natriumaufnahme mit der Nahrung von 2 g.
InteraktionenEs wurden keine Studien zur Erfassung von Wechselwirkungen durchgeführt.
Aufgrund der potenziellen Hemmwirkung von Ravulizumab auf die komplementabhängige Zytotoxizität von Rituximab kann Ravulizumab die erwarteten pharmakodynamischen Wirkungen von Rituximab mindern.
Siehe Abschnitt "Dosierung/Anwendung" für Hinweise im Falle einer gleichzeitigen PE, PP- oder IVIg-Behandlung.
Eine chronische Behandlung mit intravenösem humanem Immunglobulin (IVIg) kann den Recycling-Mechanismus des endosomalen neonatalen Fc-Rezeptors (FcRn) von monoklonalen Antikörpern, wie Ravulizumab, beeinträchtigen und dadurch die Ravulizumab-Konzentrationen im Serum verringern.
Schwangerschaft, StillzeitSchwangerschaft
Frauen im gebärfähigen Alter
Frauen im gebährfähigen Alter müssen während und bis zu 8 Monate nach der Behandlung eine zuverlässige Verhütungsmethode anwenden.
Schwangerschaft
Bisher liegen keine klinischen Daten zur Anwendung von Ravulizumab bei Schwangeren vor.
Es wurden keine nichtklinischen reproduktionstoxikologischen Studien mit Ravulizumab durchgeführt (siehe Abschnitt "Präklinische Daten" ). Es wurden reproduktionstoxikologische Studien an Mäusen mithilfe des murinen Surrogatmoleküls BB5.1 durchgeführt, in denen die Auswirkung der C5-Blockade auf das Reproduktionssystem bewertet wurde. In diesen Studien wurden keine spezifischen Testprodukt-bezogenen Reproduktionstoxizitäten nachgewiesen. Humanes IgG passiert bekanntlich die Plazentaschranke und demzufolge kann Ravulizumab potentiell eine terminale Komplementinhibition im fetalen Kreislauf verursachen. Es liegen keine ausreichenden tierexperimentellen Studien in Bezug auf eine Reproduktionstoxizität vor (siehe Abschnitt "Präklinische Daten" ).
Bei Schwangeren kann die Anwendung von Ravulizumab nach einer Nutzen-Risiko-Analyse in Betracht gezogen werden.
Stillzeit
Es ist nicht bekannt, ob Ravulizumab in die Muttermilch übergeht. Bei an Mäusen mithilfe des murinen Surrogatmoleküls BB5.1 durchgeführten nichtklinischen reproduktionstoxikologischen Studien wurden an Jungtieren keine unerwünschten Wirkungen festgestellt, die auf die Aufnahme von Milch von behandelten Muttertieren zurückzuführen wären.
Ein Risiko für den Säugling kann nicht ausgeschlossen werden.
Da viele Arzneimittel und Immunglobuline in die menschliche Muttermilch übergehen und bei gestillten Säuglingen das Potenzial für schwerwiegende unerwünschte Reaktionen besteht, sollte das Stillen während und bis 8 Monate nach der Behandlung mit Ravulizumab unterbrochen werden.
Fertilität
Es wurden keine spezifischen nicht-klinischen Studien zur Fertilität mit Ravulizumab durchgeführt.
Bei an Mäusen mithilfe eines murinen Surrogatmoleküls (BB5.1) durchgeführten nicht-klinischen reproduktionstoxikologischen Studien wurden keine unerwünschten Auswirkungen auf die Fertilität der behandelten Weibchen bzw. Männchen festgestellt.
Wirkung auf die Fahrtüchtigkeit und auf das Bedienen von MaschinenUltomiris hat keinen oder einen zu vernachlässigenden Einfluss auf die Verkehrstüchtigkeit und die Fähigkeit zum Bedienen von Maschinen.
Unerwünschte WirkungenZusammenfassung des Sicherheitsprofils
Das Sicherheitsprofil von Ultomiris basiert auf noch laufenden und abgeschlossenen klinischen Studien mit insgesamt 804 Patienten, die mit Ultomiris behandelt wurden. Die häufigsten Nebenwirkungen von Ravulizumab sind Kopfschmerz (28,2 %), Infektion der oberen Atemwege (19,9 %), Nasopharyngitis (19,5 %), Diarrhö (16,9 %), Fieber (16,4 %), Übelkeit (13,7 %), Arthralgie (13,2 %), Ermüdung/Fatigue (13,1 %), Rückenschmerzen (12,6 %), Abdominalschmerzen (11,8 %) und Schwindelgefühl (10,1 %). Die schwerwiegendsten Nebenwirkungen sind Meningokokkeninfektion (0,7 %) einschliesslich Meningokokken-Sepsis, Meningokokken-Enzephalitis und Meningokokkeninfektion (siehe Abschnitt "Warnhinweise und Vorsichtsmassnahmen" ) und disseminierte Gonokokkeninfektion (0,1 %).
Tabellarische Auflistung der Nebenwirkungen
In Tabelle 8 sind die Nebenwirkungen aus klinischen Studien sowie aus Beobachtungen nach Markteinführung aufgeführt.
Die Nebenwirkungen sind nach Systemorganklassen gemäss MedDRA-Datenbank und Häufigkeit gemäss der folgenden Konvention aufgeführt: sehr häufig (≥ 1/10); häufig (≥ 1/100, < 1/10); gelegentlich (≥ 1/1.000, < 1/100); selten (≥ 1/10.000, < 1/1.000); sehr selten (< 1/10.000) und nicht bekannt (Häufigkeit auf Grundlage der verfügbaren Daten nicht abschätzbar).
Innerhalb jeder Häufigkeitskategorie sind die Nebenwirkungen nach abnehmendem Schweregrad aufgeführt.
Tabelle 8: Nebenwirkungen aus klinischen Studien und nach Markteinführung
|
MedDRA-Systemorganklasse
|
Sehr häufig
(≥ 1/10)
|
Häufig
(≥ 1/100, < 1/10)
|
Gelegentlich (≥ 1/1.000, < 1/100)
| |
Infektionen und parasitäre Erkrankungen
|
Infektion der oberen Atemwege,
Nasopharyngitis
|
Harnwegsinfektion
|
Meningokokkeninfektion a,
disseminierte
Gonokokkeninfektion b
| |
Erkrankungen des Immunsystems
|
|
Überempfindlichkeit d
|
Anaphylaktische Reaktion c,
| |
Erkrankungen des Nervensystems
|
Kopfschmerz
Schwindelgefühl
|
|
| |
Erkrankungen des Gastrointestinaltrakts
|
Diarrhö, Übelkeit, Abdominalschmerz
|
Erbrechen, Dyspepsie
|
| |
Erkrankungen der Haut und des Unterhautgewebes
|
|
Urtikaria, Pruritus, Ausschlag,
|
| |
Skelettmuskulatur-, Bindegewebs- und Knochenerkrankungen
|
Rückenschmerzen, Arthralgie
|
Myalgie,
Muskelspasmen
|
| |
Allgemeine Erkrankungen und Beschwerden am Verabreichungsort
|
Fieber, Ermüdung/Fatigue
|
Grippeähnliche Erkrankung, Schüttelfrost, Abgeschlagenheit
|
| |
Verletzung, Vergiftung und durch Eingriffe bedingte Komplikationen
|
|
Reaktion im Zusammenhang mit einer Infusion
|
|
a Meningokokkeninfektion umfasst die bevorzugten Begriffe [Preferred Terms (PT)] Meningokokkeninfektion, Meningokokken-Sepsis und Meningokokken-Enzephalitis
b Gonokokkeninfektion umfasst disseminierte Gonokokkeninfektion
c Schätzungen auf der Grundlage von Erfahrungen nach der Markteinführung
d Überempfindlichkeit ist ein Sammelbegriff für den bevorzugten Begriff Arzneimittelüberempfindlichkeit mit verbundener Kausalität und den bevorzugten Begriff Überempfindlichkeit
Beschreibung ausgewählter Nebenwirkungen
Meningokokkeninfektion/-Sepsis/Enzephalitis
Die Impfung verringert zwar das Risiko von Meningokokkeninfektionen, schliesst es jedoch nicht vollständig aus. In klinischen Studien entwickelten < 1% der Patienten während der Behandlung mit Ravulizumab schwere Meningokokkeninfektionen. Alle waren erwachsene und geimpfte Patienten mit PNH oder NMOSD. Beachten Sie den Abschnitt "Warnhinweise und Vorsichtsmassnahmen" bzgl. Informationen zur Vorbeugung und zur Behandlung bei Verdacht auf Meningokokkeninfektion. Meningokokkeninfektionen zeigten sich bei mit Ravulizumab-behandelten Patienten als Meningokokken-Sepsis und -Enzephalitis. Die Patienten sollten über die Anzeichen und Symptome einer Meningokokken-Septikämie sowie über die Notwendigkeit einer unverzüglichen ärztlichen Behandlung informiert werden.
Immunogenität
Die Behandlung mit jedem therapeutischen Protein kann eine Immunreaktion induzieren. In Studien mit erwachsenen PNH-Patienten (N = 475), in einer Studie an Kindern und Jugendlichen mit PNH (N = 13), in Studien bei aHUS (N = 89), in einer Studie bei gMG (N=86) und in einer Studie bei NMOSD (N = 58) wurde im Zusammenhang mit Ravulizumab nur über zwei Fälle (0,3 %) mit Bildung von therapiebedingten Anti-Wirkstoff-Antikörpern berichtet (1 erwachsener PNH-Patient und 1 erwachsener aHUS-Patient). Diese Anti-Wirkstoff-Antikörper waren transient und niedrig-titrig und korrelierten nicht mit dem klinischen Ansprechen oder unerwünschten Ereignissen.
Kinder und Jugendliche
Paroxysmale nächtliche Hämoglobinurie (PNH)
Bei den in die pädiatrische PNH-Studie (ALXN1210 PNH 304) aufgenommenen Kindern und Jugendlichen mit PNH (N = 13, im Alter von 9 bis 17 Jahren) schien das Sicherheitsprofil dem bei erwachsenen PNH-Patienten ähnlich zu sein. Die häufigsten Nebenwirkungen, die bei Kindern und Jugendlichen mit PNH gemeldet wurden, waren Abdominalschmerz, Übelkeit, Nasopharyngitis und Kopfschmerz, die bei 3 Patienten auftraten (23.1 %).
Atypisches hämolytisch-urämisches Syndrom (aHUS)
Bei Kindern und Jugendlichen mit Anzeichen eines aHUS (N = 34) (im Alter von 10 Monaten bis unter 18 Jahren), die an der Studie ALXN1210_aHUS_312 teilnahmen, schien das Sicherheitsprofil von Ravulizumab ähnlich zu sein wie das von erwachsenen Patienten mit Anzeichen eines aHUS. Die Sicherheitsprofile in den verschiedenen pädiatrischen Alters-Untergruppen scheinen vergleichbar zu sein. Die Sicherheitsdaten für Patienten unter 2 Jahren beschränken sich auf vier Patienten. Die häufigste bei pädiatrischen Patienten gemeldete Nebenwirkung war Fieber (32.3%).
Generalisierte Myasthenia gravis (gMG)
Ravulizumab wurde nicht an Kindern und Jugendlichen mit gMG untersucht.
Neuromyelitis-optica-Spektrum-Erkrankungen (NMOSD)
Ravulizumab wurde nicht an Kindern und Jugendlichen mit NMOSD untersucht.
Meldung des Verdachts auf Nebenwirkungen
Die Meldung des Verdachts auf Nebenwirkungen nach der Zulassung ist von grosser Wichtigkeit. Sie ermöglicht eine kontinuierliche Überwachung des Nutzen-Risiko-Verhältnisses des Arzneimittels. Angehörige von Gesundheitsberufen sind aufgefordert, jeden Verdacht einer neuen oder schwerwiegenden Nebenwirkung über das Online-Portal ElViS (Electronic Vigilance System) anzuzeigen. Informationen dazu finden Sie unter www.swissmedic.ch.
ÜberdosierungEs wurden keine Fälle von Überdosierung berichtet.Bei Patienten, bei denen es zu einer Überdosierung kommt, muss die Infusion sofort unterbrochen werden und eine engmaschige Überwachung erfolgen. erfolgen.
Eigenschaften/WirkungenATC-Code
Pharmakotherapeutische Gruppe: Immunsuppressiva, selektive Immunsuppressiva, ATC-Code: L04AA43
Wirkungsmechanismus
Ravulizumab ist ein monoklonaler IgG2/4K-Antikörper, der spezifisch an das Komplementprotein C5 bindet und dadurch dessen Spaltung in C5a (das proinflammatorische Anaphylatoxin) und C5b (die initiierende Untereinheit des Membranangriffskomplexes [MAC oder C5b-9]) hemmt und die Bildung des C5b-9 verhindert. Ravulizumab erhält die frühen Komponenten der Komplementaktivierung, die von wesentlicher Bedeutung für die Opsonisierung von Mikroorganismen und die Elimination (Clearance) von Immunkomplexen sind.
Pharmakodynamik
Nach der Ravulizumab-Behandlung wurde sowohl bei zuvor nicht mit Komplementinhibitoren behandelten erwachsenen und pädiatrischen Patienten mit PNH als auch bei mit Eculizumab vorbehandelten Patienten mit PNH in Phase-3-Studien eine sofortige, vollständige und anhaltende Hemmung von freiem Serum-C5 (Konzentration von < 0,5 µg/ml) am Ende der ersten Infusion beobachtet und über den gesamten 26-wöchigen Behandlungszeitraum aufrechterhalten, und zwar bei allen Patienten. Eine sofortige und vollständige Hemmung von freiem C5 im Serum wurde auch bei erwachsenen und pädiatrischen Patienten mit aHUS, bei erwachsenen Patienten mit gMG und bei erwachsenen Patienten mit NMOSD am Ende der ersten Infusion und während des 26wöchigen Behandlungszeitraums beobachtet.
Umfang und Dauer des pharmakodynamischen Ansprechens bei Patienten mit PNH, aHUS, gMG oder NMOSD waren bei Ravulizumab expositionsabhängig. Konzentrationen von freiem C5 von weniger als 0,5 µg/ml korrelierten mit einer maximalen intravasalen Hämolysekontrolle und einer vollständigen Hemmung des terminalen Komplements. Bei gMG führt die Aktivierung des terminalen Komplements zu MAC-Ablagerungen an den neuromuskulären Verbindungsstellen und zu einer Beeinträchtigung der neuromuskulären Übertragung. Bei NMOSD führt eine durch Autoantikörper gegen AQP4 verursachte unkontrollierte Aktivierung des terminalen Komplements zur Entstehung der MAC- und C5a-abhängigen Entzündung, die Astrozytennekrose und eine erhöhte Durchlässigkeit der Blut-Hirn-Schranke sowie die Schädigung der umgebenden Gliazellen und Neuronen zur Folge hat.
Klinische Wirksamkeit
Paroxysmale nächtliche Hämoglobinurie (PNH)
Die Sicherheit und Wirksamkeit von Ravulizumab bei erwachsenen Patienten mit PNH wurden in zwei offenen, randomisierten, aktiv kontrollierten Phase-3-Studien untersucht:
- einer Studie mit erwachsenen Patienten mit PNH, die zuvor nicht mit Komplementinhibitoren behandelt worden waren,
- einer Studie mit erwachsenen Patienten mit PNH, die klinisch stabil waren, nachdem sie mindestens in den 6 Monaten zuvor mit Eculizumab behandelt worden waren.
Ravulizumab wurde gemäss dem empfohlenen, in Abschnitt "Dosierung/Anwendung" beschriebenen Dosierungsschema (4 Infusionen von Ravulizumab über 26 Wochen) angewendet, während Eculizumab gemäss dem zugelassenen Dosierungsschema von Eculizumab 600 mg wöchentlich in den ersten 4 Wochen und 900 mg alle 2 Wochen (15 Infusionen über 26 Wochen) verabreicht wurde.
Die Patienten wurden vor bzw. zu Beginn der Behandlung mit Ravulizumab bzw. Eculizumab gegen Meningokokkeninfektion geimpft oder erhielten bis 2 Wochen nach der Impfung eine prophylaktische Behandlung mit entsprechenden Antibiotika.
Zwischen der Ravulizumab- und der Eculizumab-Behandlungsgruppe bestanden in keiner der beiden Phase-3-Studien nennenswerte Unterschiede bei den demografischen bzw. bei Studienbeginn vorliegenden Merkmalen. Die 12-monatige Transfusionshistorie war in beiden Phase-3-Studien in der Ravulizumab- und der Eculizumab-Behandlungsgruppe ähnlich.
Studie mit erwachsenen PNH-Patienten, die zuvor nicht mit Komplementinhibitoren behandelt worden waren (ALXN1210-PNH-301)
Die Studie mit zuvor nicht mit Komplementinhibitoren behandelten Patienten war eine 26-wöchige, multizentrische, offene, randomisierte, aktiv kontrollierte Phase-3-Studie, die mit 246 Patienten durchgeführt wurde, die vor Studieneintritt nicht mit Komplementinhibitoren behandelt worden waren, gefolgt von einer Langzeit-Verlängerungsphase, in der alle Patienten Ravulizumab erhielten. Geeignete Patienten für diese Studie mussten eine hohe Krankheitsaktivität, definiert als LDH-Wert ≥ 1,5 × ULN (Upper Limit of Normal/oberer Grenzwert) beim Screening sowie das Vorhandensein von einem oder mehreren der folgenden PNH-bedingten Anzeichen oder Symptome innerhalb von 3 Monaten vor dem Screening aufweisen: Ermüdung/Fatigue, Hämoglobinurie, Abdominalschmerz, Kurzatmigkeit (Dyspnoe), Anämie (Hämoglobin < 10 g/dl), ein zurückliegendes schwerwiegendes unerwünschtes vaskuläres Ereignis (einschliesslich Thrombose), Dysphagie oder Erektionsstörung; oder eine zurückliegende PNH-bedingte Transfusion von Erythrozytenkonzentraten.
Mehr als 80 % der Patienten in beiden Behandlungsgruppen hatten innerhalb von 12 Monaten vor Studieneintritt eine Transfusion erhalten. Die Mehrheit der Studienpopulation aus der Studie mit zuvor nicht mit Komplementinhibitoren behandelten Patienten war zu Studienbeginn stark hämolytisch; 86,2 % der eingeschlossenen Patienten wiesen im Zusammenhang mit PNH einen erhöhten LDH-Wert ≥ 3 × ULN auf, was ein direktes Mass für die intravaskuläre Hämolyse darstellt.
Die Tabelle 9 zeigt die Merkmale der PNH-Patienten, die in die Studie mit zuvor nicht mit Komplementinhibitoren behandelten Patienten aufgenommen wurden, bei Studienbeginn; zwischen den Behandlungsarmen wurden keine offensichtlichen, klinisch bedeutsamen Unterschiede beobachtet.
Tabelle 9: Merkmale bei Studienbeginn in der Studie bei zuvor nicht mit Komplementinhibitoren behandelten Patienten
|
Parameter
|
Statistik
|
Ravulizumab
(N = 125)
|
Eculizumab
(N = 121)
| |
Alter (in Jahren) bei PNH-Diagnose
|
Mittelwert (SD)
Median
Min.; Max.
|
37,9 (14,90)
34,0
15; 81
|
39,6 (16,65)
36,5
13; 82
| |
Alter (in Jahren) bei der ersten Infusion in der Studie
|
Mittelwert (SD)
Median
Min.; Max.
|
44,8 (15,16)
43,0
18; 83
|
46,2 (16,24)
45,0
18; 86
| |
Geschlecht (n, %)
|
männlich
weiblich
|
65 (52,0)
60 (48,0)
|
69 (57,0)
52 (43,0)
| |
LDH-Werte vor der Behandlung
|
Mittelwert (SD)
|
1633,5 (778,75)
|
1578,3 (727,06)
| |
Median
|
1513,5
|
1445,0
| |
Anzahl Patienten mit Transfusionen von Erythrozytenkonzentraten in den 12 Monaten vor der ersten Dosis
|
n (%)
|
103 (82,4)
|
100 (82,6)
| |
Einheiten von in den 12 Monaten vor der ersten Dosis transfundierten Erythrozytenkonzentraten
|
Gesamtwert
|
925
|
861
| |
Mittelwert (SD)
|
9,0 (7,74)
|
8,6 (7,90)
| |
Median
|
6,0
|
6,0
| |
Gesamt-PNH-Erythrozyten-Klongrösse
|
Median
|
33,6
|
34,2
| |
Gesamt-PNH-Granulozyten-Klongrösse
|
Median
|
93,8
|
92,4
| |
Patienten mit PNH-bedingten Symptomen und Erkrankungena vor Studienbeginn
|
n (%)
|
121 (96,8)
|
120 (99,2)
| |
Anämie
|
|
103 (82,4)
|
105 (86,8)
| |
Hämaturie oder Hämoglobinurie
|
|
81 (64,8)
|
75 (62,0)
| |
Aplastische Anämie
|
|
41 (32,8)
|
38 (31,4)
| |
Niereninsuffizienz
|
|
19 (15,2)
|
11 (9,1)
| |
Myelodysplastisches Syndrom
|
|
7 (5,6)
|
6 (5,0)
| |
Schwangerschaftskomplikation
|
|
3 (2,4)
|
4 (3,3)
| |
Sonstigeb
|
|
27 (21,6)
|
13 (10,7)
|
a Basierend auf Krankengeschichte.
b „Sonstige“ wie auf dem Prüfbogen angegeben beinhaltete Thrombozytopenie, chronische Nierenerkrankung und Panzytopenie sowie eine Reihe weiterer Symptome und Erkrankungen.
Die koprimären Endpunkte waren Transfusionsvermeidung und Hämolyse, direkt gemessen an der Normalisierung der LDH-Werte (LDH-Werte ≤ 1 × ULN; der ULN für LDH ist 246 U/l). Die wichtigen sekundären Endpunkte umfassten die prozentuale Veränderung der LDH-Werte von der Baseline, die Veränderung der Lebensqualität (FACIT-Fatigue-Score), den Anteil an Patienten mit Durchbruchhämolyse und den Anteil an Patienten mit stabilisiertem Hämoglobinspiegel.
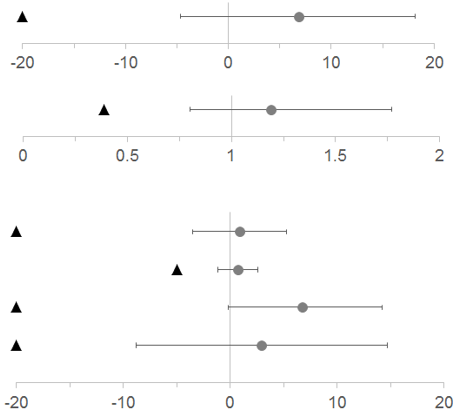
Bei beiden koprimären Endpunkten, Vermeidung der Transfusion von Erythrozytenkonzentraten gemäss den im Prüfplan spezifizierten Richtlinien und LDH-Normalisierung von Tag 29 bis Tag 183, und bei allen vier wichtigen sekundären Endpunkten war Ravulizumab im Vergleich zu Eculizumab nicht unterlegen (Abbildung 1).
Abbildung 1: Analyse der koprimären und sekundären Endpunkte – Full Analysis Set (Studie mit zuvor nicht mit Komplementinhibitoren behandelten Patienten)
|
|
|
Ravulizumab
(N = 125)
|
Eculizumab
(N = 121)
|
Differenz (95 %-KI)
| |
Transfusionsvermeidung (%)
|
|
|
73,6
|
66,1
|
6,8 (-4,7; 18,1)
| |
|
|
|
| |
LDH-Normalisierung
|
|
|
Odds Ratio (95 %-KI)
| |
(Odds Ratio)
|
53,6
|
49,4
|
1,19 (0,80; 1,77)
| |
|
|
|
| |
|
|
|
Differenz (95 %-KI)
| |
LDH-Veränderung gegenüber Baseline (%)
|
-76,8
|
-76,0
|
0,8 (-3,6; 5,2)
| |
Veränderung des FACIT-Fatigue-Scores
|
7,1
|
6,4
|
0,7 (-1,2; 2,6)
| |
Durchbruchhämolyse (%)
|
4,0
|
10,7
|
6,7 (-0,2; 14,2)
| |
Hämoglobin-Stabilisierung (%)
|
68,0
|
64,5
|
2,9 (-8,8; 14,6)
| |
|
|
|
| |
|
Zugunsten von Eculizumab
|
Zugunsten von Ravulizumab
|
|
|
|
Hinweis: Schwarze Dreiecke zeigen die Nichtunterlegenheitsgrenzen an, graue Punkte zeigen Punktschätzungen an.
Hinweis: LDH = Lactat-Dehydrogenase; KI = Konfidenzintervall; FACIT = Functional Assessment of Chronic Illness Therapy.
Die abschliessende Wirksamkeitsanalyse der Studie bezog alle Patienten ein, die jemals mit Ravulizumab (n = 244) behandelt worden waren und die mediane Behandlungsdauer betrug 1423 Tage. Die abschliessende Analyse bestätigte, dass das im primären Auswertungszeitraum beobachtete Ansprechen auf die Ravulizumab-Behandlung über die gesamte Studiendauer hinweg anhielt.
Studie mit erwachsenen PNH-Patienten, die zuvor mit Eculizumab behandelt wurden (ALXN1210-PNH-302)
Die Studie bei zuvor mit Eculizumab behandelten Patienten war eine 26-wöchige, multizentrische, offene, randomisierte, aktiv kontrollierte Phase-3-Studie, die mit 195 PNH-Patienten, die klinisch stabil waren (LDH ≤ 1,5 x ULN), nachdem sie mindestens in den 6 Monaten zuvor mit Eculizumab behandelt worden waren, durchgeführt wurde, gefolgt von einer Langzeit-Verlängerungsphase, in der alle Patienten Ravulizumab erhielten.
Die Krankengeschichte in Bezug auf PNH war in der Ravulizumab- und der Eculizumab-Behandlungsgruppe ähnlich. Die 12-monatige Transfusionshistorie war in der Ravulizumab- und der Eculizumab-Behandlungsgruppe ähnlich, und mehr als 87 % der Patienten in beiden Behandlungsgruppen hatten innerhalb von 12 Monaten vor Studieneintritt keine Transfusion erhalten. Die mittlere Gesamt-PNH-Erythrozyten-Klongrösse betrug 60,05 %, die mittlere Gesamt-PNH-Granulozyten-Klongrösse betrug 83,30 % und die mittlere Gesamt-PNH-Monozyten-Klongrösse betrug 85,86 %.
Die Tabelle 10 zeigt die Merkmale bei Studienbeginn der PNH-Patienten, die in die Studie bei zuvor mit Eculizumab behandelten Patienten aufgenommen wurden; zwischen den Behandlungsarmen wurden keine offensichtlichen, klinisch bedeutsamen Unterschiede beobachtet.
Tabelle 10: Merkmale bei Studienbeginn in der Studie bei zuvor mit Eculizumab behandelten Patienten
|
Parameter
|
Statistik
|
Ravulizumab
(N = 97)
|
Eculizumab
(N = 98)
| |
Alter (in Jahren) bei PNH-Diagnose
|
Mittelwert (SD)
Median
Min., Max.
|
34,1 (14,41)
32,0
6, 73
|
36,8 (14,14)
35,0
11, 74
| |
Alter (in Jahren) bei der ersten Infusion in der Studie
|
Mittelwert (SD)
Median
Min., Max.
|
46,6 (14,41)
45,0
18, 79
|
48,8 (13,97)
49,0
23, 77
| |
Geschlecht (n, %)
|
männlich
weiblich
|
50 (51,5)
47 (48,5)
|
48 (49,0)
50 (51,0)
| |
LDH-Werte vor der Behandlung
|
Mittelwert (SD)
|
228,0 (48,71)
|
235,2 (49,71)
| |
Median
|
224,0
|
234,0
| |
Anzahl Patienten mit Transfusionen von Erythrozytenkonzentraten/Vollblut in den 12 Monaten vor der ersten Dosis
|
n (%)
|
13 (13,4)
|
12 (12,2)
| |
Einheiten von in den 12 Monaten vor der ersten Dosis transfundiertem Erythrozytenkonzentrat/Vollblut
|
Gesamtwert
|
103
|
50
| |
Mittelwert (SD)
|
7,9 (8,78)
|
4,2 (3,83)
| |
Median
|
4,0
|
2,5
| |
Patienten mit PNH-bedingten Symptomen und Erkrankungena vor Studienbeginn
|
n (%)
|
90 (92,8)
|
96 (98,0)
| |
Anämie
|
|
64 (66,0)
|
67 (68,4)
| |
Hämaturie oder Hämoglobinurie
|
|
47 (48,5)
|
48 (49,0)
| |
Aplastische Anämie
|
|
34 (35,1)
|
39 (39,8)
| |
Niereninsuffizienz
|
|
11 (11,3)
|
7 (7,1)
| |
Myelodysplastisches Syndrom
|
|
3 (3,1)
|
6 (6,1)
| |
Schwangerschaftskomplikation
|
|
4 (4,1)
|
9 (9,2)
| |
Sonstigeb
|
|
14 (14,4)
|
14 (14,3)
|
a Basierend auf Krankengeschichte.
b Die Kategorie „Sonstige“ umfasste Neutropenie, Nierenfunktionsbeeinträchtigung und Thrombopenie sowie eine Reihe weiterer Symptome und Erkrankungen.
Der primäre Endpunkt war Hämolyse, gemessen an der prozentualen Veränderung der LDH-Werte gegenüber Baseline. Die sekundären Endpunkte umfassten den Anteil an Patienten mit Durchbruchhämolyse, die Lebensqualität (FACIT-Fatigue-Score), die Transfusionsvermeidung und den Anteil an Patienten mit stabilisiertem Hämoglobinspiegel.
In Hinblick auf den primären Endpunkt, die prozentuale Veränderung der LDH-Konzentration von Baseline bis Tag 183, und bei allen vier wichtigen sekundären Endpunkten war Ravulizumab im Vergleich zu Eculizumab nicht unterlegen (Abbildung 2).
Abbildung 2: Analyse des primären und der sekundären Endpunkte – Full Analysis Set (Studie bei zuvor mit Eculizumab behandelten Patienten)
|
|
|
Ravulizumab
(N=97)
|
Eculizumab
(N = 98)
|
Differenz (95 %-KI)
| |
|
|

|
|
|
| |
LDH-Veränderung gegenüber Baseline (%)
|
-0,8
|
8,4
|
9,2 (-0,4; 18,8)
| |
|
|
|
| |
Durchbruchhämolyse (%)
|
0
|
5,1
|
5,1 (-8,9; 19,0)
| |
Veränderung des FACIT-Fatigue-Scores
|
2,0
|
0,5
|
1,5 (-0,2; 3,2)
| |
|
|
|
| |
Transfusionsvermeidung (%)
|
87,6
|
82,7
|
5,5 (-4,3; 15,7)
| |
Hämoglobin-Stabilisierung (%)
|
76,3
|
75,5
|
1,4 (-10,4; 13,3)
| |
|
|
|
| |
|
|
|
| |
|
Zugunsten von Eculizumab
|
Zugunsten von Ravulizumab
|
|
|
|
Hinweis: Schwarze Dreiecke zeigen die Nichtunterlegenheitsgrenzen an, graue Punkte zeigen Punktschätzungen an.
Hinweis: LDH = Lactat-Dehydrogenase; KI = Konfidenzintervall.
Die abschliessende Wirksamkeitsanalyse der Studie bezog alle Patienten ein, die jemals mit Ravulizumab behandelt worden waren (n = 192) und eine mittlere Behandlungsdauer von 968 Tagen hatten. Die abschliessende Analyse bestätigte, dass das im primären Auswertungszeitraum beobachtete Ansprechen auf die Ravulizumab-Behandlung über die gesamte Studiendauer hinweg anhielt.
Atypisches hämolytisch-urämischen Syndrom (aHUS)
Studie an erwachsenen Patienten mit aHUS (ALXN1210-aHUS-311)
Die Studie an Erwachsenen war eine multizentrische, einarmige klinische Phase-3-Studie bei Patienten mit dokumentiertem aHUS, die vor dem Eintritt in diese Studie noch keine Behandlung mit einem Komplement-Inhibitor erhalten hatten und Anzeichen einer thrombotischen Mikroangiopathie (TMA) aufwiesen. Die Studie bestand aus einem 26wöchigen Zeitraum für die Erstbeurteilung und die Patienten hatten die Möglichkeit, an einem Verlängerungszeitraum von bis zu 4,5 Jahren teilzunehmen.
Es wurden insgesamt 58 Patienten mit dokumentiertem aHUS aufgenommen. Die Einschlusskriterien schlossen Patienten aus, die mit TMA infolge thrombotischer thrombozytopenischer Purpura (TTP) bzw. hämolytisch-urämischem Syndrom in Zusammenhang mit dem Shiga-Toxin von Escherichia coli (STEC HUS) vorstellig wurden. Zwei Patienten wurden aufgrund der bestätigten Diagnose eines STEC HUS aus dem vollständigen Analyseset ausgeschlossen. Zu Studienbeginn zeigten 93 % der Patienten extrarenale (kardiovaskuläre, pulmonale, zentralnervöse, gastrointestinale, die Haut oder Skelettmuskulatur betreffende) Anzeichen oder Symptome eines aHUS.
Tabelle 11 zeigt die demographischen Merkmale und Ausgangsmerkmale von 56 erwachsenen Patienten, die in Studie ALXN1210aHUS-311 aufgenommen wurden und das vollständige Analyseset bildeten.
Tabelle 11: Ausgangsmerkmale in der Studie an Erwachsenen
|
Parameter
|
Statistik
|
Ravulizumab
(N = 56)
| |
Alter bei Erstinfusion (Jahre)
|
Mittel (SD)
Min., Max.
|
42,2 (14,98)
19,5; 76,6
| |
Geschlecht
Männlich
|
n (%)
|
19 (33,9)
| |
Ethnie a
Asiatisch
Weißhäutig
Sonstige
|
n (%)
|
15 (26,8)
29 (51,8)
12 (21,4)
| |
Transplantation in der Vorgeschichte
|
n (%)
|
8 (14,3)
| |
Thrombozyten (109/l) im Blut
|
n
Median (Min., Max.)
|
56
95,25 (18; 473)
| |
Hämoglobin (g/l) im Blut
|
n
Median (Min., Max.)
|
56
85,00 (60,5; 140)
| |
LDH (U/l) im Serum
|
n
Median (Min., Max.)
|
56
508,00 (229,5; 3249)
| |
eGFR (ml/min/1,73 m2)
|
n (%)
Median (Min., Max.)
|
55
10,00 (4; 80)
| |
Dialyse-Patienten
|
N (%)
|
29 (51,8)
| |
Patientinnen post partum
|
N (%)
|
8 (14,3)
|
Hinweis: Die Prozentangaben basieren auf der Gesamtzahl von Patienten.
Abkürzungen: eGFR = geschätzte glomeruläre Filtrationsrate; LDH = Laktatdehydrogenase; Max. = Maximum; Min. = Minimum.
Der primäre Endpunkt war das vollständige Ansprechen der TMA während des 26wöchigen Zeitraums für die Erstbeurteilung, belegt durch eine Normalisierung der hämatologischen Parameter (Thrombozytenzahl ≥ 150 x 109/l und LDH ≤ 246 U/l) und eine Verbesserung des Serumkreatinins um ≥ 25 % gegenüber dem Ausgangswert. Die Patienten mussten jedes Kriterium für ein vollständiges Ansprechen der TMA bei 2 verschiedenen Beurteilungen im Abstand von mindestens 4 Wochen (28 Tagen) und bei jeder zwischenzeitlichen Messung erfüllen.
Ein vollständiges Ansprechen der TMA wurde bei 30 der 56 Patienten (53,6 %) während des 26wöchigen Zeitraums für die Erstbeurteilung beobachtet, wie es in Tabelle 12 gezeigt ist.
Tabelle 12: Analyse des vollständigen Ansprechens der TMA und der Komponenten des vollständigen Ansprechens der TMA während des 26wöchigen Zeitraums für die Erstbeurteilung (ALXN1210aHUS-311)
|
|
Summe
|
Responder
| |
n
|
Anteil (95 %-KI)a
| |
Vollständiges Ansprechen der TMA
|
56
|
30
|
0,536 (0,396; 0,675)
| |
Komponenten des vollständigen Ansprechens der TMA
|
|
|
| |
Normalisierung der Thrombozytenzahl
|
56
|
47
|
0,839 (0,734; 0,944)
| |
Normalisierung der LDH
|
56
|
43
|
0,768 (0,648; 0,887)
| |
≥25 %ige Verbesserung des Serumkreatinins gegenüber dem Ausgangswert
|
56
|
33
|
0,589 (0,452; 0,727)
| |
Normalisierung der Blutwerte
|
56
|
41
|
0,732 (0,607; 0,857)
|
a Die 95 %-KI für den Anteil basierten auf der asymptotischen Gaußschen Approximationsmethode mit Kontinuitätskorrektur.
Abkürzungen: KI = Konfidenzintervall; LDH = Laktatdehydrogenase; TMA = thrombotische Mikroangiopathie.
Vier weitere Patienten zeigten ein vollständiges Ansprechen der TMA, das nach dem 26wöchigen Zeitraum für die Erstbeurteilung bestätigt wurde (das vollständige Ansprechen der TMA wurde an Tag 169, 302, 401 und 407 festgestellt). Somit zeigten insgesamt 34 von 56 Patienten ein vollständiges Ansprechen der TMA (60,7 %; 95 %-KI: 47,0 %, 74,4 %). Die Zahl des Ansprechens einzelner Komponenten erhöhte sich auf 48 Patienten (85,7 %; 95 %-KI: 75,7 %, 95,8 %) bei der Normalisierung der Thrombozytenzahl, auf 47 Patienten (83,9 %; 95 %-KI: 73,4 %, 94,4%) bei der Normalisierung der LDH und auf 35 Patienten (62,5 %; 95 %-KI: 48,9 %, 76,1 %) bei der Besserung der Nierenfunktion.
Ein vollständiges Ansprechen der TMA wurde innerhalb eines medianen Zeitraums von 86 Tagen (7 bis 169 Tage) erzielt. Eine Zunahme der durchschnittlichen Thrombozytenzahl wurde bald nach Behandlungsbeginn mit Ravulizumab beobachtet, wobei ein Anstieg von 118,52 × 109/l zu Studienbeginn auf 240,34 × 109/l an Tag 8 festgestellt wurde. Der Wert blieb bei allen anschliessenden Besuchsterminen während des Zeitraums für die Erstbeurteilung (26 Wochen) über 227 × 109/l. Ebenso sank der mittlere LDH-Wert während der ersten 2 Behandlungsmonate gegenüber dem Ausgangswert und blieb für die Dauer des Erstbeurteilungszeitraums (26 Wochen) erhalten.
Von den Patienten, die mit einer chronischen Nierenerkrankung in Stadium 5 vorstellig wurden, zeigten 67,6 % (23/34) eine Besserung der chronischen Nierenerkrankung um 1 oder mehrere Stadien. Das Stadium der chronischen Nierenerkrankung besserte sich weiterhin bei vielen Patienten (19/30), nachdem während des 26wöchigen Zeitraums für die Erstbeurteilung ein vollständiges Ansprechen der TMA erreicht wurde. Von den 29 dialysepflichtigen Patienten bei Eintritt in die Studie konnten 17 die Dialysebehandlung bis zum Ende des verfügbaren Nachbeobachtungszeitraums absetzen, während 6 von 27 Patienten, die zu Studienbeginn keine Dialysebehandlung erhielten, bei der letzten verfügbaren Nachuntersuchung eine Dialysebehandlung bekamen. Tabelle 13 fasst die sekundären Wirksamkeitsergebnisse von Studie ALXN1210aHUS-311 zusammen.
Tabelle 13: Sekundäres Wirksamkeitsergebnis für Studie ALXN1210aHUS-311
|
Parameter
|
Studie ALXN1210aHUS-311
(N = 56)
| |
Hämatologische Parameter bei TMA, Tag 183
Thrombozyten (109/L) im Blut
Mittelwert (SD)
Median
LDH (U/l) im Serum
Mittelwert (SD)
Median
|
Beobachteter Wert (n = 48)
237,96 (73,528)
232,00
194,46 (58,099)
176,50
|
Veränderung gegenüber dem Ausgangswert (n = 48)
114,79 (105,568)
125,00
-519,83 (572,467)
-310,75
| |
Anstieg des Hämoglobins um ≥ 20 g/l gegenüber dem Ausgangswert mit einem bestätigenden Ergebnis bis zum Ende des Zeitraums für die Erstbeurteilung
m/n
Anteil (95 %-KI)**
|
40/56
0,714 (0,587; 0,842)
| |
Veränderung des CKD-Stadiums gegenüber dem Ausgangswert, Tag 183
Verbesserunga
m/n
Anteil (95 %-KI)*
Verschlechterungb
m/n
Anteil (95 %-KI)*
|
32/47
0,681 (0,529; 0,809)
2/13
0,154 (0,019; 0,454)
| |
eGFR (ml/min/1,73 m2), Tag 183
Mittelwert (SD)
Median
|
Beobachteter Wert (n = 48)
51,83 (39,162)
40,00
|
Veränderung gegenüber dem Ausgangswert (n = 47)
34,80 (35,454)
29,00
|
Hinweis: n: Anzahl von Patienten mit verfügbaren Daten für eine bestimmte Untersuchung bei dem Besuchstermin an Tag 183. m: Anzahl von Patienten, die ein bestimmtes Kriterium erfüllen. Das Stadium der chronischen Nierenerkrankung (CKD) wird anhand der Klassifikation der National Kidney Foundation für Stadien der chronischen Nierenerkrankung (Chronic Kidney Disease Stage) bestimmt. Stadium 5 gilt als schlechteste Kategorie, während Stadium 1 die beste Kategorie ist. Der Ausgangswert wird anhand der letzten verfügbaren eGFR vor Behandlungsbeginn ermittelt. Verbesserung/Verschlechterung: im Vergleich zum CKD-Stadium zu Studienbeginn. *Die 95 %-Konfidenzintervalle (95 %-KI) basieren auf dem exakten Clopper-Pearson-Konfidenzintervall. aSchließt Patienten mit CKD- Stadium 1 zu Studienbeginn aus, weil bei ihnen keine Besserung möglich ist. bSchließt Patienten mit Stadium 5 zu Studienbeginn aus, da bei ihnen keine Verschlechterung möglich ist.
Abkürzungen: eGFR = geschätzte glomeruläre Filtrationsrate;; LDH = Laktatdehydrogenase; TMA = thrombotische Mikroangiopathie.
Generalisierte Myasthenia gravis (gMG)
Studie an erwachsenen Patienten mit gMG
Die Wirksamkeit und Sicherheit von Ravulizumab bei erwachsenen Patienten mit gMG wurden in einer randomisierten, doppelblinden, placebokontrollierten Multizenterstudie der Phase III (ALXN1210-MG-306) untersucht. Die an dieser Studie teilnehmenden Patienten konnten anschliessend in eine nicht verblindete-Verlängerungsphase überführt werden, in der alle Patienten Ravulizumab erhielten.
Patienten mit gMG (Diagnosestellung vor mindestens 6 Monaten) und positivem Serumtest auf Azetylcholinrezeptor (AchR)-Antikörper, klinischer Klassifikationsklasse II bis IV gemäss MGFA (Myasthenia Gravis Foundation of America) und einer Restsymptomatik, die durch einen Myasthenia Gravis Activities of Daily Living (MG-ADL) Gesamtscore ≥ 6 belegt wurde, wurden zu einer Behandlung mit entweder Ravulizumab (N = 86) oder Placebo (N = 89) randomisiert. Patienten mit immunsupprimierenden Therapien (Kortikosteroide, Azathioprin, Cyclophosphamid, Cyclosporin, Methotrexat, Mycophenolatmofetil oder Tacrolimus) konnten diese vorbestehende Therapie während der gesamten Dauer der Studie fortsetzen. Zusätzlich war eine Notfalltherapie (einschliesslich hochdosierter Kortikosteroide, PE/PP oder IVIg) erlaubt, falls ein Patient eine klinische Verschlechterung gemäss Definition im Studienprotokoll zeigte.
Insgesamt 162 Patienten (92,6 %) beendeten den 26-wöchigen, randomisierten, kontrollierten Zeitraum der Studie ALXN1210-MG-306. Die Merkmale der Patienten zu Studienbeginn sind in Tabelle 14 zusammengestellt. Die überwiegende Mehrheit der in die Studie aufgenommenen Patienten (97 %) waren in den letzten zwei Jahren vor Eintritt in die Studie mit mindestens einer immunmodulatorischen Therapie, einschliesslich Immunsuppressiva, PE/PP oder IVIg behandelt worden.
Tabelle 14: Merkmale zu Studieneginn von Studie ALXN1210-MG-306
|
Parameter
|
Statistik
|
Placebo
(N = 89)
|
Ravulizumab
(N = 86)
| |
Geschlecht
Männlich
Weiblich
|
n (%)
|
44 (49,4)
45 (50,6)
|
42 (48,8)
44 (51,2)
| |
Alter bei Erstinfusion des Studienmedikaments (Jahre)
|
Mittel (SD)
(Min., Max.)
|
53,3 (16,05)
(20, 82)
|
58,0 (13,82)
(19, 79)
| |
Ältere Patienten (≥ 65 Jahre) bei Eintritt in die Studie
|
n (%)
|
24 (27,0)
|
30 (34,9)
| |
Dauer der MG seit Diagnosestellung (Jahre)
|
Mittel (SD)
(Min., Max.)
Median
|
10,0 (8,90)
(0,5; 36,1)
7,6
|
9,8 (9,68)
(0,5; 39,5)
5,7
| |
MG-ADL-Ausgangsscore
|
Mittel (SD)
(Min., Max.)
Median
|
8,9 (2,30)
(6,0; 15,0)
9,0
|
9,1 (2,62)
(6,0; 24,0)
9,0
| |
QMG-Ausgangsscore
|
Mittel (SD)
(Min., Max.)
Median
|
14,5 (5,26)
(2,0; 27,0)
14,0
|
14,8 (5,21)
(6,0; 39,0)
15,0
| |
MGFA-Klassifikation zu Studienbeginn
Klasse II (leichte Schwäche)
Klasse III (moderate Schwäche)
Klasse IV (stark ausgeprägte Schwäche)
|
n (%)
|
39 (44)
45 (51)
5 (6)
|
39 (45)
41 (48)
6 (7)
| |
Etwaige frühere Intubationen seit Diagnosestellung (MGFA-Klasse V)
|
n (%)
|
9 (10,1)
|
8 (9,3)
| |
Anzahl Patienten mit früherer MG-Krise seit Diagnosestellunga
|
n (%)
|
17 (19,1)
|
21 (24,4)
| |
Anzahl stabiler immunsupprimierender Therapienb bei Studieneintritt
0
1
≥ 2
|
n (%)
|
8 (9,0)
34 (38,2)
47 (52,8)
|
10 (11,6)
40 (46,5)
36 (41,9)
|
a Angaben zu früheren MG-Krisen wurden bei Aufnahme der Anamnese erfasst und nicht nach der Definition im klinischen Prüfplan bewertet.
b Immunsuppressive Therapien umfassen Kortikosteroide, Azathioprin, Cyclophosphamid, Cyclosporin, Methotrexat, Mycophenolatmofetil oder Tacrolimus.
Abkürzungen: Max. = Maximum; Min. = Minimum; MG = Myasthenia gravis; MG-ADL = Myasthenia Gravis Activities of Daily Living; MGFA = Myasthenia Gravis Foundation of America; QMG = Quantitative Myasthenia Gravis; SD = Standardabweichung
Der primäre Endpunkt war die Veränderung des MG-ADL-Gesamtscores gegenüber Studienbeginn bis Woche 26.
Die sekundären Endpunkte, die ebenfalls die Veränderungen gegenüber Studienbeginn bis Woche 26 bewerteten, umfassten die Veränderung des Quantitative-Myasthenia-Gravis-(QMG-)Gesamtscores, den Anteil von Patienten mit Verbesserungen von mindestens 5 bzw. 3 Punkten bei den QMG- und MG-ADL-Gesamtscores sowie Veränderungen bei den Bewertungen der Lebensqualität.
Ravulizumab zeigte einen statistisch signifikanten Unterschied des MG-ADL-Gesamtscores im Vergleich zu Placebo. Der primäre und die sekundären Endpunkte sind in Tabelle 15 zusammengestellt.
Tabelle 15: Analyse des primären und der sekundären Wirksamkeitsendpunkte
|
Wirksamkeits-endpunkte in Woche 26
|
Placebo
(N = 89)
LS- Mittelwert (SEM)
|
Ravulizumab
(N = 86)
LS-Mittelwert (SEM)
|
Statistik für den Vergleich
|
Behandlungs-effekt
(95 %-KI)
|
p-Wert
(mit Mixed Effect Repeated Measures)
| |
MG-ADL
|
-1,4 (0,37)
|
-3,1 (0,38)
|
Unterschied der Veränderung zur Baseline
|
-1,6 (-2,6; -0,7)
|
0,0009
| |
QMG
|
-0,8 (0,45)
|
-2,8 (0,46)
|
Unterschied der Veränderung zur Baseline
|
-2,0 (-3,2; -0,8)
|
0,0009
| |
MG-QoL15r
|
-1,6 (0,70)
|
-3,3 (0,71)
|
Unterschied der Veränderung zur Baseline
|
-1,7 (-3,4; 0,1)
|
0,0636
| |
Neuro-QoL-Fatigue
|
-4,8 (1,87)
|
-7,0 (1,92)
|
Unterschied der Veränderung zur Baseline
|
-2,2 (-6,9; 2,6)
|
0,3734a
|
a Der Endpunkt wurde nicht formal auf statistische Signifikanz getestet; ein nominaler p-Wert wurde ermittelt.
Abkürzungen: KI = Konfidenzintervall; LS = kleinste Quadrate; MG-ADL = Myasthenia Gravis Activities of Daily Living; MG-QoL15r = überarbeitete Lebensqualitätsskala für Myasthenia Gravis mit 15 Items; Neuro-QoL-fatigue = Neurologische Lebensqualität, Fatigue; QMG = Quantitative Myasthenia Gravis; SEM = Standardfehler des Mittelwerts.
In der Studie ALXN1210-MG-306 war ein klinischer Responder nach dem MG-ADL-Gesamtscore definiert als ein Patient mit einer Verbesserung um mindestens 3 Punkte. Der Anteil der klinischen Responder in Woche 26 betrug 56,7 % unter Ravulizumab gegenüber 34,1 % unter Placebo (nominal p=0,0049). Ein klinischer Responder war nach dem QMG-Gesamtscore definiert als ein Patient mit einer Verbesserung um mindestens 5 Punkte. Der Anteil der klinischen Responder in Woche 26 lag bei 30,0 % unter Ravulizumab gegenüber 11,3 % unter Placebo (p=0,0052).
Tabelle 16 zeigt eine Übersicht über die Patienten mit klinischer Verschlechterung und die Patienten, die im Verlauf des 26-wöchigen randomisierten kontrollierten Zeitraums eine Notfallbehandlung benötigten.
Tabelle 16: Klinische Verschlechterung und Notfalltherapie
|
Parameter
|
Statistik
|
Placebo
(N = 89)
|
Ravulizumab
(N = 86)
| |
Gesamtzahl von Patienten mit klinischer Verschlechterung
|
n (%)
|
15 (16,9)
|
8 (9,3)
| |
Gesamtzahl von Patienten mit Bedarf für eine Notfalltherapiea
|
n (%)
|
14 (15,7)
|
8 (9,3)
|
a Die Notfalltherapie umfasste ein hochdosiertes Kortikosteroid, Plasmaaustausch/Plasmapherese oder intravenöses Immunglobulin.
Zum Zeitpunkt der Analyse waren 150 der 158 Patienten, die in die offene Verlängerungsphase aufgenommen wurden, noch an der Studie beteiligt.
Bei Patienten, die während des randomisierten kontrollierten Behandlungszeitraums anfangs ULTOMIRIS erhielten und auch während der ersten 34 Wochen der offenen Verlängerungsphase weiterhin mit ULTOMIRIS behandelt wurden, hielt die Behandlungswirkung an (Abbildung 3). Bei Patienten, die während des 26-wöchigen randomisierten kontrollierten Behandlungszeitraums zunächst Placebo erhielten und während der offenen Verlängerungsphase eine Behandlung mit ULTOMIRIS begannen, war ein rasches und andauerndes Ansprechen auf die Behandlung zu beobachten (Abbildung 3).
Abbildung 3: Veränderung des MG-ADL-Gesamtscores (A) und des QMG-Gesamtscores (B) gegenüber Baseline im randomisierten kontrollierten Behandlungszeitraum bis einschliesslich Woche 60 (Mittelwert und 95 %-KI)
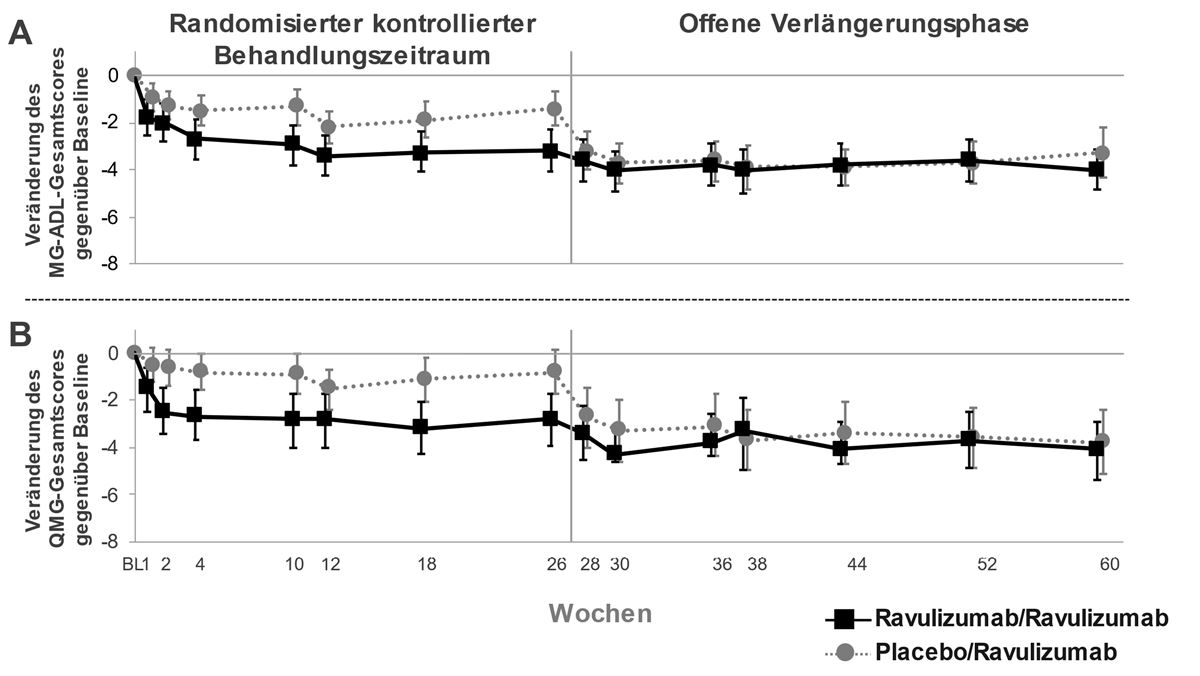
Abkürzungen: KI = Konfidenzintervall; MG-ADL = Myasthenia Gravis Activities of Daily Living; QMG = Quantitative Myasthenia Gravis
In der offenen Verlängerungsphase der Studie hatten die behandelnden Ärzte die Möglichkeit, die immunsuppressiven Therapien anzupassen. Von den Patienten, die in der offenen Verlängerungsphase über eine Dauer von 34 Wochen beobachtet wurden, reduzierten 28,0 % der Patienten ihre tägliche Kortikosteroiddosis und 6,2 % der Patienten beendeten die Kortikosteroidtherapie. Der häufigste Grund für die Anpassung der Kortikosteroidtherapie war eine Besserung der MG-Symptome während der Behandlung mit Ravulizumab.
Neuromyelitis-optica-Spektrum-Erkrankungen (NMOSD)
Studie an erwachsenen Patienten mit NMOSD
Die Wirksamkeit von Ravulizumab bei erwachsenen NMOSD-Patienten mit positiven Anti-AQP4-Antikörpern wurde in einer globalen offenen Studie ALXN1210-NMO-307 untersucht.
In Studie ALXN1210-NMO-307 wurden 58 erwachsene NMOSD-Patienten mit positivem Serumtest auf Anti-AQP4-Antikörper, mindestens 1 Schub in den letzten 12 Monaten vor dem Screeningszeitraum sowie einem Expanded Disability Status Scale (EDSS) Score von ≤ 7 eingeschlossen. Eine vorherige Behandlung mit immunsuppressiven Therapien (IST) war für die Aufnahme in die Studie nicht erforderlich und 51,7 % der Patienten erhielten eine Monotherapie mit Ravulizumab. Patienten, die eine etablierte IST (d. h. Kortikosteroide, Azathioprin, Mycophenolat-Mofetil, Tacrolimus) erhielten, durften mit der Anforderung einer gleichbleibenden Dosierung bis zum Erreichen von Woche 106 der Studie, die Therapie in Kombination mit Ravulizumab fortsetzen. Darüber hinaus war eine Soforttherapie zur Rückfallbehandlung (einschliesslich hochdosierter Kortikosteroide, PE/PP und IVIg) erlaubt, falls ein Patient während der Studie einen Schub zeigte.
Eingeschlossene Patienten waren im Median 47.4 (18-74) Jahre alt und vorwiegend weiblich (90%). Das mediane Alter bei der ersten klinischen Manifestation der NMOSD war 42.5 Jahre, zwischen 16 bis 73 Jahre. Die Merkmale zu Studienbeginn sind in Tabelle 17 dargestellt.
Table 17: Krankheitsanamnese und Ausgangscharakteristika in Studie ALXN1210-NMO-307
|
Variable
|
Statistik
|
ALXN1210-NMO-307 Ravulizumab
(N = 58)
| |
Zeitraum von der ersten klinischen Manifestation der NMOSD bis zur Anwendung der ersten Dosis des Studienmedikaments (Jahre)
|
Mittelwert (SD)
|
5,2 (6,38)
| |
Median
|
2,0
| |
Min., Max.
|
0,19; 24,49
| |
Anamnestische annualisierte Schubrate (ARR) innerhalb von 24 Monaten vor dem Screening
|
Mittelwert (SD)
|
1,87 (1,59)
| |
Median
|
1,44
| |
Min., Max.
|
0,5; 6,9
| |
HAI-Ausgangsscore
|
Mittelwert (SD)
|
1,2 (1,42)
| |
Median
|
1,0
| |
Min., Max.
|
0; 7
| |
EDSS-Ausgangsscore
|
Mittelwert (SD)
|
3,30 (1,58)
| |
Median
|
3,25
| |
Min., Max.
|
0,0; 7,0
| |
Jegliche frühere Anwendung von Rituximab
|
n (%)
|
21 (36,2)
| |
Anzahl der Patienten, die bei Studienbeginn ausschliesslich stabile Kortikosteroide erhielten
|
n (%)
|
12 (20,7)
| |
Anzahl der Patienten, die bei Studienbeginn keine IST erhalten
|
n (%)
|
30 (51,7)
|
Abkürzungen: ARR = annualized relapse rate; EDSS = Expanded Disability Status Scale; HAI = Hauser Ambulation Index; IST = immunsuppressive Therapie; Max. = maximum; Min. = minimum; NMOSD = Neuromyelitis-optica-Spektrum-Erkrankungen; SD = Standardabweichung (standard deviation).
Der primäre Endpunkt der Studie ALXN1210-NMO-307 war die Zeit bis zum ersten in der Studie aufgetretenen Schub, der von einem unabhängigen Entscheidungsgremium bestätigt wurde. Bei den mit Ravulizumab behandelten Patienten wurde während des primären Behandlungszeitraums kein festgestellter Schub in der Studie beobachtet. Alle mit Ravulizumab behandelten Patienten waren während der medianen Nachbeobachtungszeit von 90,93 Wochen schubfrei. Die mit Ravulizumab behandelten Patienten zeigten konsistentes schubfreies Ergebnis hinsichtlich des primären Endpunkts mit oder ohne begleitender IST-Behandlung.
Ravulizumab wurde nicht für die Akutbehandlung von Schüben bei NMOSD-Patienten untersucht.
Sicherheit und Wirksamkeit bei pädiatrischen Patienten
Paroxysmale nächtliche Hämoglobinurie (PNH)
Studie an pädiatrischen Patienten mit PNH (ALXN1210-PNH-304)
Die pädiatrische Studie (ALXN1210-PNH-304) ist eine multizentrische, offene Phase-3-Studie, welche an Kindern und Jugendlichen mit PNH durchgeführt wurde, die entweder zuvor mit Eculizumab behandelt worden waren, oder die zuvor nicht mit einem Komplementinhibitor vorbehandelt waren.
Gemäß den Zwischenergebnissen schlossen insgesamt 13 pädiatrische PNH-Patienten die Behandlung mit Ravulizumab während des primären Auswertungszeitraums (26 Wochen) von Studie ALXN1210-PNH-304 ab. Fünf der 13 Patienten waren noch nie mit einem Komplementinhibitor behandelt worden und 8 Patienten erhielten vor Studieneintritt eine Behandlung mit Eculizumab.
Die meisten Patienten waren bei der ersten Infusion zwischen 12 und 17 Jahre alt (Durchschnittsalter: 14,4 Jahre), während 2 Patienten unter 12 Jahren (11 Jahre und 9 Jahre alt) waren. Acht der 13 Patienten waren weiblich. Das Durchschnittsgewicht zu Studienbeginn betrug 56 kg, Bereich: 37 bis 72 kg. Tabelle 18 zeigt die Krankheitsvorgeschichte und die Merkmale der in Studie ALXN1210-PNH-304 aufgenommenen pädiatrischen Patienten zu Studienbeginn.
Tabelle 18: Krankheitsvorgeschichte und Merkmale zu Studienbeginn (vollständiges Analyseset)
|
Variable
|
Nicht mit Komplementinhibitor vorbehandelte Patienten
(N = 5)
|
Mit Eculizumab vorbehandelte Patienten
(N = 8)
| |
Gesamt-PNH-Erythrozyten-Klongröße (%)
|
(N = 4)
|
(N = 6)
| |
Median (Min., Max.)
|
40,05 (6,9; 68,1)
|
71,15 (21,2; 85,4)
| |
Gesamt-PNH-Granulozyten-Klongröße (%)
|
|
| |
Median (Min., Max.)
|
78,30 (36,8; 99,0)
|
91,60 (20,3; 97,6)
| |
Anzahl Patienten mit pRBC/Vollbluttransfusionen innerhalb von 12 Monaten vor der ersten Dosis, n (%)
|
2 (40,0)
|
2 (25,0)
| |
Anzahl pRBC/Vollblut-Transfusionen innerhalb von 12 Monaten vor der ersten Dosis
|
|
| |
Insgesamt
|
10
|
2
| |
Median (Min.; Max.)
|
5,0 (4; 6)
|
1,0 (1; 1)
| |
Transfundierte pRBC/Vollblut-Einheiten innerhalb von 12 Monaten vor der ersten Dosis
|
|
| |
Insgesamt
|
14
|
2
| |
Median (Min., Max.)
|
7,0 (3; 11)
|
2,0 (2; 2)
| |
Patienten mit PNH-assoziierten Erkrankungen vor Einholung der Einwilligungserklärung nach Aufklärung über die Studie, n (%)
|
5 (100)
|
8 (100)
| |
Anämie
|
2 (40,0)
|
5 (62,5)
| |
Hämaturie oder Hämoglobinurie
|
2 (40,0)
|
5 (62,5)
| |
Aplastische Anämie
|
3 (60,0)
|
1 (12,5)
| |
Niereninsuffizienz
|
2 (40,0)
|
2 (25,0)
| |
Sonstigea
|
0
|
1 (12,5)
| |
LDH-Spiegel vor der Behandlung (E/l)
|
|
| |
Median (Min., Max.)
|
588,50 (444; 2.269,7)
|
251,50 (140,5; 487)
|
a Andere mit PNH assoziierte Erkrankungen wurden als „Nieren- und Milzinfarkte“ und als „multiple Läsionen die auf einen embolischen Prozess hindeuten“ beschrieben.
Hinweis: Die prozentualen Angaben basieren auf der Gesamtzahl von Patienten in jeder Kohorte.
Abkürzungen: LDH = Laktatdehydrogenase; Max. = Maximum; Min. = Minimum; PNH = paroxysmale nächtliche Hämoglobinurie; pRBC = Erythrozytenkonzentrat (packed red blood cell); RBC = Erythrozyt.
Die Patienten erhielten an Tag 1 eine Initialdosis Ravulizumab auf der Grundlage des Körpergewichts, gefolgt von einer Erhaltungstherapie an Tag 15 und danach einmal alle 8 Wochen (q8W) für Patienten mit einem Gewicht ≥ 20 kg oder einmal alle 4 Wochen (q4W) für Patienten mit einem Körpergewicht < 20 kg. Bei Patienten, die bei Eintritt in die Studie eine Behandlung mit Eculizumab erhielten, war Tag 1 der Studienbehandlung 2 Wochen nach der letzten Dosis Eculizumab des Patienten geplant.
Das auf dem Körpergewicht basierende Dosierungsschema von Ravulizumab bewirkte eine sofortige, vollständige und anhaltende Hemmung des terminalen Komplementsystems während des gesamten 26-wöchigen Zeitraums für die Erstbeurteilung, unabhängig davon, ob sie mit Eculizumab vorbehandelt worden waren oder nicht. Nach Beginn der Ravulizumab-Behandlung wurden sofort nach der ersten Dosis therapeutische Steady-State-Serumkonzentrationen von Ravulizumab erreicht und über den gesamten 26-wöchigen Zeitraum für die Erstbeurteilung in beiden Kohorten aufrechterhalten. In der Studie traten keine Durchbruchhämolyse-Ereignisse auf und bei keinem Patienten lagen die Konzentrationen von freiem C5-Protein nach der Baseline über 0,5 µg/ml. Die mittlere prozentuale Veränderung des LDH-Wertes gegenüber Baseline betrug -47,91 % an Tag 183 in der Kohorte ohne vorherige Behandlung mit einem Komplementinhibitor und blieb in der Kohorte mit Eculizumab-Vorbehandlung während des 26wöchigen Zeitraums für die Erstbeurteilung stabil. Sechzig Prozent (3/5) der Patienten ohne vorherige Behandlung mit einem Komplementinhibitor und 75 % (6/8) der Patienten mit Eculizumab-Vorbehandlung erreichten bis Woche 26 eine Hämoglobinstabilisierung. Eine Transfusionsvermeidung wurde von 84,6 % (11/13) der Patienten während des 26-wöchigen Zeitraums für die Erstbeurteilung erreicht.
Diese Zwischenergebnisse zur Wirksamkeit sind in Tabelle 19 unten zusammengestellt.
Tabelle 19: Ergebnisse für die Wirksamkeit in der klinischen Studie an pädiatrischen Patienten mit PNH (ALXN1210-PNH-304) - 26-wöchiger Zeitraum für die Erstbeurteilung
|
Endpunkt
|
Ravulizumab
(zuvor nicht mit Komplementinhibitoren behandelt, N = 5)
|
Ravulizumab
(Umstellung, mit Komplementinhibitoren vorbehandelt, N = 8)
| |
LDH- prozentuale Veränderung gegenüber Baseline
Mittelwert (SD)
|
-47,91 (52,716)
|
4,65 (44,702)
| |
Transfusionsvermeidung
Prozentualer Anteil (95-%-KI)
|
60,0 (14,66; 94,73)
|
100,0 (63,06; 100,00)
| |
Hämoglobinstabilisierung
Prozentualer Anteil (95-%-KI)
|
60,0 (14,66; 94,73)
|
75 (34,91; 96,81)
| |
Durchbruchhämolyse (%)
|
0
|
0
|
Abkürzungen: LDH = Laktatdehydrogenase
Die Langzeitergebnisse für die Wirksamkeit bis zum Studienende über eine Behandlungsdauer von im Median 915 Tagen zeigten ein anhaltendes Ansprechen der Behandlung bei pädiatrischen Patienten mit PNH.
Ausgehend von diesen Zwischenergebnissen scheint die Wirksamkeit von Ravulizumab bei pädiatrischen PNH-Patienten ähnlich zu sein wie die bei erwachsenen PNH-Patienten beobachtete.
Atypisches hämolytisch-urämischen Syndrom (aHUS)
Die Anwendung von Ultomiris bei pädiatrischen Patienten zur Behandlung eines aHUS wird durch die Ergebnisse einer klinischen Studie an Kindern und Jugendlichen untermauert (insgesamt 31 Patienten mit dokumentiertem aHUS wurden aufgenommen. 28 Patienten im Alter von 10 Monaten bis 17 Jahren wurden in das vollständige Analyseset eingeschlossen).
Studie an pädiatrischen Patienten mit aHUS (ALXN1210-aHUS-312)
Bei dieser pädiatrischen Studie handelt es sich um eine 26wöchige, fortlaufende, multizentrische, einarmige Phase-3-Studie an Kindern und Jugendlichen.
Insgesamt wurden 21 Patienten ohne Eculizumab-Vorbehandlung mit der dokumentierten Diagnose eines aHUS und Anhaltspunkten für eine TMA in die Studie aufgenommen; davon wurden 18 in das vollständige Analyseset eingeschlossen. Die Einschlusskriterien schlossen Patienten aus, die mit einer TMA aufgrund von TTP und STEC-HUS vorstellig wurden. Zwei Patienten erhielten eine Einzeldosis und ein Patient erhielt 2 Dosen; die Patienten brachen die Behandlung aber dann ab und wurden aus dem vollständigen Analyseset ausgeschlossen, weil das aHUS nicht bestätigt war. Das mittlere Körpergewicht zu Studienbeginn betrug insgesamt 22,2 kg; die Mehrheit der Patienten befand sich zu Studienbeginn in der Gewichtskategorie ≥ 10 bis < 20 kg. Die meisten Patienten (72,2 %) wiesen vor der Behandlung extrarenale (kardiovaskuläre, pulmonale, zentralnervöse, gastrointestinale, die Haut oder Skelettmuskulatur betreffende) Zeichen oder Symptome eines aHUS zu Studienbeginn auf. Zu Studienbeginn hatten 33,3 % (n = 6) der Patienten eine CKD in Stadium 5.
Insgesamt wurden 10 Patienten, die von Eculizumab zu Ravulizumab wechselten und eine dokumentierte aHUS-Diagnose sowie Anzeichen einer TMA aufwiesen, in die Studie aufgenommen. Es musste ein klinisches Ansprechen auf Eculizumab vorliegen, bevor die Patienten in die Studie aufgenommen wurden (d. h. LDH < 1,5 x ULN und Thrombozytenzahl ≥ 150.000/µl und eGFR > 30 ml/min/l,73 m²). Demzufolge gibt es keine Daten über die Anwendung von Ravulizumab bei Patienten, die nicht auf Eculizumab ansprechen.
Tabelle 20 zeigt die Ausgangsmerkmale von pädiatrischen Patienten, die in Studie ALXN1210aHUS-312 aufgenommen wurden.
Tabelle 20: Demographische Merkmale und Ausgangsmerkmale in Studie ALXN1210aHUS-312
|
Parameter
|
Statistik
|
Ravulizumab
(ohne Vorbehandlung, N = 18)
|
Ravulizumab
(Behandlungs-wechsel, N = 10)
| |
Alterskategorie bei Erstinfusion (Jahre)
Geburt bis < 2 Jahre
2 bis < 6 Jahre
6 bis < 12 Jahre
12 bis < 18 Jahre
|
n (%)
|
2 (11,1)
9 (50,0)
5 (27,8)
2 (11,1)
|
1 (10,0)
1 (10,0)
1 (10,0)
7 (70,0)
| |
Geschlecht
Männlich
|
n (%)
|
8 (44,4)
|
9 (90,0)
| |
Ethniea
Ureinwohner Nordamerikas (Indianer) oder Alaskas
Asiatisch
Schwarz oder Afroamerikaner
Weißhäutig
Unbekannt
|
n (%)
|
1 (5,6)
5 (27,8)
3 (16,7)
9 (50,0)
1 (5,6)
|
0 (0,0)
4 (40,0)
1 (10,0)
5 (50,0)
0 (0,0)
| |
Transplantation in der Vorgeschichte
|
n (%)
|
1 (5,6)
|
1 (10,0)
| |
Thrombozyten (109/l) im Blut
|
Median (Min., Max.)
|
51,25 (14; 125)
|
281,75 (207; 415,5)
| |
Hämoglobin (g/l)
|
Median (Min., Max.)
|
74,25 (32; 106)
|
132,0 (114,5; 148)
| |
LDH (U/l)
|
Median (Min., Max.)
|
1963,0 (772; 4985)
|
206,5 (138,5; 356)
| |
eGFR (ml/min/1,73 m2)
|
Median (Min., Max.)
|
22,0 (10; 84)
|
99,75 (54; 136,5)
| |
Dialysepflichtigkeit zu Studienbeginn
|
n (%)
|
6 (33,3)
|
0 (0,0)
|
Hinweis: Die prozentualen Anteile basieren auf der Gesamtzahl der Patienten.
a Auf die Patienten können mehrere Ethnien zutreffen.
Abkürzungen: aHUS = atypisches hämolytisch-urämischen Syndrom; eGFR = geschätzte glomeruläre Filtrationsrate; LDH = Laktatdehydrogenase; Max. = Maximum; Min. = Minimum.
Der primäre Endpunkt war das vollständige Ansprechen der TMA während des 26wöchigen Zeitraums für die Erstbeurteilung, festgestellt anhand der Normalisierung der hämatologischen Parameter (Thrombozyten ≥ 150 x 109/l und LDH ≤ 246 U/l) sowie eine Besserung des Serumkreatinins von ≥ 25 % gegenüber dem Ausgangswert. Die Patienten mussten alle Kriterien für ein vollständiges Ansprechen der TMA bei 2 verschiedenen Beurteilungen im Abstand von mindestens 4 Wochen (28 Tagen) und bei jeder zwischenzeitlichen Messung erfüllen.
Ein vollständiges Ansprechen der TMA wurde bei 14 der 18 nicht vorbehandelten Patienten (77,8 %) während des 26wöchigen Zeitraums für die Erstbeurteilung festgestellt, wie es in Tabelle 21 gezeigt ist.
Tabelle 21: Vollständiges Ansprechen der TMA und Analyse der Komponenten des vollständigen Ansprechens der TMA während des 26-wöchigen Zeitraums für die Erstbeurteilung (ALXN1210aHUS-312)
|
|
Summe
|
Responder
| |
n
|
Anteil (95 %-KI)a
| |
Vollständiges Ansprechen der TMA
|
18
|
14
|
0,778 (0,524; 0,936)
| |
Komponenten des vollständigen Ansprechens der TMA
|
|
|
| |
Normalisierung der Thrombozytenzahl
|
18
|
17
|
0,944 (0,727; 0,999)
| |
Normalisierung von LDH
|
18
|
16
|
0,889 (0,653; 0,986)
| |
Verbesserung des Serumkreatinins um ≥ 25 % gegenüber dem Ausgangswert
|
18
|
15
|
0,833 (0,586; 0,964)
| |
Normalisierung der Blutwerte
|
18
|
16
|
0,889 (0,653; 0,986)
|
Hinweis: 1 Patient schied nach Behandlung mit 2 Dosen Ravulizumab aus der Studie aus.
a Die 95 %-Konfidenzintervalle (95 %-KI) für den Anteil basieren auf der asymptotischen Gaußschen Approximationsmethode mit Kontinuitätskorrektur.
Abkürzungen: KI = Konfidenzintervall; LDH = Laktatdehydrogenase; TMA = thrombotische Mikroangiopathie.
Das vollständige Ansprechen der TMA während des Zeitraums für die Erstbeurteilung wurde in einer medianen Zeitdauer von 30 Tagen (15 bis 97 Tage) erzielt. Bei allen Patienten mit vollständigem Ansprechen der TMA blieb das Ansprechen während des gesamten Zeitraums für die Erstbeurteilung erhalten, wobei kontinuierliche Verbesserungen der Nierenfunktion beobachtet wurden. Nach Beginn der Ravulizumab-Behandlung war rasch ein Anstieg der mittleren Thrombozytenzahl mit einer Zunahme von 60,50 × 109/l zu Studienbeginn auf 296,67 × 109/l an Tag 8 zu beobachten, die bei allen anschließenden Besuchsterminen im Zeitraum für die Erstbeurteilung (26 Wochen) über 296 × 109/l lag.
Drei weitere Patienten zeigten ein vollständiges Ansprechen der TMA, das nach dem 26wöchigen Zeitraum für die Erstbeurteilung bestätigt wurde (das vollständige Ansprechen der TMA wurde an Tag 291, 297 und 353 festgestellt); somit zeigten 17 der 18 pädiatrischen Patienten (94,4 %) (95 %-KI: 72,7 %; 99,9 %) ein vollständiges Ansprechen der TMA. Das Ansprechen individueller Komponenten erhöhte sich auf 17 von 18 Patienten (94,4 %; 95 %-KI: 72,7 %, 99,9 %) für die Normalisierung der Thrombozytenzahl, auf 17 von 18 Patienten (94,4 %; 95 %-KI: 72,7 %, 99,9 %) für die LDH-Normalisierung und auf 17 von 18 Patienten (94,4 %; 95 %-KI: 72,7 %, 99,9 %) für die Besserung der Nierenfunktion.
Alle 6 Patienten, die bei Eintritt in die Studie dialysepflichtig waren, konnten die Dialysebehandlung absetzen. Bei fünf dieser Patienten war dies bereits spätestens an Tag 43 möglich. Kein Patient begann während der Studie eine Dialysebehandlung. Der größte Teil der Patientenpopulation (15/17) zeigte eine Besserung der CKD um 1 oder mehrere Stadien bis Tag 183; 14 Patienten zeigten eine Besserung um 2 oder mehr Stadien. Tabelle 22 fasst die sekundären Wirksamkeitsergebnisse für Studie ALXN1210aHUS-312 zusammen.
Tabelle 22: Sekundäre Wirksamkeitsergebnisse von Studie ALXN1210aHUS-312
|
Parameter
|
Studie ALXN1210aHUS-312
(N = 18)
| |
Hämatologische Parameter bei TMA, Tag 183
Thrombozyten (109/l)im Blut
Mittelwert (SD)
Median
LDH (U/l) im Serum
Mittelwert (SD)
Median
|
Beobachteter Wert (n = 17)
304,94 (75,711)
318,00
262,41 (59,995)
247,00
|
Veränderung gegenüber dem Ausgangswert (n = 17)
245,59 (91,827)
247,00
-2044,13 (1328,059)
-1851,50
| |
Anstieg des Hämoglobins um ≥ 20 g/l gegenüber dem Ausgangswert mit bestätigtem Ergebnis während des Zeitraums für die Erstbeurteilung
m/N
Anteil (95 %-KI)*
|
16/18
0,889 (0,653; 0,986)
| |
Veränderung des CKD-Stadiums gegenüber dem Ausgangswert, Tag 183
Verbesserunga
m/n
Anteil (95 %-KI)*
Verschlechterungb
m/n
Anteil (95 %-KI)*
|
15/17
0,882 (0,636; 0,985)
0/11
0,000 (0,000; 0,285)
| |
eGFR (ml/min/1,73 m2), Tag 183
Mittelwert (SD)
Median
|
Beobachteter Wert (n = 17)
108,5 (56,87)
108,0
|
Veränderung gegenüber dem Ausgangswert (n = 17)
85,4 (54,33)
80,0
|
Hinweis: n: Anzahl von Patienten mit verfügbaren Daten für eine bestimmte Untersuchung beim Besuchstermin an Tag 183. m: Anzahl von Patienten, die ein bestimmtes Kriterium erfüllen. Das Stadium der chronischen Nierenerkrankung (CKD) wird anhand der Klassifikation der National Kidney Foundation für Stadien der chronischen Nierenerkrankung (Chronic Kidney Disease Stage) bestimmt. Stadium 1 wird als die beste Kategorie betrachtet, während Stadium 5 als schlechteste Kategorie gilt. Der Ausgangswert wird anhand der letzten verfügbaren eGFR vor Behandlungsbeginn ermittelt. Verbesserung/Verschlechterung: Im Vergleich zum CKD-Stadium zu Studienbeginn.
*Die 95 %-Konfidenzintervalle (95 %-KI) basieren auf dem exakten Clopper-Pearson-Konfidenzintervall. .
a Verbesserung schließt Patienten mit Stadium 1 zu Studienbeginn aus, da bei ihnen keine Besserung möglich ist; bVerschlechterung schließt Patienten mit Stadium 5 zu Studienbeginn aus, da bei ihnen keine Verschlechterung möglich ist.
Abkürzungen: eGFR = geschätzt glomeruläre Filtrationsrate; LDH = Laktatdehydrogenase; TMA = thrombotische Mikroangiopathie.
Bei Patienten, die mit Eculizumab vorbehandelt wurden, führte die Umstellung auf Ravulizumab zu einer Aufrechterhaltung der Krankheitskontrolle, wie an stabilen hämatologischen und renalen Parametern abzulesen ist, ohne offenbare Auswirkungen auf die Sicherheit.
Die Wirksamkeit von Ravulizumab bei der Behandlung von aHUS scheint bei pädiatrischen Patienten ähnlich zu sein wie bei erwachsenen Patienten.
PharmakokinetikAbsorption
Da die Art der Anwendung von Ravulizumab eine intravenöse Infusion und die Darreichungsform eine Lösung ist, gilt die verabreichte Dosis als zu 100 % bioverfügbar. Die Zeit bis zum Erreichen der maximalen Konzentration (tmax) dauert voraussichtlich bis zum Ende der Infusion oder bis kurz nach Ende der Infusion. Therapeutische Steady-State-Arzneimittelkonzentrationen werden bereits nach der ersten Dosis erreicht.
Distribution
Das mittlere (Standardabweichung [SD]) zentrale Volumen und Verteilungsvolumen im Gleichgewichtszustand (Steady State) bei erwachsenen und pädiatrischen Patienten mit PNH und aHUS sowie bei erwachsenen Patienten mit gMG oder NMOSD ist in Tabelle 23 dargestellt.
Metabolismus
Als monoklonaler Immunglobulin-G (IgG)-Antikörper wird Ravulizumab voraussichtlich auf die gleiche Weise wie jedes endogene IgG verstoffwechselt (über Abbauwege in kleine Peptide und Aminosäuren zerlegt) und unterliegt einer ähnlichen Elimination. Ravulizumab enthält nur natürlich vorkommende Aminosäuren und hat keine bekannten aktiven Metabolite.
Elimination
Die Mittelwerte (SD) für die terminale Eliminationshalbwertszeit bzw. die Clearance von Ravulizumab bei erwachsenen und pädiatrischen Patienten mit PNH oder aHUS und erwachsenen Patienten mit gMG oder NMOSD sind in Tabelle 23 dargestellt.
Tabelle 23: Parameter für das geschätzte zentrale Volumen sowie für die Verteilung, Biotransformation und Elimination nach Behandlung mit Ravulizumab
|
|
Erwachsene und pädiatrische Patienten mit PNH
|
Erwachsene und pädiatrische Patienten mit aHUS
|
Erwachsene Patienten mit gMG
|
Erwachsene Patienten mit NMOSD
| |
Geschätztes zentrales Volumen (Liter)
Mittelwert (SD)
|
Erwachsene: 3,44 (0,65)
Kinder und Jugendliche: 2,87 (0,60)
|
Erwachsene: 3,25 (0,61)
Kinder und Jugendliche: 1,14 (0,51)
|
3,42 (0,756)
|
2,91 (0,571)
| |
Verteilungsvolumen im Steady State (Liter)
Mittelwert (SD)
|
5,30 (0,9)
|
5,22 (1,85)
|
5,74 (1,16)
|
4,77 (0,819)
| |
Terminale Eliminations-Halbwertszeit (Tage)
Mittelwert (SD)
|
49,6 (9,1)
|
51,8 (16,2)
|
56,6 (8,36)
|
64,3 (11,0)
| |
Clearance (Liter/Tag)
Mittelwert (SD)
|
0,08 (0,022)
|
0,08 (0,04)
|
0,08 (0,02)
|
0,05 (0,016)
|
Abkürzungen: aHUS = atypisches hämolytisch-urämisches Syndrom; gMG = generalisierte Myasthenie gravis; NMOSD = Neuromyelitis-optica-Spektrum-Erkrankungen; PNH = paroxysmale nächtliche Hämoglobinurie; SD = Standardabweichung.
Linearität/Nicht Linearität
Über den untersuchten Bereich der Dosierung und des Dosierungsschemas hinweg wies Ravulizumab eine dosisproportionale und zeitlich lineare Pharmakokinetik (PK) auf.
Kinetik spezieller Patientengruppen
Körpergewicht
Das Körpergewicht ist eine signifikante Kovariable bei Patienten mit PNH, aHUS, gMG oder NMOSD, die zu einer geringeren Bioverfügbarkeit bei schwereren Patienten führt. Die körpergewichtsbasierte Dosierung ist in Abschnitt "Dosierung/Anwendung" , Tabelle 1, Tabelle 2 und Tabelle 3 angegeben.
Es wurden keine spezifischen Studien zur Untersuchung der Pharmakokinetik von Ravulizumab in Bezug auf Geschlecht, ethnische Herkunft, Alter (geriatrische Patienten), Vorliegen einer Leber- oder Nierenfunktionsbeeinträchtigung durchgeführt. Auf Basis einer pharmakokinetischen Populationsanalyse wurde bei den untersuchten gesunden Probanden und Patienten mit PNH, aHUS, gMG oder NMOSD jedoch keine Auswirkung von Geschlecht, Alter, ethnischer Herkunft sowie Vorliegen einer Leber- oder Nierenfunktionsbeeinträchtigung auf die PK von Ravulizumab festgestellt, weshalb Dosisanpassungen als nicht erforderlich angesehen werden.
Die Pharmakokinetik von Ravulizumab wurde bei aHUS-Patienten mit unterschiedlich ausgeprägten Nierenfunktionsbeeinträchtigungen, einschließlich dialysepflichtiger Patienten, untersucht. In diesen Subpopulationen von Patienten, darunter auch Patienten mit Proteinurie, wurden keine Unterschiede in Bezug auf die pharmakokinetischen Parameter festgestellt.
Präklinische DatenEs wurden keine reproduktionstoxikologischen Studien an Tieren mit Ravulizumab durchgeführt, es wurden jedoch reproduktionstoxikologische Studien an Mäusen mit einem murinen Surrogat-Antikörper zur Hemmung der Komplementaktivierung, BB5.1, durchgeführt. In den reproduktionstoxikologischen Studien an Mäusen mit dem murinen Ersatzantikörper wurden keine eindeutigen behandlungsbezogenen Auswirkungen oder unerwünschten Wirkungen beobachtet. Bei der Exposition von Muttertieren gegenüber dem Antikörper während der Organogenese wurden zwei Fälle von Retinadysplasie und ein Fall von Nabelhernie unter 230 Nachkommen von Muttertieren, die den höheren Antikörperdosen (etwa dem Vierfachen der für den Menschen empfohlenen Höchstdosis von Ravulizumab, basierend auf einem Körpergewichtsvergleich) ausgesetzt waren, beobachtet; die Exposition erhöhte jedoch nicht den Fetusverlust oder die neonatale Sterblichkeit.
Es wurden keine tierexperimentellen Studien zur Bewertung des genotoxischen und karzinogenen Potenzials von Ravulizumab durchgeführt.
Basierend auf nicht-klinischen Studien an Mäusen mithilfe des murinen Surrogatmoleküls BB5.1 lassen die präklinischen Daten keine besonderen Gefahren für den Menschen erkennen.
Sonstige HinweiseInkompatibilitäten
Dieses Arzneimittel darf nicht mit anderen Arzneimitteln gemischt werden.
Bei der Verdünnung sollte als Verdünnungsmittel nur Natriumchlorid 9 mg/ml (0,9 %) Injektionslösung verwendet werden.
Haltbarkeit
Das Arzneimittel darf nur bis zu dem auf der Packung mit „EXP“ bezeichneten Datum verwendet werden.
Haltbarkeit nach Anbruch
Ultomiris 300 mg/3 ml und 1100 mg/11 ml
Nach Verdünnung sollte das Arzneimittel sofort verwendet werden. Es wurde jedoch nachgewiesen, dass das verdünnte Arzneimittel bis zu 24 Stunden bei 2–8 °C und bis zu 4 Stunden bei Raumtemperatur chemisch und physikalisch stabil ist.
Ultomiris 300 mg/30 ml
Nach Verdünnung sollte das Arzneimittel sofort verwendet werden. Es wurde jedoch nachgewiesen, dass das verdünnte Arzneimittel bis zu 24 Stunden bei 2–8 °C und bis zu 6 Stunden bei Raumtemperatur chemisch und physikalisch stabil ist.
Besondere Lagerungshinweise
Im Kühlschrank (2-8°C) lagern.
Nicht einfrieren.
Den Behälter im Umkarton aufbewahren, um den Inhalt vor Licht (und/oder Feuchtigkeit) zu schützen.
Ausser Reichweite von Kindern aufbewahren.
Aufbewahrungsbedingungen nach Verdünnung des Arzneimittels siehe Abschnitt "Haltbarkeit nach Anbruch" .
Hinweise für die Handhabung
Jede Durchstechflasche ist nur für den Einmalgebrauch bestimmt.
Ultomiris 300 mg/3 ml und 1100 mg/11 ml
Ultomiris muss auf eine Endkonzentration von 50 mg/ml verdünnt werden.
Die üblichen aseptischen Bedingungen sind zu beachten.
Ultomiris wie folgt zubereiten:
1. Die Anzahl der zu verdünnenden Durchstechflaschen wird basierend auf dem Körpergewicht des Patienten und der verordneten Dosis ermittelt, siehe Abschnitt "Dosierung/Anwendung" .
2. Vor der Verdünnung sollte die Lösung in den Durchstechflaschen visuell überprüft werden; die Lösung sollte frei von Partikeln und Präzipitat sein. Nicht verwenden, wenn Hinweise auf Partikel oder Präzipitat vorliegen.
3. Die berechnete Arzneimittelmenge wird aus der entsprechenden Anzahl Durchstechflaschen entnommen und in einem Infusionsbeutel mit Natriumchlorid 9 mg/ml (0,9 %) Injektionslösung als Verdünnungsmittel verdünnt. Siehe nachfolgende Referenztabellen zur Verabreichung. Das Produkt vorsichtig mischen. Nicht schütteln.
4. Nach Verdünnung beträgt die endgültige Konzentration der zu infundierenden Lösung 50 mg/ml.
5. Die zubereitete Lösung sollte sofort nach der Zubereitung verabreicht werden, ausser sie wird bei 2-8 °C gelagert. Wird die verdünnte Lösung bei 2-8 °C gelagert, muss sie vor der Verabreichung Raumtemperatur erreichen. Nicht als intravenöse Druck- oder Bolusinjektion verabreichen. Die Mindest-Infusionsdauer ist Tabelle 4 und Tabelle 5 zu entnehmen. Die Infusion muss durch ein 0,2-µm-Filter verabreicht werden.
6. Wird das Arzneimittel nicht sofort nach der Verdünnung verwendet, dürfen die Lagerungszeiten bei 2 °C – 8 °C bzw. bei Raumtemperatur nicht mehr als 24 Stunden bzw. 4 Stunden betragen, wobei die voraussichtliche Infusionsdauer zu berücksichtigen ist.
Tabelle 24: Referenztabelle zur Verabreichung der Initialdosis von Ultomiris 300 mg/3 ml und 1100 mg/11 ml
|
Körpergewicht (kg)a
|
Initialdosis (mg)
|
Ultomiris-Volumen (ml)
|
Volumen des NaCl-Verdünnungsmittelsb (ml)
|
Gesamtvolumen (ml)
| |
≥ 10 bis < 20
|
600
|
6
|
6
|
12
| |
≥ 20 bis < 30
|
900
|
9
|
9
|
18
| |
≥ 30 bis < 40
|
1200
|
12
|
12
|
24
| |
≥ 40 bis < 60
|
2.400
|
24
|
24
|
48
| |
≥ 60 bis < 100
|
2.700
|
27
|
27
|
54
| |
≥ 100
|
3.000
|
30
|
30
|
60
|
a Körpergewicht zum Behandlungszeitpunkt.
b Ultomiris sollte nur mit Natriumchlorid 9 mg/ml (0,9 %) Injektionslösung verdünnt werden.
Tabelle 25: Referenztabelle zur Verabreichung der Erhaltungsdosis von Ultomiris 300 mg/3 ml und 1100 mg/11 ml
|
Körpergewicht (kg)a
|
Erhaltungsdosis (mg)
|
Ultomiris-Volumen (ml)
|
Volumen des NaCl-Verdünnungsmittelsb (ml)
|
Gesamtvolumen (ml)
| |
≥ 10 bis < 20
|
600
|
6
|
6
|
12
| |
≥ 20 bis < 30
|
2100
|
21
|
21
|
42
| |
≥ 30 bis < 40
|
2700
|
27
|
27
|
54
| |
≥ 40 bis < 60
|
3.000
|
30
|
30
|
60
| |
≥ 60 bis < 100
|
3.300
|
33
|
33
|
66
| |
≥ 100
|
3.600
|
36
|
36
|
72
|
a Körpergewicht zum Behandlungszeitpunkt.
b Ultomiris sollte nur mit Natriumchlorid 9 mg/ml (0,9 %) Injektionslösung verdünnt werden.
Tabelle 26: Referenztabelle für die Ergänzungsdosis von Ultomiris 300 mg/3 ml und 1.100 mg/11 ml Konzentrat zur Herstellung einer Infusionslösung
|
Körpergewicht (kg)a
|
Ergänzungs-dosis (mg)
|
ULTOMIRIS
Volumen (ml)
|
Volumen der NaCl-Verdünnungs-lösungb (ml)
|
Gesamtvolumen (ml)
| |
≥ 40 bis < 60
|
600
|
6
|
6
|
12
| |
1200
|
12
|
12
|
24
| |
1500
|
15
|
15
|
30
| |
≥ 60 bis < 100
|
600
|
6
|
6
|
12
| |
1500
|
15
|
15
|
30
| |
1800
|
18
|
18
|
36
| |
≥ 100
|
600
|
6
|
6
|
12
| |
1500
|
15
|
15
|
30
| |
1800
|
18
|
18
|
36
|
a Körpergewicht zum Behandlungszeitpunkt
b Ultomiris sollte nur mit Natriumchlorid 9 mg/ml (0,9 %) Injektionslösung verdünnt werden.
Ultomiris 300 mg/30 ml
Ultomiris muss auf eine Endkonzentration von 5 mg/ml verdünnt werden.
Die üblichen aseptischen Bedingungen sind zu beachten.
Ultomiris wie folgt zubereiten:
1. Die Anzahl der zu verdünnenden Durchstechflaschen wird basierend auf dem Körpergewicht des Patienten und der verordneten Dosis ermittelt, siehe Abschnitt "Dosierung/Anwendung" .
2. Vor der Verdünnung sollte die Lösung in den Durchstechflaschen visuell überprüft werden; die Lösung sollte frei von Partikeln und Präzipitat sein. Nicht verwenden, wenn Hinweise auf Partikel oder Präzipitat vorliegen.
3. Die berechnete Arzneimittelmenge wird aus der entsprechenden Anzahl Durchstechflaschen entnommen und in einem Infusionsbeutel mit Natriumchlorid 9 mg/ml (0,9 %) Injektionslösung als Verdünnungsmittel verdünnt. Siehe nachfolgende Referenztabellen zur Verabreichung. Das Produkt vorsichtig mischen. Nicht schütteln.
4. Nach Verdünnung beträgt die endgültige Konzentration der zu infundierenden Lösung 5 mg/ml.
5. Die zubereitete Lösung sollte sofort nach der Zubereitung verabreicht werden, ausser sie wird bei 2-8 °C gelagert. Wird die verdünnte Lösung bei 2-8 °C gelagert, muss sie vor der Verabreichung Raumtemperatur erreichen. Nicht als intravenöse Druck- oder Bolusinjektion verabreichen. Die Mindest-Infusionsdauer ist Tabelle 6 und Tabelle 7 zu entnehmen. Die Infusion muss durch ein 0,2-µm-Filter verabreicht werden.
6. Wird das Arzneimittel nicht sofort nach der Verdünnung verwendet, dürfen die Lagerungszeiten bei 2 °C – 8 °C bzw. bei Raumtemperatur nicht mehr als 24 Stunden bzw. 6 Stunden betragen, wobei die voraussichtliche Infusionsdauer zu berücksichtigen ist.
Tabelle 27: Referenztabelle zur Verabreichung der Initialdosis von Ultomiris 300 mg/30 ml
|
Körpergewicht (kg)a
|
Initialdosis (mg)
|
Ultomiris-Volumen (ml)
|
Volumen des NaCl-Verdünnungsmittelsb (ml)
|
Gesamtvolumen (ml)
| |
≥ 10 bis < 20
|
600
|
60
|
60
|
120
| |
≥ 20 bis < 30
|
900
|
90
|
90
|
180
| |
≥ 30 bis < 40
|
1200
|
120
|
120
|
240
| |
≥ 40 bis < 60
|
2.400
|
240
|
240
|
480
| |
≥ 60 bis < 100
|
2.700
|
270
|
270
|
540
| |
≥ 100
|
3.000
|
300
|
300
|
600
|
a Körpergewicht zum Behandlungszeitpunkt.
b Ultomiris sollte nur mit Natriumchlorid 9 mg/ml (0,9 %) Injektionslösung verdünnt werden.
Tabelle 28: Referenztabelle zur Verabreichung der Erhaltungsdosis von Ultomiris 300 mg/30 ml
|
Körpergewicht (kg)a
|
Erhaltungsdosis (mg)
|
Ultomiris-Volumen (ml)
|
Volumen des NaCl-Verdünnungsmittelsb (ml)
|
Gesamtvolumen (ml)
| |
≥ 10 bis < 20
|
600
|
60
|
60
|
120
| |
≥ 20 bis < 30
|
2100
|
210
|
210
|
420
| |
≥ 30 bis < 40
|
2700
|
270
|
270
|
540
| |
≥ 40 bis < 60
|
3.000
|
300
|
300
|
600
| |
≥ 60 bis < 100
|
3.300
|
330
|
330
|
660
| |
≥ 100
|
3.600
|
360
|
360
|
720
|
a Körpergewicht zum Behandlungszeitpunkt.
b Ultomiris sollte nur mit Natriumchlorid 9 mg/ml (0,9 %) Injektionslösung verdünnt werden.
Tabelle 29: Referenztabelle für die Ergänzungsdosis von Ultomiris 300 mg/30 ml Konzentrat zur Herstellung einer Infusionslösung
|
Körpergewicht (kg)a
|
Ergänzungsdosis (mg)
|
ULTOMIRIS
Volumen (ml)
|
Volumen der NaCl-Verdünnungslösungb (ml)
|
Gesamtvolumen (ml)
| |
≥ 40 bis < 60
|
600
|
60
|
60
|
120
| |
1200
|
120
|
120
|
240
| |
1500
|
150
|
150
|
300
| |
≥ 60 bis < 100
|
600
|
60
|
60
|
120
| |
1500
|
150
|
150
|
300
| |
1800
|
180
|
180
|
360
| |
≥ 100
|
600
|
60
|
60
|
120
| |
1500
|
150
|
150
|
300
| |
1800
|
180
|
180
|
360
|
a Körpergewicht zum Behandlungszeitpunkt.
b Ultomiris sollte nur mit Natriumchlorid 9 mg/ml (0,9 %) Injektionslösung verdünnt werden.
Nicht verwendetes Arzneimittel oder Abfallmaterial ist entsprechend den nationalen Anforderungen zu beseitigen.
Zulassungsnummer67278 (Swissmedic)
PackungenUltomiris 300 mg/3 ml (100 mg/ml) Konzentrat zur Herstellung einer Infusionslösung
3 ml steriles Konzentrat in einer Typ-I-Glas-Durchstechflasche enthält 300 mg Ravulizumab (A)
Ultomiris 1100 mg/11 ml (100 mg/ml) Konzentrat zur Herstellung einer Infusionslösung
11 ml steriles Konzentrat in einer Typ-I-Glas-Durchstechflasche enthält 1100 mg Ravulizumab (A)
Ultomiris 300 mg/30 ml (10 mg/ml) Konzentrat zur Herstellung einer Infusionslösung
30 ml steriles Konzentrat in einer Typ-I-Glas-Durchstechflasche enthält 300 mg Ravulizumab (A)
ZulassungsinhaberinAlexion Pharma GmbH
Neuhofstrasse 34,
6340 Baar
Schweiz
Stand der InformationApril 2025
|