ZusammensetzungWirkstoffe
Selpercatinib
Hilfsstoffe
Mikrokristalline Cellulose, hochdisperses Siliciumdioxid
Zusammensetzung der Kapselhülle von Retsevmo 40 mg
Gelatine, Titandioxid (E171), schwarzes Eisenoxid (E172)
Zusammensetzung der Kapselhülle von Retsevmo 80 mg
Gelatine, Titandioxid (E171), Brillantblau FCF (E133)
Zusammensetzung der schwarzen Druckfarbe auf den Retsevmo-Kapseln
Schellack, Propylenglycol, Kaliumhydroxid, schwarzes Eisenoxid (E172)
Darreichungsform und Wirkstoffmenge pro EinheitHartkapsel.
Retsevmo 40 mg Hartkapseln:
Jede Hartkapsel enthält 40 mg Selpercatinib.
Graue undurchsichtige Kapsel, Grösse 2, mit schwarzem Aufdruck "Lilly" , "3977" und "40 mg" auf dem Kapselkörper.
Retsevmo 80 mg Hartkapseln:
Jede Hartkapsel enthält 80 mg Selpercatinib.
Hellblaue undurchsichtige Kapsel, Grösse 0, mit schwarzem Aufdruck "Lilly" "2980" und "80 mg" auf dem Kapselkörper.
Indikationen/AnwendungsmöglichkeitenNicht befristet zugelassene Indikationen
Retsevmo als Monotherapie ist indiziert
zur Behandlung von Erwachsenen mit metastasiertem RET-fusionspositivem nicht-kleinzelligem Lungenkarzinom (NSCLC), die eine systemische Therapie benötigen und bei denen es nach einer vorherigen Behandlung zu einer Progression gekommen ist (siehe "Klinische Wirksamkeit" ).
zur Behandlung von Erwachsenen mit fortgeschrittenem RET-fusionspositivem papillären Schilddrüsenkarzinom, die eine systemische Therapie benötigen und bei denen es nach einer vorherigen Behandlung einschliesslich radioaktivem Jod zu einer Progression gekommen ist (siehe "Klinische Wirksamkeit" )
zur Behandlung von Erwachsenen und Jugendlichen ab einem Alter von 12 Jahren mit fortgeschrittenem RET-mutiertem medullärem Schilddrüsenkarzinom (MTC) (siehe "Warnhinweise und Vorsichtsmassnahmen" und "Eigenschaften/Wirkungen" ).
Befristet zugelassene Indikation
Retsevmo als Monotherapie ist indiziert zur Erstlinienbehandlung von Erwachsenen mit metastasiertem RET-fusionspositivem nicht-kleinzelligem Lungenkarzinom (NSCLC) (siehe "Eigenschaften/Wirkungen" ).
Die Wirksamkeit und Sicherheit von Retsevmo wurde bei Patienten mit weiteren onkogenen Treibermutationen nicht untersucht (siehe "Warnhinweise und Vorsichtsmassnahmen" ).
Aufgrund einer zum Zeitpunkt der Begutachtung des Gesuches unvollständigen Dokumentation, wird/werden diese Indikation/en befristet zugelassen (Art. 9a Heilmittelgesetz). Die befristete Zulassung ist zwingend an die zeitgerechte Erfüllung von Auflagen gebunden. Nach deren Erfüllung kann die befristete Zulassung in eine Zulassung ohne besondere Auflagen überführt werden.
Dosierung/AnwendungTherapieeinleitung
Die Behandlung mit Retsevmo sollte von in der Anwendung von Krebstherapeutika erfahrenen Ärzten eingeleitet und überwacht werden.
Das Vorhandensein einer RET-Genfusion (NSCLC und nicht-medulläres Schilddrüsenkarzinom) oder Mutation (MTC) muss vor Beginn der Therapie mit Retsevmo durch einen validierten Test für RET-Fusionen und RET-Mutationen bestätigt werden (siehe "Klinische Wirksamkeit" ).
Übliche Dosierung
Die empfohlene Dosis Retsevmo nach Körpergewicht beträgt:
-Weniger als 50 kg: 120 mg
-50 kg oder mehr: 160 mg
Retsevmo wird zweimal täglich oral eingenommen (etwa alle 12 Stunden) bis zur Progression oder inakzeptablen Toxizität.
Dosisanpassung aufgrund unerwünschter Wirkungen/Interaktionen
Die Kontrolle einiger Nebenwirkungen kann eine Unterbrechung der Behandlung und/oder Dosisreduktionen erfordern. Anpassungen der Dosis von Retsevmo sind in Tabelle 1 zusammengefasst.
Tabelle 1. Empfehlungen zur Anpassung der Dosis von Retsevmo bei Nebenwirkungen
|
Dosisanpassung
|
Patienten mit einem Gewicht von weniger als 50 kg
|
Patienten mit einem Gewicht von 50 kg oder mehr
| |
Erste Dosisreduktion
|
80 mg oral zweimal täglich
|
120 mg oral zweimal täglich
| |
Zweite Dosisreduktion
|
40 mg oral zweimal täglich
|
80 mg oral zweimal täglich
| |
Dritte Dosisreduktion
|
40 mg oral einmal täglich
|
40 mg oral zweimal täglich
|
Bei Patienten, die Retsevmo nach drei Dosisreduktionen weiterhin nicht vertragen, ist Retsevmo endgültig abzusetzen.
Tabelle 2 zeigt die bei Nebenwirkungen empfohlenen Dosisanpassungen.
Tabelle 2. Empfohlene Dosierungen für Patienten mit Dosisanpassung aufgrund von Nebenwirkungen
|
Nebenwirkung
|
Schweregrad
|
Dosisanpassung
| |
Hepatotoxizität
(siehe "Warnhinweise und Vorsichtsmassnahmen" )
|
Grad 3
oder
Grad 4
|
-Unterbrechung von Selpercatinib und Überwachung von AST/ALT einmal wöchentlich bis zur Besserung auf Grad 1 oder den Ausgangswert.
-Fortsetzung mit einer um 2 Stufen reduzierten Dosis und Kontrolle von AST/ALT einmal wöchentlich bis 4 Wochen nach Erreichen der Dosis, die vor Beginn der AST- oder ALT-Erhöhung Grad 3 oder 4 angewendet wurde.
-Nach mindestens 2 Wochen ohne Wiederauftreten von AST- oder ALT-Erhöhungen wird die Dosis um 1 Dosierungsstufe erhöht, dann nach mindestens 4 Wochen ohne Wiederauftreten Erhöhung auf die Dosis, die vor Beginn der AST- oder ALT-Erhöhung Grad 3 oder 4 angewendet wurde.
-Falls trotz Dosisanpassungen erneut ALT- oder AST-Erhöhungen Grad 3 oder 4 auftreten, ist Selpercatinib endgültig abzusetzen.
| |
Hypertonie
(siehe "Warnhinweise und Vorsichtsmassnahmen" )
|
Grad 3
|
-Unterbrechung von Selpercatinib bei Hypertonie Grad 3, wenn diese trotz optimaler antihypertensiver Therapie anhält.
-Wiederaufnahme mit reduzierter Dosis, wenn die Hypertonie kontrolliert ist.
| |
Grad 4
|
-Absetzen von Selpercatinib.
| |
Verlängerung des QT-Intervalls
(siehe "Warnhinweise und Vorsichtsmassnahmen" )
|
Grad 3
|
-Unterbrechung von Selpercatinib bis zur Besserung auf den Ausgangswert oder Grad 0 oder 1
-Wiederaufnahme mit reduzierter Dosis.
| |
Grad 4
|
-Absetzen von Selpercatinib.
| |
Interstitielle Lungenerkrankung/Pneumonitis
|
Grad 2
|
-Die Anwendung von Selpercatinib bis zum Abklingen unterbrechen. Zum Fortsetzen die Dosis den Angaben in Tabelle 1 entsprechend verringern.
Bei rezidivierender ILD/Pneumonitis Selpercatinib dauerhaft absetzen.
| |
|
Grad 3 oder 4
|
-Bei Grad 3 oder 4 ist Selpercatinib abzusetzen
| |
Blutungen
(siehe "Warnhinweise und Vorsichtsmassnahmen" )
|
Grad 3
oder
Grad 4
|
-Unterbrechung von Selpercatinib bis zur Besserung auf den Ausgangswert oder Grad 0 oder 1.
-Absetzen von Selpercatinib bei schweren oder lebensbedrohlichen Blutungen.
| |
Überempfindlichkeit
(siehe "Warnhinweise und Vorsichtsmassnahmen" )
|
Alle Grade
|
-Unterbrechung von Selpercatinib bis zur Besserung des Ereignisses. Einleitung einer Steroidtherapie.
-Fortsetzung mit einer um 3 Stufen reduzierten Dosis und bei Fortsetzung der Steroidtherapie.
-Jede Woche Erhöhung um 1 Dosisstufe bis zum Erreichen der Dosis, die vor dem Beginn der Überempfindlichkeit angewendet wurde, dann schrittweise Reduktion der Steroidtherapie.
| |
Andere Nebenwirkungen (siehe "Unerwünschte Wirkungen" )
|
Grad 3
oder
Grad 4
|
-Unterbrechung von Selpercatinib bis zur Besserung auf den Ausgangswert oder Grad 0 oder 1.
-Fortsetzung mit reduzierter Dosis.
|
Hypertonie
Wenn eine medizinisch relevante Hypertonie mit einer antihypertensiven Therapie nicht kontrolliert werden kann, muss Selpercatinib endgültig abgesetzt werden (siehe "Warnhinweise und Vorsichtsmassnahmen" ).
CYP-Inhibitoren
Die gleichzeitige Anwendung von Selpercatinib mit starken und moderaten CYP3A-Inhibitoren ist zu vermeiden. Falls die gleichzeitige Anwendung mit einem starken oder moderaten CYP3A-Inhibitor nicht vermieden werden kann, muss die Selpercatinib-Dosis entsprechend der Empfehlungen in Tabelle 3 reduziert werden. Nach dem Absetzen des CYP3A-Inhibitors nach 3 bis 5 Halbwertszeiten wird Selpercatinib mit der Dosis fortgesetzt, die vor dem Beginn der Behandlung mit dem CYP3A-Inhibitor verwendet wurde (siehe "Interaktionen" ).
Tabelle 3. Dosisempfehlung bei begleitender Anwendung von starken oder moderaten CYP3A-Inhibitoren
|
Derzeitige Retsevmo-Dosierung
|
Empfohlene Retsevmo-Dosierung
| |
Moderater CYP3A-Inhibitor
|
Starker CYP3A-Inhibitor
| |
120 mg oral zweimal täglich
|
80 mg oral zweimal täglich
|
40 mg oral zweimal täglich
| |
160 mg oral zweimal täglich
|
120 mg oral zweimal täglich
|
80 mg oral zweimal täglich
|
CYP-Induktoren
Die gleichzeitige Anwendung von starken und moderaten CYP-Induktoren ist zu vermeiden (siehe "Interaktionen" ).
Wirkstoffe, die den gastrischen pH beeinflussen
Die gleichzeitige Gabe von Selpercatinib mit Protonenpumpeninhibitoren, H2-Rezeptor-Antagonisten oder lokal wirksamen Antazida sollte vermieden werden. Ist eine gleichzeitige Gabe jedoch nicht zu vermeiden, sind die folgenden Einnahme-Empfehlungen zu beachten.
Protonenpumpeninhibitoren
Bei gleichzeitiger Anwendung eines Protonenpumpeninhibitors muss die Einnahme zusammen mit einer Mahlzeit erfolgen (siehe "Interaktionen" ).
H2-Rezeptor-Antagonisten
Retsevmo muss 2 Stunden vor einem gleichzeitig angewendeten H2-Rezeptor-Antagonisten eingenommen werden (siehe "Interaktionen" ).
Lokal wirksame Antazida
Selpercatinib muss 2 Stunden vor oder 2 Stunden nach der Gabe eines lokal wirksamen Antazidums eingenommen werden (siehe "Interaktionen" )
Patienten mit Leberfunktionsstörungen
Bei Patienten mit milder oder moderater Leberfunktionsstörung (Gesamtbilirubin < ULN (upper limit of normal) mit AST > ULN oder Gesamtbilirubin > 1- bis 3-fach ULN, unabhängig vom AST) ist eine Dosisanpassung nicht erforderlich. Patienten mit schwerer Leberfunktionsstörung [Gesamtbilirubin > 3- bis 10-fach ULN, unabhängig vom AST) sollten eine reduzierte Dosierung von 80 mg zweimal täglich erhalten (siehe "Pharmakokinetik" )
Bei Patienten mit schwerer Leberfunktionsstörung ist die empfohlene Dosierung von Retsevmo entsprechend den Empfehlungen in Tabelle 4 zu reduzieren.
Tabelle 4. Empfohlene Retsevmo-Dosierung bei schwerer Leberfunktionsstörung
|
Derzeitige Retsevmo-Dosierung
|
Empfohlene Retsevmo-Dosierung
| |
120 mg oral zweimal täglich
|
80 mg oral zweimal täglich
| |
160 mg oral zweimal täglich
|
80 mg oral zweimal täglich
|
Patienten mit Nierenfunktionsstörungen
Bei Patienten mit leichter (geschätzte glomeruläre Filtrationsrate (eGFR) berechnet auf Grundlage von Modification of Diet in Renal Disease (MDRD) ≥60 und <90 ml/min/1.73 m2), moderater (eGFR ≥30 und <60 ml/min/1.73 m2) oder schwerer (eGFR ≥15 und <30 ml/min/1.73 m2) Nierenfunktionsstörung ist eine Dosisanpassung nicht erforderlich. Bei Patienten mit Nierenerkrankung im Endstadium (End-Stage Renal Disease) oder bei Dialyse liegen keine Daten vor (siehe "Pharmakokinetik" ).
Ältere Patienten
Dosisanpassungen aufgrund des Alters sind nicht erforderlich (siehe "Pharmakokinetik" ).
Kinder und Jugendliche
Retsevmo soll bei Kindern unter 12 Jahren nicht verwendet werden.
Bei Kindern oder Jugendlichen mit RET-fusionspositivem NSCLC oder Schilddrüsenkarzinom liegen keine Daten vor.
Für RETmutiertes MTC liegen sehr begrenzte Daten bei Kindern oder Jugendlichen unter 18 Jahren vor (siehe "Pharmakodynamik" , "Unerwünschte Wirkungen" und "Klinische Wirksamkeit" ). Bei den meisten pädiatrischen Patienten wurde eine Dosis von 92 mg/m2 Körperoberfläche untersucht. Die Dosierung von Retsevmo erfolgt in Abhängigkeit des Körpergewichts (siehe "Dosierung Anwendung" und "Pharmakokinetik" ).
Bei jugendlichen Patienten mit offenen Wachstumsfugen sind die Wachstumsfugen zu überwachen. Die Unterbrechung oder das Absetzen der Therapie ist zu erwägen, je nach Schweregrad von etwaigen Anomalien an der Wachstumsfuge und individueller Nutzen-Risiko-Abwägung (siehe "Warnhinweise und Vorsichtsmassnahmen" ).
Art der Anwendung
Zur oralen Anwendung.
Die Kapsel ist als Ganzes zu schlucken (die Kapseln dürfen vor dem Schlucken nicht geöffnet, zerbrochen oder gekaut werden) und kann mit oder ohne Nahrungsmittel eingenommen werden.
Die Patienten sollten die Dosis jeden Tag ungefähr zu den gleichen Uhrzeiten einnehmen. Eine versäumte Dosis soll nicht eingenommen werden, sofern nicht mindestens 6 Stunden bis zur nächsten geplanten Dosis verbleiben. Der Patient sollte angewiesen werden, bei Erbrechen oder Vergessen der Einnahme von Retsevmo die nächste Dosis zur geplanten Zeit einzunehmen; es darf keine zusätzliche Dosis eingenommen werden.
KontraindikationenÜberempfindlichkeit gegenüber dem Wirkstoff oder einem der unter "Zusammensetzung" angeführten Hilfsstoffe.
Warnhinweise und VorsichtsmassnahmenInterstitielle Lungenerkrankung/Pneumonitis
Bei Patienten, die Selpercatinib erhielten, wurde interstitielle Lungenerkrankung und/oder Pneumonitis gemeldet, einschliesslich schwerer, lebensbedrohlicher und tödlich verlaufender Erkrankung (siehe "Unerwünschte Wirkungen" ). Patienten sind auf pulmonale Symptome zu überwachen, die auf ILD/Pneumonitis hindeuten. Bei Patienten mit akuten oder sich verstärkenden respiratorischen Symptomen, die auf eine ILD hinweisen (z.B. Dyspnoe, Husten und Fieber), ist die Anwendung von Selpercatinib zu unterbrechen und der Patient unverzüglich hinsichtlich des Vorliegens einer ILD zu untersuchen. Je nach Schweregrad der ILD/Pneumonitis kann eine Unterbrechung, Dosisreduktion oder ein endgültiges Absetzen der Behandlung mit Selpercatinib erforderlich sein (siehe "Dosierung/Anwendung" ).
Tumorlysesyndrom (TLS)
Unter Behandlung mit Selpercatinib wurden Fälle von TLS beobachtet. Zu den Risikofaktoren eines TLS gehören eine hohe Tumorlast, vorbestehende chronische Niereninsuffizienz, Oligurie, Dehydratation, Hypotonie und saurer Urin. Diese Patienten sollten engmaschig überwacht und nach klinischer Indikation behandelt werden, eine geeignete Prophylaxe einschliesslich Flüssigkeitszufuhr sollte in Betracht gezogen werden.
Erhöhte ALT/AST-Werte
Bei Patienten, die Selpercatinib im Rahmen von klinischen Studien erhielten, wurden ALT-Erhöhungen Grad ≥3 und AST-Erhöhungen Grad ≥3 berichtet (siehe "Unerwünschte Wirkungen" ).
Gleichzeitige Erhöhungen von AST/ALT (≥3x ULN) und Bilirubin (≥2x ULN) ohne Befund einer Cholestase (erhöhte Serum-ALP) wurden bei 6 Patienten im klinischen Programm beobachtet.
Die ALT- und AST-Werte sind zu überwachen vor dem Beginn der Behandlung mit Selpercatinib, alle 2 Wochen während der ersten 3 Behandlungsmonate, danach monatlich und nach klinischer Indikation. Je nach Schweregrad ist die Behandlung mit Retsevmo zu unterbrechen, die Dosis zu reduzieren oder Retsevmo endgültig zu beenden (siehe "Dosierung/Anwendung" ).
Hypertonie
Bei Patienten, die Selpercatinib im Rahmen von klinischen Studien erhielten, wurde über Hypertonie berichtet (siehe "Unerwünschte Wirkungen" ). Vor Beginn der Behandlung mit Selpercatinib muss der Blutdruck des Patienten kontrolliert sein. Bei Patienten mit unkontrolliertem Blutdruck darf eine Behandlung mit Selpercatinib nicht begonnen werden. Der Blutdruck ist vor Beginn der Behandlung mit Selpercatinib zu optimieren. Während der Behandlung mit Selpercatinib ist der Blutdruck nach 1 Woche zu kontrollieren, danach mindestens monatlich und nach klinischer Indikation. Wenn angemessen, ist eine antihypertensive Behandlung zu beginnen oder anzupassen. Je nach Schweregrad ist die Behandlung mit Retsevmo zu unterbrechen, die Dosis zu reduzieren oder Retsevmo endgültig zu beenden (siehe "Dosierung/Anwendung" ).
Verlängerung des QT-Intervalls
Bei Patienten, die Selpercatinib im Rahmen von klinischen Studien erhielten, wurde über eine Verlängerung des QT-Intervalls berichtet (siehe "Eigenschaften/Wirkungen" ). Torsades de pointes, Ereignisse Grad ≥3 oder klinisch relevante Arrhythmien während der Behandlung, ventrikuläre Tachykardie, Kammerflimmern oder Kammerflattern wurden nicht berichtet. Fatale Ereignisse wie plötzlicher Tod und Herzstillstand wurden bei Patienten mit relevanter kardialer Vorgeschichte berichtet. Bei Patienten mit Erkrankungen wie kongenitalem Long-QT-Syndrom oder erworbenem Long-QT-Syndrom oder anderen klinischen Zuständen, die für Herzrhythmusstörungen prädisponieren, muss Selpercatinib mit Vorsicht angewendet werden.
Die klinischen Studien mit Selpercatinib schlossen Patienten aus bei einem QTc-Intervall >470 ms zu Studienbeginn, klinisch relevanter aktiver kardiovaskulärer Erkrankung oder kürzlichem Myokardinfarkt.
Patienten mit relevantem Risiko für die Entwicklung einer QTc-Verlängerung, einschliesslich Patienten mit bekanntem Long-QT-Syndrom, klinisch relevanter Bradyarrhythmie und schwerer oder unkontrollierter Herzinsuffizienz sind zu überwachen.
Bei allen Patienten müssen Elektrokardiogramm, Elektrolyte und TSH zu Beginn, nach 1 Woche Selpercatinib-Behandlung, mindestens monatlich in den ersten 6 Monaten Selpercatinib-Behandlung und anschliessend alle 3 Monate kontrolliert werden, mit Anpassung der Häufigkeit auf Grundlage von Risikofaktoren einschliesslich Diarrhoe. Eine Hypokaliämie, Hypomagnesiämie und Hypocalcämie sind vor Beginn und während der Behandlung mit Selpercatinib zu korrigieren. Bei klinischer Indikation und/oder bei QTc-Verlängerungen Grad ≥3 sollte an einen Spezialisten überwiesen werden.
Die Anwendung in Kombination mit anderen Arzneimitteln, die bekanntermassen das QTc-Intervall verlängern, sollte vermieden werden. Das QT-Intervall ist häufiger zu kontrollieren, wenn Selpercatinib gleichzeitig mit starken oder moderaten CYP3A-Inhibitoren oder mit Arzneimitteln, die bekanntermassen das QTc-Intervall verlängern, angewendet wird. Je nach Schweregrad ist die Behandlung mit Selpercatinib zu unterbrechen, die Dosis zu reduzieren oder Selpercatinib endgültig zu beenden (siehe "Dosierung/Anwendung" ).
Hypothyreose
Hypothyreose wurde bei Patienten berichtet, die in klinischen Studien Selpercatinib erhalten haben (siehe "Unerwünschte Wirkungen" ). Alle Patienten sollen während der Behandlung mit Selpercatinib eng auf Anzeichen und Symptome einer Schilddrüsenfunktionsstörung beobachtet werden. Die Schilddrüsenfunktion ist vor und regelmässig während der Behandlung mit Selpercatinib zu überwachen. Patienten, die eine Schilddrüsenfunktionsstörung entwickeln, sind medizinisch angemessen zu behandeln.
Gegebenenfalls sprechen Patienten jedoch nicht ausreichend auf die Substitution mit Levothyroxin (T4) an, da Selpercatinib die Umwandlung von Levothyroxin in Triiodthyronin (T3) hemmen kann und eine Ergänzung mit Triiodthyronin erforderlich sein kann.
Blutungen
Schwerwiegende Blutungen, einschliesslich Blutungsereignisse mit Todesfolge, können unter Selpercatinib auftreten. Selpercatinib muss bei Patienten mit schweren oder lebensbedrohlichen Blutungen dauerhaft abgesetzt werden (siehe "Dosierung/Anwendung" ).
Überempfindlichkeit
Bei Patienten unter Selpercatinib wurde Überempfindlichkeit berichtet, wobei die Mehrheit der Fälle bei NSCLC-Patienten mit einer vorangegangenen Anti-PD1/PD-L1-Immuntherapie beobachtet wurde. Zu den Anzeichen und Symptomen einer Überempfindlichkeit gehören Fieber, Ausschlag, Arthralgie oder Myalgie mit begleitender Abnahme der Thrombozyten oder Transaminitis.
Wenn eine Überempfindlichkeit auftritt, ist Retsevmo zu unterbrechen und eine Behandlung mit Corticosteroiden in einer Dosierung von 1 mg/kg einzuleiten. Nach Besserung des Ereignisses ist Selpercatinib bei reduzierter Dosis fortzusetzen, bei Verträglichkeit kann die Selpercatinib-Dosis jede Woche um 1 Dosisstufe erhöht werden, bis die vor Beginn der Überempfindlichkeit angewendete Dosis erreicht ist (siehe "Dosierung/Anwendung" ). Die Steroide werden fortgesetzt, bis der Patient die Zieldosis erreicht hat, und werden dann schrittweise abgesetzt. Bei erneuter Überempfindlichkeit ist Selpercatinib endgültig abzusetzen.
Risiko einer beeinträchtigten Wundheilung
Eine beeinträchtigte Wundheilung kann bei Patienten auftreten, die mit Arzneimitteln behandelt werden, die den VEGF-Signalweg inhibieren (vascular endothelial growth factor, VEGF). Selpercatinib hat daher das Potential, die Wundheilung zu beeinträchtigen.
Selpercatinib ist mindestens 7 Tage vor einer geplanten Operation abzusetzen. Nach grossen Operationen darf über mindestens 2 Wochen und bis zur adäquaten Wundheilung keine Anwendung erfolgen. Die Sicherheit der Wiederaufnahme einer Behandlung mit Selpercatinib nach Besserung von Wundheilungskomplikationen ist nicht belegt.
Pädiatrische Population
Daten bei Kindern und Jugendlichen sind begrenzt.
Epiphysenlösung des Femurkopfes bei jugendlichen Patienten
Epiphysenlösung (Slipped Capital Femoral Epiphysis/Slipped Upper Femoral Epiphysis, SCFE/SUFE) wurde bei jugendlichen Patienten berichtet, die Selpercatinib erhalten haben (siehe "Unerwünschte Wirkungen" ). Patienten sind auf Symptome, die auf eine SCFE/SUFE hindeuten, zu überwachen und medizinisch und chirurgisch angemessen zu behandeln.
Kontrazeption bei Frauen und Männern
Frauen im gebärfähigen Alter müssen während der Behandung und für mindestens zwei Wochen nach der letzten Gabe von Selpercatinib eine sehr zuverlässige Methode zur Empfängnisverhütung anwenden. Männer mit Partnerinnen im gebärfähigen Alter müssen eine zuverlässige Methode zur Empfängnisverhütung während und für mindestens zwei Wochen nach der letzten Gabe von Selpercatinib anwenden.
Fertilität
Basierend auf Ergebnissen aus nicht-klinischen Sicherheitsstudien kann Retsevmo die männliche und weibliche Fertilität beeinträchtigen (siehe "Präklinische Daten" ). Männer und Frauen sollten sich vor der Behandlung bezüglich der Aufrechterhaltung der Fertilität beraten lassen.
Begleitende onkogene Treibermutationen (driver mutations)
Die Wirksamkeit und Sicherheit von Selpercatinib bei Patienten mit bekannten onkogenen Treiberveränderungen ist nicht belegt. Folgende Treibermutationen wurden in der Libretto-001 Studie ausgeschlossen:
-NSCLC: EGFR- oder MET-Mutationen, ALK- oder ROS-Rearrangement, aktivierende Mutation KRAS
-MTC: ALK- oder RAS-Rearrangement
-Schilddrüsenkarzinom (ausser MTC): BRAF-Mutation oder aktivierende RAS-Mutation
InteraktionenWirkung von Selpercatinib auf andere Arzneimittel
Sensitive CYP2C8-Substrate
Die gleichzeitige Anwendung von Selpercatinib mit sensitiven CYP2C8-Substraten kann die Plasmakonzentration der CYP2C8-Substrate erhöhen, was zum vermehrten Auftreten von Nebenwirkungen führen kann, wenn bereits geringere Erhöhungen der CYP2C8 Substrat Konzentrationen zu vermehrten Auftreten von Nebenwirkungen führen können. Wenn eine kombinierte Verabreichung nicht vermieden werden kann, sollten zur möglichen Anpassung der Dosierung die Fachinformation des Kombinationspartners beachtet werden.
Selpercatinib erhöhte die Cmax und AUC von Repaglinid (ein Substrat von CYP2C8) um etwa 188% und 91%. Daher sollte die gemeinsame Anwendung mit sensitiven CYP2C8 Substraten (z.B. Enzalutamid, Paclitaxel, Repaglinid, Torasemid, Sorafenib, Buprenorphin, Selexipag, Dasabuvir und Montelukast) vermieden werden.
Sensitive CYP3A4-Substrate
Die gleichzeitige Anwendung von Selpercatinib mit sensitiven CYP3A4-Substraten kann die Plasmakonzentration der CYP3A4 Substrate erhöhen, was zu vermehrten Auftreten von Nebenwirkungen führen kann, wenn bereits geringere Erhöhungen der CYP3A4 Substrat Konzentrationen zu vermehrten Auftreten von Nebenwirkungen führen können. Wenn eine kombinierte Verabreichung nicht vermieden werden kann, sollte zur möglichen Anpassung der Dosierung die Fachinformation des Kombinationspartners beachtet werden.
Selpercatinib erhöhte die Cmax und AUC von Midazolam (ein Substrat von CYP3A4) um etwa 39% und 54%. Daher sollte die gemeinsame Anwendung mit sensitiven CYP3A4 Substraten (z.B. Alfentanil, Avanafil, Darifenacin, Darunavir, Midazolam, Naloxegol, Simvastatin, Tipranavir, Triazolam, Vardenafil) vermieden werden.
Wirkung anderer Arzneimittel auf Selpercatinib
Der Metabolismus von Sepercatinib erfolgt über CYP3A4. Arzneimittel, die die Enzymaktivität von CAP3A4 beeinflussen, können daher die Pharmakokinetik von Selpercatinib verändern.
Starke CYP3A4-Inhibitoren
Substanzen, die die Plasmakonzentration von Selpercatinib erhöhen können
Selpercatinib wird vorwiegend durch CYP3A4 metabolisiert. Die gleichzeitige Anwendung von Selpercatinib mit einem starken CYP3A4-Inhibitor kann die Selpercatinib-Plasmakonzentration erhöhen (siehe "Dosierung/Anwendung" ).
Itraconazol steigerte die Cmax und AUC von Selpercatinib um 30% und 130% im Vergleich zu Selpercatinib alleine. Wenn gleichzeitig starke CYP3A- und/oder P-gp-Inhibitoren einschliesslich, aber nicht begrenzt auf Ketoconazol, Itraconazol, Voriconazol, Ritonavir, Saquinavir und Posaconazol anzuwenden sind, sollte die Dosis von Selpercatinib reduziert werden (siehe "Dosierung/Anwendung" ).
Starke CYP3A4-Induktoren
Substanzen, die die Plasmakonzentration von Selpercatinib verringern können
Die gleichzeitige Anwendung von Selpercatinib mit einem starken CYP3A4-Induktor kann die Plasmakonzentrationen von Selpercatinib verringern (siehe "Dosierung/Anwendung" ).
Die gleichzeitige Anwendung mit Rifampicin, einem starken CYP3A4-Induktor, führte zu einer Abnahme der Cmax und AUC von Selpercatinib um etwa 87% und 70% im Vergleich zu Selpercatinib alleine, daher sollte die gleichzeitige Anwendung mit starken CYP3A4-Induktoren einschliesslich, aber nicht begrenzt auf Carbamazepin, Phenobarbital, Phenytoin, Rifabutin, Rifampicin und Johanniskraut (Hypericum perforatum) vermieden werden.
Andere Interaktionen
Gleichzeitige Anwendung mit Arzneimitteln, die den pH-Wert des Magens beeinflussen
Selpercatinib weist eine pH-abhängige Löslichkeit auf, wobei die Löslichkeit mit zunehmendem pH-Wert sinkt und zu einem Verlust der Wirksamkeit führen kann (siehe "Dosierung/Anwendung" ).
Bei gleichzeitigen, mehreren täglichen Anwendungen von Ranitidin (ein H2-Rezeptor-Antagonist), welche 2 Stunden nach Selpercatinib verabreicht wurden, wurden keine klinisch relevanten Veränderungen der Pharmakokinetik von Selpercatinib beobachtet.
Gleichzeitige Anwendung mit Protonenpumpeninhibitoren
Bei gleichzeitigen, mehreren täglichen Anwendungen von Omeprazol (ein Protonenpumpeninhibitor) sanken die AUC0-INF und Cmax von Selpercatinib, wenn Selpercatinib nüchtern eingenommen wurde. Die gleichzeitige mehrfache tägliche Anwendung von Omeprazol veränderte die AUC0-INF und Cmax von Selpercatinib nicht signifikant, wenn Retsevmo mit einer Mahlzeit angewendet wurde.
Im nüchternen Zustand führte die gleichzeitige Anwendung von Omeprazol zu einer 69% bis 88% niedrigeren allgemeinen und maximalen Exposition gegenüber Selpercatinib im Vergleich zur alleinigen Anwendung von Selpercatinib.
Gleichzeitige Anwendung mit Arzneimitteln, die Substrate von Transportern sind
Selpercatinib hemmt den Nierentransporter Multidrug and toxin extrusion protein 1 (MATE1). In vivo können Interaktionen von Selpercatinib mit klinisch relevanten Substraten von MATE1, wie etwa Kreatinin, auftreten.
Selpercatinib hemmt P-Glycoprotein (P-gp) und Breast Cancer Resistance Protein (BCRP) in vitro.
In vivo erhöhte Selpercatinib Cmax und AUC des P-gp-Substrats Dabigatran um 43% und 38%. Daher ist bei Einnahme eines sensitiven P-gp-Substrats (z.B. Fexofenadin, Dabigatranetexilat, Colchicin, Saxagliptin) Vorsicht geboten, und insbesondere bei solchen mit enger therapeutischer Breite (z.B. Digoxin) (siehe "Pharmakokinetik" ).
Selpercatinib ist in vitro ein Substrat von P-gp und BCRP.
Selpercatinib ist in vitro ein Substrat von P-Glykoprotein (P-gp) und Breast Cancer Resistance Protein (BCRP), jedoch scheinen diese Transporter die orale Absorption von Selpercatinib nicht zu limitieren, da die orale Bioverfügbarkeit 73% beträgt und die Exposition durch gleichzeitige Gabe des P-gp-Inhibitors Rifampicin minimal gesteigert wurde (Anstieg der AUC0-24 und Cmax von Selpercatinib um etwa 6.5% und 19%).
Pädiatrische Population
Die Interaktionsstudien wurden nur bei Erwachsenen durchgeführt.
In vitro Daten
CYP-Enzyme: Selpercatinib ist kein Inhibitor oder Induktor von CYP1A2, CYP2B6, CYP2C9, CYP2C19 oder CYP2D6 bei klinisch relevanten Selpercatinib Konzentrationen.
Transporter-Systeme: Selpercatinib ist ein Inhibitor von MATE1, P-gp und BCRP, jedoch nicht von OAT1, OAT3, OCT1, OCT2, OATP1B1, OATP1B3, BSEP und MATE2-K bei klinisch relevanten Selpercatinib-Konzentrationen. Selpercatinib kann die Kreatininserumkonzentrationen durch eine verringerte renale tubuläre Kreatininsekretion ansteigen lassen, durch Hemmung von MATE1. Selpercatinib ist ein Substrat von P-gp and BCRP, aber nicht von OAT1, OAT3, OCT1, OCT2, OATP1B1, OATP1B3, MATE1 oder MATE2-K.
Schwangerschaft, StillzeitEmpfängnisverhütung
Frauen im gebärfähigen Alter müssen während der Behandlung und während mindestens 2 Wochen nach der letzten Anwendung von Selpercatinib eine zuverlässige Empfängnisverhütungsmethode anwenden. Männer mit Partnerinnen im gebärfähigen Alter müssen während der Behandlung und bis mindestens 2 Wochen nach der letzten Anwendung von Selpercatinib eine zuverlässige Empfängnisverhütungsmethode anwenden.
Schwangerschaft
Es liegen keine Daten zur Anwendung von Selpercatinib bei schwangeren Frauen vor. Tierstudien haben eine Reproduktionstoxizität gezeigt (siehe "Präklinische Daten" ). Während der Schwangerschaft darf das Arzneimittel nur dann angewendet werden, wenn der potentielle Nutzen das potentielle Risiko für den Fötus rechtfertigt. Schwangere Frauen sollten über das Risiko für den Fötus aufgeklärt werden.
Stillzeit
Es ist nicht bekannt, ob Selpercatinib in die menschliche Muttermilch übergeht. Ein Risiko für Neugeborene/Säuglinge kann nicht ausgeschlossen werden. Wegen der potenziellen Gefahr für den Säugling sollten Frauen während der Behandlung mit Retsevmo und bis mindestens 2 Wochen nach der letzten Anwendung nicht stillen.
Fertilität
Die Auswirkung von Selpercatinib auf die menschliche Fertilität ist nicht bekannt. Basierend auf Ergebnissen aus nicht-klinischen Sicherheitsstudien kann Retsevmo die männliche und weibliche Fertilität beeinträchtigen (siehe "Präklinische Daten" ).
Wirkung auf die Fahrtüchtigkeit und auf das Bedienen von MaschinenEs wurden keine Studien zur Bestimmung der Auswirkungen von Selpercatinib auf die Fahrtüchtigkeit oder die Fähigkeit zum Bedienen von Maschinen durchgeführt. Aufgrund fehlender Studien zur Beurteilung der Fahrtüchtigkeit und der Fähigkeit zum Bedienen von Maschinen während der Behandlung mit Selpercatinib sollten die Patienten darauf hingewiesen werden, beim Lenken eines Fahrzeugs oder beim Bedienen von Maschinen vorsichtig zu sein, falls sie sich während der Behandlung mit Retsevmo müde oder schwindlig fühlen (siehe "Unerwünschte Wirkungen" ).
Unerwünschte WirkungenZusammenfassung des Sicherheitsprofils
Sofern nicht anders beschrieben die Inzidenz von Nebenwirkungen, die in zwei offenen, multizentrischen Dosissteigerungsstudien Phase 1/2 (LIBRETTO-001 und LIBRETTO-121) und zwei offenen, multizentrischen, randomisierten Vergleichsstudien Phase 3 (LIBRETTO-431 und LIBRETTO-531) bei Patienten mit Selpercatinib berichtet wurden, wurden kombiniert ausgewertet und hier zusammengefasst.
Die häufigsten unerwünschten Wirkungen (>20%) bei mit Selpercatinib behandelten Patienten sind oödeme, Diarrhoe, Fatigue, Hypertonie, Mundtrockenheit, erhöhte Aspartataminotransferase (AST), erhöhte Alaninaminotransferase (ALT), Hautausschlag, Bauchschmerzen, Obstipation, Übelkeit, erhöhtes Kreatinin, Kopfschmerzen, Erbrechen, Hämorrhagie und verminderter Appetit.
Die häufigsten schwerwiegenden Nebenwirkungen (≥1.0%) sind Pneumonie (6.0%), Hämorrhagie (2.9%), Bauchschmerzen (2.3%), vermindertes Natrium im Blut (2.1%), Diarrhoe (1.6%), Erbrechen (1.6%), Pyrexie (1.5%), erhöhtes Kreatinin im Blut (1.5%), Überempfindlichkeit (1.4%), Harnwegsinfektionen (1.4%), Übelkeit (1.2%), erhöhte ALT-Werte (1.0.%), erhöhte AST-Werte (1.0%) und Fatigue (1.0%).
Unabhängig von der Zuordnung brachen 9 % der Patienten die Behandlung mit Retsevmo aufgrund von während der Therapie aufgetretenen unerwünschten Ereignissen endgültig ab.
Die Nebenwirkungen, die am häufigsten (bei 3 oder mehr Patienten) zum endgültigen Abbruch führten, waren erhöhte ALT-Werte (0.6%), Fatigue (0.5%), erhöhte AST-Werte (0.4%), Pneumonie (0.3 %), erhöhte Bilirubin-Werte im Blut (0.3%), Elektrokardiogramm QT verlängert (0.2 %), Hämorrhagie( 0.2%), Überempfindlichkeit (0.2%), Thrombozytenzahl erniedrigt (0.2%).
Nachfolgend werden die Nebenwirkungen beschrieben, die bei 1241 Patienten in den Studien LIBRETTO-001, LIBRETTO-121, LIBRETTO-431 und Study LIBRETTO-531 unter Retsevmo aufgetreten sind.
Die Nebenwirkungen werden nach den Systemorganklassen des MedDRA-Systems aufgelistet.
Die Häufigkeiten sind folgendermassen definiert: sehr häufig (≥1/10); häufig (≥1/100 bis <1/10); gelegentlich (≥1/1000 bis <1/100); selten (≥1/10'000 bis <1/1000); sehr selten (<1/10'000) und nicht bekannt (Häufigkeit auf Grundlage der verfügbaren Daten nicht abschätzbar).
Infektionen und parasitäre Erkrankungen
Sehr häufig: Harnwegsinfektionena (17.7%, Grad ≥3: 2.1%), Pneumonieb (11.8%, Grad ≥3: 6.3%).
Erkrankungen des Immunsystemsc
Häufig: Überempfindlichkeitd
Endokrine Erkrankungen
Sehr häufig: Hypothyreose (15.6%)
Stoffwechsel- und Ernährungsstörungen
Sehr häufig: Verminderter Appetit (20.4%, Grad ≥3: 0.7%), Magnesium erniedrigtt (11.8%, Grad ≥3: 0.3%), Calcium erniedrigt (16.3%, Grad ≥3: 2.5%), Albumin erniedrigt (15.1%, Grad ≥3: 0.9%), Natrium erniedrigt (14.7%, Grad ≥3 6.9 %), Kalium erniedrigt (11.0 %, Grad ≥3 2.2 %).
Gelegentlich: Tumorlysesyndrom
Erkrankungen des Nervensystems
Sehr häufig: Kopfschmerzene (26.5%, Grad ≥3: 1.3%), Schwindelf (18.2%, Grad ≥3: 0.4%)
Herzerkrankungen
Sehr häufig: QT-Verlängerung im Elektrokardiogrammg (19.7%, Grad ≥3: 5.6%)
Gefässerkrankungen
Sehr häufig: Hypertonieh (43.3%, Grad ≥3: 20.0%), Blutungeni (23.0%, Grad ≥3: 3.2%).
Erkrankungen der Atemwege, des Brustraums und Mediastinums
Häufig: Chylothorax, interstitielle Lungenerkrankung/Pneunomitisj
Gastrointestinale Erkrankungen
Sehr häufig: Diarrhoek (46.7%, Grad ≥3: 4.9%), Mundtrockenheitl (40.9% Grad ≥3: 0.1%), Bauchschmerzenm (33.4%, Grad ≥3: 2.4%), Obstipation (30.6%, Grad ≥3: 0.9%), Übelkeit (29.3%, Grad ≥3: 1.6%), Erbrechenn (23.4%, Grad ≥3: 2.2%), Stomatitiso (16.7%, Grad ≥3: 0.3%).
Häufig: Chylöser Aszitesp
Erkrankungen der Haut und des Unterhautgewebes
Sehr häufig: Hautausschlagq (34.7%, Grad ≥3: 1.1%)
Skelettmuskulatur-, Bindegewebs- und Knochenerkrankungen
Häufig: Epiphysenlösung (Slipped Capital Femoral Epiphysis/Slipped Upper Femoral Epiphysis) (6.4%) wurde bei mit Selpercatinib behandelten Kindern und Jugendlichen beobachtet (n=47).
Erkrankungen der Geschlechtsorgane und der Brustdrüse
Häufig: Erektile Dysfunktion (7.7%, Grad ≥3: 0.3%).
Allgemeine Erkrankungen und Beschwerden am Verabreichungsort
Sehr häufig: Ödemer (47.9%, Grad ≥3: 1.2%), Fatigues (44.0%, Grad ≥3: 4.0%), Pyrexie (18.4%, Grad ≥3: 0.5%).
Leber- und Gallenerkrankungent
Sehr häufig: ALT-Erhöhung (38.3%, Grad ≥3: 12.9%), AST-Erhöhung (39.4%, Grad ≥3: 8.7%), Gesamtbilirubin erhöht (17.9%, Grad ≥3: 1.6%), alkalische Phosphatase erhöht (14.6%, Grad ≥3: 2.1%).
Erkrankungen der Nieren und Harnweget
Sehr häufig: Kreatinin-Erhöhung (27.5%, Grad ≥3: 2.3%)
Erkrankungen des Blutes und des Lymphsystemst
Sehr häufig: Lymphozytenzahl erniedrigt (12.1%, Grad ≥3: 4.9%), Thrombozytenzahl erniedrigt (16.8%, Grad ≥3: 2.9%), Hämoglobin erniedrigt (14.7%, Grad ≥3: 2.9%), Neutrophilenzahl erniedrigt (13.1%, Grad ≥3: 2.4%), Leukozytenzahl erniedrigt (16.3%, Grad ≥3: 1.4%).
a Harnwegsinfektionen umfasst Harnwegsinfektion, Zystitis, Urosepsis, Escherichia-Infektion der Harnwege, Escherichia-Pyelonephritis, Niereninfektion, Nitrit im Urin, Pyelonephritis, Urethritis, bakterielle Harnwegsinfektion und urogenitale Pilzinfektion
b Pneumonie umfasst Pneumonie, Lungeninfektion, Aspirationspneumonie, Empyem, Lungenkonsolidierung, Pleurainfektion, bakterielle Pneumonie, Staphylokokken-Pneumonie, atypische Pneumonie, Lungenabszess, Pneumocystis-jirovecii-Pneumonie, Pneumokokken-Pneumonie, Respiratorisches-synzytial-Virus-Pneumonie, infektiöser Pleuraerguss und Virus-Pneumonie.
c Überempfindlichkeitsreaktionen waren durch einen makulopapulösen Hautausschlag gekennzeichnet, dem häufig Fieber mit assoziierten Arthralgien/Myalgien vorausging während des ersten Behandlungszyklus des Patienten (typischerweise zwischen Tag 7 und 21)
d Überempfindlichkeit umfasst Arzneimittelüberempfindlichkeit und Überempfindlichkeit
e Kopfschmerzen umfasst Kopfschmerzen, Sinus-Kopfschmerzen und Spannungskopfschmerzen
f Schwindel umfasst Schwindel, Vertigo, Präsynkope und posturaler Schwindel.
g QT-Verlängerung im Elektrokardiogramm umfasst QT-Verlängerung im Elektrokardiogramm und QT-Intervall-Abweichungen im Elektrokardiogramm.
h Hypertonie umfasst Hypertonie und erhöhter Blutdruck.
i Blutungen umfasst Epistaxis, Hämoptysis, Bluterguss, Hämaturie, rektale Blutungen, vaginale Blutungen, zerebrale Blutungen, traumatische Hämatome, Blut im Urin, Konjunktiva-Blutungen, Ekchymose, Zahnfleischbluten, Hämatochezie, Petechien, Blutblasen, spontane Hämatome, Hämatome der Bauchwand, anale Blutungen, Angina bullosa haemorrhagica, disseminierte intravaskuläre Koagulation, Augenblutungen, Magenblutungen, gastrointestinale Blutungen, intrakranielle Blutungen, subkutane Blutungen, hämorrhoidale Blutungen, Leberhämatome, intra-abdominale Blutungen, Mundblutungen, Ösophagusblutungen, Beckenhämatome, periorbitale Hämatome, periorbitale Blutungen, pharyngeale Blutungen, Lungenkontusion, Purpura, retroperitoneale Hämatome, Hautblutungen, subarachnoidale Blutungen, intestinale Divertikelblutung, Augenhämatome, Hämatemesis, Blutungen, hämorrhagischer Schlaganfall, Leberblutungen, Larynxblutungen, Blutungen des unteren Gastrointestinaltrakts, Melaena, Menorrhagie, positiver Test auf okkultes Blut, Blutungen nach Eingriffen, postmenopausale Blutungen, retinale Blutungen, Sklerablutungen, subdurale Blutungen, traumatischer Hämatothorax, Tumorblutungen, Blutungen des oberen Gastrointestinaltrakts, uterine Blutungen, Hämatome an der Punktionsstelle von Gefässen, Hämarthrose und Hämatom, arterielle Blutungen, Augenkontusion, Hämothorax, subdurale Hämatome, Hodenblutung, Trachealblutung, International Normalized Ratio erhöht, aktivierte partielle Thromboplastinzeit verlängert, Koagulopathie, Hämoglobin verringert, Fibrin-D-Dimer erhöht und Anzahl roter Blutkörperchen verringert.
j Interstitielle Lungenerkrankung/Pneumonitis umfasst interstitielle Lungenerkrankung, Pneumonitis, Strahlenpneumonitis, restriktive pulmonale Erkrankung, akutes Atemnotsyndrom (Acute Respiratory Distress Syndrome), Alveolitis, Bronchiolitis, Langerhans-Zell-Histiozytose, Strahlenschädigung der Lunge, zystische Lungenerkrankung, Lungeninfiltration und Lungenopaleszenz.
k Diarrhoe umfasst Diarrhoe, Analinkontinenz, Stuhldrang, häufiger Stuhlgang und gastrointestinale Hypermotilität.
l Mundtrockenheit umfasst Mundtrockenheit und trockene Mundschleimhaut.
m Bauchschmerzen umfassen Bauchschmerzen, Oberbauchschmerzen, Bauchbeschwerden, Unterbauchschmerzen und gastrointestinale Schmerzen.
n Erbrechen umfasst Erbrechen, Würgen und Aufstossen.
o Stomatitis umfasst Stomatitis, Mund-Ulzera, Mukosaentzündung und orale Mukosa-Blasen
p Chyloser Aszites umfasst chyloser Aszites und Aszites chylos (MedDRA LLTs).
q Hautausschlag umfasst Hautausschlag, makulopapulöser Hautausschlag, Dermatitis, Hautexfoliation, makulöser Hautausschlag, erythematöser Hautausschlag, Urtikaria, allergische Dermatitis, exfoliativer Hautausschlag, papulöser Hautausschlag, morbilliformer Hautausschlag, pruritischer Hautausschlag, vesikulärer Hautausschlag, Schmetterlingsausschlag, follikulärer Hautausschlag, generalisierter Hautausschlag, pustulöser Hautausschlag und Hautreaktion
r Ödeme umfassen periphere Ödeme, Gesichtsödeme, periorbitale Ödeme, Gesichtsschwellungen, lokalisierte Ödeme, periphere Schwellungen; generalisierte Ödeme, Augenlidödeme, Augenschwellungen, Lymphödeme, genitale Ödeme, Skrotalschwellungen, Angioödeme, Augenödeme, Ödeme; Skrotalödeme, Hautödeme, Schwellungen, orbitale Ödeme, Hodenschwellungen, vulvovaginale Schwellungen, Orbitalschwellung, Penisödem, periorbitale Schwellung und Augenlidschwellung
s Fatigue umfasst Fatigue, Asthenie und Unwohlsein.
t Auf Basis von Laboruntersuchungen. Der prozentuale Anteil errechnet sich auf Basis der Anzahl Patienten mit Ausgangswert und mindestens einem nachfolgenden Wert im Nenner.
Beschreibung ausgewählter Nebenwirkungen
Transaminasen-Anstieg (AST/ALT-Erhöhung)
Auf Basis von Laboruntersuchungen wurden ALT- und AST-Erhöhungen in alle Studien (bis Mai 2024) bei 38.3% und 39.4% der Patienten berichtet. ALT- oder AST-Erhöhungen Grad 3 oder 4 wurden von 12.9% und 8.7% der Patienten berichtet. ALT wurde bei 1% der Patienten als schwerwiegend berichtet, und AST wurde bei 1% der Patienten als schwerwiegend berichtet.
Die mediane Zeit bis zum ersten Auftreten betrug: AST-Erhöhung 4.7 Wochen (Bereich: 0.7, 227.9), ALT-Erhöhung 4.4 Wochen (Bereich: 0.9, 186.1) in der LIBRETTO-001-Studie (bis Januar 2023); AST-Erhöhung 5.1 Wochen (Bereich: 0.7, 88.1), ALT-Erhöhung 5.1 Wochen (Bereich: 0.7, 110.9) in der LIBRETTO-431-Studie (bis Mai 2023), AST-Erhöhung 6.1 Wochen (Bereich: 0.1, 85.1), ALT-Erhöhung 6.1 Wochen (Bereich: 0.1, 85.1) in der LIBRETTO-531-Studie (bis Mai 2023).
Bei Patienten, die eine ALT- oder AST-Erhöhung Grad 3 oder 4 entwickeln, wird eine Dosisanpassung empfohlen (siehe "Dosierung/Anwendung" ).
Verlängerung des QT-Intervalls
Bei den 837 Patienten der LIBRETTO-001-Studie (bis Januar 2023), die ein EKG hatten, ergab eine Auswertung der Daten, dass 8.1% der Patienten nach Studienbeginn einen maximalen QTcF-Wert >500 ms und 21.6% der Patienten einen maximalen Anstieg des QTcF-Intervalls >60 ms gegenüber dem Ausgangswert aufwiesen. Bei 156 Patienten der LIBRETTO-431-Studie (bis Mai 2023) mit vorhandenen EKGs hatten 5.1% der Patienten nach Studienbeginn einen maximalen QTcF-Wert >500 msec, und 16.7% der Patienten wiesen einen maximalen Anstieg des QTcF-Intervalls >60 ms gegenüber dem Ausgangswert auf. Bei 191 Patienten der LIBRETTO-531-Studie (bis Mai 2023) mit vorhandenen EKGs hatten 3.7% der Patienten nach Studienbeginn einen maximalen QTcF-Wert >500 msec, und 17.8% der Patienten wiesen einen maximalen Anstieg des QTcF-Intervalls >60 ms gegenüber dem Ausgangswert auf.
In allen Studien (bis Mai 2024) LIBRETTO-001, LIBRETTO-431 LIBRETTO-531 und LIBRETTO-121 gab es keine Berichte über Torsades de pointes, Ereignisse Grad ≥3 oder unter Behandlung aufgetretene klinisch relevante Ereignisse mit Arrhythmie, ventrikulärer Tachykardie, Kammerflimmern oder Kammerflattern. Fatale Ereignisse mit plötzlichem Tod und Herzstillstand wurden bei Patienten mit relevanter kardialer Vorgeschichte berichtet. Über alle Studien hinweg brachen zwei Patienten (0.2%) die Behandlung mit Selpercatinib aufgrund von QT-Verlängerung ab.
Eine Unterbrechung der Behandlung mit Retsevmo oder eine Dosisanpassung kann erforderlich sein (siehe "Dosierung/Anwendung" und "Warnhinweise und Vorsichtsmassnahmen" ).
Hypertonie
In allen Studien (bis Mai 2024) Hypertonien wurden bei 43% der Patienten berichtet, Ereignisse Grad ≥3 bei 20.0% der Patienten.
Der mediane maximale Anstieg des systolischen Blutdrucks gegenüber dem Ausgangswert bei den 837 Patienten der LIBRETTO-001-Studie (Stand Januar 2023), bei denen der Blutdruck gemessen wurde, betrug 32 mmHg (Bereich: -15, +100). Die Ergebnisse für den diastolischen Blutdruck waren ähnlich, jedoch war das Ausmass der Anstiege geringer. In der LIBRETTO-001-Studie behielten nur 10.3% der Patienten ihren Grad zu Studienbeginn während der Behandlung bei, bei 40.7% zeigte sich eine Verschiebung mit Anstieg um 1 Grad, bei 38.5% um 2 Grade und bei 9.8% um 3 Grade.
Insgesamt berichteten 19.68% in der LIBRETTO-001, 20.3% in der LIBRETTO-431 (Stand Mai 2023) und 19.2% in der LIBRETTO-531 (Stand Mai 2023) während der Behandlung eine Hypertonie Grad 3 (definiert als maximaler systolischer Blutdruck von mehr als 160 mmHg). Eine während der Behandlung aufgetretene Hypertonie Grad 4 wurde bei 0.1% der Patienten in der LIBRETTO-001-Studie (Stand Mai 2023) berichtet, in den Studien LIBRETTO-431 und LIBRETTO-531 (Stand Mai 2023) gab es keine Berichte.
Bei zwei Patienten (0.2%) kam es in der LIBRETTO-001-Studie (Stand Januar 2023) aufgrund von Hypertonie zu einem endgültigen Therapieabbruch, und bei keinem Patienten der Studien LIBRETTO-431 und LIBRETTO-531 (Stand Mai 2023).
Für Patienten, die eine Hypertonie entwickeln, werden Dosisanpassungen empfohlen (siehe "Dosierung/Anwendung" ). Die Behandlung mit Selpercatinib muss bei einer medizinisch relevanten Hypertonie unterbrochen werden, bis der Blutdruck mit entsprechender Therapie wieder unter Kontrolle ist. Die Behandlung sollte mit der nächstniedrigeren Dosis wieder aufgenommen werden, wenn dies klinisch indiziert ist.
Erhöhtes Kreatinin
Bei gesunden Probanden führte die Anwendung von Selpercatinib 160 mg oral zweimal täglich zu einem Anstieg der Kreatinin-Serumkonzentration von 18% nach 10 Tagen. Wenn anhaltende Erhöhungen der Kreatinin-Serumkonzentrationen beobachtet werden, sind für die Nierenfunktion alternative Marker in Erwägung zu ziehen (siehe "Eigenschaften/Wirkungen" ).
Zusätzliche Informationen zu besonderen Patientengruppen
Pädiatrische Population
Es gab 3 Patienten <18 Jahre (Bereich 15-17) mit RET-mutiertem MTC in der LIBRETTO-001-Studie. Es gab 11 Patienten <18 Jahre (Bereich 6-17) mit RET-fusionspositivem Schilddrüsenkarzinom und 12 Patienten <18 Jahre (Bereich 2-17) mit RET-mutiertem MTC in der LIBRETTO-121. Es gab 1 Patient mit einem Alter von 12 Jahren mit RET-mutiertem MTC in der LIBRETTO-531-Studie.
Von den insgesamt 33 Patienten in LIBRETTO-121 berichteten 97% unerwünschte Ereignisse während der Behandlung (TEAE). Die häufigsten unerwünschten Ereignisse (≥25%) waren Diarrhoe, Pyrexie, Übelkeit, Bauchschmerzen, Kopfschmerzen, erhöhter AST-Wert, erhöhter ALT-Wert, Coronavirus-Infektion, Husten, Epistaxis und Erbrechen.
TEAE Grad ≥3 traten bei 57.6% der Patienten auf. Die häufigsten unerwünschten Ereignisse Grad ≥3 waren Gewichtszunahme (12.1%), Erbrechen (9.1%), ALT-Anstieg (6.1%), Obstipation (6.1%), Hypokaliämie (6.1%) und Abnahme der Neutrophilenzahl (6.1%).
Schwerwiegende unerwünschte Ereignisse (SAE) traten bei 42.4% der Patienten auf. Das häufigste schwerwiegende unerwünschte Ereignis war Erbrechen (6.1%). Alle anderen SAEs wurden von jeweils 1 Patient berichtet. Fatale Ereignisse wurden nicht berichtet.
Daten aus Tierstudien deuten auf ein potenzielles Risiko für Wachstumsstörungen bei Kindern, die ihre endgültige Körpergrösse nicht erreicht haben (siehe "Präklinische Daten" ).
Bei 3 pädiatrischen Patienten, die in klinischen Studien mit Selpercatinib behandelt wurden, wurde über eine Epiphysenverlagerung des oberen Oberschenkelknochens (SCFE/SUFE) berichtet (siehe "Warnhinweise und Vorsichtsmassnahmen" ).
Ältere Patienten
Von den in der LIBRETTO-001-Studie mit Selpercatinib behandelten Patienten waren 24.7% der Patienten ≥65-74 Jahre, 8.6% waren 75-84 Jahre und 1.0% ≥85 Jahre alt. Das Sicherheitsprofil bei älteren Patienten (≥65 Jahre) entspricht jenem bei jüngeren Patienten (<65 Jahre).
Die Meldung des Verdachts auf Nebenwirkungen nach der Zulassung ist von grosser Wichtigkeit. Sie ermöglicht eine kontinuierliche Überwachung des Nutzen-Risiko-Verhältnisses des Arzneimittels. Angehörige von Gesundheitsberufen sind aufgefordert, jeden Verdacht einer neuen oder schwerwiegenden Nebenwirkung über das Online-Portal ElViS (Electronic Vigilance System) anzuzeigen. Informationen dazu finden Sie unter www.swissmedic.ch.
ÜberdosierungDie Symptome einer Überdosierung sind nicht bekannt. Bei vermuteter Überdosierung sollte unterstützende Behandlung erfolgen.
Eigenschaften/WirkungenATC-Code
L01EX22
Wirkungsmechanismus
Selpercatinib aus der Gruppe der kleinen Moleküle ist ein Inhibitor der RET (Rearranged during transfection)-Rezeptor-Tyrosinkinase. Selpercatinib hemmte den RET-Wildtyp und mehrere mutierte RET-Isoformen, sowie VEGFR1 und VEGFR3 mit IC50-Werten im Bereich von 0.92 nM bis 67.8 nM. In anderen Enzymtests hemmte Selpercatinib ausserdem bei höheren, klinisch noch erreichbaren Konzentrationen FGFR 1, 2 und 3. Selpercatinib war 250-fach selektiver für RET als für 98% von ~300 Kinasen, einschliesslich VEGFR2, dies wurde in präklinischen Studien untersucht. In Zelltests hemmte Selpercatinib RET bei einer etwa 60-fach niedrigeren Konzentrationen als FGFR1 und 2 und bei etwa 8-fach niedrigeren Konzentrationen als VEGFR3.
Bestimmte Punktmutationen der RET oder chromosomale Neuanordnungen mit In-frame-Fusionen der RET mit verschiedenen Partnern können zu konstitutiv aktivierten, chimeren RET Fusionsproteinen führen, die als onkogene Treiber fungieren können, indem sie die Zellproliferation von Tumorzelllinien fördern. In Tumormodellen zeigte Selpercatinib in vitro und in vivo antitumorale Aktivität in Zellen, die infolge von Genfusionen und Genmutation einschliesslich CCDC6 RET, KIF5B RET, RET V804M und RET M918T eine konstitutive Aktivierung des RET-Protein erworben haben. Darüber hinaus zeigte Selpercatinib antitumorale Aktivität bei Mäusen mit intrakranialer Implantation eines von Patienten stammenden RET-fusionspositiven Tumors.
Pharmakodynamik
Kardiale Elektrophysiologie
In einer eingehenden QT-Studie mit positiver Kontrolle bei 32 gesunden Probanden zeigte eine Expositions-Response-Analyse, dass supra-therapeutische Konzentrationen zu einem Anstieg des QTc >20 ms führen könnten.
Verlängerungen des QT-Intervalls wurden bei Patienten unter Selpercatinib berichtet. Daher können bei Patienten Unterbrechungen der Behandlung oder Dosisanpassungen erforderlich sein (siehe "Dosierung/Anwendung" und "Warnhinweise und Vorsichtsmassnahmen" ).
Klinische Wirksamkeit
Die Wirksamkeit von Retsevmo wurde bei erwachsenen Patienten mit fortgeschrittenem RET-fusionspositivem NSCLC sowie bei Patienten mit RET-mutiertem MTC und RET-fusionspositivem Schilddrüsenkarzinom mit oder ohne vorausgegangene Standard-Erstlinientherapie untersucht, die in die multizentrische, offene, einarmige klinische Studie der Phase 1/2 LIBRETTO-001 aufgenommen worden waren.
Patienten in den Phase 1- und Phase 2-Abschnitten wiesen einen Progress unter Standardtherapie auf, vertrugen die Standardtherapie nicht, oder es gab keine Standardtherapie.
Patienten mit ZNS-Metastasen waren einschlussfähig, sofern sie stabil waren, während Patienten mit symptomatischen primären ZNS-Tumoren, Metastasen, leptomeningealer Karzinomatose oder Rückenmarkkompression ausgeschlossen wurden. Patienten mit bekannter primärer Treiberveränderung ausser RET, klinisch signifikanter aktiver Herz-Kreislauf-Erkrankung oder Herzinfarkt in der Anamnese, QTcF-Intervall > 470 ms wurden ausgeschlossen.
Im Phase-2-Abschnitt der Studie erhielten die Patienten Retsevmo 160 mg oral zweimal täglich bis zum Auftreten einer nicht-akzeptablen Toxizität oder bis zur Progression. Das Vorhandensein einer RET-Gen-Veränderung wurde prospektiv in lokalen Laboratorien mithilfe von Next Generation Sequencing (NGS), Polymerase-Kettenreaktion (PCR) oder Fluorescence In Situ Hybridization (FISH) bestimmt. Die wichtigen Wirksamkeitsendpunkte waren die Gesamtansprechrate (overall response rate, ORR) und die Ansprechdauer (duration of response, DOR), die durch ein verblindetes unabhängiges Komitee (blinded independent review committee, BIRC) auf Basis von RECIST v1.1 bestimmt wurden.
Therapienaives RET-fusionspositives nicht-kleinzelliges Lungenkarzinom
LIBRETTO-431
Die Wirksamkeit von Retsevmo bei RETfusionspositivem NSCLC wurde in der Studie LIBRETTO-431 untersucht, eine multizentrische, randomisierte, offene Phase 3 Studie zum Vergleich von Selpercatinib zu platinbasierter Pemetrexed-Therapie, mit oder ohne Pembrolizumab, bei Patienten mit fortgeschrittenem oder metastasiertem, RET-fusionspositivem NSCLC. Eingeschlossen wurden erwachsene Patienten mit histologisch bestätigtem, nicht-resezierbarem, lokal fortgeschrittenem oder metastasiertem NSCLC ohne vorherige systemische Therapie der metastasierten Erkrankung. Patienten mit adjuvanter oder neoadjuvanter Therapie wurden ebenfalls eingeschlossen, wenn die letzte Gabe der systemischen Behandlung mindestens 6 Monate vor der Randomisierung abgeschlossen war. Patienten mit plattenepithelialer Histologie und Patienten mit kardiovaskulärer Erkrankung, Myokardinfarkt in den letzten 6 Monaten, QTc-Intervall >470 ms oder einer Begleitmedikation, von der bekannt ist, dass sie QTc-Verlängerung verursacht, wurden ausgeschlossen. Die Patienten erhielten 160 mg Selpercatinib zweimal täglich (Anfangsdosis) oder eine platinbasierte Therapie und Pemetrexed, mit oder ohne Pembrolizumab. Die Feststellung einer RET-Genveränderung erfolgte prospektiv unter Verwendung von NGS (Next-Generation Sequencing) oder PCR (Polymerase-Kettenreaktion). Die Patienten wurden stratifiziert nach geographischer Region (Ostasien vs. andere), Status hinsichtlich der durch den Prüfarzt bewerteten ZNS-Metastasierung zu Studienbeginn (fehlend oder unbekannt vs. vorhanden), und ob der Prüfarzt (vor der Randomisierung) eine Behandlung des Patienten mit oder ohne Pembrolizumab beabsichtigt hatte.
Der primäre Endpunkt war das progressionsfreie Überleben (progression-free survival, PFS) auf Basis von RECIST 1.1 durch ein verblindetes unabhängiges Komitee (blinded, independent review committee, BICR). Das PFS wurde sequentiell zuerst in der Population derjenigen Patienten ermittelt, bei welchen der Prüfarzt die Anwendung von Pembrolizumab beabsichtigt hatte (ITT-Pembrolizumab), wenn sie in die Kontrollgruppe randomisiert wurden, und dann in der gesamten ITT-Population. Zu den fehlerkontrollierten sekundären Wirksamkeitsendpunkten gehörte das Gesamtüberleben (overall survival, OS).
Das mediane Alter der Patienten in der ITT-Population betrug 62.5 Jahre (Bereich 31 bis 87 Jahre). 54.8% der Patienten waren Frauen. 40.2% der Patienten waren weisser, 57.4% asiatischer, 0.8% schwarzer Abstammung. 67.4% hatten nie geraucht. In der ITT-Population wiesen 93.5% eine metastasierte Erkrankung auf, 19.5% der Patienten hatten ZNS-Metastasen zu Studienbeginn. Der berichtete ECOG-Performance-Status betrug 0-1 (96.9%) oder 2 (3.1%). Die häufigsten Fusionspartner waren KIF5B (46.0%), gefolgt von CCDC6 (9.6%).
Von den 261 Patienten, die in die Intention-to-treat (ITT)-Population der LIBRETTO-431-Studie eingeschlossen und randomisiert wurden, wurden 212 in die ITT-Pembrolizumab-Population stratifiziert. In der ITT-Pembrolizumab-Population erhielten 129 Patienten Selpercatinib, während 83 eine platinbasierte Pemetrexed-Chemotherapie mit Pembrolizumab erhielten.
Das mediane Alter der Patienten in der ITT-Pembrolizumab-Population betrug 61.5 Jahre (Bereich 31 bis 84 Jahre). 53.3% der Patienten waren Frauen. 41.3% der Patienten waren weisser, 56.3% asiatischer und 1% schwarzer Abstammung. 67.9% haben nie geraucht. In der ITT-Pembrolizumab-Population hatten 93% eine metastasierte Erkrankung, 20.3% der Patienten hatten ZNS-Metastasen zu Studienbeginn. Der berichtete ECOG-Performance-Status betrug 0-1 (96.7%) oder 2 (3.3%). Der häufigste Fusionspartner war KIF5B (44.8%), gefolgt von CCDC6 (9.9%). Die Studie erreichte die primären Endpunkte einer Verbesserung des PFS in der ITT-Pembrolizumab-Population und in der ITT-Population.
Zum Zeitpunkt der zuvor geplanten Wirksamkeitszwischenauswertung (Stand Mai 2023) in der ITT-Population betrug das mediane progressionsfreie Überleben 24.84 Monate (95% Konfidenzintervall [CI]: 17.31, nicht schätzbar [NE]) mit Selpercatinib und 11.17 Monate (95% CI: 8.77, 16.76) im Kontrollarm, bei einer Hazard-Ratio (HR) für Progress oder Tod, 0.482 (95% CI: 0.331, 0.700; p=0.0001).
Zum Zeitpunkt einer zuvor geplanten Wirksamkeitszwischenauswertung (Stand Mai 2023) in der ITT-Pembrolizumab-Population betrug das mediane progressionsfreie Überleben 24.84 Monate (95% Konfidenzintervall [CI], 16.89 bis NE) mit Selpercatinib und 11.17 Monate (95% CI, 8.77 bis 16.76) mit Kontrollbehandlung (Hazard-Ratio für Progress oder Tod, 0.465; 95% CI, 0.309 bis 0.699; P=0.0002).
Das OS war zum Zeitpunkt der primären PFS-Analyse nicht reif. Zum Zeitpunkt einer interim deskriptiven Zwischenanalyse des OS (01 May 2024) (43 % der vorab festgelegten OS-Ereignisse, die für die endgültige Analyse erforderlich sind) wurden in der ITT-Population 75 Ereignisse in beiden Armen beobachtet (49 (31% der ITT ) im Selpercatinib-Arm und 26 (25%) im Kontrollarm) und die HR betrug 1.259 ([95 % KI: 0.777-2.040]).
Abbildung 1. LIBRETTO-431: Kaplan-Meier-Diagramm des OS (BICR-Bewertung, ITT-Population)
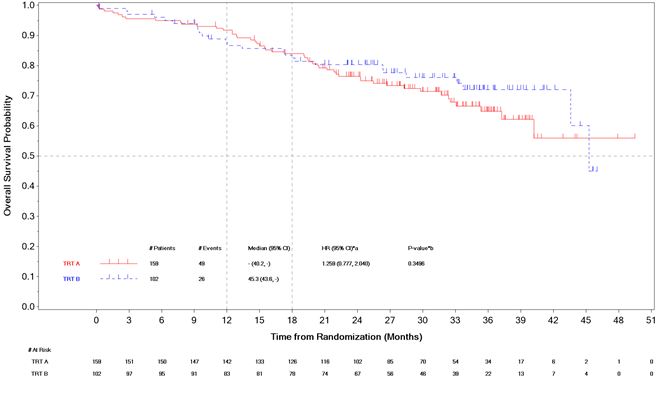
Abbreviations: CI = confidence interval; HR = hazard ratio; TRT A = Selpercatinib; TRT B = Carboplatin or Cisplatin+Pemetrexed+/-Pembrolizumab.
*a HR - IWRS stratified hazard ratio from Cox proportional hazard model and 95% CI of TRT A versus TRT B.
*b Log-rank IWRS stratified p-value(2-sided) for comparison of TRT A versus TRT B
Cut-off datum: 01 Mai 2024.
Von 68 Patienten im Kontrollarm, bei denen die Krankheit fortschritt, erhielten 50 Patienten (74 %) Selpercatinib bei der Progression. Von den 71 Patienten im Selpercatinib-Arm, bei denen es zu einem Fortschreiten der Krankheit kam, erhielten 16 (23 %) eine Chemotherapie und/oder eine Therapie mit Immuncheckpoint-Inhibitoren und 44 (62 %) setzten die Behandlung mit Selpercatinib fort.
Vorbehandeltes RET-fusionspositives nicht-kleinzelliges Lungenkarzinom
Insgesamt hatten 247 Patienten mit RET-Fusions-positivem NSCLC, welche in der LIBRETTO-001 eingeschlossen wurden, zuvor eine Platin-basierte Chemotherapie erhalten. Das mediane Alter lag bei 61 Jahren (Bereich 23 bis 81 Jahre). 56.7% der Patienten waren weiblich. 43.7% der Patienten waren Weisse, 47.8% waren Asiaten, 4.9% waren Schwarz und 66.8% waren Nie-Raucher. Die meisten Patienten (98.8%) hatten bei Studieneinschluss eine metastasierte Erkrankung, und 31.2% hatten nach Aussage des Prüfarztes bei Baseline ZNS-Metastasen. Der ECOG-Performance-Status wurde mit 0-1 (97.1%) oder 2 (2.8 %) angegeben. Der häufigste Fusionspartner war KIF5B (61.9%), gefolgt von CCDC6 (21.5%) und dann NCOA4 (2.0%). Zwei Patienten hatten ein lokal fortgeschrittenes NSCLC, ein Patient hatte ein plattenepitheliales NSCLC. Die mediane Anzahl vorangegangener systemischer Therapien lag bei 2 (Bereich 1 bis 15), und 43.3% (n=107/247) hatten 3 oder mehr systemische Behandlungen erhalten. Zu den vorausgegangenen Behandlungen gehörten eine Anti-PD1/PDL1-Therapie (58.3%), Multikinase-Inhibitoren (MKI) (31.6%) und Taxane (34.8%); 41.3% hatten eine andere systemische Therapie.
Zum Stichtag 13. Januar 2023 in der für die Wirksamkeit geeigneten Patientenpopulation (Patienten, die zuvor eine Platin-basierten Chemotherapie erhalten und mindestens 6 Monate Beobachtung in der LIBRETTO-001 Studie abgeschlossen hatten, n=247) betrug die objektive Ansprechrate (objective response rate, ORR) 61.5% (95% CI: 55.2, 67.6) und die mediane Ansprechdauer 31.6 Monate (95% CI: 20.4, 42.3) bei einer medianen Beobachtungszeit von 39.52 Monaten.
Vandetanib- und Cabozantinib-naive Patienten mit RET-mutiertem medullärem Schilddrüsenkarzinom
LIBRETTO-531
Die Wirksamkeit von Retsevmo bei RET-mutiertem MTC wurde in der LIBRETTO-531 untersucht, eine multizentrische, randomisierte, offene Vergleichsstudie Phase 3 zum Vergleich von Selpercatinib mit Cabozantinib oder Vandetanib nach Entscheidung des Prüfarztes, bei Patienten mit progressivem, fortgeschrittenem, Kinase-Inhibitor-naivem, RET-mutiertem MTC. Eingeschlossen wurden erwachsene oder jugendliche Patienten mit histologisch bestätigtem, nicht-resezierbarem, lokal fortgeschrittenem oder metastasiertem MTC ohne vorausgegangene Behandlung mit einem Kinase-Inhibitor.
Patienten mussten zum Screening eine radiologisch progressive Erkrankung nach RECIST 1.1 aufweisen, im Vergleich zu einem Bild, das in den letzten 14 Monaten aufgenommen wurde. Patienten mit kardiovaskulärer Erkrankung, Myokardinfarkt in den letzten 6 Monaten, QTc-Intervall >470 ms oder einer Begleitmedikation, von der bekannt ist, dass sie QTc-Verlängerung verursacht, wurden ausgeschlossen. Die Patienten erhielten 160 mg Selpercatinib zweimal täglich (Anfangsdosis) oder nach Wahl des Prüfarztes Cabozantinib (140 mg einmal täglich) oder Vandetanib (300 mg einmal täglich). Die RET-Genveränderung wurde prospektiv unter Verwendung von NGS (Next-Generation Sequencing) oder PCR (Polymerase-Kettenreaktion) ermittelt. Die Patienten wurden stratifiziert nach RET-Mutation (M918T vs. andere) und beabsichtigter Behandlung, bei Randomisierung in den Kontrollarm (Cabozantinib vs. Vandetanib). Primärer Wirksamkeitsendpunkt war das PFS nach RECIST 1.1 durch BIRC. Wesentliche sekundäre Wirksamkeitsendpunkte umfassten das therapieversagenfreie Überleben (treatment-failure-free survival, TFFS) und die Verträglichkeit im Vergleich. Zu den anderen sekundären Wirksamkeitsendpunkten gehörte das OS.
Von den 291 Patienten der Intention-to-treat (ITT)-Population, die in die LIBRETTO-531-Studie eingeschlossen und randomisiert wurden, wurden 193 in den Arm Selpercatinib randomisiert, 98 in den Kontrollarm. Von den 98 in den Kontrollarm randomisierten Patienten wurden 73 für Cabozantinib stratifiziert, und 25 für Vandetanib. Das mediane Alter der Patienten in der ITT-Population betrug 55 Jahre (Bereich: 12 bis 84 Jahre). 37.1% der Patienten waren Frauen. 69.4% der Patienten waren weisser, 27.7% asiatischer und 2.9% schwarzer Abstammung.
Bei Studieneinschluss hatten die meisten Patienten (77%) eine metastasierte Erkrankung. 8.2 % der Patienten befanden sich im Stadium M0 (d.h. keine Fernmetastasen zum Zeitpunkt der Studienaufnahme).
Der ECOG-Performance-Status betrug 0-1 (98.3%) oder 2 (1%). Die häufigste Mutation war M918T (62.5%).
Die Studie erreichte den primären Endpunkt einer Verbesserung des PFS in der ITT-Population (cut off Datum 22 May 2023).
Bei einer medianen Beobachtungszeit von 12 Monaten war das mediane progressionsfreie Überleben mit Selpercatinib auf Basis des verblindeten, unabhängigen, zentralen Reviews nicht erreicht, und betrug 16.8 Monate (95% Konfidenzintervall [CI], 12.22 bis 25.10) in der Kontrollgruppe (Hazard-Ratio für Erkrankungsprogression oder Tod, 0.280; 95% CI, 0.165 bis 0.475; P<0.0001).
Die mediane Dauer des therapieversagenfreien Überlebens (TFFS) mit Selpercatinib war noch nicht erreicht, und betrug 13.9 Monate in der Kontrollgruppe (27 Ereignisse im Selpercatinib-Arm, 37 Ereignisse im Kontrollarm, (HR 0.254; 95% CI: 0.153, 0.423; p<0.0001)).
Bei einer späteren OS-Auswertung mit Stichtag 11. März 2024 waren in den zwei Armen 26 Ereignisse beobachtet worden, 10 mit Selpercatinib und 16 im Kontrollarm. und die HR betrug 0.275 (95% CI: 0.124, 0.608). Die PFS HR in dieser Auswertung betrug 0.202 (95% CI: 0.128, 0.320).
Wirksamkeit bei pädiatrischen Patienten
Die Evidenz für die Wirksamkeit und Sicherheit von Selpercatinib in der jugendlichen Population ist limitiert. Nur ein Patient unter 18 Jahren wurde in LIBRETTO-531 mit einem therapienaiven MTC eingeschlossen und es liegen insgesamt Resultate von 3 Patienten aus der LIBRETTO-001 vor, von denen 2 mit therapienaiven MTC. Weiter sind Resultate von insgesamt 14 pädiatrischen Patienten mit MTC aus der Phase-1/2-studie LIBRETTO-121 eingeschlossen. Die ORR für Patienten mit MTC in LIBRETTO-121 betrug 42.9% (95% KI: 17,7%-71.1%) in der RET-mutierten MTC-Population (n=14) und 60% (95% CI 26.2, 87.8) in der RET-Fusions-positiven Schilddrüsenkrebspopulation (n=10), verglichen mit der ORR bei Erwachsenen (77.5% bei RET-mutierten MTC-Patienten (n=117) und 89.2% bei RET-fusionspositiven Schilddrüsenkrebspatienten (n=58)).
Vorbehandeltes RET-fusionspositives Schilddrüsenkarzinom
Von den Patienten mit RETfusionspositivem Schilddrüsenkarzinom, die zuvor mit einer anderen systemischen Therapie als radioaktivem Iod behandelt worden waren und in die LIBRETTO-001 Studie eingeschlossen wurden, konnten (am Stichtag 13. Januar 2023) 41 Patienten über mindestens 6 Monate beobachtet werden und die Wirksamkeit wurde als auswertbar angesehen. Alle 41 Patienten hatten zuvor eine eine systemische Therapie erhalten, und 31 Patienten waren zudem mit radioaktivem Jod behandelt worden.
Bei den 41 vorbehandelten Patienten des primären Analysenset zeigten sich folgende Histologien: papillar (n=31), schwach differenziert (n=5), anaplastisch (n=4) und Hürthle-Zell (n=1).
Das mediane Alter betrug 58 Jahre (Bereich 25 bis 88 Jahre). 43.9% der Patienten waren männlich. 58.5% der Patienten waren weisser, 29.3% asiatischer und 7.3% schwarzer Abstammung. Der ECOG-Performance-Status betrug 0-1 (92.7%) oder 2 (7.3%). 100% der Patienten hatten eine metastasierte Erkrankung.
Die Patienten hatten zuvor im Median 3 systemische Therapien erhalten (Bereich: 1-7). Zu den häufigsten vorausgegangenen Therapien gehörten radioaktives Jod (73.2%), MKI (85.4%) und 9.8% hatten andere systemische Therapien erhalten.
Im primären Analysensatz, welcher 41 vorbehandelte Patienten mit RET-fusionspositivem Schilddrüsenkarzinom umfasste, betrug die objektive Ansprechrate (ORR) 85.4% (95% CI: 70.8, 94.4) und die mediane Ansprechdauer 26.7 Monate (95% CI: 12.1-NB) bei einer medianen Beobachtungszeit von 33.87 Monaten.
In der schlecht differenzierten Schilddrüsenkarzinom-Population (n=5) betrug die ORR 100 % (95 %-KI: 47, 82,100). In der anaplastischen Schilddrüsenkarzinom-Population (n=4) betrug die ORR 75 % (19, 41, 99, 37). Der einzige Patient mit Hurthle-Zell- Schilddrüsenkarzinom zeigte ein partielles Ansprechen.
Vorbehandeltes RET-mutiertes medulläres Schilddrüsenkarzinom
Von den Patienten mit RETmutiertem MTC, die in die LIBRETTO-001 Studie eingeschlossen wurden, wurden 152 Patienten zuvor mit Cabozantinib und/oder Vandetanib behandelt und konnten über mindestens 6 Monate beobachtet werden, sodass die Wirksamkeit als auswertbar angesehen wurde. Das mediane Alter betrug 58 Jahre (Bereich 17 Jahre bis 90 Jahre); 1 Patient (0.7%) hatte ein Alter von <18 Jahren. 63.8% der Patienten waren Männer. 90.1% der Patienten waren weisser, 1.3% asiatischer und 1.3% schwarzer Abstammung. Der ECOG-Performance-Status betrug 0-1 (92.7%) oder 2 (7.2%). 98.0% der Patienten hatten eine metastasierte Erkrankung. 100% (n = 152) der Patienten hatten zuvor eine systemische Therapie mit median 2 vorherigen systemischen Therapien erhalten, und 27.6% (n=42) hatten 3 oder mehr vorherige systemische Therapien erhalten. Die häufigste Mutation war M918T (65.1%), gefolgt von extrazellulären Cystein-Mutationen (15.8%).
Bei den 152 zuvor behandelten Patienten mit RET-mutiertem MTC betrug die objektive Ansprechrate (ORR) 77.6% (70.2, 84.0) und die mediane Ansprechdauer betrug 45.3 Monate (95% CI: 33.6, NB) bei einer medianen Beobachtungszeit von 38.3 Monaten.
Kinder und Jugendliche
Die Evidenz für die Wirksamkeit und Sicherheit von Selpercatinib in der jugendlichen Population ist limitiert. Es wurden Resultate von insgesamt 14 pädiatrischen Patienten mit MTC aus der Phase-1/2-studie LIBRETTO-121 vorgelegt. LIBRETTO-121 ist eine laufende Phase-1/2 Studie bei pädiatrischen Patienten mit einem fortgeschrittenen soliden oder primären ZNS-Tumor, welche eine aktivierende RET-Veränderung aufweisen.
Die objektive Ansprechrate (ORR) in der RET-mutierten MTC-Population (n=14) in LIBRETTO-121 betrug 42.9% (95% KI: 17,7%-71.1%).
PharmakokinetikDie Pharmakokinetik von Selpercatinib wurde bei Patienten mit lokal fortgeschrittenen oder metastasierten soliden Tumoren unter Anwendung von 160 mg zweimal täglich beurteilt, sofern nicht anders angegeben. Im Steady-state stiegen AUC und Cmax von Selpercatinib in einem Dosierungsbereich von 20 mg einmal täglich bis 240 mg zweimal täglich dosisproportional.
Das Steady-state wurde nach etwa 7 Tagen erreicht und die mediane Akkumulationsrate nach Anwendung von 160 mg zweimal täglich betrug 3.4. Die mittlere Cmax im Steady-state von Selpercatinib [Variationskoeffizient (CV%)] betrug 2,980 (53%) ng/ml und die AUC0 24h betrug 51,600 (58%) ng*h/ml.
In vitro durchgeführte Studien zeigen, dass Selpercatinib in klinisch relevanten Konzentrationen CYP1A2, CYP2B6, CYP2C9, CYP2C19 oder CYP2D6 nicht inhibiert oder induziert.
Absorption
Nach einer oralen Dosis von 160 mg wurde Retsevmo rasch absorbiert mit einer Tmax von etwa 2 Stunden. Die geometrische mittlere absolute orale Bioverfügbarkeit betrug 73% (Bereich: 60-82%).
Einfluss von Nahrungsmitteln
Im Vergleich zur AUC und Cmax von Selpercatinib im nüchternen Zustand war nach oraler Anwendung einer einzelnen Dosis von 160 mg bei gesunden Probanden zusammen mit einer fettreichen Mahlzeit die AUC von Selpercatinib um 9% erhöht und die Cmax um 14% verringert
Diese Veränderungen wurden als klinisch nicht relevant angesehen. Daher kann Selpercatinib mit oder ohne Mahlzeit eingenommen werden.
Distribution
Das mittlere (CV%) Verteilungsvolumen (Vss/F) von Selpercatinib nach oraler Anwendung von Selpercatinib bei erwachsenen Patienten beträgt 191 (69%) l. Selpercatinib wird in vitro zu 96% an menschliche Plasmaproteine gebunden, wobei die Bindung unabhängig von der Konzentration ist. Das Blut-Plasma-Konzentrationsverhältnis beträgt 0.7.
Metabolismus
Selpercatinib wird vorwiegend durch CYP3A4 metabolisiert. Nach oraler Anwendung einer einzelnen [14C]-radiomarkierten 160-mg-Dosis Selpercatinib bei gesunden Probanden bildete unverändertes Selpercatinib 86% der gemessenen radioaktiven Komponenten im Plasma.
Elimination
Nach oraler Anwendung von Selpercatinib bei erwachsenen Patienten beträgt die mittlere (CV%) Clearance (CL/F) von Selpercatinib 6.0 (49%) l/h, und die Halbwertszeit beträgt 22 Stunden. Nach oraler Anwendung einer einzelnen [14C]-radiomarkierten 160-mg-Dosis Selpercatinib bei gesunden Probanden wurden 69% (14% unverändert) der verabreichten Radioaktivität in der Faeces und 24% (11.5% unverändert) im Urin gefunden.
Kinetik spezieller Patientengruppen
Alter, Geschlecht und Körpergewicht
Alter (Bereich: 15 Jahre bis 90 Jahre) und Geschlecht hatten keine klinisch bedeutsame Wirkung auf die Pharmakokinetik von Retsevmo.
Leberfunktionsstörungen
Die AUC0-INF von Selpercatinib stieg um 7%, 32% und 77% bei Patienten mit leichter (Gesamtbilirubin weniger als oder gleich ULN mit AST grösser als ULN oder Gesamtbilirubin grösser als 1 bis 1.5x ULN bei jeglicher AST), moderater (Gesamtbilirubin grösser als 1.5 bis 3x ULN bei jeglicher AST), und schwerer (Gesamtbilirubin grösser als 3 bis 10x ULN bei jeglicher AST) Leberfunktionsstörung im Vergleich zu Probanden mit normaler Leberfunktion.
Die klinischen Daten zur Sicherheit von Selpercatinib bei Patienten mit schwerer Leberfunktionsstörung sind begrenzt. Daher werden für Patienten mit schwerer Leberfunktionsstörung Dosisanpassungen empfohlen (siehe "Dosierung/Anwendung" ).
Nierenfunktionsstörungen
In einer klinisch-pharmakologischen Studie blieb die Exposition (AUC) nach einer Einzeldosis Selpercatinib 160 mg bei Patienten mit leichter, moderater oder schwerer Nierenfunktionsstörung unverändert. Patienten mit Nierenerkrankung im Endstadium (End stage renal disease) und Dialyse wurden nicht untersucht.
Pädiatrische Patienten
Auf Grundlage begrenzter pharmakokinetischer Daten waren Cmax und AUC bei Jugendlichen im Alter von 12-18 Jahren und Erwachsenen ähnlich. Ähnliche Expositionen wurden in der pädiatrischen Population von LIBRETTO 121 bei einer Dosis von 92 mg/m2 im Vergleich zur erwachsenen Population in LIBRETTO-001 bei einer Dosierung von 160 mg beobachtet.
Präklinische DatenSicherheitspharmakologie / Toxizität nach wiederholter Gabe
Studien mit wiederholter Gabe zur Charakterisierung der Toxizität wurden bei Ratten und Minischweinen durchgeführt. Zielorgane der Toxizität bei Ratten sowie Minischweinen waren das hämatopoetische System, lymphatische Gewebe, Zunge, Pankreas, Epiphysenfugen und männliches Reproduktionsgewebe. Im Allgemeinen waren die Toxizitäten an diesen Organen reversibel; hiervon ausgenommen war die testikuläre Toxizität. Eine reversible Toxizität wurde an Ovarien und Gastrointestinaltrakt nur bei Minischweinen beobachtet; bei hohen Dosierungen bei Minischweinen verursachte die gastrointestinale Toxizität Morbidität bei Expositionen, die allgemein niedriger waren als die Expositionen, die bei Menschen unter empfohlener Dosierung gemessen wurden. In einer Studie bei Minischweinen zeigten weibliche Tiere eine leichte, reversible QTc-Verlängerung von etwa 12% im Vergleich zu Kontrollen und 7% im Vergleich zu den Werten vor Gabe der Dosis. Nur bei Ratten beobachtete Zielorgane der Toxizität waren Schneidezähne, Leber, Vagina, Lungen, Brunner-Drüsen und verschiedene andere von Mineralisierung betroffene Gewebe im Zusammenhang mit Hyperphosphatämie. Diese Toxizitäten, die nur in diesen Organen bei Ratten auftraten, waren reversibel.
Gentoxizität
Selpercatinib ist in in vitro-Bakterien-Rückmutationstests (Ames-Tests) mit oder ohne metabolische Aktivierung nicht mutagen, und es war bei einem in vitro-Mikronukleustest an menschlichen peripheren Lymphozyten mit oder ohne metabolische Aktivierung nicht klastogen. Selpercatinib war positiv im in vivo-Mikronukleustest bei Ratten unter hohen Dosen mit etwa dem 11-Fachen über der menschlichen Cmax bei empfohlener Dosis von 160 mg zweimal täglich.
In einem in vivo bei Ratten durchgeführten Mikronukleustest war Selpercatinib positiv bei Konzentrationen >7-fach der Cmax bei Menschen unter einer Dosis von 160 mg zweimal täglich. In einem in vitro an menschlichen peripheren Blutlymphozyten durchgeführten Mikronukleustest wurde ein mehrdeutiges Ansprechen beobachtet, bei einer Konzentration, die etwa dem 485-Fachen der Cmax einer bei Menschen verwendeten Dosis entspricht.
Kanzerogenität
In einer Studie zur Kanzerogenität von Selpercatinib, die über 2 Jahre bei Ratten durchgeführt wurde, wurden bei einigen weiblichen Tieren vaginale Tumore beobachtet, bei Plasmaexpositionen ähnlich jenen bei erwachsenen Patienten mit einer Dosis von 160 mg zweimal täglich. Im Reproduktionstrakt weiblicher Ratten wurden keine präneoplastischen Veränderungen beobachtet. Die klinische Relevanz dieser Befunde ist unbekannt. Bei männlichen Ratten in dieser Studie war Selpercatinib nicht kanzerogen.
In einer Studie über 6 Monate bei männlichen und weiblichen Mäusen war Selpercatinib nicht kanzerogen.
Reproduktionstoxizität
In einer Studie zur embryofetalen Entwicklung führte eine tägliche orale Gabe von Selpercatinib in Dosierungen von mehr als oder gleich 100 mg/kg [etwa dem 3.6-Fachen der menschlichen geometrischen mittleren Exposition basierend auf der Fläche unter der Kurve (AUC) bei klinischer Dosierung von 160 mg zweimal täglich] an trächtige Ratten während der Organogenese zu einem 100%-igen Postimplantationsverlust. Bei einer Dosis von 50 mg/kg [etwa dem 1.5-Fachen der menschlichen geometrischen mittleren Exposition basierend auf der AUC bei klinischer Dosierung von 160 mg zweimal täglich] kam es bei 6 von 8 Weibchen zu einer 100%-igen frühen Resorption; bei den übrigen 2 Weibchen wurden in den 2 Würfen überwiegend frühe Resorptionen und nur 3 lebensfähige Feten beobachtet. Die 3 lebensfähigen Feten wiesen ein verringertes fetales Körpergewicht auf; die 2 Feten aus dem einen Wurf wiesen einen kurzen Schwanz auf, und der einzige Fetus aus dem anderen Wurf wies eine kleine Schnauze und ein lokalisiertes fetales Ödem an Hals und Thorax auf.
Ergebnisse von bei Ratten und Minischweinen durchgeführten Studien weisen darauf hin, dass Selpercatinib die Fertilität von Männern und Frauen beeinträchtigen kann.
In einer Fertilitätsstudie bei männlichen Ratten wurden keine Wirkungen von Selpercatinib auf die Paarung oder Fertilität beobachtet. Allerdings wurde bei allen Dosierungen eine dosisabhängige Verminderung von Keimzellen und Spermatidretention in den Hoden sowie vermehrte Zelldebris in den Nebenhoden beobachtet. Diese Wirkungen waren mit reduziertem Organgewicht, reduzierter Spermienmotilität und einem Anstieg der Anzahl veränderter Spermien unter der höchsten Dosierung verbunden, bei welcher die AUC-basierte Exposition etwa dem 2.3-Fachen der klinischen Exposition bei der für Menschen empfohlenen Dosis entspricht. Die mikroskopischen Befunde in der Fertilitätsstudie bei männlichen Ratten standen im Einklang mit den Wirkungen in Studien mit wiederholter Gabe bei Ratten und Minischweinen, in welcher eine dosisabhängige, nicht-reversible testikuläre Degeneration mit einem reduziertem luminalem Sperma in den Nebenhoden verbunden war, bei AUC-basierten Expositionen entsprechend dem 0.1- bis 0.4-Fachen der klinischen Exposition bei der für Menschen empfohlenen Dosis.
In einer Studie zur Fertilität und frühen embryonalen Entwicklung bei weiblichen Ratten zeigten sich keine Wirkungen von Selpercatinib auf die Paarung oder die Fertilität. Allerdings wurde nur bei der hohen Dosis eine Reduzierung der Anzahl von Östruszyklen mit Anstieg des prä-koitalen Intervalls beobachtet, sowie ein Anstieg der Anzahl toter Embryos, ein erhöhter Postimplantationsverlust und eine Reduktion der Anzahl lebender Embryos. Diese Wirkungen wurden bei einer AUC-basierten Exposition beobachtet, die etwa der klinischen Exposition bei der für Menschen empfohlenen Dosis entspricht. In Studien mit wiederholter Gabe bei weiblichen Ratten wurden reversible vaginale Schleimbildung mit einzelner Zellverhornung und veränderten Östruszyklen festgestellt, bei klinisch relevanter Exposition auf Basis der AUC. Bei weiblichen Minischweinen wurden verkleinerte Gelbkörper und/oder Gelbkörperzysten beobachtet, bei AUC-basierten Expositionen entsprechend dem 0.07- bis 0.3-Fachen der klinischen Exposition bei der für Menschen empfohlenen Dosis.
Toxizitätsprüfungen mit juvenilen Tieren
Die Skelettveränderungen wurden bei Expositionen beobachtet, die ungefähr der Exposition bei Erwachsenen mit der empfohlenen Dosis von 160 mg zweimal täglich entsprachen. Juvenile und adoleszente Ratten, sowie adoleszente Minischweine mit offenen Wachstumsfugen, denen Selpercatinib verabreicht wurde, zeigten mikroskopische Veränderungen wie Hypertrophie, Hyperplasie und Dysplasie des Wachstumsfugenknorpels (Epiphysenfuge). In der Studie an juvenilen Ratten waren diese Veränderungen der Wachstumsfuge mit einer verringerten Femurlänge und einer Verringerung der Knochenmineraldichte verbunden.
Sonstige HinweiseHaltbarkeit
Das Arzneimittel darf nur bis zu dem auf dem Behälter mit "EXP" bezeichneten Datum verwendet werden.
Besondere Lagerungshinweise
Nicht über 30°C lagern
Behälter in der Verpackung aufbewahren, um den Inhalt vor Feuchtigkeit zu schützen.
Ausser Reichweite von Kindern aufbewahren.
Zulassungsnummer67862 (Swissmedic)
PackungenRetsevmo 40 mg: 56 Hartkapseln (A)
Retsevmo 80 mg: 56, 112 Hartkapseln (A)
Es werden möglicherweise nicht alle Packungsgrössen in den Verkehr gebracht.
ZulassungsinhaberinEli Lilly (Suisse) SA, 1214 Vernier/GE.
Stand der InformationMärz 2025
|