Eigenschaften/WirkungenATC-Code
L04AF03
Wirkungsmechanismus
Upadacitinib ist ein selektiver und reversibler Inhibitor von JAK1. Januskinasen (JAKs) sind wichtige intrazelluläre Enzyme, die Signale von Zytokinen oder Wachstumsfaktoren weiterleiten, die an einer Vielzahl von zellulären Prozessen, wie Entzündungsantworten, Hämatopoese und Immunüberwachung, beteiligt sind. Die JAK-Enzymfamilie umfasst vier Mitglieder – JAK1, JAK2, JAK3 und TYK2 –, die paarweise Signaltransduktoren und Transkriptionsaktivatoren (STATs) phosphorylieren und aktivieren. Diese Phosphorylierung moduliert wiederum die Genexpression und Zellfunktion. JAK1 ist für Signalwege von Entzündungszytokinen von Bedeutung, während JAK2 für die Reifung von Erythrozyten wichtig ist und JAK3-Signale eine Rolle im Rahmen der Immunüberwachung und Lymphozytenfunktion spielen.
Upadacitinib inhibiert JAK1 stärker als JAK2 oder JAK3. In zellbasierten Assays zur Wirkstärke, die mit pharmakodynamischen Antworten in vivo korrelierten, zeigte Upadacitinib eine 50- bis 70fach grössere Selektivität für JAK1 als für JAK2 und eine > 100fach grössere Selektivität für JAK1 gegenüber JAK3.
Pharmakodynamik
Hemmung der durch IL-6 induzierten STAT3-Phosphorylierung und durch IL-7 induzierten STAT5-Phosphorylierung
Bei gesunden Probanden führte die Verabreichung von Upadacitinib (Formulierung mit sofortiger Freisetzung) zu einer dosis- und konzentrationsabhängigen Inhibierung der durch IL-6 (JAK1/JAK2) induzierten STAT3-Phosphorylierung und durch IL-7 (JAK1/JAK3) induzierten STAT5-Phosphorylierung im Vollblut. Die maximale Inhibierung wurde eine Stunde nach Verabreichung beobachtet und fiel bis zum Ende des Verabreichungsintervalls wieder auf nahezu den Ausgangswert ab.
Lymphozyten
Bei Patienten mit rheumatoider Arthritis ging die Behandlung mit Upadacitinib mit einem geringen, vorübergehenden Anstieg der mittleren ALC gegenüber des Ausgangswerts bis zu Woche 36 einher, der unter weiterer Behandlung allmählich ganz oder nahezu auf die Ausgangswerte zurückging.
Immunglobuline
Bei Patienten mit rheumatoider Arthritis wurden unter Upadacitinib geringe Abnahmen der mittleren IgG- und IgM-Spiegel gegenüber des Ausgangswerts in der kontrollierten Phase beobachtet; die Mittelwerte zu Beginn und allen anderen Besuchen lagen jedoch innerhalb des üblichen Referenzbereichs.
hsCRP und andere Entzündungsmarker
Bei Patienten mit rheumatoider Arthritis war die Behandlung mit Upadacitinib bereits nach Woche 1 mit einem erheblichen Rückgang der mittleren hsCRP-Spiegel gegenüber dem Ausgangswert verbunden, der während der weiteren Behandlung erhalten blieb.
Bei Patienten mit Morbus Crohn wurden nach der Induktionsbehandlung mit Upadacitinib Verringerungen von hsCRP und fäkalem Calprotectin (FCP) beobachtet. Die Verringerung von hsCRP und FCP wurde bis Woche 52 der Erhaltungsstudie beibehalten.
Kardiale Elektrophysiologie
Die Wirkung von Upadacitinib auf das QTc-Intervall wurde bei Studienteilnehmern geprüft, die Einzel- und Mehrfachdosen von Upadacitinib erhielten.
Das 2.5-Fache der durchschnittlichen Exposition der maximalen therapeutischen Dosis von 45 mg einmal täglich ergab keinen klinisch relevanten Effekt auf das QTc-Intervall.
Impfstudien
Der Einfluss von RINVOQ auf das humorale Ansprechen nach Verabreichung eines adjuvantierten rekombinanten Glycoprotein-E-Herpes-Zoster-Impfstoffs wurde bei 93 Patienten älter als 50 Jahre mit rheumatoider Arthritis unter dauerhafter Behandlung (mediane Behandlungsdauer 3,9 Jahre) mit RINVOQ 15 mg untersucht. 98 % der Patienten (n = 91) erhielten gleichzeitig Methotrexat. 49 % der Patienten erhielten bei Baseline orale Kortikosteroide. Die Impfung führte unabhängig von der Ko-Medikation nach 16 Wochen (4 Wochen nach der zweiten Impfdosis) bei 88 % (95 %-KI: 81,0, 94,5) der mit RINVOQ 15 mg behandelten Patienten zu einem mindestens 4fachen Anstieg der Anti-Glycoprotein-E-Antikörperkonzentration gegenüber dem Ausgangswert vor der Impfung. Inwieweit diese Impfantwort einen Schutz vor Infektionen bzw. Reaktivierungen erlaubt, ist unklar.
Der Einfluss von RINVOQ auf das humorale Ansprechen nach Verabreichung von Prevenar 13®, eines inaktivierten 13-wertigen Pneumokokken-Konjugat-Impfstoffs wurde bei 111 Patienten mit rheumatoider Arthritis unter durchgehender Behandlung mit RINVOQ 15 mg (n=87) oder 30 mg (n=24) untersucht. 97% der Patienten (n=108) erhielten gleichzeitig Methotrexat. Die Impfung führte bei 67,5% (95% KI: 57,4, 77,5) und 56,5% (95% KI: 36,3, 76,8) der Patienten, die mit RINVOQ 15 mg bzw. 30 mg behandelt wurden, zu einem mindestens 2-fachen Anstieg der Antikörperkonzentration gegenüber dem Ausgangswert vor der Impfung bei mindestens 6 der einzelnen Pneumokokken-Antigene des Impfstoffes. Inwieweit diese Impfantwort einen Schutz vor Infektionen erlaubt, ist unklar.
Klinische Wirksamkeit
Rheumatoide Arthritis
Die Wirksamkeit und Sicherheit von RINVOQ 15 mg einmal täglich wurde in fünf randomisierten, doppelblinden, multizentrischen Phase-III-Studien bei Patienten mit mittelschwerer bis schwerer aktiver rheumatoider Arthritis, die die ACR/EULAR-Klassifikationskriterien von 2010 erfüllten, untersucht (siehe Tabelle 5). Patienten, die 18 Jahre und älter waren, konnten an den Studien teilnehmen. Das Vorliegen von mindestens 6 druckschmerzempfindlich und 6 geschwollenen Gelenken sowie der Nachweis einer systemischen Entzündung auf Basis der hsCRP-Erhöhung waren bei Baseline erforderlich. Vier Studien schlossen Langzeitfortsetzungsphasen ein mit einer Dauer bis zu 5 Jahren und eine Studie (SELECT-COMPARE) schloss eine Langzeitfortsetzungsphase ein mit einer Dauer bis zu 10 Jahren.
Tabelle 5: Zusammenfassung der klinischen Studien
|
Studienname
|
Population
(n)
|
Behandlungsarme
|
Wesentliche Endpunkte
| |
SELECT-EARLY
|
MTXnaiva
(947)
|
·15 mg Upadacitinib
·30 mg Upadacitinib
·MTX
Monotherapie
|
Primärer Endpunkt:
·ACR50 in Woche 12
Wichtige Sekundäre Endpunkte:
·Klinische Remission (DAS28-CRP < 2,6) in Woche 24
·Niedrige Krankheitsaktivität (DAS28-CRP ≤3,2) in Woche 12
·Δ Körperliche Funktionsfähigkeit (HAQ-DI) in Woche 12
·Radiologische Progression (ΔmTSS) in Woche 24
·SF-36 PCS
| |
SELECT-MONOTHERAPY
|
MTX-IRb
(648)
|
·Upadacitinib 15 mg
·Upadacitinib 30 mg
·MTX
Monotherapie
|
Primärer Endpunkt:
·ACR20 in Woche 14
Wichtige Sekundäre Endpunkte:
·Niedrige Krankheitsaktivität (DAS28-CRP ≤3,2) in Woche 14
·Klinische Remission (DAS28-CRP < 2,6) in Woche 14
·Δ Körperliche Funktionsfähigkeit (HAQ-DI) in Woche 14
·SF-36 PCS
·Morgensteifigkeit
| |
SELECT-NEXT
|
csDMARD-IRc
(661)
|
·Upadacitinib 15 mg
·Upadacitinib 30 mg
·Placebo
In Kombination mit csDMARDs
|
Primärer Endpunkt:
·ACR20 in Woche 12
Wichtige Sekundäre Endpunkte:
·Niedrige Krankheitsaktivität (DAS28-CRP ≤3,2) in Woche 12
·Klinische Remission (DAS28-CRP < 2,6) in Woche 12
·Δ Körperliche Funktionsfähigkeit (HAQ-DI) in Woche 12
·SF-36 PCS
·Morgensteifigkeit
·FACIT-F
| |
SELECT-COMPARE
|
MTX-IRd
(1629)
|
·15 mg Upadacitinib
·Placebo
·40 mg Adalimumab
In Kombination mit MTX
|
Primärer Endpunkt:
·ACR20 in Woche 12
Wichtige Sekundäre Endpunkte:
·Klinische Remission (DAS28-CRP < 2,6) in Woche 12
·Niedrige Krankheitsaktivität (DAS28-CRP ≤3,2) in Woche 12
·ACR50 vs. Adalimumab in Woche 12
·Δ Körperliche Funktionsfähigkeit (HAQ-DI) in Woche 12
·Radiologische Progression (ΔmTSS) in Woche 26
·SF-36 PCS
·Morgensteifigkeit
·FACIT-F
| |
SELECT-BEYOND
|
bDMARD-IRe
(499)
|
·15 mg Upadacitinib
·30 mg Upadacitinib
·Placebo
In Kombination mit csDMARDs
|
Primärer Endpunkt:
·ACR20 in Woche 12
Wichtige Sekundäre Endpunkte:
·Niedrige Krankheitsaktivität (DAS28-CRP ≤3,2) in Woche 12
·Δ Körperliche Funktionsfähigkeit (HAQ-DI) in Woche 12
·SF-36 PCS
| |
Abkürzungen: ACR20 (oder 50) = Verbesserung um ≥20 % (oder ≥50 %) gemäss American College of Rheumatology; bDMARD = biologic disease-modifying antirheumatic drug; CR = Clinical Response, CRP = Creaktives Protein; DAS28 = Disease Activity Score 28 joints, mTSS = modifizierter Total-Sharp-Score; csDMARD = konventionelle synthetische krankheitsmodifizierende Antirheumatika; HAQ-DI = Health Assessment Questionnaire Disability Index; IR = inadequate responder; MTX = Methotrexat
a MTXnaive Patienten oder Behandlung mit nicht mehr als drei wöchentlichen MTX-Dosen
b Patienten mit unzureichendem Ansprechen auf MTX
c Patienten mit unzureichendem Ansprechen auf csDMARDs; Patienten mit Vorbehandlung mit höchstens einem bDMARD konnten teilnehmen (max. 20 % der Zahl an Studienteilnehmern insgesamt), wenn sie nur eine begrenzte Zeit (< 3 Monate) behandelt worden waren oder die Behandlung mit dem bDMARD wegen Unverträglichkeit abbrechen mussten
d Patienten mit unzureichendem Ansprechen auf MTX; Patienten mit Vorbehandlung mit höchstens einem bDMARD (ausser Adalimumab) konnten teilnehmen (max. 20 % der Zahl an Studienteilnehmern insgesamt), wenn sie nur eine begrenzte Zeit (< 3 Monate) behandelt worden waren oder die Behandlung mit dem bDMARD wegen Unverträglichkeit abbrechen mussten
e Patienten mit unzureichendem Ansprechen auf oder Unverträglichkeit gegen mindestens ein bDMARD
|
Klinisches Ansprechen
Remission und niedrige Krankheitsaktivität
In allen Studien erreichte ein grösserer Patientenanteil unter RINVOQ 15 mg eine niedrige Krankheitsaktivität (DAS28-CRP ≤3,2) und klinische Remission (DAS28-CRP < 2,6) im Vergleich zu Placebo, MTX oder Adalimumab (Tabelle 6). Im Vergleich zu Adalimumab wurden bereits nach 8 Wochen höhere Ansprechraten erzielt, die bis Woche 48 anhielten. Ebenso wurde ein stärkeres Ansprechen für andere Masse der Krankheitsaktivität beobachtet, einschliesslich einer Remission definiert als CDAI ≤2,8, SDAI ≤3,3 und Boolesche Remission. Insgesamt waren die Ansprechraten in Bezug auf niedrige Krankheitsaktivität und klinische Remission über alle Patientenpopulationen hinweg mit oder ohne MTX vergleichbar. Nach drei Jahren blieben 297 von 651 (45,6 %) ursprünglich mit RINVOQ 15 mg randomisierten Patienten bzw. 111 von 327 (33,9 %) ursprünglich mit Adalimumab randomisierten Patienten in der Studie SELECT-COMPARE und 216 von 317 (68,1 %) ursprünglich mit RINVOQ 15 mg randomisierten Patienten bzw. 149 von 315 (47,3 %) ursprünglich mit MTX-Monotherapie randomisierten Patienten in der Studie SELECT-EARLY bei ihrer Behandlung. Bei den Patienten, die ihre ursprünglich zugewiesene Behandlung beibehielten, blieben die niedrige Krankheitsaktivität und die klinische Remission über 3 Jahre erhalten.
ACR-Ansprechen
In allen Studien erzielten mehr mit RINVOQ 15 mg behandelte Patienten im Vergleich zu Placebo, MTX oder Adalimumab ein ACR20, ACR50 und ACR70 Ansprechen nach 12 Wochen (Tabelle 6). Bei allen Parametern war die Dauer bis zum Eintreten der Wirkung kurz, wobei höhere ACR20 Ansprechraten bereits nach einer Woche verzeichnet wurden. Es wurde ein anhaltendes Ansprechen beobachtet (mit oder ohne MTX) und die Ansprechraten bezüglich ACR20/50/70 konnten bei Patienten, die ihre ursprünglich zugewiesene Behandlung beibehielten, über 3 Jahre aufrechterhalten werden.
Die Behandlung mit RINVOQ 15 mg, allein oder in Kombination mit csDMARDs, führte im Vergleich zu Placebo, einer MTX-Monotherapie oder Adalimumab zu einer grösseren Verbesserung in allen einzelnen ACR-Komponenten, einschliesslich der Anzahl druckschmerzempfindlicher und geschwollener Gelenke, der allgemeinen Beurteilung durch den Arzt (Physician Global Assessment) und den Patienten (Patient Global Assessment), des HAQ-DI, der Schmerzbeurteilung und des hsCRP (Tabelle 7).
In der Studie SELECT-COMPARE erreichte ein grösserer Patientenanteil unter RINVOQ 15 mg im Vergleich zu Adalimumab in Woche 12 bis Woche 48 ein ACR20/50/70 Ansprechen (Tabelle 6).
Tabelle 6: Ansprechen und Remission
|
Studie
|
SELECT
EARLY
MTXnaiv
|
SELECT
MONO
MTX-IR
|
SELECT
NEXT
csDMARD-IR
|
SELECT
COMPARE
MTX-IR
|
SELECT
BEYOND
bDMARD-IR
| |
|
MTX
|
UPA
15 mg
|
MTX
|
UPA
15 mg
|
PBO
|
UPA
15 mg
|
PBO
|
UPA
15 mg
|
ADA
40 mg
|
PBO
|
UPA
15 mg
| |
N
|
314
|
317
|
216
|
217
|
221
|
221
|
651
|
651
|
327
|
169
|
164
| |
Woche
|
| |
ACR20 (% Patienten)
| |
12a/14b
|
54
|
76g
|
41
|
68e
|
36
|
64e
|
36
|
71e,i
|
63
|
28
|
65e
| |
24c/26d
|
59
|
79g
|
|
|
|
|
36
|
67g,i,
|
57
|
|
| |
48
|
57
|
74g
|
|
|
|
|
|
65i
|
54
|
|
| |
ACR50 (% Patienten)
| |
12a/14b
|
28
|
52e
|
15
|
42g
|
15
|
38g
|
15
|
45g,h
|
29
|
12
|
34g
| |
24c/26d
|
33
|
60g
|
|
|
|
|
21
|
54g,i
|
42
|
|
| |
48
|
43
|
63g
|
|
|
|
|
|
49i
|
40
|
|
| |
ACR70 (% Patienten)
| |
12a/14b
|
14
|
32g
|
3
|
23g
|
6
|
21g
|
5
|
25g,i
|
13
|
7
|
12
| |
24c/26d
|
18
|
44g
|
|
|
|
|
10
|
35g,i,
|
23
|
|
| |
48
|
29
|
51g
|
|
|
|
|
|
36i
|
23
|
|
| |
LDA DAS28-CRP ≤3,2 (% Patienten)
| |
12a/14b
|
28
|
53f
|
19
|
45e
|
17
|
48e
|
14
|
45e,i
|
29
|
14
|
43e
| |
24c26d
|
32
|
60g
|
|
|
|
|
18
|
55g,i
|
39
|
|
| |
48
|
39
|
59g
|
|
|
|
|
|
50i
|
35
|
|
| |
CR DAS28-CRP < 2,6 (% Patienten)
| |
12a/14b
|
14
|
36g
|
8
|
28e
|
10
|
31e
|
6
|
29e,i
|
18
|
9
|
29g
| |
24c26d
|
18
|
48f
|
|
|
|
|
9
|
41g,i
|
27
|
|
| |
48
|
29
|
49g
|
|
|
|
|
|
38i
|
28
|
|
| |
SDAI ≤3,3 (% Patienten)
| |
12a14b
|
6
|
16g
|
1
|
14g
|
3
|
10g
|
3
|
12g,i
|
7
|
5
|
9
| |
24c/26d
|
9
|
28g
|
|
|
|
|
5
|
24g,i
|
14
|
|
| |
48
|
16
|
32g
|
|
|
|
|
|
25i
|
17
|
|
| |
CDAI ≤2,8 (% Patienten)
| |
12a/14b
|
6
|
16g
|
1
|
13g
|
3
|
9g
|
3
|
13g,i
|
8
|
5
|
8
| |
24c/26d
|
11
|
28g
|
|
|
|
|
6
|
23g,i
|
14
|
|
| |
48
|
17
|
32g
|
|
|
|
|
|
25i
|
17
|
|
| |
Boolesche Remission (% Patienten)
| |
12a/14b
|
6
|
13g
|
1
|
9g
|
4
|
10g
|
2
|
10g,i
|
4
|
2
|
7g
| |
24c/26d
|
7
|
24g
|
|
|
|
|
4
|
18g,i
|
10
|
|
| |
48
|
13
|
28g
|
|
|
|
|
|
21i
|
15
|
|
| |
Abkürzungen: ACR20 (oder 50 oder 70) = Verbesserung ≥20 % (oder ≥50 % oder ≥70 %) gemäss American College of Rheumatology; ADA = Adalimumab; bDMARD = biologic disease-modifying anti-rheumatic drug; CDAI = Clinical Disease Activity Index; CR = Clinical Remission; CRP = Creaktives Protein; csDMARD = conventional synthetic disease-modifying anti-rheumatic drug; DAS28 = Disease Activity Score 28 joints; IR= inadequate responder; LDA = Low Disease Activity; MTX = Methotrexat; PBO = Placebo; SDAI = Simple Disease Activity Index; UPA = Upadacitinib
a SELECT-NEXT, SELECT-EARLY, SELECT-COMPARE, SELECT-BEYOND
b SELECT-MONOTHERAPY
c SELECT-EARLY
d SELECT-COMPARE
e p ≤0,001 Upadacitinib im Vergleich zu Placebo oder MTX
f p ≤0,01 Upadacitinib im Vergleich zu Placebo oder MTX
g Upadacitinib im Vergleich zu Placebo oder MTX (diese Vergleiche sind für Multiples Testen nicht kontrolliert)
h p ≤0,001 Upadacitinib im Vergleich zu Adalimumab
i Upadacitinib im Vergleich zu Adalimumab (diese Vergleiche sind für Multiples Testen nicht kontrolliert)
|
Tabelle 7: Komponenten des ACR-Ansprechens (mittlere Veränderung gegenüber dem Ausgangswert)a
|
Studie
|
SELECT
EARLY
MTX-naiv
|
SELECT
MONO
MTX-IR
|
SELECT
NEXT
csDMARD-IR
|
SELECT
COMPARE
MTX-IR
|
SELECT
BEYOND
bDMARD-IR
| |
|
MTX
|
UPA
15
mg
|
MTX
|
UPA
15
mg
|
PBO
|
UPA
15
mg
|
PBO
|
UPA
15
mg
|
ADA
40
mg
|
PBO
|
UPA
15
mg
| |
N
|
314
|
317
|
216
|
217
|
221
|
221
|
651
|
651
|
327
|
169
|
164
| |
Woche
|
| |
Anzahl druckschmerzempfindlicher Gelenke (0-68)
| |
12b/
14c
|
-13
|
-17h
|
-11
|
-15h
|
-8
|
-14h
|
-10
|
-16h,l
|
-14
|
-8
|
-16h
| |
24d/
26e
|
-16
|
-19h
|
|
|
|
|
-9
|
-18h,I
|
-15
|
|
| |
Anzahl geschwollener Gelenke (0-66)
| |
12b/
14c
|
-10
|
-12h
|
-8
|
-11h
|
-6
|
-9h
|
-7
|
-11h,l
|
-10
|
-6
|
-11h
| |
24d/
26e
|
-12
|
-14h
|
|
|
|
|
-6
|
-12h,l
|
-11
|
|
| |
Schmerzenf
| |
12b/
14c
|
-25
|
-36h
|
-14
|
-26h
|
-10
|
-30h
|
-15
|
-32h,j
|
-25
|
-10
|
-26h
| |
24d/
26e
|
-28
|
-40h
|
|
|
|
|
-19
|
-37h,l
|
-32
|
|
| |
Allgemeine Beurteilung durch den Patientenf (Patient Global Assessment)
| |
12b/
14c
|
-25
|
-35h
|
-11
|
-23h
|
-10
|
-30h
|
-15
|
-30 h,l
|
-24
|
-10
|
-26h
| |
24d/
26e
|
-28
|
-39h
|
|
|
|
|
-18
|
-36h,l
|
-30
|
|
| |
Körperliche Funktionsfähigkeit (HAQ-DI)g
| |
12b/
14c
|
-0.5
|
-0.8i
|
-0.3
|
-0.7i
|
-0.3
|
-0.6i
|
-0.3
|
-0.6i,k
|
-0.5
|
-0.2
|
-0.4i
| |
24d/
26e
|
-0.6
|
-0.9h
|
|
|
|
|
-0.3
|
-0.7h,l
|
-0.6
|
|
| |
Allgemeine Beurteilung durch den Arztf (Physician Global Assessment)
| |
12b/
14c
|
-35
|
-46h
|
-26
|
-40h
|
-23
|
-38h
|
-25
|
-39h
|
-36
|
-26
|
-39h
| |
24d/
26e
|
-45
|
-50h
|
|
|
|
|
-27
|
-45h,l
|
-41
|
|
| |
hsCRP (mg/l)
| |
12b/
14c
|
-10.6
|
-17.5h
|
-1.1
|
-10.2h
|
-0.4
|
-10.1h
|
-1.7
|
-12.5h,l
|
-9.2
|
-1.1
|
-11.0h
| |
24d/
26e
|
-11.6
|
-18.4h
|
|
|
|
|
-1.5
|
-13.5h,l
|
-10.3
|
|
| |
Abkürzungen: ACR = American College of Rheumatology; ADA = Adalimumab; bDMARD = biologic disease-modifying anti-rheumatic drug; CRP = Creaktives Protein; csDMARD = conventional synthetic disease-modifying anti-rheumatic drug; HAQ-DI = Health Assessment Questionnaire Disability Index; IR = inadequate responder; MTX = Methotrexat; PBO = Placebo; UPA = Upadacitinib
a Daten angegeben als Mittelwerte
b SELECT-NEXT, SELECT-EARLY, SELECT-COMPARE, SELECT-BEYOND
c SELECT-MONOTHERAPY
d SELECT-EARLY
e SELECT-COMPARE
f Visuelle Analogskala: 0 = bester Wert, 100 = schlechtester Wert
g Health Assessment Questionnaire-Disability Index: 0 = bester Wert, 3 = schlechtester Wert; 20 Fragen; 8 Kategorien: Ankleiden und Körperpflege, Aufstehen, Essen, Gehen, Hygiene, Greifen, Festhalten und andere Tätigkeiten.
h Upadacitinib im Vergleich zu Placebo oder MTX (diese Vergleiche sind für multiples Testen nicht kontrolliert)
i p ≤0,001 Upadacitinib im Vergleich zu Placebo oder MTX
j p ≤0,001 Upadacitinib im Vergleich zu Adalimumab
k p ≤0,01 Upadacitinib im Vergleich zu Adalimumab
l Upadacitinib im Vergleich zu Adalimumab (diese Vergleiche sind für multiples Testen nicht kontrolliert)
|
Radiologisches Ansprechen
Die Hemmung der Progression struktureller Gelenkschäden wurde anhand des modifizierten Total-Sharp-Score (mTSS) und seiner Komponenten, dem Score für die Erosion und dem Score für Gelenkspaltverschmälerung, in Woche 26 und Woche 48 (SELECT-COMPARE) sowie in Woche 24 (SELECT-EARLY) untersucht.
Die Behandlung mit RINVOQ 15 mg führte im Vergleich zu Placebo in Woche 26 und Woche 48 in der Studie SELECT-COMPARE und als Monotherapie im Vergleich zu MTX in Woche 24 in der Studie SELECT-EARLY zu einer signifikant grösseren Hemmung der Progression struktureller Gelenkschäden (Tabelle 8). Statistisch signifikante Ergebnisse wurden auch beim Score für die Erosion und dem Score für Gelenkspaltverschmälerung erzielt. Der Anteil an Patienten ohne radiologische Progression (mTSS-Veränderung ≤0) war unter RINVOQ 15 mg im Vergleich zu Placebo in Woche 26 und Woche 48 (SELECT-COMPARE) und im Vergleich zu MTX in Woche 24 (SELECT-EARLY) signifikant grösser. Die Hemmung der Progression struktureller Gelenkschäden konnte in Patienten, die ihre ursprünglich zugewiesene Behandlung mit RINVOQ 15 mg beibehielten, in beiden Studien über 96 Wochen aufrechterhalten werden (basierend auf den verfügbaren Ergebnissen von 327 Patienten in der Studie SELECT-COMPARE und 238 Patienten in der Studie SELECT-EARLY).
Tabelle 8: Radiologische Veränderungen
|
Studie
|
SELECT
EARLY
MTXnaiv
|
SELECT-
COMPARE
MTX-IR
| |
Behandlungsgruppe
|
MTX
|
UPA
15 mg
|
PBOa
|
UPA
15 mg
|
ADA
40 mg
| |
Modifizierter Total-Sharp-Score, mittlere Veränderung gegenüber dem Ausgangswert
| |
Woche 24b/26c
|
0,7
|
0,1f
|
0,9
|
0,2e
|
0,1
| |
Woche 48
|
|
|
1,7
|
0,3e
|
0,4
| |
Score für die Erosion, mittlere Veränderung gegenüber dem Ausgangswert
| |
Woche 24b/26c
|
0,3
|
0,1e
|
0,4
|
0e
|
0
| |
Woche 48
|
|
|
0,8
|
0,1e
|
0,2
| |
Score für die Gelenkspaltverschmälerung, mittlere Veränderung gegenüber dem Ausgangswert
| |
Woche 24b/26c
|
0,3
|
0,1g
|
0,6
|
0,2e
|
0,1
| |
Woche 48
|
|
|
0,8
|
0,2e
|
0,2
| |
Anteil der Patienten ohne radiologische Progressiond
| |
Woche 24b/26c
|
77,7
|
87,5f
|
76,0
|
83,5f
|
86,8
| |
Woche 48
|
|
|
74,1
|
86,4e
|
87,9
| |
Abkürzungen: ADA = Adalimumab; IR = inadequate responder; MTX = Methotrexat; PBO = Placebo; UPA = Upadacitinib
a Alle Placebodaten in Woche 48 mittels linearer Extrapolation abgeleitet
b SELECT-EARLY
c SELECT-COMPARE
d Keine Progression definiert als mTSS-Veränderung ≤0.
e p ≤0,001 Upadacitinib im Vergleich zu Placebo oder MTX
f p ≤0,01 Upadacitinib im Vergleich zu Placebo oder MTX
g p ≤0,05 Upadacitinib im Vergleich zu Placebo oder MTX
|
Körperliche Funktionsfähigkeit und gesundheitsbezogene Ergebnisse
Die Behandlung mit RINVOQ 15 mg, allein oder in Kombination mit csDMARDs, führte zu einer signifikanten Verbesserung der körperlichen Funktionsfähigkeit im Vergleich zu allen Vergleichspräparaten (Placebo, MTX, Adalimumab), gemessen mittels des HAQ-DI. Verbesserungen wurden in den Studien SELECT-NEXT und SELECT-BEYOND im Vergleich zu Placebo bereits nach Woche 1 beobachtet und konnten bis zu 60 Wochen aufrechterhalten werden.
In allen Studien führte die Behandlung mit RINVOQ 15 mg, allein oder in Kombination mit csDMARDs, zu einer signifikant grösseren Schmerzlinderung im Vergleich zu allen Vergleichspräparaten; dies wurde auf einer visuellen Analogskala von 0–100 nach 12/14 Wochen gemessen und das Ansprechen konnte bis zu 48–60 Wochen aufrecht erhalten werden. Bereits in Woche 1 wurde im Vergleich zu Placebo bzw. in Woche 4 im Vergleich zu Adalimumab eine grössere Schmerzlinderung beobachtet.
In allen Studien führte die Behandlung mit RINVOQ 15 mg zu einer signifikant grösseren Verbesserung der mittleren Dauer und der Schwere der Morgensteifigkeit im Vergleich zu Placebo oder MTX.
Verbesserungen des HAQ-DI und Schmerzlinderung wurden gemäss verfügbaren Ergebnissen der Studien SELECT-COMPARE und SELECT-EARLY bei Patienten, die ihre ursprünglich zugewiesene Behandlung mit RINVOQ 15 mg beibehielten, über 3 Jahre aufrechterhalten.
In allen Studien wurde im Vergleich zu Placebo oder MTX eine grössere Verbesserung auf der körperlichen Summenskala des Fragebogens zum Gesundheitszustand, SF-36, verzeichnet. In den Studien SELECT-EARLY, SELECT-MONOTHERAPY und SELECT-COMPARE wurde im Vergleich zu Placebo oder MTX bei mit RINVOQ 15 mg behandelten Patienten eine signifikant grössere Verbesserung auf der psychischen Summenskala und in allen 8 Teilbereichen des SF-36 verzeichnet.
Fatigue wurde in den Studien SELECT-EARLY, SELECT-NEXT und SELECT- COMPARE anhand der Scores auf der Skala zur Fatigue in der Funktionsbeurteilung bei Therapie chronischer Erkrankungen (FACIT-F) gemessen. Die Behandlung mit RINVOQ 15 mg führte im Vergleich zu Placebo, MTX oder Adalimumab zu einer Verbesserung der Fatigue.
Die RA-assoziierte Arbeitsunfähigkeit wurde bei Patienten mit einem Anstellungsverhältnis in den Studien SELECT-NEXT und SELECT-COMPARE anhand der Skala zur Arbeitsunfähigkeit bei RA (RA-WIS) beurteilt. Die Behandlung mit RINVOQ 15 mg führte im Vergleich zu Placebo zu einer signifikant stärkeren Verringerung der Arbeitsunfähigkeit.
Psoriasis-Arthritis
Die Wirksamkeit und Sicherheit von RINVOQ 15 mg einmal täglich wurden in zwei randomisierten, doppelblinden, multizentrischen, placebokontrollierten Phase-III-Studien bei Patienten im Alter von mindestens 18 Jahren mit mittelschwerer bis schwerer aktiver Psoriasis-Arthritis beurteilt (Tabelle 9). Bei allen Patienten lag die aktive Psoriasis-Arthritis auf Grundlage der CASPAR-Kriterien (Classification Criteria for Psoriatic Arthritis) mindestens 6 Monate lang vor; weiterhin hatten sie mindestens 3 druckschmerzempfindliche Gelenke und mindestens 3 geschwollene Gelenke sowie eine aktive Plaque-Psoriasis oder eine Vorgeschichte mit Plaque-Psoriasis. Bei beiden Studien konnte eine vorherige Behandlung mit cDMARD unverändert fortgesetzt werden. Die Studien beinhalteten langfristige Fortsetzungen um bis zu 5 Jahre (SELECT-PsA 1) und 3 Jahre (SELECT PsA 2).
Tabelle 9: Zusammenfassung der klinischen Studien
|
Studienname
|
Population
(n)
|
Behandlungsarme
|
Wesentliche Endpunkte
| |
SELECT-
PsA 1
|
Non- bDMARD-IRa
(1705)
|
·15 mg Upadacitinib
·30 mg Upadacitinib
·Placebo
·40 mg Adalimumab
|
Primärer Endpunkt:
·ACR20 in Woche 12
| |
Wichtige sekundäre Endpunkte:
·MDA in Woche 24
·Abheilung der Enthesis (LEI=0) und Daktylitis (LDI=0) in Woche 24
·PASI 75 in Woche 16
·sIGA in Woche 16
·SAPS in Woche 16
·Radiologische Progression (ΔmTSS) in Woche 24
·Δ Körperliche Funktionsfähigkeit (HAQ-DI) in Woche 12
·SF-36 PCS in Woche 12
·FACIT-F in Woche 12
·ACR20, Schmerz und Δ Körperliche Funktionsfähigkeit (HAQ-DI) im Vergleich zu Adalimumab in Woche 12
| |
SELECT-
PsA 2
|
bDMARD-IRb
(642)
|
·15 mg Upadacitinib
·30 mg Upadacitinib
·Placebo
|
Primärer Endpunkt:
·ACR20 in Woche 12
| |
Wichtige sekundäre Endpunkte:
·MDA in Woche 24
·PASI 75 in Woche 16
·sIGA in Woche 16
·SAPS in Woche 16
·Δ Körperliche Funktionsfähigkeit (HAQ-DI) in Woche 12
·SF-36 PCS in Woche 12
·FACIT-F in Woche 12
| |
Abkürzungen: ACR20 = Verbesserung um ≥20% gemäss American College of Rheumatology; bDMARD = biologic disease-modifying anti-rheumatic drug; FACIT-F = Functional Assessment of Chronic Illness Therapy-Fatigue score; HAQ-DI = Health Assessment Questionnaire-Disability Index; IR = inadequate responder (unzureichendes Ansprechen); MDA = minimal disease activity; mTSS = modifizierter Total-Sharp-Score; PASI = Psoriasis Area and Severity Index; SAPS = Self-Assessment of Psoriasis Symptoms; SF-36 PCS = Short Form (36) Health Survey (SF-36) Physical Component Summary; sIGA = static Investigator Global Assessment of psoriasis
a Patienten mit einem unzureichenden Ansprechen auf oder einer Intoleranz gegenüber mindestens einem nicht-biologischen DMARD
b Patienten mit einem unzureichenden Ansprechen auf oder einer Intoleranz gegenüber mindestens einem bDMARD
|
Klinisches Ansprechen
In beiden Studien erreichte ein signifikant grösserer Patientenanteil unter RINVOQ 15 mg ein ACR20 Ansprechen im Vergleich zu Placebo in Woche 12 (Tabelle 10, Abbildung 1). In SELECT-PsA 1 erreichte RINVOQ 15 mg eine Nichtunterlegenheit im Vergleich zu Adalimumab im Patientenanteil mit ACR20 Ansprechen in Woche 12. Ein höherer Patientenanteil unter RINVOQ 15 mg erreichte ein ACR50- und ACR70-Ansprechen in Woche 12 im Vergleich zu Placebo. Die Dauer bis zum Eintreten der Wirkung war über alle Messungen schnell, mit höheren Ansprechraten bereits in Woche 2 für ACR20.
Die Behandlung mit RINVOQ 15 mg führte zu Verbesserungen bei den einzelnen ACR-Komponenten, einschliesslich der Anzahl druckschmerzempfindlicher/schmerzhafter und geschwollener Gelenke, der allgemeinen Beurteilung durch den Patienten (Patient Global Assessment) und den Arzt (Physician Global Assessment), des HAQ-DI, der Schmerzbeurteilung und des hsCRP im Vergleich zu Placebo (Tabelle 11). Die Behandlung mit RINVOQ 15 mg erreichte grössere Verbesserungen der Schmerzen im Vergleich zu Adalimumab in Woche 24.
In beiden Studien wurden konsistente Ansprechraten allein oder in Kombination mit nicht-biologischen DMARDs für primäre und wichtige sekundäre Endpunkte beobachtet.
Die Wirksamkeit von RINVOQ 15 mg zeigte sich unabhängig von den beurteilten Subgruppen, einschliesslich Baseline-BMI, Baseline-hsCRP und der Anzahl der vorherigen nicht-biologischen DMARDs (≤1 oder >1).
Abbildung 1. Prozentzahl der Patienten, die ACR20 in SELECT-PsA 1 erreicht haben
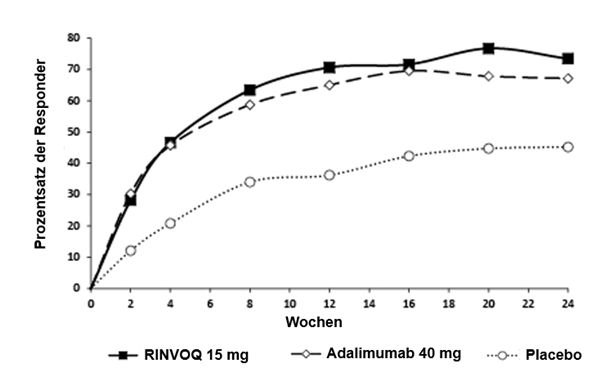
Tabelle 10: Klinisches Ansprechen
|
Studie
|
SELECT-PsA 1
Non-bDMARD-IR
|
SELECT-PsA 2
bDMARD-IR
| |
Behandlungsgruppe
|
PBO
|
UPA
15 mg
|
ADA
40 mg
|
PBO
|
UPA
15 mg
| |
N
|
423
|
429
|
429
|
212
|
211
| |
ACR20 (% der Patienten)
| |
Woche 12
|
36
|
71e
|
65
|
24
|
57e
| |
Woche 24
|
45
|
73f,g
|
67
|
20
|
59f
| |
Woche 56
|
|
74g
|
69
|
|
60
| |
ACR50 (% der Patienten)
| |
Woche 12
|
13
|
38f,g
|
38
|
5
|
32f
| |
Woche 24
|
19
|
52f,g
|
44
|
9
|
38f
| |
Woche 56
|
|
60g
|
51
|
|
41
| |
ACR70 (% der Patienten)
| |
Woche 12
|
2
|
16f,g
|
14
|
1
|
9f
| |
Woche 24
|
5
|
29f,g
|
23
|
1
|
19f
| |
Woche 56
|
|
41g
|
31
|
|
24
| |
MDA (% der Patienten)
| |
Woche 12
|
6
|
25f,g
|
25
|
4
|
17f
| |
Woche 24
|
12
|
37e,g
|
33
|
3
|
25e
| |
Woche 56
|
|
45g
|
40
|
|
29
| |
Abheilung der Enthesitis (LEI = 0; % der Patienten)a
| |
Woche 12
|
33
|
47f,g
|
47
|
20
|
39f
| |
Woche 24
|
32
|
54e,g
|
47
|
15
|
43f
| |
Woche 56
|
|
59g
|
54
|
|
43
| |
Abheilung der Daktylitis (LDI = 0; % der Patienten)b
| |
Woche 12
|
42
|
74f,g
|
72
|
36
|
64f
| |
Woche 24
|
40
|
77f,g
|
74
|
28
|
58f
| |
Woche 56
|
|
75g
|
74
|
|
51
| |
PASI 75 (% der Patienten)c
| |
Woche 16
|
21
|
63e,g
|
53
|
16
|
52e
| |
Woche 24
|
27
|
64f.g
|
59
|
19
|
54f
| |
Woche 56
|
|
65g
|
61
|
|
52
| |
PASI 90 (% der Patienten)c
| |
Woche 16
|
12
|
38f,g
|
39
|
8
|
35f
| |
Woche 24
|
17
|
42f,g
|
45
|
7
|
36f
| |
Woche 56
|
|
49g
|
47
|
|
41
| |
PASI 100 (% der Patienten)c
| |
Woche 16
|
7
|
24f,g
|
20
|
6
|
25f
| |
Woche 24
|
10
|
27f,g
|
28
|
5
|
22f
| |
Woche 56
|
|
35g
|
31
|
|
27
| |
sIGA 0/1 (% der Patienten)d
| |
Woche 16
|
11
|
42e,g
|
39
|
9
|
37e
| |
Woche 24
|
12
|
45f,g
|
41
|
10
|
33f
| |
Woche 56
|
|
52g
|
47
|
|
33
| |
Abkürzungen: ACR20 (oder 50 oder 70) = Verbesserung um ≥20% (oder ≥50% oder ≥70%) gemäss American College of Rheumatology; ADA = Adalimumab; bDMARD = biologic disease-modifying anti-rheumatic drug; IR = inadequate responder (unzureichendes Ansprechen); MDA = minimal disease activity; MTX = Methotrexat; PASI 75 (oder 90 oder 100) = Verbesserung um ≥75% (oder ≥90% oder 100%) gemäss Psoriasis Area and Severity Index; PBO = Placebo; sIGA = static Physician Global Assessment; UPA= Upadacitinib
Patienten, die die randomisierte Behandlung abgesetzt haben oder bei denen Daten in der Woche der Beurteilung fehlten, galten als Non-Responder in den Analysen. In Bezug auf MDA, die Abheilung der Enthesitis und die Abheilung der Daktylitis in Woche 24 und Woche 56 galten Patienten die in Woche 16 eine Rescue Therapie erhielten, als Nicht-Responder in den Analysen.
a Bei Patienten mit Enthesitis bei Baseline (n = 241, 270 bzw. 265 für SELECT-PsA 1 und n = 144 bzw. 133 für SELECT-PsA 2)
b Bei Patienten mit Daktylitis bei Baseline (n = 126, 136 bzw. 127 für SELECT-PsA 1 und n = 64 bzw. 55 für SELECT-PsA 2)
c Bei Patienten mit BSA Psoriasis ≥3% bei Baseline (n = 211, 214 bzw. 211 für SELECT-PsA 1 und n = 131 bzw. 130 für SELECT-PsA 2)
d Bei Patienten mit sIGA ≥2 bei Baseline (n = 313, 322 bzw. 330 für SELECT-PsA 1 und n = 163 bzw. 171 für SELECT-PsA 2)
e p ≤0,001 Upadacitinib im Vergleich zu Placebo
f Upadacitinib im Vergleich zu Placebo (diese Vergleiche sind für multiples Testen nicht kontrolliert)
g Upadacitinib im Vergleich zu Adalimumab (diese Vergleiche sind für multiples Testen nicht kontrolliert)
|
Tabelle 11: Komponenten des ACR-Ansprechens (mittlere Änderung gegenüber dem Ausgangswert)
|
Studie
|
SELECT-PsA 1
Non-bDMARD-IR
|
SELECT-PsA 2
bDMARD-IR
| |
Behandlungsgruppe
|
PBO
|
UPA
15 mg
|
ADA
40 mg
|
PBO
|
UPA
15 mg
| |
N
|
423
|
429
|
429
|
212
|
211
| |
Anzahl der druckschmerzempfindlichen/schmerzhaften Gelenke (0–68)
| |
Woche 12
|
-7,1
|
-11,3d,e
|
-10,3
|
-6,2
|
-12,4d
| |
Woche 24
|
-9,2
|
-13,7d,e
|
-12,5
|
-6,6
|
-14,0d
| |
Anzahl der geschwollenen Gelenke (0–66)
| |
Woche 12
|
-5,3
|
-7,9d,e
|
-7,6
|
-4,8
|
-7,1d
| |
Woche 24
|
-6,3
|
-9,0d,e
|
-8,6
|
-5,6
|
-8,3d
| |
Beurteilung des Schmerzes durch den Patientena
| |
Woche 12
|
-0,9
|
-2,3d
|
-2,3
|
-0,5
|
-1,9d
| |
Woche 24
|
-1,4
|
-3,0d,e
|
-2,6
|
-0,7
|
-2,2d
| |
Allgemeine Beurteilung durch den Patienten (Patient Global Assessment)a
| |
Woche 12
|
-1,2
|
-2,7d,e
|
-2,6
|
-0,6
|
-2,3d
| |
Woche 24
|
-1,6
|
-3,4d,e
|
-2,9
|
-0,8
|
-2,6d
| |
Körperliche Funktionsfähigkeit (HAQ-DI)b
| |
Woche 12
|
-0,14
|
-0,42c
|
-0,34
|
-0,10
|
-0,30c
| |
Woche 24
|
-0,19
|
-0,51d,e
|
-0,39
|
-0,08
|
-0,33d
| |
Allgemeine Beurteilung durch den Arzt (Physician Global Assessment)a
| |
Woche 12
|
-2,1
|
-3,6d,e
|
-3,4
|
-1,4
|
-3,1d
| |
Woche 24
|
-2,8
|
-4,3d,e
|
-4,1
|
-1,8
|
-3,8d
| |
hsCRP (mg/l)
| |
Woche 12
|
-1,3
|
-7,1d,e
|
-7,6
|
0,3
|
-6,6d
| |
Woche 24
|
-2,1
|
-7,6d,e
|
-7,3
|
-0,9
|
-6,3d
| |
Abkürzungen: ACR = American College of Rheumatology; ADA = Adalimumab; hsCRP = C-reaktives Protein; HAQ-DI = Health Assessment Questionnaire-Disability Index; IR = inadequate responder (unzureichendes Ansprechen); PBO = Placebo; UPA = Upadacitinib
a Numerische Ratingskala (NRS): 0 = bester Wert, 10 = schlechtester Wert
b Health Assessment Questionnaire-Disability Index: 0 = bester Wert, 3 = schlechtester Wert; 20 Fragen; 8 Kategorien: Ankleiden und Körperpflege, Aufstehen, Essen, Gehen, Hygiene, Greifen, Festhalten und Aktivitäten
c p ≤0,001 Upadacitinib im Vergleich zu Placebo
d Upadacitinib im Vergleich zu Placebo (diese Vergleiche sind für multiples Testen nicht kontrolliert)
e Upadacitinib im Vergleich zu Adalimumab (diese Vergleiche sind für multiples Testen nicht kontrolliert)
|
In beiden Studien wurden die Ansprechraten für ACR20/50/70, MDA, PASI 75/90/100, sIGA, die Abheilung der Enthesitis und Daktylitis bei mit RINVOQ 15 mg behandelten Patienten bis Woche 56 aufrechterhalten.
Radiologisches Ansprechen
Bei SELECT-PsA 1 wurde in Woche 24 die Hemmung der Progression struktureller Schäden radiologisch beurteilt und als Veränderung gegenüber dem Ausgangswert anhand des modifizierten Total-Sharp-Scores (mTSS) und seinen Komponenten, dem Score für die Erosion und dem Score für Gelenkspaltverschmälerung, dargestellt.
Die Behandlung mit RINVOQ 15 mg führte im Vergleich zu Placebo in Woche 24 zu einer signifikant grösseren Inhibierung der Progression struktureller Gelenkschäden (Tabelle 12). Statistisch signifikante Ergebnisse wurden auch beim Score für die Erosion und dem Score für Gelenkspaltverschmälerung erzielt. Der Anteil an Patienten ohne radiologische Progression (mTSS-Veränderung ≤0,5) war unter RINVOQ 15 mg im Vergleich zu Placebo in Woche 24 höher.
Tabelle 12: Radiologische Veränderungen in SELECT-PsA 1
|
Behandlungsgruppe
|
PBO
|
UPA
15 mg
|
ADA
40 mg
| |
Modifizierter Total-Sharp-Score, mittlere Veränderung gegenüber dem Ausgangswert
| |
Woche 24
|
0,25
|
-0,04c
|
0,01
| |
Woche 56a
|
0,44
|
-0,05d
|
-0,06
| |
Score für die Erosion, mittlere Veränderung gegenüber dem Ausgangswert
| |
Woche 24
|
0,12
|
-0,03d
|
0,01
| |
Woche 56a
|
0,30
|
-0,03d
|
-0,05
| |
Score für die Gelenkspaltverschmälerung, mittlere Veränderung gegenüber dem Ausgangswert
| |
Woche 24
|
0,10
|
-0,00d
|
-0,02
| |
Woche 56a
|
0,14
|
-0,03d
|
-0,03
| |
Anteil der Patienten ohne radiologische Progressiona
| |
Woche 24
|
92
|
96d
|
95
| |
Week 56a
|
89
|
97d
|
94
| |
Abkürzungen: ADA = Adalimumab; PBO = Placebo; UPA= Upadacitinib
a Alle Placebodaten in Woche 56 wurden mittels linearer Extrapolation abgeleitet
b «Ohne Progression» definiert als mTSS-Veränderung ≤0,5
c p ≤0,001 Upadacitinib im Vergleich zu Placebo
d Upadacitinib im Vergleich zu Placebo (diese Vergleiche sind für multiples Testen nicht kontrolliert)
|
Körperliche Funktionsfähigkeit und gesundheitsbezogene Ergebnisse
In beiden Studien führte die Behandlung mit RINVOQ 15 mg bei Patienten zu einer signifikanten Verbesserung der körperlichen Funktionsfähigkeit gegenüber dem Ausgangswert im Vergleich zu Placebo gemäss HAQ-DI in Woche 12 (Tabelle 11); dies wurde bis Woche 56 aufrechterhalten.
Der Anteil der HAQ-DI-Responder (≥0,35 Verbesserung gegenüber dem Ausgangswert bezüglich des HAQ-DI-Scores) in Woche 12 bei SELECT-PsA 1 und SELECT-PsA 2 lag bei 58% bzw. 45% bei Patienten, die RINVOQ 15 mg erhielten, bei 33% bzw. 27% bei Patienten, die Placebo erhielten, und bei 47% bei Patienten, die Adalimumab erhielten (SELECT-PsA 1).
Die gesundheitsbezogene Lebensqualität wurde gemäss SF-36 beurteilt. In beiden Studien wurde bei Patienten unter RINVOQ 15 mg im Vergleich zu Placebo eine signifikant grössere Verbesserung auf der körperlichen Summenskala gegenüber dem Ausgangswert in Woche 12 verzeichnet. Eine grössere Verbesserung wurde auch im Vergleich zu Adalimumab beobachtet. Es wurde im Vergleich zu Placebo eine grössere Verbesserung auf der psychischen Summenskala und in allen 8 Teilbereichen des SF-36 (körperliche Funktionsfähigkeit, körperlicher Schmerz, Vitalität, soziale Funktionsfähigkeit, körperliche Rolle, allgemeine Gesundheit, emotionale Rolle und psychische Gesundheit) verzeichnet. Die Verbesserungen gegenüber dem Ausgangswert wurden in beiden Studien bis Woche 56 aufrechterhalten.
In beiden Studien wurde bei Patienten unter RINVOQ 15 mg im Vergleich zu Placebo eine signifikant grössere Verbesserung der Müdigkeit, gemessen anhand der Scores auf der Skala zur Fatigue in der Funktionsbeurteilung bei Therapie chronischer Erkrankungen (FACIT F), gegenüber dem Ausgangswert in Woche 12 verzeichnet. Die Verbesserungen gegenüber dem Ausgangswert wurden in beiden Studien bis Woche 56 aufrechterhalten.
Es wurde in beiden Studien bei Patienten unter RINVOQ 15 mg im Vergleich zu Placebo und Adalimumab eine grössere Verbesserung der von Patienten gemeldeten Psoriasis-Symptome, gemessen anhand der Self-Assessment of Psoriasis Symptoms (SAPS), in Woche 16 verzeichnet. Die Verbesserungen gegenüber dem Ausgangswert wurden in beiden Studien bis Woche 56 aufrechterhalten.
Mit RINVOQ 15 mg behandelte Psoriasis-Arthritis Patienten mit Spondylitis zeigten Verbesserungen gegenüber dem Ausgangswert bezüglich der Scores des Bath Ankylosing Spondylitis Disease Activity Index (BASDAI) und der Ankylosing Spondylitis Disease Activity (ASDAS) im Vergleich zu Placebo in Woche 24. Die Verbesserungen gegenüber dem Ausgangswert wurden in beiden Studien bis Woche 56 aufrechterhalten.
Ankylosierende Spondylitis
Die Wirksamkeit und Sicherheit von RINVOQ 15 mg einmal täglich wurden in zwei randomisierten, doppelblinden, multizentrischen, placebokontrollierten Studien bei Patienten im Alter von mindestens 18 Jahren mit aktiver ankylosierender Spondylitis auf Grundlage des Bath Ankylosing Spondylitis Disease Activity Index (BASDAI) ≥4 und der Beurteilung der Gesamt-Rückenschmerzen durch den Patienten bei einem Score ≥4 beurteilt (Tabelle 13). Beide Studien umfassten eine offene Fortsetzung um bis zu 2 Jahre nach einer doppelblinden, placebokontrollierten Periode über 14 Wochen.
Tabelle 13: Zusammenfassung der klinischen Studie
|
Studienname
|
Population
(n)
|
Behandlungsarme
|
Wesentliche Endpunkte
| |
SELECT-AXIS 1
|
NSAID-IRa,b bDMARD-naiv
(187)
|
·15 mg Upadacitinib
·Placebo
|
Primärer Endpunkt:
·ASAS40 in Woche 14
| |
Wichtige sekundäre Endpunkte in Woche 14:
·ASAS-Teilremission
·BASDAI 50
·ASDAS-CRP
·BASFI (Funktionsfähigkeit)
·SPARCC MRI-Score (Wirbelsäule)
·AS Qualtiy of Life
·BASMI (Mobilität der Wirbelsäule)
·MASES (Enthesitis)
·WPAI
·ASAS Health Index
| |
SELECT-AXIS 2
|
bDMARD-IRa,c
(420)
|
·15 mg Upadacitinib
·Placebo
|
Primärer Endpunkt:
·ASAS40 in Woche 14
| |
Wichtige sekundäre Endpunkte in Woche 14:
·ASAS-Teilremission
·BASDAI50
·ASDAS-CRP
·BASFI (Funktionsfähigkeit)
·SPARCC-MRI-Score (Wirbelsäule)
·AS Quality of Life
·BASMI (Mobilität der Wirbelsäule)
·MASES (Enthesitis)
·ASAS Health Index
·ASAS20
·ASDAS inaktive Erkrankung
·Gesamt-Rückenschmerzen
·Nächtliche Rückenschmerzen
·ASDAS niedrige Krankheitsaktivität
| |
Abkürzungen: ASAS40 = Verbesserung um ≥40 % gemäss Assessment of SpondyloArthritis international Society; ASAS HI = ASAS Health Index; ASDAS-CRP = Ankylosing Spondylitis Disease Activity Score C-Reactive Protein; ASQoL = AS Quality of Life Questionnaire; BASDAI = Bath Ankylosing Spondylitis Disease Activity Index; BASFI = Bath Ankylosing Spondylitis Functional Index; BASMI = Bath Ankylosing Spondylitits Metrology Index; bDMARD = biologic disease-modifying anti-rheumatic drug; IR = inadequate responder; MASES = Maastricht Ankylosing Spondylitis Enthesitis Score; NSAID = Nonsteroidal Anti-inflammatory Drug; SPARCC MRI = Spondyloarthritis Research Consortium of Canada Magnetic Resonance Imaging; WPAI = Work Productivity and Activity Impairment
a Patienten mit einem unzureichenden Ansprechen auf mindestens zwei NSAIDs oder Intoleranz gegenüber NSAIDs oder bei denen NSAIDs kontraindiziert sind.
b Bei Baseline erhielten ungefähr 16 % der Patienten ein begleitendes csDMARD.
c Bei Baseline zeigten 77,4 % entweder gegenüber einem TNF-Inhibitor oder einem Interleukin-17-Inhibitor (IL-17i) eine mangelnde Wirksamheit; bei 30,2 % lag eine Unverträglichkeit vor; bei 12,9 % waren zuvor zwei bDMARDs verabreicht worden, zeigten jedoch keine mangelnde Wirksamkeit gegenüber diesen.
|
Klinisches Ansprechen
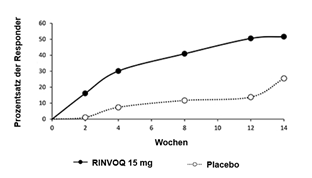
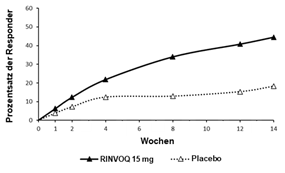
In beiden Studien erreichte ein signifikant grösserer Patientenanteil unter RINVOQ 15 mg ein ASAS40-Ansprechen im Vergleich zu Placebo in Woche 14 (51,6% vs. 25,5%; p<0,001 für SELECT-AXIS 1 und 44,5% vs. 18,2%; p<0,001 für SELECT-AXIS 2) (Tabelle 14, Abbildung 2 und 3). Höhere Ansprechraten für ASAS40 zeigten sich ab Woche 2 in SELECT-AXIS 1 (16,1% vs. 1,1%; nomineller p-Wert <0,001) und ab Woche 4 in SELECT-AXIS 2 (21,8% vs. 12,4%; nomineller p-Wert = 0,01).
Die Behandlung mit RINVOQ 15 mg führte in beiden Studien zu Verbesserungen bei den einzelnen ASAS-Komponenten (allgemeine Beurteilung der Krankheitsaktivität durch den Patienten, Beurteilung der Gesamt-Rückenschmerzen, Entzündungen und Funktionen) und andere Messungen der Krankheitsaktivität, einschliesslich hsCRP, in Woche 14 im Vergleich zu Placebo.
Die Wirksamkeit von RINVOQ 15 mg zeigte sich in beiden Studien unabhängig von den beurteilten Subgruppen, einschliesslich Geschlecht, Baseline-BMI, Dauer der Symptome der ankylosierenden Spondylitis, Baseline-hsCRP und in SELECT-AXIS 2 auch vorherige Anwendung von bDMARDs.
|
Abbildung 2: Prozentzahl der Patienten, die in SELECT-AXIS 1 ASAS40 erreicht haben
|
Abbildung 3: Prozentzahl der Patienten, die in SELECT-AXIS 2 ASAS40 erreicht haben
| |
|
|
|
|
Tabelle 14: Klinisches Ansprechen
|
Studie
|
SELECT-AXIS 1
bDMARD-naiv
|
SELECT-AXIS 2
bDMARD-IR
| |
Behandlungsgruppe
|
PBO
|
UPA 15 mg
|
PBO
|
UPA 15 mg
| |
N
|
94
|
93
|
209
|
211
| |
ASAS40 (% der Patienten)
| |
Woche 14
|
25,5
|
51,6a
|
18,2
|
44,5a
| |
ASAS20 (% der Patienten)
| |
Woche 14
|
40,4
|
64,5c
|
38,3
|
65,4a
| |
ASAS Teilremission (% der Patienten)
| |
Woche 14
|
1,1
|
19,4a
|
4,3
|
17,5a
| |
BASDAI 50 (% der Patienten)
| |
Woche 14
|
23,4
|
45,2b
|
16,7
|
43,1a
| |
ASDAS-CRP (Veränderung gegenüber dem Ausgangswert)
| |
Woche 14
|
-0,54
|
-1,45a
|
-0,49
|
-1,52a
| |
ASDAS inaktive Erkrankung (% der Patienten)
| |
Woche 14
|
-
|
-
|
1,9
|
12,8a
| |
ASDAS niedrige Krankheitsaktivität (% der Patienten)
| |
Woche 14
|
-
|
-
|
10,1
|
44,1a
| |
Abkürzungen: ASAS20 (oder 40) = Verbesserung um ≥20% (oder ≥40%) gemäss Assessment of SpondyloArthritis international Society; ASDAS-CRP = Ankylosing Spondylitis Disease Activity Score C-Reactive Protein; BASDAI = Bath Ankylosing Spondylitis Disease Activity Index; IR= inadequate responder; PBO = Placebo; UPA= Upadacitinib
a p ≤0,001 Upadacitinib im Vergleich zu Placebo
b p ≤0,01 Upadacitinib im Vergleich zu Placebo
c Upadacitinib im Vergleich zu Placebo (dieser Vergleich ist für multiples Testen nicht kontrolliert)
Für binäre Endpunkte basieren die Ergebnisse in Woche 14 auf der Non-Responder-Imputation (SELECT-AXIS 1) und auf der Non-Responder-Imputation in Verbindung mit der multiplen Imputation (SELECT-AXIS 2). Für kontinuierliche Endpunkte basieren die Ergebnisse in Woche 14 auf der mittleren Veränderung der least squares gegenüber dem Ausgangswert unter Verwendung gemischter Modelle für die Analyse wiederholter Messungen.
|
Die Ansprechraten für ASAS40, ASAS20, ASAS Teilremission, BASDAI 50, ASDAS inaktive Erkrankung, ASDAS niedrige Krankheitsaktivität, die Veränderung von ASDAS-CRP gegenüber dem Ausgangswert und hsCRP bei Patienten, die mit RINVOQ 15 mg behandelt wurden, wurden in beiden Studien bis Woche 104 beibehalten.
Körperliche Funktionsfähigkeit und gesundheitsbezogene Ergebnisse
In beiden Studien führte die Behandlung mit RINVOQ 15 mg bei Patienten zu einer signifikanten Verbesserung (p = 0,001 für SELECT-AXIS 1 und p <0,0001 für SELECT-AXIS 2) der körperlichen Funktionsfähigkeit im Vergleich zu Placebo, gezeigt gemäss BASFI-Score-Änderung zur Baseline in Woche 14 (Rinvoq 15 mg -2,29 in SELECT-AXIS 1 und -2,26 in SELECT-AXIS 2 vs. Placebo -1,30 und -1,09). Die Standardabweichung der Veränderung innerhalb der Gruppe im Vergleich zur Baseline in Woche 14 betrug in SELECT-AXIS 1 2,44 für Patienten, welche mit RINVOQ 15 mg behandelt wurden, und 2,05 für Patienten, welche mit Placebo behandelt wurden, und in SELECT-AXIS 2 2,28 für Patienten, welche mit RINVOQ 15 mg behandelt wurden, und 1,67 für Patienten, welche mit Placebo behandelt wurden.
In SELECT-AXIS 1 führte die Behandlung mit RINVOQ 15 mg bei Patienten zu einer grösseren Verbesserung der Rückenschmerzen beurteilt anhand der Gesamt-Rückenschmerz-Komponente des ASAS-Ansprechens im Vergleich zu Placebo in Woche 14.
Eine Verbesserung der nächtlichen Rückenschmerzen im Vergleich zu Placebo in Woche 14 wurde gezeigt, welche bereits ab Woche 2 festgestellt wurde.
In SELECT-AXIS 2 führte die Behandlung mit RINVOQ 15 mg bei Patienten zu signifikanten Verbesserungen der Gesamt- und nächtlichen Rückenschmerzen im Vergleich zu Placebo in Woche 14. Ein Ansprechen wurde bereits in Woche 1 bei Gesamt-Rückenschmerzen und in Woche 2 bei nächtlichen Rückenschmerzen beobachtet.
In beiden Studien wurden Verbesserungen auch bei peripheren Schmerzen und Schwellungen (gemessen anhand der BASDAI-Frage 3 zu allgemeinen Schmerzen in den Gelenken mit Ausnahme des Nackens, Rückens oder der Hüften) im Vergleich zu Placebo in Woche 14 beobachtet.
In beiden Studien wurden bei Patienten unter RINVOQ 15 mg die Ansprechraten von Woche 14 im BASFI, bei den Gesamt-Rückenschmerzen und bei den nächtlichen Rückenschmerzen bis Woche 104 aufrechterhalten.
In SELECT-AXIS 2 führte die Behandlung mit RINVOQ 15 mg im Vergleich zu Placebo bei Patienten zu im Vergleich zur Baseline signifikanten Verbesserungen der gesundheitsbezogenen Lebensqualität und der allgemeinen Gesundheit, gemessen anhand des ASQoL-Fragebogens und des ASAS-Gesundheitsindex in Woche 14. Die Verbesserungen beim ASQoL-Fragebogen und im ASAS-Gesundheitsindex wurden bis Woche 104 aufrechterhalten.
Enthesitis
In SELECT-AXIS 2 führte die Behandlung mit RINVOQ 15 mg bei Patienten mit bereits bestehender Enthesitis zu einer signifikanten Verbesserung der Enthesitis im Vergleich zu Placebo, gemessen anhand der Veränderung des MASES gegenüber Baseline in Woche 14. Die Verbesserung der Enthesitis wurde bis Woche 104 aufrechterhalten.
Beweglichkeit der Wirbelsäule
In SELECT-AXIS 2 führte die Behandlung mit RINVOQ 15 mg bei Patienten zu einer signifikanten Verbesserung der Beweglichkeit der Wirbelsäule im Vergleich zu Placebo, gemessen anhand der Veränderung des Bath Ankylosing Spondylitis Metrology Index (BASMI) gegenüber Baseline in Woche 14. Die Verbesserung des BASMI wurde bis Woche 104 aufrechterhalten.
In SELECT-AXIS 1 wurden bei Patienten unter RINVOQ 15 mg bei vordefinierten sekundären Endpunkten von BASMI, MASES, ASQoL, ASAS HI und WPAI im Vergleich zur Baseline in Woche 14 Verbesserungen im Vergleich zu Placebo beobachtet, die jedoch in den multiplizitätsbereinigten Analysen statistisch nicht signifikant waren. Die in Woche 14 beobachteten Ansprechraten wurden bei Patienten unter RINVOQ 15 mg bis Woche 104 aufrechterhalten.
Objektive Messung von Entzündungen
Anzeichen von Entzündungen wurden per MRI beurteilt und als Veränderung gegenüber dem Ausgangswert im SPARCC-Score für die Wirbelsäule wiedergegeben. In beiden Studien wurde in Woche 14 eine signifikante Verbesserung der Entzündungsanzeichen in der Wirbelsäule bei Patienten unter RINVOQ 15 mg im Vergleich zu Placebo verzeichnet. Die in Woche 14 beobachteten Ansprechraten im Hinblick auf Entzündung, ermittelt per MRI, wurden bis Woche 104 aufrechterhalten.
Riesenzellarteriitis
Die Wirksamkeit und Sicherheit von RINVOQ 15 mg einmal täglich wurden in SELECT-GCA, einer randomisierten, doppelblinden, multizentrischen, placebokontrollierten Phase-III-Studie bei Patienten im Alter von 50 Jahren und älter mit neu auftretender oder rezidivierender Riesenzellarteriitis untersucht. Patienten mit einem Rezidiv unter einer vorherigen IL6-Inhibitor-Therapie waren von der Studie ausgeschlossen. SELECT-GCA war eine 52wöchige Studie mit 428 Patienten, die im Verhältnis 2:1:1 randomisiert wurden und einmal täglich entweder Upadacitinib (RINVOQ) 15 mg, Upadacitinib 7,5 mg oder Placebo erhielten. Alle Patienten erhielten eine Hintergrundtherapie mit Kortikosteroiden. Die beiden Behandlungsarme mit Upadacitinib befolgten eine vorgegebene ausschleichende Kortikosteroidtherapie mit dem Ziel von 0 mg in Woche 26. Der Placeboarm befolgte eine vorgegebene ausschleichende Kortikosteroidtherapie mit dem Ziel von 0 mg in Woche 52. Der primäre Endpunkt war der Anteil der Patienten, die in Woche 52 eine anhaltende Remission erreichten, definiert als das Fehlen von Anzeichen und Symptomen einer Riesenzellarteriitis von Woche 12 bis Woche 52 und die Einhaltung der im Studienprotokoll festgelegten ausschleichenden Kortikosteroidtherapie. Die Studie beinhaltete eine 52wöchige Fortsetzungsphase für eine Gesamtstudiendauer von bis zu 2 Jahren.
Klinisches Ansprechen
Upadacitinib 7,5 mg und eine 26wöchige ausschleichende Kortikosteroidtherapie zeigten keine statistisch signifikante Differenz im primären Endpunkt im Vergleich zu Placebo und einer 52wöchigen ausschleichenden Kortikosteroidtherapie (die entsprechenden Daten sind nicht weiter aufgeführt).
Von 209 Patienten, die zur Behandlung mit Upadacitinib 15 mg randomisiert wurden, hatten 61 Patienten eine aktive rezidivierende Riesenzellarteriitis.
Upadacitinib 15 mg und eine 26wöchige ausschleichende Kortikosteroidtherapie zeigten eine Überlegenheit in Bezug auf das Erreichen einer kortikosteroidfreien, anhaltenden Remission in Woche 52 im Vergleich zu Placebo und einer 52wöchigen ausschleichenden Kortikosteroidtherapie (Tabelle 15).
Tabelle 15. Klinisches Ansprechen der Population mit rezidivierender RZA in der Studie SELECT-GCA
|
Behandlungsarm
|
PBO + 52wöchige ausschleichende Kortikosteroidtherapie
N = 36
|
UPA 15 mg + 26wöchige ausschleichende Kortikosteroidtherapie
N = 61
|
Behandlungsunterschied
(95 %-KI)
| |
Anhaltende Remission in Woche 52a
|
22,2%
|
42,3%
|
20,1% (1,7; 38,6)
| |
Anhaltende komplette Remission in Woche 52b
|
8,3%
|
32,7%
|
24,4% (9,5; 39,2)
| |
Abkürzungen: BSG = Blutsenkungsgeschwindigkeit; hsCRP = hochsensitives Creaktives Protein; PBO = Placebo; RZA = Riesenzellarteriitis; UPA = Upadacitinib
a Anhaltende Remission wird definiert als das Erreichen eines Fehlens von Anzeichen und Symptomen einer RZA von Woche 12 bis Woche 52 und die Einhaltung der im Studienprotokoll festgelegten ausschleichenden Kortikosteroidtherapie.
b Anhaltende komplette Remission wird definiert als das Erreichen eines Fehlens von Anzeichen und Symptomen einer RZA von Woche 12 bis Woche 52, einer Normalisierung der BSG (auf ≤30 mm/h; falls BSG > 30 mm/h und Erhöhung nicht auf RZA zurückzuführen, kann dieses Kriterium dennoch erfüllt werden) von Woche 12 bis Woche 52, einer Normalisierung des hsCRP auf < 1 mg/dl ohne Erhöhung auf ≥1 mg/dl (bei 2 aufeinanderfolgenden Besuchen) von Woche 12 bis Woche 52 und die Einhaltung der im Studienprotokoll festgelegten ausschleichenden Kortikosteroidtherapie.
|
Gesundheitsbezogene Ergebnisse
Fatigue wurde anhand des FACIT-Fatigue-Scores beurteilt. Patienten, die mit Upadacitinib 15 mg und einer 26wöchigen ausschleichenden Kortikosteroidtherapie behandelt wurden, zeigten in Woche 52 eine grössere Verbesserung des FACIT-Fatigue-Scores gegenüber Baseline (3,7; 95%-CI: 0,88 – 6,54) als Patienten unter Placebo und einer 52wöchigen ausschleichenden Kortikosteroidtherapie (-4,1; 95%-CI: -8,16 – 0,02).
Atopische Dermatitis
Die Wirksamkeit und Sicherheit von RINVOQ 15 mg und 30 mg einmal täglich wurden in drei randomisierten, doppelblinden, multizentrischen Phase-III-Studien (MEASURE UP 1, MEASURE UP 2 und AD UP) bei insgesamt 2584 Patienten (im Alter von mindestens 12 Jahren) bewertet (Tabelle 16). RINVOQ wurde bei 344 jugendlichen und 2240 erwachsenen Patienten mit mittelschwerer bis schwerer atopischer Dermatitis (AD) untersucht, die nicht genügend durch topische Medikamente kontrolliert wurden. Bei Baseline musste bei den Patienten Folgendes vorliegen: ein Investigator Global Assessment-(vIGA-AD-)Score ≥3 bei der allgemeinen AD-Bewertung (Erythem, Induration/Papulation und Nässen/Verkrustung) auf einer Skala mit zunehmendem Schweregrad von 0 bis 4, ein Eczema Area and Severity Index(EASI)-Score ≥16 (zusammengesetzter Score, der das Ausmass und den Schweregrad des Erythems, Ödems/der Papulation, Kratzer und Lichenifikation an 4 verschiedenen Körperstellen erfasst), eine minimale Beteiligung der Körperoberfläche (BSA) von ≥10 % und ein durchschnittlicher wöchentlicher Score auf der Worst Pruritus Numerical Rating Scale (NRS) von ≥4.
In allen drei Studien haben die Patienten über einen Zeitraum von 16 Wochen RINVOQ einmal täglich 15 mg bzw. 30 mg oder ein entsprechendes Placebo erhalten. In der AD UP-Studie haben die Patienten zusätzlich topische Kortikosteroide (TCS) als Begleitmedikation erhalten. Nach Abschluss des doppelblinden Abschnitts wurden die Patienten, welche ursprünglich RINVOQ erhielten, bis Woche 136 mit der gleichen Dosis weiterbehandelt. Die Patienten in der Placebo-Gruppe wurden im Verhältnis 1:1 erneut randomisiert und erhielten bis Woche 136 RINVOQ 15 mg oder 30 mg.
Tabelle 16. Zusammenfassung der klinischen Studie
|
Studienname
|
Behandlungsarme
|
Wichtige Ergebnismessungen
| |
MEASURE UP 1
und
MEASURE UP 2
|
·Upadacitinib 15 mg
·Upadacitinib 30 mg
·Placebo
|
Co-primäre Endpunkte nach Woche 16:
·EASI 75
·vIGA-AD 0/1
| |
Wichtige sekundäre Endpunkte (nach Woche 16, wenn nicht anders vermerkt)
·EASI 90/100
·EASI 75 nach Woche 2
·% Änderung des EASI
·% Änderung des SCORAD
·Verbesserung des Worst Pruritus NRS ≥4 in Woche 1 und 16
·Verbesserung des Worst Pruritus NRS ≥4 an Tag 2 (30 mg); Tag 3 (15 mg)
·% Änderung der Worst Pruritus NRS
·EASI-Anstieg ≥6,6 Punkte (Aufflammen) während des doppelblinden Abschnitts
·ADerm-SS TSS-7 Verbesserung ≥28
·ADerm-SS Hautschmerzen Verbesserung ≥4
·ADerm-IS Schlaf Verbesserung ≥12
·ADerm-IS Gemütszustand Verbesserung ≥11
·ADerm-IS Tägliche Aktivitäten Verbesserung ≥14
·POEM Verbesserung ≥4
·HADS-A < 8 und HADS-D < 8
·DLQI 0/1
·DLQI Verbesserung ≥4
| |
AD UP
|
·Upadacitinib 15 mg + TCS
·Upadacitinib 30 mg +TCS
·Placebo +TCS
|
Co-primäre Endpunkte nach Woche 16:
·EASI 75
·vIGA-AD 0/1
Wichtige sekundäre Endpunkte (nach Woche 16, wenn nicht anders vermerkt)
·EASI 75 nach Woche 2 und 4
·EASI 90 nach Woche 4 und 16
·EASI 100 (30 mg)
·% Änderung des EASI
·Verbesserung des Worst Pruritus NRS ≥4 nach Woche 1, 4 und 16
·% Änderung des Worst Pruritus NRS
| |
Abkürzungen: SCORAD = SCORing Atopic Dermatitis, POEM: Patient Oriented Eczema Measure, DLQI: Dermatology Life Quality Index, HADS: Hospital Anxiety and Depression Scale, ADerm-SS = Atopic Dermatitis Symptom Scale, ADerm-IS: Atopic Dermatitis Impact Scale
|
Baseline Charakteristika
In den Monotherapie Studien (MEASURE UP 1 und 2) hatten 50,0% der Patienten einen Baseline vIGA-AD Score von 3 (moderat) und 50,0% der Patienten einen vIGA-AD Score von 4 (schwer). Die mittlere Baseline des EASI Score war 29,3 und die mittlere Baseline der wöchentlichen Worst Pruritus NRS war 7,3. In den Monotherapie-Studien betrug das Durchschnittsalter über alle Behandlungsgruppen 33,8 Jahre, das Durchschnittskörpergewicht 74,8 kg, 44,9% waren Frauen, 67,3% waren Weisse, 22,9% waren Asiaten und 6,3% waren Schwarze.
In der Studie mit begleitend verabreichten TCS (AD UP), hatten 47,1% der Patienten einen Baseline vIGA-AD Score von 3 (moderat) und 52,9% der Patienten hatten einen vIGA-AD Score von 4 (schwer). Die mittlere Baseline des EASI Score war 29,7 und die mittlere Baseline des wöchentlichen Worst Pruritus NRS war 7,2. In der AD UP-Studie betrug das Durchschnittsalter in allen Behandlungsgruppen 34,1 Jahre, das Durchschnittskörpergewicht 75,5 kg, 39,3% waren Frauen, 71,8% waren Weisse, 20,5% waren Asiaten und 5,5% waren Schwarze.
Klinisches Ansprechen
Monotherapie-Studien (MEASURE UP 1 UND MEASURE UP 2)
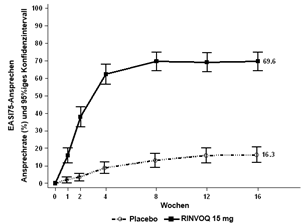
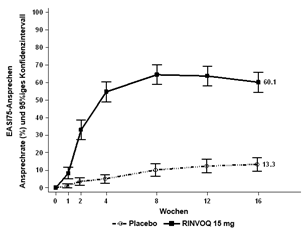
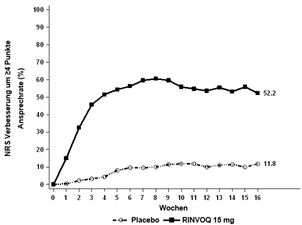
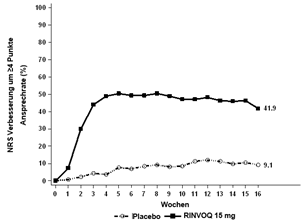
In den MEASURE UP-Studien erreichte ein signifikant höherer Anteil der mit RINVOQ 15 mg behandelten Patienten nach Woche 16 ein vIGA-AD-Ansprechen von 0 oder 1 und einen EASI 75 im Vergleich zu Placebo (Tabelle 17). Es wurde bei RINVOQ 15 mg im Vergleich zu Placebo eine schnelle Verbesserung des Hautbildes (gemäss EASI 75 bereits nach Woche 2) erreicht (p < 0,001).
Bei einem signifikant höheren Anteil der mit RINVOQ 15 mg behandelten Patienten wurde nach Woche 16 eine klinisch bedeutungsvolle Verbesserung des Juckreizes (als ≥4-Punkte-Verringerung des Worst Pruritus NRS definiert) im Vergleich zu Placebo erreicht. Es wurde eine schnelle Verbesserung des Juckreizes (als ≥4-Punkte-Verringerung des Worst Pruritus NRS nach Woche 1 definiert) bei RINVOQ 15 mg im Vergleich zu Placebo (p < 0,001) erreicht; Unterschiede wurden dabei bereits zwei Tage nach Beginn der Behandlung mit RINVOQ 15 mg (Tag 3, p < 0,001) beobachtet.
Bei einem signifikant tieferen Anteil der mit RINVOQ 15 mg behandelten Patienten trat während der ersten 16 Wochen Behandlung im Vergleich zu Placebo (p < 0,001) ein Aufflammen der Erkrankung auf, definiert als klinisch bedeutungsvolle Verschlechterung der Krankheit (Anstieg des EASI um ≥6,6).
Abbildung 4 und Abbildung 5 zeigen den Anteil der Patienten, die ein EASI-75-Ansprechen erreicht haben bzw. den Anteil der Patienten mit einer ≥4-Punkte-Verbesserung des Worst Pruritus NRS bis zu Woche 16.
Tabelle 17: Wirksamkeitsergebnisse der RINVOQ-Monotherapie-Studien nach Woche 16
|
Studie
|
MEASURE UP 1
|
MEASURE UP 2
| |
Behandlungsgruppe
|
PBO
|
UPA
15 mg
|
PBO
|
UPA
15 mg
| |
Anzahl der randomisierten Teilnehmer
|
281
|
281
|
278
|
276
| |
% Responder
| |
vIGA-AD 0/1a,b
|
8,4%
|
48,1%f
|
4,7%
|
38,8%f
| |
EASI 75a
|
16,3%
|
69,6%f
|
13,3%
|
60,1%f
| |
EASI 90 a
|
8,1%
|
53,1%f
|
5,4%
|
42,4%f
| |
EASI 100 a
|
1,8 %
|
16,7%f
|
0,7%
|
14,1%f
| |
Worst Pruritus NRSc
(≥4-Punkte-Verbesserung)
|
11,8%
N=272
|
52,2%f
N=274
|
9,1%
N=274
|
41,9%f
N=270
| |
Worst Pruritus NRS 0 oder 1d
|
5,5%
N=275
|
36,6%g
N=279
|
4,3%
N=277
|
26,9%g
N=275
| |
Mittlere prozentuale Änderung (SE)e
| |
EASI
|
-40,7% (2,28)
|
-80,2%f (1,91)
|
-34,5% (2,59)
|
-74,1%f (2,20)
| |
SCORAD
|
-32,7% (2,33)
|
-65,7%f (1,78)
|
-28,4% (2,50)
|
-57,9%f (2,01)
| |
Worst Pruritus NRS
|
-26,1% (5,41)
|
-62,8%f (4,49)
|
-17,0% (2,73)
|
-51,2%f (2,34)
| |
Abkürzungen: UPA= Upadacitinib (RINVOQ); PBO = Placebo
a Basierend auf der Anzahl randomisierter Patienten
b Responder wurde als Patient mit vIGA-AD 0 oder 1 («klar» oder «fast klar») mit einer Verringerung um ≥2 Punkte auf einer Ordnungsskala von 0 bis 4 definiert
c N = Anzahl der Patienten, deren Worst Pruritus NRS bei Baseline bei ≥4 ist
d N = Anzahl der Patienten, deren Worst Pruritus NRS bei Baseline bei > 1 ist
e % Veränderung = mittlere prozentuale Veränderung der «least squares» gegenüber dem Ausgangswert
f für multiples Testen kontrollierter p < 0,001 Upadacitinib im Vergleich zu Placebo
g nominal p < 0,001 Upadacitinib im Vergleich zu Placebo
|
Abbildung 4: Anteil der Patienten, die ein EASI-75-Ansprechen in den Monotherapie-Studien erreicht haben
|
MEASURE UP 1
|
MEASURE UP 2
| |
|
|
|
|
Abbildung 5: Anteil der Patienten mit einer ≥4-Punkte-Verbesserung des Worst Pruritus NRS in den Monotherapie-Studien
|
MEASURE UP 1
|
MEASURE UP 2
| |
|
|
|
|
Das Behandlungsansprechen in Subgruppen (Gewicht, Alter, Geschlecht, ethnische Herkunft und vorherige Behandlung mit Immunsuppressiva) waren in beiden Studien mit den Ergebnissen in der allgemeinen Studienpopulation konsistent.
Bei beiden Studien wurden die Ergebnisse nach Woche 16 bis Woche 52 bei mit RINVOQ 15 mg behandelten Patienten weiterhin beobachtet.
Studie zu begleitend verabreichten TCS (AD UP)
In der AD UP-Studie erreichte ein signifikant höherer Anteil der mit RINVOQ 15 mg plus TCS behandelten Patienten nach Woche 16 ein vIGA-AD-Ansprechen mit 0 oder 1 und ein EASI-75-Ansprechen im Vergleich zu Placebo plus TCS (Tabelle 18). Es wurde im Vergleich zu Placebo plus TCS eine schnelle Verbesserung des Hautbildes (gemäss EASI 75 nach Woche 2) erreicht (p < 0,001). Zusätzlich wurde im Vergleich zu Placebo plus TCS nach Woche 4 eine höhere EASI-90-Ansprechrate erreicht (p < 0,001).
Bei einem signifikant höheren Anteil an mit RINVOQ 15 mg plus TCS behandelten Patienten wurde nach Woche 16 eine klinisch bedeutungsvolle Verbesserung des Juckreizes (als ≥4-Punkte-Verringerung des Worst Pruritus NRS definiert) im Vergleich zu Placebo erreicht. Es wurde im Vergleich zu Placebo eine schnelle Verbesserung des Juckreizes (als eine ≥4-Punkte-Verringerung des Worst Pruritus NRS bereits nach Woche 1 definiert) erreicht (p < 0,001).
Abbildung 6 und Abbildung 7 zeigen den Anteil der Patienten, die ein EASI-75-Ansprechen erreicht haben bzw. den Anteil der Patienten mit einer ≥4-Punkte-Verbesserung des Worst Pruritus NRS bis zu Woche 16.
Tabelle 18: Wirksamkeitsergebnisse von RINVOQ mit begleitenden TCS nach Woche 16
|
Behandlungsgruppe
|
Placebo + TCS
|
UPA 15 mg + TCS
| |
Anzahl der randomisierten Teilnehmer
|
304
|
300
| |
% Responder
| |
vIGA-AD 0/1a,b
|
10,9%
|
39,6%f
| |
EASI 75a
|
26,4%
|
64,6%f
| |
EASI 90a
|
13,2%
|
42,8%f
| |
EASI 100a
|
1,3%
|
12,0% g
| |
Worst Pruritus NRSc
(≥4-Punkte-Verbesserung)
|
15,0%
N=294
|
51,7%f
N=288
| |
Worst Pruritus NRS 0 oder 1d
|
7,3%
N=300
|
33,1%g
N=296
| |
Mittlere prozentuale Änderung (SE)e
| |
EASI
|
-45,9% (2,16)
|
-78,0%f (1,98)
| |
SCORAD
|
-33,6% (1,90)
|
-61,2%g (1,70)
| |
Worst Pruritus NRS
|
-25,1% (3,35)
|
-58,1%f (3,11)
| |
Abkürzungen: UPA= Upadacitinib (RINVOQ); PBO = Placebo
a Basierend auf der Anzahl randomisierter Patienten
b Responder wurde als Patient mit vIGA-AD 0 oder 1 («klar» oder «fast klar») mit einer Verringerung um ≥2 Punkte auf einer Ordnungsskala von 0 bis 4 definiert
c N = Anzahl der Patienten, deren Worst Pruritus NRS bei Baseline bei ≥4 ist
d N = Anzahl der Patienten, deren Worst Pruritus NRS bei Baseline bei > 1 ist
e % Veränderung = mittlere prozentuale Veränderung der least squares gegenüber dem Ausgangswert
f für multiples Testen kontrolliert p < 0,001 Upadacitinib + TCS im Vergleich zu Placebo + TCS
g nominal p < 0,001 Upadacitinib + TCS im Vergleich zu Placebo + TCS
|
Abbildung 6: Anteil der Patienten, die ein EASI-75-Ansprechen in der AD UP-Studie erreicht haben
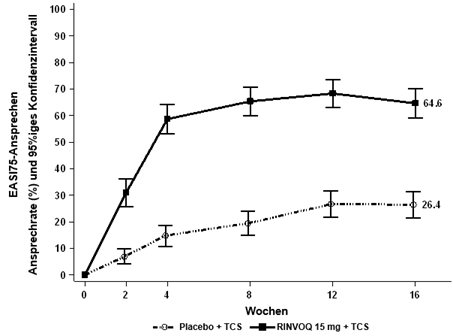
Abbildung 7: Anteil der Patienten mit einer ≥4-Punkte-Verbesserung des Worst Pruritus NRS in der AD UP-Studie
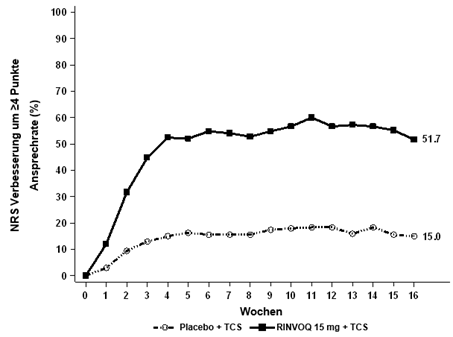
Die Behandlungswirkungen in Subgruppen (Gewicht, Alter, Geschlecht, ethnische Herkunft und vorherige Behandlung mit Immunsuppressiva) waren in der AD UP-Studie mit den Ergebnissen in der allgemeinen Studienpopulation konsistent.
Patienten, die mit RINVOQ 15 mg behandelt wurden, hatten signifikant mehr Tage ohne TCS-Anwendung bei einem gleichzeitigen EASI-75-Ansprechen (Mittelwert: 33,5 Tage) über einen Zeitraum von 16 Wochen im Vergleich zur Placebogruppe (Mittelwert: 7,9 Tage).
Die Ergebnisse nach Woche 16 wurden bis Woche 52 bei mit RINVOQ 15 mg behandelten Patienten weiterhin beobachtet.
Lebensqualität / von Patienten berichtete Ergebnisse
In den MEASURE UP-Studien berichtete ein signifikant höherer Anteil der mit RINVOQ 15 mg behandelten Patienten nach Woche 16 klinisch bedeutungsvolle Linderungen der AD-Symptome und Verringerungen der AD-Auswirkung auf die gesundheitsbezogene Lebensqualität im Vergleich zu Placebo (Tabelle 19). Ein signifikant höherer Anteil der mit RINVOQ behandelten Patienten erreichten nach Woche 16 klinisch bedeutungsvolle Verringerungen des Schweregrads der AD-Symptome gemäss ADerm SS TSS-7 und ADerm SS Hautschmerzen im Vergleich zu Placebo. Ein höherer Anteil der mit RINVOQ behandelten Patienten erreichte nach Woche 16 klinisch bedeutungsvolle Verringerungen der von Patienten berichteten Auswirkungen von AD auf den Schlaf, tägliche Aktivitäten und den Gemütszustand gemäss ADerm IS-Domänen-Scores im Vergleich zu Placebo. Im Vergleich zu Placebo erreichte in gleicher Weise ein höherer Anteil der mit RINVOQ behandelten Patienten nach Woche 16 klinisch bedeutungsvolle Verbesserungen der AD-Symptomhäufigkeit und gesundheitsbezogenen Lebensqualität gemäss POEM und DLQI.
Symptome von Angst und Depressionen gemäss HADS-Score waren signifikant verringert; bei Patienten mit Ausgangswerten auf der HADS-Angst- oder HADS-Depressionssubskala ≥8 (Cut-off-Wert für Angstzustände oder Depressionen) erreichte ein höherer Anteil der Patienten in der mit RINVOQ 15 mg nach Woche 16 HADS-Angst- und HADS-Depressions-Scores < 8 im Vergleich zu Placebo (Tabelle 19).
Tabelle 19: Von Patienten berichtete Ergebnisse der RINVOQ-Monotherapie-Studien nach Woche 16
|
Studie
|
MEASURE UP 1
|
MEASURE UP 2
| |
Behandlungsgruppe
|
PBO
|
UPA
15 mg
|
PBO
|
UPA
15 mg
| |
Anzahl der randomisierten Teilnehmer
|
281
|
281
|
278
|
276
| |
% Responder
| |
ADerm-SS TSS-7
(≥28-Punkte-Verbesserung)a,b
|
15,0%
N=226
|
53,6%h
N=233
|
12,7%
N=244
|
53,0%h
N=230
| |
ADerm-SS Hautschmerzen
(≥4-Punkte-Verbesserung)a
|
15,0%
N=233
|
53,6% h
N=237
|
13,4%
N=247
|
49,4%h
N=237
| |
ADerm-IS Schlaf
(≥12-Punkte-Verbesserung)a,c
|
13,2%
N=220
|
55,0%h
N=218
|
12,4%
N=233
|
50,2%h
N=219
| |
ADerm-IS Tägliche Aktivitäten
(≥14-Punkte-Verbesserung) a,d
|
20,3%
N=197
|
65,0%h
N=203
|
18,9%
N=227
|
57,0%h
N=207
| |
ADerm-IS Gemütszustand
(≥11-Punkte-Verbesserung)a,d
|
19,8%
N=212
|
62,6%h
N=227
|
16,7%
N=234
|
57,0%h
N=228
| |
DLQI
(DLQI 0/1)f
|
4,4%
N=252
|
30,3%h
N=258
|
4,7%
N=257
|
23,8%h
N=252
| |
DLQI
(≥4-Punkte-Verbesserung)a
|
29,0%
N=250
|
75,4%h
N=254
|
28,4%
N=250
|
71,7%h
N=251
| |
POEM
(≥4-Punkte-Verbesserung)a
|
22,8%
N=276
|
75,0%h
N=278
|
28,7%
N=268
|
70,9%h
N=268
| |
HADS
(HADS-A < 8 und HADS-D < 8)g
|
14,3%
N=126
|
45,5%h
N=145
|
11,4%
N=140
|
46,0%h
N=137
| |
Abkürzungen: UPA= Upadacitinib (RINVOQ); PBO = Placebo
Die festgelegten Grenzwerte entsprechen dem geringsten klinisch relevanten Unterschied (MCID) und wurden zur Bestimmung des Ansprechens verwendet.
a N = Anzahl der Patienten, deren Ausgangswert grösser oder gleich MCID ist.
b ADerm-SS TSS-7 bewertet das Jucken beim Schlafen und im Wachzustand, Hautschmerzen, Hautrisse, durch Hautrisse verursachten Schmerz, trockene Haut und Hautschuppung aufgrund von AD.
c ADerm-IS Schlaf bewertet die Schwierigkeit beim Einschlafen, Auswirkung auf den Schlaf und das Aufwachen in der Nacht aufgrund von AD.
d ADerm-IS Tägliche Aktivitäten bewertet die Auswirkung von AD auf Haushalts- Aktivitäten, körperliche Aktivitäten, soziale Aktivitäten und die Konzentration.
e ADerm-IS Gemütszustand bewertet das Selbstbewusstsein, emotionalen Stress und Traurigkeit aufgrund von AD.
f N ist die Anzahl der Patienten, deren Ausgangs-DLQI-Score > 1 beträgt.
g N ist die Anzahl der Patienten, deren Ausgangs-HADS-A- oder HADS-D-Score ≥8 beträgt.
h für multiples Testen kontrolliert p < 0,001 Upadacitinib im Vergleich zu Placebo
|
Colitis ulcerosa
Die Wirksamkeit und Sicherheit von RINVOQ wurde in drei multizentrischen, doppelblinden, placebokontrollierten Phase-III-Studien untersucht: zwei Replikatinduktionsstudien – UC-1 (U-ACHIEVE Induction) und UC-2 (U-ACCOMPLISH) – und eine Erhaltungsstudie UC-3 (U-ACHIEVE Maintenance). Zusätzlich wurde die Wirksamkeit und Sicherheit von RINVOQ in einer Langzeitstudie, UC-4 (U-ACTIVATE), beurteilt.
Die Krankheitsaktivität basierte auf dem adaptierten Mayo-Score (aMS, Mayo-Scoring-System ohne die allgemeine Beurteilung durch den Arzt), der zwischen 0 und 9 lag und drei Subscores mit einem Wert von 0 (normal) bis 3 (äusserst schwer) umfasst: Subscore zur Stuhlfrequenz (SFS), Subscore zur Darmblutung (RBS) und einen zentral überprüften endoskopischen Subscore (ES).
Tabelle 20: Zusammenfassung der klinischen Studien
|
Name der Studie
|
Population (n)
|
Behandlungsarme
|
Wichtige Endpunkte
| |
Induktion
| |
U-ACHIEVE
(UC-1)
U-ACCOMPLISH (UC-2)
|
Versagen der Biologikatherapie*
(246/473)
Kein Versagen der Biologikatherapie+(227/473)
Versagen der Biologikatherapie
(262/515)
Kein Versagen der Biologikatherapie (253/515)
|
·Upadacitinib 45 mg
·Placebo
|
Primärer Endpunkt:
·Klinische Remission gemäss adaptiertem Mayo-Score in Woche 8
| |
Sekundäre Endpunkte in Woche 8 oder wie angegeben:
·Endoskopische Verbesserung
·Endoskopische Remission
·Klinisches Ansprechen
·Klinisches Ansprechen in Woche 2
·Histologisch-endoskopische Verbesserung der Mukosa
·Kein Stuhldrang
·Keine Bauchschmerzen
·Histologische Verbesserung
·Veränderung des Gesamtscores auf der IBDQ gegenüber Baseline
·Mukosaheilung
·Veränderung des FACIT-F gegenüber Baseline
| |
Erhaltung
| |
U-ACHIEVE
(UC-3)
|
Versagen der Biologikatherapie
(225/451)
Kein Versagen der Biologikatherapie (226/451)
|
·Upadacitinib 15 mg
·Upadacitinib 30 mg
·Placebo
|
Primärer Endpunkt:
·Klinische Remission gemäss adaptiertem Mayo-Score in Woche 52
| |
Sekundäre Endpunkte in Woche 52:
·Endoskopische Verbesserung
·Aufrechterhaltung der klinischen Remission
·Kortikosteroidfreie klinische Remission
·Aufrechterhaltung der endoskopischen Verbesserung
·Endoskopische Remission
·Aufrechterhaltung des klinischen Ansprechens
·Histologisch-endoskopische Verbesserung der Mukosa
·Veränderung des IBDQ-Gesamtscores gegenüber Baseline
·Mukosaheilung
·Kein Stuhldrang
·Keine Bauchschmerzen
·Veränderung des FACIT-F gegenüber Baseline
| |
*Versagen der Biologikatherapie: unzureichendes Ansprechen, Verlust des Ansprechens oder Unverträglichkeit gegenüber einer vorherigen Biologikatherapie
+Kein Versagen der Biologikatherapie: unzureichendes Ansprechen, Verlust des Ansprechens oder Unverträglichkeit gegenüber einer konventionellen Therapie, aber kein Versagen der Biologikatherapie
Abkürzungen: IBDQ: Fragebogen zu chronisch entzündlichen Darmerkrankungen (Inflammatory Bowel Disease Questionnaire), FACIT-F: Fragebogen zur Fatigue in der Funktionsbeurteilung bei Therapie chronischer Krankheiten (Functional Assessment of Chronic Illness Therapy-Fatigue)
|
Induktionsstudien (UC-1 und UC-2)
In den Studien UC-1 und UC-2 wurden 979 erwachsene und 9 jugendliche Patienten (473 bzw. 515 Patienten) im Verhältnis 2:1 randomisiert und 8 Wochen lang mit RINVOQ 45 mg einmal täglich oder Placebo behandelt und in die Auswertung zur Wirksamkeit eingeschlossen. Alle in Frage kommenden Patienten hatten eine mittelschwere bis schwere aktive Colitis ulcerosa, definiert als aMS von 5 bis 9 mit einem ES von 2 oder 3, und ein vorheriges Therapieversagen, einschliesslich unzureichendem Ansprechen, Verlust des Ansprechens oder Unverträglichkeit gegenüber einer vorherigen konventionellen und/oder Biologikatherapie. Patienten mit einer vorherigen JAK-Inhibitor-Therapie waren von den Studien ausgeschlossen.
Von diesen 979 erwachsenen Patienten hatten 500 unzureichend auf die Behandlung mit einem oder mehreren Biologika angesprochen oder diese nicht vertragen (vorheriges Versagen einer Biologikatherapie)(246 bzw. 254 Patienten). Bei Baseline erhielten 46 % bzw. 47 % der Patienten Kortikosteroide, 1 % bzw. 1 % Methotrexat und 57 % bzw. 61 % Aminosalicylate. Die gleichzeitige Anwendung von Thiopurinen war in den Studien nicht erlaubt. Die Krankheitsaktivität war bei 59 % bzw. 59 % der Patienten mittelschwer (aMS ≤7) und bei 41 % bzw. 41 % schwer (aMS > 7).
Die Resultate des primären Endpunkts der klinischen Remission und der sekundären Endpunkte in Woche 8 bei erwachsenen Patienten mit vorherigem Versagen einer Biologikatherapie sind in Tabelle 21 aufgeführt. Die gepoolten Ergebnisse des klinischen Ansprechens im Zeitverlauf pro paMS bei UC-1 und UC-2 sind in Abbildung 8 dargestellt.
Tabelle 21: Anteil der erwachsenen Patienten mit vorherigem Versagen einer Biologikatherapie, die in Woche 8 die primären und sekundären Endpunkte zur Wirksamkeit in den Induktionsstudien UC-1 und UC-2 erreichten
|
|
UC-1
(U-ACHIEVE)
|
UC-2
(U-ACCOMPLISH)
| |
Endpunkt
|
PBO
N = 78
|
UPA
45 mg
N = 168
|
Behandlungsunterschied
(95 %-KI)
|
PBO
N = 86
|
UPA
45 mg
N = 168
|
Behandlungsunterschied
(95 %-KI)
| |
Krankheitsaktivität und UC-Symptome
| |
Klinische Remissiona
|
0,4 %
|
17,9 %
|
17,5 %***
(11,4; 23,6)
|
2,5 %
|
29,9 %
|
27,3 %***
(19,6; 35,1)
| |
Klinisches Ansprechenb
|
12,8 %
|
64,4 %
|
51,6 %***
(41,2; 61,9)
|
19,9 %
|
69,0 %
|
49,1 %***
(38,1; 60,1)
| |
Kein Stuhldrang
|
14,1 %
|
40,2 %
|
26,1 %***
(15,4; 36,8)
|
17,4 %
|
50,0 %
|
32,6 %***
(21,5 43,6)
| |
Keine Bauchschmerzen
|
20,5 %
|
42,6 %
|
22,1 %***
(10,4; 33,8)
|
18,6 %
|
53,0 %
|
34,4 %***
(23,2; 45,5)
| |
Endoskopische und histologische Bewertung
| |
Endoskopische Remissionc
|
0
|
8,9 %
|
8,9 %**
(4,6; 13,3)
|
1,2 %
|
12,5 %
|
11,3 %**
(5,8; 16,8)
| |
Endoskopische Verbesserungd
|
1,7 %
|
27,0 %
|
25,3 %***
(17,8; 32,7)
|
5,0 %
|
37,0 %
|
32,0 %***
(23,3; 40,7)
| |
Histologische Verbesserunge
|
17,5 %
|
51,0 %
|
33,5 %***
(22,1; 44,9)
|
21,0 %
|
58,9 %
|
37,9 %***
(26,4; 49,4)
| |
Histologisch-endoskopische Mukosaverbesserungf
|
1,4 %
|
22,7 %
|
21,3 %***
(14,4; 28,2)
|
4,8 %
|
30,4 %
|
25,6 %***
(17,3; 34,0)
| |
Mukosaheilungg
|
0
|
6,5 %
|
6,5 %*
(2,8; 10,3)
|
1,2 %
|
8,9 %
|
7,8 %*
(2,9; 12,6)
| |
Lebensqualität
| |
Veränderung des FACIT-F-Scores gegenüber Baseline
|
N = 65
1,3
|
N = 154
9,6
|
8,3***
(5,66; 11,01)
|
N = 76
3,3
|
N = 154
9,8
|
6,6***
(4,15; 8,99)
| |
Veränderung des Gesamtscores auf der IBDQ gegenüber Baseline
|
N = 65
14,6
|
N = 155
55,7
|
41,1***
(31,53; 50,64)
|
N = 76
17,4
|
N = 156
53,5
|
36,1***
(27,57; 44,72)
| |
Abkürzung: PBO = Placebo
*p ≤0.05
**p ≤0.01
***p ≤0,001, Behandlungsunterschied (95 %-KI)
a Gemäss aMS: SFS ≤1 und nicht grösser als bei Baseline, RBS = 0, ES ≤1 ohne Friabilität
b Gemäss aMS: Verringerung um ≥2 Punkte und ≥30 % gegenüber Baseline und Verringerung des RBS um ≥1 gegenüber Baseline oder ein absoluter RBS-Wert von ≤1
c ES von 0
d ES ≤1 ohne Friabilität
e Verringerung des Geboes-Score gegenüber Baseline. Die histologischen Ergebnisse wurden anhand des Geboes-Scores beurteilt.
f ES ≤1 ohne Friabilität und Geboes-Score ≤3,1 (weist auf eine Neutrophileninfiltration in < 5 % der Krypten hin, keine Zerstörung der Krypten und keine Erosionen, Ulzerationen oder Granulationsgewebe).
g ES = 0, Geboes-Score < 2 (weist darauf hin, dass keine Neutrophile in Krypten oder der Lamina propria vorliegen und die Eosinophilen nicht erhöht sind, keine Zerstörung der Krypten und keine Erosionen, Ulzerationen oder Granulationsgewebe)
|
Abbildung 8: Anteil der erwachsenen Patienten mit vorherigem Versagen einer Biologikatherapie, die ein klinisches Ansprechen gemäss paMS (SFS und RBS) erreichten, im Zeitverlauf in den Induktionsstudien UC-1 und UC-2
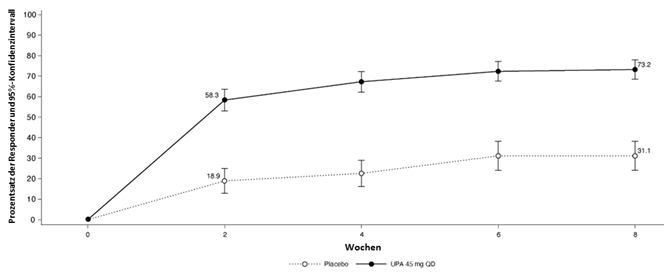
Verlängerte Induktionsphase
Insgesamt 124 erwachsene Patienten in UC-1 und UC-2, die nach 8wöchiger Behandlung mit RINVOQ 45 mg einmal täglich kein klinisches Ansprechen erreichten, traten in eine 8wöchige, offene, verlängerte Induktionsphase ein. Von den Patienten mit vorherigem Versagen einer Biologikatherapie, welche die Behandlung mit RINVOQ 45 mg einmal täglich für weitere 8 Wochen (insgesamt 16 Wochen) erhielten (82 Patienten), erreichten 48,7 % der Patienten ein klinisches Ansprechen gemäss aMS. Von den Patienten, die auf die Behandlung mit RINVOQ 45 mg einmal täglich über 16 Wochen ansprachen, zeigten unter einer Erhaltungstherapie mit RINVOQ 15 mg und 30 mg einmal täglich 36,4 % der Patienten (N=11) bzw. 86,7 % der Patienten (N=15) weiterhin ein klinisches Ansprechen gemäss aMS. Nach der 16-wöchigen Behandlung mit RINVOQ 45 mg einmal täglich, erreichten 20,0 % der Patienten (N=15) unter einer Erhaltungstherapie mit RINVOQ 15 mg bzw. 41,2 % der Patienten (N=17) unter einer Erhaltungstherapie mit RINVOQ 30 mg einmal täglich eine klinische Remission gemäss aMS in Woche 52.
Erhaltungsstudie (UC-3)
In die Auswertung zur Wirksamkeit für UC-3 wurden 223 erwachsene Patienten mit vorherigem Versagen einer Biologikatherapie aufgenommen, die mit einer 8wöchigen Induktionstherapie mit RINVOQ 45 mg einmal täglich ein klinisches Ansprechen gemäss aMS erreichten. Die Patienten wurden randomisiert und erhielten bis zu 52 Wochen lang einmal täglich RINVOQ 15 mg, RINVOQ 30 mg oder Placebo.
Primärer Endpunkt war die klinische Remission in Woche 52. Die primären und sekundären Endpunkte sind in Tabelle 22 aufgeführt. Die symptomatische Remission pro paMS im Zeitverlauf ist in Abbildung 9 dargestellt.
Tabelle 22: Anteil der erwachsenen Patienten mit vorherigem Versagen einer Biologikatherapie, welche die primären und sekundären Endpunkte zur Wirksamkeit in Woche 52 in der Erhaltungsstudie UC-3 erreichten
|
Endpunkt
|
PBO
N = 80
|
UPA
15 mg
N = 71
|
UPA
30 mg
N = 72
|
Behandlungsunterschied
15 mg vs. PBO (95 %-KI)
|
Behandlungsunterschied
30 mg vs. PBO
(95 %-KI)
| |
Krankheitsaktivität und Colitis ulcerosa-Symptome
| |
Klinische Remissiona
|
7,6 %
|
40,5 %
|
49,8 %
|
32,9 %**
(20,0; 45,8)
|
42,2 %**
(29,1; 55,4)
| |
Aufrechterhaltung der klinischen Remissionb
|
N = 21
14,3 %
|
N = 17
76,5 %
|
N = 20
73,0 %
|
62,2 %**
(37,1; 87,3)
|
58,7 %**
(33,6; 83,9)
| |
Kortikosteroidfreie klinische Remissionc
|
N = 21
14,3 %
|
N = 17
70,6 %
|
N = 20
73,0 %
|
56,3 %**
(30,0; 82,6)
|
58,7 %**
(33,6; 83,9)
| |
Aufrechterhaltung des klinischen Ansprechensd
|
N = 70
14,4 %
|
N = 64
60,9 %
|
N = 65
69,9 %
|
46,6 %**
(32,0; 61,1)
|
55,5 %**
(41,5; 69,6)
| |
Kein Stuhldrang
|
47,5 %
|
59,2 %
|
56,9 %
|
11,7 %
(-4,2; 27,5)
|
9,4%
(-6,4; 25,3)
| |
Keine Bauchschmerzen
|
46,3 %
|
53,5 %
|
50,0 %
|
7,3 %
(-8,7; 23,2)
|
3.7%
(-12,1; 19,6)
| |
Endoskopische und histologische Beurteilung
| |
Aufrechterhaltung der endoskopischen Verbesserunge
|
N = 31
9,7 %
|
N = 24
70,8 %
|
N = 29
60,7 %
|
61,2 %**
(40,2; 82,1)
|
51,0 %**
(30,1; 72,0)
| |
Endoskopische Remissionf
|
2,5 %
|
21,5 %
|
20,3 %
|
19,0 %**
(8,7; 29,3)
|
17,8 %**
(7,5; 28,0)
| |
Histologisch-endoskopische Mukosaverbesserungg
|
5,3 %
|
32,9 %
|
48,2 %
|
27,6 %**
(15,5; 39,8)
|
43,0 %**
(30,1; 55,8)
| |
Endoskopische Verbesserungh
|
7,9 %
|
43,3 %
|
56,9 %
|
35,4 %**
(22,3; 48,5)
|
48,9 %**
(35,6; 62,2)
| |
Mukosaheilungi
|
2,5 %
|
17,2 %
|
16,3 %
|
14,7 %*
(5,2; 24,2)
|
13,8 %*
(4,3; 23,3)
| |
Lebensqualität
| |
Veränderung des FACIT-F-Scores gegenüber Baseline
|
3,2
|
8,2
|
9,8
|
5,0*
(1,51; 8,53)
|
6,6**
(3,06; 10,10)
| |
Veränderung des Gesamtscores auf der IBDQ gegenüber Baseline
|
17,0
|
47,0
|
56,0
|
30,0**
(16,09; 43,90)
|
39,0**
(24,62; 53,30)
| |
* p ≤0.01
** p ≤0,001, Behandlungsunterschied (95 %-KI)
a Gemäss aMS: SFS ≤1 und nicht grösser als bei Baseline, RBS = 0, ES ≤1 ohne Friabilität
b Klinische Remission gemäss aMS in Woche 52 bei Patienten mit klinischer Remission am Ende der Induktionsphase
c Klinische Remission gemäss aMS in Woche 52 und Kortikosteroidfreiheit für ≥90 Tage unmittelbar vor Woche 52 bei Patienten mit klinischer Remission am Ende der Induktionstherapie
d Klinische Remission gemäss aMS in Woche 52 bei Patienten mit klinischer Remission am Ende der Induktionsphase
e Aufrechterhalten der Mukosaheilung, ES ≤1 ohne Friabilität, bei Patienten mit Mukosaheilung in der Induktionsphase
f ES-Subscore = 0
g ES ≤1 ohne Friabilität und Geboes-Score ≤3,1 (weist auf eine Neutrophileninfiltration in < 5 % der Krypten hin, keine Zerstörung der Krypten und keine Erosionen, Ulzerationen oder Granulationsgewebe).
h ES ≤1 ohne Friabilität
i ES = 0, Geboes-Score < 2 (weist darauf hin, dass keine Neutrophile in Krypten oder der Lamina propria vorliegen und die Eosinophilen nicht erhöht sind, keine Zerstörung der Krypten und keine Erosionen, Ulzerationen oder Granulationsgewebe
|
Abbildung 9: Anteil der erwachsenen Patienten mit vorherigem Versagen einer Biologikatherapie, die eine symptomatische Remission gemäss paMS (SFS ≤1 und RBS = 0) erreichten, im Zeitverlauf der Erhaltungsstudie UC-3
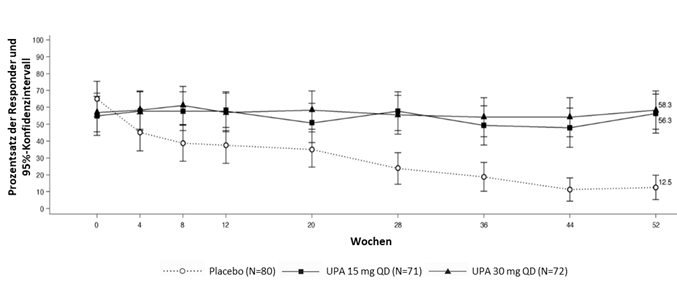
Langzeitstudie (UC-4)
Patienten, welche in UC-3 nach 1 Jahr eine klinische Remission gemäss aMS erreichten, konnten mit der gleichen Dosis in der Fortsetzungsstudie (UC-4) fortfahren. Nach insgesamt 3 Jahren zeigten 77,1 % (27/35) und 87,8 % (36/41) der erwachsenen Patienten mit vorherigem Versagen einer Biologikatherapie weiterhin eine klinische Remission und 60,0 % (9/15) und 68,4 % (13/19) der erwachsenen Patienten mit vorherigem Versagen einer Biologikatherapie weiterhin eine endoskopische Remission mit RINVOQ 15 mg bzw. RINVOQ 30 mg. Auch die Verbesserungen der Lebensqualität blieben über 3 Jahre Therapie erhalten.
Morbus Crohn
Die Wirksamkeit und Sicherheit von RINVOQ wurden in drei multizentrischen, doppelblinden, placebokontrollierten Phase-III-Studien untersucht: zwei Induktionsstudien – CD-1 (U-EXCEED) und CD-2 (U-EXCEL), gefolgt von einer 52wöchigen Erhaltungsstudie mit einer anschliessenden Langzeitstudie CD-3 (U-ENDURE). Die co-primären Endpunkte waren die klinische Remission und das endoskopische Ansprechen in Woche 12 für CD-1 und CD-2 und in Woche 52 für CD-3.
Die in Frage kommenden Patienten waren zwischen 18 und 75 Jahre alt und hatten mittelschweren bis schweren aktiven Morbus Crohn (MC), definiert als eine durchschnittliche tägliche Stuhlfrequenz (SF) mit sehr weichem oder flüssigem Stuhl von ≥4 und/oder einen durchschnittlichen täglichen Score für Bauchschmerzen (BS) von ≥2 und einen von einem zentralen Prüfer bestätigten einfachen endoskopischen Score für MC (Simple Endoscopic Score for CD, SES-CD) von ≥6 bzw. ≥4 bei isolierter Erkrankung des Ileums, abgesehen von einer vorliegenden Verengungskomponente.
Induktionsstudien (CD-1 und CD-2)
In den Studien CD-1 und CD-2 wurden 1'021 Patienten (495 bzw. 526 Patienten) im Verhältnis 2:1 randomisiert und 12 Wochen lang mit RINVOQ 45 mg einmal täglich oder Placebo behandelt.
In CD-1 hatten alle Patienten ein vorheriges Versagen einer Biologikatherapie. Von diesen Patienten hatten 61 % (301/495) ein unzureichendes Ansprechen auf oder eine Unverträglichkeit gegenüber zwei oder mehr Biologikatherapien.
In CD-2 hatten 45 % (239/526) der Patienten ein vorheriges Versagen einer Biologikatherapie, und 55 % (287/526) hatten ein unzureichendes Ansprechen auf oder eine Unverträglichkeit gegenüber einer Behandlung mit konventionellen Therapien, aber nicht mit Biologika (ohne vorheriges Versagen der Biologikatherapie).
Nachfolgend werden nur Studienresultate für Patienten mit vorherigem Versagen einer Biologikatherapie präsentiert. Bei Baseline erhielten in CD-1 und CD-2 34 % bzw. 44 % dieser Patienten Kortikosteroide, 7 % bzw. 3 % Immunmodulatoren und 15 % bzw. 12 % Aminosalicylate.
In beiden Studien begannen die Patienten, die bei Baseline Kortikosteroide erhielten, ab Woche 4 mit einer ausschleichenden Behandlung mit Kortikosteroiden.
Klinische Krankheitsaktivität und Symptome
In CD-1 und CD-2 erreichte in Woche 12 im Vergleich zu Placebo ein signifikant grösserer Anteil an Patienten mit vorherigem Versagen einer Biologikatherapie unter RINVOQ 45 mg den koprimären Endpunkt der klinischen Remission (Tabelle 23). In beiden Studien trat die Wirksamkeit im Vergleich zu Placebo bereits in Woche 2 ein, wobei ein signifikant grösserer Anteil der Patienten, die mit RINVOQ 45 mg einmal täglich behandelt wurden, ein klinisches Ansprechen (CR-100) erreichte. Ein signifikant grösserer Anteil der Patienten erreichte in Woche 4 im Vergleich zu Placebo ein klinisches Ansprechen.
In den Studien CD-1 und CD-2 erreichte ein signifikant grösserer Anteil der mit RINVOQ 45 mg behandelten Patienten mit vorherigem Versagen einer Biologikatherapie (51 % bzw. 59 %) im Vergleich zu Placebo (27 % bzw. 25 %) ein klinisches Ansprechen gemäss CDAI in Woche 12.
In beiden Studien verzeichneten Patienten, die RINVOQ 45 mg erhielten, in Woche 12 im Vergleich zu Placebo eine signifikant grössere Verbesserung der Fatigue gegenüber Baseline, gemessen anhand des FACIT-F-Scores.
Endoskopische Beurteilung
In CD-1 und CD-2 erreichte in Woche 12 im Vergleich zu Placebo ein signifikant grösserer Anteil an Patienten mit vorherigem Versagen einer Biologikatherapie, die mit RINVOQ 45 mg behandelt wurden, den koprimären Endpunkt des endoskopischen Ansprechens (Tabelle 23). In den Studien CD-1 und CD-2 erreichte ein grösserer Anteil der mit RINVOQ 45 mg behandelten Patienten mit vorherigem Versagen einer Biologikatherapie (17 % bzw. 16 %) im Vergleich zu Placebo (0 % bzw. 1 %) eine Mukosaheilung (SES-CD-Teilscore für Ulzerationen auf der Oberfläche von 0 bei Patienten mit SES-CD-Teilscore für Ulzerationen auf der Oberfläche ≥1 bei Baseline) in Woche 12.
In CD-1 und CD-2 erreichte im Vergleich zu Placebo (0 % bzw. 1 %) ein grösserer Anteil an Patienten mit vorherigem Versagen einer Biologikatherapie, die mit RINVOQ 45 mg behandelt wurden (14% bzw. 12 %) einen SES-CD von 0-2.
Tabelle 23: Anteil der Patienten mit vorherigem Versagen einer Biologikatherapie, welche die primären und weiteren Endpunkte zur Wirksamkeit in den Induktionsstudien CD-1 und CD-2 erreichten
|
Studie
|
CD-1
(U-EXCEED)
|
CD-2
(U-EXCEL)
| |
Behandlungsgruppe
|
PBO
N = 171
|
UPA
45 mg
n = 324
|
Behandlungs-unterschied (95 %-KI)
|
PBO
N = 78
|
UPA
45 mg
n = 161
|
Behandlungs-unterschied (95 %-KI)
| |
Koprimäre Endpunkte in Woche 12
| |
Klinische Remissiona
|
14 %
|
40 %
|
26 %
(19, 33)*
|
14 %
|
47 %
|
33 %
(22; 44)*
| |
Endoskopisches Ansprechenb
|
4 %
|
35 %
|
31 %
(25, 37)*
|
9 %
|
38 %
|
29 %
(19, 39)*
| |
Weitere Endpunkte in Woche 12
| |
Klinische Remission gemäss CDAIc
|
21 %
|
39 %
|
18 %
(10, 26)*
|
16 %
|
44 %
|
28 %
(17; 39)*
| |
Kortikosteroidfreie klinische Remissiona,d
|
N=60
7%
|
N=108
37%
|
30%
(19, 41)*
|
N=36
6 %
|
N=70
39 %
|
33 %
(19; 47)*
| |
Endoskopische Remissione
|
2 %
|
19 %
|
17 %
(12, 22)*
|
4 %
|
21 %
|
17 %
(9; 24)*
| |
Abkürzung: PBO = Placebo; UPA = Upadacitinib
* p ≤0,001, UPA im Vergleich zu PBO, Behandlungsunterschied (95 %-KI)
a Durchschnittliche tägliche sehr weiche oder flüssige SF ≤2,8 und BS ≤1,0 und beide nicht grösser als bei Baseline
b Verringerung des SES-CD > 50 % gegenüber Baseline der Induktionsstudie (bzw. bei Patienten mit einem SES-CD von 4 bei Baseline der Induktionsstudie eine Verringerung um mindestens 2 Punkte gegenüber Baseline der Induktionsstudie)
c CDAI < 150
d Absetzen der Steroidbehandlung und Erreichen einer klinischen Remission bei Patienten, die bei Baseline Steroide erhielten
e SES-CD von ≤4 und mindestens 2 Punkte niedriger als bei Baseline und kein Teilscore einer einzelnen Variable > 1
|
Erhaltungsstudie (CD-3)
In die primäre Auswertung zur Wirksamkeit für CD-3 wurden 502 Patienten aufgenommen, die mit einer 12wöchigen Induktionstherapie mit RINVOQ 45 mg einmal täglich ein klinisches Ansprechen gemäss SF/BS (≥30 % Verringerung der durchschnittlichen täglichen SF mit sehr weichem oder flüssigem Stuhl und/oder ≥30 % Verringerung der durchschnittlichen täglichen BS gegenüber Baseline und beide nicht grösser als bei Baseline) erreichten. Die Patienten wurden erneut randomisiert und erhielten über 52 Wochen eine Erhaltungstherapie mit RINVOQ 15 mg oder 30 mg einmal täglich bzw. Placebo; sie kamen somit auf eine Gesamtbehandlungsdauer von mindestens 64 Wochen. Von den erneut randomisierten Responder in der primären Auswertung hatten 377 Patienten ein vorheriges Versagen einer Biologikatherapie. Nachfolgend werden nur Studienresultate für Patienten mit vorherigem Versagen einer Biologikatherapie präsentiert.
Klinische Krankheitsaktivität und Symptome
Ein signifikant grösserer Anteil der Patienten, die mit RINVOQ 15 mg bzw. 30 mg behandelt wurden, erreichte in Woche 52 im Vergleich zu Placebo den koprimären Endpunkt einer klinischen Remission nach SF/BS (Abbildung 10, Tabelle 24).
Abbildung 10: Anteil an Patienten mit vorherigem Versagen einer Biologikatherapie, die eine klinische Remission in der Erhaltungsstudie CD-3 erreichten
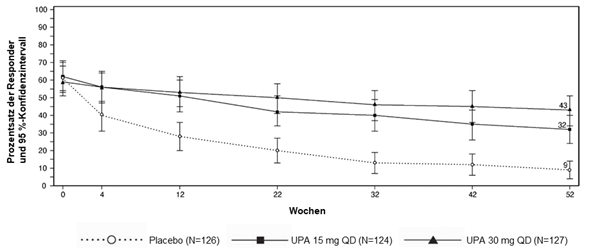
Patienten, die RINVOQ 30 mg erhielten, verzeichneten in Woche 52 im Vergleich zu Placebo eine signifikant grössere Verbesserung der Fatigue gegenüber Baseline, gemessen anhand des FACIT-F-Scores.
Tabelle 24: Anteil der Patienten mit vorherigem Versagen einer Biologikatherapie, welche die primären und weiteren Endpunkte zur Wirksamkeit in Woche 52 in der Erhaltungsstudie CD-3 (U-ENDURE) erreichten
|
Behandlungsgruppe
|
PBO+
N = 126
|
UPA
15 mg
n = 124
|
UPA
30 mg
n = 127
|
Behandlungsunterschied 15 mg vs. PBO (95 %-KI)
|
Behandlungsunterschied 30 mg vs. PBO (95 %-KI)
| |
Koprimäre Endpunkte
| |
Klinische Remissiona
|
9 %
|
32 %
|
43 %
|
24 %
(14; 33)**
|
34 %
(24; 44)**
| |
Endoskopisches Ansprechenb
|
4 %
|
23 %
|
39 %
|
19 %
(11; 27)**
|
35%
(26; 44)**
| |
Weitere Endpunkte
| |
Klinische Remission gemäss CDAIc
|
12 %
|
34 %
|
45 %
|
22 %
(12; 32)**
|
33 %
(23; 43)**
| |
Kortikosteroidfreie klinische Remissiona,d
|
9 %
|
32 %
|
41 %
|
24 %
(14; 33)**
|
32 %
(22; 42)**
| |
Aufrechterhaltung der klinischen Remissiona,e
|
N=77
13 %
|
N=77
44 %
|
N=75
60 %
|
31 %
(18; 45)**
|
47 %
(34; 60)**
| |
Endoskopische Remissionf
|
2 %
|
16 %
|
27 %
|
14 %
(7; 21)**
|
24 %
(16; 33)**
| |
Tiefe Remissiona,f,g
|
2 %
|
13 %
|
21 %
|
11 %
(4; 17)*
|
19 %
(11; 26)**
| |
Abkürzung: PBO = Placebo; UPA = Upadacitinib
+ Der Placeboarm bestand aus Patienten, die mit RINVOQ 45 mg am Ende der Induktionsstudie ein klinisches Ansprechen gemäss SF/BS erreicht hatten. Sie wurden zu Beginn der Erhaltungstherapie zu Placebo randomisiert.
* p ≤0,01, UPA im Vergleich zu PBO, angepasster Behandlungsunterschied (95 %-KI)
** p ≤0,001, UPA im Vergleich zu PBO, angepasster Behandlungsunterschied (95 %-KI)
a Durchschnittliche tägliche sehr weiche oder flüssige SF ≤2,8 und BS ≤1,0 und beide nicht grösser als bei Baseline
b Verringerung des SES-CD > 50 % gegenüber Baseline der Induktionsstudie (bzw. bei Patienten mit einem SES-CD von 4 bei Baseline der Induktionsstudie eine Verringerung um mindestens 2 Punkte gegenüber Baseline der Induktionsstudie)
c CDAI < 150
d Kortikosteroidfrei für 90 Tage vor Woche 52 und Erreichen einer klinischen Remission. In der Untergruppe von Patienten, die bei Baseline der Induktionsstudie mit Kortikosteroiden behandelt wurden, waren 35 % (N = 46) im Behandlungsarm mit RINVOQ 15 mg, 40 % (N = 50) im Behandlungsarm mit RINVOQ 30 mg und 2 % (N = 50) im Placeboarm für 90 Tage vor Woche 52 kortikosteroidfrei und in klinischer Remission.
e Definiert als Erreichen einer klinischen Remission in Woche 52 bei Patienten mit klinischer Remission zu Beginn der Erhaltungsstudie.
f SES-CD ≤4 und mindestens 2 Punkte niedriger als bei Baseline und kein Teilscore einer einzelnen Variable > 1
g Klinische Remission und endoskopische Remission
|
Patienten, die in Woche 12 der Induktionsphase mit RINVOQ in CD-1 und CD-2 nicht in klinischer Remission gemäss SF/BS waren, erhielten weitere 12 Wochen RINVOQ 30 mg einmal täglich (122 Patienten). Patienten, welche die verlängerte Behandlung erhielten, hatten eine längere durchschnittliche Krankheitsdauer (12,3 Jahre), einen höheren Anteil von vorherigem Biologika-Versagen (79,5 %) und häufiger ein Versagen von mindestens drei Biologika-Therapien (42,3 %) als die gesamte Patientenpopulation, welche in den Induktionsstudien eingeschlossen wurde. Von den Patienten mit vorherigem Versagen einer Biologikatherapie (97 Patienten) erreichten 23 % eine klinische Remission und 54 % ein klinisches Ansprechen in Woche 24. Von den Patienten, die auf die erweiterte Behandlungsphase ansprachen und weiterhin eine Erhaltungstherapie mit RINVOQ 30 mg erhielten, erreichten in Woche 52 18 % eine klinische Remission und 15 % eine endoskopische Remission.
Endoskopische Beurteilungen
Von den Patienten mit vorherigem Versagen einer Biologikatherapie in CD-3 erreichte in Woche 52 im Vergleich zu Placebo ein signifikant grösserer Anteil an Patienten, die mit RINVOQ 15 mg bzw. 30 mg behandelt wurden, den koprimären Endpunkt des endoskopischen Ansprechens (Tabelle 24). Ein grösserer Anteil der Patienten, die mit RINVOQ 15 mg und 30 mg (12 % bzw. 20 %) behandelt wurden, erreichte im Vergleich zu Placebo (2 %) eine Mukosaheilung (SES-CD-Teilscore für Ulzerationen auf der Oberfläche von 0 bei Patienten mit einem SES-CD-Teilscore für Ulzerationen auf der Oberfläche von ≥1 bei Baseline) in Woche 52. Ein grösserer Anteil der Patienten, die mit RINVOQ 15 mg und 30 mg (11 % bzw. 18 %) behandelt wurden, erreichte im Vergleich zu Placebo (2 %) in Woche 52 einen SES-CD von 0-2. Eine kortikosteroidfreie endoskopische Remission von Patienten, die bei Baseline Steroide erhielten, wurde in Woche 52 im Vergleich zu Placebo (0 %) von einem grösseren Anteil an Patienten erreicht, die mit RINVOQ 15 mg bzw. 30 mg (15 % bzw. 24 %) behandelt wurden.
Notfallbehandlung
In CD-3 kamen Patienten mit unzureichendem Ansprechen oder einem Verlust des Ansprechens in der Erhaltungsphase für eine Notfallbehandlung mit RINVOQ 30 mg infrage. Von den Patienten mit vorherigem Versagen einer Biologikatherapie, die in den Behandlungsarm mit RINVOQ 15 mg randomisiert worden waren und eine Notfallbehandlung mit RINVOQ 30 mg erhielten, erreichten 12 bzw. 24 Wochen nach Einleitung der Notfallbehandlung 83 % bzw. 88 % ein klinisches Ansprechen gemäss SF/BS und 42 % bzw. 55 % eine klinische Remission. Von den Patienten, die in den Placeboarm randomisiert worden waren und eine Notfallbehandlung mit RINVOQ 30 mg erhielten, erreichten 12 bzw. 24 Wochen nach Einleitung der Notfallbehandlung 87 % bzw. 93 % ein klinisches Ansprechen gemäss SF/BS und 52 % bzw. 55 % eine klinische Remission.
Gesundheitsbezogene Ergebnisse und Lebensqualität
In CD-1 und CD-2 erreichten die mit RINVOQ behandelten Patienten in Woche 12 im Vergleich zu Placebo eine grössere Verbesserung gegenüber Baseline beim IBDQ-Gesamtscore, in allen IBDQ-Bereichsscores, u.a. Darmsymptome, systemische Symptome, emotionale und soziale Funktionsfähigkeit. Diese Verbesserungen wurden bei Patienten, die mit RINVOQ 15 mg und 30 mg behandelt wurden in CD-3 bis Woche 52 aufrechterhalten.
|