ZusammensetzungWirkstoffe
Konzentrat proteolytischer Enzyme angereichert aus Bromelain (Bromelain aus dem Stamm)
Die proteolytischen Enzyme sind eine Mischung von Enzymen aus dem Stamm von Ananas comosus (Ananas-Pflanze).
Hilfsstoffe
NexoBrid-Pulver
Ammoniumsulfat
Essigsäure
Gel
Carbomer 980
Dinatriumphosphat
Natriumhydroxid
Wasser für Injektionszwecke
Darreichungsform und Wirkstoffmenge pro EinheitPulver und Gel zur Herstellung eines Gels.
Das Pulver ist gebrochen weiss bis leicht hellbraun. Das Gel ist durchsichtig und farblos.
Eine Durchstechflasche enthält 5 g Konzentrat proteolytischer Enzyme angereichert aus Bromelain; nach dem Mischen entspricht dies 0,09 g/g Konzentrat proteolytischer Enzyme angereichert aus Bromelain (bzw. 5 g/55 g Gel).
Indikationen/AnwendungsmöglichkeitenNexoBrid wird zur Entfernung des Verbrennungsschorfs (Eschar) bei Patienten aller Altersklassen mit tiefen thermischen Verletzungen (Grad IIb – III; "deep partial" und "full thickness" ) angewendet.
Dosierung/AnwendungNexoBrid darf ausschliesslich durch geschultes medizinisches Fachpersonal in speziellen Verbrennungszentren angewendet werden.
Erwachsene
2 g NexoBrid-Pulver in 20 g Gel werden auf 1 % der Gesamtkörperoberfläche (TBSA, Total Body Surface Area) eines Erwachsenen bzw. rund 180 cm2 mit einer Gelschichtdicke von 1,5 bis 3 mm aufgetragen.
5 g NexoBrid-Pulver in 50 g Gel werden auf 2,5 % der Gesamtkörperoberfläche (TBSA, Total Body Surface Area) eines Erwachsenen bzw. rund 450 cm2 mit einer Gelschichtdicke von 1,5 bis 3 mm aufgetragen.
NexoBrid darf nicht auf mehr als 15 % der TBSA aufgetragen werden (siehe auch Rubrik "Warnhinweise und Vorsichtsmassnahmen: Koagulopathie" ).Anwendung in der Pädiatrie
Kinder und Jugendliche (von der Geburt bis zum Alter von 18 Jahren)
Bei Pädiatrie-Patienten im Alter von 4 bis 18 Jahren darf NexoBrid nicht auf mehr als 15 % der TBSA aufgetragen werden.
Bei Pädiatrie-Patienten im Alter von 0 bis 3 Jahren darf NexoBrid nicht auf mehr als 10 % der TBSA aufgetragen werden.
NexoBrid soll für eine Dauer von 4 Stunden auf der Brandwunde belassen werden. Es liegen nur sehr begrenzte Daten zur Anwendung von NexoBrid auf Bereichen vor, von denen sich Verbrennungsschorf nach der ersten Anwendung nicht abgelöst hat.
Eine zweite Anwendung wird nicht empfohlen.
Rückverfolgbarkeit
Um die Rückverfolgbarkeit von biologischen Arzneimitteln sicherzustellen, wird empfohlen, Handelsname und Chargennummer bei jeder Behandlung zu dokumentieren.
Patienten mit Leberfunktionsstörungen
Es liegen keine Daten zur Anwendung von NexoBrid bei Patienten mit eingeschränkter Leberfunktion vor. Diese Patienten sollten sorgfältig überwacht werden.
Patienten mit Nierenfunktionsstörungen
Es liegen keine Daten zur Anwendung von NexoBrid bei Patienten mit eingeschränkter Nierenfunktion vor. Diese Patienten sollten sorgfältig überwacht werden.
Ältere Patienten
Die Erfahrung mit NexoBrid bei älteren Patienten (>65 Jahre) ist begrenzt. Bei der Nutzen-/Risiko-Bewertung ist zu beachten, dass bei älteren Patienten häufiger Begleiterkrankungen oder eine begleitende Behandlung mit anderen Arzneimitteln vorliegen. Es ist keine Dosisanpassung erforderlich.
Art der Anwendung
Anwendung auf der Haut.
Vor der Anwendung muss das Pulver mit dem Gel vermengt werden, um ein homogenes Gel herzustellen. Für Hinweise zur Herstellung von NexoBrid-Gel siehe Rubrik "Sonstige Hinweise/Hinweise für die Handhabung" .
NexoBrid soll auf einen sauberen, keratinfreien (vorherige Entfernung von Blasen) und feuchten Wundbereich aufgetragen werden.
Jede NexoBrid-Durchstechflasche, das Gel bzw. das rekonstituierte Gel dürfen jeweils nur für ein und denselben Patienten verwendet werden.
Vor der Applikation von NexoBrid müssen andere auf der Wunde befindliche topische Arzneimittel (wie Sulfadiazin-Silber oder Povidon-Iod) entfernt werden, und die Wunde muss gereinigt werden da ein mit Arzneimitteln und deren Rückständen gesättigter Eschar die Wirkung und Wirksamkeit des Gels herabsetzen würde.
Vorbereitung des Patienten und der Wunde
Die mit NexoBrid behandelte Wundfläche darf insgesamt nicht mehr als 15 % der TBSA betragen (siehe auch Rubrik "Warnhinweise und Vorsichtsmassnahmen" , Koagulopathie).
-Das enzymatische Debridement ist eine schmerzhafte Prozedur und bedarf einer adäquaten Analgesie und/oder Anästhesie. Es muss eine Schmerzbehandlung erfolgen, wie sie bei grossen Verbandswechseln üblich ist. Diese sollte mindestens 15 Minuten vor Applikation von NexoBrid beginnen.
-Die Wunde muss gründlich gereinigt und die oberflächliche Keratinschicht oder Blasen müssen vom Wundbereich entfernt werden, da Keratin den direkten Kontakt zwischen dem Verbrennungsschorf und NexoBrid und damit die Eschar-Entfernung durch NexoBrid verhindert.
-Für die Dauer von 2 Stunden muss eine in antibakterieller Lösung (z.B. Chlorhexidin oder Natriumhypochlorit (Dakin-Lösung) 0,05–0,5 %, hypertone Kochsalzlösung 5-10 %) getränkte Wundkompresse aufgelegt werden. Sulfadiazin-Silber oder Povidon-Iod sollten nicht verwendet werden.
-Vor der Applikation von NexoBrid sind topisch applizierte Antiseptika zu entfernen. Reste von antibakteriellen Arzneimitteln können die Wirkung von NexoBrid herabsetzen, da die Wirksamkeit reduziert wird.
-Der Bereich, von dem der Verbrennungsschorf abgelöst werden soll, muss mit einer haftenden sterilen Paraffin-Salbe (adhäsive Barriere) umrandet werden. Tragen Sie die Salbe hierzu einige Zentimeter ausserhalb des zu behandelnden Bereichs auf (mit Hilfe eines Spenders). Die Paraffin-Schicht darf nicht in Kontakt mit dem zu behandelnden Bereich kommen, um zu verhindern, dass sie den Verbrennungsschorf bedeckt und dadurch den direkten Kontakt zwischen dem Schorf und NexoBrid verhindern würde.Um eine mögliche Reizung abgeschürfter Hautbereiche durch versehentlichen Kontakt mit NexoBrid sowie mögliche Blutungen vom Wundbett zu vermeiden, sollten akute Wundbereiche wie Schnittwunden oder Escharotomie-Schnitte mit einer Schicht steriler Fettsalbe oder Schutzsalbe (z. B. Vaseline) geschützt werden.
-Die Brandwunde muss mit steriler isotonischer Natriumchloridlösung [9 mg/ml (0,9 %)] besprüht und während der Applikation von NexoBrid feucht gehalten werden.
Applikation von NexoBrid
-Bereich befeuchten, indem sterile Kochsalzlösung auf den zu behandelnden Bereich gesprüht wird, der von der sterilen Fettsalbe als adhäsive Barriere umrandet wird.
-NexoBrid muss innerhalb von 15 Minuten nach dem Einmischen des Pulvers in das Gel (siehe Rubrik "Sonstige Hinweise/Hinweise für die Handhabung" ) auf die befeuchtete Verbrennungswunde aufgetragen werden, in einer Schicht von 1,5 bis 3 Millimetern Dicke.
-Anschliessend muss die Wunde mit einem sterilen okklusiven Folienverband abgedeckt werden, der an der sterilen Paraffin-Salbe anhaftet, die wie oben beschrieben aufgetragen wurde (siehe Vorbereitung des Patienten und der Wunde). Das NexoBrid-Gel muss den gesamten Okklusivverband ausfüllen. Achten Sie besonders darauf, dass keine Luft unter dem Okklusivverband verbleibt. Indem Sie den Okklusivverband an den Kontaktstellen mit der adhäsiven Barriere leicht andrücken, stellen Sie sicher, dass der okklusive Folienverband und die sterile Paraffin-Salbe aneinanderhaften und NexoBrid vollständig auf dem behandelten Bereich verbleibt.
-Die Wunde mit dem Folienverband muss mit einem lockeren, dicken und bauschigen Verband bedeckt werden, der durch eine Binde fixiert wird.
-Der Verband muss für 4 Stunden auf der Wunde verbleiben.
Entfernen von NexoBrid
-Die Entfernung dieses Arzneimittels ist eine schmerzhafte Prozedur und bedarf einer adäquaten Analgesie und/oder Anästhesie. Der Patient muss eine angemessene präventive Analgesie mindestens 15 Minuten vor der Entfernung des Gels erhalten.
-Nach 4-stündigem Einwirken von NexoBrid ist der Okklusivverband mit aseptischer Technik abzunehmen.
-Die Paraffin-Salbe ist mit einem sterilen stumpfen Instrument (z. B. einem Zungenspatel) zu entfernen.
-Der aufgelöste Verbrennungsschorf muss mit einem sterilen stumpfen Instrument von der Wunde entfernt werden.
-Danach muss die Wunde zunächst gründlich mit einem grossen Stück trockenen sterilen Verbandsmulls oder einem Tuch und anschliessend mit in steriler isotonischer Natriumchloridlösung [9 mg/ml (0,9 %)] getränktem sterilem Verbandsmull/Tuch abgewischt werden. Der behandelte Bereich muss so lange abgerieben werden, bis eine rosa Oberfläche mit punktförmigen Blutungen oder weissliches Gewebe zu sehen ist. Durch das Reiben lässt sich anhaftender nicht aufgelöster Verbrennungsschorf in Bereichen, in denen er verblieben ist, nicht entfernen.
-Anschliessend muss für weitere 2 Stunden eine in antibakterieller Lösung (z.B. Chlorhexidin oder Natriumhypochlorit (Dakin-Lösung) 0,05–0,5 %, hypertone Kochsalzlösung 5-10 %) getränkte Wundkompresse aufgelegt werden.
Wundversorgung nach dem Debridement
-Die Wundfläche muss sofort nach dem Debridement mit einem vorübergehenden oder permanenten Hautersatz gedeckt oder mit einem Verband bedeckt werden, um eine Austrocknung und/oder die Bildung von Pseudoschorf ( "Pseudoeschar" ) und/oder Infektionen zu vermeiden.
-Vor der vorübergehenden oder permanenten Deckung mit Hautersatz muss ein durchtränkter "Wetto-dry" -Verband auf die frisch debridierte Wundfläche aufgelegt werden.
-Vor Aufbringen des Transplantats oder primären Verbands muss das debridierte Wundbett gesäubert und angefrischt werden, z. B. durch Bürsten oder Abschaben, um ein Anhaften zu ermöglichen.
-Wundbereiche mit tiefer und drittgradiger Verbrennung ( "full thickness" ) sollen so früh wie möglich nach dem Debridement mit einem autologen Hauttransplantat versorgt werden. Ebenso ist bei tief-dermalen Wunden ( "deep partial thickness" ; Grad IIb) zu erwägen, diese bald nach dem Debridement mit einem permanenten Hautersatz (z. B. autologes Hauttransplantat) zu versorgen (siehe Rubrik "Warnhinweise und Vorsichtsmassnahmen" ).
KontraindikationenÜberempfindlichkeit gegen den Wirkstoff, Ananas oder Papaya/Papain (siehe auch Rubrik "Warnhinweise und Vorsichtsmassnahmen" ) oder einen der in der Rubrik "Hilfsstoffe" genannten sonstigen Bestandteile.
Warnhinweise und VorsichtsmassnahmenBrandwunden, für die NexoBrid nicht empfohlen wird
Die Anwendung von NexoBrid wird nicht empfohlen bei:
penetrierenden Brandwunden, bei denen es während des Debridements zu einer Exposition von körperfremden Materialien (z. B. Implantate, Schrittmacher oder Shunts) und/oder von lebenswichtigen Strukturen (z. B. grössere Gefässe, Augen) kommt oder kommen könnte;
chemischen Brandwunden;
-Anwendung in Hohlräumen wie Peritoneal- und Pleurahöhlen;
mit radioaktiven oder anderen Gefahrstoffen kontaminierten Wunden, um unvorhersehbare Reaktionen mit NexoBrid und ein erhöhtes Risiko einer Ausbreitung der gesundheitsschädlichen Substanz zu verhindern;
-Brandwunden an den Füssen von Diabetikern mit okklusiver Gefässerkrankung;
elektrischen Verbrennungen.
Systemische Resorption
Das Konzentrat proteolytischer Enzyme angereichert aus Bromelain wird von den Wundflächen in den systemischen Kreislauf resorbiert (siehe Rubrik "Pharmakokinetik" ).
Die pharmakokinetischen Daten zu Patienten, bei denen mehr als 15% der Gesamtkörperoberfläche betroffen sind, sind begrenzt. Aus Gründen der Sicherheit (siehe auch Rubrik "Warnhinweise und Vorsichtsmassnahmen: Koagulopathie" ) darf dieses Arzneimittel bei Erwachsenen2 und pädiatrischen Patienten von 4 bis 18 Jahren3 nicht auf mehr als 15 % der Gesamtkörperoberfläche (TBSA) aufgetragen werden.
Bei pädiatrischen Patienten von 0 bis 3 Jahren darf dieses Arzneimittel nicht auf mehr als 10 % der Gesamtkörperoberfläche (TBSA)3 aufgetragen werden.
Anwendung bei Patienten mit kardiopulmonaler oder pulmonaler Erkrankung
NexoBrid sollte bei Patienten mit kardiopulmonaler oder pulmonaler Erkrankung, einschliesslich Patienten mit pulmonalem Verbrennungstrauma oder Verdacht auf pulmonales Verbrennungstrauma, mit Vorsicht angewendet werden.
Anwendung bei Patienten mit Varizen
Dieses Arzneimittel sollte in Bereichen mit Varizen mit Vorsicht angewendet werden, um eine Erosion der Venenwände und ein Blutungsrisiko zu verhüten.
Brandwunden, für die nur begrenzte oder keine Erfahrung vorliegt.
Es gibt keine Erfahrung mit der Anwendung von NexoBrid bei Verbrennungen im Bereich des Perineums und der Genitalien.
Es liegen begrenzte Daten zur Anwendung von NexoBrid bei Verbrennungen im Gesicht vor. Es liegen Berichte in der Literatur zur erfolgreichen Anwendung von NexoBrid bei Verbrennungen im Gesicht vor. Verbrennungschirurgen, die keine Erfahrung mit der Anwendung dieses Arzneimittels haben, sollten nicht mit der Anwendung bei Gesichtsverbrennungen beginnen. NexoBrid muss bei diesen Patienten mit Vorsicht angewendet werden. Die Augen müssen während der Behandlung von Verbrennungen im Gesicht sorgfältig geschützt werden; dazu wird eine für die Anwendung am Auge geeignete Fettsalbe auf die Augen und eine Paraffin-Salbe als adhäsive Barriere um die Augen herum aufgetragen, um die Augen zu isolieren; die Augen werden dann mit einem okklusiven Film abgedeckt.
Vorbeugung von Wundkomplikationen
Bei der Anwendung von NexoBrid sind die allgemeinen Prinzipien der Wundbehandlung von Verbrennungen zu beachten. Dazu gehört eine adäquate Abdeckung der exponierten Wundfläche (siehe Rubrik "Dosierung/Anwendung" .
In Studien zu NexoBrid wurden Wunden mit sichtbaren Dermisresten einer Spontanepithelialisierung überlassen. In mehreren Fällen blieb eine adäquate Abheilung aus, und zu einem späteren Zeitpunkt wurde eine Hauttransplantation notwendig. Dies führte zu einer Verzögerung des Wundverschlusses, die mit einem erhöhten Risiko für Wundkomplikationen verbunden sein kann. Daher sollen Wunden mit Bereichen drittgradiger Verbrennung („full thickness“) bzw. Wundbereiche mit tiefen Verbrennungen, so früh wie möglich nach dem NexoBrid-Debridement mit einem autologen Hauttransplantat versorgt werden (zu Studienergebnissen siehe Rubrik "Klinische Wirksamkeit" ). Ebenso ist bei Verbrennungen Grad IIb („deep partial thickness“) sorgfältig zu erwägen, diese früh nach dem NexoBrid-Debridement mit einer permanenten Hautdeckung (z. B. einem autologen Hauttransplantat) zu versorgen (siehe auch die Rubriken "Dosierung/Anwendung" und "Unerwünschte Wirkungen" ).
Wie auch nach einem operativen Debridement sollte der mit NexoBrid behandelte Bereich sofort durch vorübergehenden oder permanenten Hautersatz gedeckt oder mit einem Verband bedeckt werden, um eine Austrocknung und/oder die Bildung von "Pseudoeschar" und/oder Infektionen zu vermeiden. Vor permanenter (z. B. durch ein autologes Hauttransplantat) oder vorübergehender Deckung durch Hautersatz (z. B. ein Allotransplantat) eines frisch debridierten Bereichs ist darauf zu achten, dass das Wundbett gesäubert und angefrischt wird, z. B. durch Bürsten oder Abschaben, um ein Anhaften zu ermöglichen.
Schutz der Augen
Direkter Augenkontakt muss verhütet werden. Bei Gefahr von Augenkontakt sollten die Augen des Patienten mit einer fettenden Augensalbe geschützt werden.
Sollte NexoBrid in die Augen gelangen, müssen die betroffenen Augen mindestens 15 Minuten lang mit viel Wasser gespült werden. Eine Augenärztliche Untersuchung wird vor und nach dem Debridement empfohlen.
Überempfindlichkeitsreaktionen, Hautkontakt, Inhalation
Bei wiederholter Exposition eines Patienten mit Bromelain-haltigen Präparaten zu einem späteren Zeitpunkt ist das Sensibilisierungspotential von NexoBrid (einem proteinhaltigen Produkt) zu beachten. Die Anwendung von NexoBrid bei nachfolgenden Verbrennungen wird nicht empfohlen.
Es wurden bei Patienten infolge eines NexoBrid-Debridements schwerwiegende allergische Reaktionen einschliesslich Anaphylaxie (die sich als Ausschlag, Erythem, Hypotonie, Tachykardie äusserte) gemeldet (siehe Rubrik "Unerwünschte Wirkungen" ). In diesen Fällen wurde ein kausaler Zusammenhang mit NexoBrid als möglich erachtet, obwohl eine Beziehung auf Begleitmedikamente nicht ausgeschlossen werden konnte.
In der Literatur wurden allergische Reaktionen auf Bromelain beschrieben (wie anaphylaktische Reaktionen und andere Reaktionen vom Soforttyp, die sich als Bronchospasmus, Angioödem, Urtikaria und Schleimhaut- bzw. gastrointestinale Reaktionen manifestierten). Im Rahmen einer Studie, in der die Menge an Schwebeteilchen während der NexoBrid-Gelzubereitung untersucht wurde, wurde keine berufsbedingte Gefahr identifiziert. Jedoch sind geeignete Massnahmen beim Umgang mit dem Präparat für das Wund-Debridement (u. a. das Tragen von Handschuhen, Schutzkittel und Schutzmaske) erforderlich.
Vor der Anwendung muss eine Allergieanamnese erstellt werden (siehe Rubriken "Kontraindikationen" und "Hinweise für die Handhabung" .
Bei Hautkontakt von NexoBrid sollte das Gel mit Wasser abgewaschen werden, um das Risiko einer Hautsensibilisierung zu verringern (siehe Rubrik "Hinweise für die Handhabung" ).
Kreuzsensibilisierung
In der Literatur wurde eine Kreuzsensibilisierung zwischen Bromelain und Papaya/Papain sowie Latex-Proteinen (Latex-Frucht-Syndrom), Bienengift und Olivenbaumpollen beschrieben.
Analgesie
Das enzymatische Debridement ist eine schmerzhafte Prozedur und darf nur nach einer adäquaten Analgesie und/oder Anästhesie durchgeführt werden.
Koagulopathie
Nach oraler Verabreichung von Bromelain wurden in der Literatur als mögliche Wirkungen eine Verringerung der Thrombozytenaggregation und des Fibrinogen-Plasmaspiegels sowie eine mässige Verlängerung der partiellen Thromboplastin- und Prothrombinzeit beschrieben. In-vitro-Daten und Daten aus tierexperimentellen Untersuchungen weisen darauf hin, dass Bromelain darüber hinaus die Fibrinolyse fördern kann. Während der klinischen Entwicklung von NexoBrid gab es keine Hinweise auf eine vermehrte Blutungsneigung oder Blutungen am Ort des Debridements.
Die Behandlung sollte nicht bei Patienten mit unkontrollierten Gerinnungsstörungen angewendet werden. NexoBrid sollte mit Vorsicht bei Patienten angewendet werden, die unter einer Therapie mit Antikoagulanzien oder anderen Arzneimitteln stehen, die die Blutgerinnung beeinflussen, sowie bei Patienten mit niedrigen Thrombozytenzahlen oder erhöhtem Blutungsrisiko anderer Ursache, z. B. aufgrund eines peptischen Ulkus oder einer Sepsis, mit Vorsicht angewendet werden.
Die Patienten sind auf mögliche Zeichen einer Gerinnungsstörung und Anzeichen von Blutungen zu kontrollieren.
Überwachung
Neben der routinemässigen Überwachung von Verbrennungspatienten (z. B. Vitalzeichen/ Volumen-/ Flüssigkeits-/ Elektrolytstatus, grosses Blutbild, Albumin-Serumkonzentration und Leberwerte) müssen bei mit NexoBrid behandelten Patienten folgende Parameter überwacht werden:
-Anstieg der Körpertemperatur.
-Zeichen lokaler oder systemischer entzündlicher oder infektiöser Prozesse.
-Störungen, die durch eine analgetische Prämedikation (z. B. Magendilatation, Übelkeit und Risiko von plötzlichem Erbrechen, Obstipation) oder Antibiotika-Prophylaxe (z. B. Durchfall) ausgelöst oder verstärkt werden könnten.
-Hinweise auf lokale oder systemische allergische Reaktionen.
-Potenzielle Auswirkungen auf die Hämostase (siehe oben).
Entfernung von topisch applizierten antibakteriellen Arzneimitteln vor Applikation von NexoBrid
Vor der Applikation von NexoBrid müssen alle topisch applizierten Antiseptika entfernt werden. Reste dieser Arzneimittel können die Wirkung von NexoBrid herabsetzen, da seine Wirksamkeit reduziert wird.
InteraktionenEs wurden keine Studien zur Erfassung von Wechselwirkungen mit NexoBrid durchgeführt.
Arzneimittel mit Einfluss auf die Gerinnung
Nach oraler Verabreichung von Bromelain wurden als mögliche Wirkungen eine Verringerung der Thrombozytenaggregation und Fibrinogen-Plasmaspiegel sowie eine mässige Verlängerung der partiellen Thromboplastin- und Prothrombinzeit beschrieben. In-vitro-Daten und Daten aus tierexperimentellen Untersuchungen weisen darauf hin, dass Bromelain darüber hinaus die Fibrinolyse fördern kann. Daher ist bei gleichzeitiger Verordnung von Arzneimitteln, die die Blutgerinnung beeinflussen können, Vorsicht geboten, und es sind entsprechende Kontrollen erforderlich (siehe auch Rubrik "Warnhinweise und Vorsichtsmassnahmen" ).
Substrate von CYP2C8 und CYP2C9
Resorbiertes NexoBrid ist ein Inhibitor von Cytochrom-P450-2C8 (CYP2C8) und Cytochrom-P450-2C9 (CYP2C9). Dieser Umstand ist zu berücksichtigen, wenn NexoBrid bei Patienten angewendet wird, die CYP2C8-Substrate (wie Amiodaron, Chloroquin, Fluvastatin, Paclitaxel, Pioglitazon, Repaglinid und Torasemid) und CYP2C9-Substrate (wie Ibuprofen, Losartan, Celecoxib, Warfarin und Phenytoin) erhalten.
Topische antibakterielle Wirkstoffe
Topisch applizierte antibakterielle Wirkstoffe (wie Sulfadiazin-Silber oder Povidon-Iod) können die Wirksamkeit von NexoBrid verringern (siehe Rubrik "Warnhinweise und Vorsichtsmassnahmen" ).
Fluorouracil und Vincristin
Bromelain kann die Wirkungen von Fluorouracil und Vincristin verstärken. Die Patienten sollten im Hinblick auf eine erhöhte Toxizität überwacht werden.
ACE-Hemmer
Bromelain kann die blutdrucksenkende Wirkung von Angiotensin-Converting-Enzyme (ACE)-Hemmern verstärken und einen stärkeren Blutdruckabfall als erwartet zur Folge haben. Bei Patienten, die ACE-Hemmer erhalten, sollte der Blutdruck überwacht werden.
Benzodiazepine, Barbiturate, Narkotika und Antidepressiva
Bromelain kann die durch bestimmte Arzneimittel (z. B. Benzodiazepine, Barbiturate, Narkotika und Antidepressiva) verursachte Benommenheit verstärken. Dieser Aspekt sollte bei der Dosierung derartiger Arzneimittel berücksichtigt werden.
Pädiatrische Patienten
Für Kinder und Jugendliche wurden keine Studien zur Erfassung von Wechselwirkungen durchgeführt.
Schwangerschaft, StillzeitSchwangerschaft
Bisher liegen keine Erfahrungen mit der Anwendung von NexoBrid bei Schwangeren vor.
Es liegen keine ausreichenden tierexperimentellen Studien vor, die eine adäquate Beurteilung des Potentials von NexoBrid für eine Beeinflussung der embryonalen/fetalen Entwicklung zulassen (siehe Rubrik "Präklinische Daten" ).
Da die sichere Anwendung von NexoBrid in der Schwangerschaft noch nicht erwiesen ist, wird die Anwendung von NexoBrid während der Schwangerschaft nicht empfohlen.
Stillzeit
Es ist nicht bekannt, ob das Konzentrat proteolytischer Enzyme angereichert aus Bromelain oder Metaboliten in die Muttermilch übergehen. Ein Risiko für das Neugeborene / Kind kann nicht ausgeschlossen werden. Das Stillen soll ab dem Zeitpunkt der erstmaligen Applikation von NexoBrid für mindestens 4 Tage unterbrochen werden.
Fertilität
Es wurden keine Studien zu den Auswirkungen von NexoBrid auf die Fertilität durchgeführt.
Wirkung auf die Fahrtüchtigkeit und auf das Bedienen von MaschinenNicht relevant.
Unerwünschte WirkungenIm Rahmen der klinischen Entwicklung wurden 536 Patienten mit NexoBrid in neun klinischen Studien behandelt, wovon drei randomisierte und kontrollierte offene Phase-3-Studien waren,.
Die am häufigsten beschriebenen Nebenwirkungen nach Anwendung von NexoBrid bei Erwachsenen in der Gruppe der mit diesem Arzneimittel behandelten Patienten aus den Studien MW2004, MW2005, MW2008 und MW2010 (insgesamt 203 Patienten) sind Pyrexie (bei 13,3 % der mit Nexo-Brid behandelten Patienten) und Schmerzen (3,9 %).
Die am häufigsten beschriebenen Nebenwirkungen nach Anwendung bei Kindern und Jugendlichen in der Gruppe der mit diesem Arzneimittel behandelten Patienten aus den Studien MW2004, MW2008 und MW2012 (insgesamt 89 Patienten) sind Pyrexie und Schmerzen (Inzidenz 16,9 % bzw. 7,9 %).
Die schwerwiegenden Nebenwirkungen, die bei mehr als einem Patienten (≥ 1,1 %) auftraten, waren nach Vorzugsbenennung: Sepsis und bakterielle Wundinfektion.
Die Nebenwirkungen bis zu 3 Monate nach Wundverschluss werden unten aufgeführt.
Bei den Häufigkeitsangaben zu Nebenwirkungen werden folgende Kategorien zugrunde gelegt:
Sehr häufig (≥1/10)
Häufig (≥1/100, <1/10)
Gelegentlich (≥1/1.000, <1/100)
Selten (≥1/10.000, <1/1.000)
Sehr selten (<1/10.000)
Nicht bekannt (Häufigkeit auf Grundlage der verfügbaren Daten nicht abschätzbar).
Die im Folgenden angegebenen Häufigkeiten von Nebenwirkungen beziehen sich auf die Anwendung von NexoBrid zur Eschar-Entfernung bei tiefen Verbrennungen (Grad IIb und III, "deep partial" und "full thickness" ) im Rahmen von Behandlungsprotokollen, die eine lokale Antiseptika-Behandlung, die empfohlene Analgesie/Anästhesie und die Bedeckung des Wundbereichs mit einem Okklusivverband über 4 Stunden (um NexoBrid nach Applikation auf der Wunde zu halten) vorsahen.
Infektionen und parasitäre Erkrankungen
Häufig: Wundinfektion einschliesslich Zellulitis
Erkrankungen des Immunsystems
Häufig: Nicht schwerwiegende allergische Reaktionen wie Hautausschlag
Nicht bekannt: Schwerwiegende allergische Reaktionen einschliesslich Anaphylaxie
Herzerkrankungen
Häufig: Tachykardie
Erkrankungen der Haut und des Unterhautzellgewebes
Häufig: Wundkomplikationen (einschliesslich Austrocknung der Wunde, Vertiefung der Wunde, Wiedereröffnung der Wunde, Transplantatverlust/Transplantatversagen), lokaler Hautausschlag, lokaler Pruritus
Gelegentlich: Intradermales Hämatom
Allgemeine Erkrankungen und Beschwerden am Verabreichungsort
Sehr häufig: Pyrexie/Hyperthermie
Häufig: Lokale Schmerzen
Beschreibung spezifischer unerwünschter Wirkungen und Zusatzinformationen
Immunogenität
Die DETECT-Studie umfasste Immunogenitätstests von Proben, die vor und zu verschiedenen Zeitpunkten nach der Behandlung mit NexoBrid untersucht wurden (Baseline, Tag 28, Tag 56, 6 Monate und 24 Monate).
Zu Studienbeginn (Baseline) waren 39,4 % (26/66) der getesteten Personen Antikörper-positiv gegen NexoBrid, was möglicherweise auf eine frühere Sensibilisierung gegenüber aus Ananas stammenden Proteinen und Glykoproteinen mit kreuzreagierenden Kohlenhydratdeterminanten zurückführen könnte.
Die Inzidenz der während der Behandlung entwickelten Antikörper betrug 92,4 % (61/66), wovon 62,3 % (38/61) der Patienten, die zu Studienbeginn negativ waren, und 37,7 % (23/61), die zu Studienbeginn positiv waren und einen mindestens vierfachen Anstieg der Antikörpertiter nach der Behandlung erfuhren.
Der Zeitpunkt des Auftretens von Antikörpern während der Behandlung (erhöhte Antikörpertiter) stimmte mit einer affinitätsgereiften Immunantwort auf ein xenogenes Protein überein (100 % Serokonversion nach 4 Wochen), und die Antwort war anhaltend (mindestens 24 Monate).
Es gab keinen offensichtlichen Zusammenhang zwischen dem maximalen Antikörpertiter nach der Behandlung und entweder der Gesamtdosis von NexoBrid (in Gramm) oder der mit TBSA behandelte Dosis. Es gab keinen offensichtlichen Zusammenhang zwischen der Wirksamkeit (vollständige Eschar-Entfernung) und dem Antikörpertiter vor oder nach der Behandlung. Es konnte kein eindeutiger Zusammenhang zwischen dem Vorhandensein von Antikörpern vor der Behandlung und dem Auftreten von Überempfindlichkeitsreaktionen hergestellt werden.
Pädiatrische Population
Die Daten aus klinischen Studien an pädiatrischen Patienten (von der Geburt bis zum Alter von 18 Jahren) umfassen die Anwendung dieses Arzneimittels in einer kontrollierten Studie gegen TdR (MW2012), in der 69 Patienten mit diesem Arzneimittel behandelt wurden (Altersspanne: von der Geburt bis zum Alter von 18 Jahren; siehe Abschnitt "Klinische Wirksamkeit" für die Altersverteilung) sowie die Anwendung bei pädiatrischen Patienten in den Studien MW2004 und MW2008, an denen 17 bzw. 3 pädiatrische Patienten teilnahmen (Altersspanne: 4 bis 17 Jahre).
Insgesamt ähnelt das Sicherheitsprofil bei pädiatrischen Patienten dem bei Erwachsenen. Angesichts der geringen Anzahl der in den verschiedenen Altersgruppen gemeldeten Nebenwirkungen ist es nicht möglich, gültige Schlussfolgerungen hinsichtlich möglicher altersbedingter Unterschiede im Sicherheitsprofil zu ziehen.
Meldung des Verdachts auf Nebenwirkungen
Die Meldung des Verdachts auf Nebenwirkungen nach der Zulassung ist von grosser Wichtigkeit. Sie ermöglicht eine kontinuierliche Überwachung des Nutzen-Risiko-Verhältnisses des Arzneimittels. Angehörige von Gesundheitsberufen sind aufgefordert, jeden Verdacht einer neuen oder schwerwiegenden Nebenwirkung über das Online-Portal ElViS (Electronic Vigilance System) anzuzeigen. Informationen dazu finden Sie unter www.swissmedic.ch.
ÜberdosierungIm Rahmen einer klinischen Studie führte die Behandlung mit dem Konzentrat proteolytischer Enzyme angereichert aus Bromelain in einem Pulver:Gel-Verhältnis von 1:5 (0,16 g je Gramm des fertigen Gels) bei Patienten mit tiefen Verbrennungen (Grad IIb – III) nicht zu signifikant anderen Sicherheitsergebnissen als die Behandlung mit dem Konzentrat proteolytischer Enzyme angereichert aus Bromelain in einem Pulver:Gel-Verhältnis von 1:10 (0,09 g je Gramm des fertigen Gels).
Begrenzte Daten aus klinischen Studien und der Anwendung nach Markteinführung von NexoBrid in einer Konzentration von 0,09 g pro 1 g gemischtem Gel zeigten kein erhöhtes Risiko, wenn NexoBrid in zwei Anwendungen mit einer TBSA > 15 % (bis zu 30 % TBSA) verwendet wurde.
Eigenschaften/WirkungenATC-Code
D03BA03
Pharmakotherapeutische Gruppe: Präparate zur Behandlung von Wunden und Geschwüren, proteolytische Enzyme.
Wirkungsmechanismus/Pharmakodynamik
Das Konzentrat proteolytischer Enzyme angereichert aus Bromelain ist ein Präparat für das Wund-Debridement und wird topisch aufgetragen, um bei tiefen Verbrennungen (Grad IIb – III) den Verbrennungsschorf (Eschar) zu entfernen.
Die Enzym-Mischung in NexoBrid löst den Verbrennungsschorf auf Brandwunden auf. Die für diese Wirkung verantwortlichen Bestandteile wurden noch nicht identifiziert. Hauptbestandteil ist Stamm-Bromelain.
Klinische Wirksamkeit
Die Wirksamkeit und Sicherheit von NexoBrid zur Eschar-Entfernung bei thermischen tiefen Verbrennungen (DPT und/oder FT) wurden in drei multizentrischen, randomisierten, kontrollierten Phase-3-Studien beurteilt (MW2010, MW2004 und MW2012).
DETECT-Studie (MW2010) - Phase 3b
Bei dieser Studie handelte es sich um eine randomisierte, kontrollierte, Prüfarzt-verblindete, dreiarmige Studie mit dem Ziel, die NexoBrid-Behandlung mit der Gelvehikel-Kontrolle (Placebo) und der Standardbehandlung bei hospitalisierten erwachsenen Patienten mit thermischen Verbrennungen (DPT und/oder FT) von 3 bis 30 % der TBSA und einer Gesamtfläche an Brandwunden von nicht mehr als 30 % der TBSA zu vergleichen.
Insgesamt wurden 175 Probanden randomisiert (Intendto-Treat-Kohorte) in einem Verhältnis von 3:3:1 (NexoBrid:Standardtherapie:Gelvehikel), und 169 Probanden erhielten eine Behandlung. Die Patienten im Behandlungsarm der Standardtherapie erhielten je nach Ermessen der Prüfärzte eine chirurgische und/oder nicht chirurgische Standardtherapie.
Insgesamt waren die demografischen Daten der Probanden und die Ausgangsmerkmale der Wunde in den Studienarmen vergleichbar. Die Altersspanne lag in der mit NexoBrid behandelten Gruppe bei 18 bis 75 Jahren, in der Standardtherapiegruppe bei 18 bis 72 Jahren und in der Gelvehikel-Gruppe bei 18 bis 70 Jahren. Das Durchschnittsalter in allen 3 Gruppen betrug 41 Jahre, und 65 %, 79 % bzw. 60 % der Probanden in den Gruppen NexoBrid, Standardtherapie bzw. Gelvehikel (Placebo) waren männlich. Die Zielwunde (TW) war der Verbrennungsbereich, der mit NexoBrid, Standardtherapie oder Gel Vehikel behandelt werden sollte (Eschar-Entfernung). Auf den Patienten bezogen war die mittlere Grösse der TW-Fläche in der NexoBrid-Behandlungsgruppe 6,28 % TBSA, in der Standardtherapiegruppe 5,91 % TBSA und in der Gelvehikel-Gruppe 6,53 % TBSA (durchschnittlich 1,7 Zielwunden pro Patient).
Primärer Endpunkt war die Inzidenz der vollständigen (> 95 %) Eschar-Entfernung im Vergleich zum Gelvehikel. Zu den sekundären Endpunkten gehörten die Zeit bis zur vollständigen Eschar-Entfernung, die Inzidenz der chirurgischen Exzision und der mit dem Debridement verbundene Blutverlust im Vergleich zur Standardtherapie. Als Sicherheitsendpunkte wurden die Zeit bis zum vollständigen Wundverschluss, die Langzeit-Kosmetik und die Funktion (gemessen mit dem Modified Vancouver Scar Scale (MVSS)) nach dem 12-monatigen Nachbeobachtungszeitraum analysiert.
Die Studienergebnisse zeigten, dass die Inzidenz der vollständigen Eschar-Entfernung in der Nexo-Brid-Gruppe signifikant höher als in der Gelvehikel-Kontrolle war.
Inzidenz der vollständigen Eschar-Entfernung in der DETECT-Studie
NexoBrid (ER/N) Gelvehikel (ER/N) p-Wert
Inzidenz der vollständigen Eschar-Entfernung 93, 3% 70/75 4,0 % (1/25) p < 0,0001
ER = Eschar-Entfernung
Im Vergleich zur Standardtherapie führte NexoBrid zu einer signifikanten Verringerung der Inzidenz der chirurgischen Eschar-Entfernung (tangentiale/kleine/Avulsion/Versajet-Exzision und/oder Dermabrasion), der Zeit bis zur vollständigen Eschar-Entfernung und des tatsächlichen Blutverlusts (siehe Tabelle unten).
Inzidenz der chirurgischen Eschar-Exzision, Zeit bis zur vollständigen Eschar-Entfernung und Blutverlust in der DETECT-Studie
NexoBrid (N=75) Standardtherapie p-Wert
(N=75)
Inzidenz der chirurgischen 4,0 % (3) 72,0 % (54) p < 0,0001
Exzision (Anzahl der Probanden)
Mediane Zeit bis zur vollständigen 1,0 Tag 3,8 Tag p < 0,0001
Eschar-Entfernung
Mit der Eschar-Entfernung 14,2 ±512,4 ml 814,5 ±1020,3 ml p < 0,0001
zusammenhängender Blutverlust a
a Tatsächlicher Blutverlust, berechnet nach der in McCullough 2004 beschriebenen Methode:
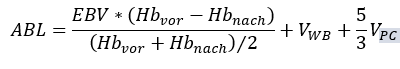
EBV = Geschätztes Blutvolumen wird mit 70 cm3/kg*Gewicht (kg) angenommen; (Hbvor – Hbnach) = Veränderung des Hb-Werts während der Eschar-Entfernung; VWB= Volumen [mL] des während der Eschar-Entfernung transfundierten Vollbluts; VPC= Volumen [mL] des während der Eschar-Entfernung transfundierten Erythrozyten-Konzentrats.
Langfristige Ergebnisse (12 und 24 Monate nach Wundverschluss)
Die Phase-3-Studie (DETECT) umfasste eine Langzeitbeobachtung zur Beurteilung der Langzeit-Kosmetik und der Funktion bei Nachuntersuchungen nach 12 und 24 Monaten. Nach 12 Monaten zeigte die Bewertung der Narbe anhand der Modified Vancouver Scar Scale (MVSS) vergleichbare Ergebnisse zwischen den verschiedenen Behandlungen mit NexoBrid, TdR und Gelvehikel mit durchschnittlichen Werten von 3,70, 5,08 bzw. 5,63. Nach 24 Monaten lagen die Durchschnittswerte auf der MVSS-Skala bei 3,04, 3,30 bzw. 2,93. Statistische Analysen zeigten die Nichtunterlegenheit (Nichtunterlegenheitsmarge von 1,9 Punkten) der Behandlung mit NexoBrid gegenüber TdR und zeigten, dass die Behandlung mit NexoBrid im Vergleich zu TdR 24 Monate nach der Wundheilung keine klinisch signifikanten schädlichen Auswirkungen auf Kosmetik und den funktionellen Zustand der Verbrennungsnarbe hat.
Die Messungen der Funktionalität und Lebensqualität (QoL) nach 12 und 24 Monaten waren in den verschiedenen Behandlungsgruppen ähnlich. Die Durchschnittswerte auf der LEFS-Skala (Lower Extremity Functional Scale), die durchschnittlichen QuickDASH-Werte, die Bewertungen des Bewegungsumfangs (ROM) sowie die langfristige Lebensqualität, gemessen anhand der visuellen Analogskala (VAS) EQ-5D und der BSHS-B-Skala (Burn Specific Health Scale-Brief), waren in den verschiedenen Behandlungsgruppen ähnlich
Studie MW2004 Phase 3
Bei der Stude MW2004 handelte sich um eine randomisierte, multizentrische, multinationale, unverblindete, konfirmatorische Phase-III-Studie, in der NexoBrid im Vergleich zur Standardtherapie bei hospitalisierten Patienten mit tiefen Verbrennungen (Grad IIb und/oder III; "deep partial" und/oder "full thickness" ) mit einer Ausdehnung von 5 bis 30 % der TBSA (bei denen aber insgesamt nicht mehr als 30 % der TBSA von Verbrennungen betroffen waren) untersucht wurde. Die mittlere behandelte TW-Fläche als prozentualer Anteil der TBSA betrug für dieses Arzneimittel 5,1 ± 3,5 % und für die Standardtherapie 5,2 ± 3,4 %.
Die Standardtherapie bestand je nach üblicher Praxis im jeweiligen Prüfzentrum in einer primären chirurgischen Exzision und/oder einem nicht-chirurgischen Debridement mit topischen Arzneimitteln, die eine Mazeration oder Autolyse des Schorfs induzieren sollten.
Die Altersspanne betrug in der Gruppe der mit NexoBrid behandelten Patienten 4,4 bis 55,7 Jahre und bei den mit der Standardtherapie behandelten Patienten 5,1 bis 55,7 Jahre.
Die Wirksamkeit der Schorfentfernung wurde über die Bestimmung des prozentualen Anteils der noch mit Eschar bedeckten Wundfläche beurteilt, bei der eine weitere Entfernung mittels Exzision oder Dermabrasion erforderlich war, sowie über den prozentualen Anteil der Wunden (Anzahl der Wunden), bei denen der Verbrennungsschorf operativ entfernt werden musste.
Der Effekt auf die Zeitdauer bis zur erfolgreichen Eschar-Entfernung wurde bei Patienten mit erfolgreichem Debridement (Entfernung von mindestens 90 % des Verbrennungsschorfs bezogen auf alle Wunden dieses Patienten) über die Zeitspanne zwischen Verletzung bzw. Einwilligungserklärung und erfolgreicher Eschar-Entfernung untersucht.
Die beiden primären Endpunkte der Wirksamkeitsanalyse waren:
der prozentuale Anteil der Verbrennungswunden Grad IIb ( "deep partial thickness" ), bei denen eine Exzision oder Dermabrasion erforderlich wurde, und
der prozentuale Anteil der Verbrennungswunden Grad IIb ( "deep partial thickness" ), die mit einem autologen Hauttransplantat versorgt wurden.
Dieser Endpunkt lässt sich nur bei Verbrennungen Grad IIb ohne Bereiche mit Verbrennung Grad III („full thickness“), bestimmen, da bei Letzteren immer eine Deckung mit einem Hauttransplantat erforderlich ist.
Im Folgenden sind die Wirksamkeitsergebnisse dieser Studie kombiniert für alle Altersgruppen zusammengefasst.
NexoBrid Standardtherapie p-Wert
Grad IIb Verbrennungen ( "deep
partial thickness" ), bei denen
eine Exzision/Dermabrasion
(Operation) erforderlich war
Anzahl der Wunden 106 88
% der Wunden, bei denen eine 15,1% 62,5% <0,0001
Operation erforderlich war
% der mittels Exzision oder 5,5% ± 14,6 52,0% ± 44,5 <0,0001
Dermabrasion behandelten
Wundfläche1 (Mittelwert± SD)
Mit einem Autotransplantat
gedeckte Grad IIb Verbrennungen (
"deep partial thickness" ) *
Anzahl der Wunden 106 88
% der mit einem Autotransplantat 17,9% 34,1% 0,0099
gedeckten Wunden
% der mit einem Autotransplantat 8,4% ± 21,3 21,5% ± 34,8 0,0054
gedeckten Wundfläche (Mittelwert±
SD)
Grad IIb Verbrennungen ( "deep
partial thickness" ), bei denen
eine Exzision/Dermabrasion
(Operation) erforderlich war
Anzahl der Wunden 163 170
% der Wunden, bei denen eine 24,5% 70,0% <0,0001
Operation erforderlich war
% der mittels Exzision oder 13,1% ± 26,9 56,7% ± 43,3 <0,0001
Dermabrasion behandelten
Wundfläche1 (Mittelwert± SD)
Zeit bis zum vollständigen
Wundverschluss (Zeit ab
Einwilligungserklärung)
Anzahl der Patienten 2 70 78
Tage bis zum Verschluss der 36,2 ± 18,5 28,8 ± 15,6 0,0185
letzten Wunde (Mittelwert ± SD)
Zeit bis zur erfolgreichen
Eschar-Entfernung
Anzahl der Patienten 67 73
Tage (Mittelwert ± SD) seit 2,2 ± 1,4 8,7 ± 5,7 <0,0001
Verletzung
Tage (Mittelwert± SD) seit 0,8 ± 0,8 6,7 ± 5,8 <0,0001
Einwilligungserklärung
Patienten, bei denen keine 7 8
erfolgreiche Eschar-Entfernung
beschrieben wurde
1 Bestimmt bei der ersten operativen Versorgung, wenn mehr als eine Operation erfolgte. 2 Alle randomisierten Patienten, für die Daten zum vollständigen Wundverschluss vorlagen.
*Dieser Endpunkt lässt sich nur bei Verbrennungen Grad IIb ohne Bereiche mit Grad III bestimmen, da bei Letzteren immer eine Deckung mit einem Hauttransplantat erforderlich ist.
SD: Standardabweichung.
Langfristige Ergebnisse
In einer multizentrischen, nicht-interventionellen Studie mit verblindeter Bewertung wurde die langfristige Narbenbildung und die Lebensqualität von Erwachsenen und Kindern untersucht, die an der MW2004-Studie teilgenommen hatten. Die darin enthaltene Population von 89 Patienten, darunter 72 Erwachsene und 17 pädiatrische Patienten (< 18 Jahre), war repräsentativ für die Population der MW2004-Studie.
Die Bewertung der Narbenbildung anhand der MVSS-Skala nach 2 bis 5 Jahren ergab vergleichbare Ergebnisse zwischen den Studiengruppen, mit einem durchschnittlichen Gesamtwert von 3,12 für dieses Arzneimittel und 3,38 für TdR (p = 0,88).
Die Lebensqualität wurde bei Erwachsenen anhand des SF-36-Fragebogens bewertet. Die Durchschnittswerte für die verschiedenen Parameter waren in beiden Gruppen ähnlich. Der Gesamtwert für die körperliche Komponente (51,1 bzw. 51,3) und der Gesamtwert für die mentale Komponente (51,8 bzw. 49,1) waren in beiden Gruppen vergleichbar.
Dauer bis zum vollständigen Wundverschluss
In der DETECT-Studie (MW2010) betrug die geschätzte mediane Dauer nach der Kaplan-Meier-Methode bis zum vollständigen Wundverschluss im NexoBrid-Behandlungsarm 27 Tage, bzw. 28 Tage im Standardtherapie-Behandlungsarm. Der p-Wert von 0,0003 bestätigte die Nichtunterlegenheit (7-Tage-Nichtunterlegenheitsgrenze) des NexoBrid-Behandlungsarms im Vergleich zur Standardtherapie. Die laut effektiven Daten berechnete mediane Dauer bis zum vollständigen Wundverschluss betrug 23 Tagen im NexoBrid-Behandlungsarm sowie im Standardtherapie-Behandlungsarm.
Die Ergebnisse der gepoolten Daten zum Wundverschluss aus beiden Phase-III-Studien unterstützen die Nichtunterlegenheit von NexoBrid im Vergleich zur Standardtherapie auf der Grundlage einer Nichtunterlegenheitsgrenze von 7 Tagen. Basierend auf den gepoolten Daten der DETECT-Studie und der Studie MW2004 war die Dauer bis zum vollständigen Wundverschluss in der NexoBrid-Gruppe etwas länger als in der Standardtherapiegruppe, wenn sie nach der Kaplan-Meier-Methode geschätzt wurde (Median 30,0 Tage vs. 25,0 Tage) oder anhand der effektiven Daten berechnet wurde (Mittelwert 31,7 Tage NexoBrid vs. 29,8 Tage Standardtherapie). Nach der Nichtunterlegenheitsanalyse war die Zeit bis zum vollständigen Wundverschluss mit NexoBrid weniger als 7 Tage länger als mit der Standardtherapie (p für Nichtunterlegenheit = 0,0006).
Pädiatrische Studie MW2012 (CIDS)
Es handelte sich um eine randomisierte (1:1), offene, gegen TdR kontrollierte Parallelgruppenstudie mit 145 hospitalisierten Patienten (0–18 Jahre) mit tiefen Verbrennungen zweiten Grades oder Verbrennungen dritten Grades, die 1 % bis 30 % der gesamten Körperoberfläche betrafen (durchschnittliche Fläche der PC: 5,57 % der KBA),. Die Patienten wurden randomisiert und erhielten entweder dieses Arzneimittel (2 g Pulver in 20 g Gel für 180 cm² über 4 Stunden) oder TdR (chirurgische und/oder nicht-chirurgische Massnahmen zur Entfernung von Wundschorf). Die Studie umfasste drei koprimäre Endpunkte: die mediane Zeit bis zur vollständigen Entfernung des Eschar, den Prozentsatz der chirurgisch exzidierten Wundfläche sowie das ästhetische Erscheinungsbild und den Funktionszustand der Haut 12 Monate nach Wundverschluss (Score auf der Modified Vancouver Scar Scale). Die demografischen Daten und die wichtigsten Ergebnisse sind in der folgenden Tabelle dargestellt.
Insgesamt wurden 145 Patienten randomisiert und in die FAS-Population (Full Analysis Set) aufgenommen: 72 in der mit diesem Arzneimittel behandelten Gruppe und 73 in der TdR-Gruppe. Von diesen Patienten wurden 139 (95,9 %) behandelt und in die SAS-Population (Safety Analysis Set) aufgenommen: 69 (95,8 %) in der mit dem Arzneimittel behandelten Gruppe und 70 (95,9 %) in der TdR-Gruppe.
Die Altersverteilung lautete wie folgt (dieses Arzneimittel gegenüber TdR): 0–11 Monate: 4 gegenüber 4, 12–23 Monate: 19 gegenüber 18, 24 Monate–3 Jahre: 15 gegenüber 15, 4–11 Jahre: 25 gegenüber 25 und 12–18 Jahre: 9 gegenüber 11.
Insgesamt waren Alter, ethnische Zugehörigkeit, Körpergrösse, Gewicht und Body-Mass-Index (BMI) der Patienten in den Behandlungsgruppen vergleichbar. Auf Patientenebene betrug der durchschnittliche Prozentsatz der TBSA der PC 5,85 % für die Patienten in der mit diesem Medikament behandelten Gruppe gegenüber 5,30 % in der TdR-Gruppe.
Wirksamkeitsergebnisse:
Im Vergleich zu TdR verkürzte die Behandlung mit dem Arzneimittel die Zeit bis zur vollständigen Entfernung des Eschar und reduzierte den durchschnittlichen Prozentsatz der chirurgisch exzidierten Wundfläche zur Entfernung des Eschar signifikant. Patienten, die mit diesem Arzneimittel behandelt wurden, benötigten weniger chirurgische Exzisionen als Patienten, die TdR erhielten (siehe Tabelle).
NexoBrid (N=72) TdR (N=73) p-Wert
Alter (Durchschnitt, 5,71 (4,84) 5,83 (4,91)
Standardabweichung)
52
Ergebnisse
Dauer bis zur
vollständigen
Eschar-Entfernung
Median, Tage (FAS) 0,99 5,99 0,0008
Prozentualer Anteil
des chirurgisch
entfernten Wundberei
chs (FAS)
Durchschnitt ± 1,5 ± 12,1 48,1 ± 46,6 < 0,0001
Standardabweichung
(FAS)
MVSS-Score nach 12
Monaten
Durchschnitt ± 3,83 ± 2,88 4,86 ± 3,26 < 0,0001 (Nachgewies
Standardabweichung ene Nichtunterlegenh
(FAS) eit)
Inzidenz der chirurg
ischen Exzision (%)
Anteil und Anzahl 8,33 64,38
der Patienten, die
eine chirurgische
Exzision zur Entfern
ung des Eschars
benötigen (FAS)*
Durchschnittliche
Dauer bis zur
Narbenbildung der
letzten Wunde –
Beobachtungsdaten
(Tage)
Durchschnitt, 28,65 ± 16,56 27,74 ± 18,15
Standardabweichung
(FAS)
*In einer Subgruppenanalyse nach Altersgruppen wurde die Überlegenheit dieses Arzneimittels gegenüber TdR in jeder Altersgruppe systematisch nachgewiesen
Die durchschnittliche Schwankung des Hämoglobinspiegels nach Eingriff zur Eschar-Entfernung war sowohl auf Patienten- als auch auf Interventionsebene bei den mit dem Arzneimittel behandelten Patienten geringer als bei den Patienten, die mit TdR behandelt wurden.
Zeit bis zum vollständigen Wundverschluss
Die nach dem Kaplan-Meier-Verfahren geschätzte mediane Zeit bis zum vollständigen Wundverschluss (> 95 %) auf PC-Ebene war in der mit diesem Arzneimittel und in der mit TdR behandelten Gruppe vergleichbar. In der zusammengefassten Erwachsenenpopulation betrug die nach der Kaplan-Meier-Analyse (geclusterte Daten der PC eines Patienten) geschätzte mediane Zeit bis zum vollständigen Wundverschluss (dieses Arzneimittel [N = 280] gegenüber TdR [N = 179]): 32 (95 % KI: 29,0 – 34,0) Tage gegenüber 28 (95 % KI: 24,0 – 29,0) Tagen. In der zusammengefassten pädiatrischen Population war die Zeit bis zum vollständigen Wundverschluss (> 95 %) an der PC in der mit dem Arzneimittel behandelten Gruppe und der mit TdR behandelten Gruppe vergleichbar. Die nach der Kaplan-Meier-Analyse geschätzte mediane Zeit betrug (dieses Arzneimittel [N = 89] gegenüber TdR [N = 86]): 31 (95 % KI: 27,0 – 36,0) Tage gegenüber 31 (95 % KI: 24,0 – 37,0) Tagen67.
Die Ergebnisse beider Populationen bestätigen die Nichtunterlegenheit dieses Arzneimittels gegenüber TdR, basierend auf einer Nichtunterlegenheitsmarge von 7 Tagen.
Langfristige Ergebnisse (12 Monate)
Hinsichtlich des kosmetischen Ergebnisses und des Funktionszustands gemäss Bewertung nach der MVSS-Skala nach 12 Monaten wurde die Nichtunterlegenheit der Behandlung mit dem Arzneimittel im Vergleich zur TdR (p-Wert < 0,0001) mit einer Nichtunterlegenheitsmarge von 1,9 nachgewiesen.
PharmakokinetikErwachsenenpopulation
Absorption
Pharmakokinetische Analysen wurden an einer Untergruppe von NexoBrid-Patienten durchgeführt, die an der Studie MW2008 und der Studie MW201 (DETECT) teilgenommen hatten, wobei dieselbe bioanalytische Methode angewendet wurde.
Nach der topischen Anwendung von NexoBrid wurde bei allen Patienten eine systemische Serumexposition festgestellt. Im Allgemeinen wird NexoBrid schnell resorbiert, mit einem medianen Tmax-Wert von 4,0 Stunden (Dauer der Applikation). Die NexoBrid-Exposition wurde mit quantifizierbaren Serumkonzentrationen bis 48 Stunden nach der Applikation der Dosis beobachtet.
Die Expositionsergebnisse aus den Studien MW2008 und MW2010 sind in der nachstehenden Tabelle aufgeführt.
In jeder Studie waren die Cmax und die dosisnormalisierte Cmax, nach der ersten und zweiten Anwendung vergleichbar. Der Vergleich zwischen der AUC0-4 und dosisnormalisierten AUC0-4- bei der ersten Anwendung gegenüber der zweiten Anwendung zeigt, dass die Exposition nach der zweiten Anwendung etwas höher war, aber vergleichbar (weniger als 2-fach). Nicht bei allen Patienten lagen Werte über 4 Stunden vor, sodass die AUCder-Werte für einige Patienten nur 4 Stunden der Exposition abdecken, während sie für andere Patienten 48 Stunden der Exposition abbilden.
In diesen beiden Studien gab es eine statistisch signifikante Korrelation zwischen den Serum-Cmax- und -AUC0–4-Werten in Abhängigkeit von der Dosis oder dem prozentualen Anteil an der TBSA, was auf eine Dosisoder Behandlungsbereich- abhängige Zunahme der Exposition hindeutet. Die Tiefe der mit NexoBrid behandelten Wunde hat einen vernachlässigbaren Einfluss auf die systemische Exposition.
Zusammenfassung der PK-Parameter gemessen in allen Patienten der Studien MW2008 und MW2010 (erste Anwendung)
Studien ID N Tmax Median (Bereich Cmax (ng/mL) Cmax/Dosis(ng/mL/g) AUC0-4(h*ng/mL/g) AUC0-4/Dosis(h*ng/mL AUCder(h*ng/mL) AUCder/Dosis(h*ng/mL
) (h) /g) /g)
Studie MW2008
13a 4,0 (0,50 – 4,1) 800±640(Min=222) 44,7±36,6 1930±648 103±48,8 2760±2870 149±147
(Max=2440)
Studie MW2010
21 4,0 (0,50 – 12) 200±184 (Min=30.7) 16,4±11,9 516±546 39,8±29,7 2500±2330 215±202
(Max=830)
* Werte werden angegeben als Mittel ± SD, mit Ausnahme von Tmax, angegeben als Median (Min-Max).
a n=8 für AUC0–4 und AUC0–4/dosis
AUCder = Fläche unter der Kurve bis zum letzten messbaren Zeitpunkt; AUC0–4 = Fläche unter der Konzentrations-Zeit-Kurve vom Zeitpunkt Null bis zum Zeitpunkt 4 h; Cmax = maximale beobachtete Konzentration; Tmax=Zeitpunkt, zu dem die maximale Konzentration beobachtet wurde.
Distribution
Einem Bericht aus der Literatur zufolge bindet Bromelain im Plasma zu etwa 50 % an die humanen Plasma-Antiproteasen α2-Makroglobulin und α1-Antichymotrypsin.
Metabolismus
Keine Daten vorhanden.
Elimination
In Studie MW2008-09-03 betrug die mediane Eliminationshalbwertszeit 12 ± 4,4 Stunden. Bei der nach 72 Stunden durchgeführten Untersuchung wurden bei der Mehrheit der Patienten keine messbaren Konzentrationen festgestellt.
Kinetik spezieller Patientengruppen
Kinder und Jugendliche
Explorative pharmakokinetische Analysen wurden in einer PK-Teilstudie der MW2012-Studie (CIDS) durchgeführt. Diese Analysen bezogen sich auf die Daten zur Serumkonzentration des Arzneimittels im Verhältnis zur Zeit.
Blutproben für die PK-Analyse wurden von 16 Patienten entnommen, die mit dem Arzneimittel behandelt wurden. Alle Patienten erhielten eine einmalige Gabe des Arzneimittels.
Bei allen 16 Patienten, von denen PK-Proben verfügbar waren, wurden Anzeichen einer systemischen Serumexposition beobachtet. Die Konzentrationen stiegen relativ schnell an, mit medianen Tmax-Werten zwischen 2 und 4 Stunden, was dem Zeitraum der topischen Anwendung entspricht.
Es wurde eine Korrelation zwischen der systemischen Exposition gegenüber dem Arzneimittel und der topisch applizierten Dosis festgestellt.
Die Expositionsergebnisse entnehmen Sie der folgenden Tabelle.
Zusammenfassung der bei den Patienten der Studie MW2012 gemessenen PK-Parameter
(Altersgruppe, N Tmax Median (Spanne Cmax (ng/mL) Cmax/Dosis /ng/mL/g) AUC0-4 (h*ng/mL) AUC0-4/ Dosis AUCder (h*ng/mL) AUCder /Dosis
Jahre) ) (h) (h*ng/mL) (h*ng/mL/g)
< 2 2 2,00 200 66,7 476 159 876 292
4-11 5 4,0 (2,0-4,0) 205 ± 169 32,8 ± 23,9 416 ± 259 67,9 ± 44,7 2 240 ± 2 220 366 ± 350
12-18 5 4,0 (2,0-4,0) 180 ± 114 19,2 ± 7,50 499 ± 315 53,3 ± 20,4 1 560 ± 887 174 ± 67,4
*Zehn Patienten wurden in die wichtigsten PK-Analysen einbezogen.
Elimination
Nach 48 Stunden wurde bei der Mehrheit der Patienten keine messbare Konzentration des Arzneimittels festgestellt. Nach 72 Stunden wurde bei keinem Patienten mehr eine messbare Konzentration festgestellt.
Präklinische DatenToxizität bei wiederholter Gabe
Eine einmalige intravenöse Infusion einer aus NexoBrid-Pulver hergestellten Lösung wurde von Minischweinen in einer Dosis von bis zu 12 mg/kg gut vertragen (womit Plasmaspiegel erzielt wurden, die dem 2,5fachen Wert nach Applikation der vorgesehenen klinischen Dosis für 15 % der TBSA beim Menschen entsprachen). Dagegen erwiesen sich höhere Dosen als eindeutig toxisch und verursachten in mehreren Geweben eine Hämorrhagie. Wiederholte intravenöse Injektionen von Dosen von bis zu 12 mg/kg jeden dritten Tag wurden von Minischweinen über die ersten vier Injektionen gut vertragen, während nach den verbleibenden zwei Injektionen klinische Zeichen einer schweren Intoxikation (z. B. Blutungen in mehreren Organen) beobachtet wurden. Diese Auswirkungen waren auch nach der Erholungszeit von 2 Wochen noch erkennbar.
Genotoxizität
Nexobrid zeigte bei der üblichen Reihe von In-vitro-Tests (bakterieller Mutagenitätstest [mit Salmonella thyphimurium], Chromosomenaberrations-test an V79-Zellen des chinesischen Hamsters), und In-vivo-Test (Maus Knochenmark Mikronukleus Test) kein genotoxisches Potential.
Kanzerogenität
Studien zur Kanzerogenität wurden mit NexoBrid nicht durchgeführt.
Entwicklungstoxizität
In Studien zur embryo-fetalen Entwicklung an Kaninchen und Ratten ergaben sich nach intravenöser Verabreichung von NexoBrid keine Hinweise auf eine indirekte und direkte toxische Wirkung auf den sich entwickelnden Embryo/Feten. Allerdings war die Exposition des Muttertiers deutlich niedriger als sie maximal im klinischen Bereich angegeben wird (10–500-fach niedriger als die AUC beim Menschen, 3–50-fach niedriger als die Cmax beim Menschen). Da NexoBrid von den Muttertieren schlecht vertragen wurde, wird diesen Studien keine Relevanz für die Bewertung des Risikos beim Menschen beigemessen.
Toxizitätsanalyse bei Jungtieren
Die toxikologischen Ergebnisse des Arzneimittels bei jungen Minischweinen waren mit denen bei erwachsenen vergleichbar. Die topische Anwendung des Arzneimittels (0,09 g/g) bei jungen Schweinen (im Alter von 2 Monaten) führte nach der Anwendung auf Verbrennungen in einer für die Anwendung beim Menschen relevanten Rezeptur und Dosierung zu keinen nennenswerten lokalen oder systemischen toxikologischen Wirkungen78. Nach wiederholten intravenösen Injektionen von Dosen von 4, 8 und 12 mg/kg alle drei Tage an junge Minischweine wurden nach der fünften Dosis am Tag 10 in allen Dosisgruppen entsprechende Veränderungen beobachtet. Die Beobachtungen umfassten Krämpfe und Hautrötungen sowie eine verminderte Aktivität, Atembeschwerden und Ataxie bei einigen Tieren.
An Tag 10 nach der Verabreichung der Dosis wurde bei den behandelten Tieren eine Tendenz zur Verlängerung der QT- und QTc-Intervalle beobachtet. Diese Werte wurden nach den oben beschriebenen wichtigen klinischen Beobachtungen ermittelt.
Lokale Toxizität
NexoBrid verursachte nach Applikation auf intakte Haut von Minischweinen keine signifikanten Reizungen, während es nach Applikation auf beschädigte (abgeschürfte) Haut schwere Reizungen und Schmerzen hervorrief.
Sonstige HinweiseInkompatibilitäten
Vor der Applikation von NexoBrid müssen auf der Wunde befindliche topisch applizierte Arzneimittel (wie Sulfadiazin-Silber oder Povidon-Iod) entfernt werden und die Wunde muss gereinigt werden. Reste von antibakteriellen Arzneimitteln können die Wirkung von NexoBrid herabsetzen, da die Wirksamkeit reduziert wird.
Das Arzneimittel darf nicht mit anderen Arzneimitteln gemischt werden.
Haltbarkeit
Das Arzneimittel darf nur bis zu dem auf der Packung und den Fläschchen mit „EXP“ bezeichneten Datum verwendet werden.
Unter mikrobiologischen Gesichtspunkten und angesichts der Tatsache, dass die enzymatische Aktivität des Präparats nach der Mischung fortlaufend abnimmt, soll das rekonstituierte Präparat unmittelbar nach der Zubereitung verabreicht werden (innerhalb von 15 Minuten).
Besondere Lagerungshinweise
Kühl lagern und transportieren (2 °C – 8 °C).
Aufrecht lagern, damit das Gel am Boden der Flasche verbleibt. In der Originalverpackung aufbewahren, um den Inhalt vor Licht zu schützen.
Nicht einfrieren.
Ausser Reichweite von Kindern aufbewahren.
Hinweise für die Handhabung
Es gibt Berichte über eine berufliche Exposition mit Bromelain, die zu einer Sensibilisierung führte. Die Sensibilisierung wurde möglicherweise durch Inhalation von Bromelain-Pulver verursacht. Mögliche allergische Reaktionen auf Bromelain umfassen anaphylaktische Reaktionen und andere Reaktionen vom Soforttyp, die sich als Bronchospasmus, Angioödem, Urtikaria und Schleimhaut- bzw. gastrointestinale. Beim Einmischen von NexoBrid-Pulver in das Gel ist eine sachgemässe Handhabung, einschliesslich des Tragens von Handschuhen und Schutzkleidung sowie einer Augenschutzbrille und einer chirurgischen Maske, erforderlich (siehe auch Rubrik "Warnhinweise / Vorsichtsmassnahmen" ). Das Pulver nicht einatmen.
Versehentlicher Kontakt mit den Augen muss vermieden werden. Sollte NexoBrid in die Augen gelangen, müssen die betroffenen Augen mindestens 15 Minuten lang mit viel Wasser gespült werden. Bei Hautkontakt muss NexoBrid mit Wasser abgewaschen werden.
Zubereitung von NexoBrid-Gel (Mischen von Pulver und Gel)
-NexoBrid-Pulver und das Gel sind steril. Das Einmischen des Pulvers in das Gel muss mit aseptischer Technik erfolgen.
-Beim Öffnen der Durchstechflasche mit dem Pulver vorsichtig den Aluminium-Schnappdeckel abnehmen und den Gummistopfen entfernen. Das Pulver nicht einatmen. Geeignete Massnahmen beim Umgang mit dem Präparat für das Wund-Debridement (u. a. das Tragen von Handschuhen, Schutzkittel und Schutzmaske) sind erforderlich.
-Beim Öffnen der Flasche mit dem Gel ist darauf zu achten, dass sich der manipulationssichere Ring vom Flaschendeckel trennt. Sollte der manipulationssichere Ring bereits vor dem Öffnen vom Flaschendeckel getrennt gewesen sein, muss die Flasche mit dem Gel verworfen und eine andere, neue Gel-Flasche verwendet werden.
-Das Pulver wird dann in die dazugehörige Flasche mit dem Gel gegeben.
-Pulver und Gel müssen gründlich gemischt werden, bis eine homogene, leicht hellbraune bis bräunliche Mischung entsteht. In der Regel müssen Pulver und Gel hierfür 1 bis 2 Minuten gemischt werden.
-Das Gel sollte am Bett des Patienten zubereitet werden.
Nicht verwendetes Arzneimittel oder Abfallmaterial ist entsprechend den nationalen Anforderungen zu beseitigen.
Zulassungsnummer68012 (Swissmedic)
PackungenJede Packung enthält eine Durchstechflasche mit Pulver und eine Flasche mit Gel.
NexoBrid 5 g [A]
ZulassungsinhaberinTriskel Integrated Services, Le Grand-Saconnex-Genève
Stand der InformationApril 2025
|