CompositionPrincipes actifs
Insuline dégludec 100 unités/ml* et liraglutide 3.6 mg/ml* (* produits par génie génétique dans Saccharomyces cerevisiae).
Excipients
Phénol, glycérol, acétate de zinc dihydraté, acide chlorhydrique, hydroxyde de sodium (contient au maximum 0.0416 mmol/ml de sodium), eau pour préparations injectables.
Forme pharmaceutique et quantité de principe actif par unitéXultophy est une solution isotonique incolore et limpide.
1 ml de solution contient 100 unités d'insuline dégludec et 3.6 mg de liraglutide.
Un stylo prérempli contient 3 ml, ce qui correspond à 300 unités d'insuline dégludec et 10.8 mg de liraglutide.
Indications/Possibilités d’emploiXultophy est utilisé en association avec des hypoglycémiants pour traiter les adultes atteints de diabète sucré de type 2 insuffisamment contrôlé, en complément d'un régime alimentaire et d'une activité physique (pour les résultats des études sur les associations étudiées dans les essais cliniques et les effets sur les événements cardiovasculaires, voir "Propriétés/Effets" ).
Posologie/Mode d’emploiXultophy est une association d'insuline dégludec et de liraglutide à administrer par injection sous-cutanée une fois par jour. Xultophy doit être administré une fois par jour, si possible toujours à la même heure.
La dose de Xultophy doit être ajustée en fonction des besoins individuels du patient. Il est recommandé d'optimiser le contrôle glycémique en ajustant la dose en fonction de la glycémie à jeun.
Comme pour toutes les insulines, un ajustement de la dose peut être nécessaire si le patient augmente son activité physique, modifie son régime alimentaire habituel ou en cas de maladies concomitantes.
Il est conseillé aux patients qui oublient une dose de se l'administrer dès qu'ils s'en rendent compte, puis de reprendre leur schéma posologique habituel à raison d'une fois par jour. Toutefois, un intervalle minimum de 8 heures entre deux injections devra toujours être respecté. Cet intervalle s'applique aussi quand il n'est pas possible d'administrer la dose au moment habituel de la journée.
L'unité de dosage de Xultophy est exprimée en doses unitaires. Une dose unitaire contient 1 unité d'insuline dégludec et 0.036 mg de liraglutide. Avec le stylo prérempli, il est possible d'administrer de 1 à 50 doses unitaires en une injection, par paliers de 1 unité. La dose journalière maximale de Xultophy est de 50 doses unitaires (50 unités d'insuline dégludec/1.8 mg de liraglutide). L'affichage des doses sur le stylo indique le nombre de doses unitaires.
Utilisation en association avec des hypoglycémiants oraux
La dose initiale journalière recommandée de Xultophy est de 10 doses unitaires (10 unités d'insuline dégludec/0.36 mg de liraglutide).
Xultophy peut être ajouté à un traitement existant par metformine ou à un traitement par metformine et une sulfonylurée. Si Xultophy est administré en association avec un traitement par sulfonylurée, il faut envisager de réduire la dose de sulfonylurée (voir sous "Mises en garde et précautions" ).
Passage d'un agoniste du récepteur du GLP-1
Avant le début d'un traitement avec Xultophy, l'agoniste du récepteur du GLP-1 devrait être arrêté. Lors du passage d'un agoniste du récepteur du GLP-1, la dose initiale recommandée de Xultophy est de 16 doses unitaires (16 unités d'insuline dégludec/0.6 mg de liraglutide) (voir "Mises en garde et précautions" et "Propriétés/Effets" ). La dose initiale recommandée ne doit pas être dépassée. Lors du passage d'un agoniste de longue durée d'action du récepteur du GLP-1 (p.ex. lors d'une administration hebdomadaire), l'effet prolongé devrait être pris en compte. Le traitement avec Xultophy devrait être instauré au moment où la dose suivante de l'agoniste du récepteur du GLP-1 serait administrée. Une surveillance étroite de la glycémie est recommandée au cours de la transition et dans les semaines qui suivent.
Passage de toute insulinothérapie contenant une composante d'insuline basale
Tout traitement par un autre régime insulinique doit être arrêté avant de commencer un traitement par Xultophy. En cas de passage de toute autre insulinothérapie contenant une composante d'insuline basale, la dose initiale recommandée de Xultophy est de 16 doses unitaires (16 unités d'insuline dégludec et 0.6 mg de liraglutide) (voir "Mises en garde et précautions" et "Propriétés/Effets" ). La dose initiale recommandée ne devra pas être dépassée. Une surveillance étroite de la glycémie est recommandée au cours de la transition et dans les semaines qui suivent.
Si la dose nécessaire pour éviter toute hypoglycémie doit être abaissée au-dessous de 16 doses unitaires, l'indication du traitement combiné devra être reconsidérée.
Instructions spéciales pour la posologie
Patients âgés (≥65 ans)
Xultophy peut être utilisé chez les patients âgés. Le contrôle de la glycémie doit être intensifié et la dose individuelle d'insuline doit être adaptée (voir sous "Pharmacocinétique" ).
Patients présentant des troubles de la fonction rénale
En cas d'utilisation de Xultophy chez des patients présentant une insuffisance rénale légère, modérée ou sévère, le contrôle de la glycémie doit être intensifié et la dose d'insuline doit être adaptée au cas par cas. L'utilisation de Xultophy chez des patients présentant une insuffisance rénale au stade terminal ne peut pas être recommandée (voir sous "Pharmacocinétique" ).
Patients présentant des troubles de la fonction hépatique
En cas d'utilisation de Xultophy chez des patients présentant une insuffisance hépatique, le contrôle de la glycémie doit être intensifié et la dose d'insuline doit être adaptée de façon individuelle (voir sous "Pharmacocinétique" ).
Enfants et adolescents
L'utilisation de Xultophy chez les enfants et les adolescents de moins de 18 ans est déconseillée. Aucune étude concernant les patients de moins de 18 ans n'a été menée.
Mode d'administration
Xultophy est réservé à l'administration sous-cutanée. Xultophy ne doit pas être administré par voie intraveineuse ou intramusculaire.
Xultophy est administré par voie sous-cutanée par injection dans la cuisse, le haut du bras ou la paroi abdominale. Le site d'injection devrait être changé pour chaque injection au sein d'une même région du corps, afin de diminuer le risque de lipodystrophie et d'amyloïdose cutanée (voir "Mises en garde et précautions" et "Effets indésirables" ). Pour connaître les autres remarques concernant la manipulation, voir "Remarques concernant la manipulation" dans la rubrique "Remarques particulières" .
Contre-indicationsHypersensibilité à l'un des principes actifs ou aux deux, ou à l'un des excipients selon la composition.
Mises en garde et précautionsXultophy ne doit être utilisé ni chez des patients présentant un diabète de type 1, ni dans le traitement de l'acidocétose diabétique.
Hypoglycémie
Pour le diabétique traité à l'insuline, il existe en principe un risque d'hypoglycémie légère ou sévère. Celle-ci peut atténuer la capacité de concentration et est notamment susceptible de compromettre la sécurité du patient lors de la conduite d'un véhicule ou de l'utilisation de machines. Ces phénomènes se présentent en particulier au début du traitement, lors d'un changement de préparation ou de repas irréguliers, ainsi que plus généralement quand le métabolisme est mal ou non équilibré, ce qui peut causer de fortes fluctuations de la glycémie et, entre autres, une hypoglycémie. Il faut rappeler au patient que la consommation d'alcool accroît encore ce risque (par inhibition de la gluconéogenèse hépatique). En outre, les patients ayant déjà été atteints d'hypoglycémie sévère présentent un risque accru de récidive. Afin que le patient soit en mesure de prévenir suffisamment tôt une hypoglycémie, il y a lieu de lui recommander de se munir constamment de sucre de raisin, de sucre en morceaux ou d'un équivalent. Il doit aussi porter sur lui sa carte de diabétique.
Une hypoglycémie peut survenir quand la dose de Xultophy est supérieure à ce qui est nécessaire. L'omission d'un repas ou un exercice physique important non prévu peut entraîner une hypoglycémie.
En cas d'association avec une sulfonylurée, il est possible de réduire le risque d'hypoglycémie en diminuant la dose de sulfonylurée.
Les maladies concomitantes des reins, du foie ou les maladies qui affectent les surrénales, l'hypophyse ou la glande thyroïde peuvent nécessiter des modifications de la dose de Xultophy.
Chez les patients présentant un état métabolique nettement amélioré (p.ex. en raison d'une insulinothérapie intensifiée), il arrive, dans certaines circonstances, que les symptômes annonciateurs de l'hypoglycémie soient modifiés. Ces patients doivent être prévenus de ce problème. Chez les patients présentant un diabète depuis longtemps, les symptômes d'alerte habituels pourraient disparaître.
Comme pour les autres insulines basales, l'effet prolongé de Xultophy peut retarder la récupération après une hypoglycémie.
Les β-bloquants peuvent affaiblir ou masquer les symptômes d'alerte habituels de l'hypoglycémie. La consommation de marijuana peut éventuellement provoquer une aggravation de la tolérance au glucose. (D'autres substances illégales n'ont pas été testées; pour les autres interactions, voir "Interactions" ).
Hyperglycémie
Une dose insuffisante et/ou l'interruption du traitement antidiabétique peut mener à une hyperglycémie et, dans certains cas, à un coma hyperosmolaire. Dans le cas où Xultophy devait être arrêté, il faudrait s'assurer que des instructions pour une autre médication antidiabétique soient suivies. De plus, les maladies concomitantes, en particulier les infections, peuvent entraîner une hyperglycémie et, de ce fait, augmenter les besoins en traitement antidiabétique.
En général, les premiers symptômes de l'hyperglycémie apparaissent progressivement, en quelques heures ou quelques jours. Il s'agit d'une sensation de soif, de mictions d'un volume important, de nausées, de vomissements, de fatigue, de sécheresse et de rougeurs cutanées, de sécheresse buccale, de perte d'appétit et d'odeur acétonique de l'haleine. Il faut indiquer au patient de consulter le médecin immédiatement, dès les premiers signes d'une hyperglycémie.
L'administration d'une insuline d'action rapide est recommandée dans les situations d'hyperglycémie sévère.
Les réactions hyperglycémiques non traitées peuvent conduire à un coma hyperosmolaire/une acidocétose diabétique, potentiellement mortels.
Affections de la peau et du tissu sous-cutané
Les patients doivent avoir pour instruction de changer régulièrement de site d'injection afin de réduire le risque de lipodystrophie et d'amyloïdose cutanée. Il existe un risque potentiel de retard de l'absorption de l'insuline et de mauvais contrôle de la glycémie après des injections d'insuline aux endroits où se produisent ces réactions. Il a été rapporté que le changement soudain du site d'injection vers une zone non affectée provoquait une hypoglycémie. Après un changement de site d'injection d'une zone affectée à une zone non affectée, il est recommandé de surveiller la glycémie et d'envisager un ajustement de la dose des médicaments antidiabétiques.
Association de la pioglitazone et de médicaments contenant de l'insuline
Des cas d'insuffisance cardiaque ont été rapportés lorsque la pioglitazone était utilisée en association avec des médicaments contenant de l'insuline, en particulier chez les patients présentant des facteurs de risque de survenue d'une insuffisance cardiaque. Ce doit être pris en compte si un traitement associant la pioglitazone et Xultophy est envisagé. Si une telle association est instaurée, il sera nécessaire de surveiller, chez ces patients, la survenue de signes et de symptômes d'insuffisance cardiaque, de prise de poids et d'œdème. La pioglitazone devra être arrêtée si une aggravation des symptômes d'insuffisance cardiaque survient.
Affections oculaires
Une intensification du traitement par insuline, entrant dans la composition de Xultophy, avec une amélioration soudaine de l'équilibre glycémique, peut être associée à une aggravation transitoire de la rétinopathie diabétique, tandis que l'amélioration de l'équilibre glycémique à long terme diminue le risque de progression de la rétinopathie diabétique.
Anticorps
L'administration de Xultophy peut induire la formation d'anticorps contre l'insuline dégludec et/ou le liraglutide. Dans de rares cas, la présence de tels anticorps peut nécessiter un ajustement de la dose de Xultophy afin de corriger une tendance à l'hyperglycémie ou à l'hypoglycémie. Très peu de patients ont développé des anticorps spécifiques contre l'insuline dégludec, des anticorps présentant une réaction croisée contre l'insuline humaine ou des anticorps contre le liraglutide lors du traitement par Xultophy. La formation d'anticorps n'a pas été associée à une diminution de l'efficacité de Xultophy.
Pancréatite
Une pancréatite aiguë a été observée avec l'utilisation d'agonistes des récepteurs du GLP-1. Il faut indiquer aux patients quels sont les symptômes caractéristiques d'une pancréatite aiguë. Si l'on suspecte une pancréatite, Xultophy doit être arrêté; si une pancréatite aiguë est confirmée, le traitement par Xultophy ne doit pas être repris. Une augmentation ponctuelle des enzymes pancréatiques au cours du traitement par Xultophy (sans symptômes caractéristiques) ne traduit pas nécessairement la présence d'une pancréatite aiguë (voir sous "Effets indésirables" ).
Maladies thyroïdiennes
Dans des études cliniques portant sur les agonistes du récepteur du GLP-1, dont le liraglutide, des événements indésirables touchant la thyroïde (p.ex. goitre), en particulier chez les patients présentant une affection préexistante de la thyroïde, ont été rapportés. C'est pourquoi Xultophy devrait être utilisé avec prudence chez ces patients.
Aspiration liée à une anesthésie générale ou à une sédation profonde
Des cas d'aspiration pulmonaire ont été rapportés chez des patients recevant des agonistes des récepteurs du GLP-1 soumis à une anesthésie générale ou à une sédation profonde, qui avaient pourtant bien respecté les recommandations de jeûne préopératoire. Par conséquent, le risque accru de contenu résiduel gastrique dû au retard de vidange gastrique doit être pris en considération avant d'effectuer des interventions sous anesthésie générale ou sédation profonde.
Maladie inflammatoire de l'intestin et gastroparèse diabétique
On ne dispose d'aucune expérience avec Xultophy chez des patients atteints d'une maladie inflammatoire de l'intestin et de gastroparèse diabétique. C'est pourquoi l'emploi de Xultophy chez ces patients ne peut pas être recommandé.
Déshydratation
Des signes et symptômes de déshydratation, dont des troubles de la fonction rénale et une insuffisance rénale aiguë, ont été signalés dans des études cliniques portant sur les agonistes du récepteur du GLP-1, dont le liraglutide, un composant de Xultophy. Les patients traités par Xultophy doivent être informés du risque de déshydratation dû aux effets indésirables gastro-intestinaux et prendre les mesures nécessaires pour éviter une telle déshydratation.
Prévention d'erreurs de médication
Le patient doit être instruit du fait qu'il doit contrôler l'étiquette du stylo avant chaque injection, afin d'éviter une confusion accidentelle entre Xultophy et d'autres médicaments antidiabétiques à injecter.
Groupes de patients non étudiés
Le remplacement d'une dose d'insuline basale <20 et >50 unités par Xultophy n'a pas été étudié.
On ne dispose que d'expériences limitées chez les patients présentant une insuffisance cardiaque de classe I-II selon la New York Heart Association (NYHA) et Xultophy doit par conséquent être utilisé avec précaution chez ses patients. On ne dispose d'aucune expérience clinique chez les patients présentant une insuffisance cardiaque de classe IV de la New York Heart Association (NYHA), c'est pourquoi l'utilisation de Xultophy est par conséquent déconseillée chez ces patients.
Autres composants
Ce médicament contient moins de 1 mmol de sodium (23 mg) par dose, c.-à-d. qu'il est essentiellement "sans sodium" .
InteractionsInteractions pharmacodynamiques
Aucune étude sur les interactions n'a été menée concernant Xultophy.
Certains médicaments peuvent influencer le métabolisme du glucose.
1. Le besoin en Xultophy peut être réduit en présence de substances qui améliorent l'efficacité de l'insuline (en augmentant la sensibilité à l'insuline), qui stimulent la sécrétion insulinique, qui inhibent la gluconéogenèse hépatique ou qui modifient l'absorption intestinale du glucose. En présence d'une quantité inchangée d'insuline, il existe de ce fait un risque accru d'hypoglycémie en cas de prise/d'utilisation simultanée de:
antidiabétiques oraux; inhibiteurs de l'ECA (p.ex. captopril et énalapril); antiarythmiques tels que le disopyramide; α-bloquants et clonidine; ISRS; fenfluramine; IMAO; antidépresseurs tricycliques; salicylates et (rarement) autres AINS; fibrates; tétracyclines; pentamidine (hypoglycémie; parfois suivie d'une hyperglycémie); antimalariens (quinine, chloroquine, méfloquine); sulfonamides (p.ex. cotrimoxazol); cimétidine et ranitidine.
2. Le besoin en Xultophy peut être augmenté lors de la prise/l'utilisation des substances ou groupes de substances suivants:
contraceptifs oraux et autres composés estrogéniques ou progestatifs; corticostéroïdes et ACTH; hormone de croissance (somatotropine); danazol; hormones thyroïdiennes; sympathicomimétiques (particulièrement les β2-sympathicomimétiques tels que la ritodrine, le salbutamol, la terbutaline, mais également certains sympathicomimétiques α-sélectifs ainsi que non sélectifs comme l'épinéphrine); diazoxide; acide nicotinique et ses dérivés; chlorpromazine (surtout à doses élevées) et autres dérivés de la phénothiazine; diurétiques (p.ex. diurétiques à base de thiazide, d'indapamide et de furosémide); substances antirétrovirales; substances immunosuppressives (ciclosporine, tacrolimus, sirolimus), antipsychotiques atypiques.
3. L'efficacité de Xultophy peut être accrue ou réduite, en fonction de la dose, lors de l'utilisation des substances suivantes:
dérivés du lanréotide, dérivés d'octréotide, dérivés salicylés, sels de lithium (rarement).
Les β-bloquants peuvent aggraver l'insulinorésistance mais aussi, dans certains cas, entraîner une hypoglycémie. Il existe en outre un risque de diminution ou de masquage des symptômes d'alerte de l'hypoglycémie.
L'alcool peut intensifier ou réduire l'effet hypoglycémiant de Xultophy.
Interactions pharmacocinétiques
Évaluation in vitro des interactions médicamenteuses avec l'insuline dégludec
L'affinité de l'insuline dégludec pour l'albumine sérique humaine à des taux thérapeutiques/physiologiques pertinents n'a pas été influencée par les médicaments se liant habituellement aux protéines plasmatiques.
Évaluation in vitro des interactions médicamenteuses avec le liraglutide
Le liraglutide a montré un très faible potentiel d'interactions pharmacocinétiques avec d'autres principes actifs associés au cytochrome P450 (CYP) et à une liaison aux protéines plasmatiques.
Évaluation in vivo des interactions médicamenteuses avec le liraglutide
Le léger retard de la vidange gastrique, dû au liraglutide, pourrait être susceptible d'influencer l'absorption de médicaments administrés par voie orale de façon concomitante. Les études d'interaction ne montrent pas de retard clinique significatif de l'absorption.
Quelques patients traités par liraglutide souffrent de diarrhées sévères. Les diarrhées peuvent altérer l'absorption de médicaments administrés par voie orale en même temps.
Anticoagulants
Aucune étude d'interaction n'a été menée. Une interaction cliniquement significative avec des principes actifs qui présentent une faible solubilité ou un indice thérapeutique étroit, par exemple la warfarine, ne peut pas être exclue. Chez les patients utilisant des anticoagulants, il est recommandé d'intensifier la surveillance du RIN (Rapport International Normalisé) au début d'un traitement par Xultophy.
Paracétamol
Après l'administration d'une dose unique de 1000 mg de paracétamol, le liraglutide n'a provoqué aucune modification de l'exposition systémique du paracétamol. La Cmax du paracétamol a connu une diminution de 31% et le tmax moyen a été retardé de 15 minutes. Aucune adaptation de la dose n'est nécessaire en cas d'administration concomitante de paracétamol.
Atorvastatine
Après l'administration d'une dose unique de 40 mg d'atorvastatine, le liraglutide n'a provoqué aucune modification cliniquement significative de l'exposition systémique de l'atorvastatine. Par conséquent, aucune adaptation de la dose d'atorvastatine n'est nécessaire lorsqu'elle est administrée en même temps que le liraglutide. La Cmax de l'atorvastatine, administrée en même temps que le liraglutide, a connu une diminution de 38% et le tmax moyen a été retardé, passant d'une à trois heures.
Griséofulvine
Après l'administration d'une dose unique de 500 mg de griséofulvine, le liraglutide n'a provoqué aucune modification de l'exposition systémique de la griséofulvine. La Cmax de la griséofulvine a connu une augmentation de 37% et le tmax moyen est resté inchangé. Aucune adaptation de la dose de griséofulvine et d'autres médicaments à faible solubilité et à haute perméabilité n'est nécessaire.
Digoxine
L'administration concomitante de liraglutide et d'une dose unique de 1 mg de digoxine a provoqué une diminution de 16% de l'AUC de la digoxine. La Cmax de la digoxine a diminué de 31% et le tmax moyen a été retardé d'une heure à une heure et demie. Ces résultats indiquent qu'aucune adaptation de la dose de digoxine n'est nécessaire.
Lisinopril
L'administration concomitante de liraglutide et d'une dose unique de 20 mg de lisinopril a provoqué une diminution de 15% de l'AUC du lisinopril. La Cmax du lisinopril a diminué de 27% et le tmax moyen a été retardé de six à huit heures. Ces résultats indiquent qu'aucune adaptation de la dose de lisinopril n'est nécessaire.
Contraceptifs oraux
Après l'administration d'une dose unique d'un contraceptif oral, le liraglutide a provoqué une baisse de 12% et 13% de la Cmax de l'éthinylestradiol et du lévonorgestrel. Le tmax des deux principes actifs a été retardé d'une heure et demie. On n'a relevé aucun effet cliniquement significatif sur l'exposition systémique de l'éthinylestradiol et du lévonorgestrel. On peut donc s'attendre à ce que l'effet contraceptif ne soit pas altéré en cas d'administration concomitante du liraglutide.
Grossesse, allaitementGrossesse
Il n'existe pas d'expérience clinique concernant l'utilisation de Xultophy, de l'insuline dégludec ou du liraglutide chez la femme enceinte. La prise de Xultophy doit être interrompue chez les femmes qui souhaitent tomber enceintes ou qui sont tombées enceintes lors du traitement.
Les études sur la reproduction animale n'ont mis en évidence aucune différence entre l'insuline dégludec et l'insuline humaine en termes d'embryotoxicité ou d'effets tératogènes (voir sous "Données précliniques" ). Dans les études animales portant sur le liraglutide, on a observé une toxicité sur la reproduction (voir sous "Données précliniques" ). Le risque potentiel pour l'être humain n'est pas connu.
Allaitement
Il n'existe pas d'expérience clinique concernant l'utilisation de Xultophy pendant l'allaitement. On ignore si l'insuline dégludec ou le liraglutide sont excrétés dans le lait maternel.
Chez les rats, l'insuline dégludec était sécrétée dans le lait maternel; la concentration dans le lait maternel était plus faible que dans le plasma. Des études d'expérimentation animale ont montré que de petites quantités de liraglutide et de métabolites de structure similaire passent dans le lait maternel. En raison du manque d'expérience, Xultophy ne doit pas être utilisé pendant l'allaitement.
Fertilité
Il n'existe pas d'expérience clinique avec Xultophy en ce qui concerne la fertilité. Si la patiente prévoit une grossesse, le traitement avec Xultophy doit être interrompu.
Effet sur l’aptitude à la conduite et l’utilisation de machinesLes capacités de concentration et de réaction du patient pourraient être altérées suite à une hypoglycémie. Ceci peut constituer un risque dans les situations où ces facultés sont indispensables (p.ex. la conduite automobile ou l'utilisation de machines).
Les patients doivent être informés des précautions à prendre afin d'éviter toute hypoglycémie pendant la conduite de véhicules. Ceci est particulièrement important chez les patients peu ou mal familiarisés avec les signes précurseurs d'hypoglycémie ou sujets à de fréquents épisodes hypoglycémiques. Dans de telles circonstances, l'aptitude à conduire des véhicules doit être réévaluée.
Effets indésirablesRésumé du profil de sécurité:
Xultophy, qui a été testé dans le programme de développement clinique chez environ 1'900 patients, n'a montré aucune augmentation de l'accumulation des effets indésirables spécifiques par rapport aux deux composants isolés insuline dégludec et liraglutide.
Les effets indésirables les plus fréquemment rapportés lors du traitement par Xultophy étaient l'hypoglycémie et les événements indésirables gastro-intestinaux (voir sous "Description de certains effets indésirables" ).
Liste tabulée des effets indésirables
Les effets indésirables répertoriés dans le Tableau 1 ci-dessous sont rassemblés selon la classe de systèmes d'organes MedDRA et les fréquences. Les fréquences des effets indésirables ont été définies de la manière suivante: très fréquents (≥1/10), fréquents (≥1/100, <1/10), occasionnels (≥1/1'000, <1/100), rares (≥1/10'000, <1/1'000), très rares (<1/10'000) et fréquence inconnue (ne peut être estimée sur la base des données disponibles).
Tableau 1: Effets indésirables dans les études contrôlées de phase III et dans les rapports post-marketing
Classe de systèmes Fréquence Effet indésirable
d'organes selon
MedDRA
Affections du Occasionnel Urticaire
système immunitaire
Occasionnel Hypersensibilité
Rare* Réaction anaphylactique
Rare* Angio-œdème
Troubles du métaboli Très fréquent Hypoglycémie
sme et de la nutriti
on
Fréquent Perte d'appétit
Occasionnel Déshydratation
Affections du Fréquent Vertiges†
système nerveux
Occasionnel Dysgueusie
Affections gastro-in Fréquent Nausées, diarrhées, vomissements,
testinales constipation, dyspepsie, gastrite,
douleurs abdominales, reflux
gastro-œsophagien
Occasionnel Éructation, ballonnements
Très rare* Pancréatite (y compris pancréatite
nécrosante)
Fréquence inconnue Retard de la vidange gastrique†
Obstruction intestinale†a
Affections hépatobil Occasionnel Lithiase biliaire, cholécystite
iaires
Non connu Augmentation des concentrations
d’enzymes hépatiques†,
hyperbilirubinémie†, cholestase†,
hépatite†
Affections de la Occasionnel Éruption cutanée, prurit, lipodystrophie
peau et du tissu acquise
sous-cutané
Non connu Amyloïdose cutanée†
Troubles généraux Fréquent Réactions au site d'injection
et anomalies au
site d'administratio
n
Cas isolés Œdème périphérique
Examens Fréquent Lipase augmentée
Fréquent Amylase augmentée
Occasionnel Hausse de la fréquence cardiaque
* Fréquence basée sur des études contrôlées de phase 3a à long terme avec Victoza, car aucun rapport d'études contrôlées de phase 3 avec Xultophy n'est disponible.
† Effets indésirables à un médicament à partir des rapports de post-commercialisation
a Terme groupé couvrant les événements indésirables obstruction intestinale, iléus et obstruction de l'intestine grêle
Description de certains effets indésirables
Hypoglycémie
L'hypoglycémie peut survenir lorsque la dose de Xultophy est trop élevée par rapport aux besoins. L'hypoglycémie sévère peut entraîner une perte de connaissance et/ou des convulsions et peut causer une altération transitoire ou définitive des fonctions cérébrales, voire le décès. Les symptômes de l'hypoglycémie surviennent habituellement de manière soudaine. Ils peuvent inclure: sueurs froides, pâleur et froideur cutanées, épuisement, nervosité ou tremblement, anxiété, fatigue ou faiblesse inhabituelles, confusion, difficultés de concentration, somnolence, sensation de faim excessive, troubles visuels, céphalées, nausées et palpitations.
Réactions allergiques
Des cas de réactions allergiques (qui se traduisent par des signes et symptômes tels que de l'urticaire, une éruption cutanée, un prurit et/ou un gonflement de la langue et du visage) ont été rapportés en association avec l'insuline dégludec et le liraglutide, qui sont les deux composants de Xultophy.
Seuls quelques cas de réactions anaphylactiques accompagnées de symptômes supplémentaires, comme une chute de la tension artérielle, des palpitations, une détresse respiratoire et un œdème, ont été signalés après la commercialisation du liraglutide. Les réactions anaphylactiques peuvent être potentiellement mortelles.
Affections gastro-intestinales
Des effets indésirables gastro-intestinaux comme des nausées, des diarrhées, des vomissements, une constipation, une dyspepsie, une gastrite, des douleurs abdominales, une éructation, des flatulences, un reflux gastro-œsophagien, des ballonnements et une perte d'appétit ont été rapportés chez des patients sous Xultophy. Ces effets indésirables gastro-intestinaux peuvent être fréquents au début d'un traitement par Xultophy mais deviennent généralement de plus en plus rares dans les jours ou les semaines qui suivent quand le traitement est poursuivi.
Lipodystrophie
Une lipodystrophie (notamment lipohypertrophie, lipoatrophie) peut survenir au niveau du site d'injection. Une rotation continue des sites d'injection au sein d'une même région peut aider à diminuer le risque de développer ces réactions.
Réactions au site d'injection
Des réactions au site d'injection (notamment hématomes au site d'injection, douleurs, hémorragies, érythèmes, nodules, gonflements, décolorations, prurit et chaleur) se sont produites chez des patients traités par Xultophy. Ces réactions sont habituellement légères et transitoires et disparaissent généralement lors de la poursuite du traitement.
Troubles de la peau et du tissu sous-cutané
La lipodystrophie (y compris la lipohypertrophie, la lipoatrophie) et l'amyloïdose cutanée peuvent se produire au site d'injection et retarder l'absorption locale de l'insuline. Le changement régulier du site d'injection dans la zone d'injection respective peut contribuer à réduire ou à prévenir ces réactions (voir "Mises en garde et précautions" ).
Augmentation de la fréquence cardiaque
Dans les études cliniques avec Xultophy ainsi que dans les études cliniques avec le liraglutide, une augmentation moyenne de la fréquence cardiaque de 2 à 3 battements par minute a été observée par rapport aux valeurs initiales. Une étude sur la sécurité cardiovasculaire à grande échelle (étude LEADER) a montré que l'augmentation de la fréquence cardiaque due au liraglutide n'augmente pas le risque d'événements cardiovasculaires.
L'annonce d'effets secondaires présumés après l'autorisation est d'une grande importance. Elle permet un suivi continu du rapport bénéfice-risque du médicament. Les professionnels de santé sont tenus de déclarer toute suspicion d'effet secondaire nouveau ou grave via le portail d'annonce en ligne ElViS (Electronic Vigilance System). Vous trouverez des informations à ce sujet sur www.swissmedic.ch.
SurdosageLes données disponibles concernant le surdosage de Xultophy sont limitées.
Une hypoglycémie peut survenir quand la dose de Xultophy est supérieure à ce qui est nécessaire.
-Les épisodes d'hypoglycémie légère peuvent être traités par l'administration orale de glucose ou d'autres aliments contenant du sucre. On conseille donc aux patients d'avoir toujours sur eux des aliments contenant du glucose.
-Les épisodes d'hypoglycémie sévère, au cours desquels le patient n'est pas capable de s'auto-traiter, peuvent être traités par administration intramusculaire, sous-cutanée ou intranasale de glucagon par une personne formée à cet effet, ou par administration intraveineuse de glucose par le personnel médical. Si le patient ne répond pas au glucagon dans un délai de 10 à 15 minutes, du glucose devra être administré par voie intraveineuse. Dès que le patient a repris connaissance, une prise orale de glucides est recommandée afin de prévenir une rechute.
Propriétés/EffetsCode ATC
A10AE56
Mécanisme d'action
Xultophy est un médicament combiné composé d'insuline dégludec et de liraglutide, possédant un mécanisme d'action complémentaire permettant d'améliorer la glycémie.
L'insuline dégludec est une insuline basale qui forme des multi-hexamères solubles après injection sous-cutanée, ce qui entraîne un dépôt à partir duquel l'insuline dégludec est lentement absorbée en continu dans la circulation. Cela entraîne un effet hypoglycémiant faible et stable de l'insuline dégludec, qui ne varie que très peu d'un jour à l'autre.
L'insuline dégludec se lie spécifiquement au récepteur de l'insuline humaine et a donc les mêmes effets pharmacologiques que l'insuline humaine.
L'effet hypoglycémiant de l'insuline dégludec est dû à la liaison de l'insuline aux récepteurs des cellules musculaires et adipeuses, facilitant ainsi l'assimilation du glucose, et à l'inhibition simultanée de la production hépatique de glucose.
Le liraglutide est un "glucagon-like peptide-1" (GLP-1) présentant une homologie de séquence de 97% avec le GLP-1 humain, qui se lie au récepteur du GLP-1 (GPL-1R) et qui l'active. Dans le cas de l'administration sous-cutanée, le délai existant avant le pic d'action est dû à trois mécanismes: l'auto-association, qui entraîne un ralentissement de la résorption, la liaison à l'albumine, ainsi que l'augmentation de la stabilité enzymatique par rapport aux enzymes dipeptidyl-peptidase-4 (DPP-4) et endopeptidase neutre (NEP), qui provoque un allongement de la demi-vie plasmatique.
L'effet du liraglutide est médié par une interaction spécifique avec les récepteurs du GLP-1, qui mène à une augmentation du taux d'AMPc. Le liraglutide stimule la sécrétion d'insuline de façon glucose-dépendante. Le liraglutide diminue également la sécrétion excessive de glucagon de façon glucose-dépendante. Par conséquent, en cas de glycémie élevée, la sécrétion d'insuline est stimulée et la sécrétion de glucagon est freinée. À l'inverse, en cas d'hypoglycémie, le liraglutide diminue la sécrétion d'insuline mais n'a pas d'effet sur la sécrétion de glucagon. Le mécanisme hypoglycémiant induit, en outre, un léger retard de la vidange gastrique. Grâce à des mécanismes permettant de réduire la sensation de faim et l'absorption d'énergie, le liraglutide entraîne une diminution du poids et de la masse de graisse corporels.
Le GLP-1 est un régulateur physiologique de l'appétit et de la prise alimentaire. Les récepteurs du GLP-1 (GLP-1R) sont présents dans diverses régions du cerveau impliquées dans la régulation de l'appétit. Des études chez l'animal ont montré qu'après administration périphérique, le liraglutide est absorbé dans les régions du cerveau impliquées dans la régulation de l'appétit, p.ex. l'hypothalamus. L'activation des récepteurs du GLP-1 dans ces régions du cerveau a augmenté la satiété et diminué les principaux signaux de la faim, ce qui a entraîné une perte de poids.
Les récepteurs du GLP-1 se trouvent également à des endroits précis du cœur, du système vasculaire, du système immunitaire et des reins. Des études effectuées chez l'homme et l'animal ont montré que l'activation de ces récepteurs par le liraglutide peut induire des effets cardiovasculaires et microvasculaires, y compris une réduction des inflammations. Dans des études effectuées chez l'animal, le liraglutide a inhibé le développement de l'athérosclérose.
Par ailleurs, des études effectuées chez l'animal ont montré que le liraglutide induisait une réduction significative des lésions de la plaque de l'aorte. Cependant, le liraglutide n'a montré aucune inhibition sur le développement ultérieur de la plaque de l'aorte ni aucun effet sur les plaques existantes. De plus, le liraglutide a induit une diminution de l'inflammation des tissus environnants et a eu un effet positif sur les lipides plasmatiques.
Pharmacodynamique
La durée d'action de l'insuline dégludec et du liraglutide permet d'administrer Xultophy une fois par jour indépendamment des heures de repas.
Xultophy améliore le contrôle glycémique en diminuant de façon prolongée la glycémie à jeun et postprandiale après tous les repas. Ces effets permettent de contrôler la glycémie pendant toute la journée, y compris au moment des pics glycémiques postprandiaux.
Efficacité clinique
La sécurité et l'efficacité de Xultophy ont été évaluées dans six études de phase 3 randomisées, contrôlées et menées en groupes parallèles chez des adultes atteints de diabète sucré de type 2 et divers prétraitements antihyperglycémiques. Les thérapies comparatives étaient l'insuline basale, la thérapie avec l'agoniste des récepteurs GLP-1, le placebo et la thérapie selon le schéma bolus-basal. La durée de l'étude était de ≥26 semaines, avec 199 à 833 patients dans le groupe Xultophy. Xultophy a été titré deux fois par semaine dans toutes les études (voir tableau 2). Le même algorithme de titrage a été appliqué dans le groupe de comparaison avec l'insuline basale.
Tableau 2: titration de Xultophy et de l'insuline basale
Glucose plasmatique mesuré avant le Adaptation de la dose(deux fois
petit-déjeuner* par semaine)
mmol/l mg/dl Xultophy (doses
unitaires)
<4.0 <72 -2
4.0–5.0 72–90 0
>5.0 >90 +2
* Glucose plasmatique mesuré par le patient
Xultophy utilisé en association avec la metformine
L'efficacité et la sécurité de Xultophy par rapport à l'insuline dégludec et le liragultide, administrés tous une fois par jour, ont été évaluées dans le cadre d'une étude treat-to-target, ouverte, contrôlée, randomisée, de 26 semaines, chez des patients atteints du diabète de type 2, avec une prolongation de 26 semaines. La dose initiale de Xultophy et d'insuline dégludec se montait à 10 doses unitaires ou 10 unités, la dose ayant été titrée deux fois par semaine conformément au tableau 2 ci-dessus.
Chez les patients sous liraglutide, la dose a été augmentée de manière fixe, avec une dose initiale de 0.6 mg et une hausse hebdomadaire de la dose de 0.6 mg, jusqu'à l'obtention d'une dose d'entretien de 1.8 mg. La dose maximale de Xultophy était de 50 doses unitaires, alors qu'aucune dose maximale n'a été fixée pour l'insuline dégludec. Une partie des patients (140/833) a reçu de la pioglitazone en plus de Xultophy et de la metformine.
Les principaux résultats de l'étude figurent dans le Tableau 3 et sur la Figure 1.
Tableau 3: Résultats d'une étude de 26 semaines portant sur Xultophy chez des patients présentant un contrôle glycémique insuffisant sous metformine seule ou associée à de la pioglitazone
Traitement précédent
par met ± pioglitazon
e
Xultophy Insuline dégludec Liraglutide
N 833 413 414
HbA1c (%)
Valeur de base → fin de l'étude 8.3 → 6.4 8.3 → 6.9 8.3 → 7.0
Modification moyenne -1.91 -1.44 -1.28
Différence estimée -0.47AB[-0.58; -0.64AB[-0.75;
-0.36] -0.53]
Nombre de cas confirmés 1.80 (31.9%) 2.57 (38.6%) 0.22 (6.8%)
d'hypoglycémie* par patient-année
d'exposition (pourcentage de
patients)
Nombre estimé 0.68AC[0.53; 0.87] 7.61B[5.17; 11.21]
Poids corporel (kg)
Valeur de base → fin de l'étude 87.2 → 86.7 87.4 → 89.0 87.4 → 84.4
Modification moyenne -0.5 1.6 -3.0
Différence estimée -2.22AB[-2.64; 2.44B[2.02; 2.86]
-1.80]
Augmentation postprandiale du
glucose (mmol/l) Test au moment
des repas (moyenne sur 4 heures)
Valeur de base → fin de l'étude 4.11 → 3.22 4.12 → 3.95 4.12 → 3.36
Modification moyenne -0.87 -0.17 -0.78
Différence estimée -0.71AC[-1.17; -0.09[-0.56; 0.37]
-0.26]
Dose à la fin de l'étude
Insuline dégludec (unités) 38 53 -
Liraglutide (mg) 1.4 - 1.8
Différence estimée, dose -14.90AB[-17.14;
d'insuline dégludec -12.66]
La valeur de base, la fin de l'étude et la modification moyenne sont considérées comme "Last observation carried forward" (LOCF). L'intervalle de confiance de 95% figure entre crochets: [].
* L'hypoglycémie confirmée était définie par une hypoglycémie sévère (épisode nécessitant qu'une tierce personne aide le patient) et/ou une hypoglycémie légère (glucose plasmatique <3.1 mmol/l, quels que soient les symptômes).
** Le test réalisé au moment des repas a été mené lors d'une étude partielle comprenant 260 patients dont la glycémie n'était pas suffisamment contrôlée par le traitement par metformine ± pioglitazone (131 patients ont été traités par Xultophy, 64 par l'insuline dégludec et 65 par le liraglutide).
A Critères avec supériorité confirmée de Xultophy vs substance de comparaison
B p<0.0001
C p<0.05
Le pourcentage d'hypoglycémies confirmées était plus faible avec Xultophy qu'avec l'insuline dégludec, quel que soit le contrôle glycémique. Voir Figure 1.
Fig.1a
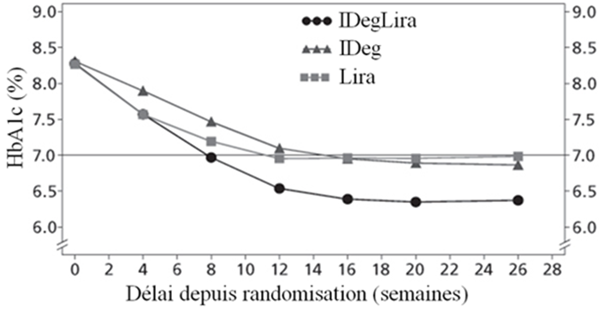
Fig. 1b

Fig. 1c

Fig. 1d

IDegLira =Xultophy, IDeg = insuline dégludec, Lira = liraglutide, taux obs. = taux observé, PYE = patient-année d'exposition
Figure 1: Taux moyen de l'HbA1c (%) selon le nombre de semaines de traitement (Fig. 1a), nombre d'hypoglycémies confirmées par patient-année d'exposition par rapport au taux moyen de l'HbA1c (%) (Fig. 1b), modification moyenne du poids corporel selon le nombre de semaines de traitement (Fig. 1c) et nombre moyen cumulé d'événements d'hypoglycémie selon le nombre de semaines de traitement (Fig. 1d)
Le taux par année-patient sous exposition (pourcentage des patients) d'hypoglycémies sévères, définies comme étant des épisodes ayant nécessité de l'aide de tierces personnes, était de 0.01 (2 patients sur 825) pour Xultophy, de 0.01 (2 patients sur 412) pour l'insuline dégludec et de 0.00 (0 patients sur 412) pour le liraglutide.
Le taux d'hypoglycémies nocturnes était semblable sous le traitement par Xultophy et par l'insuline dégludec malgré le taux de l'HbA1c plus faible pour Xultophy par rapport à l'insuline dégludec.
Globalement, les patients traités par Xultophy présentaient moins d'effets indésirables gastro-intestinaux que les patients traités par liraglutide mais plus que ceux sous insuline dégludec. Cette différence pourrait être due à une hausse plus lente de la dose de la composante liraglutide au début du traitement avec Xultophy par rapport à une thérapie au liraglutide seul.
Données à long terme (52 semaines)
L'efficacité et la sécurité de Xultophy se sont maintenues pendant les 52 semaines de traitement. La diminution d'HbA1c entre la valeur de base et la semaine 52 était, pour Xultophy, de 1.84% et la différence entre les traitements a été estimée à -0.65% [-0.76; -0.53]IC à 95% par rapport au liraglutide et à -0.46% [-0.57; -0.34]IC à 95% par rapport à l'insuline dégludec. Le poids corporel a diminué de 0.4 kg, la différence entre Xultophy et l'insuline dégludec étant estimée à -2.80 kg. Le taux d'hypoglycémies confirmées était de 1.8 événements par patient-année pour Xultophy et de 2.8 événements pour l'insuline dégludec avec une différence entre les traitements estimée de 0.63 [0.50; 0.79]IC à 95%.
Utilisation de Xultophy en association avec une sulfonylurée seule ou combinée avec de la metformine
L'efficacité et la sécurité de Xultophy, administré en association avec une sulfonylurée seule ou combinée avec de la metformine, ont fait l'objet d'une étude "treat-to-target" de 26 semaines, randomisée, en double aveugle, contrôlée par placebo et comprenant 435 patients, dont 289 étaient traités par Xultophy. 259 patients sur 289 ont reçu Xultophy plus de la metformine plus des sulfonylurées, 30 patients sur 289 Xultophy plus des sulfonylurées sans metformine. La dose initiale de Xultophy était de 10 doses unitaires et la dose a été titrée deux fois par semaine, comme indiqué dans le Tableau 2 ci-dessus, avec une cible de titration située entre 4 et 6 mmol/l.
La diminution du taux initial d'HbA1c était de -1.45%, passant ainsi de 7.9% à 6.4% pour Xultophy; la diminution du taux initial d'HbA1c était de -0.46%, passant ainsi de 7.9% à 7.4% pour le placebo et la différence entre les traitements a été estimée à -1.02% [-1.18; -0.87] IC à 95% (voir Figure 2).
Fig. 2a

Fig. 2b

Fig. 2c

IDegLira = Xultophy
Figure 2: HbA1c moyenne (%) selon le nombre de semaines de traitement (Fig. 2a), nombre cumulé moyen d'événements d'hypoglycémie (Fig. 2b) et modification moyenne du poids corporel selon le nombre de semaines de traitement (Fig. 2c) chez des patients présentant un contrôle glycémique insuffisant sous SU±Met
41.7% des patients sous Xultophy et 17.1% des patients sous placebo ont subi au moins un épisode d'hypoglycémie confirmée au cours de l'étude. Cela correspond à un taux d'hypoglycémie estimé de 3.52 par patient-année sous Xultophy vs. 1.35 sous placebo. Les patients traités par Xultophy ont connu une prise de poids corporel moyenne de 0.5 kg tandis que les patients du groupe placebo ont perdu, en moyenne, 1.0 kg (différence estimée entre traitements: 1.48 kg; [0.90; 2.06]IC à 95%).
La dose moyenne de Xultophy était de 28 doses unitaires à la fin de l'étude, ce qui correspond à 28 unités d'insuline dégludec et 1.0 mg de liraglutide.
En ajout aux iSGLT2 seuls ou en association avec la metformine: Xultophy par rapport à l'insuline glargine U100
Dans un essai en ouvert comparant l'efficacité et la sécurité de Xultophy et de l'insuline glargine U100 en ajout aux iSGLT2 ± ADO, Xultophy était supérieur à l'insuline glargine en termes de réduction de l'HbA1c moyenne après 26 semaines. Une réduction de 1.9% (de 8.2% à 6.3%) a été obtenue avec Xultophy et de 1.7% (de 8.4% à 6.7%) avec l'insuline glargine, correspondant à une différence de traitement estimée à -0.36% [-0.50; -0.21].
Comparativement à la valeur initiale, Xultophy n'a pas entraîné de changement au niveau du poids corporel moyen alors qu'une augmentation du poids moyen de 2,0 kg a été observée chez les patients traités par l'insuline glargine (différence de traitement estimée à -1.92 kg [IC à 95%: -2.64; -1.19]). Le pourcentage de patients ayant présenté une hypoglycémie sévère ou symptomatique confirmée par une glycémie était de 12.9% dans le groupe Xultophy et de 19.5% dans le groupe de l'insuline glargine (le ratio estimé était de 0.42 [IC à 95%: 0.23; 0.75]). La dose journalière moyenne d'insuline à la fin de l'essai était de 36 unités pour les patients traités avec Xultophy et de 54 unités pour les patients traités avec l'insuline glargine.
Xultophy en remplacement d'une thérapie par des agonistes du récepteur du GLP-1
L'efficacité et la sécurité de Xultophy (une fois par jour) par rapport à une thérapie inchangée à un agoniste du récepteur du GLP-1 (posologie selon la notice d'emballage) ont été examinées dans le cadre d'une étude randomisée de 26 semaines, menée en ouvert, treat-to-target, chez des patients atteints du diabète de type 2 qui présentaient un contrôle glycémique insuffisant sous un agoniste du récepteur du GLP-1 et la metformine (Met) seule (74.2%) ou en association avec la pioglitazone (2.5%), des sulfonylurées (21.2%) ou les deux.
La dose initiale de Xultophy et de l'insuline dégludec était de 16 doses unitaires (16 unités d'insuline dégludec et 0.6 mg de liraglutide) et la dose a été titrée deux fois par semaine selon le tableau 2. Les patients du bras de l'agoniste du récepteur du GLP-1 ont poursuivi leur traitement aux agonistes du récepteur du GLP-1 comme avant l'étude.
Les résultats de l'étude sont présentés dans le tableau 4 et la figure 3.
Tableau 4: Résultats d'une étude de 26 semaines avec Xultophy chez des patients atteints du diabète de type 2 avec un contrôle glycémique insuffisant sous des agonistes du récepteur du GLP-1
Traitement antérieur
à l'agoniste du
récepteur du GLP-1
Xultophy Agoniste du récepteu
r du GLP-1
N 292 146
HbA1c (%)
Valeur initiale → fin de l'étude 7.8 → 6.4 7.7 → 7.4
Modification moyenne -1.3 -0.3
Différence évaluée -0.94AB[-1.11; -0.78]
Patients (%) ayant obtenu une HbA1c
<7%
Tous les patients 75.3 35.6
Odds-Ratio estimé 6.84B[4.28; 10.94]
Patients (%) ayant obtenu une HbA1c
<6.5%
Tous les patients 63.0 22.6
Odds-Ratio estimé 7.53B[4.58; 12.38]
Nombre d'hypoglycémies confirmées* 2.82 (32.0%) 0.12 (2.8%)
par patient- année sous exposition
(fraction en pour-cent de patients)
Nombre estimé 25.36B[10.63; 60.51]
Poids corporel (en kg)
Valeur initiale → fin de l'étude 95.6 → 97.5 95.5 → 94.7
Modification moyenne 2.0 -0.8
Différence estimée -2.89B[2.17; 3.62]
GAJ (mmol/l)
Valeur initiale → fin de l'étude 9.0 → 6.0 9.4 → 8.8
Modification moyenne -2.98 -0.60
Différence estimée -2.64B[-3.03; -2.25]
Dose à la fin de l'étude
Insuline dégludec (unités) 43
Liraglutide (mg) 1.6 La dose de l'agoniste du récepteur du
GLP-1 a été maintenue sans être
modifiée par rapport à la valeur
initiale
Différence estimée, dose d'insuline
dégludec
Valeur initiale, fin de l'étude et modification moyenne ont été notées comme „Last observation carried forward” (LOCF). L'intervalle de confiance à 95% est indiqué entre "[]" .
* Hypoglycémie confirmée définie comme hypoglycémie sévère (épisode nécessitant l'aide d'une tierce personne) et/ou hypoglycémies modérées (glucose plasmatique <3.1 mmol/l, indépendamment de symptômes).
A Critères d'évaluation avec supériorité confirmée de Xultophy vs. substance de comparaison
B p<0.001

IDegLira=Xultophy, GLP-1 RA= Agoniste des récepteurs du GLP-1
Figure 3: HbA1c (%) moyenne en fonction du nombre de semaines de traitement chez des patients atteints du diabète sucré de type 2 avec contrôle glycémique insuffisant sous agonistes du récepteur du GLP-1.
Le nombre d'hypoglycémies sévères confirmées par patient-année sous exposition (fraction en pourcent de patients) était de 0.01 (1 patient sur 291) pour Xultophy et de 0.00 (0 patients sur 199) pour les agonistes du récepteur du GLP-1.
Intensification de l'insulinothérapie basale par le passage à Xultophy
Comparaison avec la thérapie selon le schéma bolus basal
L'efficacité et la sécurité de Xultophy par rapport à un schéma insulinique basal-bolus composé d'insuline glargine 100 U en combinaison avec de l'insuline asparte prise au moment des repas principaux ont été étudiées dans un essai randomisé, mené en ouvert, "treat-to-treat" , d'une durée de 26 semaines, chez des patients diabétiques de type 2 n'ayant pas obtenu un contrôle glycémique adéquat sous insuline glargine 100 U (20 à 50 unités) et metformine. La dose initiale de Xultophy était de 16 doses unitaires. Dans le bras de l'essai du schéma basal-bolus, la dose initiale d'insuline glargine 100 U correspondait à la dose quotidienne avant l'étude et la dose initiale d'insuline asparte liée au repas était de 4 unités avant les repas principaux. Les doses de Xultophy et d'insuline glargine 100 U ont été titrées deux fois par semaine selon le tableau 2, tandis que l'insuline asparte liée au repas a été titrée deux fois par semaine selon les mesures de la glycémie effectuées par les patients (SMPG) durant les trois jours précédents. La dose quotidienne maximale pour Xultophy était de 50 doses unitaires, tandis qu'il n'y avait pas de dose maximale pour l'insuline glargine 100 U et l'insuline asparte.
Les principaux résultats de l'essai sont mentionnés dans le Tableau 5 et la Figure 4.
Dans le groupe Xultophy ou du schéma basal-bolus, après 26 semaines de traitement, 57.6% et 33.5% des patients respectivement ont atteint une valeur cible d'HbA1c de <7% sans épisodes hypoglycémiques symptomatiques graves ou confirmés (p<0.0001).

IDegLira = Xultophy, IGlar + IAsp = insuline glargine 100 U + insuline asparte
Figure 4 HbA1c moyenne (%) par semaine de traitement chez des patients diabétiques de type 2 n'ayant pas obtenu un contrôle glycémique adéquat sous insuline glargine 100 U
Le taux d'hypoglycémies sévères par patient-année d'exposition était de 0.02 (3 patients sur 252) pour Xultophy et de 0.08 (4 patients sur 253) pour le schéma basal-bolus.
Le taux d'évènements d'hypoglycémie grave, symptomatique et nocturne sévère ou confirmée était statistiquement significativement plus faible avec Xultophy qu'avec le schéma basal-bolus (taux lié au traitement estimé à 0.08, p <0.0001).
Tableau 5: Résultats d'un essai d'une durée de 26 semaines avec Xultophy chez des patients atteints de diabète sucré de type 2 n'ayant pas obtenu un contrôle glycémique adéquat sous insuline glargine 100 U
Xultophy Basal-bolus (insuline glargine +
insuline asparte)
n 252 254
HbA1c (%)
Valeur initiale → Fin de l'étude 8.2→6.7 8.2→6.7
Changement moyen -1.49 -1.48
Différence estimée -0.02A [-0.16;-0.12]
Patients (%) ayant obtenu une HbA1c <7%
Tous les patients 66.0 67.0
Odds ratio estimé 0.91 [0.62; 1.33]
Patients (%) ayant obtenu une HbA1c <6,5%
Tous les patients 49.6 44.6
Odds ratio estimé 1.26 [0.88; 1.82]
Taux d'hypoglycémies* par patient-année
d'exposition (pourcentage de patients)
Taux estimé 1.07 (19.8%) 8.17 (52.6%)0.11B [0.08; 0.17]
Poids corporel (kg)
Valeur initiale → Fin de l'étude 87.2→85.8 88.2→90.7
Changement moyen -0.9 2.6
Différence estimée -3.57B [-4.19; -2.95]
Glycémie à jeun (mmol/l)
Valeur initiale → Fin de l'étude 8.5→6.1 8.3→6.4
Changement moyen -2.35 -1.88
Différence estimée -0.31 [-0.67; 0.05]
Dose à la fin de l'étude
Insuline dégludec (unités) 40
Liraglutide (mg) 1.4
Insuline glargine (unités) 52
Insuline asparte (unités) 32
Différence estimée, dose totale d'insuline -44.5 [-48.3; -40.7]
Différence estimée, dose d'insuline basale -12.6 [-14.9; -10.3]
Aucune imputation pour les données manquantes. L'intervalle de confiance de 95% figure entre crochets: [].
* L'hypoglycémie symptomatique grave ou confirmée par la glycémie se définit comme un épisode nécessitant l'aide d'une tierce personne qui administre activement des glucides ou du glucagon ou qui effectue d'autres actions correctives, ou comme un épisode confirmé par une glycémie de <3.1 mmol/l (56 mg/dl) et associé aux symptômes typiques de l'hypoglycémie.
A Non-infériorité confirmée de Xultophy par rapport au produit de comparaison (intervalle de 0.3%). p <0.0001
B Supériorité confirmée de Xultophy par rapport au produit de comparaison. p <0.0001.
Comparaison avec l'insulinothérapie basale optimisée
L'efficacité et la sécurité de Xultophy ont été étudiées par rapport à l'insuline glargine, administrés tous deux une fois par jour, dans un essai randomisé, mené en ouvert, "treat-to-target" , d'une durée de 26 semaines, chez des patients diabétiques de type 2 n'ayant pas obtenu un contrôle glycémique adéquat sous insuline glargine (20 à 50 unités) et metformine. La dose initiale de Xultophy était de 16 doses unitaires et la dose initiale de l'insuline glargine était égale à la dose quotidienne antérieure à l'essai. Une titration de la dose dans chaque bras a eu lieu deux fois par semaine conformément au Tableau 2. La dose maximale autorisée était de 50 doses unitaires pour Xultophy tandis qu'il n'y avait pas de dose maximale pour l'insuline glargine.
Les principaux résultats de l'essai sont mentionnés dans le Tableau 5 et la Figure 5.
54.3% des patients traités avec Xultophy ont atteint le taux cible de HbA1c <7% sans épisode d'hypoglycémie confirmée, contre 29.4% des patients traités avec l'insuline glargine (odds ratio à 3.24, p <0.001).

Figure 5 - HbA1c moyenne (%) selon la semaine de traitement chez des patients diabétiques de type 2 n'ayant pas obtenu un contrôle glycémique adéquat sous insuline glargine
Le taux d'hypoglycémies sévères par patient-année d'exposition (pourcentage de patients) était de 0.00 (0 patient sur 278) avec Xultophy et de 0.01 (1 patient sur 279) avec l'insuline glargine. Le taux d'évènements d'hypoglycémie nocturne était significativement plus bas avec Xultophy par rapport à l'insuline glargine (taux lié au traitement estimé à 0.17, p <0.001).
L'efficacité et la sécurité de Xultophy ont été comparées à celles de l'insuline dégludec (tous les deux administrés une fois par jour) dans une étude "treat-to-target" randomisée, en double aveugle, de 26 semaines. Cette étude comprenait des patients présentant un diabète de type 2 et un contrôle glycémique insuffisant sous insuline basale (20 à 40 unités) et metformine (Met) seule ou associée à des sulfonylurées/glinides. L'insuline basale et les sulfonylurées/glinides ont été arrêtées au moment de la randomisation.
La dose initiale de Xultophy et d'insuline dégludec était de 16 doses unitaires ou, respectivement, 16 unités, et la dose a été titrée deux fois par semaine comme indiqué dans le Tableau 2. La dose maximale autorisée était de 50 doses unitaires pour Xultophy et de 50 unités pour l'insuline dégludec.
Les résultats concernant le critère d'évaluation principal de l'étude, ainsi que la dose d'insuline et l'incidence de l'hypoglycémie, sont résumés dans le Tableau 5 et la Figure 6.
Un total de 48.7% des patients a obtenu un taux-cible de l'HbA1c <7% sans épisodes hypoglycémiques confirmés; cette fraction était significativement plus importante que chez les patients traités par l'insuline dégludec (15.6%, odds ratio 5.57; p<0.0001).
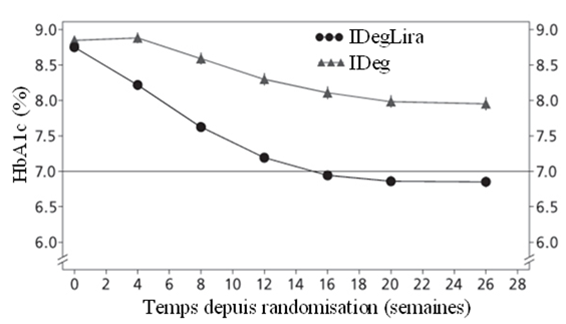
Figure 6: HbA1c moyen (%) selon le nombre de semaines de traitement chez des patients présentant un contrôle glycémique insuffisant sous insuline basale.
Tableau 6 - Résultats de deux essais d'une durée de 26 semaines avec Xultophy chez des patients diabétiques de type 2 n'ayant pas obtenu un contrôle glycémique adéquat sous insuline glargine (gauche) ou sous insuline basale (droite)
Traitement antérieur Traitement antérieur
par insuline , par insuline
glargine basale (NPH, insulin
e détémir, insuline
glargine)
Xultophy Insuline glargine, Xultophy Insuline dégludec,
sans limitation des 50 unités au maximum
doses autorisées
N 278 279 199 199
HbA1c (%)
Début→Fin de l'étude 8.4→6.6 8.2→7.1 8.7→6.9 8.8→8.0
Changement moyen -1.81 -1.13 -1.90 -0.89
Différence estimée -0.59AB[-0.74; -1.05AB[-1.25;
-0.45] -0.84]
Patients (%) atteign
ant un taux de
HbA1c <7%
Tous les patients 71.6 47.0
Odds ratio estimé 3.45B[2.36; 5.05]
Taux d'hypoglycémies 2.23 (28.4%) 5.05 (49.1%) 1.53 (24.1%) 2.63 (24.6%)
confirmées par
patient-année
d'exposition (pource
ntage de patients)
Taux estimé 0.43AB [0.30;0.61] 0.66 [0.39; 1.13]
Poids (kg)
Début→Fin de l'étude 88.3→86.9 87.3→89.1
Changement moyen -1.4 1.8
Différence estimée -3.20AB [-3.77;
-2.64]
Glycémie à jeun
(mmol/l)
Début→Fin de l'étude 8.9→6.1 8.9→6.1
Changement moyen -2.83 -2.77
Différence estimée -0.01 [-0.35;0.33]
Dose à la fin de
l'étude
Insuline (unités) 41 66D 45 45
Liraglutide (mg) 1.5 - 1.7 -
Différence estimée, -25.47B [-28.90;-22. -0.02 [-1.88; 1.84]
dose d'insuline 05]
basale
La valeur de base, la fin de l'étude et la modification moyenne sont considérées comme "Last observation carried forward" (LOCF). L'intervalle de confiance de 95% figure entre crochets: [].
A Critères d'évaluation avec supériorité confirmée de Xultophy par rapport au produit de comparaison
B p <0.0001
C p <0.05
D La dose moyenne d'insuline glargine antérieure à l'essai était de 32 unités
Sécurité cardiovasculaire
Aucune étude cardiovasculaire (CV) n'a été réalisée spécifiquement pour la préparation combinée Xultophy. La sécurité cardiovasculaire de ses deux composants a été démontrée dans les études CV associées, LEADER et DEVOTE. D'autres analyses post-hoc ont porté sur l'effet chez les patients de ces deux études qui ont été traités simultanément avec du liraglutide et une insuline à action prolongée (y compris l'insuline dégludec). Les résultats de ces analyses post-hoc confirment la transférabilité des résultats positifs de LEADER et de DEVOTE à Xultophy (sécurité et bénéfice cardiovasculaire).
Liraglutide (Victoza)
LEADER - Analyse primaire
L'étude LEADER (Liraglutide Effect and Action in Diabetes Evaluation of Cardiovascular Outcome Results) a examiné l'incidence des événements cardiovasculaires graves (MACE: décès cardiovasculaire, infarctus du myocarde non mortel, accident vasculaire cérébral non mortel) chez 9340 patients atteints de diabète sucré de type 2 et présentant un risque élevé d'événements cardiovasculaires. Après randomisation (1:1), les patients ont reçu, en plus du traitement standard, soit jusqu'à 1.8 mg de liraglutide par jour (4668) soit un placebo (4672) (durée moyenne du traitement environ 3.5 ans). Le critère d'évaluation principal était le délai avant la première occurrence d'un MACE. Le rapport des risques estimé jusqu'au premier MACE a été considérablement réduit par le liraglutide à la dose étudiée (rapport des risques 0.87 [0.78; 0.97] IC à 95%.
Une réduction de l'HbA1c par rapport aux valeurs initiales à 36 mois a été observée avec le liraglutide par rapport au placebo, en plus du traitement standard (-1.16% vs -0.77%; différence de traitement estimée [ETD] -0.40% [-0.45; -0.34]).
LEADER - Analyse post-hoc
L'analyse primaire LEADER a été répétée pour 2 118 patients recevant de l'insuline à action prolongée de base afin de déterminer l'applicabilité des résultats de l'analyse primaire pour l'association du liraglutide et de l'insuline à action prolongée. Les caractéristiques de base de ce sous-groupe étaient comparables à celles de la population totale. Les résultats de l'analyse post-hoc pour la période précédant la première analyse MACE étaient conformes à l'analyse primaire LEADER (RR estimé [IC à 95%] dans le sous-groupe: 0.82 [0.66; 1.03]).
Insuline dégludec (Tresiba)
DEVOTE – Analyse primaire
DEVOTE (Trial Comparing Cardiovascular Safety of Insulin Degludec versus Insulin Glargine in Patients with Type 2 Diabetes at High Risk of Cardiovascular Events) était un essai clinique randomisé en double aveugle qui comparait la sécurité cardiovasculaire de l'insuline dégludec à celle de l'insuline glargine (100 unités/ml) chez 7637 patients atteints de diabète sucré de type 2 et présentant un risque élevé d'événements cardiovasculaires. La durée de l'étude était "événementielle" , c'est-à-dire que l'étude était menée jusqu'à ce que le nombre d'événements soit ≥633. La durée moyenne du traitement était de 1.83 an, la durée d'observation moyenne étant de 1.99 an.
L'analyse primaire couvrait la période allant de la randomisation à la première occurrence d'un événement cardiovasculaire grave (MACE: décès d'origine cardiovasculaire, infarctus du myocarde non mortel ou accident vasculaire cérébral non mortel). L'analyse primaire a montré un RR estimé à [IC à 95%] pour l'insuline dégludec par rapport à l'insuline glargine de 0.91 [0.78; 1.06].
Le taux d'HbA1c initial était de 8.4% dans les deux groupes de thérapie et après 2 ans, l'HbA1c était de 7.5% pour l'insuline dégludec et l'insuline glargine.
DEVOTE - Analyse post-hoc
Afin de déterminer la transférabilité des résultats de l'analyse primaire à l'association d'une insuline à action prolongée (y compris l'insuline dégludec) avec du liraglutide, une analyse post-hoc a été effectuée pour le sous-groupe de patients qui étaient déjà traités par du liraglutide dans le groupe de référence.
Les caractéristiques de base des 436 patients traités avec le liraglutide de référence étaient semblables à celles des 7 201 patients traités avec un traitement de référence sans liraglutide. L'analyse post-hoc du délai avant le premier MACE a révélé un RR estimé à [IC à 95%] 0.65 [0.45; 0.96] chez les patients traités avec du liraglutide comparativement aux patients non traités avec du liraglutide.
Évaluation microvasculaire
Dans l'étude LEADER, les événements microvasculaires comprenaient la néphropathie et la rétinopathie. L'analyse du temps avant le premier événement microvasculaire sous liraglutide par rapport au placebo a montré un RR de 0.84 [0.73, 0.97]. Le RR pour le liraglutide par rapport au placebo était de 0.78 [0.67, 0.92] pour la période précédant la première occurrence de néphropathie et de 1.15 [0.87, 1.52] pour la période précédant la première occurrence de rétinopathie.
PharmacocinétiqueLes paragraphes suivants traitent des propriétés pharmacocinétiques de Xultophy mais n'incluent pas les données relatives à l'administration unique d'insuline dégludec ou de liraglutide.
Absorption
Après l'administration de Xultophy, l'exposition systémique de l'insuline dégludec était équivalente à celle de l'insuline dégludec seule, alors que la Cmax était supérieure de 12%.
Après l'administration de Xultophy, l'exposition systémique du liraglutide était inférieure de 11% à celle observée après l'administration du liraglutide seul et la Cmax était inférieure de 23%. Cependant, ces différences n'ont pas été considérées comme étant cliniquement significatives car le traitement par Xultophy a été débuté et titré individuellement, selon les valeurs glycémiques cible des patients.
L'exposition de l'insuline dégludec et du liraglutide augmente proportionnellement à la dose de Xultophy sur l'ensemble de la fourchette posologique.
Le profil pharmacocinétique de Xultophy correspond à la posologie d'une fois par jour et la concentration à l'état d'équilibre de l'insuline dégludec et du liraglutide a été atteinte au bout de 2 à 3 jours avec une administration quotidienne de la dose.
Distribution
L'insuline dégludec et le liraglutide se lient en grande partie aux protéines plasmatiques (>99% et >98%).
Métabolisme
Insuline dégludec
La dégradation de l'insuline dégludec est semblable à celle de l'insuline humaine. Tous les métabolites formés sont inactifs.
Liraglutide
Dans les 24 heures suivant l'administration d'une dose unique de [3H]-liraglutide à des sujets sains, le principal composant plasmatique était le liraglutide intact. Deux métabolites plasmatiques mineurs ont été détectés dans le plasma (≤9% et ≤5% de la radioactivité plasmatique totale). Le liraglutide est métabolisé de la même manière que les grosses protéines et aucun organe en particulier n'a été identifié comme étant la voie d'élimination principale.
Élimination
La demi-vie de l'insuline dégludec est d'environ 25 heures et celle du liraglutide d'environ 13 heures.
Cinétique pour certains groupes de patients
Patients âgés
L'âge n'a pas eu d'influence cliniquement significative sur la pharmacocinétique de l'insuline dégludec et du liraglutide après l'administration de Xultophy.
Sexe
Le sexe n'a pas eu d'influence cliniquement significative sur la pharmacocinétique de l'insuline dégludec et du liraglutide après l'administration de Xultophy.
Origine ethnique
L'origine ethnique n'a pas eu d'influence cliniquement significative sur la pharmacocinétique de l'insuline dégludec et du liraglutide après l'administration de Xultophy.
Troubles de la fonction rénale
Insuline dégludec
Il n'existe aucune différence concernant les propriétés pharmacocinétiques de l'insuline dégludec entre les sujets sains et les patients présentant une insuffisance rénale.
Liraglutide
L'exposition au liraglutide était plus basse chez les patients présentant une insuffisance rénale que chez les personnes ayant une fonction rénale normale. L'exposition au liraglutide a diminué de 33%, 14%, 27% et 26% chez les patients présentant respectivement une insuffisance rénale légère (clairance de la créatinine, Clcr de 50 à 80 ml/min), modérée (Clcr de 30 à 50 ml/min), sévère (Clcr <30 ml/min) et une insuffisance rénale terminale nécessitant une dialyse.
De même, dans un essai clinique de 26 semaines chez des patients atteints du diabète sucré de type 2 et d'insuffisance rénale modérée (Clcr de 30 à 59 ml/min), l'exposition au liraglutide a diminué de 26% par rapport à une étude séparée chez des patients atteints de diabète sucré de type 2 présentant une insuffisance rénale normale ou légère.
Troubles de la fonction hépatique
Insuline dégludec
Il n'existe aucune différence au niveau des propriétés pharmacocinétiques de l'insuline dégludec entre les sujets sains et les patients présentant une insuffisance hépatique.
Liraglutide
La pharmacocinétique du liraglutide a été évaluée chez des patients présentant un degré variable d'insuffisance hépatique dans un essai en dose unique. L'exposition au liraglutide a diminué de 13 à 23% chez les patients qui présentaient une insuffisance hépatique légère à modérée par rapport aux sujets sains. L'exposition était significativement diminuée (44%) chez les patients atteints d'une insuffisance hépatique sévère (score de Child-Pugh >9).
Enfants et adolescents
Aucune étude concernant Xultophy n'a été menée chez l'enfant et l'adolescent de moins de 18 ans.
Données précliniquesLe programme de développement préclinique pour l'insuline dégludec/liraglutide comprenait des études pivots de toxicité sur le traitement combiné portant sur une seule espèce pertinente (rats Wistar) et allant jusqu'à 90 jours. La tolérance locale a été étudiée chez le lapin et le cochon.
Les données précliniques de sécurité issues des études de toxicologie en administration répétée n'ont pas révélé d'éléments préoccupants pour la sécurité humaine.
Dans les deux études, les réactions locales observées sur les tissus des lapins et des cochons se limitaient à des réactions inflammatoires légères.
Aucune étude portant sur la carcinogénicité, la mutagénèse ou les effets de l'association insuline dégludec/liraglutide sur la fertilité n'a été menée.
Les données ci-après se fondent sur des études portant sur l'insuline dégludec et le liraglutide utilisés séparément.
Insuline dégludec
Les données précliniques issues des études de pharmacologie de sécurité, de toxicité en administration répétée, de carcinogénicité, et des fonctions de reproduction n'ont pas révélé d'éléments préoccupants pour la sécurité humaine.
Le rapport entre pouvoir mitogène et potentiel métabolique de l'insuline dégludec est semblable à celui de l'insuline humaine.
Liraglutide
Les données précliniques issues des études conventionnelles de pharmacologie de sécurité, toxicologie en administration répétée ou génotoxicité n'ont pas révélé de risque particulier pour l'homme.
Des tumeurs non létales des cellules C de la thyroïde ont été observées lors d'études de carcinogénicité sur 2 ans chez le rat et la souris. Chez le rat, aucune dose sans effet nocif observé NOAEL (no adverse effect level) n'a été déterminée pour ces tumeurs. Ces résultats décrits chez les rongeurs sont dus à un mécanisme non génotoxique, spécifique, médié par les récepteurs du GLP-1, auquel les rongeurs, contrairement aux singes et à l'homme, sont particulièrement sensibles. La pertinence de ces résultats pour l'être humain est probablement faible mais ne peut pas être complètement exclue. Aucun autre type de tumeur liée au traitement n'a été identifié.
Les études effectuées chez l'animal n'ont mis en évidence aucun effet délétère direct sur la fertilité mais une légère augmentation des morts embryonnaires précoces a été observée à la dose la plus élevée. L'utilisation de liraglutide en milieu de gestation a entraîné une perte de poids maternel et une diminution de la croissance fœtale, avec des effets ambigus sur la cage thoracique chez le rat et une modification du squelette chez le lapin. Chez les rats exposés au liraglutide, on a observé un ralentissement de la croissance néonatale. Dans le groupe recevant des doses élevées, cet effet a persisté après le sevrage. Il n'est pas établi si le retard de croissance des jeunes rats est imputable à une consommation de lait réduite due à un effet direct du GLP-1 ou à une baisse de la production de lait maternel induite par une réduction de l'apport calorique.
Remarques particulièresIncompatibilités
Certaines substances mélangées à Xultophy peuvent entraîner une dégradation des principes actifs.
Xultophy ne doit pas être mélangé avec des solutions de perfusion.
Ce médicament ne doit pas être mélangé avec d'autres médicaments.
Stabilité
24 mois. Le médicament ne doit pas être utilisé au-delà de la mention "EXP" sur l'emballage.
Stabilité après ouverture
Après la première utilisation, le médicament peut être conservé pendant 21 jours à une température maximale de 30 °C ou au réfrigérateur (2–8 °C). Le médicament doit être jeté 21 jours après la première ouverture. En cas de non-utilisation, conserver le capuchon sur le stylo, à l'abri de la lumière.
Remarques concernant le stockage
Avant la première utilisation: à conserver au réfrigérateur (2–8 °C). Maintenir à distance d'éléments réfrigérants. Ne pas congeler. En cas de non-utilisation, conserver le capuchon sur le stylo, à l'abri de la lumière.
Tenir hors de portée et de la vue des enfants.
Remarques concernant la manipulation
Le stylo prérempli Xultophy est conçu pour être administré avec les aiguilles NovoTwist ou NovoFine d'une longueur maximale de 8 mm et d'une épaisseur de 32 G au minimum.
Le stylo prérempli Xultophy ne doit être utilisé que par une seule personne.
Xultophy ne doit pas être utilisé si la solution n'est pas limpide et incolore.
Xultophy ne doit pas être utilisé s'il a été congelé.
Le patient doit jeter l'aiguille après chaque injection.
Tout médicament non utilisé et tout déchet doivent être éliminés conformément à la réglementation en vigueur.
Consulter la notice pour des instructions détaillées d'utilisation.
Numéro d’autorisation65041 (Swissmedic).
Présentation3 stylos préremplis (3× 3 ml) [B]
Titulaire de l’autorisationNovo Nordisk Pharma AG, Kloten
Domicile: Zürich
Mise à jour de l’informationAvril 2025
|