Propriétés/EffetsCode ATC
R03DX11
Mécanisme d'action
Le tézépélumab est un anticorps monoclonal humain dirigé contre la lymphopoïétine stromale thymique (anticorps anti-TSLP, IgG2λ), qui se lie à la TSLP humaine et empêche son interaction avec le récepteur hétérodimérique TSLP. La TSLP, une cytokine issue des cellules épithéliales, est impliquée dans la cascade inflammatoire et joue un rôle dans le déclenchement et la persistance de l'inflammation des voies respiratoires et des muqueuses en cas d'asthme et de CRSwNP. La TSLP régule l'immunité sur l'épithélium en exerçant une influence sur les cellules dendritiques, sur d'autres cellules innées et adaptatives, sur les processus inflammatoires ainsi que sur l'hyperréactivité bronchique. En outre, il a été mis en évidence que la TLSP peut déployer des effets indirects sur les structures cellulaires des voies respiratoires (p.ex. fibroblastes et muscles lisses des voies respiratoires), et qu'on retrouve des concentrations accrues d'ARNm de TLSP et de protéine TLSP dans les voies respiratoires des patients atteints d'asthme ainsi que dans le tissu de polypes nasaux. En présence d'asthme et de CRSwNP, des déclencheurs allergiques et non allergiques induisent la production de TLSP. Le blocage de la TLSP par le tézépélumab exerce une influence sur un large spectre de biomarqueurs et de cytokines qui sont associés aux inflammations (p.ex. éosinophiles dans le sang, IgE, FeNO, IL-5 et IL-13).
Pharmacodynamique
Effet sur les éosinophiles dans le sang, les biomarqueurs inflammatoires et les cytokines
Dans les études cliniques sur l'asthme, l'administration de 210 mg SC de tézépélumab toutes les 4 semaines a induit une réduction des biomarqueurs et des cytokines inflammatoires par rapport aux valeurs initiales en comparaison avec le placebo; cet effet s'est instauré après 2 semaines et une réduction durable du nombre d'éosinophiles dans le sang, de la valeur de la FeNO et des concentrations sériques d'IL-5 et d'IL-13 a été observée jusqu'à la semaine 52. Le tézépélumab a entraîné une réduction croissante des concentrations sériques d'IgE totales, et les valeurs ont diminué continuellement au cours du traitement.
Dans une étude sur la CRSwNP, l'administration de 210 mg SC de tézépélumab toutes les 4 semaines a induit une réduction des biomarqueurs inflammatoires (nombre d'éosinophiles dans le sang et IgE sériques) qui correspondait à la réduction observée dans les études sur l'asthme.
Effet sur les éosinophiles dans la sous-muqueuse des voies respiratoires
Dans une étude clinique, l'administration de 210 mg SC de tézépélumab toutes les 4 semaines a diminué de 89 % le nombre d'éosinophiles dans la sous-muqueuse, par comparaison avec une réduction de 25 % avec le placebo. La réduction était constante, indépendamment des valeurs initiales des biomarqueurs inflammatoires.
Immunogénicité
Des anticorps anti-médicament (AAM) ont été décelés à tout moment pendant la durée totale de 52 semaines de l'étude NAVIGATOR chez 26 (4,9 %) des 527 patients asthmatiques traités par le tézépélumab au schéma posologique recommandé. Parmi ces 26 patients, 10 (1,9 % des patients traités par le tézépélumab) ont développé des anticorps au cours du traitement, et 1 patient (0,2 % des patients traités par le tézépélumab) a développé des anticorps neutralisants. Les titres d'AAM étaient généralement faibles et souvent mesurables uniquement de manière transitoire. Il y avait trop peu de patients présentant des AAM ou des anticorps neutralisants liés au traitement pour évaluer les effets sur la pharmacocinétique, la pharmacodynamie, l'efficacité et la sécurité de TEZSPIRE.
Le profil d'immunogénicité du tézépélumab s'est maintenu dans le cadre de l'étude DESTINATION chez les patients initialement inclus dans l'étude NAVIGATOR présentant un asthme sévère (n = 415) pendant 76 semaines de traitement; il s'est maintenu également pendant 104 semaines chez les patients qui ont été par la suite inclus dans la phase de suivi prolongée de l'étude DESTINATION (n = 289).
Chez les patients atteints de CRSwNP (WAYPOINT), 5 (3 %) des 164 patients qui recevaient 210 mg de tézépélumab SC toutes les 4 semaines pendant la phase de traitement de 52 semaines ont développé des AAM au cours du traitement. Des anticorps neutralisants ont été décelés chez 1 patient présentant des AAM. Bien que les AAM n'aient eu aucun effet perceptible sur la pharmacocinétique, la pharmacodynamie, l'efficacité et la sécurité de TEZSPIRE, le nombre de patients ayant développé des AAM n'était pas suffisamment important pour effectuer une évaluation formelle en cas de CRSwNP.
Efficacité clinique
Asthme
L'efficacité de TEZSPIRE a été évaluée lors de trois études cliniques randomisées, contrôlées contre placebo, réalisées en double aveugle et en groupes parallèles (études PATHWAY, NAVIGATOR et SOURCE) pendant une durée de 48 à 52 semaines chez un total de 1761 patients dès l'âge de 12 ans. Une valeur initiale minimale d'éosinophiles dans le sang ou d'autres biomarqueurs de l'inflammation (p.ex. FeNO ou IgE) n'était pas requise pour l'inclusion des patients dans les trois études.
L'étude PATHWAY était une étude d'une durée de 52 semaines évaluant l'effet sur les exacerbations ayant inclus un total de 550 patients (dès l'âge de 18 ans) atteints d'asthme sévère non contrôlé qui ont été randomisés pour recevoir soit un traitement par 70 mg SC de TEZSPIRE administré toutes les 4 semaines, soit un traitement par 210 mg SC de TEZSPIRE administré toutes les 4 semaines, soit un traitement de 280 mg SC de TEZSPIRE administré toutes les 2 semaines, soit un placebo. Les patients devaient avoir eu au moins 2 exacerbations de l'asthme ayant nécessité un traitement oral ou systémique par des corticostéroïdes ou 1 exacerbation de l'asthme ayant conduit à une hospitalisation lors des 12 derniers mois.
L'étude NAVIGATOR était une étude d'une durée de 52 semaines évaluant l'effet sur les exacerbations ayant inclus un total de 1061 patients (adultes et adolescents à partir de 12 ans) atteints d'asthme sévère non contrôlé qui ont été randomisés pour recevoir soit un traitement par 210 mg SC de TEZSPIRE administré toutes les 4 semaines, soit un placebo. Les patients devaient avoir eu au moins 2 exacerbations de l'asthme ayant nécessité un traitement oral ou systémique par des corticostéroïdes ou ayant conduit à une hospitalisation lors des 12 derniers mois.
Lors des études PATHWAY et NAVIGATOR, les patients devaient présenter un score d'au moins 1,5 au questionnaire de contrôle de l'asthme (ACQ-6) lors de la sélection et une valeur initiale réduite de la fonction pulmonaire (VEMS pré-bronchodilatateur inférieur à 80 % de la valeur prédite chez les adultes et inférieur à 90 % de la valeur prédite chez les adolescents). Les patients devaient prendre un traitement régulier par des corticostéroïdes inhalés (CSI) à moyennes ou hautes doses et au moins un traitement complémentaire de contrôle de l'asthme, en association ou non avec des corticostéroïdes oraux (CSO). Les patients poursuivaient leur traitement de base de l'asthme pendant toute la durée de l'étude.
Un total de 150 patients asthmatiques (à partir de 18 ans) ont été randomisés dans l'étude SOURCE, d'une durée de 48 semaines, évaluant la diminution de l'utilisation des CSO. Les patients inclus nécessitaient un traitement quotidien par un CSO (7,5 mg à 30 mg par jour) en complément de l'utilisation régulière de CSI à hautes doses et de bêta-agonistes à longue durée d'action (LABA), en association ou non avec un traitement complémentaire de contrôle de l'asthme. Les patients devaient avoir eu au moins une exacerbation au cours des 12 mois précédents. Après une phase de 8 semaines d'optimisation de la dose de CSO, les patients ont reçu des doses de 210 mg de tézépélumab SC administrées toutes les 4 semaines ou un placebo pendant une durée totale de 48 semaines. Pendant l'étude, les patients ont continué de recevoir le traitement de base de l'asthme qu'ils prenaient jusqu'alors; leur dose de CSO a toutefois été réduite toutes les 4 semaines (semaine 4 à 40) pendant la phase de réduction des CSO, tant que le contrôle de l'asthme était maintenu. La phase de réduction des CSO a été suivie d'une phase d'entretien de 8 semaines au cours de laquelle les patients devaient maintenir la dose de CSO atteinte à la semaine 40.
Les données démographiques et les caractéristiques de départ de ces 3 études figurent dans le Tableau 2 ci-dessous.
Tableau 2: Données démographiques
et caractéristiques de départ des
études sur l'asthme
PATHWAY n = 550 NAVIGATOR n = 1059 SOURCE n = 150
Âge moyen (ans) (ET) 52 (12) 50 (16) 53 (12)
Femmes (%) 66 64 63
Caucasiens (%) 92 62 84
Noirs ou Afro-américains (%) 3 6 1
Asiatique (%) 3 28 15
Hispaniques ou Latinos (%) 1 15 16
Patients n'ayant jamais fumé (%) 81 80 74
Utilisation de CSI à hautes doses 49 75 99
(%)
Utilisation de CSO (%) 9 9 100
Nombre moyen d'exacerbations 2,4 (1,2) 2,8 (1,4) 2,0 (1,5)
l'année précédente (ET)
Durée moyenne de l'asthme 17 (12) 22 (16) 23 (15)
(années) (ET)
Valeur initiale moyenne en % de 60 (13) 63 (18) 54 (18)
la valeur prédite du VEMS (ET)
Réversibilité moyenne du VEMS 23 (20) 15 (15) 15 (15)
post-bronchodilatateur (%) (ET)
Valeur initiale moyenne du nombre 371 (353) 340 (403) 242 (180)
d'EOS dans le sang (cellules/µl)
(ET)
Statut allergique positif (%)a 43 64 39
FeNO moyenne (ppb) (ET) 35 (39) 44 (41) 41 (39)
ACQ-6 moyen (ET) 2,7 (0,8) 2,8 (0,8) 2,5 (1,1)
a Statut allergique positif défini par un résultat positif au test de recherche sérique d'IgE spécifique à l'un des aéro-allergènes pérennes dans le profil FEIA.
ACQ-6: Asthma Control Questionnaire 6 (questionnaire du contrôle de l'asthme); EOS: éosinophiles; FEIA: Fluorescent-Enzyme IimmunoAssay (dosage immunoenzymatique par fluorescence); FeNO: Fractional Exhaled Nitric Oxide (fraction du monoxyde d'azote expiré); VEMS: volume expiratoire maximal par seconde; CSI: corticostéroïde inhalé; IgE: Immunoglobuline E; CSO: corticostéroïde oral; ppb: parts per billion (parts par milliard); ET: écart type.
Les résultats résumés ci-après se réfèrent au schéma posologique recommandé de 210 mg SC de tézépélumab administré toutes les 4 semaines.
Exacerbations
Le critère principal d'évaluation des études PATHWAY et NAVIGATOR était le taux d'exacerbations de l'asthme cliniquement significatives pendant 52 semaines. Les exacerbations de l'asthme cliniquement significatives étaient définies par une aggravation de l'asthme nécessitant l'utilisation ou l'augmentation de la dose de corticostéroïdes oraux ou systémiques pendant au moins 3 jours ou l'administration d'une injection unique de corticostéroïdes à effet retard et/ou une consultation au service donnant lieu à l'administration de corticostéroïdes oraux ou systémiques et/ou à une hospitalisation.
Chez les patients inclus dans les études PATHWAY et NAVIGATOR traités par TEZSPIRE, une réduction significative du taux annualisé d'exacerbations de l'asthme a été observée en comparaison avec le placebo (Tableau 3). En outre, les patients traités par TEZSPIRE présentaient moins d'exacerbations nécessitant une consultation au service des urgences et/ou une hospitalisation que les patients sous placebo. De plus, le pourcentage de patients n'ayant présenté aucune exacerbation de l'asthme pendant le traitement d'une durée de 52 semaines était plus important sous TEZSPIRE que sous placebo.
Tableau 3: Taux
d'exacerbations
cliniquement signifi
catives pendant 52
semaines lors de
l'étude PATHWAY et
de l'étude NAVIGATOR
PATHWAY NAVIGATOR
TEZSPIRE n = 137 Placebo n = 138 TEZSPIRE n = 528 Placebo n = 531
Taux annualisé
d'exacerbations de
l'asthme
Taux 0,20 0,72 0,93 2,10
Rapport des taux 0,29 (0,16–0,51) 0,44 (0,37–0,53)
(IC à 95 %)
Valeur p < 0,001 < 0,001
Le taux d'exacerbations ayant nécessité une hospitalisation/une admission au service des urgences était de 0,03 chez les patients recevant TEZSPIRE contre 0,18 chez les patients sous placebo (rapport des taux 0,15; IC à 95 % 0,04–0,58, p = 0,005) pour l'étude PATHWAY et 0,06 contre 0,28 (rapport des taux 0,21; IC à 95 % 0,12–0,37, p < 0,001) pour l'étude NAVIGATOR. Des résultats similaires ont été observés pour la réduction du taux d'exacerbations ayant nécessité une seule hospitalisation (0,02 contre 0,14 [rapport des taux 0,14; IC à 95 % 0,03–0,71, p = 0,017]) pour l'étude PATHWAY et 0,03 contre 0,19 (rapport des taux 0,15; IC à 95 % 0,07–0,33, p < 0,001) pour l'étude NAVIGATOR.
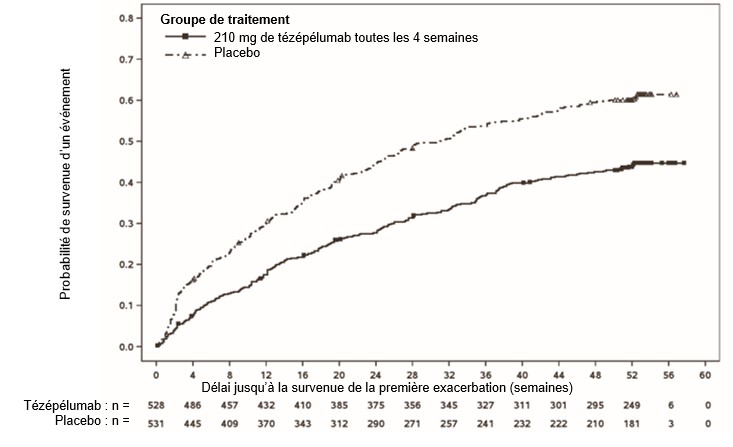
Le délai jusqu'à la survenue de la première exacerbation était plus long chez les patients sous TEZSPIRE que chez les patients sous placebo lors de l'étude NAVIGATOR (Figure 1). Des résultats similaires ont été observés lors de l'étude PATHWAY.
Figure 1: Courbes d'incidence cumulée de Kaplan-Meier pour le délai jusqu'à la survenue de la première exacerbation pendant 52 semaines, étude NAVIGATOR
Analyse en sous-groupes
Au cours de l'étude NAVIGATOR, TEZSPIRE a induit une réduction du taux d'exacerbations de l'asthme tant dans la population globale que dans le sous-groupe de patients présentant des valeurs initiales des éosinophiles dans le sang < 300 cellules/µl.
Figure 2: Rapport du taux annualisé d'exacerbations de l'asthme sur 52 semaines pour différentes valeurs initiales des biomarqueurs dans l'étude NAVIGATOR
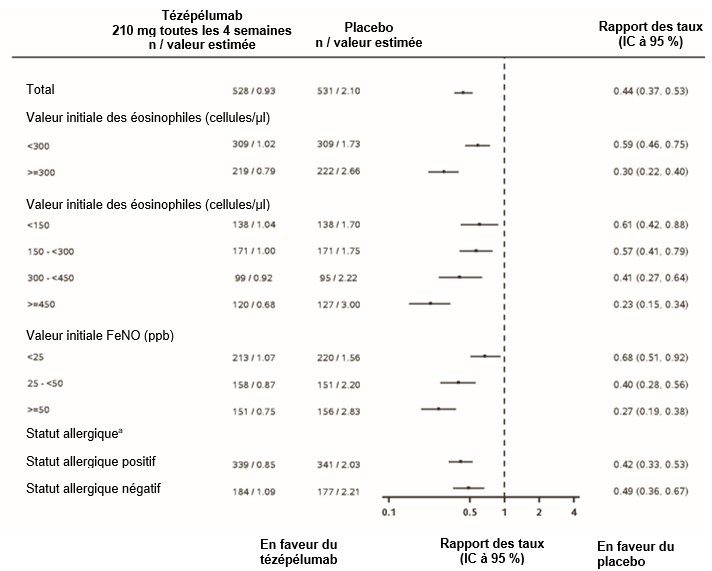
aStatut allergique défini comme le résultat des IgE sériques spécifiques à l'un des aéroallergènes pérennes dans le profil FEIA.
Fonction pulmonaire
La variation du VEMS par rapport à la valeur initiale a été évaluée en tant que critère d'évaluation secondaire des études PATHWAY et NAVIGATOR. En comparaison avec le placebo, TEZSPIRE a induit une amélioration cliniquement significative de la variation moyenne du VEMS par rapport à la valeur initiale lors des deux études (Tableau 4).
Tableau 4: Variation
moyenne du VEMS
pré-bronchodilatateu
r vs valeur initiale
après 52 semaines
lors des études
PATHWAY et NAVIGATOR
PATHWAY NAVIGATOR
TEZSPIRE n = 133* Placebo n = 138* TEZSPIRE n = 527* Placebo n = 531*
Variation moyenne 0,08 -0,06 0,23 0,10
des MC vs valeur
initiale (l)
Différence moyenne 0,13 (0,03–0,23) 0,13 (0,08–0,18)
des MC par rapport
au placebo (l) (IC
à 95 %)
Valeur p 0,009 †* < 0,001
* Nombre de patients contribuant à l'analyse complète (AC) présentant au moins une variation par rapport à la valeur initiale.
†* Valeur p nominale
IC: intervalle de confiance; VEMS: volume expiratoire maximal par seconde; MC: moindres carrés.
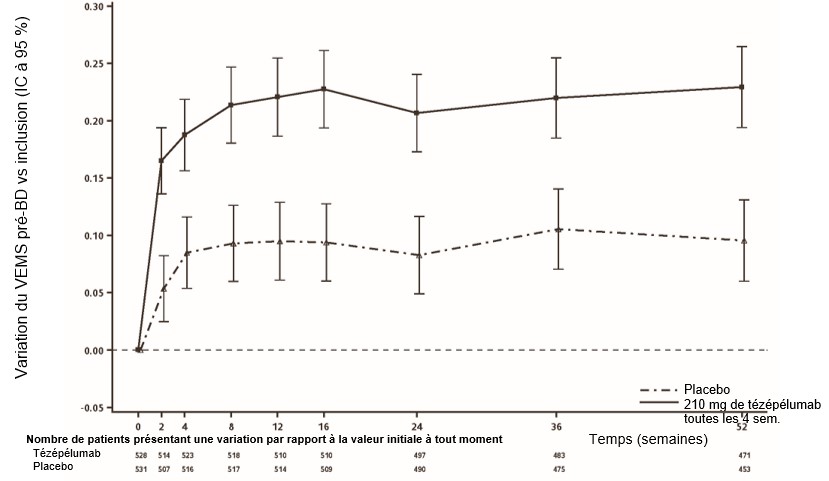
Lors de l'étude NAVIGATOR, une amélioration de la valeur du VEMS a été constatée seulement 2 semaines après l'instauration du traitement et s'est maintenue jusqu'à la semaine 52 (Figure 3).
Figure 3: Variation moyenne (IC à 95 %) du VEMS pré-bronchodilatateur (l) vs valeur initiale lors de l'étude NAVIGATOR
Auto-évaluations des patients
Les variations du score du questionnaire du contrôle de l'asthme (Asthma Control Questionnaire 6, ACQ-6) et du questionnaire standardisé sur la qualité de vie des personnes asthmatiques dès l'âge de 12 ans (Standardised Asthma Quality of Life Questionnaire, AQLQ[S]+12) par rapport aux valeurs initiales ont été évaluées en tant que critères d'évaluation secondaires lors des études PATHWAY et NAVIGATOR. Les résultats de l'étude NAVIGATOR figurent dans le Tableau 5. Dans les deux études, des améliorations de l'ACQ-6 et de l'AQLQ(S)+12 ont été observées seulement 2 et 4 semaines respectivement après le début de l'administration de TEZSPIRE et se sont maintenues jusqu'à la semaine 52.
Davantage de patients sous TEZSPIRE que sous placebo ont présenté une amélioration cliniquement significative de l'ACQ-6 et de l'AQLQ(S)+12 lors des deux études. Une amélioration cliniquement significative (taux de répondants) de l'ACQ-6 et de l'AQLQ(S)+12 était définie par une amélioration du score de 0,5 à la fin de l'étude. Lors de l'étude 2, le taux de répondants ACQ-6 était de 86 % pour TEZSPIRE contre 77 % pour le placebo (odds ratio = 1,99; IC à 95 % 1,43–2,76) et le taux de répondants AQLQ(S)+12 était de 78 % pour TEZSPIRE contre 72 % pour le placebo (odds ratio = 1,36; IC à 95 % 1,02–1,82). Des résultats similaires ont été observés lors de l'étude PATHWAY.
Les scores moyens hebdomadaires consignés dans le journal de l'asthme (Asthma Symptom Diary, ASD) ont également été évalués en tant que critère d'évaluation secondaire lors de l'étude NAVIGATOR. La sévérité de la respiration sifflante, de l'essoufflement, de la toux et de l'oppression thoracique a été évaluée deux fois par jour (matin et soir). Les réveils nocturnes et l'activité ont été évalués tous les jours. Le score ASD total a été calculé comme étant la moyenne de 10 items. Davantage de patients sous TEZSPIRE que sous placebo ont présenté une amélioration cliniquement significative du score ASD. Une amélioration cliniquement significative (taux de répondants) était définie par une amélioration du score d'au moins 0,5 à la fin de l'étude. Le taux de répondants ASD était de 58 % pour TEZSPIRE contre 51 % pour le placebo (odds ratio = 1,68; IC à 95 % 1,12–2,53).
Tableau 5 Résultats
de l'AQLQ(S)+12,
l'ACQ-6 et l'ASD
après 52 semaines
lors de l'étude
NAVIGATOR
n* Variation moyenne Différence par Valeur p
des MC vs valeur rapport au placebo
initiale (IC à 95 %)
Score AQLQ(S)+12
total
TEZSPIRE 524 1,48 0,33 (0,20–0,47) < 0,001
Placebo 526 1,14
Score ACQ-6
TEZSPIRE 527 -1,53 -0,33 (-0,46–-0,20) < 0,001
Placebo 531 -1,20
ASD
TEZSPIRE 525 -0,70 -0,11 (-0,19–-0,04) 0,004
Placebo 531 -0,59
* Nombre de patients contribuant à l'analyse complète (AC) présentant au moins une variation par rapport à la valeur initiale.
ACQ-6: Asthma Control Questionnaire 6 (questionnaire du contrôle de l'asthme); AQLQ(S)+12: questionnaire standardisé sur la qualité de vie des personnes asthmatiques âgées de 12 ans et plus; ASD: Asthma Symptom Diary (journal de l'asthme); IC: intervalle de confiance; MC: moindres carrés.
Réduction des corticostéroïdes oraux
L'effet de TEZSPIRE sur la réduction du traitement d'entretien par CSO a été évalué lors de l'étude SOURCE. Le critère principal d'évaluation était la réduction de la dose finale de CSO, classée par catégories de pourcentage, après 48 semaines par rapport à la valeur initiale (réduction ≥90 %, réduction ≥75 % à < 90 %, réduction ≥50 % à < 75 %, réduction > 0 % à < 50 % et aucune modification ou une augmentation) en maintenant le contrôle de l'asthme. En comparaison avec le placebo, numériquement plus de patients sous TEZSPIRE ont obtenu une réduction de la dose d'entretien de CSO par rapport à la valeur initiale sans perte de contrôle de l'asthme (odds ratio cumulé = 1,28; IC à 95 % 0,69–2,35); la différence n'était toutefois statistiquement pas significative.
Figure 4: Pourcentage de réduction de la dose quotidienne finale de CSO à la semaine 48 en fonction des différents biomarqueurs de base, étude SOURCE
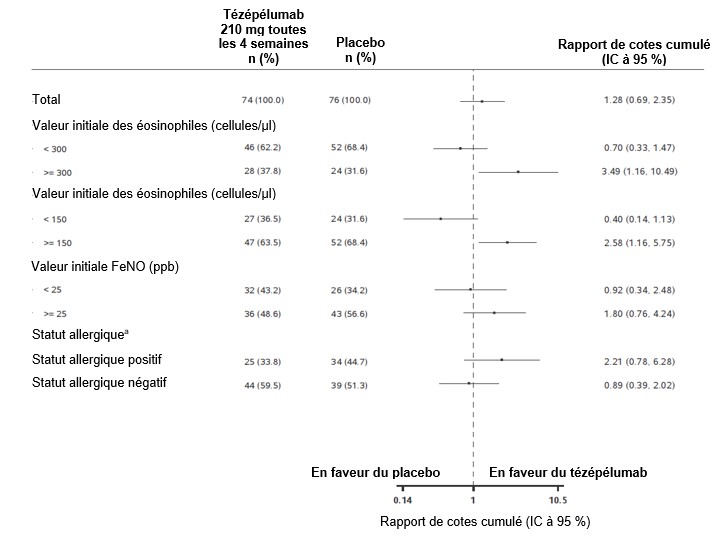
aStatut allergique défini comme le résultat des IgE sériques spécifiques à l'un des aéroallergènes pérennes dans le profil FEIA.
Les critères d'évaluation secondaires de l'étude SOURCE, dont le taux annualisé d'exacerbations de l'asthme, la variation du VEMS pré-bronchodilatateur par rapport à la valeur initiale ainsi que l'ACQ-6 et l'AQLQ(S)+12, n'ont pas mis en évidence de différences statistiquement significatives avec TEZSPIRE en comparaison avec le placebo.
Rhinosinusite chronique avec polypose nasale (CRSwNP)
L'efficacité de TEZSPIRE a été évaluée au cours d'une étude en groupes parallèles, randomisée, en double aveugle, multicentrique et contrôlée contre placebo (WAYPOINT) menée sur une durée traitement de 52 semaines auprès de 408 patients âgés d'au moins 18 ans qui suivaient le traitement standard pour une CRSwNP. Une administration au-delà de 52 semaines n'a pas été étudiée chez CRSwNP.
Cette étude incluait des patients présentant une CRSwNP symptomatique malgré un traitement par corticostéroïdes systémiques au cours des 12 derniers mois et/ou une chirurgie nasosinusienne, ou présentant des contre-indications/intolérances à l'égard de ces deux approches.
Les patients ont reçu 210 mg de TEZSPIRE ou le placebo par injection SC toutes les 4 semaines pendant 52 semaines en plus du traitement intranasal de la CRSwNP par des corticostéroïdes.
Les données démographiques et les caractéristiques de départ de l'étude WAYPOINT figurent dans le Tableau 6 ci-dessous.
Tableau 6: Données démographiques et caractéristiques de départ de l'étude WAYPOINT
WAYPOINT n = 408a
Âge moyen (ans) (ET) 50 (14)
Hommes (%) 65
Durée moyenne de la CRSwNP (années) (ET) 13 (10)
Patients avec ≥ 1 antécédent de chirurgie (%) 71
Patients ayant utilisé des corticostéroïdes contre la CRSwNP l’année 58
précédente (%)
NPS total moyenb (ET), plage 0–8 6,1 (1,2)
NCS bihebdomadaire moyenb, c (ET), plage 0–3 2,6 (0,5)
Perte d’odorat bihebdomadaire moyenneb, d (ET), plage 0–3 2,9 (0,4)
Score total SNOT-22 moyenb (ET), plage 0–110 69 (18)
Nombre moyen d’éosinophiles dans le sang (cellules/µl) (ET) 358 (238)
Nombre moyen d’IgE totales (UI/ml) (ET) 176 (285)
Asthmee (%) 61
NSAR-ERD/AERD (%) 17
Rhinite allergique (%) 14
a Nombre de patients (n) = 407 pour le NPS total moyen; n = 406 pour le NCS bihebdomadaire moyen et la perte d'odorat bihebdomadaire moyenne; n = 404 pour le score total LMK-CT moyen et le nombre moyen d'éosinophiles dans le sang; n = 389 pour le nombre moyen d'IgE totales.
b Un score élevé indique une sévérité plus importante de l'affection ou des symptômes.
c Évalué dans le cadre du journal des symptômes pour la polypose nasale (Nasal Polyposis Symptom Diary, NPSD).
d Évalué au moyen du score relatif aux problèmes affectant l'odorat dans le NPSD.
e Inclut les patients atteints d'asthme, d'AERD ou de NSAR-ERD. Un diagnostic d'asthme a également été posé chez l'ensemble des patients atteints d'AERD ou de NSAR-ERD de ce sous-groupe, sauf 3. AERD: maladie respiratoire exacerbée par l'aspirine; CRSwNP: rhinosinusite chronique avec polypose nasale; CT: tomodensitométrie; IgE: immunglobuline E; UI: unités internationales; LMK: Lund-Mackay; NCS: Nasal congestion score; NPS: Nasal polyp score; NSAR-ERD: maladie respiratoire exacerbée par les AINS; ET: écart-type; SNOT-22: Sino-Nasal Outcome Test à 22 items.
Les critères d'évaluation coprimaires de l'efficacité étaient la variation du score de polypose nasale (NPS) total, évalué par endoscopie nasale après 52 semaines par rapport aux valeurs initiales au moyen d'une classification par des évaluateurs indépendants en aveugle, ainsi que la variation du score de congestion nasale (NCS) bihebdomadaire moyen par rapport aux valeurs initiales selon l'évaluation effectuée dans le cadre du journal des symptômes de polypose nasale (NPSD) à la semaine 52. Le NPS total était classé au moyen d'une échelle catégorielle (0–8). La congestion nasale était évaluée quotidiennement par les patients sur une échelle catégorielle de sévérité allant de 0 à 3. Les valeurs p non ajustées sont indiquées pour l'étude WAYPOINT.
Dans le cadre de l'étude WAYPOINT, on a observé une efficacité statistiquement significative en ce qui concerne l'amélioration du NPS total et du NCS bihebdomadaire moyen à la semaine 52 (voir Tableau 7).
Tableau 7: Résultats des critères d'évaluation coprimaires après 52 semaines dans l'étude WAYPOINT
TEZSPIRE (n = 203) Placebo (n = 205) Différence de la
moyenne LS vs
placebo (IC à 95 %)
Scores Moyenne à l'inclusio Variation de la Moyenne à l'inclusio Variation de la
n moyenne LS n moyenne LS
NPS 6,1 -2,46 6,1 -0,38 -2,08 (-2,40,
-1,76) p<0,0001a
NCS 2,59 -1,74 2,55 -0,70 -1,04 (-1,21,
-0,87) p<0,0001a
a Les valeurs p non ajustées sont indiquées. Variation de la moyenne LS: variation de la moyenne des moindres carrés par rapport à la valeur initiale; une réduction du score indique une amélioration; NCS: Nasal congestion score; NPS: Nasal polyp score.
Figure 5: Variation de la moyenne LS du score de polypose nasale total jusqu'à la semaine 52 par rapport aux valeurs initiales
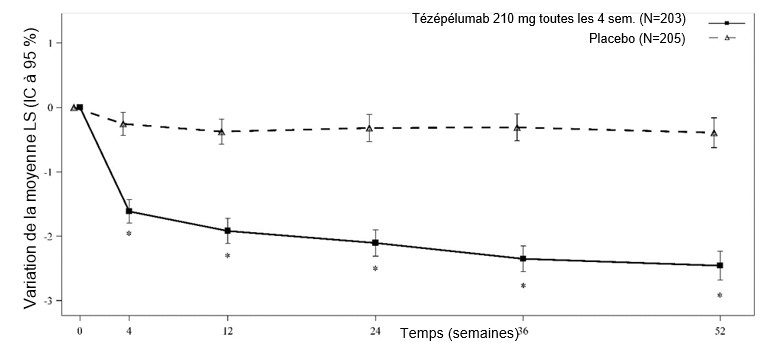
* Indique une valeur p non ajustée <0,01 pour la comparaison du traitement tézépélumab 210 mg toutes les 4 semaines versus placebo.
Après 52 semaines, l'amélioration du critère d'évaluation primaire du NPS total par rapport au placebo était cohérent chez les patients avec et sans antécédent de chirurgie nasosinusienne ainsi que chez les patients avec et sans asthme concomitant.
Figure 6: Variation de la moyenne LS du score de congestion nasale bihebdomadaire moyen jusqu'à la semaine 52 par rapport aux valeurs initiales
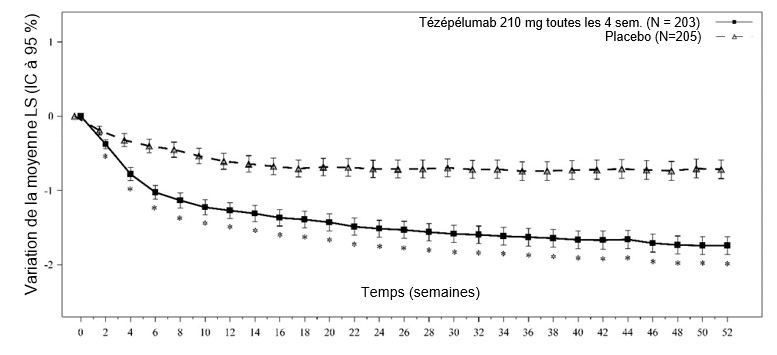
* Indique une valeur p non ajustée <0,01 pour la comparaison du traitement tézépélumab 210 mg toutes les 4 semaines versus placebo.
Après 52 semaines, l'amélioration du critère d'évaluation primaire du NCS total par rapport au placebo était cohérent chez les patients avec et sans antécédent de chirurgie nasosinusienne ainsi que chez les patients avec et sans asthme concomitant.
TEZSPIRE a amélioré significativement la perte d'odorat par rapport au placebo. Après 52 semaines, la différence de la moyenne LS pour la perte d'odorat (évaluée par le score NPDS bihebdomadaire moyen relatif aux problèmes affectant l'odorat) était de -1,01 [IC à 95 %: -1,18, -0,83; p<0,0001] dans le groupe TEZSPIRE par rapport au groupe placebo. Une amélioration significative de la perte d'odorat chez les patients traités par TEZSPIRE par rapport aux patients qui recevaient le placebo pouvait déjà être observée dès la première évaluation après 2 semaines.
TEZSPIRE a montré une amélioration significative des symptômes nasosinusaux, mesurés au moyen du score SNOT-22, après 52 semaines comparativement au placebo (différence de la moyenne LS -27,44 [IC à 95 %: -32,51, -22,37; p<0,0001]).
Sur 52 semaines, TEZSPIRE a réduit significativement le nombre de patients nécessitant une chirurgie nasosinusienne ou des corticostéroïdes systémiques de 92 % par rapport au placebo (Hazard Ratio: 0,08; IC à 95 %: 0,03, 0,16; p<0,0001).
|