Propriétés/EffetsCode ATC
L04AC16
Mécanisme d'action
Le guselkumab est un anticorps monoclonal (AcM) IgG1λ humain qui se lie de façon sélective via les sites de liaison à l'antigène à l'interleukine 23 (IL-23) avec une spécificité et une affinité élevées. L'IL-23 agit notamment sur la différenciation, l'expansion et la survie de certaines sous-populations de lymphocytes T (p. ex.lymphocytes Th17) et de certaines sous-populations de cellules de l'immunité innée, ainsi que sur la libération des cytokines proinflammatoires IL-17A, IL-17F et IL-22. Chez l'homme, il a été montré que le blocage sélectif de l'IL-23 permet de normaliser la production de ces cytokines.
Dans les modèles in vitro, il a été montré que le guselkumab inhibe la bioactivité de l'IL-23 en bloquant son interaction avec le récepteur de surface cellulaire de l'IL-23, perturbant ainsi la signalisation, l'activation et la cascade cytokinique médiées par l'IL-23.
Dans le psoriasis, la maladie de Crohn et la colite ulcéreuse, les cellules myéloïdes qui expriment le récepteur Fc-gamma 1 (CD64) se sont révélées être la source principale d'IL-23 dans les tissus enflammés. Pour le guselkumab, le blocage de l'IL-23 et la liaison au CD64 ont été démontrés in vitro. Ces résultats indiquent que le guselkumab est en mesure de neutraliser l'IL-23 dans les sites présentant une inflammation cellulaire.
Pharmacodynamique
Psoriasis en plaques
Les taux d'IL-23 sont élevés dans la peau des patients atteints de psoriasis en plaques. Le guselkumab exerce ses effets cliniques sur le psoriasis en plaques en inhibant la voie cytokinique de l'IL-23 par modulation de profils d'expression de gènes dans les zones cutanées concernées. Ces effets locaux entraînent une réduction de l'épaisseur de l'épiderme et de la concentration des lymphocytes T. Par ailleurs, lors des études de phase II et de phase III, une réduction des taux sériques d'IL-17A, d'IL-17F et d'IL-22 a été observée chez les patients atteints de psoriasis en plaques traités par le guselkumab comparés au groupe placebo.
Arthrite psoriasique
Dans les études de phase III conduites chez des patients présentant une arthrite psoriasique, les taux sériques de protéines de phase aiguë, protéine C-réactive, sérum amyloïde A et IL-6 ainsi que les taux de cytokines effectrices des Th17 (IL-17A, IL-17F et IL-22) étaient élevés au début de l'étude. Le guselkumab a abaissé la concentration de ces protéines dans les 4 semaines suivant l'instauration du traitement. Le guselkumab a réduit la concentration de ces protéines jusqu'à la semaine 24 par rapport à la valeur initiale et au placebo.
Efficacité clinique
Psoriasis en plaques
L'efficacité et la sécurité du guselkumab ont été étudiées au cours de quatre études de phase III randomisées, en double aveugle, contrôlées versus placebo et/ou comparateur actif, menées chez des patients adultes atteints de psoriasis en plaques modéré à sévère, candidats à une photothérapie ou à un traitement systémique. Deux études (VOYAGE 1 et VOYAGE 2) ont évalué l'efficacité et la sécurité du guselkumab versus placebo et adalimumab chez 1829 patients adultes. Dans l'étude VOYAGE 2, l'arrêt du guselkumab et la reprise du traitement ont été en outre étudiés à la semaine 28 chez les patients répondeurs, par rapport à la poursuite du traitement en continu. Les patients qui avaient déjà été traités par le guselkumab ou l'adalimumab, ainsi que les patients présentant un psoriasis érythrodermique, un psoriasis en gouttes ou un psoriasis pustuleux ont été exclus des études VOYAGE 1 et VOYAGE 2. Une autre étude (NAVIGATE) a évalué l'efficacité et la sécurité du guselkumab versus ustékinumab chez 268 patients adultes ayant présenté une réponse insuffisante à l'ustékinumab.
L'étude clinique (ORION) visait à évaluer l'efficacité, la sécurité, la PK, l'immunogénicité, les possibilités d'utilisation et l'acceptation du guselkumab administré avec le stylo prérempli.
VOYAGE 1 et VOYAGE 2
Les patients randomisés dans le groupe guselkumab ont reçu 100 mg aux semaines 0 et 4, puis toutes les 8 semaines jusqu'à la semaine 48 (VOYAGE 1) ou jusqu'à la semaine 20 (VOYAGE 2). Les patients randomisés dans le groupe adalimumab ont reçu 80 mg à la semaine 0 et 40 mg à la semaine 1, puis 40 mg toutes les deux semaines jusqu'à la semaine 48 (VOYAGE 1) ou jusqu'à la semaine 23 (VOYAGE 2). Dans les deux études, les patients randomisés dans le groupe placebo ont reçu 100 mg de guselkumab aux semaines 16 et 20, puis toutes les 8 semaines. Dans l'étude VOYAGE 1, tous les patients, y compris ceux qui avaient été randomisés dans le groupe sous adalimumab à la semaine 0, ont reçu 100 mg de guselkumab en ouvert à la semaine 52, puis toutes les 8 semaines. Dans l'étude VOYAGE 2, les patients qui avaient été randomisés dans le groupe guselkumab à la semaine 0 et présentaient une amélioration du PASI (Psoriasis Area and Severity Index) d'au moins 90% (réponse PASI 90) à la semaine 28 ont été re-randomisés soit pour poursuivre le traitement par le guselkumab toutes les 8 semaines (traitement d'entretien), soit pour recevoir le placebo (arrêt du traitement). Les patients appartenant au dernier groupe (nouvelle randomisation après placebo) ont à nouveau été traités par le guselkumab après la perte d'au moins 50% de l'amélioration de leur PASI à la semaine 28 (posologie au moment de la reprise du traitement ainsi que 4 semaines plus tard, puis toutes les 8 semaines). Les patients du groupe guselkumab sans réponse PASI 90 ont poursuivi le traitement par le guselkumab. Chez les patients qui avaient été randomisés dans le groupe sous adalimumab et qui présentaient une réponse PASI 90 à la semaine 28, le traitement a été arrêté et un traitement par le guselkumab a été instauré en cas de perte d'au moins 50% de l'amélioration du PASI à la semaine 28. Les patients qui avaient été randomisés dans le groupe sous adalimumab à la semaine 0 et qui n'avaient pas obtenu de réponse PASI 90 ont reçu le guselkumab aux semaines 28 et 32, puis toutes les 8 semaines. Tous les participants de l'étude ont reçu le guselkumab en ouvert toutes les 8 semaines à partir de la semaine 76.
Dans les études VOYAGE 1 et 2, les caractéristiques de la maladie à l'inclusion étaient homogènes au sein des populations des études, avec respectivement une moyenne de surface corporelle atteinte (SCA) de 22% et 24%, une médiane de score PASI à l'inclusion de 19 dans les deux études, un score IGA à l'inclusion (Investigator's Global Assessment, évaluation globale par l'investigateur) respectivement jugé «modéré» ou «sévère» chez 74,6% et 75,5% ou 25,1% et 24,5% des patients. 19% et 18% des patients avaient des antécédents d'arthrite psoriasique.
Parmi l'ensemble des patients inclus dans VOYAGE 1 et 2, respectivement 32% et 29% étaient naïfs à la fois de traitement systémique conventionnel et de traitement biologique, 54% et 57% avaient déjà reçu précédemment une photothérapie et 62% et 64% avaient déjà reçu un traitement systémique conventionnel. Dans les deux études, 21% des patients avaient déjà reçu précédemment un traitement biologique, parmi lesquels 11% avaient reçu au moins un anti-TNFα (facteur de nécrose tumorale alpha) et environ 10% un anti-IL-12/IL-23.
Un traitement topique ou systémique associé ou une photothérapie concomitante contre le psoriasis n'étaient pas autorisés dans l'étude.
L'efficacité du guselkumab a été évaluée sur la base de l'atteinte cutanée globale, de l'atteinte localisée du cuir chevelu, des mains, des pieds et des ongles, ainsi que de la qualité de vie. Les co-critères principaux d'évaluation dans les études VOYAGE 1 et 2 étaient le pourcentage de patients ayant obtenu un score IGA de type «blanchi» ou «lésion minime» (IGA 0/1) et une réponse PASI 90 à la semaine 16 versus placebo (voir le tableau 1).
Effets sur les symptômes cutanés
Le traitement par le guselkumab a entraîné des améliorations significatives des paramètres de l'activité de la maladie par rapport au placebo à la semaine 16 et par rapport à l'adalimumab aux semaines 16 et 48. Les principaux résultats d'efficacité sont présentés dans le tableau 1 ci-dessous.
Tableau 1: Résumé des réponses cliniques observées lors des études VOYAGE 1 et VOYAGE 2
|
|
Nombre de patients (%)
| |
Placebo (n = 174)
|
VOYAGE 1 Guselkumab (n = 329)
|
Adalimumab (n = 334)
|
Placebo (n = 248)
|
VOYAGE 2 Guselkumab (n = 496)
|
Adalimumab (n = 248)
| |
Semaine 16
| |
PASI 75
|
10 (5,7)
|
300 (91,2)a
|
244 (73,1)b
|
20 (8,1)
|
428 (86,3)a
|
170 (68,5)b
| |
PASI 90
|
5 (2,9)
|
241 (73,3)c
|
166 (49,7)b
|
6 (2,4)
|
347 (70,0)c
|
116 (46,8)b
| |
IGA 0/1
|
12 (6,9)
|
280 (85,1)c
|
220 (65,9)b
|
21 (8,5)
|
417 (84,1)c
|
168 (67,7)b
| |
IGA 0
|
2 (1,1)
|
157 (47,7)a
|
88 (26,3)d
|
2 (0,8)
|
215 (43,3)a
|
71 (28,6)d
| |
Semaine 48
| |
PASI 75
|
-
|
289 (87,8)
|
209 (62,6)e
|
-
|
-
|
-
| |
PASI 90
|
-
|
251 (76,3)
|
160 (47,9)b
|
-
|
-
|
-
| |
IGA 0/1
|
-
|
265 (80,5)
|
185 (55,4)b
|
-
|
-
|
-
| |
IGA 0
|
-
|
166 (50,5)
|
86 (25,7)b
|
-
|
-
|
-
| |
a
p < 0,001 pour la comparaison entre le guselkumab et le placebo.
b p < 0,001 pour la comparaison entre le guselkumab et l'adalimumab sur les critères secondaires majeurs d'évaluation.
c p < 0,001 pour la comparaison entre le guselkumab et le placebo sur les co-critères principaux d'évaluation.
d Des comparaisons entre le guselkumab et l'adalimumab n'ont pas été effectuées.
e p < 0,001 pour la comparaison entre le guselkumab et l'adalimumab.
|
Des améliorations statistiquement significatives du psoriasis des ongles, de l'atteinte du cuir chevelu et de l'atteinte palmoplantaire ont également été observées à la semaine 16 par rapport au placebo.
Réponse au cours du temps
Le guselkumab a montré une efficacité d'apparition rapide, avec un pourcentage d'amélioration du score PASI significativement plus élevé comparé au placebo dès la semaine 2 (p < 0,001), avec une différence maximale atteinte autour de la semaine 20 (VOYAGE 1 et 2) et se maintenant jusqu'à la semaine 48 (VOYAGE 1).
Figure 1: Pourcentage de patients ayant obtenu une réponse PASI 90 lors des différentes visites jusqu'à la semaine 48 (patients randomisés à la semaine 0) dans l'étude VOYAGE 1

Dans l'étude VOYAGE 1, le taux de réponse PASI 90 chez les patients sous traitement continu par le guselkumab a été maintenu pendant la période en ouvert de la semaine 52 à la semaine 252. Chez les patients qui avaient été randomisés dans le groupe sous adalimumab à la semaine 0 et qui sont passés au guselkumab à la semaine 52, le taux de réponse PASI 90 a augmenté entre la semaine 52 et la semaine 76 comprise, puis a été maintenu pendant la période en ouvert jusqu'à la semaine 252.
L'efficacité et la sécurité du guselkumab ont été démontrées indépendamment de l'âge, du sexe, de l'appartenance ethnique, du poids corporel, de la localisation des plaques, du score de sévérité PASI à l'inclusion, de la présence concomitante d'une arthrite psoriasique et de la prise d'un traitement antérieur biologique. Le guselkumab s'est avéré efficace chez les patients naïfs de traitement systémique conventionnel, chez les patients naïfs de traitement biologique et chez les patients précédemment exposés à un traitement biologique.
Arrêt, puis reprise du traitement
Dans l'étude VOYAGE 2, à la semaine 48, 88,6% des patients ayant reçu un traitement d'entretien par le guselkumab présentaient une réponse PASI 90 versus 36,8% des patients ayant arrêté le traitement à la semaine 28 (p < 0,001). Une perte de la réponse PASI 90 a été observée dès 4 semaines après l'arrêt du traitement par le guselkumab avec un délai médian de perte de la réponse PASI 90 d'environ 15 semaines. Quatre-vingts pour cent des patients ayant arrêté le traitement par le guselkumab et l'ayant repris ultérieurement ont obtenu une rémission PASI 90 lors de l'évaluation réalisée 20 semaines après le début de la reprise du traitement. Dans l'étude VOYAGE 2, respectivement 36% et 41% des 95 patients qui avaient été randomisés dans le groupe sous guselkumab et qui n'avaient pas obtenu de réponse PASI 90 à la semaine 28 ont obtenu une réponse PASI 90 après 20 et 44 semaines de poursuite du traitement par le guselkumab.
Résultats du traitement concernant différentes régions atteintes
Dans les études VOYAGE 1 et 2, à la semaine 16, des améliorations significativement plus importantes de l'atteinte du cuir chevelu (ss-IGA), des mains et des pieds (hf-PGA) et des ongles (NAPSI, f-PGA) ont été observées chez les patients traités par le guselkumab par rapport aux patients du groupe placebo.
Qualité de vie liée à la santé/résultats rapportés par les patients
Dans les études VOYAGE 1 et 2, à la semaine 16, des améliorations significativement plus importantes de la qualité de vie liée à la santé, mesurée à l'aide du Dermatology Life Quality Index (DLQI), et des symptômes (démangeaisons, douleurs, brûlures, picotements et tiraillements cutanés) et signes (sécheresse cutanée, fissures, desquamation, exfoliation, rougeurs et saignements) du psoriasis, rapportés par les patients dans le carnet de suivi Psoriasis Symptoms and Signs Diary (PSSD) ont été observées chez les patients traités par le guselkumab par rapport aux patients ayant reçu le placebo.
NAVIGATE
L'étude NAVIGATE a évalué l'efficacité du guselkumab chez des patients ayant présenté une réponse insuffisante à l'ustékinumab à la semaine 16 (c.-à-d. n'ayant pas de réponse de type «blanchi» ou «lésion minime», définie par un score IGA ≥2). Les patients ne devaient pas avoir reçu un traitement antérieur par le guselkumab et/ou l'ustékinumab. Tous les patients ont reçu un traitement par l'ustékinumab en ouvert aux semaines 0 et 4. À la semaine 16, 268 patients présentant un score IGA ≥2 ont été randomisés pour poursuivre le traitement par l'ustékinumab toutes les 12 semaines ou pour débuter un traitement par le guselkumab aux semaines 16 et 20, puis toutes les 8 semaines. Les caractéristiques à l'inclusion des patients randomisés étaient similaires à celles des patients des études VOYAGE 1 et 2.
12 semaines après la randomisation, la proportion de patients ayant obtenu un score IGA 0/1 et une amélioration ≥2 points a été plus élevée dans le groupe guselkumab que dans le groupe ustékinumab (31,1% vs 14,3%; p = 0,001), de même que la proportion de patients ayant obtenu une réponse PASI 90 (48% vs 23%; p < 0,001). Aucune donnée n'est disponible sur le passage inverse du guselkumab à l'ustékinumab.
ORION
L'étude ORION visait à évaluer l'efficacité, la sécurité, la PK, l'immunogénicité, les possibilités d'utilisation et l'acceptation du guselkumab administré avec le stylo prérempli. Dans cette étude, 78 patients atteints de psoriasis en plaques modéré à sévère ont été randomisés pour recevoir soit TREMFYA (100 mg aux semaines 0 et 4, puis toutes les 8 semaines) soit un placebo. La population de l'étude ORION était comparable à celle des études VOYAGE 1 et 2. L'efficacité, mesurée par le score IGA (0,1) et le PASI 90 à la semaine 16, était comparable dans les trois études ORION, VOYAGE 1 et 2. L'acceptation par les patients et la sécurité d'emploi du stylo ont été établies.
ECLIPSE
L'efficacité et la sécurité du guselkumab ont également été évaluées dans une étude en double aveugle versus sécukinumab. Les patients ont été randomisés pour recevoir le guselkumab (n = 534; 100 mg aux semaines 0 et 4, puis toutes les 8 semaines) ou le sécukinumab (n = 514; 300 mg aux semaines 0, 1, 2, 3, 4, puis toutes les 4 semaines). La dernière dose était administrée à la semaine 44 dans les deux groupes de traitement.
Le guselkumab était supérieur au sécukinumab en ce qui concerne le critère d'évaluation principal, la réponse PASI 90 à la semaine 48 (84,5% contre 70,0%, p < 0,001).
Arthrite psoriasique (AP)
Il a été établi que le guselkumab améliore les signes et les symptômes, la fonction physique et la qualité de vie liée à la santé.
DISCOVER 1 et DISCOVER 2
L'efficacité et la sécurité du guselkumab ont été évaluées, en comparaison avec un placebo, au cours de deux études de phase III, randomisées, en double aveugle et contrôlées contre placebo (DISCOVER 1 et DISCOVER 2), conduites chez des patients adultes atteints d'AP active (≥3 articulations enflées, ≥3 articulations douloureuses à la pression et un taux de protéine Créactive (CRP) de ≥0,3 mg/dl dans l'étude DISCOVER 1, et ≥5 articulations enflées, ≥5 articulations douloureuses à la pression et un taux de CRP de ≥0,6 mg/dl dans l'étude DISCOVER 2) malgré l'administration d'un traitement par un (cs)DMARD conventionnel synthétique, par l'aprémilast ou par un antirhumatismal non stéroïdien. L'AP des patients inclus dans ces études a été diagnostiquée sur la base des critères de classification pour l'arthrite psoriasique [CASPAR], avec une durée médiane de 4 ans. Des patients présentant différentes sous-formes d'AP – y compris l'arthrite polyarticulaire sans nodules rhumatoïdes (40%), la spondylarthrite avec arthrite périphérique (30%), l'arthrite périphérique asymétrique (23%), l'atteinte des articulations interphalangiennes distales (7%) et l'arthrite mutilante (1%) – ont été inclus dans les deux études. En raison du faible nombre de patients à l'étude présentant une arthrite mutilante (n = 6), aucune déclaration pertinente ne peut être faite à propos de l'efficacité et de la sécurité dans ce sous-groupe. Respectivement plus de 65% et 42% des patients présentaient une enthésite ou une dactylite au début de l'étude, et plus de 75% des patients présentaient ≥3% de la surface corporelle (SC) affectée par le psoriasis. Respectivement 381 et 739 patients ont été évalués dans les études DISCOVER 1 et DISCOVER 2; ces patients ont été traités soit par 100 mg de guselkumab aux semaines 0 et 4, puis toutes les 8 semaines (q8s), soit par 100 mg de guselkumab q4s, soit par un placebo. Dès la semaine 24, les sujets qui avaient reçu le placebo ont été traités par 100 mg de guselkumab toutes les 4 semaines dans les deux études. Environ 58% des patients des deux études ont reçu des doses stables de MTX en continu (≤25 mg/semaine).
Dans les deux études, plus de 90% des patients avaient déjà reçu précédemment un csDMARD. Dans l'étude DISCOVER 1, 31% des patients avait été traités antérieurement par un anti-TNFα biologique. Dans l'étude DISCOVER 2, aucun patient n'avait été traité au préalable par un agent biologique.
Signes et symptômes
À la semaine 24, le traitement par le guselkumab a entraîné des améliorations significatives des indicateurs de l'activité de la maladie, comparé au placebo. Le critère d'évaluation principal dans les deux études était le pourcentage de patients présentant une réponse ACR 20 de l'American College of Rheumatology à la semaine 24. Dans l'intervalle des traitements d'entretien par 100 mg de guselkumab respectivement toutes les 4 et 8 semaines, aucune différence cliniquement significative n'a été observée en matière d'efficacité. Le tableau 2 montre les événements principaux portant sur l'efficacité.
Tableau 2: Réponse clinique dans les études DISCOVER 1 et DISCOVER 2
|
|
DISCOVER 1 i
|
DISCOVER 2 j
| |
Placebo
|
100 mg q8s
|
Placebo
|
100 mg q8s
| |
(n = 126)
|
(n = 127)
|
(n = 246)
|
(n = 248)
| |
Réponse ACR20
| |
Semaine 16
|
25,40%
|
52,0% b
|
33,70%
|
55,2% g
| |
Semaine 24
|
22,20%
|
52,0% a
|
32,90%
|
64,1% a
| |
Réponse ACR50
| |
Semaine 16
|
12,70%
|
22,8% d
|
9,30%
|
28,6% g
| |
Semaine 24
|
8,70%
|
29,9% b
|
14,20%
|
31,5% g
| |
Réponse ACR70
| |
Semaine 24
|
5,60%
|
11,8% d
|
4,10%
|
18,5% g
| |
DAS 28 (CRP)-LS mean Modification par rapport à la valeur initiale
| |
Semaine 24 c
|
-0,7
|
-1,43 b
|
-0,97
|
-1,59 b
| |
a
p < 0,001 (critère d'évaluation principal)
b p < 0,001 (critère d'évaluation secondaire important)
c p = 0,006 (critère d'évaluation secondaire important)
d statistiquement non significatif p = 0,086 (critère d'évaluation secondaire important)
f nominal p < 0,012
g Aucun examen spécifique n'a été réalisé dans le cadre de la procédure de test hiérarchisée, nominal p < 0,001 (critère d'évaluation secondaire important)
i incluant ××> 90% des patients avec un traitement antérieur par un csDMARD
j incluant 31% des patients avec un traitement antérieur par un anti-TNFα biologique
|
Chez les patients atteints d'AP active et présentant ≥3% de la SC affectée ainsi qu'un score IGA ≥2 au début de l'étude, des amélioration significatives de la maladie cutanée ont été observées dans le groupe guselkumab-q8s, comparé au groupe placebo, tout comme cela avait été observé dans les études sur le psoriasis VOYAGE 1 et VOYAGE 2.
Réponse au cours du temps
Dans l'étude DISCOVER 2, une réponse ACR20 plus importante a été obtenue dès la semaine 4 dans les deux groupes traités par le guselkumab par rapport au placebo, et la différence entre les traitements s'est accentuée au cours du temps jusqu'au terme de la semaine 24 (figure 2).
Figure. 2: Réponse ACR20 lors des visites médicales jusqu'au terme de la semaine 24 dans l'étude DISCOVER 2
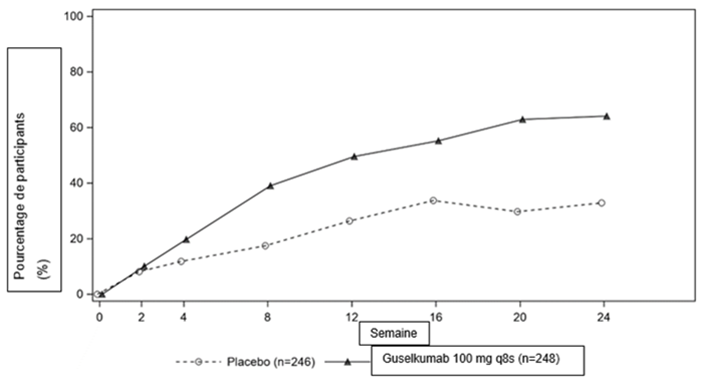
Dans l'étude DISCOVER 2, la réponse ACR20 s'est maintenue de la semaine 24 à la semaine 52 chez les patients ayant reçu un traitement continu par le guselkumab à la semaine 24. Chez les patients ayant reçu un traitement continu par le guselkumab à la semaine 52, la réponse ACR20 s'est maintenue de la semaine 52 à la semaine 100.
La réponse observée était comparable dans les groupes traités par le guselkumab, indépendamment de l'utilisation concomitante d'un csDMARD, y compris le MTX (DISCOVER 1 et 2). Par ailleurs, l'analyse de l'âge, du sexe, de l'ethnie, du poids corporel, de l'utilisation antérieure d'un csDMARD (DISCOVER 1 et 2) et de l'administration antérieure d'un anti-TNFα biologique (DISCOVER 1) n'a révélé aucune différence entre ces sous-groupes en matière de réponse au guselkumab.
La réponse clinique s'est maintenue jusqu'à la semaine 52 dans l'étude DISCOVER 1 et jusqu'à la semaine 100 dans l'étude DISCOVER 2, comme le montrent les taux de réponse selon ACR 20/50/70 et DAS 28 (CRP).
Le nombre de patients atteints d'AP avec une arthrite mutilante était trop faible pour permettre une évaluation pertinente.
Dans les études DISCOVER 1 et 2 des améliorations se sont produites pour tous les critères des scores ARC, y compris pour l'évaluation de la douleur par le patient.
L'évaluation de la dactylite et de l'enthésite a reposé sur les données groupées des patients présentant une dactylite ou une enthésite dans les études DISCOVER 1 et 2 (n = 160 et n = 230 respectivement). Le pourcentage de participants présentant une dactylite à l'inclusion et une rémission de la dactylite à la semaine 24 était plus élevé dans les groupes traités par le guselkumab q8s (59,4%, nominal p < 0,001) que dans le groupe placebo (42,2%). Le pourcentage de participants présentant une enthésite à l'inclusion et une rémission de l'enthésite à la semaine 24 était plus élevé dans les groupes traités par le guselkumab q8s (49,6%, nominal p < 0,001) que dans le groupe placebo (29,4%). À la semaine 52, la rémission de la dactylite (81,2%) et la rémission de l'enthésite (62,7%) se sont maintenues dans le groupe traité par le guselkumab q8s. Dans l'étude DISCOVER 2, la rémission de la dactylite et la rémission de l'enthésite se sont maintenues jusqu'à la semaine 100.
Réponse radiographique
Au cours de l'étude DISCOVER 2, l'inhibition de la progression des lésions structurelles a été évaluée radiographiquement et présentée comme une variation du score de van der Heijde-Sharp (vdH-S) modifié par rapport à la valeur initiale. À la semaine 24, le groupe traité par le guselkumab q8s a présenté une progression moindre sur le plan numérique que le groupe placebo (modification des moyennes des moindres carrés [least square mean change] par rapport à la valeur initiale, respectivement de 0,52 et de 0,95, p = 0,068). À la semaine 52 et à la semaine 100, la modification moyenne par rapport à la valeur initiale a été de respectivement de 0,97 et 1,5 dans le groupe guselkumab q8s.
Fonction physique et qualité de vie liée à la santé
Au cours des études DISCOVER 1 et 2, les patients traités par le guselkumab ont présenté une amélioration significative (p < 0,001) de la fonction physique à la semaine 24, évaluée à l'aide du questionnaire pour l'évaluation de la limitation physique (Health Assessment Questionnaire-Disability Index, HAQ-DI), par rapport au placebo. Les améliorations du HAQ-DI se sont maintenues de la semaine 24 jusqu'à la semaine 52 dans l'étude DISCOVER 1 et jusqu'à la semaine 100 dans l'étude DISCOVER 2.
Colite ulcéreuse (CU)
L'efficacité et la sécurité de TREMFYA ont été examinée dans deux études de phase III multicentriques, randomisées, en double aveugle, contrôlées par placebo (étude d'induction QUASAR et étude d'entretien QUASAR) auprès de patients adultes atteints de colite ulcéreuse active modérée à sévère qui avaient répondu de manière insuffisante ou n'avaient pas répondu aux corticostéroïdes, aux immunomodulateurs classiques, aux traitements par agents biologiques (inhibiteur de TNF, védolizumab) et/ou à un inhibiteur de Janus kinase (JAK), ou qui ne les avaient pas tolérés. De plus, l'efficacité et la sécurité de TREMFYA ont été évaluées dans une étude de phase IIb randomisée, en double aveugle, contrôlée par placebo visant à déterminer la dose d'induction (étude QUASAR de détermination de la dose d'induction).
L'activité de la maladie a été évaluée au moyen du score Mayo modifié (mMS), un score Mayo à 3 composantes (0–9), qui consiste en la somme des sous-scores suivants (0 à 3 pour chaque sous-score): fréquence des selles (SFS), hémorragies rectales (RBS) et résultats d'une endoscopie évaluée de manière centrale (ES). La colite ulcéreuse active modérée à sévère a été définie comme un mMS allant de 5 à 9, un RBS > 1 et un ES de 2 (défini par un érythème marqué, l'absence de profil vasculaire, une friabilité et/ou une érosion) ou un ES de 3 (défini par des hémorragies spontanées et des ulcérations).
Étude d'induction QUASAR: QUASAR-IS
Dans l'étude d'induction QUASAR-IS, les patients ont été randomisés selon un rapport de 3:2 et ont reçu soit TREMFYA 200 mg soit un placebo sous forme de perfusion intraveineuse aux semaines 0, 4 et 8. Au total, 701 patients ont été évalués. Au début de l'étude, le mMS médian était de 7, 35,5% des patients présentant au début de l'étude un mMS de 5 à 6 et 64,5% un mMS de 7 à 9. Au début de l'étude, 67,9% des patients avaient un ES de 3. L'âge médian était de 39 ans (18 à 79 ans), 43,1% étaient de sexe féminin et 72,5% étaient d'origine caucasienne, 21,4% des Asiatiques, 1% des Noirs, 0,1% des habitants indigènes américains ou d'Alaska et 0,1% appartenaient à différents groupes ethniques.
Les patients inclus pouvaient prendre des doses stables d'aminosalicylés oraux, de méthotrexate, de 6-MP, d'AZA et/ou de corticostéroïdes oraux. Au début du traitement, 72,5% des patients recevaient des aminosalicylés, 20,8% des immunomodulateurs (MTX, 6-MP ou AZA) et 43,1% des corticostéroïdes. Les traitements concomitants par des agents biologiques ou inhibiteurs de JAK n'étaient pas autorisés.
Au total, chez 49,1% des patients, un traitement par au moins un agent biologique et/ou un inhibiteur de JAK avait antérieurement échoué. Parmi ces patients, respectivement 88%, 54% et 18% n'avaient pas répondu à un traitement antérieur par inhibiteur de TNF, védolizumab ou inhibiteur de JAK. 47% n'avaient pas répondu à 2 ou plus de ces traitements. Au total, 48,4% des patients n'avaient pas encore été traités par un agent biologique ou un inhibiteur de JAK, et 2,6% avaient déjà reçu un agent biologique ou un inhibiteur de JAK auparavant et y avaient répondu.
Le critère d'évaluation principal était la rémission clinique, définie par le mMS à la semaine 12. Parmi les critères d'évaluation secondaires à la semaine 12, on trouvait la rémission symptomatique, la guérison endoscopique (amélioration endoscopique), la réponse clinique, la guérison histologique-endoscopique de la muqueuse (amélioration histologique-endoscopique de la muqueuse), l'amélioration des symptômes de fatigue et la rémission IBDQ (voir tableau 3).
À la semaine 12, il y avait significativement plus de patients en rémission clinique dans le groupe traité par TREMFYA que dans le groupe placebo.
Tableau 3: Proportion de patients ayant atteint les critères d'évaluation de l'efficacité à la semaine 12 dans l'étude QUASAR IS
|
Critère d'évaluation
|
Placebo
(n = 280)
|
TREMFYA
200 mg perfusion intraveineusea
(n = 421)
|
Différence entre les traitements
(IC à 95%)
| |
Rémission cliniqueb
| |
Population globale
|
22 (8%)
|
95 (23%)
|
15% (10%, 20%)c
| |
Rémission symptomatiquef
| |
Population globale
|
58 (21%)
|
210 (50%)
|
29% (23%, 36%)c
| |
Guérison endoscopique (amélioration endoscopique)g
| |
Population globale
|
31 (11%)
|
113 (27%)
|
16% (10%, 21%)c
| |
Réponse cliniqueh
| |
Population globale
|
78 (28%)
|
259 (62%)
|
34% (27%, 41%)c
| |
Guérison histologique-endoscopique de la muqueuse (amélioration histologique-endoscopique de la muqueuse)i
| |
Population globale
|
21 (8%)
|
99 (24%)
|
16% (11%, 21%)c
| |
Amélioration des symptômes de fatiguej
| |
Population globale
|
60 (21%)
|
173 (41%)
|
20% (13%, 26%)c
| |
Rémission IBDQk
| |
Population globale
|
83 (30%)
|
216 (51%)
|
22% (15%, 29%)c
| |
a
TREMFYA 200 mg sous forme de perfusion intraveineuse aux semaines 0, 4 et 8.
b Sous-score de fréquence des selles de 0 ou 1, non augmenté par rapport à la valeur initiale, sous-score d'hémorragies rectales de 0 et sous-score endoscopique de 0 ou 1 sans friabilité détectable à l'endoscopie.
c p < 0,001, différence corrigée entre les traitements (IC à 95%) sur la base de la méthode de Cochran-Mantel-Haenszel (ajustée pour les facteurs de stratification: le statut d'échec thérapeutique pour les agents biologiques et/ou les inhibiteurs de JAK, ainsi que l'utilisation concomitante de corticostéroïdes au début de l'étude).
f Sous-score de fréquence des selles de 0 ou 1, qui n'a pas augmenté par rapport à la valeur initiale au début de la phase d'induction et sous-score d'hémorragies rectales de 0.
g Sous-score endoscopique de 0 ou 1 sans friabilité à l'endoscopie.
h Baisse du score Mayo modifié de ≥30% et ≥2 points par rapport à la valeur initiale au début de la phase d'induction, avec soit une diminution du sous-score d'hémorragies rectales de ≥1 point par rapport à la valeur initiale soit un sous-score d'hémorragies rectales de 0 ou 1.
i Combinaison de guérison histologique (amélioration histologique) [infiltration de neutrophiles dans < 5% des cryptes, absence de dégradation des cryptes et absence d'érosion, d'ulcération et de tissu de granulation selon la classification de Geboes] et guérison endoscopique (amélioration endoscopique), comme défini ci-dessus.
j La fatigue a été estimée à l'aide du formulaire abrégé de fatique PROMIS 7a. L'amélioration des symptômes de fatigue a été définie par une amélioration d'au moins 7 points par rapport à la valeur initiale, ce qui est considéré comme cliniquement significatif.
k Nombre total de points au Questionnaire relatif aux maladies inflammatoires de l'intestin (Inflammatory Bowel Disease Questionnaire, IBDQ) ≥170.
|
Étude d'entretien QUASAR MS
Dans l'étude d'entretien (QUASAR MS), 568 patients ont été examinés. Ces patients avaient obtenu une réponse clinique à la semaine 12 après l'administration intraveineuse de TREMFYA pendant l'étude QUASAR IS ou l'étude de détermination de la dose d'induction QUASAR. Ces patients ont été randomisés et ont reçu un traitement d'entretien par voie sous-cutanée par TREMFYA 100 mg toutes les 8 semaines, par TREMFYA 200 mg toutes les 4 semaines ou par placebo pendant 44 semaines.
Le critère d'évaluation principal était la rémission clinique, définie par le mMS à la semaine 44. Les critères d'évaluation secondaires à la semaine 44 comprenaient entre autres la rémission symptomatique, la guérison endoscopique (amélioration endoscopique), la rémission clinique sans corticostéroïdes, la guérison histologique-endoscopique de la muqueuse (amélioration histologique-endoscopique de la muqueuse), l'amélioration des symptômes de fatigue et la rémission IBDQ (voir tableau 4).
À la semaine 44, significativement plus de patients des deux groupes traités par TREMFYA étaient en rémission clinique que dans le groupe placebo.
Tableau 4: Proportion de patients ayant atteint les critères d'évaluation de l'efficacité à la semaine 44 dans l'étude QUASAR MS
|
Critère d'évaluation
|
Placebo
n = 190
|
TREMFYA 100 mg Injection sous-cutanée toutes les 8 semainesa
n = 188
|
TREMFYA 200 mg Injection sous-cutanée toutes les 4 semainesb
n = 190
|
Différence de traitement
vs placebo
(IC à 95%)
| |
TREMFYA 100 mg
|
TREMFYA 200 mg
| |
Rémission cliniquec
| |
Population globaled
|
36 (19%)
|
85 (45%)
|
95 (50%)
|
25%
(16%, 34%)e
|
30%
(21%, 38%)e
| |
Rémission symptomatiqueh
| |
Population globaled
|
71 (37%)
|
132 (70%)
|
131 (69%)
|
32%
(23%, 41%)e
|
31%
(21%, 40%)e
| |
Rémission clinique sans corticostéroïdesi
| |
Population globaled
|
35 (18%)
|
85 (45%)
|
93 (49%)
|
26%
(17%, 34%)e
|
29%
(20%, 38%)e
| |
Guérison endoscopique (amélioration endoscopique)j
| |
Population globaled
|
36 (19%)
|
93 (49%)
|
98 (52%)
|
30%
(21%, 38%)e
|
31%
(22%, 40%)e
| |
Guérison histologique-endoscopique de la muqueuse (amélioration histologique-endoscopique de la muqueuse)k
| |
Population globaled
|
32 (17%)
|
82 (44%)
|
91 (48%)
|
26%
(17%, 34%)e
|
30%
(21%, 38%)e
| |
Réponse cliniquel
| |
Population globaled
|
82 (43%)
|
146 (78%)
|
142 (75%)
|
34%
(25%, 43%)e
|
31%
(21%, 40%)e
| |
Maintien de la rémission clinique à la semaine 44 chez les patients ayant obtenu une rémission clinique 12 semaines après l'induction
| |
Population globaled
|
20/59 (34%)
|
40/66 (61%)
|
50/69 (72%)
|
26%
(9%, 43%)m
|
38%
(23%, 54%)e
| |
Normalisation endoscopiquen
| |
Population globaled
|
29 (15%)
|
65 (35%)
|
64 (34%)
|
18%
(10%, 27%)e
|
17%
(9%, 25%)e
| |
Amélioration des symptômes de fatigueo
| |
Population globaled
|
56 (29%)
|
95 (51%)
|
82 (43%)
|
20%
(11%, 29%)e
|
13
(3%, 22%)m
| |
Rémission IBDQp
| |
Population globaled
|
71 (37%)
|
121 (64%)
|
122 (64%)
|
26%
(17%, 36%)e
|
26%
(16%, 35%)e
| |
a
TREMFYA 100 mg en injection sous-cutanée toutes les 8 semaines après le traitement d'induction
b TREMFYA 200 mg en injection sous-cutanée toutes les 4 semaines après le traitement d'induction
c Sous-score de fréquence des selles de 0 ou 1, pas augmenté par rapport à la valeur initiale, sous-score d'hémorragies rectales de 0
d Patients ayant obtenu une réponse clinique à la semaine 12 après l'administration intraveineuse de TREMFYA dans l'étude d'induction QUASAR ou l'étude de détermination de la dose d'induction QUASAR.
e p < 0,001, différence corrigée entre les traitements (IC à 95%) sur la base de la méthode de Cochran-Mantel-Haenszel (ajustée pour les facteurs de stratification).
h Sous-score de fréquence des selles de 0 ou 1, qui n'a pas augmenté par rapport à la valeur initiale de l'induction et sous-score d'hémorragies rectales de 0.
i Aucun traitement par corticostéroïdes pendant au moins 8 semaines avant la semaine 44 et satisfaction des critères pour une rémission clinique à la semaine 44.
j Sous-score endoscopique de 0 ou 1 sans friabilité à l'endoscopie.
k Combinaison de guérison histologique (amélioration histologique) [infiltration de neutrophiles dans < 5% des cryptes, absence de dégradation des cryptes et absence d'érosion, d'ulcération et de tissu de granulation selon la classification de Geboes] et de guérison endoscopique (amélioration endoscopique), comme défini ci-dessus.
l Baisse du score Mayo modifié de ≥30% et ≥2 points par rapport à la valeur initiale de l'induction, avec une diminution du sous-score d'hémorragies rectales de ≥1 point par rapport à la valeur initiale ou un sous-score d'hémorragies rectales de 0 ou 1.
m p < 0,01, différence corrigée entre les traitements (IC à 95%) sur la base de la méthode de Cochran-Mantel-Haenszel (ajustée pour les facteurs de stratification).
n Sous-score endoscopique de 0.
o La fatigue a été estimée à l'aide du Formulaire abrégé de fatigue PROMIS 7a. L'amélioration des symptômes de fatigue a été définie par une amélioration d'au moins 7 points par rapport à la valeur initiale, ce qui est considéré comme cliniquement significatif.
p Nombre total de points du Questionnaire des maladies inflammatoires de l'intestin (Inflammatory Bowel Disease Questionnaire; IBDQ) ≥170.
|
Dans les études QUASAR IS et QUASAR MS, l'efficacité et la sécurité de TREMFYA ont été analysées systématiquement indépendamment de l'âge, du sexe, de la couleur de peau, du poids corporel et du traitement antérieur par un agent biologique ou par un inhibiteur de JAK. TREMFYA était actif chez les patients qui n'avaient pas encore été traités par un agent biologique ou un inhibiteur de JAK, ainsi que chez les patients chez lesquels un traitement antérieur par agent biologique ou inhibiteur de JAK avait échoué.
Résultats des sous-groupes relatifs à la posologie
Dans l'étude QUASAR MS, les patients présentant une haute charge inflammatoire ont montré après la fin du traitement d'induction un bénéfice supplémentaire de TREMFYA 200 mg par voie sous-cutanée toutes les 4 semaines comparé à 100 mg par voie sous-cutanée toutes les 8 semaines. Chez les patients présentant un taux de CRP > 3 mg/l, des différences numériques cliniquement significatives entre les deux groupes de dose de TREMFYA ont été observées après la fin de la phase d'induction pour les critères d'évaluation suivants à la semaine 44: rémission clinique (48% pour 200 mg toutes les 4 semaines contre 30% pour 100 mg toutes les 8 semaines), maintien de la rémission clinique (88% pour 200 mg toutes les 4 semaines contre 50% pour 100 mg toutes les 8 semaines), rémission clinique sans corticostéroïdes (46% pour 200 mg toutes les 4 semaines contre 30% pour 100 mg toutes les 8 semaines), guérison endoscopique (amélioration endoscopique) (52% pour 200 mg toutes les 4 semaines contre 35% pour 100 mg toutes les 8 semaines), et guérison histologique-endoscopique de la muqueuse (amélioration histologique-endoscopique de la muqueuse) (46% pour 200 mg toutes les 4 semaines contre 29% pour 100 mg toutes les 8 semaines).
Efficacité après perte de la réponse à TREMFYA
Dix-neuf patients ayant tous reçu TREMFYA 100 mg toutes les 8 semaines par voie sous-cutanée et chez lesquels une première perte de la réponse (10%) est survenue entre la semaine 8 et la semaine 32 de l'étude QUASAR MS ont reçu des schémas posologiques de TREMFYA en aveugle de 200 mg de TREMFYA par voie sous-cutanée toutes les 4 semaines, et 11 de ces patients (58%) ont obtenu une réponse symptomatique et 5 patients (26%) ont obtenu une rémission symptomatique après 12 semaines.
Maladie de Crohn
L'efficacité et la sécurité de TREMFYA ont été évaluées dans trois études incluant des patients adultes atteints de la maladie de Crohn active modérée à sévère et répondant insuffisamment ou pas du tout à des corticostéroïdes oraux, des immunomodulateurs conventionnels (AZA, 6-MP, MTX) et/ou un traitement par un agent biologique (inhibiteur de TNF ou védolizumab) ou ne tolérant pas ceux-ci. Il s'agissait de deux études de concept identique, d'une durée de 48 semaines, multicentriques, randomisées, en double aveugle, contrôlées par placebo et agent biologique (ustékinumab) en groupes parallèles (induction intraveineuse et traitement d'entretien sous-cutané (SC), GALAXI 2 et GALAXI 3 [NCT03466411]) et d'une étude d'une durée de 48 semaines, multicentrique, randomisée, en double aveugle, contrôlée par placebo, en groupe parallèles (induction sous-cutanée (SC) et entretien sous-cutané (SC): GRAVITI [NCT05197049]). Le concept des trois études était de type «Threat-Through»: les patients randomisés sous TREMFYA conservaient cette attribution du traitement pendant la totalité de l'étude.
Études GALAXI 2 et GALAXI 3
Dans les études GALAXI 2 et GALAXI 3, la maladie de Crohn active modérée à sévère était définie par un indice d'activité de la maladie de Crohn (CDAI) de ≥220 et ≤450, ainsi qu'un Simple Endoscopic Score for CD (SES-CD) de ≥6 (ou ≥4 chez les patients avec maladie isolée de l'iléum).
Dans les études GALAXI 2 et GALAXI 3, les patients ont été randomisés selon un rapport de 2:2:2:1 dans les groupes suivants: TREMFYA 200 mg en induction intraveineuse aux semaines 0, 4 et 8, suivi du traitement d'entretien par voie sous-cutanée par TREMFYA 200 mg toutes les 4 semaines, TREMFYA 200 mg en induction intraveineuse aux semaines 0, 4 et 8, suivi du traitement d'entretien par voie sous-cutanée par TREMFYA 100 mg toutes les 8 semaines, un groupe de contrôle actif par agent biologique (ustékinumab) ainsi qu'un groupe placebo. Les patients qui ne répondaient pas au placebo ont reçu également l'agent biologique de contrôle à partir de la semaine 12.
Au total, 1021 patients ont été évalués: n = 508 dans GALAXI 2 et n = 513 dans GALAXI 3. L'âge médian était de 34 ans (18 à 83 ans), 42,4% étaient de sexe féminin, et 74,3% étaient d'origine caucasienne, 21,3% des Asiatiques et 1,5% des Noirs ou Afro-Américains.
Dans l'étude GALAXI 2, un traitement par au moins un agent biologique avait précédemment échoué chez 52,8% des patients, 41,9% des patients n'avaient jamais reçu d'agent biologique et 5,3% avaient précédemment reçu un agent biologique sans échec. Au début de l'étude, 37,4% des patients recevaient des corticostéroïdes oraux et 29,9% des patients, des immunomodulateurs conventionnels. Les patients sous corticostéroïdes oraux ont été maintenus stables du début à la semaine 12. À partir de la semaine 12, une diminution progressive obligatoire a été réalisée, sauf lorsqu'elle n'était pas réalisable sur le plan médical.
Dans l'étude GALAXI 3, un traitement par au moins un agent biologique avait précédemment échoué chez 51,9% des patients, 41,5% des patients n'avaient jamais reçu d'agent biologique et 6,6% avaient précédemment reçu un agent biologique sans échec. Au début de l'étude, 36,1% des patients recevaient des corticostéroïdes oraux et 30,2% des patients des immunomodulateurs conventionnels.
Dans les études GALAXI 2 et GALAXI 3, les co-critères d'évaluation principaux combinés étaient (1) réponse clinique à la semaine 12 et rémission clinique à la semaine 48, ou (2) réponse clinique à la semaine 12 et réponse endoscopique à la semaine 48, chaque fois par comparaison avec le placebo (tableau 5). Parmi les critères d'évaluation secondaires, on trouvait les résultats cliniques et endoscopiques à court et à long terme (semaine 12 et jusqu'à la semaine 48), chacun par comparaison avec le placebo (tableaux 6 et 7). Pour les critères d'évaluation combinés dans GALAXI 2 et GALAXI 3, les patients devaient atteindre les deux composantes du critère d'évaluation.
Tableau 5: Proportion de patients ayant atteint les critères d'évaluation principaux de l'efficacité dans les études GALAXI 2 et GALAXI 3
|
GALAXI 2
| |
Critère d'évaluation
|
Placebo
(n = 76)
|
TREMFYA
Induction intraveineuse
100 mg Injection sous-cutanée toutes les 8 semainesa
(n = 143)
|
TREMFYA
Induction intraveineuse
200 mg Injection sous-cutanée toutes les 4 semainesb
(n = 146)
|
Différence de traitement par rapport au placebo
(IC à 95%)c
| |
TREMFYA
100 mg
|
TREMFYA 200 mg
| |
Réponse cliniqued à la semaine 12 et rémission cliniquee à la semaine 48
| |
Population globale
|
9 (12%)
|
70 (49%)
|
80 (55%)
|
38%
(27%, 49%)f
|
43%
(32%, 54%)f
| |
Réponse cliniqued à la semaine 12 et réponse endoscopiqueg à la semaine 48
| |
Population globale
|
4 (5%)
|
56 (39%)
|
56 (38%)
|
34%
(24%, 43%)f
|
33%
(24%, 42%)f
| |
GALAXI 3
| |
Critère d'évaluation
|
Placebo
(n = 72)
|
TREMFYA
Induction intraveineuse
100 mg Injection sous-cutanée toutes les 8 semainesa
(n = 143)
|
TREMFYA
Induction intraveineuse
200 mg Injection sous-cutanée toutes les 4 semainesb
(n = 150)
|
Différence de traitement par rapport au placebo
(IC à 95%)c
| |
TREMFYA
100 mg
|
TREMFYA
200 mg
| |
Réponse cliniqued à la semaine 12 et rémission cliniquee à la semaine 48
| |
Population globale
|
9 (13%)
|
67 (47%)
|
72 (48%)
|
34%
(23%, 45%)f
|
35%
(24%, 46%)f
| |
Réponse cliniqued à la semaine 12 et réponse endoscopiqueg à la semaine 48
| |
Population globale
|
4 (6%)
|
48 (34%)
|
54 (36%)
|
28%
(19%, 37%)f
|
31%
(21%, 40%)f
| |
a
TREMFYA 200 mg sous forme de perfusion intraveineuse à la semaine 0, à la semaine 4 et à la semaine 8, suivi de TREMFYA 100 mg en injection sous-cutanée toutes les 8 semaines, jusqu'à la semaine 48.
b TREMFYA 200 mg sous forme de perfusion intraveineuse à la semaine 0, à la semaine 4 et à la semaine 8, suivi de TREMFYA 200 mg en injection sous-cutanée toutes les 4 semaines, jusqu'à la semaine 48.
c La différence de traitement corrigée et l'IC sont basés sur le test de différence de risque global en utilisant la pondération de strate de Mantel-Haenszel et l'estimation de variance de Sato. Les variables de stratification utilisées étaient le score CDAI (≤300 ou > 300) et le score SES-CD (≤12 ou > 12) au début du traitement, l'échec antérieur d'agents biologiques (Oui ou Non) ainsi que la prise de corticostéroïdes au début du traitement (Oui ou Non). IC = intervalle de confiance.
d La réponse clinique est définie par une diminution du score CDAI de ≥100 points par rapport à la valeur initiale ou un score CDAI < 150.
e La rémission clinique est définie par un score CDAI < 150.
f p < 0,001
g La réponse endoscopique est définie par une amélioration de ≥50% du score SES-CD par rapport à la valeur initiale ou un score SES-CD ≤2.
|
Tableau 6: Proportion de patients ayant atteint les critères d'évaluation de l'efficacité à court terme dans les études GALAXI 2 et GALAXI 3
|
GALAXI 2
| |
Critère d'évaluation
|
Placebo
(n = 76)
|
TREMFYA
Induction IVa
(n = 289)
|
Différence de traitement par rapport au placebo
(IC à 95%)b
| |
Rémission cliniquec à la semaine 12
| |
Population globale
|
17 (22%)
|
136 (47%)
|
25% (14%, 36%)d
| |
Réponse endoscopiquee à la semaine 12
| |
Population globale
|
8 (11%)
|
109 (38%)
|
28% (19%, 36%)d
| |
Réponse pour la fatiguef à la semaine 12
| |
Population globale
|
22 (29%)
|
131 (45%)
|
16% (5%, 28%)g
| |
Rémission cliniquec à la semaine 12 et réponse endoscopiquee à la semaine 12
| |
Population globale
|
3 (4%)
|
62 (21%)
|
18% (11%, 24%)d
| |
GALAXI 3
| |
Critère d'évaluation
|
Placebo
(n = 72)
|
TREMFYA
Induction IVa
(n = 293)
|
Différence par rapport au placebo
(IC à 95%)b
| |
Rémission cliniquec à la semaine 12
| |
Population globale
|
11 (15%)
|
138 (47%)
|
31% (21%, 41%)d
| |
Réponse endoscopiquee à la semaine 12
| |
Population globale
|
10 (14%)
|
106 (36%)
|
22% (12%, 32%)d
| |
Réponse pour la fatiguef à la semaine 12
| |
Population globale
|
13 (18%)
|
127 (43%)
|
26% (15%, 36%)d
| |
Rémission cliniquec à la semaine 12 et réponse endoscopiquee à la semaine 12
| |
Population globale
|
2 (3%)
|
64 (22%)
|
19% (12%, 25%)d
| |
a
TREMFYA 200 mg en perfusion intraveineuse à la semaine 0, à la semaine 4 et à la semaine 8. Les deux groupes de traitement par TREMFYA ont été regroupés pour cette colonne, car ils ont obtenu le même traitement.
b La différence de traitement corrigée et l'IC sont basés sur le test de différence de risque global en utilisant la pondération de strate de Mantel-Haenszel et l'estimation de variance de Sato. Les variables de stratification utilisées étaient le score CDAI (≤300 ou > 300) et le score SES-CD (≤12 ou > 12) au début du traitement, l'échec antérieur d'agents biologiques (Oui ou Non) ainsi que la prise de corticostéroïdes au début du traitement (Oui ou Non). IC = intervalle de confiance.
c La rémission clinique est définie par un score CDAI < 150.
d p < 0,001.
e La réponse endoscopique est définie par une amélioration de ≥50% du score SES-CD par rapport à la valeur initiale ou un score SES-CD ≤2.
f La réponse de la fatigue est définie par une amélioration de ≥7 points au Formulaire abrégé de fatigue PROMIS 7a.
g p < 0,05.
|
Tableau 7 Proportion de patients ayant atteint les critères d'évaluation de l'efficacité à long terme dans les études GALAXI 2 et GALAXI 3
|
GALAXI 2
| |
Critère d'évaluation
|
Placebo
(n = 76)
|
TREMFYA
Induction intraveineuse→
100 mg Injection sous-cutanée toutes les 8 semainesa
(n = 143)
|
TREMFYA
Induction intraveineuse
200 mg Injection sous-cutanée toutes les 4 semainesb
(n = 146)
|
Différence de traitement par rapport au placebo
(IC à 95%)c
| |
TREMFYA
100 mg
|
TREMFYA
200 mg
| |
Réponse cliniqued à la semaine 12 et rémission clinique sans corticostéroïdese à la semaine 48
| |
Population globale
|
7 (9%)
|
67 (47%)
|
74 (51%)
|
39%
(28%, 49%)f
|
41%
(31%, 52%)f
| |
Réponse cliniqued à la semaine 12 et rémission endoscopiqueg à la semaine 48
| |
Population globale
|
2 (3%)
|
38 (27%)
|
48 (33%)
|
24%
(16%, 32%)f
|
30%
(21%, 39%)f
| |
GALAXI 3
| |
Critère d'évaluation
|
Placebo
(n = 72)
|
TREMFYA
Induction intraveineuse→
100 mg Injection sous-cutanée toutes les 8 semainesa
(n = 143)
|
TREMFYA
Induction intraveineuse→
200 mg Injection sous-cutanée toutes les 4 semainesb
(n = 150)
|
Différence par rapport au placebo
(IC à 95%)c
| |
TREMFYA
100 mg
|
TREMFYA
200 mg
| |
Réponse cliniqued à la semaine 12 et rémission clinique sans corticostéroïdese à la semaine 48
| |
Population globale
|
9 (13%)
|
65 (45%)
|
67 (45%)
|
33%
(22%, 44%)f
|
31%
(20%, 43%)f
| |
Réponse cliniqued à la semaine 12 et rémission endoscopiqueg à la semaine 48
| |
Population globale
|
4 (6%)
|
34 (24%)
|
34 (23%)
|
18%
(10%, 27%)f
|
17%
(8%, 25%)f
| |
a
TREMFYA 200 mg en perfusion intraveineuse à la semaine 0, à la semaine 4 et à la semaine 8, suivi de TREMFYA 100 mg en injection sous-cutanée toutes les 8 semaines jusqu'à la semaine 48.
b TREMFYA 200 mg en perfusion intraveineuse à la semaine 0, à la semaine 4 et à la semaine 8, suivi de TREMFYA 100 mg en injection sous-cutanée toutes les 4 semaines jusqu'à la semaine 48.
c La différence de traitement corrigée et l'IC sont basés sur le test de différence de risque global en utilisant la pondération de strate de Mantel-Haenszel et l'estimation de variance de Sato. Les variables de stratification utilisées étaient le score CDAI (≤300 ou > 300) et la valeur SES-CD (≤12 ou > 12) au début du traitement, l'échec antérieur d'agents biologiques (Oui ou Non) ainsi que la prise de corticostéroïdes au début du traitement (Oui ou Non). IC = intervalle de confiance.
d La réponse clinique est définie par une diminution du score CDAI de ≥100 points par rapport à la valeur initiale ou un score CDAI < 150.
e La rémission clinique sous corticostéroïdes pendant 90 jours est définie par un score CDAI < 150 et l'absence de prise de corticostéroïde au cours des 90 jours précédant la visite correspondante.
f p < 0,001.
g La rémission endoscopique (définition globale) est définie par un score SES-CD ≤4 et une diminution d'au moins 2 points par rapport à la valeur initiale. De plus, la valeur partielle ne peut être supérieure à 1 pour aucune composante.
|
Résultats des sous-groupes relatifs à la posologie
Dans les études GALAXI de phase III groupées, les patients présentant une haute charge inflammatoire ont montré après la fin du traitement d'induction un bénéfice supplémentaire de TREMFYA 200 mg par voie sous-cutanée toutes les 4 semaines par comparaison avec 100 mg par voie sous-cutanée toutes les 8 semaines. Chez les patients présentant un taux de CRP > 5 mg/l après la fin du traitement d'induction, il a été constaté une différence numérique cliniquement significative de 14 à 17 points de pourcentage concernant la rémission clinique à la semaine 48 entre les deux groupes de dose de TREMFYA (100 mg par voie sous-cutanée toutes les 8 semaines: 54% vs 200 mg par voie sous-cutanée toutes les 4 semaines: 71,0%) et concernant la réponse endoscopique à la semaine 48 (100 mg par voie sous-cutanée toutes les 8 semaines: 36% vs 200 mg par voie sous-cutanée toutes les 4 semaines: 50%).
Étude GRAVITI
Dans l'étude GRAVITI, une maladie de Crohn active modérée à sévère était définie par une maladie ayant un indice d'activité de la maladie de Crohn (CDAI) de ≥220 et ≤450, ainsi qu'un Simple Endoscopic Score for CD (SES-CD) ≥6 (ou ≥4 chez les patients avec maladie isolée de l'iléum).
Pour l'étude GRAVITI, les patients ont été randomisés selon un rapport de 1:1:1 dans les groupes suivants: TREMFYA 400 mg en induction sous-cutanée aux semaines 0, 4 et 8, suivi d'un traitement d'entretien sous-cutané par TREMFYA 200 mg toutes les 4 semaines ou TREMFYA 400 mg en induction sous-cutanée aux semaines 0, 4 et 8, suivi du traitement d'entretien sous-cutané par TREMFYA 100 mg toutes les 8 semaines, ou placebo. Tous les patients du groupe placebo qui satisfaisaient aux critères de secours ont reçu un traitement par TREMFYA 400 mg en induction sous-cutanée, suivi du traitement d'entretien sous-cutané par TREMFYA 100 mg toutes les 8 semaines. Tous les patients qui satisfaisaient aux critères de secours ont été considérés dans l'analyse comme non-répondeurs à partir du point temporel du secours.
Au total, les données de 347 patients ont été analysées. L'âge médian des patients était de 36 ans (18 à 83 ans). 41,5% étaient de sexe féminin et 66% étaient d'origine caucasienne, 21,9% des Asiatiques, 2,6% des Noirs ou Afro-Américains.
Dans l'étude GRAVITI, un traitement par au moins un agent biologique avait antérieurement échoué chez 46,4% des patients, 46,4% des patients n'avaient jamais reçu d'agent biologique et 7,2% avaient précédemment reçu un agent biologique sans échec. Au début de l'étude, 29,7% des patients recevaient des corticostéroïdes oraux et 28,5% des patients des immunomodulateurs conventionnels. Les patients sous corticostéroïdes oraux ont été maintenus stables du début à la semaine 12. À partir de la semaine 12, une diminution progressive obligatoire a été réalisée, sauf lorsqu'elle n'était pas réalisable sur le plan médical.
Dans l'études GRAVITI, les co-critères d'évaluation principaux étaient la rémission clinique à la semaine 12 et la réponse endoscopique à la semaine 12, par comparaison avec le placebo (tableau 8). Les critères d'évaluation pondérés par multiplicateur supplémentaire à la semaine 12, à la semaine 24 ou à la semaine 48 sont indiqués dans les tableaux 8 et 9.
Tableau 8: Proportion de patients ayant atteint les critères d'évaluation de l'efficacité à la semaine 12 dans l'étude GRAVITI
|
Critère d'évaluation
|
Placebo
(n = 117)
|
TREMFYA 400 mg
Injection sous-cutanéea
(n = 230)
|
Différence par rapport au placebo
(IC à 95%)b
| |
Rémission cliniquec à la semaine 12
| |
Population globale
|
25 (21%)
|
129 (56%)
|
35% (25%, 45%)d
| |
Réponse endoscopiquee à la semaine 12
| |
Population globale
|
25 (21%)
|
95 (41%)
|
20% (10%, 30%)d
| |
Réponse cliniquef à la semaine 12
| |
Population globale
|
39 (33%)
|
169 (73%)
|
40% (30%, 51%)d
| |
a
TREMFYA 400 mg en induction sous-cutanée aux semaines 0, 4 et 8.
b La différence de traitement corrigée et l'IC sont basés sur le test de différence de risque global en utilisant la pondération de strate de Mantel-Haenszel et l'estimation de variance de Sato. Les variables de stratification utilisées étaient Le score CDAI (≤300 ou > 300) et le score SES-CD (≤12 ou > 12) au début du traitement, ainsi que l'échec antérieur d'agents biologiques (Oui ou Non). IC = intervalle de confiance.
c La rémission clinique est définie par un score CDAI < 150.
d p < 0,001.
e Réponse endoscopique: amélioration de ≥50% par rapport au score SES-CD initial.
f Réponse clinique: diminution du score CDAI de ≥100 points par rapport à la valeur initiale ou un score CDAI < 150
|
Tableau 9: Proportion de patients ayant atteint les critères d'évaluation de l'efficacité à la semaine 24 et à la semaine 48 dans l'étude GRAVITI
|
Critère d'évaluation
|
Placebo
(n = 117)
|
TREMFYA
400 mg SC induction→
100 mg SC Injection q8sa
(n = 115)
|
TREMFYA
400 mg SC Induction→
200 mg SC Injection q4sb
(n = 115)
|
Différence par rapport au placebo
(IC à 95%)c
| |
TREMFYA
100 mg
|
TREMFYA
200 mg
| |
Rémission cliniqued à la semaine 24
| |
Population globale
|
25 (21%)
|
70 (61%)
|
67 (58%)
|
39%
(28%, 51%)e
|
37%
(26%, 48%)e
| |
Rémission cliniqued à la semaine 48
|
| |
Population globale
|
20 (17%)
|
69 (60%)
|
76 (66%)
|
43%
(32%, 54%)e
|
49%
(38%, 60%)e
| |
Réponse endoscopiquef à la semaine 48
|
| |
Population globale
|
8 (7%)
|
51 (44%)
|
59 (51%)
|
38%
(27%, 48%)e
|
45%
(34%, 55%)e
| |
a
TREMFYA 400 mg SC Induction aux semaines 0, 4 et 8, suivi de TREMFYA 100 mg SC toutes les 8 semaines.
b TREMFYA 400 mg SC Induction aux semaines 0, 4 et 8, suivi de TREMFYA 200 mg SC en traitement d'entretien toutes les 4 semaines.
c La différence de traitement corrigée et l'IC sont basés sur le test de différence de risque global en utilisant la pondération de strate de Mantel-Haenszel et l'estimation de variance de Sato. Les variables de stratification utilisées étaient le score CDAI (≤300 ou > 300) et le score SES-CD (≤12 ou > 12) au début du traitement, ainsi que l'échec antérieur d'agents biologiques (Oui ou Non). IC = intervalle de confiance.
d La rémission clinique est définie par un score CDAI < 150.
e p < 0,001.
f Réponse endoscopique: amélioration de ≥50% par rapport au score SES-CD initial.
|
|