ZusammensetzungWirkstoffe
Susoctocogum alfa.
Hilfsstoffe
Polysorbatum 80, Natrii chloridum, Calcii chloridum dihydricus, Saccharum, Trometamolum, Trometamolum hydrochloridum, Trinatrii citras dihydricus, Gesamtnatriumgehalt max. 4.6 mg/ml.
Lösungsmittel: Aqua ad iniectabilia.
Indikationen/AnwendungsmöglichkeitenBehandlung von Blutungsepisoden bei erwachsenen Patienten mit erworbener Hämophilie, die durch Antikörper gegen den Faktor VIII verursacht wird.
OBIZUR ist für die Behandlung angeborener Hämophilie A nicht indiziert.
Dosierung/AnwendungDie Behandlung muss unter der Überwachung eines Arztes erfolgen, der mit der Behandlung der Hämophilie vertraut ist.
Patienten mit erworbener Hämophilie A, verursacht durch Antikörper gegen Faktor VIII, sollen mit umfassenden Massnahmen gemäss lokalen institutionellen Leitlinien behandelt werden.
Eine Anfangsdosis von weniger als der empfohlenen Dosis von 200 E/kg wurde mit mangelnder Wirksamkeit in Verbindung gebracht.
Dosierung
Die Dosis, Häufigkeit und Dauer der Therapie mit OBIZUR richten sich nach dem Ort, Ausmass und Schweregrad der Blutungsepisode, nach den angestrebten Faktor VIII Spiegeln und dem klinischen Zustand des Patienten.
Die Anzahl der verabreichten Faktor VIII-Einheiten werden in Einheiten (E) angegeben, welche von einem hausinternen Standard abgeleitet werden, der mit dem WHO-Standard für Faktor VIII Produkte kalibriert wurde.
Eine Einheit (E) der Faktor VIII-Aktivität entspricht der Menge Faktor VIII in einem Milliliter normalem menschlichen Plasmas.
Die Faktor VIII-Aktivität, welche unter Verwendung des chromogenen Assays bestimmt wird, kann um 20–50 % von derjenigen, welche durch den Einstufen-Gerinnungstest bestimmt wurde, variieren.
Die empfohlene Initialdosis beträgt 200 E pro kg Körpergewicht, verabreicht als intravenöse Injektion (siehe Hinweise für die Handhabung).
Die erforderliche OBIZUR-Anfangsdosis für einen Patienten wird mit folgender Formel berechnet:
Anfangsdosis (E/kg) ÷ Produktstärke (E/Durchstechflasche) × Körpergewicht (kg) = Anzahl der Durchstechflaschen
Beispielsweise errechnet sich die Anzahl der Durchstechflaschen für die Anfangsdosis für einen 70 kg schweren Patienten wie folgt:
200 E/kg ÷ 500 E/Durchstechflasche × 70 kg = 28 Durchstechflaschen
Die Faktor VIII-Aktivität und der klinische Zustand sind 30 Minuten nach der ersten Injektion und 3 Stunden nach der OBIZUR Verabreichung zu überwachen. Die Faktor VIII-Aktivität ist jeweils direkt vor und 30 Minuten nach der Verabreichung nachfolgender Dosen zu überwachen.
Der Einstufen-Gerinnungstest für Faktor VIII wird empfohlen, da er bei der Bestimmung der Aktivität und durchschnittlichen Wiederfindungsrate von OBIZUR verwendet wurde.
Die Dosis und Häufigkeit der Verabreichung sollten auf Bestimmungen der Faktor VIII-Aktivität in angemessenen Intervallen basieren, um die empfohlene Faktor VIII-Aktivität im Blut aufrechtzuerhalten.
Anfangsphase
|
Art der Blutung
|
Ziel-Faktor VIII-Aktivität (Einheiten je dl oder % des Normalen)
|
Anfangsdosis (Einheiten je kg)
|
Nachfolgende Dosis
|
Häufigkeit und Dauer der nachfolgenden Dosierung
| |
Leichte bis mässige
oberflächliche Muskelblutung / keine neurovaskuläre Beeinträchtigung bzw. Gelenkblutung
|
> 50 %
|
200
|
Titrieren Sie nachfolgende Dosen in Abhängigkeit von der klinischen Reaktion und zur Beibehaltung der Ziel-Faktor VIII-Aktivität
|
Dosis alle 4 bis 12 Stunden; die Häufigkeit kann je nach klinischer Reaktion und gemessener Faktor VIII-Aktivität angepasst werden
| |
Grössere mässige bis schwere intramuskuläre, retroperitoneale, gastrointestinale, intrakranielle Blutung
|
> 80 %
|
Heilungsphase
Sobald die Blutung anspricht – normalerweise innerhalb der ersten 24 Stunden –, die OBIZUR-Behandlung mit einer Dosis fortsetzen, die die FVIII-Aktivität bei 30–40 % hält, bis die Blutung kontrolliert ist. Die maximale Plasma-FVIII-Aktivität darf 200 % nicht übersteigen.
Die Länge der Behandlung hängt von der klinischen Beurteilung ab.
Ältere Patienten
Das Durchschnittsalter der 29 Studienteilnehmer in der OBI-1-301 Studie betrug 70 Jahre. 19 Studienteilnehmer waren ≥65 Jahre. Es wurden keine Unterschiede im Ansprechen zwischen älteren und erwachsenen Patienten beobachtet. Die Aussagekraft dieser Beobachtung ist angesichts der geringen Anzahl von Probanden in beiden Gruppen nicht abschliessend zu beurteilen.
Dosisanpassungen und spezifische Gefahren in Zusammenhang mit der gleichzeitigen Anwendung von OBIZUR und anderen Arzneimitteln bei älteren Patienten wurden in klinischen Studien nicht untersucht.
Pädiatrie
Die Anwendung bei Kindern und Jugendlichen [von 0 (Geburt) bis 18 Jahren] mit angeborener oder in den seltenen Fällen von erworbener Hämophilie A ist zurzeit nicht genehmigt.
Art der Anwendung
Intravenöse Verabreichung.
Das totale Volumen der rekonstituierten OBIZUR Lösung sollte mit einer Geschwindigkeit von 1 bis 2 ml pro Minute verabreicht werden.
Anweisungen zur Rekonstitution des Arzneimittels vor der Anwendung, siehe Abschnitt «Hinweise für die Handhabung».
Kontraindikationen·Angeborene Hämophilie A mit Inhibitoren
·Bekannte anaphylaktische Reaktionen gegen den Wirkstoff, gegen Hamsterproteine sowie gegen einen der Hilfsstoffe.
Warnhinweise und VorsichtsmassnahmenÜberempfindlichkeit
Überempfindlichkeitsreaktionen allergischer Art sind mit OBIZUR möglich. Das Präparat enthält Spuren von Hamsterproteinen.
Brechen Sie die Behandlung ab, wenn Symptome einer Überempfindlichkeitsreaktion auftreten sollten. Patienten sollen über die frühen Anzeichen einer Überempfindlichkeitsreaktion einschliesslich Ausschlag, generalisierte Urtikaria, Engegefühl in der Brust, Keuchatmung, Hypotonie und Anaphylaxie aufgeklärt werden.
Im Falle eines Schocks sollte die medizinische Standardschocktherapie durchgeführt werden.
Bildung von neutralisierenden Antikörpern
Für die Behandlung mit OBIZUR ist es von Nutzen, den Ausgangswert des allenfalls vorhandenen anti-rpFVIII Inhibitortiters zu kennen; hierfür sollte vor Behandlungsbeginn mit OBIZUR eine Blutprobe entnommen werden. Das Testergebnis muss vor Behandlungsstart jedoch nicht vorliegen, denn aufgrund der lebensbedrohlichen Situation sollte die Behandlung mit OBIZUR sofort begonnen werden.
Die Behandlung mit OBIZUR kann mithilfe der Messung der FVIII-Aktivität überwacht werden.
Neutralisierende Antikörper (Inhibitoren) gegen porcines Faktor VIII (gemessen mittels Nijmengen-modifiziertem Bethesda Assay) wurden vor und nach der OBIZUR-Exposition festgestellt. Eine mangelnde Wirksamkeit könnte auf hemmende Antikörper gegen OBIZUR zurückzuführen sein Es wurden Inhibitorentiter von bis zu 29 Bethesda-Einheiten bei Baseline erfasst, trotzdem zeigten die Patienten eine positive Antwort auf OBIZUR. Es wird empfohlen, die Behandlung auf der klinischen Beurteilung zu basieren und nicht auf der Bestimmung von inhibierenden Antikörpern mittels Bethesda Assay.
Anamnestische Reaktionen mit Anstieg der Inhibitoren gegen humanen Faktor VIII und/oder gegen die porcine Sequenz des rekombinanten Faktors VIII wurden berichtet bei mit OBIZUR behandelten Patienten. Diese anamnestischen Anstiege können zu mangelndem Ansprechen auf OBIZUR führen. Bei Verdacht auf solche hemmenden Antikörper gegen OBIZUR und mangelndem Ansprechen sind andere therapeutische Optionen in Betracht zu ziehen.
Die Faktor VIII-Aktivität ist zu überwachen, um zu bestätigen, dass angemessene Faktor VIII-Spiegel erreicht wurden und aufrechterhalten werden, sofern dies klinisch angezeigt ist.
Es gibt keine klinischen Daten zur Entwicklung hemmender Antikörper gegen OBIZUR bei wiederholter Gabe. Deshalb sollte OBIZUR nur verabreicht werden, wenn es als klinisch erforderlich angesehen wird. Grössere kutane Purpura erfordern normalerweise keine Behandlung.
OBIZUR wird mittels rekombinanter DNA-Technologie in Babyhamster-Nierenzellen hergestellt. Antikörper gegen Babyhamster-Nierenzellprotein wurden in Patienten weder vor noch nach Anwendung mit OBIZUR gemessen.
Kardiovaskuläre Ereignisse
Bei Patienten mit bestehenden kardiovaskulären Risikofaktoren kann eine Substitutionstherapie mit FVIII das kardiovaskuläre Risiko erhöhen.
Thromboembolische Ereignisse
Eine hohe und anhaltende Faktor VIII-Aktivität im Blut kann thromboembolische Ereignisse begünstigen. Ein besonderes Risiko besteht für Patienten mit bestehenden kardiovaskulären Krankheiten und ältere Menschen.
Wenn ein Venenkatheter erforderlich ist, muss das Risiko katheterbedingter Komplikationen wie Thrombose an der Katheterstelle beachtet werden.
Überwachung der Behandlung
Die durch Chromogen-Test festgestellte Faktor VIII-Aktivität ist im Allgemeinen niedriger als die durch Einstufen-Gerinnungstest festgestellte Faktor VIII-Aktivität. Die Messung der Faktor VIII-Aktivität sollte bei einem Patienten immer auf derselben Untersuchungsmethode basieren. Der Einstufen-Gerinnungstest wird empfohlen, da er bei der Bestimmung der Aktivität und durchschnittlichen Wiederfindungsrate von OBIZUR verwendet wurde.
Rückverfolgbarkeit
Es wird empfohlen, bei jeder Verabreichung von OBIZUR an einen Patienten den Namen und die Chargennummer des Präparates zu dokumentieren, um einen Zusammenhang zwischen Patient und Produktcharge herzustellen.
Natriumgehalt
Jede Durchstechflasche enthält nach der Rekonstitution maximal 4.6 mg (198 mM) Natrium pro ml. Dies muss bei Patienten, die einer natriumkontrollierten Diät unterliegen, berücksichtigt werden.
Dieses Arzneimittel enthält 4.6 mg Natrium pro Durchstechflasche, entsprechend 0.23 % der von der WHO für einen Erwachsenen empfohlenen maximalen täglichen Natriumaufnahme mit der Nahrung von 2 g.
InteraktionenEs wurde über keine Interaktionen von OBIZUR mit anderen Arzneimitteln berichtet.
Schwangerschaft, StillzeitMit OBIZUR wurden keine Tierstudien zur Reproduktion durchgeführt. Es gibt keine Erfahrung bezüglich der Verabreichung von OBIZUR während der Schwangerschaft oder Stillzeit. Deshalb sollte OBIZUR während der Schwangerschaft und Stillzeit nur bei eindeutiger Indikationsstellung angewendet werden.
Wirkung auf die Fahrtüchtigkeit und auf das Bedienen von MaschinenOBIZUR hat keinen oder einen vernachlässigbaren Einfluss auf die Fahrtüchtigkeit oder die Fähigkeit, Maschinen zu bedienen.
Unerwünschte WirkungenZusammenfassung des Sicherheitsprofils
Überempfindlichkeit und allergische Reaktionen (welche Angioödem, Brennen und Stechen an der Injektionsstelle, Schüttelfrost, Hitzegefühl, generalisierte Urtikaria, Kopfschmerz, Nesselsucht, Hypotonie, Lethargie, Übelkeit, Unruhe, Tachykardie, Engegefühl in der Brust, Kribbeln, Erbrechen, Giemen einschliessen können) sind möglich und können zu schwerer Anaphylaxie (einschliesslich Schock) fortschreiten (siehe auch «Warnhinweise und Vorsichtsmassnahmen»).
Unerwünschte Wirkungen in klinischen Studien
In der klinischen Studie mit OBIZUR in der erworbenen Hämophilie A waren 29 erwachsene Patienten auswertbar in Bezug auf die Sicherheit.
In der klinischen Studie kam es bei 9 Patienten zu einer schwerwiegenden unerwünschten Arzneimittelwirkung. Zwei Patienten (6.9 %) bildeten hemmende Antikörper gegen porcinen FVIII (≥0.6 Bethesda-Einheiten), die vom Prüfarzt als unerwünschte Wirkung von OBIZUR beurteilt wurden. Sieben Patienten (24.1 %) entwickelten anamnestische Reaktionen mit einem Anstieg der Hemmkörper gegen humanen Faktor VIII und/oder rekombinanten Faktor VIII, porcine Sequenz, um ≥10 BE.
Die Häufigkeit der unerwünschten Wirkungen wurde ermittelt unter Verwendung der folgenden Kriterien: sehr häufig (≥10 %), häufig (≥1 % bis <10 %), gelegentlich (≥0.1 % bis <1%), selten (≥0.01 % bis <0.1 %), sehr selten (<0.01 %), nicht bekannt (Häufigkeit auf Grundlage der verfügbaren Daten nicht abschätzbar)
Untersuchungen
Häufig: Positiver Test für neutralisierende Antikörper gegen porcines Faktor VIII (siehe auch «Warnhinweise und Vorsichtsmassnahmen»)
Erkrankungen des Immunsystems
Sehr häufig: anamnestische Reaktionen (24.1 %)
Unerwünschte Wirkungen nach Markteinführung
Nach der Markteinführung wurden keine anderen als die im Abschnitt «Unerwünschte Wirkungen in klinischen Studien» aufgeführten unerwünschten Wirkungen beobachtet.
Die Meldung des Verdachts auf Nebenwirkungen nach der Zulassung ist von grosser Wichtigkeit. Sie ermöglicht eine kontinuierliche Überwachung des Nutzen-Risiko-Verhältnisses des Arzneimittels. Angehörige von Gesundheitsberufen sind aufgefordert, jeden Verdacht einer neuen oder schwerwiegenden Nebenwirkung über das Online-Portal ElViS (Electronic Vigilance System) anzuzeigen. Informationen dazu finden Sie unter www.swissmedic.ch.
ÜberdosierungDie Wirkungen von Dosierungen von OBIZUR, die über den empfohlenen Dosierungen liegen, wurden nicht beschrieben.
Eigenschaften/WirkungenATC-Code
B02BD14
Wirkungsmechanismus
OBIZUR ist ein rekombinanter Faktor VIII (ohne B-Domäne), porciner Sequenz (susoctocog alfa). Es ist ein Glykoprotein mit 1448 Aminosäuren und einem Molekulargewicht von etwa 175 kDa. Es wird durch rekombinante DNA-Technologie (rDNA) in Babyhamster-Nierenzellen (BHK) hergestellt.
Unmittelbar nach der Aufnahme in den Kreislauf des Patienten, bindet sich der Faktor VIII an den Von-Willebrand-Faktor (VWF). Der Faktor VIII/Von-Willebrand-Faktor-Komplex besteht aus zwei Molekülen (Faktor VIII und Von-Willebrand-Faktor) mit unterschiedlichen physiologischen Funktionen. Der aktivierte Faktor VIII wirkt als Co-Faktor für den aktivierten Faktor IX und beschleunigt die Bildung von aktiviertem Faktor X aus Faktor X. Der aktivierte Faktor X wandelt Prothrombin in Thrombin um. Dieses setzt dann Fibrin aus Fibrinogen frei, und die Gerinnselbildung kann erfolgen.
Die erworbene Hämophilie A ist eine seltene Gerinnungsstörung des Blutes, bei der Patienten mit normalen Faktor VIII-Genen Autoantikörper bilden, die gegen den Faktor VIII gerichtet sind. Diese Autoantikörper neutralisieren den zirkulierenden humanen Faktor VIII, so dass ein Mangel an vorhandenem Faktor VIII entsteht.
OBIZUR ersetzt temporär den inhibierten endogenen Faktor VIII, welcher für eine wirksame Hämostase gebraucht wird.
Pharmakodynamik
Klinische Wirksamkeit und Sicherheit
Die Sicherheit und Wirksamkeit von OBIZUR zur Behandlung von schwerwiegenden Blutungsereignissen in Patienten mit erworbener Hämophilie mit autoimmunen Inhibitorenantikörpern gegen humanen Faktor VIII wurde in einer prospektiven, nicht-randomisierten, offenen Studie mit 28 Patienten untersucht (18 Kaukasier, 6 Schwarze und 4 Asiaten). Die Patienten wiesen eine lebensbedrohliche Blutung und/oder eine bedrohliche Blutung einer Extremität auf, welche eine Hospitalisierung erforderte.
Nach der Beurteilung des Hauptprüfarztes zeigten 24 Stunden nach der Initialdosierung alle initialen Blutungsepisoden eine positive Reaktion auf die Behandlung. Eine positive Reaktion war eine, bei welcher die Blutung gestoppt oder vermindert wurde, mit einer klinischen Verbesserung oder mit einer Faktor VIII-Aktivität über den vorbestimmten Zielwerten. 29/29 Patienten (100 %) erhielten während der Behandlung mit OBIZUR immunsupprimierende Medikamente.
Eine positive Antwort wurde in 95 % (19/20) Patienten nach 8 Stunden und in 100 % (18/18) nach 16 Stunden beobachtet. Zusätzlich zur Antwort auf die Behandlung wurde der gesamte Behandlungserfolg durch den Prüfarzt, basierend auf seiner Fähigkeit, die Dosis und/oder Dosierungshäufigkeit von OBIZUR abzubrechen oder zu reduzieren, bestimmt. Insgesamt hatten 24/28 (86 %) eine erfolgreiche Kontrolle (Resolution) der Initialblutungsepisode. Von diesen mit OBIZUR als First-Line Therapie behandelten Patienten, definiert als kein unmittelbar vor der OBIZUR Behandlung vorausgegangener Gebrauch eines anti-hämorrhagischen Wirkstoffes, wurde bei 16/17 (94 %) ein eventueller Behandlungserfolg berichtet. 11 Patienten erhielten ein Antihämorrhagikum (z.B. rFVIIa, aktiviertes Prothrombinkomplex-Konzentrat, Tranexamsäure) vor der Behandlung mit OBIZUR. Von diesen 11 Patienten hatten 8 (73 %) einen eventuellen Behandlungserfolg.
Die mittlere Dosis pro Injektion, um die Erstblutung erfolgreich zu behandeln, betrug 133 E/kg und die mittlere Gesamtdosis 1523 E/kg für einen Median von 6 Tagen. Die mittlere Anzahl der täglichen Infusionen pro Patient betrug 1.76 (Bandbreite 0.2 bis 5.6). Während der initialen Periode von 24 Stunden wurde in den klinischen Studien eine mittlere Gesamtdosis von 493 E/kg angewendet mit einem Median von 3 Infusionen. Wenn eine Behandlung über 24 Stunden hinaus benötigt wurde, wurde eine mittlere Gesamtdosis von 1050 E/kg angewendet mit einem Median von 10.5 Infusionen (mittlere Dosis 100 E/kg), um die Blutungsepisode zu kontrollieren.
In der klinischen Studie mit OBIZUR an Patienten mit angeborener Hämophilie A mit FVIII-Inhibitoren, die sich einer Operation unterzogen, traten bei insgesamt 5 der 8 erwachsenen Patienten, die für die Sicherheitsanalyse ausgewertet werden konnten, anamnestische Reaktionen auf.
PharmakokinetikPharmakokinetische Daten von 5 Patienten mit erworbener Hämophilie A während einem Nicht-Blutungsstatus sind in Tabelle 1 aufgeführt.
|
Tabelle 1: Individuelle pharmakokinetische Daten für die Faktor VIII-Aktivität nach der Verabreichung der letzten Dosis OBIZUR von 5 Patienten mit erworbener Hämophilie. Die Patienten waren in einem Nicht-Blutungsstatus. Die Faktor VIII-Aktivität wurde mittels eines Einstufen-Gerinnungstests gemessen.
| |
Studien-teilnehmer
|
Dosis (E)
|
Dosis (E/kg)
|
Ausgangsaktivität des humanen FVIII (%)
|
t1/2 (h)
|
Tmax (h)
|
Amax (%)
|
AUC0t (%t)
|
AUC0-∞ (%t)
| |
1
|
5000
|
76,7
|
89
|
17
|
0,42
|
213
|
3124
|
4988
| |
2
|
2934
|
30,0
|
18
|
4,6
|
0,42
|
100
|
694
|
712
| |
3
|
7540
|
144,2
|
3
|
5,3
|
0,45
|
74
|
473
|
492
| |
4
|
9720
|
206,8
|
0
|
1,8
|
0,50
|
53
|
122
|
135
| |
5
|
10000
|
133,3
|
N/V
|
4,2
|
0,75
|
178
|
1583
|
1686
|
Amax = maximale beobachtete % Aktivität; AUC0-t = Fläche unter der Konzentrations-Zeit-Kurve von Zeitpunkt 0 bis zur letzten messbaren Konzentration; AUC0-∞ = Fläche unter der Konzentrations-Zeit-Kurve von der Zeit 0 extrapoliert bis unendlich; t1/2 = terminale Halbwertszeit; Tmax = Zeit der maximalen beobachteten % Aktivität
Die mittlere Recovery nach der Initialdosis von 200 E/kg, gemessen mit OSCA, betrug 1.06 ± 0.75 E/ml (Bandbreite 0.10 – 2.61).
Neutralisierende Antikörper gegen OBIZUR wurden unter Verwendung des Bethesda Assays in der Nijmegen Modifikation gemessen. 3 der in der PK Studie eingeschlossenen Patienten wiesen nachweisbare anti-porcine Faktor VIII Inhibitorentiter bei Baseline auf (≥0.6 Bethesda-Einheiten (BE)/ml), welche als kreuzreaktive anti-humane Faktor VIII Inhibitoren betrachtet werden können. 3 der 5 Patienten wiesen keine nachweisbaren anti-porcinen Faktor VIII Titer nach der Behandlung auf (<0.6 BE/ml basierend auf dem letzten gemessenen Wert), 2 Patienten wiesen nachweisbare anti-porcine Faktor VIII Titer auf (≥0.6 BE/ml).
Präklinische DatenBasierend auf konventionellen sicherheitspharmakologischen Studien oder Mehrfach-Dosis-Toxizitätsstudien zeigten präklinische Daten kein spezielles Risiko für Menschen auf.
In Studien zur Toxizität bei wiederholter Gabe stiegen jedoch Häufigkeit und Schwere von Glomerulopathien bei Affen, denen OBIZUR mit Dosen von 75, 225 und 750 E/kg/Tag intravenös verabreicht wurde, mit der Zeit an.
Konventionelle Untersuchungen zur Reproduktions- und Entwicklungstoxikologie wurden mit OBIZUR nicht durchgeführt.
Sonstige HinweiseInkompatibilitäten
Da keine Kompatibilitätsstudien durchgeführt wurden, darf dieses Arzneimittel nicht mit anderen Arzneimitteln gemischt werden.
Haltbarkeit
Das Arzneimittel darf nur bis zu dem auf dem Behälter mit «EXP» bezeichneten Datum verwendet werden. Das rekonstituierte Produkt muss sofort verwendet werden, aber auf keinen Fall später als 3 Stunden nach der Rekonstitution. Nicht verwendete rekonstituierte Lösungen sind zu verwerfen.
Besondere Lagerungshinweise
Im Kühlschrank (2–8 °C) lagern. Nicht einfrieren.
Die Behälter im Umkarton aufbewahren, um den Inhalt vor Licht zu schützen.
Ausser Reichweite von Kindern aufbewahren.
Hinweise für die Handhabung
Nach der Rekonstitution ist die Lösung klar, farblos, frei von Partikeln und hat einen pH von 6.8 bis 7.2. Die Osmolalität des Formulierungpuffers liegt zwischen 295 und 325 mOsm/L.
Das rekonstituierte Produkt ist vor der Verabreichung visuell auf Partikel und Verfärbung zu untersuchen. Lösungen mit Partikeln oder Verfärbungen dürfen nicht verabreicht werden.
Restmengen sind sachgemäss zu entsorgen.
Vorbereitung
Vor der Rekonstitution brauchen Sie folgendes:
·Berechnete Anzahl Durchstechflaschen mit OBIZUR
·Gleiche Anzahl 1 ml Fertigspritzen mit sterilem Wasser für Injektionszwecke und sterile Durchstechflaschen-Adapter
·Alkoholtupfer
·Grosse sterile Spritze, die das gesamte Volumen des rekonstituierten Präparates aufnehmen kann
Führen Sie die Rekonstitution, die Verabreichung des Produktes und die Handhabung des Verabreichungssets und der Nadeln mit Vorsicht aus.
Die unten beschriebene Vorgehensweise stellt nur eine generelle Richtlinie für die Vorbereitung und Rekonstitution von OBIZUR dar. Wiederholen Sie die folgenden Anweisungen zur Rekonstitution für jede zu verabreichende Durchstechflasche von OBIZUR.
Rekonstitution
Wenden Sie während des Rekonstitutionsprozesses eine aseptische Arbeitsweise an.
1.Die OBIZUR Durchstechflasche und die Fertigspritze mit Lösungsmittel auf Raumtemperatur bringen.
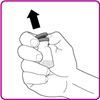
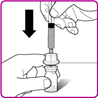
2.Die Plastikschutzkappe von der OBIZUR Durchstechflasche entfernen (Fig. A).
3.Den Gummistopfen mit Alkoholtupfer (nicht mitgeliefert) abwischen und vor Gebrauch trocknen lassen.
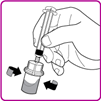
4.Die Schutzfolie der Packung der Durchstechflaschen-Adapter zurückziehen (Fig B). Den Luer-Lock (Spitze) in der Mitte des Durchstechflaschen-Adapters nicht berühren. Den Durchstechflaschen-Adapter nicht aus der Verpackung nehmen.
5.Stellen Sie die Packung mit dem Durchstechflaschen-Adapter auf eine saubere Oberfläche, so dass der Luer-Lock nach oben zeigt.
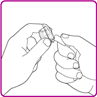
6.Schrauben Sie die fälschungssichere Kappe von der Fertigspritze (Fig. C).
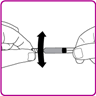
7.Während Sie die Packung mit dem Durchflaschen-Adapter festhalten, schliessen Sie die Fertigspritze auf den Durchstechflasche-Adapter an, indem Sie die Fertigspritze in den Luer-Lock in der Mitte des Durchstechflaschen-Adapters drücken und schrauben Sie diesen im Uhrzeigersinn an, bis die Fertigspritze gesichert ist. Ziehen Sie diese nicht zu stark an (Fig. D).
8.Entfernen Sie die Plastikverpackung (Fig. E).
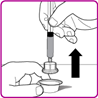
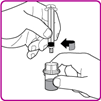
9.Stellen Sie die OBIZUR Durchstechflasche auf eine saubere, flache, harte Oberfläche. Stellen Sie den Durchstechflaschen-Adapter über die OBIZUR Durchstechflasche und drücken Sie den Filterdorn des Durchstechflaschen-Adapters fest durch die Mitte des Gummikreises der OBIZUR Durchstechflasche, bis die farblose Plastikkappe auf die Durchstechflasche einrastet (Fig. F).
10.Drücken Sie den Spritzenkolben nach unten, um das Lösungsmittel von der Spritze langsam in die Durchstechflasche zu spritzen.
11.Leicht die OBIZUR Durchstechflasche schwenken (in einer Kreisbewegung), ohne die Fertigspritze zu entfernen, bis das gesamte Pulver vollständig gelöst/rekonstituiert ist (Fig. G). Die rekonstituierte Lösung ist vor der Verabreichung visuell auf Partikel zu untersuchen. Lösungen mit Partikeln oder Verfärbungen dürfen nicht verabreicht werden.
12.Halten Sie mit einer Hand die Durchstechflasche und den Durchstechflaschen-Adapter und mit der anderen Hand fassen Sie fest den Zylinder der Fertigspritze an und schrauben Sie im Gegenuhrzeigersinn die Spritze von dem Durchstechflaschen-Adapter ab (Fig. H).
13.Verwenden Sie OBIZUR innerhalb 3 Stunden nach der Rekonstitution, wenn bei Raumtemperatur gelagert.
|
Abb. A
|
Abb. B
|
Abb. C
|
Abb. D
| |
|
|
|
|
|
|
|

| |
Abb. E
|
Abb. F
|
Abb. G
|
Abb. H
| |
|
|
|
|
|
|
|
|
Verabreichung
Nur zur intravenösen Injektion!
·Vor der Verabreichung die rekonstituierte OBIZUR Lösung immer auf Schwebeteilchen oder Verfärbung überprüfen. Die Lösung sollte klar und farblos sein. Nicht verabreichen, falls die Lösung Schwebeteilchen oder eine Verfärbung aufweist.
·OBIZUR nicht im gleichen Schlauchmaterial oder im gleichen Behälter mit anderen Arzneimittel zur Injektion verabreichen
Bei aseptischer Arbeitsweise erfolgt die Verabreichung gemäss nachfolgender Prozedere:
1.Sobald alle Durchstechflaschen rekonstituiert sind, eine grosse Spritze mit dem Durchstechflaschen-Adapter verbinden, indem die Spritzenspitze in den Luer-Lock in der Mitte des Durchstechflaschen-Adapters leicht gedrückt und durch Drehen im Uhrzeigersinn angeschraubt wird, bis die Spritze gesichert ist.
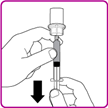
2.Die Durchstechflasche umdrehen; die Luft in der Spritze in die Durchstechflasche drücken und das rekonstituierte OBIZUR in die Spritze aufziehen (Fig. I).
Abb. I
3.Die grosse Spritze im Gegenuhrzeigersinn vom Durchstechflaschen-Adapter abschrauben und diesen Vorgang für alle rekonstituierten OBIZUR Durchstechflaschen wiederholen, bis das gesamte Volumen, das verabreicht werden soll, erreicht wird.
4.Das rekonstituierte OBIZUR intravenös mit einer Infusionsrate von 1-2 ml/min verabreichen.
Zulassungsnummer65874 (Swissmedic)
PackungenEine Packung OBIZUR enthält 1, 5 oder 10 der folgenden Komponenten (B):
·Durchstechflasche mit OBIZUR Pulver à 500 Einheiten
·Vorgefüllte Fertigspritze mit 1 ml Lösungsmittel
·Flüssigkeitstransfergerät mit integrierter Plastikspitze (Adapter)
ZulassungsinhaberinTakeda Pharma AG, 8152 Opfikon
Stand der InformationApril 2024
|