ZusammensetzungWirkstoffe
Semaglutide
Hilfsstoffe
Dinatrii phosphas dihydricus, Propylenglycolum, Phenolum, Acidum hydrochloridum/Natrii hydroxidum (q.s. ad pH), Aqua ad iniectabile q.s. ad solutionem pro 1 ml corresp natrium 0.367 mg.
Darreichungsform und Wirkstoffmenge pro EinheitInjektionslösung.
Klare, farblose oder nahezu farblose, isotonische Lösung; pH = 7.4.
Ozempic DualDose 0.25 mg oder 0.5 mg/Dosis
1 ml Lösung enthält 1.34 mg Semaglutide*. 1 Fertigpen enthält 2 mg Semaglutide* in 1.5 ml Lösung.
Ozempic FixDose 1 mg/Dosis
1 ml Lösung enthält 1.34 mg Semaglutide*. 1 Fertigpen enthält 4 mg Semaglutide* in 3 ml Lösung.
Ozempic FixDose 2 mg/Dosis
1 ml Lösung enthält 2.68 mg Semaglutide*. 1 Fertigpen enthält 8 mg Semaglutide* in 3 ml Lösung.
* Gentechnisch hergestellt durch rekombinante DNS-Technologie in Zellen von Saccharomyces cerevisiae.
Indikationen/AnwendungsmöglichkeitenOzempic wird zur Behandlung Erwachsener mit unzureichend kontrolliertem Diabetes mellitus Typ 2 ergänzend zu Diät und Bewegung angewendet:
-Als Monotherapie bei Kontraindikation oder Unverträglichkeit für Metformin.
-In Kombination mit anderen blutzuckersenkenden Arzneimitteln (siehe "Klinische Wirksamkeit" für Ergebnisse zu den in klinischen Studien untersuchten Kombinationen).
Ozempic wird angewendet zur Verzögerung des Fortschreitens einer chronischen Nierenerkrankung bei Erwachsenen mit Diabetes mellitus Typ 2 und chronischer Nierenerkrankung (siehe "Klinische Wirksamkeit" ).
Für Studienergebnisse zur kardiovaskulären Sicherheit und Mortalität bei Patienten mit Diabetes mellitus Typ 2 (siehe "Klinische Wirksamkeit" ).
Dosierung/AnwendungTherapieeinleitung
Die Anfangsdosis von Ozempic beträgt 0.25 mg einmal wöchentlich. Nach 4 Wochen sollte die Dosis auf 0.5 mg einmal wöchentlich erhöht werden.
Zur Verbesserung der Blutzuckerkontrolle bei Erwachsenen mit Diabetes mellitus Typ 2
Um die Einstellung des Blutzuckerspiegels zu verbessern kann, nach mindestens 4 weiteren Wochen mit einer Dosis von 0.5 mg einmal pro Woche, die Dosis auf 1 mg einmal pro Woche erhöht werden. Nach mindestens 4 Wochen mit einer Dosis von 1 mg einmal wöchentlich kann die Dosis auf 2 mg einmal wöchentlich erhöht werden.
Zur Verzögerung des Fortschreitens einer chronischen Nierenerkrankung bei Erwachsenen mit Diabetes mellitus Typ 2 und chronischer Nierenerkrankung
Die Dosis sollte auf die Erhaltungsdosis von 1 mg einmal wöchentlich erhöht werden, nachdem mindestens 4 Wochen lang die Dosis von 0.5 mg verabreicht wurde. Patienten mit chronischer Nierenerkrankung, die an der FLOW-Studie teilnahmen, erhielten eine Erhaltungsdosis von 1 mg (siehe "Klinische Wirksamkeit" ).
Kombinationstherapie
Wird Ozempic zusätzlich zu einer bestehenden Therapie mit Metformin und/oder Thiazolidinedion oder zu einem SGLT2-Inhibitor (Natrium-Glukose-Cotransporter-2-Hemmer) gegeben, kann die bestehende Dosis von Metformin und/oder Thiazolidinedion oder SGLT2-Inhibitor unverändert beibehalten werden. Wird Ozempic zusätzlich zu einer bestehenden Therapie mit Sulfonylharnstoffen oder Insulin gegeben, sollte eine Dosisreduktion vom Sulfonylharnstoff oder Insulin erwogen werden, um das Risiko einer Hypoglykämie zu senken. Dabei ist ausserdem zu beachten, dass eine zu rasche Verbesserung der glykämischen Kontrolle zur temporären Verschlechterung einer diabetischen Retinopathie führen kann (siehe "Warnhinweise und Vorsichtsmassnahmen" ).
Spezielle Dosierungsanweisungen
Patienten mit Leberfunktionsstörungen
Eine Dosisanpassung ist bei Patienten mit eingeschränkter Leberfunktion nicht erforderlich.
Patienten mit Nierenfunktionsstörungen
Eine Dosisanpassung ist bei Patienten mit eingeschränkter Nierenfunktion nicht erforderlich.
Ältere Patienten (≥65 Jahre alt)
Eine Dosisanpassung ist bei älteren Menschen nicht erforderlich.
Kinder und Jugendliche
Die Sicherheit und Wirksamkeit von Ozempic bei Kindern und Jugendlichen unter 18 Jahren wurde nicht untersucht.
Verspätete Dosisgabe
Falls eine Dosis ausgelassen wird, sollte diese so schnell wie möglich und innerhalb von 5 Tagen nach dem Auslassen nachgeholt werden. Falls mehr als 5 Tage verstrichen sind, sollte die ausgelassene Dosis nicht nachgeholt werden. Die nächste Dosis sollte am dafür vorgesehenen Tag gegeben werden. Weitere Informationen zur Anwendung siehe "Sonstige Hinweise" .
Art der Anwendung
Ozempic wird einmal pro Woche zu einem beliebigen Zeitpunkt und unabhängig von den Mahlzeiten angewendet.
Ozempic wird subkutan in Abdomen, Oberschenkel oder Oberarm injiziert. Die Injektionsstelle kann ohne Dosisanpassung geändert werden. Ozempic darf nicht intravenös oder intramuskulär angewendet werden.
Der Tag der wöchentlichen Anwendung kann bei Bedarf geändert werden, solange zwischen zwei Dosen mindestens 2 Tage (>48 Stunden) liegen.
KontraindikationenÜberempfindlichkeit gegenüber dem Wirkstoff oder einem der Hilfsstoffe.
Warnhinweise und VorsichtsmassnahmenOzempic sollte nicht bei Patienten mit Diabetes mellitus Typ 1 oder zur Behandlung der diabetischen Ketoazidose angewendet werden.
Aspiration im Zusammenhang mit einer Vollnarkose oder tiefer Sedierung
Bei Patienten, die GLP-1-Rezeptor-Agonisten erhielten und die sich einer Vollnarkose oder einer tiefen Sedierung unterzogen, wurden Fälle von pulmonaler Aspiration berichtet, trotz der berichteten Einhaltung der präoperativen Nüchternempfehlungen. Daher sollte vor der Durchführung von Eingriffen unter Vollnarkose oder tiefer Sedierung das erhöhte Risiko für Restmageninhalt aufgrund einer verzögerten Magenentleerung berücksichtigt werden.
Gastrointestinale Wirkungen und Dehydrierung
Die Anwendung von GLP-1-Rezeptor-Agonisten kann mit unerwünschten gastrointestinalen Wirkungen assoziiert sein. Dies sollte bei der Behandlung von Patienten mit eingeschränkter Nierenfunktion beachtet werden, da Übelkeit, Erbrechen und Diarrhö eine Dehydrierung verursachen können, was in seltenen Fällen zu einer Verschlechterung der Nierenfunktion führen kann (siehe "Unerwünschte Wirkungen" ). Patienten, die mit Semaglutide behandelt werden, müssen auf das potenzielle Dehydrierungsrisko im Zusammenhang mit gastrointestinalen Nebenwirkungen hingewiesen werden und Vorkehrungen gegen Flüssigkeitsverlust treffen.
Akute Pankreatitis
Bei Anwendung von GLP-1-Rezeptor-Agonisten wurde akute Pankreatitis beobachtet. Patienten müssen über die charakteristischen Symptome einer akuten Pankreatitis informiert werden. Wird eine Pankreatitis vermutet, ist Ozempic abzusetzen; wird eine akute Pankreatitis bestätigt, ist die Behandlung mit Ozempic nicht erneut aufzunehmen. Patienten mit Pankreatitis in der Vorgeschichte wurden in den klinischen Studien mit Semaglutide nicht behandelt. Deshalb ist bei diesen Patienten Vorsicht geboten. Sofern keine anderen Anzeichen und Symptome einer akuten Pankreatitis vorliegen, deutet eine isolierte Erhöhung der Pankreasenzyme nicht zwingend auf eine akute Pankreatitis hin.
Hypoglykämie
Patienten, die Ozempic in Kombination mit einem Sulfonylharnstoff oder Insulin erhalten, können ein erhöhtes Risiko für eine Hypoglykämie haben. Das Risiko einer Hypoglykämie kann durch Reduktion der Sulfonylharnstoff- oder der Insulin-Dosis bei Behandlungsbeginn mit Ozempic gesenkt werden.
Risiko thyreoidaler C-Zell-Tumoren
Präklinische Studien mit GLP-1-Rezeptoragonisten an Nagern legen nahe, dass GLP-1-Rezeptoragonisten möglicherweise mit einem erhöhten Risiko von fokalen Hyperplasien der thyreoidalen C-Zellen und C-Zell-Tumoren einhergehen (s. präklinische Daten).
Es ist nicht bekannt, ob beim Menschen ein Zusammenhang besteht zwischen GLP-1-Rezeptoragonisten und thyreoidalen C-Zell-Tumoren, einschliesslich des medullären Schilddrüsenkarzinoms (medullary thyroid carcinoma, MTC). Patienten mit MTC und Patienten mit multiplem endokrinem Neoplasie-Syndrom vom Typ 2 (MEN 2) in der Anamnese wurden in den klinischen Studien mit Semaglutide nicht behandelt. Vor einer Behandlung mit Ozempic ist deshalb in diesem spezifischen Kollektiv eine sorgfältige Nutzen-Risiko-Abwägung erforderlich. Der klinische Wert einer routinemässigen Überwachung des Serum-Calcitonin-Spiegels ist nicht belegt.
Diabetische Retinopathie
In einer grossen kardiovaskulären Sicherheitsstudie wurde unter der Behandlung mit Semaglutide ein im Vergleich zur Standardtherapie erhöhtes Risiko für das Auftreten von Komplikationen einer diabetischen Retinopathie beobachtet (50 [3.0 %] versus 29 [1.8 %]). Primär betroffen (>80 % der Fälle) waren Patienten, die bereits zu Behandlungsbeginn eine diabetische Retinopathie aufwiesen; weiterer Risikofaktor war die gleichzeitige Anwendung von Insulin (siehe "Unerwünschte Wirkungen" ). Semaglutide sollte in diesen Risikopatienten nur unter engmaschiger, ophthalmologischer Kontrolle eingesetzt werden. Die zu rasche Korrektur einer chronischen Hyperglykämie kann mit einer initialen Verschlechterung der diabetischen Retinopathie einhergehen, obwohl die langfristige Verbesserung der glykämischen Kontrolle das Risiko für eine diabetische Retinopathie senkt. Daher sollte zu Beginn der Behandlung mit Semaglutide zusätzlich zu Insulin eine Reduktion der Insulin-Dosis in Betracht gezogen werden.
Nichtarteriitische anteriore ischämische Optikusneuropathie (NAION)
Daten aus epidemiologischen Studien deuten auf ein erhöhtes Risiko für nichtarteriitische anteriore ischämische Optikusneuropathie (NAION) während der Behandlung mit Semaglutide hin. Es gibt kein bestimmtes Zeitintervall, in dem sich eine NAION nach Beginn der Behandlung entwickeln kann. Bei plötzlichem Verlust des Sehvermögens sollte eine augenärztliche Untersuchung erfolgen, falls eine NAION bestätigt wird, sollte die Behandlung mit Semaglutide abgebrochen werden (siehe "Unerwünschte Wirkungen" ).
Gastrointestinale (unerwünschte) Wirkungen
Nach Markteinführung wurde bei Patienten, die mit GLP-1-Rezeptor-Agonisten behandelt wurden, über akute Nierenschäden und eine Verschlechterung der chronischen Niereninsuffizienz berichtet, die manchmal eine Hämodialyse erforderlich machen können. Einige dieser Ereignisse wurden bei Patienten ohne bekannte zugrunde liegende Nierenerkrankung gemeldet. Die Mehrzahl der gemeldeten Ereignisse trat bei Patienten auf, die bereits unter Übelkeit, Erbrechen, Durchfall oder Dehydrierung litten. Die Nierenfunktion soll überwacht werden, wenn die Behandlung mit Ozempic bei Patienten, die über schwere unerwünschte Magen-Darm-Reaktionen berichten, initiiert oder auftitriert wird.
Patienten mit Gastroparese
Bei Patienten mit Gastroparese, die mit Semaglutide behandelt werden, können schwerere oder schwerwiegendere gastrointestinale unerwünschte Wirkungen auftreten. Semaglutide sollte bei diesen Patienten mit Vorsicht angewendet werden (siehe "Unerwünschte Wirkungen" ).
Hilfsstoffe
Dieses Arzneimittel enthält weniger als 1 mmol Natrium (23 mg) pro Dosis, d. h. es ist nahezu "natriumfrei" .
InteraktionenIn-vivo-Daten
Die durch Semaglutide verzögerte Magenentleerung kann die Resorption gleichzeitig oral angewendeter Arzneimittel beeinflussen. Die potenzielle Wirkung von Semaglutide auf die Resorption der unten aufgeführten, gleichzeitig oral angewendeten Arzneimittel wurde in Studien mit Semaglutide 1 mg im Steady State untersucht. Es wurden keine klinisch relevanten Arzneimittelwechselwirkungen zwischen Semaglutide und den beurteilten Arzneimitteln beobachtet. Daher ist bei gleichzeitiger Verabreichung mit Semaglutide keine Dosisanpassung erforderlich.
Orale Kontrazeptiva
Es ist nicht zu erwarten, dass Semaglutide die Wirkung oraler Kontrazeptiva herabsetzt, da die gleichzeitige Anwendung von Semaglutide mit einem oralen Kontrazeptivum (Kombinationspräparat, 0.03 mg Ethinylestradiol/0.15 mg Levonorgestrel) die Gesamtexposition von Ethinylestradiol und Levonorgestrel nicht klinisch relevant verändert hat. Die Exposition von Ethinylestradiol war nicht beeinträchtigt. Bei der Exposition von Levonorgestrel im Steady State wurde ein Anstieg von 20 % beobachtet. Cmax war bei keinem der Präparate beeinträchtigt.
Atorvastatin
Nach Gabe einer Einzeldosis Atorvastatin (40 mg) führte Semaglutide nicht zu einer Änderung der Gesamtexposition von Atorvastatin. Mit Semaglutide war die Cmax von Atorvastatin um 38 % verringert. Dies wurde als nicht klinisch relevant eingestuft.
Digoxin
Nach Gabe einer Einzeldosis Digoxin (0.5 mg) führte Semaglutide nicht zu einer Änderung der Gesamtexposition oder der Cmax von Digoxin.
Metformin
Nach Gabe von Metformin (500 mg) zweimal pro Tag über 3.5 Tage führte Semaglutide nicht zu einer Änderung der Gesamtexposition oder der Cmax von Metformin.
Warfarin und andere Cumarinderivate
Nach Gabe einer Einzeldosis Warfarin (25 mg) veränderte Semaglutide die Gesamtexposition oder die Cmax von R- und S-Warfarin nicht. Es kam auch nicht zu einer klinisch relevanten Änderung der pharmakodynamischen Wirkungen von Warfarin, gemessen an der International Normalised Ratio (INR). Jedoch wurden Fälle von INR-Senkungen bei gleichzeitiger Anwendung von Acenocoumarol und Semaglutide berichtet. Bei Einleitung einer Semaglutide-Behandlung bei Patienten, die Warfarin oder andere Cumarinderivate einnehmen, wird daher eine regelmässige Überwachung des INR empfohlen.
In-vitro-Studien
In-vitro-Studien haben für Semaglutide ein sehr geringes Potenzial für die Inhibition oder Induktion von CYP-Enzymen und für die Inhibition von Wirkstofftransportern aufgezeigt.
Schwangerschaft, StillzeitSchwangerschaft
Es liegen begrenzte Daten zur Anwendung von Semaglutide bei schwangeren Frauen vor. Tierexperimentelle Studien haben eine Reproduktionstoxizität gezeigt (siehe "Präklinische Daten" ). Semaglutide sollte während der Schwangerschaft nicht angewendet werden. Frauen im gebärfähigen Alter wird empfohlen, während der Behandlung mit Semaglutide zu verhüten. Falls eine Patientin schwanger werden möchte oder schwanger wird, sollte die Behandlung mit Semaglutide abgebrochen werden. Die Behandlung mit Semaglutide sollte aufgrund seiner langen Halbwertszeit mindestens 2 Monate vor einer geplanten Schwangerschaft abgebrochen werden (siehe "Pharmakokinetik" ).
Stillzeit
In der Muttermilch stillender Frauen wurden keine messbaren Konzentrationen von Semaglutide gefunden. Da ein Risiko für das gestillte Kind nicht ausgeschlossen werden kann, sollte Semaglutide während der Stillzeit nicht angewendet werden.
Fertilität
Die Auswirkung von Semaglutide auf die Fertilität bei Menschen ist nicht bekannt. Semaglutide hat die Fertilität männlicher Ratten nicht beeinträchtigt. Bei weiblichen Ratten wurde bei Dosen, die mit einem mütterlichen Gewichtsverlust einhergingen, eine Verlängerung des Östrus und eine geringe Abnahme der Anzahl der Ovulationen beobachtet.
Wirkung auf die Fahrtüchtigkeit und auf das Bedienen von MaschinenOzempic hat keinen oder einen zu vernachlässigenden Einfluss auf die Verkehrstüchtigkeit und die Fähigkeit zum Bedienen von Maschinen. Wenn es zusammen mit Sulfonylharnstoffen oder Insulin verwendet wird, sollten Patienten angewiesen werden, Vorsichtsmassnahmen zu treffen, um Hypoglykämien beim Lenken von Fahrzeugen und Bedienen von Maschinen zu vermeiden (siehe "Warnhinweise und Vorsichtsmassnahmen" ).
Unerwünschte WirkungenZusammenfassung des Sicherheitsprofils
In 8 klinischen Studien erhielten 4'792 Patienten Ozempic allein oder in Kombination mit anderen blutzuckersenkenden Arzneimitteln. Die Dauer der Behandlung reichte von 30 Wochen bis zu 2 Jahren.
Die in klinischen Studien am häufigsten gemeldeten unerwünschten Wirkungen waren gastrointestinale Störungen, einschliesslich Übelkeit, Durchfall und Erbrechen. Im Allgemeinen war der Schweregrad dieser Reaktionen leicht bis mittelschwer und von kurzer Dauer.
In einer 40-wöchigen Phase-3b-Studie erhielten 959 Patienten Semaglutide 1 mg oder 2 mg. Das Sicherheitsprofil von Semaglutide 2 mg war konsistent mit dem Sicherheitsprofil von Semaglutide, das in Phase-3a-Studien beobachtet wurde. Die Häufigkeit gastrointestinaler Störungen war mit Semaglutide 2 mg gegenüber Semaglutide 1 mg nur geringfügig erhöht (34.0 % vs. 30.8 %).
Tabellarische Auflistung der Nebenwirkungen
In Tabelle 1 sind unerwünschte Wirkungen aufgeführt, die in klinischen Phase-3-Studien (einschliesslich der Langzeitstudie zu kardiovaskulären Endpunkten) und Berichten nach der Markteinführung bei Patienten mit Diabetes mellitus Typ 2 berichtet wurden (weitere Informationen siehe "Eigenschaften/Wirkungen" ). Die Häufigkeiten der unerwünschten Wirkungen (ausgenommen Komplikationen der diabetischen Retinopathie, siehe Fussnote in Table 1) basieren auf den gepoolten Daten der klinischen Studien, ausgenommen der Studie zu den kardiovaskulären Ereignissen.
Liste der unerwünschten Wirkungen
Die unerwünschten Wirkungen sind untenstehend nach Systemorganklassen und Häufigkeit aufgeführt. Die Häufigkeiten sind wie folgt definiert: Sehr häufig (≥1/10); häufig (≥1/100, <1/10); gelegentlich (≥1/1'000, <1/100); selten (≥1/10'000, <1/1'000); sehr selten (<1/10'000) nicht bekannt (Häufigkeit auf Grundlage der bekannten Daten nicht abschätzbar).
Tabelle 1: Häufigkeit der unerwünschten Wirkungen von Ozempic
Systemorganklassen Häufigkeit Unerwünschte Wirkung
gemäss MedDRA
Erkrankungen des Gelegentlich Überempfindlichkeit
Immunsystems
Selten Anaphylaktische Reaktion
Stoffwechsel- und Sehr häufig Hypoglykämiea) bei
Ernährungsstörungen Anwendung mit Insulin
oder Sulfonylharnstoffen
Häufig Hypoglykämiea) bei Anwendung mit anderen oralen
Antidiabetika (OAD) Verminderter Appetit
Erkrankungen des Häufig Schwindelgefühl
Nervensystems Kopfschmerzen
Gelegentlich Dysgeusie
Augenerkrankungen Häufig Komplikationen der
diabetischen Retinopathi
eb)
Sehr selten Nichtarteriitische anteriore ischämische
Optikusneuropathie (NAION)
Herzerkrankungen Gelegentlich Erhöhte Herzfrequenz
Erkrankungen des Sehr häufig Übelkeit Durchfall
Gastrointestinaltrakts
Häufig Erbrechen Bauchschmerzen Abdominelles
Spannungsgefühl Obstipation Dyspepsie Gastritis
Gastroösophagealer Reflux Aufstossen Flatulenz
Gelegentlich Akute Pankreatitis Verzögerte Magenentleerung
Nicht bekannt Darmobstruktionc), d)
Leber- und Gallenerkra Häufig Cholelithiasis
nkungen
Gelegentlich Cholezystitis
Erkrankungen der Haut Nicht bekannt Angioedemac)
und des Unterhautgeweb
es
Allgemeine Erkrankunge Häufig Erschöpfung
n und Beschwerden am
Verabreichungsort
Gelegentlich Reaktionen an der Injektionsstelle
Untersuchungen Häufig Erhöhte Lipase Erhöhte
Amylase Gewichtsabnahme
a) Hypoglykämie definiert als schwer (erfordert die Hilfe einer anderen Person) oder symptomatisch in Kombination mit einem Blutzuckerspiegel <3.1 mmol/l
b) Komplikationen der diabetischen Retinopathie umfassen: Notwendigkeit der Photokoagulation der Netzhaut, Notwendigkeit der Behandlung mit intravitrealen Wirkstoffen, Glaskörperblutung, Einsetzen der Diabetes-bedingten Erblindung. Die Häufigkeit basiert auf der Studie zu kardiovaskulären Ergebnissen.
c) Aus Meldungen nach Markteinführung
d) Die zusammengefasste Bezeichnung umfasst die unerwünschten Ereignisse Darmobstruktion, Ileus und Dünndarmobstruktion
2-jährige Studie zu kardiovaskulären Ergebnissen und Sicherheit
In der kardiovaskulären Risikopopulation war das Nebenwirkungsprofil ähnlich dem in anderen klinischen Studien beobachteten (beschrieben in "Eigenschaften/Wirkungen" ).
Beschreibung ausgewählter Nebenwirkungen
Stoffwechsel- und Ernährungsstörungen
Sehr häufig – Hypoglykämie bei Anwendung mit Insulin (10.7 %) oder Sulfonylharnstoffen (10.4 %)
Häufig – Hypoglykämie bei Anwendung mit anderen oralen Antidiabetika (OAD)
Bei der Anwendung von Ozempic als Monotherapie wurden keine schweren Hypoglykämien beobachtet. Schwere Hypoglykämie wurde vorwiegend beobachtet, wenn Ozempic zusammen mit einem Sulfonylharnstoff (1.2 % der Teilnehmer, 0.03 Ereignisse/Patientenjahr) oder Insulin (1.5 % der Teilnehmer, 0.02 Ereignisse/Patientenjahr) verwendet wurde. Es wurden wenige Episoden (0.1 % der Teilnehmer, 0.001 Ereignisse/Patientenjahr) bei der Anwendung von Ozempic in Kombination mit anderen (als Sulfonylharnstoff) oralen Antidiabetika beobachtet.
Augenerkrankungen
Häufig – Komplikationen der diabetischen Retinopathie
In einer 2-jährigen klinischen Studie mit 3'297 Patienten mit Diabetes mellitus Typ 2 und hohem kardiovaskulären Risiko waren die Komplikationen der diabetischen Retinopathie ein Endpunkt. In dieser Studie traten Komplikationen der diabetischen Retinopathie bei Patienten, die mit Ozempic behandelt wurden, häufiger auf (3.0 %) als unter Placebo (1.8 %). Über 80 % der Patienten mit einer Komplikation der diabetischen Retinopathie hatten vor Behandlungsbeginn eine dokumentierte diabetische Retinopathie. Bei Patienten, die keine diabetische Retinopathie in der Vorgeschichte (dokumentiert) hatten, war die Anzahl der Ereignisse unter Ozempic und Placebo ähnlich.
In klinischen Studien mit einer Dauer von bis zu 1 Jahr mit 4'807 Patienten mit Diabetes mellitus Typ 2 traten unerwünschte Wirkungen im Zusammenhang mit der diabetischen Retinopathie unter Ozempic in 1.7 % der Patienten auf, unter den Vergleichspräparaten in 2.0 % der Patienten.
Sehr selten – Nichtarteriitische anteriore ischämische Optikusneuropathie (NAION)
Die Ergebnisse mehrerer grosser epidemiologischer Studien deuten darauf hin, dass die Exposition gegenüber Semaglutide bei Erwachsenen mit Diabetes mellitus Typ 2 mit einem etwa zweifachen Anstieg des relativen Risikos für die Entwicklung einer NAION verbunden ist, was etwa einem zusätzlichen Fall pro 10'000 Behandlungsjahre entspricht.
Erkrankungen des Gastrointestinaltrakts
Bei Patienten mit Gastroparese können unter Behandlung mit Semaglutide schwerere oder schwerwiegendere gastrointestinale Auswirkungen auftreten.
Sehr häufig – Übelkeit (19.9 %), Diarrhö (13.3 %)
Häufig – Erbrechen, Bauchschmerzen, abdominelles Spannungsgefühl, Obstipation, Dyspepsie, Gastritis, Gastroösophageale Refluxkrankheit, Aufstossen, Flatulenz.
Bei Patienten, die mit Ozempic 0.5 mg und 1 mg behandelt wurden, trat bei 17.0 % und 19.9 % Übelkeit, bei 12.2 % und 13.3 % Diarrhö und bei 6.4 % und 8.4 % Erbrechen auf. Die meisten Ereignisse waren leicht bis mittelschwer und von kurzer Dauer. Die Ereignisse führten bei 3.9 % und 5 % der Patienten zum Behandlungsabbruch. Die Ereignisse wurden am häufigsten in den ersten Monaten der Behandlung berichtet.
Gelegentlich – akute Pankreatitis
Die Häufigkeit von bestätigten Fällen akuter Pankreatitis, die in klinischen Phase 3a Studien gemeldet wurden, lag bei 0.3 % für Semaglutide beziehungsweise bei 0.2 % für das Vergleichspräparat. In der 2-jährigen Studie zu kardiovaskulären Endpunkten lag die Häufigkeit der bestätigten Pankreatitis Fälle bei 0.5 % für Semaglutide und 0.6 % für Placebo (siehe "Warnhinweise und Vorsichtsmassnahmen" ).
Gelegentlich – Überempfindlichkeit
In den klinischen Phase 3a Studien wurden bei 0.3 % der Patienten, die Semaglutide erhielten, Überempfindlichkeitsereignisse (z. B. Nesselsucht, Hautausschlag) berichtet, die nach Einschätzung des Prüfarztes in kausalem Zusammenhang mit der Behandlung stehen.
Unerwünschte Wirkungen nach Markteinführung
Erkrankungen der Nieren- und Harnwege: Akute Nierenschädigung (siehe "Warnhinweise und Vorsichtsmassnahmen" ).
Behandlungsabbruch aufgrund einer Nebenwirkung
Die Inzidenz der Behandlungsabbrüche aufgrund von unerwünschten Wirkungen lag bei Patienten, die mit 1 mg Ozempic behandelt wurden, bei 8.7 %. Die am häufigsten zum Abbruch führenden unerwünschten Wirkungen waren gastrointestinale Störungen.
Die Meldung des Verdachts auf Nebenwirkungen nach der Zulassung ist von grosser Wichtigkeit. Sie ermöglicht eine kontinuierliche Überwachung des Nutzen-Risiko-Verhältnisses des Arzneimittels. Angehörige von Gesundheitsberufen sind aufgefordert, jeden Verdacht einer neuen oder schwerwiegenden Nebenwirkung über das Online-Portal ElViS (Electronic Vigilance System) anzuzeigen. Informationen dazu finden Sie unter www.swissmedic.ch.
ÜberdosierungÜberdosierungen von bis zu 4 mg in einer Einzeldosis und bis zu 4 mg pro Woche wurden in klinischen Studien berichtet. Die am häufigsten berichtete Nebenwirkung war Übelkeit. Alle Patienten erholten sich komplikationslos.
Es gibt kein spezifisches Antidot für die Überdosierung mit Ozempic. Im Fall einer Überdosierung ist eine angemessene unterstützende Behandlung entsprechend den klinischen Zeichen und Symptomen des Patienten einzuleiten. Angesichts der langen Halbwertszeit von Ozempic von rund 1 Woche kann eine längere Beobachtungs- und Behandlungsperiode im Hinblick auf diese Symptome erforderlich sein (siehe "Pharmakokinetik" ).
Eigenschaften/WirkungenATC-Code
A10BJ06
Wirkungsmechanismus
Semaglutide ist ein GLP-1-Analogon mit einer Sequenzhomologie von 94 % zum humanen GLP-1. GLP-1 ist ein physiologisches Hormon, das mehrere Aufgaben bei der Glucose- und Appetitregulierung, im kardiovaskulären System und in den Nieren erfüllt. Semaglutide wirkt als ein GLP-1-Rezeptor-Agonist, der sich selektiv an den GLP-1-Rezeptor, den Zielrezeptor für natives GLP-1, bindet und diesen aktiviert. GLP-1-Rezeptoren sind im Pankreas, im Gehirn, im Herzen, im Gefässsystem, im Immunsystem und den Nieren exprimiert.
Im Vergleich zum nativen GLP-1 hat Semaglutide eine verlängerte Halbwertszeit von rund 1 Woche, weshalb es zur einmal wöchentlichen subkutanen Injektion geeignet ist. Der Hauptmechanismus der Verlängerung ist die Albuminbindung, die zur verminderten renalen Clearance und zum Schutz gegen metabolische Degradation führt. Darüber hinaus ist Semaglutide stabilisiert gegen Degradation durch das DPP-4-Enzym.
Semaglutide senkt den Blutzuckerspiegel durch die Stimulation der Insulinsekretion und die Verringerung der Glucagonsekretion, wobei beides glucoseabhängig erfolgt. Wenn der Blutzuckerspiegel hoch ist, wird die Insulinsekretion stimuliert und die Glucagonsekretion gehemmt. Der Mechanismus der Verringerung des Blutzuckerspiegels umfasst auch eine minimale Verzögerung der Magenentleerung in der frühen postprandialen Phase. Bei einer Hypoglykämie verringert Semaglutide die Insulinsekretion und beeinträchtigt die Glucagonsekretion nicht.
Semaglutide reduziert das Körpergewicht und die Körperfettmasse mittels einer verringerten Energieaufnahme. Der Mechanismus umfasst einen allgemein verminderten Appetit, der erhöhtes Sättigungs- und vermindertes Hungergefühl einschliesst. Die Insulinresistenz wird reduziert. Dies erfolgt vermutlich durch die Reduktion des Körpergewichts.
Der nephroprotektive Effekt ist nicht ausschliesslich auf kardiovaskuläre Risikofaktoren (verbesserter Glukosestoffwechsel und Lipidprofil, reduzierte systemische Entzündung und systolischer Blutdruck sowie Gewichtsverlust) zurückzuführen, sondern zusätzliche – bisher nicht aufgeklärte – Mechanismen werden angenommen.
Pharmakodynamik
Alle pharmakodynamischen Untersuchungen wurde nach 12 Behandlungswochen (einschliesslich der Dosiseskalation) im Steady State mit einmal wöchentlicher Behandlung mit Semaglutide 1 mg durchgeführt.
Nüchternblutzucker und postprandialer Blutzucker
Bei Patienten mit Diabetes mellitus Typ 2 führte die Behandlung mit Semaglutide 1 mg zur Verringerung des Blutzuckerspiegels, und zwar sowohl hinsichtlich der absoluten Veränderung gegenüber dem Ausgangswert (mmol/l), als auch der relativen Verringerung im Vergleich zum Placebo (%) hinsichtlich des Nüchternblutzuckers (1.6 mmol/l; Verminderung um 22 %), des postprandialen Blutzuckers nach 2 Stunden (4.1 mmol/l; Verminderung um 37 %), des mittleren 24-Stunden-Blutzuckerspiegels (1.7 mmol/l; Verminderung um 22 %) und der postprandialen Blutzuckerspitzen über 3 Mahlzeiten (0.6–1.1 mmol/l) im Vergleich zum Placebo.
Semaglutide verringerte den Nüchternblutzucker nach der ersten Dosis.
Betazellfunktion und Insulinsekretion
Im Vergleich zum Placebo verbesserte Semaglutide nach einem intravenösen Glucose-Bolus die Erst- und Zweitphasen-Insulinantwort um das jeweils 3- und 2-Fache. Nach einem Arginin-Stimulationstest bei Patienten mit Diabetes mellitus Typ 2 erhöhte Semaglutide im Vergleich zum Placebo die maximale sekretorische Kapazität der Betazellen. Darüber hinaus erhöhte die Behandlung mit Semaglutide im Vergleich zum Placebo die Nüchtern-Insulinkonzentration.
Glucagonsekretion
Bei Patienten mit Diabetes mellitus Typ 2 führte Semaglutide zu den folgenden relativen Verringerungen des Glucagons im Vergleich zum Placebo: Nüchternglucagon (8-21 %), postprandiale Glucagonantwort (14-15 %) und mittlerer 24-Stunden-Glucagonspiegel (12 %).
Glucoseabhängige Insulin- und Glucagonsekretion
Semaglutide senkte die hohen Blutzuckerspiegel durch die glukoseabhängige Stimulation der Insulinsekretion und die Hemmung der Glucagonsekretion. Mit Semaglutide war die Insulinsekretionsrate bei Patienten mit Diabetes mellitus Typ 2 vergleichbar mit den gesunden Teilnehmern.
Während induzierter Hypoglykämie veränderte Semaglutide im Vergleich zum Placebo nicht die gegenregulatorischen Reaktionen auf den erhöhten Glucagonspiegel und beeinträchtigte nicht die Senkung des C-Peptids bei Patienten mit Diabetes mellitus Typ 2.
Magenentleerung
Semaglutide verursachte eine geringfügige Verlängerung der frühen postprandialen Magenentleerung und verminderte dadurch die Rate, in der Glukose postprandial in den Kreislauf gelangt.
Appetit, Energieaufnahme und Auswahl von Lebensmitteln
Im Vergleich zum Placebo verminderte Semaglutide die Energiezufuhr aus 3 aufeinanderfolgenden ad libitum Mahlzeiten um 18-35 %. Dies wurde unterstützt durch die von Semaglutide induzierte Unterdrückung des Appetits sowohl im Nüchternzustand als auch postprandial, verbesserte Kontrolle des Essverhaltens, eine Reduktion der Heisshungeranfälle und eine relativ geringe Vorliebe für fettreiche Lebensmittel.
Nüchternblutfette und postprandiale Blutfette
Im Vergleich zum Placebo verringerte Semaglutide die Spiegel der Nüchterntriglyzeride und des VLDL-Cholesterins (Lipoproteine sehr niedriger Dichte) um jeweils 12 % und 21 %. Die postprandiale Triglyzerid- und VLDL-Cholesterinantwort nach einer hochgradig fettreichen Mahlzeit wurde um >40 % verringert.
Die Auswirkung von Semaglutide auf die kardiale Repolarisation wurde in einer ausführlichen QTc-Studie untersucht. Semaglutide verlängert die QTc-Intervalle bei supra-therapeutischen Dosierungen (bis zu 1.5 mg im Steady State) nicht.
Klinische Wirksamkeit
Die Wirksamkeit und Sicherheit von einmal pro Woche angewendetem Ozempic 0.5 mg und 1 mg wurden in sechs randomisierten kontrollierten klinischen Studien mit 7'215 Patienten mit Diabetes mellitus Typ 2 (davon 4'107 mit Ozempic behandelt) untersucht. Das primäre Ziel von fünf Studien (SUSTAIN 1–5) war die Beurteilung der glykämischen Wirksamkeit, während das primäre Ziel einer Studie (SUSTAIN 6) die Beurteilung der kardiovaskulären Sicherheit war.
Die Wirksamkeit und Sicherheit von Ozempic 2 mg einmal wöchentlich wurde in einer Phase-3b-Studie (SUSTAIN FORTE), in die 961 Patienten eingeschlossen wurden, untersucht.
Zusätzlich wurde die Phase-3b-Studie (SUSTAIN 7) mit 1'201 Patienten durchgeführt, um die Wirksamkeit und Sicherheit von Ozempic 0.5 mg und 1 mg einmal wöchentlich gegenüber Dulaglutide 0.75 mg bzw. 1.5 mg einmal wöchentlich zu vergleichen.
In einer Phase-3b-Studie zu renalen Ergebnissen (FLOW) mit 3'533 Patienten wurden die Auswirkungen von Semaglutide 1 mg einmal wöchentlich im Vergleich zu Placebo auf das Fortschreiten der Nierenschädigung bei Patienten mit Diabetes mellitus Typ 2 und chronischer Nierenerkrankung untersucht.
Die Phase-3b-Studie (SUSTAIN 9) mit 302 Patienten wurde durchgeführt, um die Wirksamkeit und Sicherheit von Semaglutide zusätzlich zu einer bestehenden Therapie mit einem SGLT2-Inhibitor (mit oder ohne Metformin oder Sulfonylharnstoff) bei Patienten mit Diabetes mellitus Typ 2 zu beurteilen.
Die Behandlung mit Ozempic führte im Vergleich zum Placebo und zur Behandlung mit einem aktiven Kontrollpräparat (Sitagliptin, Insulin glargin, Exenatid ER und Dulaglutide) zu anhaltenden, statistisch signifikant grösseren und klinisch bedeutsamen Senkungen des HbA1c-Werts (Abbildung 1) und des Körpergewichts für die Dauer von bis zu 2 Jahren.
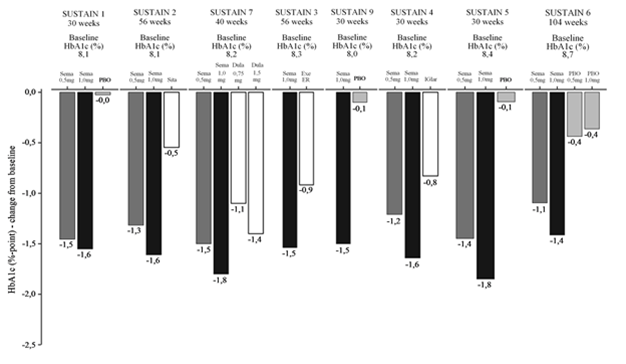
Abbildung 1: HbA1c-Wert (%), geschätzte Änderung am Ende der Behandlung gegenüber Ausgangswert in SUSTAIN 1–7+9 (Ozempic 0.5 mg dunkelgrau, Ozempic 1 mg schwarz, Vergleichspräparate weiss und Placebo hellgrau)
Alter, Geschlecht, ethnische Zugehörigkeit, BMI und Körpergewicht (kg) bei Studienbeginn, Dauer der Diabeteserkrankung und Schweregrad der Einschränkung der Nierenfunktion hatten keine Auswirkung auf die Wirksamkeit von Ozempic.
Die Ergebnisse beziehen sich jeweils auf die Behandlungsphase bei allen randomisierten Teilnehmern (Analysen basierend auf gemischten Modellen für wiederholte Messungen oder multiple Imputation).
Zusätzlich wurde eine 52-wöchige Phase-3b-Studie (SUSTAIN 11) durchgeführt, um die Wirkung von Semaglutide gegenüber Insulin aspart (jeweils als Bolus zu den drei Hauptmahlzeiten) zu untersuchen, beide als Zusatz zu Metformin und optimiertem Insulin glargin (U100).
SUSTAIN 1 – Monotherapie
In SUSTAIN 1 wurden 388 Patienten, die mit Diät und Bewegung unzureichend eingestellt waren, während 30-Wochen doppelblind mit Ozempic 0.5 mg oder Ozempic 1 mg einmal pro Woche oder mit Placebo behandelt.
Tabelle 2: SUSTAIN 1: Ergebnisse in Woche 30
Ozempic0.5 mg Ozempic1 mg Placebo
Intent-to-Treat (ITT) Population 128 130 129
(N)
HbA1c-Wert (%)
Ausgangswert (mittel) 8.1 8.1 8.0
Änderung in Woche 30 gegenüber -1.5 -1.6 0.0
dem Ausgangswert
Differenz zu Placebo [95 % KI] -1.4 [-1.7, -1.1]a -1.5 [-1.8, -1.2]a
Patienten (%), bei denen ein 74b 72b 25
HbA1c-Wert <7 % erreicht wurde
Differenz (odds ratio) zu Placebo 16.9 [8.4; 33.9] 15.7 [8.0; 30.8]
[95 % KI]
Körpergewicht (kg)
Ausgangswert (mittel) 89.8 96.9 89.1
Änderung in Woche 30 gegenüber -3.7 -4.5 -1.0
dem Ausgangswert
Differenz zu Placebo [95 % KI] -2.7 [-3.9, -1.6]a -3.6 [-4.7, -2.4]a -
a p <0.0001 (2-seitig) für Überlegenheit, angepasst hinsichtlich der Multiplizität basierend auf hierarchischer Testung von HbA1c-Wert und Körpergewicht
b p <0.0001 für Behandlungsdifferenz, nicht angepasst hinsichtlich der Multiplizität
SUSTAIN 2 – Ozempic vs Sitagliptin in Kombination mit 1–2 oralen Antidiabetika: Metformin, und/oder Thiazolidinedionen
In SUSTAIN 2 wurden 1'231 Patienten während 56 Wochen doppelblind mit Ozempic 0.5 mg, oder Ozempic 1 mg einmal pro Woche oder Sitagliptin 100 mg einmal pro Tag, jeweils in Kombination mit Metformin (94 %) und/oder Thiazolidinedionen (6 %), behandelt.
Tabelle 3: SUSTAIN 2: Ergebnisse in Woche 56
Ozempic0.5 mg Ozempic1 mg Sitagliptin100 mg
Intent-to-Treat (ITT) Population 409 409 407
(N)
HbA1c-Wert (%)
Ausgangswert (mittel) 8.0 8.0 8.2
Änderung in Woche 56 gegenüber -1.3 -1.6 -0.5
dem Ausgangswert
Differenz zu Sitagliptin [95 % KI] -0.8 [-0.9; -0.6]a -1.1 [-1.2; -0.9]a -
Patienten (%), bei denen ein 69b 78b 36
HbA1c-Wert <7 % erreicht wurde
Differenz (odds ratio) zu 4.2 [3.02; 5.74] 7.9 [5.59; 11.22]
Sitagliptin [95 % KI]
Körpergewicht (kg)
Ausgangswert (mittel) 89.9 89.2 89.3
Änderung in Woche 56 gegenüber -4.3 -6.1 -1.9
dem Ausgangswert
Differenz zu Sitagliptin [95 % KI] -2.3 [-3.1; -1.6]a -4.2 [-4.9; -3.5]a -
a p <0.0001 (2-seitig) für Überlegenheit, angepasst hinsichtlich der Multiplizität basierend auf hierarchischer Testung von HbA1c-Wert und Körpergewicht
b p <0.0001 für Behandlungsdifferenz, nicht angepasst hinsichtlich der Multiplizität
SUSTAIN 9 – Ozempic vs. Placebo als Zusatz zu einer bestehenden Therapie mit einem SGLT-2-Inhibitor ± Metformin oder SU
In einer 30-wöchigen doppelblinden, placebokontrollierten Studie wurden 302 Patienten mit Diabetes mellitus Typ 2, die mit einem SGLT2-Inhibitor mit oder ohne Metformin oder SU unzureichend eingestellt waren, zu Ozempic 1.0 mg einmal wöchentlich oder Placebo randomisiert.
Tabelle 4: SUSTAIN 9: Ergebnisse in Woche 30
Ozempic 1 mg Placebo
Intent-to-Treat (ITT)-Population (N) 151 151
HbA1c (%)
Ausgangswert (mittlerer) 8.0 8.1
Änderung gegenüber Ausgangswert in Woche 30 -1.5 -0.1
Differenz zu Placebo [95 % KI] -1.42 [-1.61, -1.24]a -
Patienten (%), die einen HbA1c-Wert <7 % erreichten 78.7 18.7
Differenz (odds ratio) zu Placebo [95 % KI] 27.32b [12.80, 58.30] -
Körpergewicht (kg)
Ausgangswert (mittlerer) 89.6 93.8
Änderung gegenüber Ausgangswert in Woche 30 -4.7 -0.9
Differenz zu Placebo [95 % KI] -3.81 [-4.70, -2.93]a -
a p <0.0001 (2-seitig) für Überlegenheit, angepasst hinsichtlich der Multiplizität basierend auf hierarchischer Testung von HbA1c-Wert und Körpergewicht
b p <0.0001 für Behandlungsdifferenz, nicht angepasst hinsichtlich der Multiplizität
SUSTAIN 7- Ozempic vs. Dulaglutid beide in Kombination mit Metformin
In einer 40-wöchigen offenen Studie wurden 1'201 Patienten mit Metformin randomisiert entweder mit Ozempic 0.5 mg oder 1 mg einmal wöchentlich oder Dulaglutid 0.75 mg oder 1.5 mg einmal wöchentlich. In der Studie wurden 0.5 mg Semaglutide mit 0.75 mg Dulaglutid und 1 mg Semaglutide mit 1.5 mg Dulaglutid verglichen.
Tabelle 5: SUSTAIN 7: Ergebnisse in Woche 40
Semaglutide0.5 mg Semaglutide1 mg Dulaglutid0.75 mg Dulaglutid1.5 mg
Intent-to-Treat 301 300 299 299
(ITT)-Population (N)
HbA1c (%)
Ausgangswert (mittle 8.3 8.2 8.2 8.2
rer)
Änderung gegenüber -1.5 -1.8 -1.1 -1.4
Ausgangswert in
Woche 40
Differenz zu Dulaglu -0.4b[-0.6, -0.2]a -0.4c[-0.6, -0.3]a - -
tid [95 % KI]
Patienten (%), die 68 79 52 67
einen HbA1c-Wert <7
% erreichten
Differenz (odds 2.5[1.68; 3.64] 2.0[1.28; 3.00]
ratio) zu Dulaglutid
[95 % KI]
Körpergewicht (kg)
Ausgangswert (mittle 96.4 95.5 95.6 93.4
rer)
Änderung gegenüber -4.6 -6.5 -2.3 -3.0
Ausgangswert in
Woche 40
Differenz zu Dulaglu -2.3b[-3.0, -1.5]a -3.6c[-4.3, -2.8]a - -
tid [95 % KI]
a p <0.0001 (2-seitig) für Überlegenheit
b 0.5 mg Ozempic gegenüber 0.75 mg Dulaglutid
c 1 mg Ozempic gegenüber 1.5 mg Dulaglutid
SUSTAIN 3 – Ozempic vs. Exenatide ER beide in Kombination mit Metformin oder Metformin mit Sulfonylharnstoff
In SUSTAIN 3 wurden 813 Patienten, die mit Metformin allein (49 %), mit Metformin plus Sulfonylharnstoffen (45 %) oder mit anderen antidiabetischen Arzneimitteln (6 %) behandelt wurden, während 56 Wochen zusätzlich mit Ozempic 1 mg einmal pro Woche oder mit Exenatid ER 2 mg einmal pro Woche, behandelt. Die Studie war nicht doppelblind.
Tabelle 6: SUSTAIN 3: Ergebnisse in Woche 56
Ozempic1 mg Exenatid ER2 mg
Intent-to-Treat (ITT) Population (N) 404 405
HbA1c-Wert (%)
Ausgangswert (mittel) 8.4 8.3
Änderung in Woche 56 gegenüber dem Ausgangswert -1.5 -0.9
Differenz zu Exenatid [95 % KI] -0.6 [-0.8; -0.4]a -
Patienten (%), bei denen ein HbA1c-Wert <7 % erreicht wurde 67b 40
Differenz (odds ratio) zu Exenatid ER [95 % KI] 3.9 [2.80; 5.38]
Körpergewicht (kg)
Ausgangswert (mittel) 96.2 95.4
Änderung in Woche 56 gegenüber dem Ausgangswert -5.6 -1.9
Differenz zu Exenatid [95 % KI] -3.8 [-4.6, -3.0]a -
a p <0.0001 (2-seitig) für Überlegenheit, angepasst hinsichtlich der Multiplizität basierend auf hierarchischer Testung von HbA1c-Wert und Körpergewicht
b p <0.0001 für Behandlungsdifferenz, nicht angepasst hinsichtlich der Multiplizität
SUSTAIN 4 – Ozempic vs Insulin Glargin beide in Kombination mit 1-2 oralen Antidiabetika: Metformin oder Metformin und Sulfonylharnstoff
In SUSTAIN 4 wurden 1'089 Patienten, die mit Metformin allein (48 %) oder mit Metformin plus Sulfonylharnstoffen (51 %) behandelt wurden, während 30-Wochen zusätzlich mit Ozempic 0.5 mg einmal pro Woche, Ozempic 1 mg einmal pro Woche oder Insulin glargin einmal pro Tag, behandelt. Die Studie war nicht doppelblind.
Tabelle 7: SUSTAIN 4: Ergebnisse in Woche 30
Ozempic0.5 mg Ozempic1 mg Insulin glargin
Intent-to-Treat (ITT) Population 362 360 360
(N)
HbA1c-Wert (%)
Ausgangswert (mittel) 8.1 8.2 8.1
Änderung in Woche 30 gegenüber -1.2 -1.6 -0.8
dem Ausgangswert
Differenz zu Insulin glargin [95 -0.4 [-0.5; -0.2]a -0.8 [-1.0; -0.7]a -
% KI]
Patienten (%), bei denen ein 57b 73b 38
HbA1c-Wert <7 % erreicht wurde
Differenz (odds ratio) zu Insulin 2.4 [1.73; 3.28] 5.8 [4.08; 8.19]
glargin [95 % KI]
Körpergewicht (kg)
Ausgangswert (mittel) 93.7 94.0 92.6
Änderung in Woche 30 gegenüber -3.5 -5.2 +1.2
dem Ausgangswert
Differenz zu Insulin glargin [95 -4.6 (-5.3; -4.0)a -6.34 [-7.0; -5.7]a -
% KI]
a p <0.0001 (2-seitig) für Überlegenheit, angepasst hinsichtlich der Multiplizität basierend auf hierarchischer Testung von HbA1c-Wert und Körpergewicht
b p <0.0001 für Behandlungsdifferenz, nicht angepasst hinsichtlich der Multiplizität
SUSTAIN 5 – Ozempic vs Placebo beide in Kombination mit Basalinsulin
In SUSTAIN 5 wurden 397 Patienten, die mit Basalinsulin mit oder ohne Metformin unzureichend eingestellt waren, zusätzlich mit Ozempic 0.5 mg einmal pro Woche, Ozempic 1 mg einmal pro Woche oder mit Placebo während 30-Wochen behandelt. Die Studie war nicht doppelblind.
Tabelle 8: SUSTAIN 5: Ergebnisse in Woche 30
Ozempic0.5 mg Ozempic1 mg Placebo
Intent-to-Treat (ITT) Population 132 131 133
(N)
HbA1c-Wert (%)
Ausgangswert (mittel) 8.4 8.3 8.4
Änderung in Woche 30 gegenüber -1.4 -1.8 -0.1
dem Ausgangswert
Differenz zu Placebo [95 % KI] -1.4 [-1.6; -1.1]a -1.8 [-2.0; -1.5]a -
Patienten (%), bei denen ein 61b 79b 11
HbA1c-Wert <7 % erreicht wurde
Differenz (odds ratio) zu Placebo 14.7 [7.43; 29.02] 34.3 [16.59; 70.83]
[95 % KI]
Körpergewicht (kg)
Ausgangswert (mittel) 92.7 92.5 89.9
Änderung in Woche 30 gegenüber -3.7 -6.4 -1.4
dem Ausgangswert
Differenz zu Placebo [95 % KI] -2.3 [-3.3; -1.3]a -5.1 [-6.1; -4.0]a -
a p <0.0001 (2-seitig) für Überlegenheit, angepasst hinsichtlich der Multiplizität basierend auf hierarchischer Testung von HbA1c-Wert und Körpergewicht
b p <0.0001 für Behandlungsdifferenz, nicht angepasst hinsichtlich der Multiplizität
SUSTAIN FORTE – Ozempic 2 mg vs. Ozempic 1 mg
In einer 40-wöchigen doppelblinden Studie wurden 961 Patienten, die mit Metformin mit oder ohne Sulfonylharnstoff (SU) nicht ausreichend eingestellt waren, randomisiert mit Ozempic 1 mg einmal wöchentlich oder Ozempic 2 mg einmal wöchentlich behandelt.
Die Behandlung mit Ozempic 2 mg führte im Vergleich zu Ozempic 1 mg zu einer statistisch überlegenen Senkung des HbA1c-Werts nach 40 Wochen Behandlung.
Tabelle 9: SUSTAIN FORTE: Ergebnisse nach 40 Wochen der Behandlung mit Ozempic 2 mg mit Metformin mit oder ohne SU
Ozempic 1 mg Ozempic 2 mg
Intent-to-Treat (ITT) Population (N) 481 480
HbA1c-Wert (%)
Ausgangswert (mittel) 8.8 8.9
Änderung in Woche 40 gegenüber dem Ausgangswert -1.9 oder -2.0 -2.2
Differenz zu Ozempic 1 mg [95 % KI] - -0.2 [-0.4; -0.1]a
Patienten (%), bei denen ein HbA1c-Wert <7 % erreicht wurde 58b 68b
Körpergewicht (kg)
Ausgangswert (mittel) 98.6 100.1
Änderung in Woche 40 gegenüber dem Ausgangswert -6.0 -6.9
Differenz zu Ozempic 1 mg [95 % KI] -0.9 [-1.7; -0.2]c
a p <0.001 (2-seitig) für Überlegenheit, angepasst hinsichtlich der Multiplizität basierend auf hierarchischer Testung von HbA1c-Wert und Körpergewicht
b p <0.0001 für Behandlungsdifferenz, nicht angepasst hinsichtlich der Multiplizität
c p <0.05 (2-seitig) für Überlegenheit, angepasst hinsichtlich der Multiplizität basierend auf hierarchischer Testung von HbA1c-Wert und Körpergewicht
SUSTAIN 11 – Semaglutide gegenüber Insulin aspart (IAsp) als Add-on bei Patienten, welche mit dem Basalinsulin Insulin glargin und Metformin keine ausreichende glykämische Kontrolle erzielen
In einer 52-wöchigen unverblindeten Studie wurden 1'748 Patienten, deren Diabetes mellitus Typ 2 nach einer 12-wöchigen Run-in Phase unter Insulin glargin und Metformin unzureichend kontrolliert war, 1:1 auf Semaglutide einmal wöchentlich (0.5 mg oder 1.0 mg) oder IAsp dreimal täglich zu den Hauptmahlzeiten randomisiert. Die durchschnittliche Dauer des Diabetes in der Studienpopulation betrug 13.4 Jahre und der mittlere HbA1c-Wert 8.6 %. Der HbA1c-Zielwert lag im Bereich von 6.5-7.5 %.
Die Behandlung mit Semaglutide war der mit IAsp nicht unterlegen. Die mittlere Differenz (Semaglutide – IAsp) in der Senkung des HbA1c [95 % KI] in Woche 52 betrug -0.29 [-0.38, -0.20]. Die HbA1c-Senkung in Woche 52 betrug -1.5 % für Semaglutide vs -1.2 % für IAsp, d. h. die glukosesenkende Wirkung von Semaglutide war numerisch grösser als die von IAsp). Die Anzahl schwerer hypoglykämischer Episoden war in beiden Behandlungsgruppen niedrig und unter Semaglutide (4 Episoden) numerisch geringer als im IAsp-Arm (7 Episoden; HR [95 % KI]: 0.6 [0.2, 2.2]).
Die Behandlung mit Semaglutide senkte das mittlere Körpergewicht bis Woche 52 um 4.1 kg, während es unter IAsp um 2.8 kg anstieg (geschätzter Behandlungsunterschied [95 % KI] zugunsten von Semaglutide: -6.99 kg [-7.41, 6.57]).
Kombination mit Sulfonylharnstoff-Monotherapie
In SUSTAIN 6 (siehe unten) wurde eine Untergruppe, die eine Sulfonylharnstoff-Monotherapie erhielt, in Woche 30 beurteilt. Bei der Ausgangsuntersuchung erhielten 123 Patienten eine Sulfonylharnstoff-Monotherapie. Der HbA1c-Wert lag bei der Ausgangsuntersuchung bei jeweils 8.2 %, 8.4 % und 8.4 % für Ozempic 0.5 mg, Ozempic 1 mg und Placebo. In der Woche 30 betrug die Änderung des HbA1c-Werts jeweils -1.6 %, -1.5 % und 0.1 % für Ozempic 0.5 mg, Ozempic 1 mg und Placebo.
Kombination mit Mischinsulin ± 1–2 OAD
In SUSTAIN 6 (siehe unten) wurde eine Untergruppe, die ein Mischinsulin (mit oder ohne 2 OAD) erhielt, in Woche 30 beurteilt. Bei der Ausgangsuntersuchung erhielten 867 Patienten ein Mischinsulin. Der HbA1c-Wert lag bei der Ausgangsuntersuchung bei jeweils 8.8 %, 8.9 % und 8.9 % für Ozempic 0.5 mg, Ozempic 1 mg und Placebo. In der Woche 30 betrug die Änderung des HbA1c-Werts -1.3 %, -1.8 % und -0.4 %, jeweils für Ozempic 0.5 mg, Ozempic 1 mg und Placebo.
Kardiovaskuläre Sicherheit
SUSTAIN 6 ist eine randomisierte, doppelblinde, klinische Studie, in der die kardiovaskuläre Sicherheit von Semaglutide 0.5 mg und Semaglutide 1 mg 1× pro Woche, mit derjenigen von Placebo bei 3'297 Patienten mit Diabetes mellitus Typ 2 verglichen wurde. In die Studie aufgenommen wurden 2'735 (83 %) Patienten mit einer vorbestehenden kardiovaskulären Erkrankung und 562 (17 %) Patienten mit einem hohen Risiko für kardiovaskuläre Ereignisse. Sowohl Semaglutide als auch Placebo wurden zusätzlich zur bereits vorhandenen antidiabetischen Therapie gegeben. Die Beobachtungsdauer betrug 2 Jahre.
Primärendpunkt war die Zeit von der Randomisierung bis zum ersten Auftreten eines schweren unerwünschten kardiovaskulären Ereignisses (MACE, major adverse cardiovascular event). MACE war definiert als Auftreten einer der drei Komponenten "kardiovaskulärer Tod" , "nicht tödlicher Herzinfarkt" oder "nicht tödlicher Schlaganfall" . Sekundärendpunkt war die Zeit von der Randomisierung bis zum ersten Auftreten eines erweiterten MACE-Endpunktes. Dieser erweiterte Endpunkt umfasste zusätzlich koronare oder periphere Revaskularisationen, instabile Angina pectoris, die eine Hospitalisierung erforderlich machte, oder Hospitalisierung wegen Herzinsuffizienz. Die Studie war als Non-inferiority-Studie konzipiert. Kriterium für den Non-Inferiority-Entscheid war die Obergrenze von 1.8 des 95 % Konfidenzintervalls für die Hazard Ratio der MACE.
Die Gesamtzahl der primären MACE-Endpunkte betrug 254, davon 108 (6.6 %) unter Semaglutide und 146 (8.9 %) unter Placebo. Die kardiovaskuläre Sicherheit von Semaglutide im Vergleich zu Placebo wurde bestätigt (Abbildung 2 und 3).
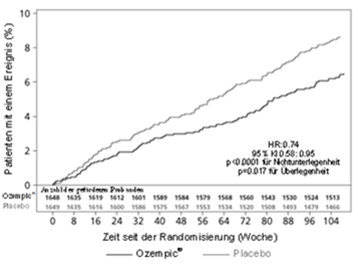
Abbildung 2: Kaplan-Meier-Plot hinsichtlich der Zeit bis zum ersten Auftreten des zusammengesetzten Endpunktes: kardiovaskulärer Tod, nicht-tödlicher Myokardinfarkt und nicht-tödlicher Schlaganfall (SUSTAIN 6)
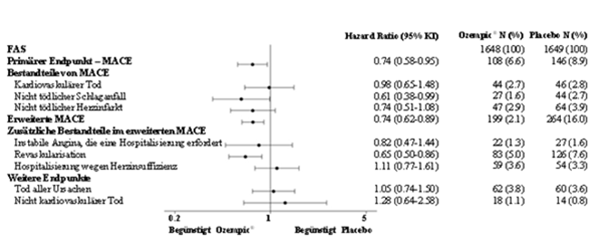
Abbildung 3: Forest-Plot: Analysen der einzelnen Arten kardiovaskulärer Ereignisse (SUSTAIN 6)
Die Differenz des HbA1c-Werts in Woche 104 gegenüber dem Ausgangswert betrug für Semaglutide 0.5 mg -1.1 % vs. -0.4 % für Placebo und -1.4 % für Semaglutide 1 mg vs. 0.4 für Placebo.
Nephrotektive Wirkung (FLOW Studie)
In einer doppelblinden Nierenendpunkt-Studie (FLOW) wurden 3'533 Patienten mit Diabetes mellitus Typ 2 und chronischer Nierenerkrankung mit einer eGFR von 50-75 ml/min/1.73 m2 und eines UACR von >300 und <5000 mg/g oder einer eGFR von 25-<50 ml/min/1.73 m2 und eines UACR von >100 und <5000 mg/g randomisiert und erhielten zusätzlich zur Standardtherapie entweder 1 mg Semaglutide einmal wöchentlich oder ein entsprechendes Placebo.
Die Studie wurde nach der geplanten Interimsanalyse basierend auf der Empfehlung des unabhängigen Data Monitoring Committee aus Wirksamkeitsgründen vorzeitig gestoppt. Die mediane Nachbeobachtungszeit betrug 40.9 Monate.
Das mittlere Alter der Population betrug 66.6 Jahre und 69.7 % waren männlich. Der mittlere BMI bei Behandlungsbeginn betrug 32.0 kg/m2. Die mittlere Diabetesdauer zu Studienbeginn betrug 17.4 Jahre und der mittlere HbA1c-Ausgangswert 7.8 % (61.5 mmol/mol). Die mittlere eGFR zu Studienbeginn betrug 47 ml/min/1.73 m2 und der mediane UACR 568 mg/g. Zu Studienbeginn wurden etwa 95 % der Patienten mit Renin-Angiotensin-Aldosteron-System-Inhibitoren und 16 % mit SGLT2-Inhibitoren behandelt.
Semaglutide war Placebo, zusätzlich zur Standardbehandlung, in der Prävention des primären kombinierten Endpunkts bestehend aus einer anhaltenden ≥50 %igen Reduktion der eGFR, dem Beginn einer anhaltenden eGFR <15 ml/min/1.73 m2, der Einleitung einer chronischen Nierenersatztherapie, Nierentod oder kardiovaskulärem Tod mit einer Hazard Ratio von 0.76 [0.66; 0.88]95 % KI überlegen, was einer relativen Risikoreduktion des Fortschreitens einer Nierenerkrankung um 24 % entspricht (siehe Abbildung 4). Die individuellen Komponenten des primären Kompositums trugen zum Behandlungseffekt bei, allerdings gab es wenige Nierentodesfälle (siehe Abbildung 5).

Abbildung 4: Kumulative Inzidenzfunktion der Zeit bis zum ersten Auftreten des primären zusammengesetzten Endpunktes: Beginn einer anhaltenden Reduktion der eGFR um ≥50 %, Beginn einer anhaltenden eGFR von <15 ml/min/1.73 m2, Einleitung einer chronischen Nierenersatztherapie, Nierentod oder kardiovaskulärer Tod (FLOW)
Semaglutide zeigte eine Überlegenheit gegenüber Placebo, zusätzlich zur Standardbehandlung, bei der Reduktion der jährlichen Änderungsrate der eGFR mit einer geschätzten Behandlungsdifferenz von 1.16 (ml/min/1.73 m2/Jahr) [0.86; 1.47]95 % KI. Die Behandlung mit Semaglutide verbesserte das Gesamtüberleben mit einer signifikanten Verringerung der Gesamtmortalität (siehe Abbildung 5).

Abbildung 5: Forest-Plot: Analyse der Zeit bis zum ersten Auftreten des primären kombinierten Endpunkts und seinen Komponenten, dem ersten Auftreten von MACE und seinen Komponenten sowie Todesfälle jeglicher Ursache (FLOW)
Ein nephroprotektiver Effekt von Semaglutide bei gleichzeitiger Behandlung mit SGLT2-Inhibitoren (SGLT2i) ist nicht gesichert. Die Subgruppenanalyse bei Patienten, die zu Studienbeginn bereits mit SGLT2i behandelt wurden, zeigte keinen nachweisbaren Vorteil hinsichtlich des zusammengesetzten primären renalen Endpunkts, basierte aber auf einer geringen Anzahl von Ereignissen. Die geschätzte Hazard Ratio betrug 1.07 [0.69; 1.67]95 % KI bei Patienten, die mit SGLT2i behandelt wurden, gegenüber 0.73 [0.63; 0.85]95 % KI bei Patienten, die nicht mit SGLT2i behandelt wurden.
PharmakokinetikAbsorption
Die Maximalkonzentration wurde 1 bis 3 Tage nach der Injektion erreicht. Nach 4-5 wöchentlichen Injektionen erreichte Exposition einen Steady State. Basierend auf pharmakokinetischen Populationsanalysen mit Daten von Patienten mit Diabetes mellitus Typ 2 betrugen die durchschnittlichen Steady State-Konzentrationen nach subkutaner Gabe von 0.5 mg und 1 mg Semaglutide jeweils ca. 16 nmol/l und 30 nmol/l.
In der Studie, in der Semaglutide 1 mg und 2 mg verglichen wurden, betrugen die mittleren Steady State-Konzentrationen 27 nmol/l und 54 nmol/l.
Die Semaglutide-Exposition nahm proportional mit den Dosen von 0.5 mg, 1 mg und 2 mg zu. Auch bei der subkutanen Gabe von Semaglutide in Abdomen, Oberschenkel oder Oberarm wurde eine vergleichbare Exposition erreicht. Die absolute Bioverfügbarkeit nach subkutaner Gabe von Semaglutide liegt bei 89 %.
Distribution
Das mittlere Verteilungsvolumen nach intravenöser Gabe von Semaglutide bei Patienten mit Diabetes mellitus Typ 2 war ca. 12.5 l. Semaglutide war stark an Plasmaalbumin gebunden (>99 %).
Metabolismus
Semaglutide wird durch die proteolytische Spaltung des Peptidrückgrats und die sequentielle Beta-Oxidation der Fettsäurenseitenketten metabolisiert. Der häufigste Plasmametabolit machte <8 % der gesamten Exposition aus und wurde als Semaglutide mit einer Trunkierung der ersten 13 Aminosäuren vom N-Terminus identifiziert.
Elimination
Die Elimination für Materialen, die mit Semaglutide im Zusammenhang stehen, erfolgte vorwiegend über Urin und Fäzes. Ca. 3 % der Dosis wurde als intaktes Semaglutide mittels Urin ausgeschieden. Die Clearance von Semaglutide bei Patienten mit Diabetes mellitus Typ 2 lag bei ca. 0.05 l/h. Mit einer Eliminationshalbwertszeit von ca. 1 Woche wird Semaglutide nach der letzten Dosis bis zu 5 Wochen im Blutkreislauf vorhanden sein.
Kinetik spezieller Patientengruppen
Die untenstehenden Informationen zum Alter, zum Geschlecht, zur ethischen Herkunft und zum Körpergewicht basieren auf populationspharmakokinetischer Analyse der Daten aus Phase-3a-Studien.
Leberfunktionsstörungen
Eine eingeschränkte Leberfunktion hatte keine Auswirkung auf die Semaglutide-Exposition. Die Pharmakokinetik von Semaglutide wurde bei Patienten mit unterschiedlichen Graden einer Leberfunktionsstörung (leicht, mittelschwer, schwer) im Vergleich zu Teilnehmer mit normaler Leberfunktion in einer Einzeldosis-Studie mit 0.5 mg Semaglutide untersucht.
Nierenfunktionsstörungen
Eine eingeschränkte Nierenfunktion hatte keine klinisch bedeutsame Auswirkung auf die Pharmakokinetik von Semaglutide, obwohl Cmax abnahm und bei Patienten mit zunehmenden Nierenfunktionsstörungen später eintrat. Dies wurde mit einer Einzeldosis von 0.5 mg Semaglutide bei Patienten mit unterschiedlichen Graden einer Nierenfunktionsstörung (leicht, mittelschwer, schwer oder dialysepflichtige Patienten) im Vergleich zu Teilnehmern mit normaler Nierenfunktion aufgezeigt. Ausgehend von Daten aus Phase-3a-Studien (populationspharmakokinetischer Analyse) wurde dies auch für Teilnehmer mit Diabetes mellitus Typ 2 sowie eingeschränkter Nierenfunktion nachgewiesen.
Ältere Patienten
Bei Patienten im Alter von 20-86 Jahre hatte das Alter keine Auswirkung auf die Pharmakokinetik von Semaglutide.
Kinder und Jugendliche
Semaglutide wurde bei Kindern und Jugendlichen nicht untersucht.
Genetische Polymorphismen
Geschlecht und ethnische Zugehörigkeit hatten keine Auswirkung auf die Pharmakokinetik von Semaglutide.
Präklinische DatenBasierend auf den konventionellen Studien zur Sicherheitspharmakologie, Toxizität bei wiederholter Gabe oder Genotoxizität lassen die präklinischen Daten keine besonderen Gefahren für den Menschen erkennen.
Karzinogenität
Nicht letale C-Zelltumoren der Schilddrüse, die bei Nagetieren beobachtet wurden, sind ein Klasseneffekt der GLP-1-Rezeptor-Agonisten. Bei 2-jährigen Karzinogenitätsstudien an Ratten und Mäusen traten bei klinisch relevanten Expositionen C-Zelltumoren der Schilddrüse auf. Die C-Zelltumore bei Nagetieren werden durch einen nicht genotoxischen, spezifisch durch den GLP-1-Rezeptor vermittelten Mechanismus verursacht, für den Nager besonders empfänglich sind. Die Relevanz für den Menschen ist wahrscheinlich gering, kann jedoch nicht komplett ausgeschlossen werden.
Reproduktionstoxizität
In Fertilitätsstudien bei Ratten beeinträchtigte Semaglutide das Paarungsverhalten oder die Fertilität männlicher Ratten nicht. Bei weiblichen Ratten wurden ein längerer Zyklus und eine geringe Verminderung der Gelbkörper (Ovulationen) bei Dosen beobachtet, die mit mütterlichem Gewichtsverlust assoziiert waren.
In embryofötalen Entwicklungsstudien bei Ratten verursachte Semaglutide Embryotoxizität unterhalb klinisch relevanter Expositionen. Semaglutide führte zu deutlichen Reduktionen des mütterlichen Körpergewichts sowie zu Reduktionen des Überlebens und des Wachstums der Embryonen. Bei Föten wurden wesentliche skelettale und viszerale Missbildungen beobachten, einschliesslich Auswirkungen auf lange Knochen, Rippen, Wirbel, Schwanz, Blutgefässe und Gehirnventrikel. Mechanistische Beurteilungen verwiesen darauf, dass die Embryotoxizität auf einer durch GLP-1-Rezeptoren vermittelten Beeinträchtigung der Nährstoffzufuhr zum Embryo durch den Dottersack der Ratte beruhte. Aufgrund der Unterschiede in Anatomie und Funktion des Dottersacks bei unterschiedlichen Spezies und aufgrund des Fehlens der GLP-1-Rezeptorexpression im Dottersack bei Nicht-Menschenaffen, gilt es als unwahrscheinlich, dass der bei Ratten beobachtete GLP-1-Rezeptor-vermittelte Mechanismus für Menschen relevant ist. Jedoch kann eine direkte Auswirkung von Semaglutide auf den Fötus nicht ausgeschlossen werden.
Im Rahmen von Studien zur Entwicklungstoxizität bei Kaninchen und Javaneraffen wurden bei klinisch relevanten Expositionen ein häufigerer Verlust von Schwangerschaften und eine leicht erhöhte Inzidenz fötaler Anomalien beobachtet. Die Befunde korrelierten mit ausgeprägtem mütterlichem Gewichtsverlust von bis zu 16 %. Es ist nicht bekannt, ob diese Wirkungen im Zusammenhang mit der verminderten Nahrungsaufnahme mütterlicherseits als direkte Auswirkung von GLP-1 stehen.
Das postnatale Wachstum und die postnatale Entwicklung wurden bei Javaneraffen beurteilt. Neugeborene waren bei der Geburt geringfügig kleiner, erholten sich jedoch während der Stillzeit.
Bei jugendlichen männlichen und weiblichen Ratten verursachte Semaglutide eine verzögerte sexuelle Reife. Diese Verzögerungen hatten weder Auswirkung auf die Fertilität und die reproduktive Kapazität beider Geschlechter, noch auf die Fähigkeit der Weibchen eine Schwangerschaft auszutragen.
Sonstige HinweiseInkompatibilitäten
Da keine Kompatibilitätsstudien durchgeführt wurden, darf dieses Arzneimittel nicht mit anderen Arzneimitteln gemischt werden.
Haltbarkeit
Das Arzneimittel darf nur bis zu dem auf dem Behälter mit "EXP" bezeichneten Datum verwendet werden.
Haltbarkeit nach Anbruch
Nach dem Anbruch 6 Wochen haltbar.
Besondere Lagerungshinweise
Vor Anbruch: Im Kühlschrank (2-8°C) lagern, nicht einfrieren, vor Licht schützen.
Nach Anbruch: Unter 30°C oder im Kühlschrank (2-8°C) lagern, nicht einfrieren.
Die Kappe auf dem Pen aufgesetzt lassen, wenn der Ozempic-Pen nicht im Gebrauch ist, um ihn vor Licht zu schützen.
Ozempic sollte vor übermässiger Hitze und Licht geschützt werden.
Die Injektionsnadel sollte nach jeder Injektion entfernt und der Ozempic-Pen ohne Nadel aufbewahrt werden. Dies verhindert verstopfte Nadeln, Kontamination, Infektion, Auslaufen der Lösung und ungenaue Dosierung.
Ausser Reichweite von Kindern aufbewahren.
Hinweise für die Handhabung
Der Patient ist anzuweisen, die Injektionsnadel nach jeder Injektion zu entsorgen und den Ozempic-Pen ohne aufgeschraubte Injektionsnadel zu lagern. Dies verhindert verstopfte Nadeln, Kontamination, Infektion, Auslaufen der Lösung und ungenaue Dosierung. Nadeln und andere Abfallmaterialien sind entsprechend den nationalen Anforderungen zu beseitigen.
Ozempic-Pen ist nur von einer Person zu verwenden.
Ozempic darf nicht verwendet werden, wenn es nicht klar und farblos aussieht.
Ozempic kann mit Nadeln bis zu einer Länge von 8 mm injiziert werden. Der Pen wurde für die Verwendung mit NovoFine oder NovoTwist Einwegnadeln entwickelt.
Zulassungsnummer66604 (Swissmedic)
PackungenEs gibt drei Varianten des Ozempic-Fertigpens:
Ozempic DualDose Injektionslösung in einem Fertigpen
-Ozempic DualDose Injektionslösung im Fertigpen ermöglicht die Abgabe von Dosen zu 0.25 mg oder 0.5 mg. Dieser Pen ist für die Dosiseskalation und die Erhaltungstherapie mit der Dosis von 0.5 mg bestimmt.
Packung zu 1 Fertigpen mit 6 NovoFine Plus Einwegnadeln (B)
Ozempic Fixdose 1 mg, Injektionslösung in einem Fertigpen
-Ozempic FixDose 1 mg/Dosis Injektionslösung in einem 3 ml Fertigpen ermöglicht ausschliesslich die Abgabe von Dosen zu 1 mg. Dieser Pen ist ausschliesslich im Rahmen der Erhaltungstherapie mit der Dosis von 1 mg zu verwenden.
Packung zu 1 Fertigpen mit 4 NovoFine Plus Einwegnadeln (B)
Ozempic FixDose 2 mg, Injektionslösung in einem Fertigpen
-Ozempic 2 mg/Dosis Injektionslösung in einem 3 ml Fertigpen ermöglicht ausschliesslich die Abgabe von Dosen zu 2 mg. Dieser Pen ist ausschliesslich im Rahmen der Erhaltungstherapie mit der Dosis von 2 mg zu verwenden. Der Pen enthält 3 ml Lösung.
Packung zu 1 Fertigpen mit 4 NovoFine Plus Einwegnadeln (B)
ZulassungsinhaberinNovo Nordisk Pharma AG, Kloten
Domizil: Zürich
Stand der InformationFebruar 2026
|