ZusammensetzungWirkstoffe
Semaglutide.
Analogon zu humanem Glucagon-like peptide-1 (GLP-1), gentechnisch hergestellt durch rekombinante DNA-Technologie in Saccharomyces cerevisiae-Zellen.
Hilfsstoffe
Wegovy FixDose
Dinatrii phosphas dihydricus, Natrii chloridum, Acidum hydrochloridum/Natrii hydroxidum (q.s. ad pH), Aqua ad iniectabile.
Das Arzneimittel enthält 0.078557 mmol Natrium pro Dosisstärke (0.25 mg, 0.5 mg, 1.0 mg) bzw. 0.11785 mmol Natrium pro Dosisstärke (1.7 mg, 2.4 mg).
Wegovy Multi FixDose
Dinatrii phosphas dihydricus, Propylenglycolum, Phenolum, Acidum hydrochloridum/Natrii hydroxidum (q.s. ad pH), Aqua ad iniectabile q.s. ad solutionem pro 0.37 ml bzw. 0.75 ml.
Das Arzneimittel enthält 0.00591 mmol (0.136 mg) Natrium pro Dosisstärke (0.25 mg, 0.5 mg) bzw. 0.01197 mmol (0.275 mg) Natrium pro Dosisstärke (1 mg, 1.7 mg, 2.4 mg).
Indikationen/AnwendungsmöglichkeitenGewichtsregulierung
Wegovy wird als Ergänzung zu einer kalorienreduzierten Ernährung und verstärkter körperlicher Aktivität zur Gewichtsregulierung angewendet bei
·Erwachsenen mit einem Ausgangs-Body-Mass-Index (BMI) von
·≥30 kg/m2 (Adipositas) oder
·≥27 kg/m2 bis < 30 kg/m2 (Übergewicht) bei Vorliegen mindestens einer gewichtsbedingten Begleiterkrankung.
·Jugendlichen ab 12 Jahren mit Adipositas gemäss den dafür international akzeptierten Grenzwerten* und einem Körpergewicht über 60 kg
*Adipositas (BMI-Perzentile ≥95) gemäss den geschlechts- und altersspezifischen BMI-Wachstumstabellen (CDC.gov) (siehe Tabelle 1).
Etablierte kardiovaskuläre Erkrankung
Wegovy ist indiziert zur Reduktion des Risikos schwerwiegender kardiovaskulärer Ereignisse bei Erwachsenen mit etablierter kardiovaskulärer Erkrankung und einem BMI ≥27 kg/m2. Die Behandlung soll in Ergänzung zur Standardtherapie für Patienten mit etablierter kardiovaskulärer Erkrankung erfolgen (siehe Abschnitt «Klinische Wirksamkeit»).
Tabelle 1: BMI-Cut-off-Punkte für Adipositas (BMI-Perzentile ≥95) nach Geschlecht und Alter für pädiatrische Patienten im Alter von 12 Jahren und älter (CDC-Kriterien)
|
Alter (Jahre)
|
95 % BMI-Perzentile (kg/m2)
| |
Männlich
|
Weiblich
| |
12
|
24.2
|
25.2
| |
12.5
|
24.7
|
25.7
| |
13
|
25.1
|
26.3
| |
13.5
|
25.6
|
26.8
| |
14
|
26.0
|
27.2
| |
14.5
|
26.4
|
27.7
| |
15
|
26.8
|
28.1
| |
15.5
|
27.2
|
28.5
| |
16
|
27.5
|
28.9
| |
16.5
|
27.9
|
29.3
| |
17
|
28.2
|
29.6
| |
17.5
|
28.6
|
30.0
|
Dosierung/AnwendungÜbliche Dosierung
Erwachsene
Die Erhaltungsdosis von 2.4 mg einmal wöchentlich wird mit einer Anfangsdosis von 0.25 mg erreicht. Um die Wahrscheinlichkeit von gastrointestinalen Symptomen zu verringern, ist die Dosis über einen Zeitraum von 16 Wochen bis zur Erhaltungsdosis von einmal wöchentlich 2.4 mg zu steigern (siehe Tabelle 2). Bei Auftreten erheblicher gastrointestinaler Symptome ist in Betracht zu ziehen, die Dosissteigerung bis zur Besserung der Symptome auszusetzen.
Höhere wöchentliche Dosen als 2.4 mg werden nicht empfohlen.
Tabelle 2: Zeitplan für die Dosissteigerung
|
Dosissteigerung
|
Wöchentliche Dosis
| |
Woche 1–4
|
0.25 mg
| |
Woche 5–8
|
0.5 mg
| |
Woche 9–12
|
1 mg
| |
Woche 13–16
|
1.7 mg
| |
Erhaltungsdosis
|
2.4 mg
|
Erwachsene in der Indikation «Gewichtsregulierung»
Falls die Patienten nach 28-wöchiger Behandlung nicht mindestens 5 % ihres ursprünglichen Körpergewichts verloren haben, muss unter Berücksichtigung des Nutzen-Risiko-Profils des einzelnen Patienten entschieden werden, ob die Behandlung fortgesetzt werden soll.
Jugendliche in der Indikation «Gewichtsregulierung»
Für Jugendliche im Alter von 12 Jahren oder älter ist das gleiche Dosiseskalationsschema wie für Erwachsene anzuwenden (siehe Tabelle 2). Die Dosis sollte bis auf 2.4 mg (Erhaltungsdosis) oder bis zum Erreichen der maximal vertragenen Dosis erhöht werden.
Falls sich der BMI der Patienten nach 28-wöchiger Behandlung nicht um mindestens 5 % verbessert hat, muss unter Berücksichtigung des Nutzen-Risiko-Profils des einzelnen Patienten entschieden werden, ob die Behandlung fortgesetzt werden soll.
Höhere wöchentliche Dosen als 2.4 mg werden nicht empfohlen.
Um die Rückverfolgbarkeit von biotechnologisch hergestellten Arzneimitteln sicherzustellen, wird empfohlen, Handelsname und Chargennummer bei jeder Behandlung zu dokumentieren.
Spezielle Dosierungsanweisungen
Patienten mit Leberfunktionsstörungen
Bei Patienten mit Einschränkung der Leberfunktion ist keine Dosisanpassung erforderlich. Die Erfahrungen mit der Anwendung von Semaglutide bei Patienten mit schwerer Einschränkung der Leberfunktion sind begrenzt. Bei der Behandlung dieser Patienten mit Semaglutide ist Vorsicht geboten (siehe «Pharmakokinetik»).
Patienten mit Nierenfunktionsstörungen
Bei Patienten mit leichter, mittelschwerer oder schwerer Einschränkung der Nierenfunktion ist keine Dosisanpassung erforderlich. Die Erfahrungen mit der Anwendung von Semaglutide bei Patienten mit schwerer Einschränkung der Nierenfunktion sind begrenzt. Bei Patienten mit terminaler Niereninsuffizienz wird die Anwendung von Semaglutide nicht empfohlen (siehe «Pharmakokinetik»).
Ältere Patienten (≥65 Jahre)
Eine Dosisanpassung ist bei älteren Menschen nicht erforderlich.
Kinder und Jugendliche
Semaglutide ist für die Anwendung bei Kindern unter 12 Jahren nicht zugelassen. Semaglutide ist zugelassen für die Behandlung von Jugendlichen ab einem Alter von 12 Jahren in der Indikation «Gewichtsregulierung».
Patienten mit anderen Grunderkrankungen
Patienten mit Diabetes Typ 2
Wegovy darf nicht in Kombination mit anderen GLP-1-Rezeptoragonisten angewendet werden.
Wenn die Behandlung mit Semaglutide begonnen wird, ist eine Dosisreduktion von gleichzeitig angewendetem Insulin oder Insulinsekretagoga (wie Sulfonylharnstoffen) zu erwägen, um das Risiko einer Hypoglykämie zu senken.
Verspätete Dosisgabe
Falls eine Dosis ausgelassen wird, sollte sie so bald wie möglich und innerhalb von 5 Tagen nach dem ursprünglichen Dosistermin verabreicht werden. Wenn mehr als 5 Tage vergangen sind, sollte die ausgelassene Dosis übersprungen werden und die nächste Dosis sollte am regulären, turnusgemässen Tag verabreicht werden. In jedem Fall können Patienten anschliessend ihr regelmässiges einmal wöchentliches Dosierungsschema wiederaufnehmen. Falls mehrere Dosen ausgelassen wurden, ist in Betracht zu ziehen, die Behandlung mit einer niedrigeren Dosis wiederaufzunehmen.
Art der Anwendung
Wegovy wird einmal wöchentlich zu einer beliebigen Tageszeit mit oder ohne einer Mahlzeit angewendet.
Wegovy soll subkutan in das Abdomen, den Oberschenkel oder den Oberarm injiziert werden. Die Injektionsstelle kann ohne Dosisanpassung geändert werden. Wegovy darf nicht intravenös oder intramuskulär angewendet werden. Der Tag der wöchentlichen Anwendung kann bei Bedarf gewechselt werden, solange die Zeit zwischen zwei Dosen mindestens 3 Tage (> 72 Stunden) beträgt. Nach der Auswahl eines neuen Verabreichungstages ist die einmal wöchentliche Dosierung fortzusetzen.
Bei der Verabreichung von Wegovy ist der Pen fest gegen die Haut zu drücken, bis der gelbe Balken sich nicht mehr bewegt. Die Injektion dauert etwa 5–10 Sekunden.
Die Patienten sind anzuhalten, vor Anwendung von Wegovy die Gebrauchsanweisung in der Packungsbeilage sorgfältig zu lesen.
KontraindikationenÜberempfindlichkeit gegenüber dem Wirkstoff oder einem der Hilfsstoffe.
Warnhinweise und VorsichtsmassnahmenRisiko thyreoidaler C-Zell-Tumoren
Präklinische Studien mit GLP-1-Rezeptoragonisten an Nagern legen nahe, dass GLP-1-Rezeptoragonisten möglicherweise mit einem erhöhten Risiko von fokalen Hyperplasien der thyreoidalen C-Zellen und C-Zell-Tumoren einhergehen (siehe «Präklinische Daten»).
Es ist nicht bekannt, ob beim Menschen ein Zusammenhang besteht zwischen GLP-1-Rezeptoragonisten und thyreoidalen C-Zell-Tumoren, einschliesslich des medullären Schilddrüsenkarzinoms (medullary thyroid carcinoma, MTC). Patienten mit MTC und Patienten mit multiplem endokrinem Neoplasie-Syndrom vom Typ 2 (MEN 2) in der Anamnese wurden in den klinischen Studien mit Semaglutide nicht behandelt. Vor einer Behandlung mit Wegovy ist deshalb in diesem spezifischen Kollektiv eine sorgfältige Nutzen-Risiko-Abwägung erforderlich. Der klinische Wert einer routinemässigen Überwachung des Serum-Calcitonin-Spiegels ist nicht belegt.
Wirkungen auf den Gastrointestinaltrakt
Die Anwendung von GLP-1-Rezeptoragonisten kann mit gastrointestinalen Nebenwirkungen verbunden sein, die eine Dehydrierung verursachen können, was zu einer Verschlechterung der Nierenfunktion führen kann. Patienten müssen auf das potenzielle Dehydrierungsrisiko im Zusammenhang mit gastrointestinalen Nebenwirkungen hingewiesen werden und Vorkehrungen gegen Flüssigkeitsverluste treffen.
Akute Pankreatitis
Akute Pankreatitis wurde unter der Anwendung von GLP-1-Rezeptoragonisten beobachtet. Patienten sollten über die charakteristischen Symptome einer akuten Pankreatitis informiert werden. Wird eine Pankreatitis vermutet, ist Wegovy abzusetzen; wird diese bestätigt, ist die Behandlung mit Wegovy nicht wieder aufzunehmen. Patienten mit Pankreatitis in der Vorgeschichte wurden in den klinischen Studien mit Semaglutide nicht untersucht. Bei Patienten, die bereits einmal an Pankreatitis erkrankt waren, ist entsprechende Vorsicht geboten.
Ohne Vorliegen weiterer Anzeichen und Symptome einer akuten Pankreatitis sind erhöhte Pankreasenzyme alleine keine Prädiktoren für akute Pankreatitis.
Patienten mit Diabetes Typ 2
Wegovy darf bei Diabetespatienten nicht als ein Ersatz für Insulin angewendet werden.
Hypoglykämie bei Patienten mit Adipositas oder Übergewicht und Diabetes Typ 2
Insulin und Sulfonylharnstoff können Hypoglykämie verursachen. Patienten, die mit Wegovy in Kombination mit einem Sulfonylharnstoff oder Insulin behandelt werden, können ein erhöhtes Risiko für eine Hypoglykämie haben. Das Risiko einer Hypoglykämie kann durch Reduktion der Sulfonylharnstoff- oder der Insulindosis bei Beginn der Behandlung mit einem GLP-1-Rezeptoragonisten gesenkt werden. Die zusätzliche Gabe von Wegovy bei Patienten, die bereits mit Insulin behandelt wurden, wurde nicht untersucht.
Diabetische Retinopathie bei Patienten mit Adipositas oder Übergewicht und Diabetes Typ 2
Bei Patienten mit diabetischer Retinopathie, die mit Insulin und Semaglutide behandelt werden, wurde ein erhöhtes Risiko für das Auftreten von Komplikationen einer diabetischen Retinopathie beobachtet. Eine rasche Verbesserung der Blutzuckerkontrolle ist mit einer vorübergehenden Verschlechterung der diabetischen Retinopathie assoziiert worden. Patienten mit diabetischer Retinopathie, die Semaglutide verwenden, sind engmaschig zu überwachen und gemäss den klinischen Richtlinien zu behandeln. Es liegen keine Erfahrungen mit Semaglutide 2.4 mg bei Patienten mit Diabetes Typ 2 mit unkontrollierter oder potenziell instabiler diabetischer Retinopathie vor.
Akute Nierenschäden
Nach Markteinführung wurde bei Patienten, die mit GLP-1-Rezeptor-Agonisten behandelt wurden, über akute Nierenschäden und eine Verschlechterung der chronischen Niereninsuffizienz berichtet, die manchmal eine Hämodialyse erforderlich machen können. Einige dieser Ereignisse wurden bei Patienten ohne bekannte zugrunde liegende Nierenerkrankung gemeldet. Die Mehrzahl der gemeldeten Ereignisse trat bei Patienten auf, die bereits unter Übelkeit, Erbrechen, Durchfall oder Dehydrierung litten. Die Nierenfunktion soll überwacht werden, wenn die Behandlung mit Wegovy bei Patienten, die über schwere unerwünschte Magen-Darm-Reaktionen berichten, initiiert oder auftitriert wird.
Nicht untersuchte Populationen
Es liegen keine Erfahrungen bei Patienten mit Herzinsuffizienz des NYHA-Stadiums IV vor. Bei Patienten im Alter ab 85 Jahren liegen nur begrenzte Erfahrungen vor.
Natriumgehalt
Dieses Arzneimittel enthält weniger als 1 mmol (23 mg) Natrium pro Dosis, d.h. es ist nahezu «natriumfrei».
InteraktionenIn vitro Studien zeigten, dass Semaglutide ein sehr geringes Potenzial für die Inhibition oder Induktion von CYP-Enzymen und für die Inhibition von Wirkstofftransportern aufweisen.
Wie andere GLP-1-Rezeptoragonisten kann auch Semaglutide die Magenentleerung verzögern und möglicherweise die Resorption gleichzeitig oral angewendeter Arzneimittel beeinflussen. Unter Semaglutide 2.4 mg wurde keine klinisch relevante Wirkung auf die Geschwindigkeit der Magenentleerung beobachtet. In klinischen Studien zur Untersuchung der Wirkung von Semaglutide 1.0 mg auf die Resorption gleichzeitig oral angewendeter Arzneimittel im Steady State wurden für die untersuchten Arzneimittel keine klinisch relevanten Arzneimittelwechselwirkungen mit Semaglutide festgestellt. Daher ist bei gleichzeitiger Verabreichung mit Semaglutide keine Dosisanpassung erforderlich.
Wirkung von Semaglutide auf andere Arzneimittel
Orale Kontrazeptiva
Eine verminderte Wirksamkeit oraler Kontrazeptiva durch Semaglutide wird nicht erwartet, da Semaglutide die Gesamtexposition von Ethinylestradiol und Levonorgestrel nicht in klinisch relevantem Masse veränderte, wenn ein orales Kombinationsarzneimittel zur Kontrazeption (0.03 mg Ethinylestradiol/0.15 mg Levonorgestrel) gemeinsam mit Semaglutide angewendet wurde. Die Exposition von Ethinylestradiol wurde nicht beeinflusst; für die Exposition von Levonorgestrel im Steady State wurde ein Anstieg um 20 % beobachtet. Die Cmax wurde für keinen der Wirkstoffe beeinflusst.
Atorvastatin
Nach Gabe einer Einzeldosis von Atorvastatin (40 mg) veränderte Semaglutide die Gesamtexposition von Atorvastatin nicht. Die Cmax von Atorvastatin war um 38 % verringert. Dies wurde als nicht klinisch relevant beurteilt.
Digoxin
Nach Gabe einer Einzeldosis von Digoxin (0.5 mg) veränderte Semaglutide die Gesamtexposition oder die Cmax von Digoxin nicht.
Metformin
Nach einer Verabreichung von 500 mg Metformin zweimal täglich über 3.5 Tage veränderte Semaglutide die Gesamtexposition oder die Cmax von Metformin nicht.
Warfarin und andere Cumarinderivate
Nach Gabe einer Einzeldosis Warfarin (25 mg) veränderte Semaglutide die Gesamtexposition oder die Cmax von R- und S-Warfarin nicht. Es kam auch nicht zu einer klinisch relevanten Änderung der pharmakodynamischen Wirkungen von Warfarin, gemessen an der International Normalised Ratio (INR). Jedoch wurden Fälle von INR-Senkungen bei gleichzeitiger Anwendung von Acenocoumarol und Semaglutide berichtet. Bei Einleitung einer Semaglutide-Behandlung bei Patienten, die Warfarin oder andere Cumarinderivate einnehmen, wird daher eine regelmässige Überwachung des INR empfohlen.
Schwangerschaft, StillzeitSchwangerschaft
Tierexperimentelle Studien haben eine Reproduktionstoxizität gezeigt (siehe «Präklinische Daten»). Es liegen nur begrenzte Daten zur Anwendung von Semaglutide bei Schwangeren vor. Daher soll Semaglutide während der Schwangerschaft nicht angewendet werden. Frauen im gebärfähigen Alter wird empfohlen, während der Behandlung mit Semaglutide eine zuverlässige Verhütungsmethode anzuwenden. Möchte eine Patientin schwanger werden oder tritt eine Schwangerschaft ein, soll Semaglutide abgesetzt werden. Aufgrund der langen Halbwertszeit (siehe «Pharmakokinetik») sollte Semaglutide mindestens 2 Monate vor einer geplanten Schwangerschaft abgesetzt werden.
Stillzeit
Bei säugenden Ratten wurde Semaglutide in die Muttermilch ausgeschieden. Ein Risiko für ein gestilltes Kind kann nicht ausgeschlossen werden. Semaglutide soll während der Stillzeit nicht angewendet werden.
Fertilität
Es ist nicht bekannt, ob Semaglutide eine Auswirkung auf die menschliche Fertilität hat. Semaglutide beeinträchtigte die Fertilität männlicher Ratten nicht. Bei weiblichen Ratten wurde bei Dosen, die mit einem mütterlichen Gewichtsverlust einhergingen, eine Verlängerung des Östrus und eine geringe Abnahme der Anzahl der Ovulationen beobachtet.
Wirkung auf die Fahrtüchtigkeit und auf das Bedienen von MaschinenSemaglutide hat keinen oder einen zu vernachlässigenden Einfluss auf die Verkehrstüchtigkeit oder die Fähigkeit zum Bedienen von Maschinen. Insbesondere im Dosissteigerungszeitraum kann aber Schwindelgefühl auftreten. Falls es zu Schwindelgefühl kommt, ist Vorsicht im Strassenverkehr und beim Bedienen von Maschinen geboten.
Patienten mit Typ 2 Diabetes mellitus
Bei Anwendung von Semaglutide in Kombination mit einem Sulfonylharnstoff oder Insulin sollten Patienten angewiesen werden, Massnahmen zur Hypoglykämievermeidung bei der Teilnahme am Strassenverkehr oder während des Bedienens von Maschinen zu ergreifen (siehe «Warnhinweise und Vorsichtsmassnahmen»).
Unerwünschte WirkungenZusammenfassung des Sicherheitsprofils
In 4 Phase-3a-Studien wurden 2'650 erwachsene Patienten mit Wegovy behandelt. Die Dauer der Studien betrug 68 Wochen. Ähnlich wie bei anderen GLP-1-Rezeptor-Agonisten waren die am häufigsten berichteten Nebenwirkungen gastrointestinale Störungen, darunter Übelkeit, Diarrhoe, Obstipation und Erbrechen.
Tabellarische Auflistung der Nebenwirkungen
In Tabelle 3 sind unerwünschte Wirkungen bei Erwachsenen aufgeführt, die in Phase-3a-Studien und Post-Marketing-Berichten bei Patienten mit Typ 2 Diabetes mellitus berichtet wurden. Die Häufigkeiten basieren auf den gepoolten Daten der Phase-3a-Studien.
Liste der unerwünschten Wirkungen
Die unerwünschten Wirkungen sind nach MedDRA-Systemorganklassen und Häufigkeit gemäss folgender Konvention, geordnet:
«sehr häufig» (≥1/10), «häufig» (≥1/100, <1/10), «gelegentlich» (≥1/1'000, <1/100), «selten» (≥1/10'000, <1/1'000), «sehr selten» (<1/10'000), «nicht bekannt» (kann aus den verfügbaren Daten nicht abgeschätzt werden).
Tabelle 3: Unerwünschte Wirkungen bei Erwachsenen aus kontrollierten Phase-3-Studien und Post-Marketing-Berichten
|
Systemorganklasse gemäss MedDRA
|
Sehr häufig
|
Häufig
|
Gelegentlich
|
Selten
|
Nicht bekannt
| |
Störungen des Immunsystems
|
|
|
|
Anaphylaktische Reaktion
|
| |
Stoffwechsel- und Ernährungsstörungen
|
|
Hypoglykämie bei Patienten mit Diabetes Typ 2a
|
|
|
| |
Erkrankungen des Nervensystems
|
Kopfschmerzenb
|
Schwindelgefühlb
Dysgeusieb,c
Dysästhesiea,c
|
|
|
| |
Augenerkrankungen
|
|
Diabetische Retinopathie bei Patienten mit Diabetes Typ 2a
|
|
|
| |
Herzerkrankungen
|
|
|
Erhöhte Herzfrequenza,c
|
|
| |
Erkrankungen des Gastrointestinaltrakts
|
Erbrechena,b
Diarrhoea,b
Obstipationa,b
Übelkeita,b
Abdominalschmerzb,c
|
Gastritisb,c
Gastroösophageale Refluxerkrankungb
Dyspepsieb
Aufstossenb
Flatulenzb
Bauch aufgetriebenb
|
Akute Pankreatitisa
Verzögerte Magenentleerung
|
|
Darmobstruktionc,d,e
| |
Leber- und Gallenerkrankungen
|
|
Cholelithiasisa
|
Erhöhte Amylasec
Erhöhte Lipasec
Cholezystitis
|
|
| |
Erkrankungen der Haut und des Unterhautgewebes
|
|
Haarausfalla
|
|
|
| |
Erkrankungen der Nieren und Harnwege
|
|
|
|
|
Akute
Nierenschäden
| |
Allgemeine Erkrankungen und Beschwerden am Verabreichungsort
|
Ermüdungb,c
|
Reaktionen an der Injektionsstellec
|
|
|
| |
a)
Siehe die folgende Beschreibung ausgewählter Nebenwirkungen
b) Hauptsächlich während der Dosiseskalation
c) Gruppierung von bevorzugten Bezeichnungen
d) Aus Meldungen nach Markteinführung
e) Die zusammengefasste Bezeichnung umfasst die unerwünschten Ereignisse Darmobstruktion, Ileus und Dünndarmobstruktion
|
Beschreibung spezifischer unerwünschter Wirkungen und Zusatzinformationen
Erkrankungen des Gastrointestinaltrakts
Sehr häufig – Übelkeit (43.9 %), Durchfall (29.7 %), Erbrechen (24.5 %), Obstipation (24.2 %)
Am häufigsten wurden die Ereignisse während der Dosiseskalation berichtet. Über 68 Wochen trat Übelkeit unter der Behandlung mit Wegovy bei 43.9 % der Patienten auf (16.1 % unter Placebo), Diarrhoe bei 29.7 % (15.9 % unter Placebo) und Erbrechen bei 24.5 % (6.3 % unter Placebo). Die meisten Ereignisse waren leicht bis mittelschwer und von kurzer Dauer. Obstipation trat bei 24.2 % der Patienten unter der Behandlung mit Wegovy auf (11.1 % unter Placebo), war leicht bis mittelschwer und von längerer Dauer.
Bei 4.3 % der Patienten führten die gastrointestinalen Ereignisse zum dauerhaften Abbruch der Behandlung mit Wegovy (0.7 % unter Placebo).
Gelegentlich – akute Pankreatitis
Die Häufigkeit von bestätigten Fällen akuter Pankreatitis, die in klinischen Studien der Phase 3a gemeldet wurden, lag bei 0.2 % für Wegovy beziehungsweise bei < 0.1 % für Placebo.
Im Rahmen der SELECT-Studie, der kardiovaskulären Ergebnisstudie, lag die Häufigkeit von bestätigten Fällen akuter Pankreatitis bei 0.2 % für Wegovy beziehungsweise bei 0.3 % für Placebo.
Häufig – akute Gallensteinerkrankung/Cholelithiasis
Cholelithiasis wurde bei 1.6 % der Patienten unter Wegovy berichtet und führte bei 0.6 % zu Cholezystitis. Cholelithiasis und Cholezystitis wurden in 1.1 % beziehungsweise in 0.3 % bei Patienten unter Placebo berichtet.
Erkrankungen der Haut und des Unterhautgewebes
Häufig – Haarausfall
Haarausfall wurde bei 2.5 % der Patienten unter Wegovy und bei 1.0 % unter Placebo berichtet. Die Ereignisse waren hauptsächlich leichter Natur und gingen bei den meisten Patienten unter fortgesetzter Behandlung zurück. Haarausfall wurde häufiger bei Patienten mit grösserem Gewichtsverlust (≥20 %) berichtet.
Herzerkrankungen
Gelegentlich – Erhöhung der Herzfrequenz
In den Phase-3a-Studien wurde unter Wegovy eine mittlere Erhöhung der Herzfrequenz um 3 Schläge pro Minute (bpm) gegenüber einem mittleren Ausgangswert von 72 bpm beobachtet. Der Anteil der Probanden mit einem maximalen Anstieg des Ruhepulses von ≥20 Schlägen pro Minute gegenüber dem Ausgangswert zu einem beliebigen Zeitpunkt während des Behandlungszeitraums betrug 26.0 % in der Wegovy Gruppe gegenüber 15.6 % in der Placebogruppe.
Erkrankungen des Nervensystems
Häufig – Dysästhesie
Eine Analyse der placebokontrollierten Phase-3a-Studien (STEP 1-4) zeigte eine erhöhte Inzidenz von Dysästhesien. Bei 2.1 % der mit Wegovy behandelten Patienten und bei 1.2 % der mit Placebo behandelten Patienten wurden Ereignisse berichtet, die mit dem klinischen Bild einer veränderten Hautempfindung zusammenhängen, wie Dysästhesie, Parästhesie, Hyperästhesie, Brennen, Allodynie und empfindliche Haut. Die Ereignisse waren leicht bis moderat ausgeprägt und die meisten Patienten erholten sich unter fortgesetzter Behandlung.
Immunogenität
Entsprechend den potenziell immunogenen Eigenschaften von protein- oder peptidhaltigen Arzneimitteln können Patienten durch die Behandlung mit Wegovy anti-Semaglutide Antikörper bilden. Der Nachweis der Antikörperbildung hängt stark von der Sensitivität und Spezifität des Assays ab. Darüber hinaus kann die beobachtete Inzidenz von Antikörper-Positivität (einschliesslich neutralisierender Antikörper) in einem Assay von mehreren Faktoren beeinflusst werden, einschliesslich Assay-Methodik, Probenhandhabung, Zeitpunkt der Probenentnahme, Begleitmedikation und Grunderkrankung. Aus diesen Gründen kann die Inzidenz von Antikörpern gegen Semaglutide in den unten beschriebenen Studien nicht direkt mit der Inzidenz von Antikörpern in anderen Studien oder bei anderen Arzneimitteln verglichen werden.
Der Anteil der Patienten, deren Test auf gegen Semaglutide gerichtete Antikörper zu einem beliebigen Zeitpunkt nach Behandlungsbeginn positiv war, war gering (2.9 %). Von den 50 mit Semaglutide behandelten Patienten, die Semaglutide-Antikörper entwickelten, bildeten 28 Patienten (1.6 % der gesamten mit Wegovy-behandelten Studienpopulation) Antikörper, die mit nativem GLP-1 kreuzreagierten. Kein Patient hatte am Ende der Studie neutralisierende Antikörper gegen Semaglutide oder Antikörper gegen Semaglutide mit endogener, GLP-1 neutralisierender Wirkung.
Stoffwechsel- und Ernährungsstörungen
Häufig – Hypoglykämie bei Patienten mit Diabetes Typ 2
In Patienten mit Typ 2 Diabetes (Studie STEP 2) wurde bei 6.2 % (0.1 Ereignisse/Patientenjahr) der mit Wegovy Behandelten gegenüber 2.5 % (0.03 Ereignisse/Patientenjahr) der mit Placebo Behandelten eine klinisch signifikante Hypoglykämie (definiert als Plasma Glukose <3.0 mmol/l (54 mg/dl)) beobachtet. Eine Episode (0.2 % der Teilnehmer, 0.002 Ereignisse/Patientenjahr) wurde als schwerwiegend eingestuft. Das Risiko einer Hypoglykämie war erhöht, wenn Wegovy zusammen mit einem Sulfonylharnstoff angewendet wurde.
In klinischen Studien mit Wegovy bei Patienten ohne Diabetes Typ 2 wurden Hypoglykämien nicht systematisch erfasst oder berichtet.
Häufig – Diabetische Retinopathie bei Patienten mit Diabetes Typ 2
In Patienten mit Diabetes Typ 2 in der Studie STEP 2 wurden wenige Episoden einer diabetischen Retinopathie (4.0 % der mit Wegovy Behandelten bzw. 2.7 % der mit Placebo Behandelten) beobachtet.
Kinder und Jugendliche
In einer klinischen Studie mit Jugendlichen von 12 Jahren bis unter 18 Jahren mit Adipositas oder Übergewicht mit mindestens einer gewichtsbedingten Begleiterkrankung erhielten 133 Patienten Wegovy. Die Dauer der Studie betrug 68 Wochen.
Insgesamt waren Häufigkeit, Art und Schwere der unerwünschten Wirkungen bei Jugendlichen vergleichbar mit denen, die bei der erwachsenen Bevölkerung beobachtetet wurden. Cholelithiasis wurde mit 3.8 % der mit Wegovy behandelten Jugendlichen häufiger als bei Erwachsenen berichtet gegenüber 0 % der mit Placebo behandelten Jugendlichen.
In der Studie wurden keine Auswirkungen auf das Wachstum oder die pubertäre Entwicklung festgestellt.
Erwachsene mit etablierter kardiovaskulärer Erkrankung
In der SELECT-Studie war das Nebenwirkungsprofil bei Erwachsenen mit etablierter kardiovaskulärer Erkrankung ähnlich wie das in den Phase-3a-Studien zur Gewichtsreduktion beobachtete.
Die Meldung des Verdachts auf Nebenwirkungen nach der Zulassung ist von grosser Wichtigkeit. Sie ermöglicht eine kontinuierliche Überwachung des Nutzen-Risiko-Verhältnisses des Arzneimittels. Angehörige von Gesundheitsberufen sind aufgefordert, jeden Verdacht einer neuen oder schwerwiegenden Nebenwirkung über das Online-Portal ElViS (Electronic Vigilance System) anzuzeigen. Informationen dazu finden Sie unter www.swissmedic.ch.
ÜberdosierungEine Überdosierung mit Semaglutide kann mit gastrointestinalen Beschwerden einhergehen, die zur Dehydratation führen. Im Fall einer Überdosierung ist der Patient auf klinische Anzeichen zu überwachen und eine angemessene unterstützende Behandlung einzuleiten.
Eigenschaften/WirkungenATC-Code
A10BJ06
Wirkungsmechanismus
Semaglutide ist ein GLP-1-Analogon mit einer Sequenzhomologie von 94 % zum humanen GLP-1. Semaglutide wirkt als GLP-1-Rezeptoragonist, der selektiv an den GLP-1-Rezeptor, das Ziel für natives GLP-1, bindet und diesen aktiviert.
Verglichen mit nativem GLP-1 weist Semaglutide eine verlängerte Halbwertszeit von ungefähr 1 Woche auf, wodurch es sich für die einmal wöchentliche subkutane Anwendung eignet. Der Hauptmechanismus der Verzögerung ist die Albuminbindung, die eine verminderte renale Clearance und den Schutz vor metabolischem Abbau zur Folge hat. Darüber hinaus ist Semaglutide gegen den Abbau durch das Enzym DDP-4 stabilisiert.
GLP-1 ist ein physiologisches Hormon, das bei der Regulierung von Appetit und Kalorienaufnahme wirkt; der GLP-1-Rezeptor ist in verschiedenen Bereichen des Gehirns vorhanden, die an der Appetitregulierung beteiligt sind. Semaglutide hat direkte Wirkungen auf Gehirnregionen, die an der homöostatischen Regulierung der Nahrungsaufnahme im Hypothalamus und Hirnstamm beteiligt sind. Semaglutide beeinflusst das hedonische Belohnungssystem über direkte und indirekte Wirkungen auf Gehirnregionen wie Septum, Thalamus und Amygdala.
Klinische Studien zeigen, dass Semaglutide die Energieaufnahme verringert, die Wahrnehmung von Sättigung, Völlegefühl und Esskontrolle erhöht, das Hungergefühl dämpft und die Häufigkeit sowie Intensität von Heisshunger verringert.
Ausserdem wurde in klinischen Studien gezeigt, dass Semaglutide den Blutzuckerspiegel glucoseabhängig durch Stimulation der Insulinsekretion und Senkung der Glucagonsekretion senkt, wenn der Blutzuckerspiegel hoch ist. Der Mechanismus der Blutzuckersenkung geht auch mit einer leicht verlangsamten Entleerung des Magens in der frühen postprandialen Phase einher. Während einer Hypoglykämie verringert Semaglutide die Sekretion von Insulin, vermindert aber nicht die Glucagonsekretion.
GLP-1-Rezeptoren sind auch im Herz, im Gefässsystem, Immunsystem und in den Nieren exprimiert.
Der Wirkmechanismus von Semaglutide zur Reduktion des kardiovaskulären Risikos ist wahrscheinlich multifaktoriell und nicht vollständig geklärt, zum Teil vermittelt durch die Wirkungen auf bekannte kardiovaskuläre Risikofaktoren (einschliesslich Blutdrucksenkung, Verbesserung des Lipidprofils, und antiinflammatorischer Effekte).
Pharmakodynamik
Appetit, Energieaufnahme und Nahrungsmittelwahl
Wegovy senkt den Appetit durch Steigerung des Sättigungs- und Völlegefühls und vermindert Hunger sowie die prospektive Nahrungsaufnahme. Nach 20 Wochen Behandlung war die Energieaufnahme bei einer Ad-libitum-Mahlzeit unter Wegovy um 35 % niedriger als unter Placebo. Dies ging einher mit einer verbesserten Esskontrolle, weniger Heisshunger (auf Milchprodukte und herzhafte Nahrungsmittel), weniger Verlangen nach Süssem und einer relativ geringeren Präferenz für fettreiche Nahrungsmittel.
Kardiale Elektrophysiologie (QTc)
Die Wirkung von Semaglutide auf die kardiale Repolarisation wurde in einer ausführlichen QTc-Studie untersucht. Semaglutide verlängerte das QTc-Intervall bei Dosen bis zu 1,5 mg im Steady-State nicht.
Die Semaglutide-Exposition bei Probanden mit Übergewicht oder Adipositas, die mit Wegovy behandelt werden, ist vergleichbar mit der Exposition, die in der Semaglutid-QTc-Studie an gesunden Probanden untersucht wurde.
Klinische Wirksamkeit
Die Wirksamkeit und Sicherheit von Wegovy für die Gewichtsregulierung in Kombination mit einer kalorienreduzierten Ernährung und verstärkter körperlicher Aktivität wurden in vier 68-wöchigen, doppelblinden, randomisierten, placebo-kontrollierten Phase-3a-Studien (STEP 1–4) untersucht. Insgesamt 4684 Patienten (2652 zur Behandlung mit Wegovy randomisiert) wurden in die Studien eingeschlossen. Darüber hinaus wurden die Wirksamkeit und Sicherheit von Semaglutide im Vergleich zu Placebo über zwei Jahre in einer doppelblinden, randomisierten, placebokontrollierten Phase-3b-Studie (STEP 5) mit 304 Patienten (152 unter Behandlung mit Semaglutide) untersucht.
Die Behandlung mit Wegovy ergab im Vergleich mit Placebo einen überlegenen, klinisch relevanten und anhaltenden Gewichtsverlust bei Patienten mit Adipositas (BMI ≥30 kg/m2) oder mit Übergewicht (BMI ≥27 kg/m2 bis < 30 kg/m2) und mindestens einer gewichtsbedingten Begleiterkrankung.
Darüber hinaus erreichte in allen Studien ein höherer Anteil der Patienten mit Semaglutide einen Gewichtsverlust von ≥5 %, ≥10 %, ≥15 % und ≥20 % als mit Placebo.
Die Behandlung mit Wegovy zeigte auch statistisch signifikante Verbesserungen des Taillenumfangs und systolischen Blutdrucks gegenüber Placebo. Darüber hinaus wirkte sich Semaglutide 2.4 mg im Vergleich zu Placebo insgesamt günstig auf die Plasmalipide und CRP (Entzündungsmarker) aus (siehe Tabelle 4 und Tabelle 5).
Die Wirksamkeit in Bezug auf die Gewichtsabnahme erwies sich unabhängig von Alter, Geschlecht, ethnischer Zugehörigkeit, den Ausgangswerten von Körpergewicht und BMI, Vorliegen eines Diabetes Typ 2 und dem Grad der Nierenfunktion.
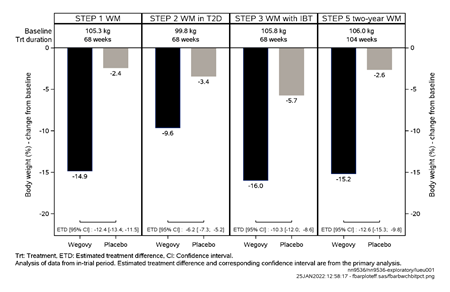
Abbildung 1 Körpergewichtveränderung (%) zwischen Studienbeginn und Woche 68 und 104
STEP 1: Gewichtsmanagement
In einer 68-wöchigen, doppelblinden Studie wurden 1 961 Patienten mit Adipositas (BMI ≥30 kg/m2) oder mit Übergewicht (BMI ≥27 kg/m2 bis < 30 kg/m2) und mindestens einer gewichtsbedingten Begleiterkrankung zur Behandlung mit Wegovy oder Placebo randomisiert. Alle Patienten hielten während der gesamten Studie eine kalorienreduzierte Diät und erhöhte körperliche Aktivität ein. Die Mehrzahl der Patienten hatte mindestens eine gewichtsbedingte Begleiterkrankung. Dazu gehörten unter anderem Prä-Diabetes (43.7 %), Dyslipidämie (37.0 %), Hypertonie (36.0 %), Osteoarthrose des Knie- oder Hüftgelenks (15.9 %), obstruktive Schlafapnoe (11.7 %), Asthma/chronisch-obstruktive Lungenerkrankung (COPD) (11.6 %), Lebererkrankung (nicht-alkoholische Fettleber (NAFLD) oder nicht-alkoholische Steatohepatitis (NASH)) (8.6 %) und polyzystisches Ovarialsyndrom (PCOS) (6.6 %).
Der Gewichtsverlust trat früh ein und setzte sich während der Studie fort. Am Behandlungsende (Woche 68) war der Gewichtsverlust verglichen mit Placebo überlegen und klinisch relevant (siehe Tabelle 4 und Abbildung 2).
Im Anschluss an die 68-wöchige Studie wurde eine 52-wöchige Verlängerung ohne Behandlung durchgeführt, welche 327 Patienten einschloss, die den Hauptstudienzeitraum mit der Erhaltungsdosis von Semaglutide oder Placebo abgeschlossen hatten. In der behandlungsfreien Zeit von Woche 68 bis Woche 120 nahm das durchschnittliche Körpergewicht in beiden Behandlungsgruppen zu. Bei den Patienten, die während des Hauptstudienzeitraums mit Semaglutide behandelt worden waren, blieb das Gewicht jedoch um 5.6 % unter dem Ausgangswert, verglichen mit 0.1 % in der Placebogruppe.
Tabelle 4: Ergebnisse einer 68-wöchigen Studie zum Vergleich von Wegovy mit Placebo bei Patienten mit Adipositas oder Übergewicht und mindestens einer gewichtsbezogenen Begleiterkrankung (STEP 1)
|
|
Wegovy
|
Placebo
| |
Full analysis set (N)
|
1'306
|
655
| |
Körpergewicht
| |
Ausgangswert (kg)
|
105.4
|
105.2
| |
Änderung (%) gegenüber Ausgangswert1,2
|
-14.9
|
-2.4
| |
Unterschied (%) gegenüber Placebo1
[95 %-KI]
|
-12.4
[-13.4; -11.5]*
|
-
| |
Änderung (kg) gegenüber Ausgangswert
|
-15.3
|
-2.6
| |
Unterschied (kg) gegenüber Placebo1
[95 %-KI]
|
-12.7
[-13.7; -11.7]
|
-
| |
Patienten (%) mit Gewichtsverlust ≥5 %3
|
83.5*
|
31.1
| |
Patienten (%) mit Gewichtsverlust ≥10 %3
|
66.1*
|
12.0
| |
Patienten (%) mit Gewichtsverlust ≥15 %3
|
47.9*
|
4.8
| |
Patienten (%) mit Gewichtsverlust ≥20 %3
|
30.2
|
1.7
| |
Taillenumfang (cm)
| |
Ausgangswert
|
114.6
|
114.8
| |
Änderung gegenüber Ausgangswert1
|
-13.5
|
-4.1
| |
Unterschied gegenüber Placebo1
[95 %-KI]
|
-9.4
[-10.3; -8.5]*
|
-
| |
Kardiometabolische Faktoren
| |
Systolischer Blutdruck (mmHg)
| |
Ausgangswert
|
126
|
127
| |
Änderung gegenüber Ausgangswert1
|
-6.2
|
-1.1
| |
Unterschied gegenüber Placebo1
[95 %-KI]
|
-5.1
[-6.3; -3.9]*
|
-
| |
Diastolischer Blutdruck (mmHg)
| |
Ausgangswert
|
80
|
80
| |
Änderung gegenüber Ausgangswert
|
-2.8
|
-0.4
| |
Unterschied gegenüber Placebo [95 %-KI]
|
-2.4
[-3.3; -1.6]
|
-
| |
Lipide
| |
Total Cholesterol
| |
Ausgangswert (mmol/l)4
|
4.9
|
5.0
| |
Änderung (%) gegenüber Ausgangswert1
|
-3.3
|
0.1
| |
Relativer Unterschied (%) gegenüber Placebo [95 %-KI]1
|
-3.3
[-4.8; -1.8]
|
-
| |
LDL Cholesterol
| |
Ausgangswert (mmol/l)
|
2.9
|
2.9
| |
Änderung (%) gegenüber Ausgangswert1
|
-2.5
|
1.3
| |
Relativer Unterschied (%) gegenüber Placebo1 [95 %-KI]
|
-3.8
[-5.9; -1.5]
|
-
| |
HDL Cholesterol
| |
Ausgangswert (mmol/l)4
|
1.3
|
1.3
| |
Änderung (%) gegenüber Ausgangswert1
|
5.2
|
1.4
| |
Relativer Unterschied (%) gegenüber Placebo1 [95 %-KI]
|
3.8
[2.2; 5.4]
|
-
| |
Triglycerides
| |
Ausgangswert (mmol/l)4
|
1.4
|
1.4
| |
Änderung (%) gegenüber Ausgangswert1
|
-21.9
|
-7.3
| |
Relativer Unterschied (%) gegenüber Placebo1 [95 %-KI]
|
-15.8
[-18.8; -12.7]
|
-
| |
CRP
| |
Ausgangswert (mg/l)
|
3.9
|
3.9
| |
Änderung (%) gegenüber Ausgangswer1
|
-52.6
|
-15.0
| |
Relativer Unterschied (%) gegenüber Placebo1 [95 %-KI]
|
-44.3
[-49.5; -38.5]
|
-
| |
Glykämischer Status
| |
Patienten (%) mit Prä-Diabetes bei Baseline
|
43.7
| |
Patienten (%) mit normoglykämischem Status bei Behandlungsende
|
84.1
|
47.8
|
* p < 0.0001 (unkorrigiert 2-seitig) für Überlegenheit.
1 Geschätzt mithilfe eines ANCOVA-Modells mit multipler Imputation auf Basis aller Daten unabhängig vom Abbruch der randomisierten Behandlung oder dem Beginn einer anderen medikamentösen Behandlung gegen Adipositas oder bariatrischer Chirurgie.
2 Während der Studie wurde die randomisierte Behandlung von 17.1 % und 22.4 % der Patienten abgebrochen, die zu Wegovy bzw. Placebo randomisiert waren. Unter der Annahme, dass alle randomisierten Patienten die Behandlung beibehielten und keine zusätzlichen Therapien gegen Adipositas erhielten, betrugen die geschätzten Änderungen des Körpergewichts von der Randomisierung bis Woche 68 -16.9 % für 2.4 mg Semaglutide und -2.4 % für Placebo, basierend auf einem gemischten Modell für wiederholte Messungen unter Einschluss aller Beobachtungen bis zum ersten Absetzen.
3 Geschätzt mit einem binären Regressionsmodell auf Basis desselben Imputationsverfahrens wie bei der primären Analyse.
4 Geometrisches Mittel.
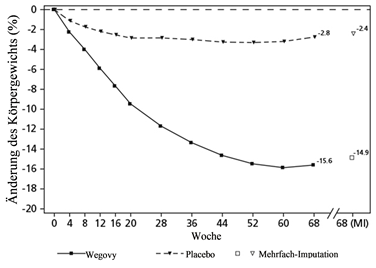
Beobachtete Werte für Patienten, die jeden geplanten Besuch absolvierten, und Schätzungen mit multiplen Imputationen (MI) für Abbrecher mit Abschlussuntersuchung («retrieved dropouts»).
Abbildung 2 STEP 1 - Mittlere Veränderung des Körpergewichts (%) von Baseline bis Woche 68
STEP 2: Gewichtsmanagement bei Patienten mit Diabetes Typ 2
In einer 68-wöchigen, doppelblinden Studie wurden 1 210 Patienten mit Übergewicht oder Adipositas (BMI ≥27 kg/m2) und Diabetes Typ 2 auf Wegovy, Semaglutide 1 mg einmal wöchentlich oder Placebo randomisiert. In die Studie wurden Patienten mit unzureichend eingestelltem Diabetes (HbA1c 7–10 %) aufgenommen, die entweder mit Diät und körperlicher Bewegung alleine oder mit 1–3 oralen Antidiabetika behandelt wurden. Alle Patienten hielten während der gesamten Studie eine kalorienreduzierte Diät und erhöhte körperliche Aktivität ein. Die Mehrzahl der Patienten hatte mindestens zwei gewichtsbedingte Begleiterkrankungen. Neben dem Diabetes Typ 2 waren dies unter anderem Hypertonie (69.8 %), Dyslipidämie (68.0 %), Osteoarthrose des Knie- oder Hüftgelenks (19.6 %), Lebererkrankung (NAFLD oder NASH) (22.6 %), obstruktive Schlafapnoe (15.1 %), Asthma/COPD (8.4 %) und PCOS (4.1 %).
Die Behandlung mit Wegovy über 68 Wochen führte zu einer überlegenen und klinisch relevanten Senkung des Körpergewichts und des HbA1c als unter Placebo (siehe Tabelle 5 und Abbildung 3).
Tabelle 5: Ergebnisse einer 68-wöchigen Studie zum Vergleich von Wegovy mit Placebo bei Patienten mit Fettleibigkeit oder Übergewicht und Diabetes Typ 2 (STEP 2)
|
|
Wegovy
|
Placebo
| |
Full analysis set (N)
|
404
|
403
| |
Körpergewicht
| |
Ausgangswert (kg)
|
99.9
|
100.5
| |
Änderung (%) gegenüber Ausgangswert1,2
|
-9.6
|
-3.4
| |
Unterschied (%) gegenüber Placebo1
[95 %-KI]
|
-6.2
[-7.3; -5.2]*
|
-
| |
Änderung (kg) gegenüber Ausgangswert
|
-9.7
|
-3.5
| |
Unterschied (kg) gegenüber Placebo1
[95 %-KI]
|
-6.1
[-7.2; -5.0]
|
-
| |
Patienten (%) mit Gewichtsverlust ≥5 %3
|
67.4*
|
30.2
| |
Patienten (%) mit Gewichtsverlust ≥10 %3
|
44.5*
|
10.2
| |
Patienten (%) mit Gewichtsverlust ≥15 %3
|
25.0*
|
4.3
| |
Patienten (%) mit Gewichtsverlust ≥20 %3
|
12.8
|
2.3
| |
Taillenumfang (cm)
| |
Ausgangswert
|
114.5
|
115.5
| |
Änderung gegenüber Ausgangswert1
|
-9.4
|
-4.5
| |
Unterschied gegenüber Placebo1 [95 %-KI]
|
-4.9
[-6.0; -3.8]*
|
-
| |
Kardiometabolische Faktoren
| |
Systolischer Blutdruck (mmHg)
| |
Ausgangswert
|
130
|
130
| |
Änderung gegenüber Ausgangswert1
|
-3.9
|
-0.5
| |
Unterschied gegenüber Placebo1 [95 %-KI]
|
-3.4
[-5.6; -1.3]**
|
-
| |
Diastolischer Blutdruck (mmHg)
| |
Ausgangswert
|
80
|
80
| |
Änderung gegenüber Ausgangswert
|
-1.6
|
-0.9
| |
Unterschied gegenüber Placebo [95 %-KI]
|
-0.7
[-2.0; 0.6]
|
-
| |
Lipide
| |
Total Cholesterol
| |
Ausgangswert (mmol/l)4
|
4.4
|
4.4
| |
Änderung (%) gegenüber Ausgangswert1
|
-1.4
|
-0.5
| |
Relativer Unterschied (%) gegenüber Placebo1 [95 %-KI]
|
-0.9
[-3.6; 2.0]
|
-
| |
LDL Cholesterol
| |
Ausgangswert (mmol/l)4
|
2.3
|
2.3
| |
Änderung (%) gegenüber Ausgangswert1
|
0.5
|
0.1
| |
Relativer Unterschied (%) gegenüber Placebo1 [95 %-KI]
|
0.4
[-4.0; 4.9]
|
-
| |
HDL Cholesterol
| |
Ausgangswert (mmol/l)4
|
1.2
|
1.1
| |
Änderung (%) gegenüber Ausgangswert1
|
6.9
|
4.1
| |
Relativer Unterschied (%) gegenüber Placebo1 [95 %-KI]
|
2.7
[0.3; 5.1]
|
-
| |
Triglycerides
| |
Ausgangswert (mmol/l)4
|
1.7
|
1.8
| |
Änderung (%) gegenüber Ausgangswert1
|
-22.0
|
-9.4
| |
Relativer Unterschied (%) gegenüber Placebo1 [95 %-KI]
|
-13.9
[-19.0; -8.4]
|
-
| |
CRP
| |
Ausgangswert (mg/l)
|
3.5
|
3.4
| |
Änderung (%) gegenüber Ausgangswer1
|
-48.9
|
-16.7
| |
Relativer Unterschied (%) gegenüber Placebo1 [95 %-KI]
|
-38.7
[-46.5; -29.8]
|
-
| |
Glykämische Faktoren
| |
HbA1c (mmol/mol (%)
| |
Ausgangswert
|
65.3 (8.1)
|
65.3 (8.1)
| |
Änderung gegenüber Ausgangswert1,2
|
-17.5 (-1.6)
|
-4.1 (-0.4)
| |
Unterschied gegenüber Placebo1 [95 %-KI]
|
-13.5
[-15.5; -11.4]
(-1.2 [-1.4; -1.1])*
|
-
-
| |
Patienten (%), die einen HbA1c-Wert < 7 %
erreichten3
|
77.4
|
26.0
| |
Patienten (%), die einen HbA1c-Wert
≤6.5 % erreichten3
|
65.9
|
15.1
|
* p < 0.001 (unkorrigiert 2-seitig) für Überlegenheit; ** p < 0.05 (unkorrigiert 2-seitig) für Überlegenheit.
1 Geschätzt mithilfe eines ANCOVA-Modells mit multipler Imputation auf Basis aller Daten unabhängig vom Abbruch der randomisierten Behandlung oder dem Beginn einer anderen medikamentösen Behandlung gegen Adipositas oder bariatrischer Chirurgie.
2 Während der Studie wurde die randomisierte Behandlung von 11.6 % und 13.9 % der Patienten abgebrochen, die zu Wegovy bzw. Placebo randomisiert waren. Unter der Annahme, dass alle randomisierten Patienten die Behandlung beibehielten und keine zusätzlichen Therapien gegen Adipositas erhielten, betrugen die geschätzten Änderungen des Körpergewichts von der Randomisierung bis Woche 68 -10.6 % für 2.4 mg Semaglutide und -3.1 % für Placebo, basierend auf einem gemischten Modell für wiederholte Messungen unter Einschluss aller Beobachtungen bis zum ersten Absetzen.
3 Geschätzt mit einem binären Regressionsmodell auf Basis desselben Imputationsverfahrens wie bei der primären Analyse.
4 Geometrisches Mittel
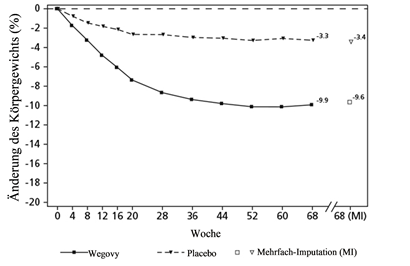
Beobachtete Werte für Patienten, die jeden geplanten Besuch absolvierten, und Schätzungen mit multiplen Imputationen (MI) für Abbrecher mit Abschlussuntersuchung («retrieved dropouts»).
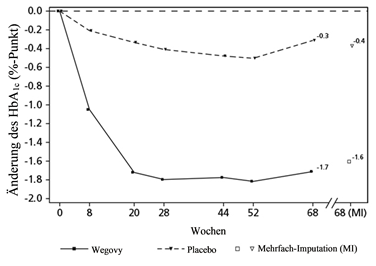
HbA1c: Hämoglobin A1c
Beobachtete Werte für Patienten, die jeden geplanten Besuch absolvierten, und Schätzungen mit multiplen Imputationen für Abbrecher mit Abschlussuntersuchung («retrieved dropouts») (RD-MI).
Abbildung 3 STEP 2 - Mittlere Veränderung des Körpergewichts (kg) und des HbA1c-Wertes (%) zwischen Studienbeginn und Woche 68
STEP 3: Gewichtsmanagement mit intensiver Verhaltenstherapie
In einer 68-wöchigen, doppelblinden Studie wurden 611 Patienten mit Adipositas (BMI ≥30 kg/m2) oder mit Übergewicht (BMI ≥27 kg/m2 bis < 30 kg/m2) und mindestens einer gewichtsbedingten Begleiterkrankung zur Behandlung mit Wegovy oder Placebo randomisiert. Während der Studie erhielten alle Patienten eine intensive Verhaltenstherapie (IBT), bestehend aus einer sehr restriktiven Diät, vermehrter körperlicher Aktivität und einer Verhaltensberatung.
Die Mehrzahl der Patienten hatte mindestens eine gewichtsbedingte Begleiterkrankung. Dazu gehörten unter anderem Prä-Diabetes (49.8 %), Hypertonie (34.7 %), Dyslipidämie (34.7 %), Osteoarthrose des Knie- oder Hüftgelenks (18.7 %), Asthma/COPD (15.1 %), obstruktive Schlafapnoe (12.6 %), Lebererkrankung (NAFLD oder NASH) (6.1 %) und PCOS (5.5 %).
Die Behandlung mit Wegovy und IBT über 68 Wochen führte zu einer überlegenen und klinisch relevanten Senkung des Körpergewichts als unter Placebo (siehe Tabelle 6).
Tabelle 6: Ergebnisse einer 68-wöchigen Studie zum Vergleich von Wegovy mit Placebo bei Patienten mit Adipositas oder Übergewicht und mindestens einer gewichtsbezogenen Begleiterkrankung unter Intensiver Verhaltenstherapie (STEP 3)
|
|
Wegovy
|
Placebo
| |
Full analysis set (N)
|
407
|
204
| |
Körpergewicht
| |
Ausgangswert (kg)
|
106.9
|
103.7
| |
Änderung (%) gegenüber Ausgangswert1,2
|
-16.0
|
-5.7
| |
Unterschied (%) gegenüber Placebo1
[95 %-KI]
|
-10.3
[-12.0; -8.6]*
|
-
| |
Änderung (kg) gegenüber Ausgangswert
|
-16.8
|
-6.2
| |
Unterschied (kg) gegenüber Placebo1
[95 %-KI]
|
-10.6
[-12.5; -8.8]
|
-
| |
Patienten (%) mit Gewichtsverlust ≥5 %3
|
84.8*
|
47.8
| |
Patienten (%) mit Gewichtsverlust ≥10 %3
|
73.0*
|
27.1
| |
Patienten (%) mit Gewichtsverlust ≥15 %3
|
53.5*
|
13.2
| |
Patienten (%) mit Gewichtsverlust ≥20 %3
|
33.9
|
3.5
| |
Taillenumfang (cm)
| |
Ausgangswert
|
113.6
|
111.8
| |
Änderung gegenüber Ausgangswert1
|
-14.6
|
-6.3
| |
Unterschied gegenüber Placebo1 [95 %-KI]
|
-8.3
[-10.1; -6.6]*
|
-
|
* p < 0.0001 (unkorrigiert 2-seitig) für Überlegenheit.
1 Geschätzt mithilfe eines ANCOVA-Modells mit multipler Imputation auf Basis aller Daten unabhängig vom Abbruch der randomisierten Behandlung oder dem Beginn einer anderen medikamentösen Behandlung gegen Adipositas oder bariatrischer Chirurgie.
2 Während der Studie wurde die randomisierte Behandlung von 16.7 % und 18.6 % der Patienten abgebrochen, die zu Wegovy bzw. Placebo randomisiert waren. Unter der Annahme, dass alle randomisierten Patienten die Behandlung beibehielten und keine zusätzlichen Therapien gegen Adipositas erhielten, betrugen die geschätzten Änderungen des Körpergewichts von der Randomisierung bis Woche 68 -17.6 % für 2.4 mg Semaglutide und -5.0 % für Placebo, basierend auf einem gemischten Modell für wiederholte Messungen unter Einschluss aller Beobachtungen bis zum ersten Absetzen.
3 Geschätzt mit einem binären Regressionsmodell auf Basis desselben Imputationsverfahrens wie bei der primären Analyse.
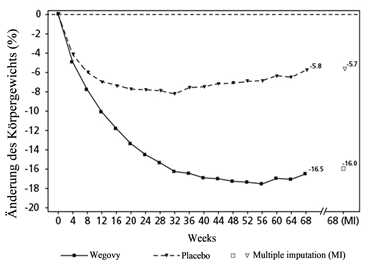
Beobachtete Werte für Patienten, die jeden geplanten Besuch abschliessen, und Schätzungen mit Mehrfach-Anrechnungen (MI) der Dropouts
Abbildung 4 STEP 3 - Mittlere Veränderung des Körpergewichts (%) von Baseline bis Woche 68
STEP 4: Langfristiges Gewichtsmanagement
In einer 68-wöchigen, doppelblinden Studie wurden 902 Patienten mit Adipositas (BMI ≥30 kg/m2) oder mit Übergewicht (BMI ≥27 kg/m2 bis < 30 kg/m2) und mindestens einer gewichtsbedingten Begleiterkrankung aufgenommen. Alle Patienten hielten während der gesamten Studie eine kalorienreduzierte Diät und erhöhte körperliche Aktivität ein. Von Woche 0 bis Woche 20 (Run-in Phase) erhielten alle Patienten Wegovy. In Woche 20 (Baseline) wurden 803 Patienten, die die Erhaltungsdosis von 2.4 mg erreicht hatten, zu Weiterbehandlung oder Umstellung auf Placebo für die restlichen 48 Wochen randomisiert.
Die Mehrzahl der Patienten hatte mindestens eine gewichtsbedingte Begleiterkrankung. Dazu gehörten unter anderem Prä-Diabetes (46.8 %), Hypertonie (37.1 %), Dyslipidämie (35.9 %), Osteoarthrose des Knie- oder Hüftgelenks (13.3 %), obstruktive Schlafapnoe (11.7 %), Asthma/COPD (11.5 %), Lebererkrankung (NAFLD oder NASH) (7.3 %) und PCOS (3.9 %).
Patienten, die in Woche 20 (Baseline) die Erhaltungsdosis von 2.4 mg erreicht hatten und 48 Wochen lang (Woche 20–68) mit Wegovy weiterbehandelt wurden, verloren weiter an Gewicht und erzielten eine überlegene und klinisch relevante Reduktion des Körpergewichts gegenüber denjenigen, die auf Placebo umgestellt worden waren (siehe Tabelle 7 und Abbildung 5). Bei den Patienten, die in Woche 20 (Baseline) auf Placebo umgestellt worden waren, nahm das Körpergewicht zwischen Woche 20 und Woche 68 dagegen wieder stetig zu. Dennoch blieb das beobachtete Körpergewicht in Woche 68 niedriger als zu Beginn der Run-in Phase (Woche 0) (siehe Abbildung 5). Patienten, die von Woche 0 (Einleitung) bis Woche 68 (Behandlungsende) mit Wegovy behandelt wurden, erreichten eine mittlere Veränderung des Körpergewichts von 17.4 %, wobei ein Gewichtsverlust von ≥5 %, ≥10 % ≥15 % oder ≥20 % von 87.8 %, 78.0 %, 62.2 % bzw. 38.6 % dieser Patienten erreicht wurde.
Tabelle 7: Ergebnisse des 48-wöchigen (Woche 20 bis Woche 68) randomisierten Zeitraum der Studie zum Vergleich von Wegovy mit Placebo bei Patienten mit Adipositas oder Übergewicht und mindestens einer gewichtsbezogenen Begleiterkrankung (STEP 4)
|
|
Wegovy
|
Placebo
| |
Full analysis set (N)
|
535
|
268
| |
Körpergewicht
| |
Ausgangswert1 (kg)
|
96.5
|
95.4
| |
Änderung (%) gegenüber Ausgangswert1,2,3
|
-7.9
|
6.9
| |
Unterschied (%) gegenüber Placebo2
[95 %-KI]
|
-14.8
[-16.0; -13.5]*
|
-
| |
Änderung (kg) gegenüber Ausgangswert1
|
-7.1
|
6.1
| |
Unterschied (kg) gegenüber Placebo2
[95 %-KI]
|
-13.2
[-14.3; -12.0]
|
-
| |
Taillenumfang (cm)
| |
Ausgangswert1
|
105.5
|
104.7
| |
Änderung gegenüber Ausgangswert1,2
|
-6.4
|
3.3
| |
Unterschied gegenüber Placebo2 [95 %-KI]
|
-9.7
[-10.9; -8.5]*
|
-
|
* p < 0.0001 (unkorrigiert 2-seitig) für Überlegenheit.
1 Ausgangswert = Woche 20.
2 Geschätzt mithilfe eines ANCOVA-Modells mit multipler Imputation auf Basis aller Daten unabhängig vom Abbruch der randomisierten Behandlung oder dem Beginn einer anderen medikamentösen Behandlung gegen Adipositas oder bariatrischer Chirurgie.
3 Während der Studie wurde die randomisierte Behandlung von 5.8 % und 11.6 % der Patienten abgebrochen, die zu Wegovy bzw. Placebo randomisiert waren. Unter der Annahme, dass alle randomisierten Patienten die Behandlung beibehielten und keine zusätzlichen Therapien gegen Adipositas erhielten, betrugen die geschätzten Änderungen des Körpergewichts von der Randomisierung bis Woche 68 -8.8 % für 2.4 mg Semaglutide und 6.5 % für Placebo, basierend auf einem gemischten Modell für wiederholte Messungen unter Einschluss aller Beobachtungen bis zum ersten Absetzen.
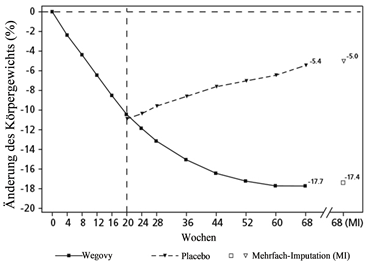
Beobachtete Werte für Patienten, die jeden geplanten Besuch absolvierten, und Schätzungen mit multiplen Imputationen (MI) für Abbrecher mit Abschlussuntersuchung («retrieved dropouts»).
Abbildung 5 STEP 4 - Mittlere Veränderung des Körpergewichts (%) zwischen Woche 0 und Woche 68
STEP 5: 2-Jahres-Daten
In einer 104-wöchigen doppelblinden Studie wurden 304 Patienten mit Adipositas (BMI ≥30 kg/m2) oder mit Übergewicht (BMI ≥27 bis < 30 kg/m2) und mindestens einer gewichtsbedingten Begleiterkrankung auf Semaglutide oder Placebo randomisiert. Alle Patienten erhielten für die Dauer der Studie eine kalorienreduzierte Diät mit erhöhter körperlicher Aktivität.
Bei Studienbeginn hatten die Patienten einen durchschnittlichen BMI von 38.5 kg/m2 und ein durchschnittliches Körpergewicht von 106.0 kg.
Die Behandlung mit Wegovy über 104 Wochen führte zu einer überlegenen und klinisch relevanten Reduktion des Körpergewichts im Vergleich zu Placebo (siehe Tabelle 8 und Abbildung 6). Das durchschnittliche Körpergewicht nahm unter Wegovy ab Studienbeginn bis zur Woche 68 ab, danach wurde ein Plateau erreicht. Unter Placebo nahm das durchschnittliche Körpergewicht weniger stark ab, und nach etwa 20 Behandlungswochen wurde ein Plateau erreicht Die mit Semaglutide behandelten Patienten erreichten eine mittlere Änderung des Körpergewichts von -15.2 %, wobei der Gewichtsverlust bei 74.7 % der Patienten ≥5 %, bei 59.2 % ≥10 % und bei 49.7 % ≥15 % betrug.
Tabelle 8: Ergebnisse einer 104-wöchigen Studie zum Vergleich von Wegovy mit Placebo bei Patienten mit Adipositas oder Übergewicht und mindestens einer gewichtsbezogenen Begleiterkrankung (STEP 5)
|
|
Wegovy
|
Placebo
| |
Full analysis set (N)
|
152
|
152
| |
Körpergewicht
| |
Ausgangswert1 (kg)
|
105.6
|
106.5
| |
Änderung (%) gegenüber Ausgangswert1,2
|
-15.2
|
-2.6
| |
Unterschied (%) gegenüber Placebo1 [95 %-KI]
|
-12.6 [-15.3; -9.8]*
|
-
| |
Änderung (kg) gegenüber Ausgangswert
|
-16.1
|
-3.2
| |
Unterschied (kg) gegenüber Placebo1 [95 %-KI]
|
-12.9 [-16.1;-9.8]
|
-
| |
Patienten (%) mit Gewichtsverlust ≥5 %3
|
74.7*
|
37.3
| |
Taillenumfang (cm)
| |
Ausgangswert
|
115.8
|
115.7
| |
Änderung gegenüber Ausgangswert1
|
-14.4
|
-5.2
| |
Unterschied gegenüber Placebo1 [95 %-KI]
|
-9.2 [-12.2; -6.2]*
|
-
| |
* p < 0.0001 (unbereinigt 2-seitig) für Überlegenheit.
1 Geschätzt anhand eines ANCOVA-Modells mit multipler Imputation auf Grundlage aller Daten, unabhängig vom Abbruch der randomisierten Behandlung, Einleitung einer anderen medikamentösen Adipositastherapie oder bariatrischer Chirurgie.
2 Während der Studie wurde die randomisierte Behandlung von 13.2 % der Patienten, die auf Semaglutide und 27.0 % der Patienten, die auf Placebo randomisiert worden waren, dauerhaft abgebrochen. Unter der Annahme, dass alle randomisierten Patienten die Behandlung beibehielten und keine zusätzlichen Adipositastherapien erhielten, betrugen die geschätzten Änderungen des Körpergewichts von der Randomisierung bis Woche 68 -16.7 % für Semaglutide und -0.6 % für Placebo, auf der Grundlage eines gemischten Modells für wiederholte Messungen, das alle Beobachtungen bis zum ersten Absetzen einschliesst.
3 Geschätzt anhand eines binären Regressionsmodells auf der Grundlage desselben Imputationsverfahrens wie in der Primäranalyse.
|
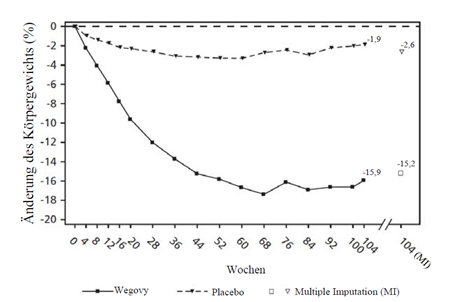
Beobachtete Werte für Patienten, die jeden geplanten Besuch absolvierten, und Schätzungen mit multiplen Imputationen (MI) für Abbrecher mit Abschlussuntersuchung («retrieved dropouts»).
Abbildung 6 STEP 5 - Mittlere Veränderung des Körpergewichts (%) zwischen Woche 0 und Woche 104
STEP 8: Semaglutide gegenüber Liraglutide
In einer 68-wöchigen, randomisierten, unverblindeten, paarweise placebokontrollierten Studie wurden 338 Patienten mit Adipositas (BMI ≥30 kg/m2) oder mit Übergewicht (BMI ≥27 bis < 30 kg/m2) und mindestens einer gewichtsbedingten Begleiterkrankung auf Wegovy einmal wöchentlich, Liraglutide 3 mg einmal täglich oder Placebo randomisiert. Wegovy einmal wöchentlich und Liraglutide 3 mg waren unverblindet, aber jede aktive Behandlungsgruppe war doppelt verblindet gegenüber Placebo, das mit der gleichen Dosierungsfrequenz verabreicht wurde. Alle Patienten erhielten für die Dauer der Studie eine kalorienreduzierte Diät mit erhöhter körperlicher Aktivität.
Bei Studienbeginn hatten die Patienten einen durchschnittlichen BMI von 37.5 kg/m2 und ein durchschnittliches Körpergewicht von 104.5 kg.
Das durchschnittliche Körpergewicht nahm unter Wegovy ab Studienbeginn bis zur Woche 68 ab. Mit Liraglutide war die Reduktion des mittleren Körpergewichts geringer (siehe Tabelle 9). 37.4 % der mit Semaglutide behandelten Patienten verloren ≥20 %, verglichen mit 7.0 % unter Liraglutide.
Tabelle 9: STEP 8: Ergebnisse einer 68-wöchigen Studie zum Vergleich von Semaglutide und
Liraglutide
|
|
Wegovy
|
Liraglutide 3 mg
| |
Full analysis set (N)
|
126
|
127
| |
Körpergewicht
| |
Ausgangswert (kg)
|
102.5
|
103.7
| |
Änderung (%) gegenüber Ausgangswert1,2,3
|
-15.8
|
-6.4
| |
Unterschied (%) zu Liraglutide1 [95 %-KI]
|
-9.4 [-12.0;-6.8]*
|
-
| |
Änderung (kg) gegenüber Ausgangswert
|
-15.3
|
-6.8
| |
Unterschied (kg) zu Liraglutide1 [95 %-KI]
|
-8.5 [-11.2;-5.7]
|
-
| |
* p < 0.005 (unbereinigt 2-seitig) für Überlegenheit.
1 Geschätzt anhand eines ANCOVA-Modells mit multipler Imputation auf Grundlage aller Daten, unabhängig vom Abbruch der randomisierten Behandlung, Einleitung einer anderen medikamentösen Adipositastherapie oder bariatrischer Chirurgie.
2 Während der Studie wurde die randomisierte Behandlung von 13.5 % der Patienten, die auf Semaglutide und 27.6 % der Patienten, die auf Liraglutide randomisiert worden waren, dauerhaft abgebrochen. Unter der Annahme, dass alle randomisierten Patienten die Behandlung beibehielten und keine zusätzlichen Adipositastherapien erhielten, betrugen die geschätzten Änderungen des Körpergewichts von der Randomisierung bis Woche 68 -16.7 % für Semaglutide und -6.7 % für Liraglutide, auf der Grundlage eines gemischten Modells für wiederholte Messungen, das alle Beobachtungen bis zum ersten Absetzen einschliesst.
3 Die Veränderung (%) gegenüber dem Ausgangswert in den gepoolten Placebogruppen betrug -1.9 %.
|
STEP 9: Gewichtsmanagement bei Patienten mit Knie-Osteoarthrose – Semaglutide
In einer 68-wöchigen doppelblinden Studie wurden 407 Patienten (Durschnittsalter 56 Jahre) mit Adipositas und moderater Knie-Osteoarthrose (OA) in einem oder beiden Knien randomisiert mit Wegovy (n=271) oder Placebo (n=136) behandelt. Zu Studienbeginn betrug das durchschnittliche Körpergewicht 108.6 kg bei einem durchschnittlichen BMI von 40.3 kg/m². Im Einklang mit den Ergebnissen früherer Studien zeigte sich ein klinisch bedeutsamer Unterschied in der Gewichtsabnahme zwischen der Behandlungsgruppen zugunsten von Wegovy (Unterschied gegenüber Placebo [95 % KI]: -10.5 % [-12.3, -8.6]). Gleichzeitig verbesserte sich der Western Ontario and McMaster Universities Osteoarthritis 3.1 Index (WOMAC), ein Mass für Knie-OA-bedingten Schmerzen, signifikant stärker in der Wegovy Gruppe im Vergleich zur Placebo Gruppe (Unterschied [95 % KI]: -14.1 [-20.0, -8.3]), wobei 59 % der Patienten in der Wegovy Gruppe eine klinisch bedeutsame Verbesserung erreichten im Vergleich zu 35 % in der Placebo Gruppe.
Wirkung auf den Körperfettanteil
In einer Unterstudie von STEP 1 (N = 140) wurde mithilfe der Dual-Röntgen-Absorptiometrie (DEXA) gezeigt, dass unter der Behandlung mit Wegovy die Körperfettmasse stärker abgebaut wurde als die fettfreie Körpermasse, was einer Verbesserung des Körperfettanteils nach 68 Wochen im Vergleich mit Placebo entspricht. Ausserdem ging mit dieser Reduktion der gesamten Fettmasse auch eine Reduktion des viszeralen Fettes einher.
Kardiovaskuläre Ergebnisse
SELECT: Kardiovaskuläre Ergebnisstudie bei Patienten mit Übergewicht oder Adipositas
SELECT war eine randomisierte, doppelblinde, placebokontrollierte, ereignisgesteuerte Studie, an der 17'604 Patienten mit etablierter kardiovaskulärer Erkrankung (67.6 % mit früherem Myokardinfarkt, 17.8 % mit früherem Schlaganfall und 4.4 % mit peripherer arterieller Erkrankung (PAD); 8.2 % mit 2 oder mehr früheren kardiovaskulären Ereignissen) und einem BMI ≥27 kg/m2 teilnahmen. Patienten mit einer Vorgeschichte von Typ-1- und Typ-2-Diabetes wurden ausgeschlossen. Die mittlere Verweildauer in der Studie betrug 41.8 Monate. Die Studienpopulation bestand aus 27.7 % Frauen und 72.3 % Männern mit einem Durchschnittsalter von 61.6 Jahren, darunter 38.2 % Patienten ≥65 Jahre (n = 6728) und 7.8 % Patienten ≥75 Jahre (n = 1366). Der durchschnittliche BMI betrug 33.3 kg/m2 und das durchschnittliche Körpergewicht betrug 96.7 kg.
Die Patienten wurden entweder auf Semaglutide mit einer anzustrebenden Maximaldosis von 2.4 mg pro Woche (n=8803) oder Placebo (n=8801) als Zusatz zu einer Standardtherapie für die kardiovaskuläre Vorerkrankung randomisiert. Zu Beginn der Studie erhielten 92.0 % der Patienten kardiovaskuläre Medikamente (70.2 % Betablocker, 45.0 % Angiotensin-Converting-Enzym (ACE)-Inhibitoren, 29.5 % Angiotensin-Rezeptor-Blocker und 26.9 % Calciumkanalblocker), 90.1 % Lipidsenker (hauptsächlich Statine 87.6 %) und 86.2 % Antithrombozytenmittel.
Zu Beginn hatten die meisten Patienten kardiovaskuläre Begleiterkrankungen, darunter 66.4 % mit HbA1c ≥5.7 % und <6.5% als Hinweis auf Prädiabetes, 24.3 % mit chronischer Herzinsuffizienz, 81.8 % mit Hypertonie, 46.8 % mit Entzündungen (hsCRP ≥2 mg/L) sowie Patienten mit leichter (48.7 %), mittelschwerer (10.4 %) oder schwerer (0.4 %) Niereninsuffizienz.
Der primäre Endpunkt war die Zeit von der Randomisierung bis zum ersten Auftreten von schwerwiegenden kardiovaskulären Ereignissen (MACE), definiert als zusammengesetzter Endpunkt bestehend aus kardiovaskulärem Tod, nicht-tödlichem Myokardinfarkt oder nicht-tödlichem Schlaganfall. Die Überlegenheit von Semaglutide 2.4 mg gegenüber Placebo wurde mit einem Hazard Ratio von 0.80 [0.72; 0.90] [95 % KI] bestätigt, was einer relativen Risikoreduktion von 20 % entspricht (siehe Abbildung 7 & 8). Die drei Bestandteile trugen zur Reduktion von MACE bei (Tabelle 10). Die kardiovaskuläre Risikoreduktion erschien weitgehend unabhängig vom Gewichtsverlust.
Die Behandlungswirkung bezüglich MACE Risikoreduktion war in den wesentlichen Subgruppen, die durch Alter, Geschlecht, Rasse, Ethnizität, Typ früherer kardiovaskulärer Erkrankung, BMI, Normoglykämie/Prädiabetes und Nierenfunktion definiert wurden, vergleichbar.
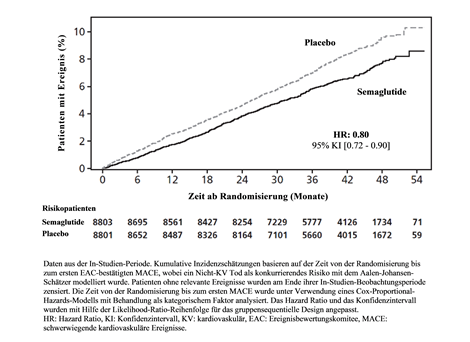
Abbildung 7 Kumulative Inzidenzfunktionsdarstellung - Zeit von der Randomisierung bis zum ersten MACE
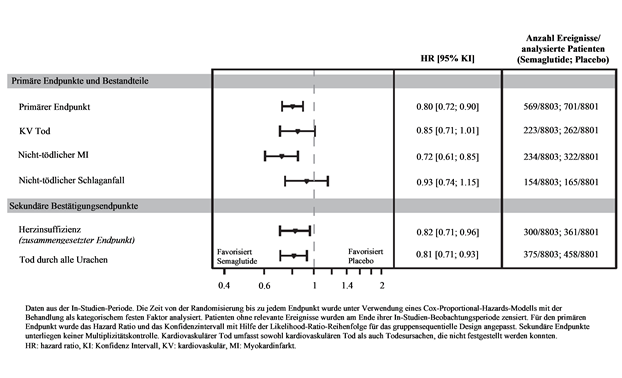
Abbildung 8 Forest Plot der Zeit von der Randomisierung bis zum ersten MACE, MACE Bestandteile und sekundäre Bestätigungsendpunkte
Tabelle 10: Zeit von der Randomisierung bis zum ersten MACE und deren Bestandteile, sekundäre Bestätigungsendpunkt KV Tod.
|
|
HR [95 % KI]
|
p-Wert
(zweiseitig)
|
Anzahl Ereignisse/ analysierte Patienten (Sema 2.4 mg)
|
Anzahl Ereignisse/ analysierte Patienten (Placebo)
| |
Primärer zusammen-gesetzter Endpunkt (MACE)
|
0.80 [0.72; 0.90]
|
<0.0001
|
569/8803
|
701/8801
| |
KV Tod
|
0.85 [0.71; 1.01]
|
0.0653
|
223/8803
|
262/8801
| |
Nicht-tödlicher MI
|
0.72 [0.61; 0.85]
|
|
234/8803
|
322/8801
| |
Nicht-tödlicher Schlaganfall
|
0.93 [0.74; 1.15]
|
|
154/8803
|
165/8801
| |
Sekundäre Bestätigungsendpunkte
| |
KV Tod1
|
0.85 [0.71; 1.01]
|
0.0653
|
223/8803
|
262/8801
| |
Herzinsuffizienz (zusammengesetzter Endpunkt)1
|
0.82 [0.71; 0.96]
|
|
300/8803
|
361/8801
| |
Tod durch alle Ursachen1
|
0.81 [0.71; 0.93]
|
|
375/8803
|
458/8801
| |
Subgruppen:
| |
KV-Erkrankung
| |
KV-Erkrankung: Nur Myokardinfarkt
|
0.78 [0.68; 0.90]
|
0.4803
|
362/5962
|
455/5944
| |
KV-Erkrankung: Nur Hirnschlag
|
0.98 [0.75; 1.27]
|
|
109/578
|
109/1556
| |
KV-Erkrankung: Nur PAD
|
0.74 [0.36; 1.48]
|
|
13/376
|
19/401
| |
KV-Erkrankung: 2 oder mehr KV-Erkrankungen
|
0.75 [0.55; 1.00]
|
|
76/718
|
100/719
| |
Geschlecht
| |
Geschlecht: Weiblich
|
0.84 [0.66; 1.07]
|
0.6324
|
126/2448
|
147/2424
| |
Geschlecht: Männlich
|
0.79 [0.70; 0.90]
|
|
443/6355
|
554/6377
|
Hinweis: Die Zeit von der Randomisierung bis zu jedem Endpunkt wurde unter Verwendung eines Cox-Proportional-Hazards-Modells mit der Behandlung als festen kategorischen Faktor analysiert. Probanden ohne Ereignis von Interesse wurden am Ende ihres In-Studien-Zeitraums zensiert. Für den primären Endpunkt wurden das HR, das Konfidenzintervall und der p-Wert unter Verwendung der Likelihood-Ratio-Reihenfolge für die gruppensequenziellen Designs angepasst. Subgruppenanalysen für den primären Endpunkt wurden in einem Cox-Proportional-Hazards-Modell mit Interaktion zwischen Behandlungsgruppe und der relevanten Subgruppe als festen Faktor analysiert. Kardiovaskulärer Tod umfasst sowohl kardiovaskulären Tod als auch Todesursachen, die nicht bestimmt werden konnten.
1Sekundärer Bestätigungsendpunkt. Nicht statistisch signifikant gemäss der vorab festgelegten Testhierarchie.
HR: hazard ratio, KI: Konfidenzinterval, p-Wert: Zweiseitiger p-Wert für den Test auf keinen Unterschied. Für die Subgruppenanalysen bezieht sich der p-Wert auf den Test auf keinen Interaktionseffekt. KV: kardiovaskulär, MACE: schwerwiegende kardiovaskuläre Ereignisse, MI: Myokardinfarkt.
Tabelle 11: SELECT – Bewertung der kardiovaskulären Risikofaktoren in Woche 104
|
|
Semaglutide
|
Placebo
| |
Full analysis set (N)
|
8803
|
8801
| |
Kardiometabolische Faktoren:
| |
Systolischer Blutdruck (mmHg)
| |
Ausgangswert
Mittelwert (SD)
|
131.0 (15.6)
|
130.9 (15.3)
| |
Änderung gegenüber Ausgangswert1
|
-3.82
|
-0.51
| |
Unterschied gegenüber Placebo [95 % KI]1
|
-3.1 [-3.75; -2.88]
|
-
| |
Diastolischer Blutdruck (mmHg)
| |
Ausgangswert
Mittelwert (SD)
|
79.4 (10.0)
|
79.2 (9.9)
| |
Änderung gegenüber Ausgangswert1
|
-1.02
|
-0.47
| |
Unterschied gegenüber Placebo [95 % KI]1
|
-0.55 [-0.83; -0.27]
|
-
| |
Herzfrequenz
| |
Ausgangswert
Mittelwert (SD)
|
68.9 (10.6)
|
68.6 (10.7)
| |
Änderung gegenüber Ausgangswert1
|
3.79
|
0.69
| |
Unterschied gegenüber Placebo [95 % KI]1
|
3.10 [2.80; 3.39]
|
-
| |
Lipide:
| |
Total Cholesterol
| |
Ausgangswert (mmol/L)
Geometrisches Mittel (CV)
|
4.03 (25.82)
|
4.04 (25.41)
| |
Änderung (%) gegenüber Ausgangswert1
|
-4.63
|
-1.92
| |
Relativer Unterschied (%) gegenüber Placebo1
|
-2.77 [-3.37; -2.16]
|
-
| |
LDL Cholesterol
| |
Ausgangswert (mmol/L)
Geometrisches Mittel (CV)
|
2.03 (43.70)
|
2.03 (43.56)
| |
Änderung (%) gegenüber Ausgangswert1
|
-5.25
|
-3.14
| |
Relativer Unterschied (%) gegenüber Placebo [95 % KI]1
|
-2.18 [-3.22; -1.12]
|
-
| |
HDL Cholesterol
| |
Ausgangswert (mmol/L)
Geometrisches Mittel (CV)
|
1.14 (25.52)
|
1.15 (25.02)
| |
Änderung (%) gegenüber Ausgangswert1
|
4.86
|
0.59
| |
Relativer Unterschied (%) gegenüber Placebo [95 % KI]1
|
4.24 [3.70; 4.79]
|
-
| |
Triglyzeride
| |
Ausgangswert (mmol/L)
Geometrisches Mittel (CV)
|
1.56 (51.75)
|
1.57 (50.84)
| |
Änderung (%) gegenüber Ausgangswert1
|
-18.34
|
-3.20
| |
Relativer Unterschied (%) gegenüber Placebo [95 % KI]1
|
-15.64 [-16.7; -14.6]
|
-
| |
Gewichtsabhängige KV-Risikofaktoren:
| |
Körpergewicht (%)
| |
Ausgangswert (kg)
Mittelwert (SD)
|
96.53 (17.52)
|
96.82 (17.80)
| |
Änderung gegenüber Ausgangswert1
|
-9.39
|
-0.88
| |
Unterschied gegenüber Placebo [95 % KI]1
|
-8.51 [-8.75; -8.27]
|
-
|
1Die Antworten wurden unter Verwendung einer ANCOVA analysiert, wobei die Behandlung als fester Faktor und der Ausgangswert als Kovariate betrachtet wurden. Vor der Analyse wurden fehlende Daten mehrfach imputiert. Das Imputationsmodell (lineare Regression) wurde für jeden Behandlungsarm separat durchgeführt und umfasste den Ausgangswert als Kovariate sowie alle Teilnehmer mit Messungen unabhängig von ihrem Behandlungsstatus in Woche 104. Das gefittete Modell wurde verwendet, um Werte für Teilnehmer ohne Messungen in Woche 104 zu imputieren. Die mittleren Schätzungen wurden entsprechend der beobachteten Ausgangsverteilung angepasst.
STEP TEENS: Gewichtsregulierung bei jugendlichen Patienten
In einer 68-wöchigen, doppelblinden Studie wurden 201 Jugendliche während der Pubertät im Alter von 12 bis < 18 Jahren mit Adipositas (n=200) oder Übergewicht und mit mindestens einer gewichtsbedingten Begleiterkrankung (n=1) 2:1 auf Semaglutide oder Placebo randomisiert. Alle Patienten erhielten für die Dauer der Studie eine kalorienreduzierte Diät mit erhöhter körperlicher Aktivität.
Am Ende der Behandlung (Woche 68) war die Verbesserung des BMI mit Semaglutide überlegen und klinisch bedeutsam im Vergleich zu Placebo (siehe Tabelle 12 und Abbildung 9). Darüber hinaus erreichte ein grösserer Anteil von Patienten einen Gewichtsverlust von ≥5 %, 10 % und ≥15 % mit Semaglutide im Vergleich zu Placebo (siehe Tabelle 12).
Tabelle 12: STEP TEENS: Ergebnisse in Woche 68
|
|
Wegovy
|
Placebo
| |
Full analysis set (N)
|
134
|
67
| |
BMI
| |
Ausgangswert (BMI)
|
37.7
|
35.7
| |
Änderung (%) gegenüber Ausgangswert BMI1, 3
|
-16.1
|
0.6
| |
Unterschied (%) zu Placebo1 [95 % KI]
|
-16.7 [-20.3; -13.2]*
|
-
| |
Ausgangswert (BMI SDS)
|
3.4
|
3.1
| |
Änderung des Ausgangswerts in BMI SDS1
|
-1.1
|
-0.1
| |
Unterschied zu Placebo1 [95 % KI]
|
-1.0 [-1.3; -0.8]
|
-
| |
Körpergewicht
| |
Ausgangswert (kg)
|
109.9
|
102.6
| |
Änderung (%) gegenüber Ausgangswert1
|
-14.7
|
2.8
| |
Unterschied (%) zu Placebo1 [95 % KI]
|
-17.4 [-21.1; -13.8]
|
-
| |
Änderung (kg) gegenüber Ausgangswert1
|
-15.3
|
2.4
| |
Unterschied (kg) zu Placebo1 [95 % KI]
|
-17.7 [-21.8; -13.7]
|
-
| |
Patienten (%) mit Gewichtsverlust ≥5 %4
|
72.5*
|
17.7
| |
Patienten (%) mit Gewichtsverlust ≥10 %4
|
61.8
|
8.1
| |
Patienten (%) mit Gewichtsverlust ≥15 %4
|
53.4
|
4.8
| |
Patienten (%) mit Gewichtsverlust ≥20 %4
|
37.4
|
3.2
| |
Taillenumfang (cm)
| |
Ausgangswert
|
111.9
|
107.3
| |
Änderung (%) gegenüber Ausgangswert1
|
-12.7
|
-0.6
| |
Unterschied zu Placebo1 [95 % KI]
|
-12.1 [-15.6; -8.7]
|
-
|
* p < 0.0001 (unbereinigt 2-seitig) für Überlegenheit.
1 Geschätzt anhand eines ANCOVA-Modells mit multipler Imputation auf Grundlage aller Daten, unabhängig vom Abbruch der randomisierten Behandlung, Einleitung einer anderen medikamentösen Adipositastherapie oder bariatrischer Chirurgie.
2 Die Zahlen beziehen sich auf Patienten ohne Diabetes Typ 2.
3 Während der Studie wurde die randomisierte Behandlung von 10.4 % der Patienten, die auf 2.4 mg Semaglutide und 10.4 % der Patienten, die auf Placebo randomisiert worden waren, dauerhaft abgebrochen. Unter der Annahme, dass alle randomisierten Patienten die Behandlung beibehielten und keine zusätzlichen Adipositastherapien erhielten, betrugen die geschätzten Änderungen des BMI von der Randomisierung bis Woche 68 -17.9 % für 2.4 mg Semaglutide und -0.6 % für Placebo, auf der Grundlage eines gemischten Modells für wiederholte Messungen, das alle Beobachtungen bis zum ersten Absetzen einschliesst.
4 Geschätzt anhand eines logistischen Regressionsmodells auf der Grundlage desselben Imputationsverfahrens wie in der Primäranalyse.
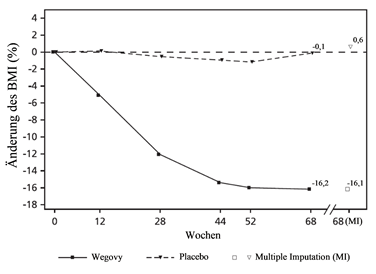
Beobachtete Werte für Patienten, die jeden Kontrolltermin wahrgenommen haben und Schätzungen mit multiplen Imputationen (MI) für erfasste Studienabbrecher
Abbildung 9 STEP TEENS - Mittlere Änderung des BMI (%) vom Ausgangswert bis Woche 68
PharmakokinetikAbsorption
Die durchschnittliche Konzentration von Semaglutide im Steady State nach der s. c. Verabreichung von 2.4 mg Semaglutide betrug, basierend auf einer populationspharmakokinetischen Analyse, bei Patienten mit Übergewicht (BMI ≥27 kg/m2 bis < 30 kg/m2) oder Adipositas (BMI ≥30 kg/m2) etwa 75 nmol/l. Die maximale Konzentration wurde etwa 24 Stunden nach der Injektion erreicht. Die Exposition gegenüber Semaglutide im Steady State stieg bei Dosen bis 2.4 mg einmal wöchentlich dosisproportional an. Bei der s. c. Verabreichung von Semaglutide in das Abdomen, den Oberschenkel oder Oberarm wurde jeweils eine ähnliche Exposition erzielt. Die absolute Bioverfügbarkeit von Semaglutide betrug 89 %.
Distribution
Das mittlere Verteilungsvolumen von Semaglutide nach s. c. Verabreichung an Patienten mit Übergewicht oder Adipositas betrug ungefähr 12.4 l. Semaglutide wird stark an Plasmaalbumin gebunden (> 99 %).
Metabolismus
Semaglutide wird durch proteolytische Spaltung des Peptid-Backbones und sequentielle beta-Oxidation der Fettsäure-Seitenkette weitgehend verstoffwechselt. Der am häufigsten vorkommende Metabolit machte weniger als 8 % der gesamten Exposition aus und wurde als Semaglutide identifiziert, das von den ersten 13 Aminosäuren des N-Terminus abgeschnitten ist.
Elimination
Die primären Ausscheidungswege für Materialen, die mit Semaglutide im Zusammenhang stehen sind Urin und Fäzes. Etwa 3 % der absorbierten Dosis wurden als intaktes Semaglutide über den Urin ausgeschieden.
Die Clearance von Semaglutide bei Patienten mit Übergewicht (BMI ≥27 kg/m2 bis < 30 kg/m2) oder Adipositas (BMI ≥30 kg/m2) betrug etwa 0.05 l/h. Bei einer Eliminationshalbwertszeit von ungefähr 1 Woche wird Semaglutide noch etwa 7 Wochen nach der letzten Dosis von 2.4 mg in der Blutbahn vorhanden sein.
Kinetik spezieller Patientengruppen
Basierend auf einer populationspharmakokinetischen Analyse haben Alter, Geschlecht, Rasse und ethnische Zugehörigkeit sowie Nierenfunktionsstörungen keinen klinisch bedeutsamen Einfluss auf die Pharmakokinetik von Semaglutide.
Körpergewicht
Das Körpergewicht wirkte sich auf die Semaglutide-Exposition aus. Ein höheres Körpergewicht war mit einer geringeren Exposition assoziiert. Die Semaglutide-Dosis von wöchentlich 2.4 mg führte in einem Bereich von 54.4–245.6 kg Körpergewicht, der in den klinischen Studien auf die erzielte Exposition untersucht wurde, zu einer adäquaten systemischen Exposition.
Leberfunktionsstörungen
Eine eingeschränkte Leberfunktion hatte keinen Einfluss auf die Semaglutide-Exposition. Die Pharmakokinetik von Semaglutide wurde in einer Studie mit einer einzelnen Dosis von 0.5 mg Semaglutide bei Patienten mit verschieden stark eingeschränkter Leberfunktion (leicht, mittelschwer, schwer) im Vergleich zu Patienten mit normaler Leberfunktion untersucht.
Nierenfunktionsstörungen
Eine Einschränkung der Nierenfunktion beeinflusste die Pharmakokinetik von Semaglutide nicht in klinisch relevantem Masse. Dies wurde anhand einer einzelnen Dosis von 0.5 mg Semaglutide bei Patienten mit verschieden stark eingeschränkter Nierenfunktion (leicht, mittelschwer, schwer oder Dialysepatienten) im Vergleich zu Patienten mit normaler Nierenfunktion nachgewiesen. Basierend auf einer populationspharmakokinetischen Analyse zeigte sich dies auch anhand der Daten aus Phase-3a-Studien von Patienten mit Übergewicht (BMI ≥27 kg/m2 bis < 30 kg/m2) oder Adipositas (BMI ≥30 kg/m2) und einer leichten bis mittelschweren Einschränkung der Nierenfunktion.
Ältere Patienten
Das Alter hatte gemäss den Daten aus den Phase-3-Studien mit Patienten im Alter von 18–86 Jahren keinen Einfluss auf die Pharmakokinetik von Semaglutide.
Kinder und Jugendliche
Die pharmakokinetischen Eigenschaften für Semaglutide wurden in einer klinischen Studie bei jugendlichen Patienten mit Adipositas oder Übergewicht und mindestens einer gewichtsbedingten Begleiterkrankung im Alter von 12 bis < 18 Jahren (124 Patienten, Körpergewicht 61.6 - 211.9 kg) bewertet. Die Semaglutide-Exposition bei Jugendlichen war ähnlich der bei Erwachsenen mit Adipositas oder Übergewicht.
Die Sicherheit und Wirksamkeit von Wegovy bei Kindern im Alter von unter 12 Jahren wurden nicht untersucht.
Genetische Polymorphismen
Geschlecht und ethnische Zugehörigkeit hatten keine Auswirkung auf die Pharmakokinetik von Semaglutide.
Präklinische DatenBasierend auf den konventionellen Studien zur Sicherheitspharmakologie, Toxizität bei wiederholter Gabe oder Genotoxizität lassen die präklinischen Daten keine besonderen Gefahren für den Menschen erkennen.
Karzinogenität
Bei Nagetieren beobachtete, nichtletale C-Zelltumoren der Schilddrüse sind ein Klasseneffekt von GLP-1-Rezeptoragonisten. In 2-jährigen Karzinogenitätsstudien bei Ratten und Mäusen verursachte Semaglutide bei klinisch relevanten Expositionen C-Zell-Tumoren der Schilddrüse. Die C-Zell-Tumoren bei Nagetieren werden durch einen nicht genotoxischen, spezifisch durch den GLP-1-Rezeptor vermittelten Mechanismus verursacht, für den Nager besonders empfänglich sind. Die Relevanz für den Menschen wird als gering eingestuft, kann jedoch nicht komplett ausgeschlossen werden.
Reproduktionstoxizität
In Fertilitätsstudien an Ratten beeinträchtigte Semaglutide das Deckverhalten oder die Fertilität männlicher Ratten nicht. Bei weiblichen Ratten wurde bei Dosen, die mit einem mütterlichen Gewichtsverlust einhergingen, eine Verlängerung des Östrus und eine geringe Abnahme der Anzahl der Corpora lutea (Ovulationen) beobachtet.
Bei embryofetalen Entwicklungsstudien an Ratten verursachte Semaglutide Embryotoxizität bei Expositionen, die unter den klinisch relevanten Werten lagen. Semaglutide verursachte deutliche Reduktionen des mütterlichen Körpergewichts und Verminderungen des Überlebens und Wachstums von Embryonen. Bei Föten wurden wesentliche skelettale und viszerale Missbildungen beobachtet, darunter Auswirkungen auf lange Knochen, Rippen, Wirbel, Schwanz, Blutgefässe und Hirnventrikel. Untersuchungen hinsichtlich des Mechanismus deuten darauf hin, dass an der Embryotoxizität eine durch den GLP-1-Rezeptor vermittelte Beeinträchtigung der Nährstoffversorgung des Embryos über den Dottersack der Ratte beteiligt ist. Aufgrund der anatomischen und funktionellen Unterschiede des Dottersacks zwischen den Spezies und aufgrund der fehlenden Expression des GLP-1-Rezeptors im Dottersack nichtmenschlicher Primaten gilt es als unwahrscheinlich, dass dieser Mechanismus für den Menschen relevant ist. Jedoch kann eine direkte Auswirkung von Semaglutide auf den Fötus nicht ausgeschlossen werden.
In Entwicklungstoxizitätsstudien mit Kaninchen und Javaneraffen wurden bei klinisch relevanten Expositionen vermehrt Aborte und eine leicht erhöhte Inzidenz fötaler Anomalien beobachtet. Die Ergebnisse fallen mit deutlichem mütterlichem Gewichtsverlust von bis zu 16 % zusammen. Es ist nicht bekannt, ob diese Effekte mit der verminderten mütterlichen Futteraufnahme als direkte Wirkung von GLP-1 zusammenhängen.
Das postnatale Wachstum und die postnatale Entwicklung wurden an Javaneraffen beurteilt. Die Neugeborenen waren bei der Geburt geringfügig kleiner, holten aber während der Stillzeit auf.
Bei jugendlichen männlichen und weiblichen Ratten verursachte Semaglutide eine verzögerte Geschlechtsreife. Diese Verzögerungen hatten keine Auswirkungen auf die Fertilität und reproduktive Kapazität beider Geschlechter oder auf die Fähigkeit der Weibchen, eine Schwangerschaft aufrechtzuerhalten.
Sonstige HinweiseInkompatibilitäten
Da keine Kompatibilitätsstudien durchgeführt wurden, darf das Arzneimittel nicht mit anderen Arzneimitteln gemischt werden.
Haltbarkeit
Das Arzneimittel darf nur bis zu dem auf der Packung mit «EXP» bezeichneten Datum verwendet werden.
Haltbarkeit nach Anbruch
Wegovy Multi FixDose kann während 42 Tagen (6 Wochen) unter 30°C oder im Kühlschrank (2-8°C) aufbewahrt werden.
Besondere Lagerungshinweise
Im Kühlschrank lagern (2-8°C). Vom Kühlelement fernhalten.
Wegovy nicht einfrieren und nicht mehr verwenden, wenn der Inhalt gefroren war.
Wegovy sollte vor übermässiger Hitze geschützt werden.
Nach Gebrauch: Entsorgen Sie Wegovy nach Gebrauch.
Ausser Reichweite von Kindern aufbewahren.
Wegovy FixDose: Den Pen im Original-Umkarton aufbewahren, um den Inhalt vor Licht zu schützen. Wegovy FixDose kann ungekühlt für bis zu 28 Tage bei einer Temperatur unter 30°C gelagert werden.
Wegovy Multi FixDose: Verschlusskappe auf dem Pen belassen, wenn er nicht benutzt wird, um den Inhalt vor Licht zu schützen.
Hinweise für die Handhabung
Wegovy FixDose: Der Pen enthält 1 Dosis.
Wegovy Multi FixDose: Der Pen enthält 4 Dosen. Die Injektionsnadel nach jeder Injektion entfernen und entsorgen. Den Pen ohne Nadel aufbewahren.
Wegovy darf nicht verwendet werden, wenn es nicht klar und farblos aussieht.
Der Pen darf nicht mehr verwendet werden, wenn der Inhalt gefroren war.
Nicht verwendetes Arzneimittel oder Abfallmaterial ist entsprechend den nationalen Anforderungen zu entsorgen.
Zulassungsnummer68238, 68798 (Swissmedic)
PackungenWegovy FixDose
Es gibt fünf Stärken des vorgefüllten Pens für Wegovy FixDose:
Wegovy FixDose 0.25 mg Injektionslösung im Fertigpen:
4 Fertigpens (B)
Wegovy FixDose 0.5 mg Injektionslösung im Fertigpen:
4 Fertigpens (B)
Wegovy FixDose 1 mg Injektionslösung im Fertigpen:
4 Fertigpens (B)
Wegovy FixDose 1.7 mg Injektionslösung im Fertigpen:
4 Fertigpens (B)
Wegovy FixDose 2.4 mg Injektionslösung im Fertigpen:
4 Fertigpens (B)
Wegovy Multi FixDose
Es gibt fünf Stärken des vorgefüllten Pens für Wegovy Multi FixDose:
Wegovy Multi FixDose 0.25 mg Injektionslösung im Fertigpen:
1 Fertigpen und 4 NovoFine Plus Einwegnadeln (B)
Wegovy Multi FixDose 0.5 mg Injektionslösung im Fertigpen:
1 Fertigpen und 4 NovoFine Plus Einwegnadeln (B)
Wegovy Multi FixDose 1 mg Injektionslösung im Fertigpen:
1 Fertigpen und 4 NovoFine Plus Einwegnadeln (B)
Wegovy Multi FixDose 1.7 mg Injektionslösung im Fertigpen:
1 Fertigpen und 4 NovoFine Plus Einwegnadeln (B)
Wegovy Multi FixDose 2.4 mg Injektionslösung im Fertigpen:
1 Fertigpen und 4 NovoFine Plus Einwegnadeln (B)
ZulassungsinhaberinNovo Nordisk Pharma AG, Kloten
Domizil: Zürich
Stand der InformationJanuar 2025
|