ZusammensetzungWirkstoffe
Brexpiprazol.
Hilfsstoffe
Tablettenkern:
0,5 mg: Laktose-Monohydrat 47,9 mg, Maisstärke, mikrokristalline Cellulose, Hydroxypropylcellulose, niedrig substituierte Hydroxypropylcellulose, Magnesiumstearat.
1 mg: Laktose-Monohydrat 47,4 mg, Maisstärke, mikrokristalline Cellulose, Hydroxypropylcellulose, niedrig substituierte Hydroxypropylcellulose, Magnesiumstearat.
2 mg: Laktose-Monohydrat 46,4 mg, Maisstärke, mikrokristalline Cellulose, Hydroxypropylcellulose, niedrig substituierte Hydroxypropylcellulose, Magnesiumstearat.
3 mg: Laktose-Monohydrat 45,5 mg, Maisstärke, mikrokristalline Cellulose, Hydroxypropylcellulose, niedrig substituierte Hydroxypropylcellulose, Magnesiumstearat.
4 mg: Laktose-Monohydrat 44,4 mg, Maisstärke, mikrokristalline Cellulose, Hydroxypropylcellulose, niedrig substituierte Hydroxypropylcellulose, Magnesiumstearat.
Tablettenüberzug:
0,5 mg: Hypromellose, Talkum, Farbstoff: Titandioxid (E171), gelbes und rotes Eisenoxid (E172).
1 mg: Hypromellose, Talkum, Farbstoff: Titandioxid (E171), gelbes Eisenoxid (E172).
2 mg: Hypromellose, Talkum, Farbstoff: Titandioxid (E171), gelbes und schwarzes Eisenoxid (E172).
3 mg: Hypromellose, Talkum, Farbstoff: Titandioxid (E171), rotes und schwarzes Eisenoxid (E172).
4 mg: Hypromellose, Talkum, Titandioxid (E171).
Darreichungsform und Wirkstoffmenge pro Einheit0,5 mg Filmtablette: 0,5 mg Brexpiprazol.
1 mg Filmtablette: 1 mg Brexpiprazol.
2 mg Filmtablette: 2 mg Brexpiprazol.
3 mg Filmtablette: 3 mg Brexpiprazol.
4 mg Filmtablette: 4 mg Brexpiprazol.
Aussehen der Filmtabletten
Alle Tabletten sind rund, flach-konvex, mit abgeschrägter Kante.
0,5 mg: hellorange, mit Prägung «BRX» und «0.5» auf einer Seite.
1 mg: hellgelb, mit Prägung «BRX» und «1» auf einer Seite.
2 mg: hellgrün, mit Prägung «BRX» und «2» auf einer Seite.
3 mg: hellviolett, mit Prägung «BRX» und «3» auf einer Seite.
4 mg: weiss, mit Prägung «BRX» und «4» auf einer Seite.
Indikationen/AnwendungsmöglichkeitenREXULTI ist indiziert zur Behandlung der Schizophrenie bei erwachsenen Patienten und Jugendlichen ab 13 Jahren.
REXULTI ist indiziert zur Behandlung von Agitiertheit bei Alzheimer-Demenz (AAD) bei erwachsenen Patienten, die auf nicht-pharmakologische Interventionen nicht ansprechen.
Dosierung/AnwendungSchizophrenie
Erwachsene
Die empfohlene Anfangsdosierung von REXULTI zur Behandlung von Patienten mit Schizophrenie ist 1 mg einmal täglich für Tag 1 bis 4.
Der empfohlene Zieldosierungsbereich beträgt 2-4 mg einmal täglich. Am Tag 5 wird auf 2 mg titriert, anschliessend am Tag 8 auf 4 mg, je nach Verträglichkeit und klinischem Ansprechen des Patienten. Die empfohlene maximale Tagesdosis beträgt 4 mg.
Jugendliche ab 13 Jahren
Die empfohlene Anfangsdosis für Brexpiprazol beträgt 0,5 mg einmal täglich an den Tagen 1 bis 4.
Die Brexpiprazol-Dosis sollte von Tag 5 bis Tag 7 auf 1 mg einmal täglich und dann am Tag 8 auf 2 mg titriert werden. Wöchentliche Dosiserhöhungen können in 1-mg-Schritten je nach klinischer Reaktion und Verträglichkeit erfolgen.
Der empfohlene Zieldosisbereich beträgt 2 mg bis 4 mg einmal täglich. Die maximal empfohlene Tagesdosis beträgt 4 mg.
Agitiertheit bei Alzheimer-Demenz
Die empfohlene Anfangsdosis für REXULTI zur Behandlung von Agitiertheit bei Alzheimer Demenz (AAD) beträgt 0,5 mg einmal täglich an Tag 1 bis Tag 7. An Tag 8 bis Tag 14 wird die Dosis auf 1 mg auftitriert, ab Tag 15 auf 2 mg.
Vor Beginn und während der REXULTI-Behandlung sollen Patienten mit AAD auf reversible Faktoren, die zu Unruhe führen können (z.B. Schmerzen, Infektionen, Polypharmazie, akutes Delir), untersucht und gegebenenfalls angemessen behandelt werden. Nichtpharmakologische Interventionen sollen sich vor Beginn der Behandlung als unwirksam erwiesen haben (siehe Klinische Wirksamkeit).
REXULTI sollte nicht als «nach Bedarf»-Behandlung von AAD eingesetzt werden.
Erhaltungstherapie
Schizophrenie
Der empfohlene Dosierungsbereich beträgt 2-4 mg/Tag. Die Erforderlichkeit der Erhaltungstherapie sowie die angemessene Dosierung müssen regelmässig überprüft werden.
Agitiertheitbei Alzheimer-Demenz
Der empfohlene Dosierungsbereich beträgt 2 bis 3 mg einmal täglich. Nach mindestens 4-wöchiger Behandlung mit 2 mg einmal täglich kann die Dosis auf die maximal empfohlene Tagesdosis von 3 mg gesteigert werden, wenn dies klinisch erforderlich erscheint. Die Wirksamkeit muss nach 12 Wochen und anschliessend regelmässig überprüft werden, um die Notwendigkeit der fortgesetzten Behandlung und die angemessene Dosierung von REXULTI festzustellen. Die Behandlung mit REXULTI ist bei ungenügender Therapieantwort zu beenden.
Art der Anwendung
REXULTI wird mit oder ohne Nahrung oral eingenommen. REXULTI darf nur als ganze Tablette eingenommen werden.
Spezielle Dosierungsanweisungen
Ältere Patienten
Die Sicherheit und Wirksamkeit von REXULTI zur Behandlung der Schizophrenie ist bei älteren Probanden (älter als 65 Jahre) nicht belegt.
Eine ähnliche Brexpiprazol Exposition wurde beobachtet, wenn REXULTI an gesunde ältere Probanden (älter als 65 Jahre) und an erwachsene Probanden (18 - 45 Jahre) verabreicht wurde (siehe Pharmakokinetik). Generell sollte die Dosierung für ältere Patienten vorsichtig und am unteren Ende des Dosierungsbereichs gewählt werden, um den häufigeren Einschränkungen der Leber-, Nieren- und Herzfunktionen, den Begleiterkrankungen sowie anderen medikamentösen Therapien Rechnung zu tragen.
Patienten mit Leberfunktionsstörungen
Bei schizophrenen Patienten mit mässigen bis schweren Leberfunktionsstörungen (Child-Pugh-Score ≥7) beträgt die maximal empfohlene Dosis 3 mg einmal täglich und 2 mg einmal täglich für Patienten mit AAD.
Patienten mit Nierenfunktionsstörungen
Bei schizophrenen Patienten mit mässiger, schwerer oder terminaler Nierenfunktionsstörung (Kreatinin-Clearance CLcr < 60 ml/min) beträgt die maximal empfohlene Dosis 3 mg einmal täglich und 2 mg einmal täglich für Patienten mit AAD.
Weitere besondere Patientengruppen
Eine Dosisanpassung von REXULTI aufgrund des Geschlechts, der ethnischen Zugehörigkeit oder von Tabakkonsum ist nicht erforderlich (siehe Pharmakokinetik).
Tabelle 1: Dosisanpassungen für langsame CYP2D6 Metabolisierer und bei gleichzeitiger Anwendung von CYP Hemmern oder Induktoren
|
|
Angepasste Dosis
| |
Langsame CYP2D6 Metabolisierer
| |
Bekannte langsame CYP2D6 Metabolisierer
|
Gabe der halben üblichen Dosis
| |
Bekannte langsame CYP2D6 Metabolisierer, die mässige/starke CYP3A4 Hemmer einnehmen
|
Gabe eines Viertels der üblichen Dosis
| |
Patienten, die CYP2D6 Hemmer und/oder CYP3A4 Hemmer einnehmen
| |
Starke CYP2D6 Hemmer
|
Gabe der halben üblichen Dosis
| |
Starke CYP3A4 Hemmer
| |
Starke/mässige CYP2D6 Hemmer mit starken/mässigen CYP3A4 Hemmern
|
Gabe eines Viertels der üblichen Dosis
| |
Patienten, die CYP3A4 Induktoren einnehmen
| |
Starke CYP3A4 Induktoren*
|
Verdoppelung der üblichen Dosis über 1–2 Wochen
|
* Wird der gleichzeitig verabreichte CYP3A4 Induktor abgesetzt, muss die Dosierung über 1-2 Wochen auf das ursprüngliche Niveau reduziert werden.
Kinder unter 13 Jahren
Rexulti ist für die Anwendung in der pädiatrischen Population unter 13 Jahren nicht zugelassen. Es kann keine Dosierungsempfehlung angegeben werden.
KontraindikationenÜberempfindlichkeit gegenüber dem Wirkstoff oder einem der Hilfsstoffe gemäss Zusammensetzung.
Warnhinweise und VorsichtsmassnahmenErhöhte Mortalität bei älteren Patienten mit demenzbedingter Psychose
Klasseneffekt: Ältere Patienten mit demenzbedingter Psychose weisen unter einer Behandlung mit Antipsychotika ein höheres Sterberisiko auf als unter Placebo.
Die Analysen von 17 Placebo-kontrollierten Studien zu demenzbedingten Psychosen (modale Dauer von 10 Wochen), überwiegend mit Patienten unter atypischen Antipsychotika, zeigten bei den medikamentös behandelten Patienten ein 1,6–1,7fach erhöhtes Sterberisiko im Vergleich zu Patienten unter Placebo. Im Verlauf einer typischen kontrollierten 10-Wochen Studie betrug die Todesrate bei den medikamentös behandelten Patienten etwa 4,5%, verglichen zu 2,6% bei Patienten unter Placebo. Bei Patienten mit Alzheimer-Demenz ist REXULTI nicht für die Behandlung einer Psychose, sondern nur für die Behandlung von Unruhe im Rahmen der Grunderkrankung zugelassen.
Suizidrisiko
Klasseneffekt: Psychotische Erkrankungen erhöhen das Risiko von Suizidversuchen. Hochrisikopatienten sollten während der medikamentösen Therapie eng überwacht und klinisch angemessen begleitet werden.
AAD-Patienten mit Anzeichen eines ernsthaften Suizidrisikos wurden im klinischen Entwicklungsprogramm REXULTI nicht untersucht.
Zerebrovaskuläre Erkrankungen
Klasseneffekt: In Placebo-kontrollierten Studien mit Risperidon, Aripiprazol und Olanzapin bei älteren Patienten mit demenzbedingter Psychose zeigte sich im Vergleich zu Placebo eine höhere Inzidenz von unerwünschten zerebrovaskulären Ereignissen (Schlaganfälle und vorübergehende Durchblutungsstörungen) einschliesslich Todesfällen.
AAD-Patienten mit bereits bestehender ZNS-Pathologie einschliesslich zerebrovaskulären Erkrankungen und gemischten Demenzformen waren vom klinischen Entwicklungsprogramm REXULTI ausgeschlossen.
Herz-Kreislauf-Erkrankungen
AAD-Patienten mit klinisch signifikanten vorbestehenden Herz-Kreislauf-Erkrankungen (einschliesslich unkontrolliertem Vorhofflimmern, Herzinsuffizienz oder ischämischer Herzkrankheit) wurden im klinischen Entwicklungsprogramm REXULTI nicht untersucht.
QT-Verlängerung
AAD-Patienten mit vorbestehendem QTcF ≥450 ms bei Männern und ≥470 ms bei Frauen und/oder einer Komedikation mit dem Potenzial, eine QT-Verlängerung zu induzieren, wurden im klinischen Entwicklungsprogramm REXULTI nicht untersucht.
Orthostatische Hypotonie und Synkope
Orthostatische Hypotonie kann mit unerwünschten Wirkungen wie Schwindel, Benommenheit und Tachykardie einhergehen. Das höchste Risiko besteht im Allgemeinen zu Behandlungsbeginn und während einer Dosiserhöhung. Patienten mit erhöhtem Risiko für diese unerwünschten Wirkungen oder für das Auftreten von Komplikationen aufgrund von Hypotonie sind Patienten mit einer Dehydratation, einer Hypovolämie, unter antihypertensiver Therapie, mit Herz-Kreislauf-Erkrankungen in der Anamnese (z.B. Herzinsuffizienz, Myokardinfarkt, Ischämie oder Überleitungsstörungen), mit zerebrovaskulären Erkrankungen in der Vorgeschichte sowie Patienten mit erstmaliger Antipsychotika- Behandlung. Eine tiefere Anfangsdosierung und eine langsamere Titration sollte bei diesen Patienten in Betracht gezogen und die orthostatischen Vitalparameter überwacht werden.
Schizophrenie
In den Placebo-kontrollierten klinischen Kurzzeitstudien mit REXULTI bei Patienten mit Schizophrenie wurde folgende Inzidenz von unerwünschten, durch orthostatische Hypotonie bedingten Ereignissen unter REXULTI versus Placebo beobachtet: Schwindel (2,3% versus 1,4%), orthostatische Hypotonie (0,4% versus 0,2%) und Synkope (0,1% versus 0%).
Agitiertheit bei Alzheimer-Demenz
AAD-Patienten mit orthostatischer Hypotonie in der Vorgeschichte wurden vom klinischen Entwicklungsprogramm REXULTI ausgeschlossen.
In den 12-wöchigen placebokontrollierten klinischen REXULTI-Studien mit fixer oder flexibler Dosis bei Patienten mit AAD (Alter 51 bis 90 Jahre) war die Inzidenz von unerwünschten Ereignissen im Zusammenhang mit orthostatischer Hypotonie bei den mit REXULTI behandelten Patienten und den mit Placebo behandelten Patienten vergleichbar, einschliesslich Schwindelgefühl (3,2% vs. 3,4%), orthostatische Hypotonie (0,5% vs. 0,5%) und Synkope (0,2% vs. 0,8%).
Venöse Thromboembolie
Klasseneffekt: Unter Antipsychotika sind Fälle von venösen Thromboembolien (VTE) gemeldet worden. Patienten, die mit Antipsychotika behandelt werden, zeigen oftmals erworbene Risikofaktoren für VTE. Mögliche Risikofaktoren für VTE sollten deshalb vor und während der Behandlung mit REXULTI gründlich abgeklärt und Vorsichtsmassnahmen ergriffen werden.
AAD-Patienten mit VTE in der Vorgeschichte wurden im klinischen Entwicklungsprogramm REXULTI nicht untersucht.
Malignes Neuroleptisches Syndrom (MNS)
Das MNS ist ein potenziell tödlicher Symptomkomplex, der in Zusammenhang mit der Einnahme von Antipsychotika, einschliesslich Brexpiprazol, auftreten kann. Klinische Manifestationen eines MNS sind: sehr hohes Fieber, Muskelrigidität, veränderte Bewusstseinslage und Anzeichen autonomer Instabilität (unregelmässiger Puls oder Blutdruck, Tachykardie, Schwitzen und Herzrhythmusstörungen). Weitere Anzeichen können ein Anstieg der Kreatinphosphokinase, Myoglobinurie (Rhabdomyolyse) und akutes Nierenversagen sein. Sollte ein Patient Anzeichen und Symptome entwickeln, die auf ein MNS hinweisen, oder hohes Fieber unklarer Genese ohne weitere klinische Manifestationen von MNS zeigen, müssen alle Antipsychotika, einschliesslich REXULTI, abgesetzt werden. Benötigt ein Patient nach der Genesung von MNS eine Antipsychotika Behandlung, muss die mögliche Wiederaufnahme der medikamentösen Therapie sorgfältig geprüft werden. Der Patient sollte sorgfältig überwacht werden, da über Rezidive von NMS berichtet wurde.
AAD-Patienten mit NMS in der Vorgeschichte wurden im klinischen Entwicklungsprogramm REXULTI nicht untersucht.
Krampfanfälle
Klasseneffekt: Wie andere Antipsychotika sollte REXULTI bei Patienten mit Krampfanfällen in der Anamnese oder bei Krankheitsbildern mit potenziell herabgesetzter Krampfschwelle mit Vorsicht angewendet werden. AAD-Patienten mit Epilepsie in der Vorgeschichte und/oder der Einnahme antiepileptischer Medikamente wurden im klinischen Entwicklungsprogramm REXULTI nicht untersucht.
Spätdyskinesien
Klasseneffekt: Unter einer Antipsychotika Behandlung kann sich ein Syndrom aus möglicherweise irreversiblen, unwillkürlichen, dyskinetischen Bewegungen entwickeln. Die höchste Prävalenz dieses Syndroms scheint bei älteren Patienten, vor allem bei älteren Frauen, zu liegen. Aufgrund von Prävalenzschätzungen kann allerdings zu Beginn einer Antipsychotika Behandlung nicht vorausgesagt werden, welche Patienten zur Entwicklung dieses Syndroms neigen.
Falls unter der Behandlung mit REXULTI Anzeichen und Symptome von Spätdyskinesien auftreten, muss eine Dosisreduktion oder das Absetzen des Arzneimittels in Erwägung gezogen werden. Die Symptome können sich anschliessend vorübergehend noch verschlechtern oder selbst erst nach Absetzen der Behandlung auftreten.
Dystonie
Klasseneffekt: Während der ersten Tage der Behandlung können bei empfindlichen Personen Dystoniesymptome, lang anhaltende abnormale Kontraktionen von Muskelgruppen, auftreten. Zu den Dystoniesymptomen zählen Spasmen der Nackenmuskulatur, die sich manchmal zu einem Engegefühl der Kehle, Schluckbeschwerden, Kurzatmigkeit und/oder einer Protrusion der Zunge verstärken können. Obwohl diese Symptome bereits bei tiefen Dosen auftreten können, treten sie häufiger und mit grösserer Intensität bei antipsychotischen Medikamenten der ersten Generation mit hoher Potenz und in höheren Dosen auf. Bei männlichen und jüngeren Patienten wird ein erhöhtes Risiko für akute Dystonie beobachtet.
Impulskontrollstörungen
Bei Patienten unter Brexpiprazol wurden Fälle von Impulskontrollstörungen inklusive Spielsucht berichtet. Patienten mit Impulskontrollstörungen in der Anamnese weisen möglicherweise ein erhöhtes Risiko auf und müssen sorgfältig überwacht werden. Es ist zu beachten, dass Symptome einer Impulskontroll-Störung bei allen Indikationen mit der zugrundeliegenden Erkrankung in Zusammenhang stehen können.
Andere ZNS-Erkrankungen als Alzheimer-Demenz (AD)
AAD-Patienten mit akutem Delirium oder einem Delirium innerhalb von 30 Tagen, einer Demenz oder anderen Gedächtnisstörungen, die nicht auf die Alzheimer-Krankheit zurückzuführen sind, waren vom klinischen Entwicklungsprogramm REXULTI ausgeschlossen. REXULTI sollte nicht zur Behandlung akuter Verwirrtheitszustände und nicht zur Behandlung von Unruhe im Zusammenhang mit anderen Erkrankungen als einer Alzheimer-Demenz angewendet werden (z.B. akutes Delir, Psychose, andere Arten von Demenz).
Hyperglykämie und Diabetes mellitus
Klasseneffekt: Bei Patienten unter Behandlung mit Antipsychotika wurde über Hyperglykämien berichtet, die in einigen Fällen sehr ausgeprägt war und mit einer Ketoazidose oder einem hyperosmolaren Koma oder Tod einherging.
Patienten, die mit Antipsychotika behandelt werden, sollten auf Anzeichen und Symptome einer Hyperglykämie überwacht werden (wie etwa Polydipsie, Polyurie, Polyphagie und Schwäche). Patienten mit Diabetes mellitus oder Risikofaktoren für Diabetes mellitus (wie etwa Fettleibigkeit oder Diabetes in der Familiengeschichte) sollten regelmässig auf eine Verschlechterung des Glukosestoffwechsels kontrolliert werden. Bei Patienten mit erheblicher therapiebedingter Hyperglykämie sollte das Absetzen von REXULTI in Betracht gezogen werden.
AAD Patienten mit instabilem oder unkontrolliertem Diabetes mellitus wurden im klinischen Entwicklungsprogramm REXULTI nicht untersucht.
Schizophrenie
In klinischen Studien mit REXULTI bei Patienten mit Schizophrenie (Studien 331-10-231 und 331-10-230 (siehe Klinische Wirksamkeit)) waren die Veränderungen des Nüchternglukosespiegels zwischen mit REXULTI und Placebo behandelten Probanden vergleichbar.
In der Langzeit-Erhaltungsstudie (Studie 331-10-232 (siehe Klinische Wirksamkeit)) waren die mittleren Veränderungen des Serumglukosespiegels vom Ausgangswert bis zum letzten Besuch für die REXULTI-Gruppe (2,11 mg/dl) und die Placebo-Gruppe (−1,62 mg/dl) ebenfalls gering, und wurden nicht als klinisch relevant betrachtet.
In den langfristigen, offenen Studien betrug die mittlere Veränderung des Nüchtern-Serumglukosespiegels vom Ausgangswert bis zum letzten Besuch 2,31 mg/dl (N = 1120).
Agitiertheit bei Alzheimer Demenz
In den 12-wöchigen placebokontrollierten Studien an Patienten (im Alter von 51 bis 90 Jahren) mit AAD war der Anteil der Patienten mit Verschiebungen des Nüchternglukosespiegels von normal (< 100 mg/dl) oder beeinträchtigt (≥100 und <126 mg/dl) zu hoch (≥126 mg/dl) bei mit REXULTI behandelten Patienten (14 %) und bei mit Placebo behandelten Patienten (14 %) ähnlich.
Von den Patienten (55 bis 90 Jahre alt) aus der 12-wöchigen placebokontrollierten klinischen Studie 331-14-213, die in eine 12-wöchige Verlängerungsstudie 331-201-00182 mit aktiver Behandlung übergingen, kam es bei 18 % der Patienten mit normalem oder beeinträchtigtem Nüchternglukoses-Ausgangswert zu einem Übergang zu einem erhöhten Nüchternglukosespiegel (>126 mg/dl).
Gewichtszunahme und Dyslipidämie
Klasseneffekt: Unter einer Behandlung mit Antipsychotika traten Stoffwechselveränderungen wie etwa eine Gewichtszunahme und eine Dyslipidämie auf. Atypische Antipsychotika können nachteilige Veränderungen des Lipidprofils verursachen. Vor oder kurz nach Beginn der antipsychotischen Medikation wird empfohlen, zu Beginn ein Fastenlipidprofil zu erstellen und während der Behandlung regelmässig zu überwachen.
Bei längerer Dauer der Brexpiprazol-Behandlung wurde eine erhöhte Häufigkeit von Gewichtszunahmen beobachtet. Eine klinische Überwachung des Gewichts wird zu Beginn und während der Behandlung empfohlen. (für Gewichtszunahme, siehe Beschreibung spezifischer unerwünschter Wirkungen und Zusatzinformationen).
Dyslipidämie
Schizophrenie
Bei Personen unter einer Behandlung mit atypischen Antipsychotika können unerwünschte Veränderungen der Lipide auftreten.
In den Studien 331-10-231 und 331-10-230 waren die Veränderungen des Nüchtern-Gesamtcholesterins, des LDL-Cholesterins und des HDL-Cholesterins vergleichbar bei Patienten unter REXULTI und unter Placebo. Tabelle 2 zeigt den Anteil der Patienten mit Veränderungen der Nüchtern-Triglyzeride.
Tabelle 2: Veränderung der Nüchtern-Triglyzeride in Studien 331-10-231 und 331-10-230
|
Anteil der Probanden mit Abweichungen von Baseline bis Post-Baseline
| |
|
Placebo
|
1 mg/Tag
|
2 mg/Tag
|
4 mg/Tag
| |
Triglyzeride
|
|
|
|
| |
Normal bis hoch
(<150 bis ≥200 mg/dl und <500 mg/dl)
|
6% (15/253)*
|
10% (7/72)*
|
8% (19/232)*
|
10% (22/226)*
| |
Normal/grenzwertig bis sehr hoch
(<200 bis ≥500 mg/dl)
|
0% (0/303)*
|
0% (0/94)*
|
0% (0/283)*
|
0.4% (1/283)*
|
* Bezeichnet n/N, wobei N=Gesamtzahl der Patienten mit einer Baseline und mindestens einer Post-Baseline Messung; n=Zahl der Patienten mit einer Abweichung.
In Studie 331-10-232 wurden keine klinisch bedeutsamen Unterschiede zwischen den Behandlungsgruppen in der Häufigkeit von potenziell klinisch relevanten Stoffwechselwerten festgestellt.
In den offenen Langzeitstudien betrug die mittlere Abweichung der Nüchtern-Triglyzeride vom Ausgangswert bis zur letzten Visite -2,14 mg/dl (N=1123).
Agitiertheit bei Alzheimer-Demenz
In den 12-wöchigen placebokontrollierten klinischen Studien bei Patienten mit AAD (Alter 51 bis 90 Jahre) war die Häufigkeit von Veränderungen des Gesamtcholesterins (von normal < 200 mg/dl nach erhöht ≥240 mg/dl), des LDL-Cholesterins (von normal <100 mg/dl nach hoch ≥160 mg/dl) und des HDL-Cholesterins (von normal ≥40 mg/dl nach niedrig <40 mg/dl) vergleichbar zwischen den mit REXULTI (6,6 %, 6,1 %, 17,1 %) und den mit Placebo behandelten Patienten (9,4 %, 5,9 %, 13,7 %). Die Häufigkeit der Veränderung der Nüchterntriglyzeridspiegel vom normalen/grenzwertigen Bereich (< 200 mg/dl) in den stark erhöhten Bereich (≥500 mg/dl) waren vergleichbar zwischen den mit REXULTI (0.4%) und den mit Placebo behandelten Patienten mit AAD (0.3%).
Bei den Patienten (Alter 55 bis 90 Jahre), die von der 12-wöchigen placebokontrollierten klinischen Studie 331-14-213 in eine 12-wöchige Verlängerungsstudie 331-201-00182 mit aktiver Behandlung übertraten, zeigten 10% der Patienten unter REXULTI eine Verschiebung des Gesamtcholesterins (Nüchternwert) vom Normbereich (< 200 mg/dl bei der Ausgangsuntersuchung) in den erhöhten Bereich (≥240 mg/dl) und 14% der Patienten unter REXULTI eine Verschiebung des HDL-Cholesterins vom Normbereich (≥40 mg/dl bei der Ausgangsuntersuchung) in den niedrigen Bereich (< 40 mg/dl). 12% der Patienten zeigten eine Verschiebung von normalen Triglyzerid-Ausgangswerten (<150 mg/dl) in den erhöhten Bereich (200 bis < 500 mg/dl).
Leukopenie, Neutropenie und Agranulozytose
Klasseneffekt: Unter einer Behandlung mit Antipsychotika wurde von Leukopenie/Neutropenie berichtet. Für andere Wirkstoffe dieser Klasse wurden Agranulozytosen (einschliesslich Todesfälle) beobachtet.
Mögliche Risikofaktoren für eine Leukopenie/Neutropenie sind unter anderem vorbestehende erniedrigte Leukozytenwerte sowie eine medikamenteninduzierte Leukopenie/Neutropenie in der Anamnese. Bei Patienten mit diesen Risikofaktoren sollte in den ersten Monaten der Therapie das vollständige Blutbild häufig kontrolliert werden. In Abwesenheit anderer ursächlicher Faktoren sollte REXULTI bei den ersten Anzeichen fallender Leukozytenzahlen abgesetzt werden.
Patienten mit Neutropenie sollten sorgfältig auf Fieber oder andere Symptome oder auf Anzeichen für Infektionen überwacht und bei Auftreten solcher Symptome unverzüglich behandelt werden. Bei Patienten mit schwerer Neutropenie (absoluter Neutrophilenwert < 1'000/mm3) sollte REXULTI abgesetzt und die Leukozytenwerte bis zur Genesung kontrolliert werden.
Thermoregulation
Klasseneffekt: Antipsychotika können die Fähigkeit des Körpers zur Senkung der Körperkerntemperatur beeinträchtigen. Bei Patienten, die dem Risiko einer Erhöhung der Körperkerntemperatur ausgesetzt sind (z.B. durch anstrengende körperliche Betätigung, extreme Hitze, Dehydratation oder gleichzeitige Behandlung mit anticholinergen Arzneimitteln) sollte REXULTI mit der entsprechenden Vorsicht verschrieben werden.
Dysphagie
Klasseneffekt: Motilitätsstörungen der Speiseröhre und Aspiration wurden unter Antipsychotika Therapie beobachtet. REXULTI und andere Antipsychotika sollten bei Patienten mit einem Risiko für eine Aspirationspneumonie mit Vorsicht angewendet werden. REXULTI wurde bei Patienten mit Dysphagie, eingeschränkter oraler Aufnahme und/oder enteralen Ernährungssonden nicht untersucht.
Prolaktin
Brexpiprazol kann den Prolaktinspiegel erhöhen. Die mit der Behandlung mit Brexpiprazol verbundenen Erhöhungen sind im Allgemeinen mild und können während der Behandlung abklingen. In seltenen Fällen kann der Effekt jedoch während der Behandlung bestehen bleiben (siehe Abschnitt « Unerwünschte Wirkungen»).
Ältere Menschen
Schizophrenie
Klinische Studien mit REXULTI umfassten eine begrenzte Zahl von Probanden ab 65 Jahren, um eine möglicherweise unterschiedliche Reaktion zu jüngeren Probanden zu ermitteln. Ältere Probanden (> 65 Jahre) zeigten eine vergleichbare systemische Brexpiprazol Exposition wie erwachsene Probanden (18–45 Jahre) (siehe Pharmakokinetik und Dosierung/Anwendung). Ältere Patienten mit demenzbedingter Psychose weisen unter Behandlung mit Antipsychotika ein höheres Sterberisiko auf als unter Placebo (siehe oben: Erhöhte Mortalität bei älteren Patienten mit demenzbedingter Psychose).
Agitierheit bei Alzheimer-Demenz
Die Gesamtzahl der Patienten im Alter von 65 Jahren und älter, die in den in den 12-wöchigen placebokontrollierten klinischen Studien zu AAD mit REXULTI behandelt wurden, betrug 556 (216 Patienten im Alter von 65 bis 74 Jahren, 273 Patienten im Alter von 75 bis 84 Jahren und 67 Patienten im Alter von 85 Jahren). In der offenen Verlängerungsstudie 331-201-00182 (NCT03594123) wurden 259 Patienten behandelt, darunter 100 Patienten im Alter von 65 bis 74 Jahren, 108 Patienten im Alter von 75 bis 84 Jahren und 22 Patienten im Alter von 85 Jahren oder älter.
In den 12-wöchigen kontrollierten klinischen Studien mit geriatrischen Patienten (65 Jahre und älter) zur Behandlung von AAD war die Häufigkeit von Stürzen (2%) und Schwindel (3%) bei mit fixer oder flexibler REXULTI-Dosis behandelten Patienten ähnlich wie bei mit Placebo behandelten Patienten (Stürze und Schwindel jeweils 3%).
Auffällige Labortestergebnisse
AAD-Patienten mit medizinisch signifikanten abnormalen Labortestergebnissen, wie z.B. erhöhte Kreatinphosphokinase-, Serumchemie-, Leber- und Schilddrüsenparameter, waren vom klinischen Entwicklungsprogramm REXULTI ausgeschlossen.
Leberfunktionsstörungen
Bei Patienten mit Leberfunktionsstörungen muss die Dosierung angepasst werden (siehe Pharmakokinetik und Dosierung/Anwendung).
Nierenfunktionsstörungen
Bei Patienten mit Nierenfunktionsstörungen muss die Dosierung angepasst werden (siehe Pharmakokinetik und Dosierung/Anwendung).
Laktose
REXULTI Filmtabletten enthalten Laktose. Patienten mit der seltenen hereditären Galaktose-Intoleranz, völligem Laktase-mangel oder Glukose-Galaktose-Malabsorption sollten dieses Arzneimittel nicht anwenden.
InteraktionenREXULTI wird in erster Linie von CYP3A4 und CYP2D6 metabolisiert. Aufgrund der Ergebnisse aus Arzneimittel-Interaktionsstudien wird bei Personen, die starke CYP2D6 oder CYP3A4 Hemmer einnehmen, eine Dosisanpassung auf die Hälfte der Erhaltungsdosis empfohlen. Basierend auf Schätzungen aus der Populations-pharmakokinetischen Analyse ist davon auszugehen, dass extensive CYP2D6 Metabolisierer, die sowohl CYP3A4 als auch CYP2D6 Hemmer erhalten, oder langsame CYP2D6 Metabolisierer, die starke CYP3A4 Hemmer erhalten, einen etwa 4–5fachen Anstieg der Brexpiprazol Konzentrationen aufweisen. Daher sollte die Dosierung von REXULTI in diesen Fällen auf einen Viertel der empfohlenen Dosis reduziert werden (siehe Dosierung/Anwendung).
Wird REXULTI gleichzeitig mit einem starken CYP3A4 Induktor (z.B. Rifampicin) angewendet, muss die Dosis um das 2fache erhöht und gemäss dem klinischen Ansprechen weiter angepasst werden (siehe Dosierung/Anwendung).
Enzyminhibitoren
Starke CYP2D6 Hemmer
Bei gleichzeitiger Anwendung einer oralen Einzeldosis von 2 mg REXULTI mit Chinidin (324 mg/Tag über 7 Tage), einem starken CYP2D6 Hemmer (in der Schweiz nicht zugelassen), stieg die area under the curve (AUC) von Brexpiprazol um 94%.
Ketoconazol und andere starke CYP3A4 Hemmer
Bei gleichzeitiger Anwendung von Ketoconazol (200 mg zweimal täglich über 7 Tage), einem starken CYP3A4 Hemmer, mit einer oralen Einzeldosis von 2 mg REXULTI stieg die AUC von Brexpiprazol um 97%.
CYP2B6 Hemmer
Die gleichzeitige Anwendung einer oralen Einzeldosis von 2 mg REXULTI mit Ticlopidin (250 mg zweimal täglich über 7 Tage), einem CYP2B6 Hemmer (in der Schweiz nicht zugelassen), hatte keinen Effekt auf die Pharmakokinetik von Brexpiprazol.
Enzyminduktoren
Rifampicin und andere CYP3A4 Induktoren
Bei gleichzeitiger Anwendung von Rifampicin (600 mg zweimal täglich über 12 Tage), einem starken CYP3A4 Induktor, mit einer oralen Einzeldosis von 4 mg REXULTI, sank die Cmax von Brexpiprazol um ca. 31% und die AUC um ca. 73%.
Andere Interaktionen
Magensäure pH-Modifikatoren
Die gleichzeitige Anwendung von Omeprazol (40 mg einmal täglich über 5 Tage), einem weit verbreiteten Protonenpumpenhemmer (PPI), mit einer oralen Einzeldosis von 4 mg REXULTI, hatte keine Wirkung auf die Absorption von Brexpiprazol.
Wirkung von REXULTI auf andere Arzneimittel
In-vitro Daten zeigten eine geringe bis gar keine Hemmung der CYP450 Isoenzyme durch Brexpiprazol. Die potenzielle in-vitro Hemmung der MDR1 (P-gp), OAT1, OAT3, OCT2, Multidrogen und Toxin Extrusion Transporter (MATE1), MATE2-K, OATP1B1, OATP1B3 und OCT1 durch Brexpiprazol wurde ebenfalls untersucht. Brexpiprazol oder sein Hauptmetabolit wurden lediglich als potenzieller Hemmer des BCRP-Efflux-Transporters von BCRP, OATP1B1, MATE1 und MATE2-K identifiziert, jedoch nicht als Hemmer der anderen getesteten Transporter.
Klinische Studien zeigen, dass orales REXULTI (2 mg/Tag, über 5 Tage) keinen Effekt hat auf die Pharmakokinetik von Dextromethorphan (ein CYP2D6-Substrat), Lovastatin (ein CYP3A4-Substrat, in der Schweiz nicht zugelassen), Bupropion (ein CYP2B6-Substrat) oder Fexofenadin (ein P-gp-Transporter-Substrat). REXULTI (6 mg Einzeldosis) hatte keinen Einfluss auf die Absorption von Rosuvastatin (ein BCRP und OATP Transporter-Substrat).
Alkohol
REXULTI wurde bei Patienten mit gleichzeitigem Alkoholkonsum nicht untersucht.
Schwangerschaft, StillzeitSchwangerschaft
Die sichere Anwendung von REXULTI vor der Empfängnis, während der Schwangerschaft oder Stillzeit wurde nicht nachgewiesen. REXULTI sollte nicht während der Schwangerschaft, von stillenden Müttern oder bei Patienten im fortpflanzungsfähigen Alter angewendet werden, die keine Verhütungsmittel anwenden.
In tierexperimentellen Reproduktionsstudien wurde keine Teratogenität beobachtet; es wurde jedoch eine erhöhte Anzahl der perinatalen Todesfälle beobachtet (siehe Präklinische Daten).
Adäquate, ausreichend kontrollierte Studien an schwangeren Frauen zur Ermittlung der Arzneimittelrisiken bei Anwendung von REXULTI wurden bisher nicht durchgeführt. Bei Neugeborenen, deren Mütter während des dritten Trimenons der Schwangerschaft Antipsychotika wie REXULTI einnahmen, besteht nach der Geburt das Risiko für extrapyramidale und/oder Entzugssymptome. Es gab Berichte von Unruhezuständen, abnorm erhöhten oder verminderten Muskeltonus, Tremor, Schläfrigkeit, Schwierigkeiten beim Atmen oder Probleme beim Füttern bei diesen Kindern. Diese Komplikationen wiesen unterschiedliche Schweregrade auf. Die Symptome waren in einigen Fällen selbstlimitierend, in anderen Fällen waren die Behandlung auf der Intensivstation und ein längerer Klinikaufenthalt erforderlich.
Die Wirkung von REXULTI auf Wehentätigkeit und Geburtsvorgang ist beim Menschen nicht bekannt.
Stillzeit
Es ist nicht bekannt, ob Brexpiprazol oder seine Metaboliten beim Menschen in die Muttermilch übergehen. Brexpiprazol wurde in die Milch von Ratten ausgeschieden. Aufgrund der möglicherweise schwerwiegenden unerwünschten Wirkungen auf den Säugling, muss entschieden werden, ob entweder das Stillen abgebrochen oder die Behandlung mit REXULTI beendet werden soll, wobei das Risiko eines Therapieabbruchs für die Mutter berücksichtigt werden soll.
Fertilität
Die Wirkung von Brexpiprazol auf die menschliche Fruchtbarkeit wurde nicht untersucht. Tierstudien haben eine verminderte weibliche Fruchtbarkeit gezeigt (siehe Präklinische Daten).
Wirkung auf die Fahrtüchtigkeit und auf das Bedienen von MaschinenWie bei anderen Antipsychotika, die potenziell das Urteilsvermögen sowie die Denk- und motorischen Fähigkeiten beeinträchtigen können, sollten Patienten zur Vorsicht beim Lenken von Fahrzeugen oder Bedienen von gefährlichen Maschinen angehalten werden, bis sichergestellt ist, dass diese Tätigkeiten für Patienten durch die REXULTI Behandlung nicht beeinträchtigt sind.
In den placebokontrollierten Kurzzeitstudien an Schizophrenie-Patienten wurde bei 5% der mit REXULTI behandelten Patienten und 4% der mit Placebo behandelten Patienten Somnolenz (einschliesslich Sedierung und vermehrtem Schlafbedürfnis) angegeben. In den unverblindeten Langzeitstudien betrug die Inzidenz von Somnolenz und ähnlichen Wirkungen 3%.
Demenz beeinträchtigt die Verkehrstüchtigkeit und die Fähigkeit zum Bedienen von Maschinen. In den 12-wöchigen placebokontrollierten klinischen Studien bei Patienten mit AAD (Alter 51 bis 90 Jahre) wurde Somnolenz (einschliesslich Sedierung) bei 4% der mit REXULTI behandelten Patienten und im Vergleich dazu bei 2% der mit Placebo behandelten Patienten berichtet.
Unerwünschte WirkungenSchizophrenie
Die am häufigsten beobachteten Nebenwirkungen (UAW) waren bei Erwachsenen Akathisie (5,6%) und Gewichtszunahme (3,9%), und bei Jugendlichen Übelkeit (6,4%), Somnolenz (4,5 %) und Akathisie (3,6 %). Die unerwünschten Reaktionen waren meist leicht bis mittelschwer und führten in der Regel nicht zu einem Studienabbruch.
Zusammenstellung der unerwünschten Wirkungen
Die Häufigkeit der mit der Brexpiprazol-Therapie verbundenen Nebenwirkungen ist nachstehend aufgeführt. Die unten aufgeführten Nebenwirkungen wurden in placebokontrollierten Kurzzeitstudien der Phasen 2 und 3 mit Erwachsenen und relevanten therapeutischen Dosen (2 mg bis 4 mg) sowie in placebokontrollierten Kurzzeitstudien der Phase 3 mit Kindern und Jugendlichen mit relevanten therapeutischen Dosen (1 mg bis 4 mg) berichtet. Alle Nebenwirkungen sind nach Systemorganklasse (SOC) und Häufigkeit aufgelistet: sehr häufig (≥1/10); häufig (≥1/100, <1/10); gelegentlich (≥1/1000, <1/100); selten (≥1/10'000, <1/1000); sehr selten (<1/10'000); nicht bekannt (kann aus den verfügbaren Daten nicht abgeschätzt werden). Die unerwünschten Wirkungen sind innerhalb der Häufigkeitskategorien nach abnehmendem Schweregrad geordnet.
Erkrankungen des Immunsystems
Häufig: Hautausschlag.
Gelegentlich: Angioödem, Urtikaria, Gesichtsschwellung.
Stoffwechsel- und Ernährungsstörungen:
Häufig: Gewichtszunahme, Erhöhung der Kreatinphosphokinase.
Psychiatrische Erkrankungen:
Häufig: Unruhe*.
Gelegentlich: Suizidversuch, Suizidgedanken.
Nicht bekannt: Spielsucht, impulsives Verhalten, Essattacken, Kaufsucht, zwanghaftes Sexualverhalten.
* Unruhe kann mit der pharmakologischen Wirkung von Brexpiprazol zusammenhängen und ist aufgeführt, obwohl die Differenz zu Placebo kleiner war als 0.5%.
Erkrankungen des Nervensystems:
Häufig: Akathisie, Tremor, Somnolenz**, Schwindel.
Gelegentlich: Parkinsonismus.
Nicht bekannt: Krampfanfälle, malignes neuroleptisches Syndrom#.
** Beinhaltet auch Sedierung und Hypersomnie
# Unerwünschte Wirkungen, die nach der Markteinführung berichtet wurden
Herzerkrankungen
Nicht bekannt: QT-Verlängerung im Elektrokardiogramm.
Gefässerkrankungen
Gelegentlich: Venöse Thromboembolie (einschliesslich Lungenembolie und tiefer Venenthrombose), orthostatische Hypotonie.
Erkrankungen der Atemwege, des Brustraums und Mediastinums:
Gelegentlich: Husten.
Erkrankungen des Gastrointestinaltrakts:
Häufig: Durchfall, Übelkeit, Mundtrockenheit, Schmerzen im Oberbauch.
Gelegentlich: Zahnkaries, Blähungen.
Erkrankungen der Haut und des Unterhautgewebes:
Häufig: Exanthem.
Skelettmuskulatur-, Bindegewebs- und Knochenerkrankungen:
Häufig: Rückenschmerzen, Schmerzen in den Extremitäten.
Gelegentlich: Myalgie.
Nicht bekannt: Rhabdomyolyse.
Allgemeine Erkrankungen und Beschwerden am Verabreichungsort:
Häufig: Schmerzen.
Schwangerschaft, Wochenbett und perinatale Erkrankungen
Nicht bekannt: Arzneimittelentzugssyndrom bei Neugeborenen.
Untersuchungen
Sehr häufig: Erhöhte Prolaktinwerte im Blut.***
Gelegentlich: Erhöhter Blutdruck, erhöhte Triglyceride im Blut, erhöhte Leberenzyme.
*** Die Einstufung erhöhter Prolaktinwerte im Blut basiert auf den Kriterien für potenziell klinisch relevante Werte (PCR) von > 1 × oberer Normwert (ULN).
Agitiertheit bei Alzheimer-Demenz
Die Sicherheit von REXULTI wurde bei 655 Patienten (Alter 51 bis 90 Jahre) mit Verdachtsdiagnose AAD untersucht, die an drei 12-wöchigen placebokontrollierten klinischen Studien teilnahmen, in denen REXULTI in Tagesdosen von ≤1 mg (157 Patienten), 2 mg (213 Patienten), 3 mg (153 Patienten) oder flexiblen Dosen zwischen 0,5 und 2 mg (132 Patienten) verabreicht wurde. Die am häufigsten beobachteten Nebenwirkungen, die in den 12-wöchigen placebokontrollierten klinischen Studien berichtet wurden, waren Schlaflosigkeit (3,7 %), Schläfrigkeit (3,4 %),Harnwegsinfektionen (2,6 %) und Diarrhoe (2,0%). Alle Ereignisse traten in der Brexpiprazol-Gruppe häufiger auf als in der Placebo-Gruppe.
Zudem wurden Sicherheitsdaten einer offenen Verlängerungsstudie berücksichtigt: Geeignete Patienten konnten zusätzlich über weitere 12-Wochen an dieser Studie (331-201-00182) mit aktiver Behandlung teilnehmen. Insgesamt wurden 259 Personen für diese Studie untersucht, aufgenommen und behandelt. In der Hauptstudie 331-14-213 (NCT03548584) erhielten 163 Personen Brexpiprazol und 96 Personen ein Placebo. Patienten, die in der Hauptstudie 331-14-213 randomisiert 2 mg/Tag oder 3 mg/Tag erhielten, behielten diese Dosis auch in der Verlängerungsstudie 331-201-00182 bei. Die am häufigsten beobachteten Nebenwirkungen, die in der Verlängerungsstudie mit aktiver Behandlung gemeldet wurden, waren Kopfschmerzen (3,5 %),Stürze (2,3 %), Somnolenz (1,9%), Schwindel (1,9%), Nasopharyngitis (1,9%) und Agitiertheit (1,5%).
Zusammenfassung der Nebenwirkungen
Die in den beschriebenen klinischen Studien zur AAD mit Brexpiprazol gemeldeten UAW sind nachstehend nach Systemorganklasse und Häufigkeit aufgeführt: sehr häufig (≥1/10), häufig (≥1/100 bis < 1/10), gelegentlich (≥1/1000 bis < 1/100), selten (≥1/10'000 bis < 1/1000), sehr selten (< 1/10'000) und nicht bekannt (Häufigkeit auf Grundlage der verfügbaren Daten nicht abschätzbar).
Infektionen und parasitäre Erkrankungen:
Häufig: Harnwegsinfektion, Pneumonie, Nasopharyngitis.
Gelegentlich: Bronchitis, Blasenentzündung, virale Infektionen der Atemwege.
Erkrankungen des Blutes und des Lymphsystems:
Gelegentlich: Anämie.
Stoffwechsel- und Ernährungsstörungen:
Häufig: Gesteigerter Appetit.
Psychiatrische Erkrankungen:
Häufig: Insomnie, Agitiertheit.
Gelegentlich: Verwirrtheit, Halluzinationen, psychomotorische Retardierung.
Erkrankungen des Nervensystems:
Häufig: Somnolenz, Kopfschmerzen, Schwindel, Bradykinese.
Gelegentlich: Akathisie, extrapyramidale Störungen.
Herzerkrankungen
Gelegentlich: verlängertes QT-Intervall im Elektrokardiogramm.
Gefässerkrankungen
Häufig: erhöhter Blutdruck.
Erkrankungen der Atemwege, des Brustraums und Mediastinums:
Häufig: Atembeschwerden.
Gelegentlich: Nasenbluten.
Erkrankungen des Gastrointestinaltrakts:
Häufig: Durchfall, trockener Mund, Übelkeit.
Gelegentlich: Speichelhypersekretion.
Erkrankungen der Haut und des Unterhautgewebes:
Gelegentlich: Exanthem.
Skelettmuskulatur-, Bindegewebs- und Knochenerkrankungen:
Häufig: Erhöhte Kreatinphosphokinase im Blut.
Gelegentlich: Muskelkrämpfe.
Allgemeine Erkrankungen und Beschwerden am Verabreichungsort:
Häufig: Asthenie, Fatigue, Gewichtszunahme.
Gelegentlich: Fieber.
Untersuchungen:
Gelegentlich: Erhöhte Laktatdehydrogenase im Blut.
Verletzung, Vergiftung und durch Eingriffe bedingte Komplikationen
Häufig: Sturz.
Ergänzende Sicherheitsdaten
Brexpiprazol wurde in zusätzlichen, in der Schweiz nicht zugelassenen Indikationen als Zusatztherapie zu Antidepressiva zur Behandlung schwerer depressiver Störungen (MDD) und als Zusatzbehandlung von ADHS untersucht. In diesen Studien fielen folgende, in den zugelassenen Indikationen Schizophrenie und AAD nicht beschriebene unerwünschte Wirkungen auf: Obstipation, Dyspepsie, Angst, Überempfindlichkeit sowie erhöhte Prolaktinspiegel berichtet.
Beschreibung spezifischer unerwünschter Wirkungen und Zusatzinformationen
Indikation: Schizophrenie bei Erwachsenen
Andere extrapyramidale Symptome einschliesslich Akathisie
In den Studien 331-10-231 (NCT01396421) und 331-10-230 (NCT01393613) betrug die Häufigkeit von EPS-verwandten Ereignissen (ohne Akathisie Ereignisse) 5,1% bei Patienten unter REXULTI versus 3,5% unter Placebo. Die Häufigkeit von Akathisie Ereignissen betrug 5,4% bei Patienten unter REXULTI versus 4,9% unter Placebo. Neu auftretende Fälle von Akathisie wurden in allen Behandlungsarmen häufiger während der ersten 3 Wochen nach Behandlungsbeginn berichtet und waren leicht bis mittelschwer.
In den Studien 331-10-231 und 331-10-230 wurden die objektiven Skalen Simpson Angus Scale (SAS) für extrapyramidale Symptome, die Barnes Akathisia Rating Scale (BARS) für Akathisie und die Abnormal Involuntary Movement Scale (AIMS) für Dyskinesie eingesetzt. Die mittleren Abweichungen vom Ausgangswert bei der Schlussvisite bei Patienten unter REXULTI auf der SAS, BARS und AIMS (-0,10; 0,02 und -0,08) waren vergleichbar mit denen bei Patienten unter Placebo (0,00; 0,01 und -0,07).Auch in Studie 331-10-232 (NCT01668797) waren die mittleren Abweichungen vom Ausgangswert bei der Schlussvisite auf der SAS, BARS und AIMS vergleichbar bei Patienten unter REXULTI und unter Placebo.
Gewichtszunahme
In den Kurzzeitstudien 331-10-231 und 331-10-230 (siehe Klinische Wirksamkeit) war der Anteil der Patienten mit einer potenziell klinisch relevanten Gewichtszunahme (≥7% des Körpergewichts) 10,5% unter REXULTI 2 mg/Tag und 10,2% unter REXULTI 4 mg/Tag, verglichen mit 4,1% unter Placebo. Die mittlere Gewichtszunahme bei der letzten Visite betrug 1,2 kg in beiden REXULTI Gruppen und 0,2 kg in der Placebo Gruppe.
In Studie 331-10-232 (siehe Klinische Wirksamkeit) betrug der Anteil der Patienten mit einer Gewichtszunahme von ≥7% bei der letzten Visite 3,1% in der REXULTI Gruppe verglichen mit 1,0% in der Placebo Gruppe.
In den offenen Schizophrenie Langzeit-Studien (331-10-237 (NCT01397786), 331-08-210 (NCT01649557), 14644B (NCT01810783)) (siehe Klinische Wirksamkeit) betrug die mittlere Abweichung des Körpergewichts vom Ausgangswert bis zur letzten Visite 1,1 kg. Der Prozentsatz von Patienten mit potentiell klinisch relevanter Gewichtszunahme (≥7%) betrug 18% und 0,4% der Probanden brachen die Behandlung aufgrund der Gewichtszunahme ab.
Indikation: Schizophrenie bei Jugendlichen ab 13 Jahren
Es wird erwartet, dass Häufigkeit, Art und Schwere der Nebenwirkungen bei Jugendlichen denen bei Erwachsenen ähneln.
Extrapyramidale Symptome (EPS)
In Kurzzeitstudien war Akathisie die am häufigsten berichtete EPS-bedingte Nebenwirkung in der Brexpiprazol-Gruppe (1 mg/Tag bis 4 mg/Tag) (3,6 %) im Vergleich zu 2,9 % in der Placebogruppe. Weitere EPS-bedingte Nebenwirkungen, die in kontrollierten Kurzzeitstudien mit pädiatrischen Patienten berichtet wurden, waren Muskelsteifigkeit (0,9 %), Hypokinesie (0,9 %) und Tremor (0,9 %).
Akathisie
Die Inzidenz von Akathisie bei mit Brexpiprazol behandelten Kindern und Jugendlichen in einer randomisierten, doppelblinden Kurzzeitstudie betrug 3,6 % gegenüber 2,9 % bei Placebo.
Die Inzidenz von Akathisie in der laufenden, offenen Langzeitstudie betrug 5,1 %. Siehe Abschnitt «Klinische Wirksamkeit».
Gewichtszunahme
In einer kontrollierten Kurzzeitstudie betrug der Prozentsatz der Teilnehmer mit klinisch signifikanter Gewichtszunahme (Zunahme des Körpergewichts um ≥7 % gegenüber dem Ausgangswert) 8,2 % in der mit Brexpiprazol behandelten Gruppe, verglichen mit 4,9 % in der Placebogruppe. Die mittlere Gewichtszunahme zwischen dem Ausgangswert und der letzten Untersuchung betrug 0,8 kg bei den mit Brexpiprazol behandelten Teilnehmern und 0,0 kg bei den mit Placebo behandelten Teilnehmern. Zur Anpassung an normales Wachstum wurden Z-Scores abgeleitet (gemessen in Standardabweichungen [SD]), die das natürliche Wachstum von Kindern und Jugendlichen durch Vergleich mit alters- und geschlechtsangepassten Bevölkerungsstandards normalisieren. Eine Z-Score-Änderung <0,5 SD wird als klinisch nicht signifikant angesehen. In dieser Studie wurde in den mit Brexpiprazol und Placebo behandelten Gruppen keine Änderung des mittleren Z-Scores zwischen dem Ausgangswert und der letzten Untersuchung beobachtet. Bei 4,5 % der mit Brexpiprazol und 3,9 % der mit Placebo behandelten Patienten war der alters- und geschlechtsbereinigte Z-Score des Körpergewichts um mindestens 0,5 SD gegenüber dem Ausgangswert erhöht. TEAEs (Gewichtszunahme) wurden bei 1,7 % aller Patienten in der Brexpiprazol-Gruppe im Vergleich zu 3,4 % in der Placebo-Gruppe berichtet.
In der offenen Langzeitstudie betrug der Anteil der Patienten mit klinisch signifikanter Gewichtszunahme (Zunahme des Körpergewichts um ≥7 % gegenüber dem Ausgangswert) bei jedem Besuch 44,6 % in der mit Brexpiprazol behandelten Gruppe. Die mittlere Veränderung des Z-Scores vom Ausgangswert bis zum letzten Besuch betrug 0,10 SD für das Körpergewicht, während 20 % der Patienten einen Anstieg des alters- und geschlechtsbereinigten Z-Scores des Körpergewichts um mindestens 0,5 SD gegenüber dem Ausgangswert aufwiesen. Gewichtszunahmebedingte unerwünschte Ereignisse (TEAE) wurden bei 11,5 % der Probanden beobachtet, während andere gewichtszunahmebedingte TEAE, wie z.B. ein erhöhter BMI und Taillenumfang, jeweils bei einem Probanden auftraten.
Prolaktin
In einer kontrollierten Kurzzeitstudie betrug die Inzidenz erhöhter Prolaktinwerte im Blut 32,4 % in der Brexpiprazol-Gruppe (2 mg bis 4 mg) gegenüber 10,2 % in der Placebo-Gruppe. In der Kurzzeitstudie traten erhöhte Prolaktinwerte häufiger bei Frauen als bei Männern auf (26,8 % vs. 24,5 %). Die Häufigkeit von Prolaktinerhöhungen > 1 × ULN betrug in der Brexpiprazol-Gruppe (2 mg bis 4 mg) 26,8 % bei Frauen gegenüber 6,3 % in der Placebo-Gruppe und 24,5 % bei Männern gegenüber 6 % in der Placebo-Gruppe. In den Langzeitstudien berichteten 1,7 % der Probanden über erhöhte Prolaktinwerte im Blut und 0,7 % der Probanden über Hyperprolaktinämie.
Somnolenz einschliesslich Sedierung und Hypersomnie
In Kurzzeitstudien betrug die Inzidenz von Somnolenz-TEAE (Sedierung, Somnolenz, Hypersomnie) in der Gruppe, die mit Brexpiprazol 2-4 mg behandelt wurde, 7,3 % im Vergleich zu 6,7 % in der Placebo-Gruppe. In einer offenen Langzeitstudie betrug die Inzidenz von Somnolenz-TEAE (Sedierung, Somnolenz, Hypersomnie) 11,9 %. Diese TEAE waren leicht bis mittelschwer.
Suizidales Verhalten
In einer kontrollierten Kurzzeitstudie wurde ein behandlungsbedingtes unerwünschtes Ereignis (TEAE), das mit suizidalem Verhalten in Zusammenhang stand, bei einem Probanden (0,9 %, ein nicht schwerwiegendes Ereignis) aus der Gruppe berichtet, die mit Brexpiprazol behandelt wurde, und bei keinem Probanden in der Placebogruppe. In einer unverblindeten Langzeitstudie wurden TEAE, die mit suizidalem Verhalten in Zusammenhang standen, bei 8 Probanden (2,7 %) berichtet (siehe Warnhinweise und Vorsichtsmassnahmen).
Indikation: Agitiertheit bei Alzheimer-Demenz
Die Inzidenz der gemeldeten EPS-bedingten Nebenwirkungen, betrug 5,3 % bei mit REXULTI behandelten Patienten gegenüber 3,1% bei mit Placebo behandelten Patienten. Die Inzidenz von Akathisie-Ereignissen bei mit REXULTI behandelten Patienten betrug 1,8 % gegenüber 0,3 % bei mit Placebo behandelten Patienten.
In den 12-wöchigen kontrollierten klinischen Studien bei AAD wurden objektiv Daten gesammelt auf der Simpson-Angus Rating Scale (SAS) für EPS, der Barnes Akathisia Rating Scale (BARS) für Akathisie und der Abnormal Involuntary Movement Skala (AIMS) für Dyskinesie.
Die mittlere Veränderung gegenüber dem Ausgangswert beim letzten Besuch war bei mit REXULTI behandelten Patienten für SAS, BARS und AIMS vergleichbar mit der bei mit Placebo behandelten Patienten.
Der Prozentsatz der Patienten, die von normal zu abnormal wechselten, war bei den mit REXULTI behandelten Patienten höher als bei Placebo für die SAS (6 % gegenüber 2 %).
Gewichtszunahme
In den 12-wöchigen placebokontrollierten klinischen Studien an Patienten (im Alter von 51 bis 90 Jahren) mit AAD (Studie 331-12-283 (NCT01862640) mit fester Dosis, Studie 331-14-213 mit fester Dosis (siehe Klinische Wirksamkeit) und Studie 331-12-284 (NCT01922258) mit flexibler Dosis betrug der Anteil der Patienten mit einer Zunahme des Körpergewichts (kg) um ≥7 % bei jedem Besuch 1,7 % in der REXULTI-Gruppe im Vergleich zu 0,8 % in der Placebogruppe.
Unter den Patienten (55 bis 90 Jahre alt), die aus der 12-wöchigen placebokontrollierten klinischen Studie 331-14-213 mit fester Dosis in eine 12-wöchige Verlängerungsstudie 331-201-00182 mit aktiver Behandlung übergingen, zeigten 3 % der Patienten eine Zunahme des Körpergewichts um ≥7 % und 3 % zeigten eine Abnahme des Körpergewichts um ≥7 % vom Ausgangswert bis zur letzten Studienvisite.
Die Meldung des Verdachts auf Nebenwirkungen nach der Zulassung ist von grosser Wichtigkeit. Sie ermöglicht eine kontinuierliche Überwachung des Nutzen-Risiko-Verhältnisses des Arzneimittels. Angehörige von Gesundheitsberufen sind aufgefordert, jeden Verdacht einer neuen oder schwerwiegenden Nebenwirkung über das Online-Portal ElViS (Electronic Vigilance System) anzuzeigen. Informationen dazu finden Sie unter www.swissmedic.ch.
ÜberdosierungBehandlung
Zur Behandlung einer Überdosierung mit REXULTI sind keine spezifischen Informationen verfügbar. Eine Magenspülung und Behandlung mit einem Emetikum können unmittelbar nach einer Überdosierung hilfreich sein. Ein EKG soll durchgeführt und bei einer Verlängerung des QT-Intervalls eine Herzkreislaufüberwachung etabliert werden. Im Übrigen sollte sich die Behandlung einer Überdosierung auf unterstützende Massnahmen, Freihalten der Atemwege, Sauerstoffzufuhr, Beatmung und Symptombehandlung fokussieren. Eine enge medizinische Überwachung und Kontrolle bis zur Genesung sind erforderlich.
Die orale Verabreichung von Aktivkohle und Sorbit (50 g/240 ml) eine Stunde nach Einnahme von REXULTI senkte die Cmax von Brexpiprazol um ca. 5% bis 23% und die AUC um 31% bis 46%. Die verfügbare Information zum therapeutischen Potenzial von Aktivkohle zur Behandlung einer REXULTI Überdosierung ist jedoch ungenügend. Obwohl keine konkreten Erfahrungen zur Wirkung der Hämodialyse vorliegen, dürfte eine Hämodialyse bei der Behandlung einer REXULTI Überdosierung aufgrund der hohen Plasmaproteinbindung von Brexpiprazol wenig hilfreich sein.
Eigenschaften/WirkungenATC-Code
N05AX16
Wirkungsmechanismus
REXULTI ist ein neuartiger, atypischer antipsychotischer Wirkstoff, dessen pharmakologische Aktivität auf der Modulation der Serotonin-Dopamin Aktivität beruht. Obwohl der genaue Wirkmechanismus von Brexpiprazol in der Behandlung der Schizophrenie oder der Agitiertheit bei Alzheimer-Demenz nicht vollständig geklärt ist, beruht die Pharmakologie von Brexpiprazol vermutlich auf einer Kombination von hoher Bindungsaffinität und funktioneller Aktivität an multiplen monoaminergen Rezeptoren. Seine modulatorische Wirkung auf die serotonergen und dopaminergen Systeme beruht auf einer Kombination von partiell agonistischer Aktivität an den serotonergen 5-HT1A und den dopaminergen D2 Rezeptoren und einer antagonistischen Aktivität an den serotonergen 5-HT2A Rezeptoren, wobei die Affinitäten zu all diesen Rezeptoren ähnlich hoch sind (Ki: 0,1–0,5 nM).
Brexpiprazol zeigt ausserdem eine antagonistische Aktivität an den noradrenergen α1B/2C Rezeptoren im gleichen sub-nanomolaren Ki-Bereich (Ki: 0,2–0,6 nM). Die partiell agonistische Aktivität am 5HT1A/D2 Rezeptor in Kombination mit der antagonistischen Aktivität an den 5-HT2A und α1B/2C Rezeptoren trägt möglicherweise zur antipsychotischen Wirkung von Brexpiprazol bei.
Pharmakodynamik
Brexpiprazol hat eine hohe Affinität (Ki <5 nM) zu multiplen monoaminergen Rezeptoren, darunter Serotonin 5-HT1A, 5-HT2A, 5-HT2B und 5-HT7, Dopamin D2 und D3 sowie noradrenerge α1A, α1B, α1D und α2C Rezeptoren. Brexpiprazol wirkt als Partialagonist an den 5-HT1A, D2 und D3 Rezeptoren und als Antagonist an den 5-HT2A, 5-HT2B, 5-HT7, α1A, α1B, α1D und α2C Rezeptoren.
An den dopaminergen D2 Rezeptoren besitzt Brexpiprazol eine schwache intrinsiche Aktivität. Brexpiprazol hat eine mässige Affinität zu Histamin H1 Rezeptoren (19 nM) und eine sehr schwache Affinität zu muscarinischen M1 Rezeptoren (67% Inhibition bei 10 µM). Die Dosis-Wirkungs-Okkupanz sowie das Verhältnis der Gehirn/Plasma Exposition wurden in präklinischen Studien invivo oder exvivo für die D2/D3, 5-HT2A, 5-HT1A, 5-HT6 und 5-HT7 Rezeptoren sowie für den 5-HT-Transporter bestimmt. Die Resultate stimmen mit den relativen invitro Bindungsaffinitäten überein und zeigen die Aktivität von Brexpiprazol an mehreren Angriffspunkten im zentralen Nervensystem bei relevanter Plasma Exposition.
In einer umfassenden QTc-Studie bei Patienten mit Schizophrenie oder schizoaffektiver Störung, bewirkte REXULTI nach 12 Tagen therapeutischer (4 mg/Tag) oder supra-therapeutischer (12 mg/Tag) Dosierung keine QTcF Verlängerung. Es wurde keine Korrelation zwischen den Brexpiprazol Konzentrationen und QTcF Verlängerung beobachtet.
Klinische Wirksamkeit
Schizophrenie
Erwachsene:
Die Wirksamkeit von REXULTI bei der Behandlung von erwachsenen Patienten mit Schizophrenie gemäss DSM-IV-TR Kriterien, wurde in zwei randomisierten, doppel-blinden, Placebo-kontrollierten 6-Wochen Studien mit fixer Dosierung (Studien 331-10-231 und 331-10-230) und einer Langzeitstudie zur Erhaltungstherapie (Studie 331-10-232) gezeigt.
Für in Frage kommende Patienten wurde eine Spitalaufnahme oder eine Verlängerung des Spitalaufenthalts zur Behandlung eines akuten Rückfalls von Schizophrenie als vorteilhaft befunden. Sie mussten im Voraus eine angemessene ambulante Behandlung mit Antipsychotika erhalten und innerhalb der letzten 12 Monate vor Studienbeginn gut auf diese Behandlung (ausser Clozapin) angesprochen haben.
In den Studien 331-10-231 und 331-10-230 wurden die Patienten auf Tagesdosen von REXULTI 2 mg oder 4 mg oder Placebo randomisiert. Patienten in den REXULTI Gruppen begannen die Behandlung mit 1 mg einmal täglich am Tag 1. Am Tag 5 wurde die REXULTI Dosis auf 2 mg einmal täglich erhöht. Am Tag 8 wurde die Dosis, abhängig vom Behandlungsarm, für die restlichen 5 Studienwochen entweder bei 2 mg belassen oder auf 4 mg einmal täglich erhöht.
Die Studien 331-10-231 und 331-10-230 umfassten auch zwei Behandlungsarme mit tiefer REXULTI Dosierung von 0.25 mg/Tag und 1 mg/Tag. Diese wurden jedoch nicht in die primäre Analyse aufgenommen.
Der primäre Wirksamkeits-Endpunkt für beide Studien war die Veränderung des Gesamtscores der Positive and Negative Syndrome Scale (PANSS) vom Ausgangswert bis Woche 6. Die PANSS umfasst 30 Items und misst die positiven (7 Items) und negativen (7 Items) Symptome der Schizophrenie sowie die allgemeine Psychopathologie (16 Items). Jedes Item wird auf einer Skala von 1 (nicht vorhanden) bis 7 (extrem) bewertet. Der PANSS Gesamt-score reicht von 30 (bestes Ergebnis) bis 210 (schlechtestes Ergebnis). Der wichtigste sekundäre Endpunkt der Studien war die Veränderung auf der Clinical Global Impression – Severity of Illness Scale (CGI S) vom Ausgangswert bis Woche 6. Die CGI S ist eine validierte, vom Arzt bewertete Skala, die den aktuellen klinischen Zustand des Patienten in Bezug auf die Symptom-schwere bewertet. Weitere sekundäre Endpunkte waren unter anderen die Personal and Social Performance Scale (PSP), eine validierte, vom Arzt bewertete Skala, welche die persönliche und soziale Funktionsfähigkeit misst.
In Studie 331-10-231 war REXULTI in beiden Dosierungen, 2 mg/Tag und 4 mg/Tag, bezüglich des PANSS-Gesamtscores gegenüber Placebo überlegen (Tabelle 3). In Studie 331-10-230 war REXULTI in einer Dosis von 4 mg/Tag im Hinblick auf den PANSS-Gesamtscore gegenüber Placebo überlegen (Tabelle 3). Für die Dosis 2 mg/Tag wurde nur in Studie 331-10-231 eine statistisch signifikante Wirksamkeit gezeigt. Aus der Untersuchung der Populationsuntergruppen auf Basis von Alter, Geschlecht und ethnischer Zugehörigkeit ergaben sich keine Hinweise auf Unterschiede bei der Ansprechempfindlichkeit.
Tabelle 3 zeigt die Ergebnisse für den primären Wirksamkeitsendpunkt für die Studien 331-10-231 und 331-10-230.
Tabelle 3: Primäre Wirksamkeitsresultate für die 6-Wochen Studien in Schizophrenie (Studien 331-10-231 und 331-10-230)
|
|
Primärer Wirksamkeitsparameter: PANSS
| |
Studie
|
Behandlungs-gruppe
|
N
|
Mittlerer Ausgangswert (SD)
|
LS mittlere Abweichung vom Ausgangswert (SE)
|
LS mittlere Differenza (95% KI)
|
p-Wert
| |
331-10-231
|
REXULTI (2 mg/Tag)*
|
180
|
95,85 (13,75)
|
-20,73 (1,55)
|
-8,7
(-13,1; -4,4)
|
<0,0001
| |
REXULTI (4 mg/Tag)*
|
178
|
94,70 (12,06)
|
-19,65 (1,54)
|
-7,6
(-12,0; -3,1)
|
0,0006
| |
Placebo
|
178
|
95,69 (11,46)
|
-12,01 (1,60)
|
--
|
-
| |
331-10-230
|
REXULTI
(2 mg/Tag)
|
179
|
96,30 (12,91)
|
-16,61 (1,49)
|
-3,1
(-7,2; 1,1)
|
0,1448
| |
REXULTI (4 mg/Tag)*
|
181
|
94,99 (12,38)
|
-20,00 (1,48)
|
-6,5
(-10,6; -2,4)
|
0,0022
| |
Placebo
|
180
|
94,63 (12,84)
|
-13,53 (1,52)
|
--
|
-
|
SD: standard deviation (Standardabweichung); SE: standard error (Standardfehler); LS: Least-Square, KI: unadjusted confidence interval (unbereinigtes Konfidenzintervall).
* REXULTI dem Placebo statistisch signifikant überlegen
a Differenz (REXULTI minus Placebo) der Least-Square mittleren Veränderung vom Ausgangswert bis Woche 6
REXULTI war in den Dosierungen 2 mg/Tag und 4 mg/Tag in den sekundären Endpunkten, CGI-S-Score und PSP-Gesamtscore, Placebo überlegen. Somit bestätigen die Ergebnisse für den CGI-S- und den PSP-Score sekundäre Endpunkte die Überlegenheit und klinisch relevante Wirkung von REXULTI im Vergleich zu Placebo.
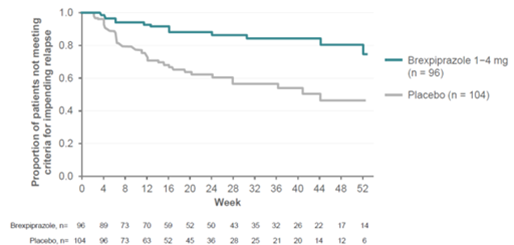
Ein vorher festgelegte Interimsanalyse zeigte bei den in die REXULTI Gruppe (1–4 mg/Tag) randomisierten Patienten eine statistisch signifikant längere Zeit bis zum drohenden Rückfall als bei den in die Placebo Gruppe randomisierten Patienten (p = 0,0008, Log-Rank Test). Die Schlussanalyse bestätigte die statistisch signifikant längere Zeit bis zum drohenden Rückfall für Patienten in der REXULTI Gruppe gegenüber der Placebo Gruppe (p < 0,0001, Log-Rank Test). Die Kaplan-Meier Kurven des kumulativen Anteils der Patienten im finalen Analyse Set, die während der doppel-blinden Behandlungsphase in den REXULTI und Placebo Gruppen einen Rückfall erlitten, sind in Abbildung 1 dargestellt. Bei der Schlussanalyse betrug die Hazard-Ratio aus dem Cox Proportional Hazard Model für den Vergleich von Placebo und REXULTI 3,42 (95% KI: 1,82; 6,41, Rückfallrate 13,54% vs. 38,46%). Somit hatten Patienten in der Placebo Gruppe ein 3,42-fach erhöhtes Risiko für einen drohenden Rückfall als Patienten in der REXULTI Gruppe.
Abbildung 1: Kaplan-Meier Kurven für die Zeit bis zum drohenden Rückfall (doppel-blinde Erhaltungsphase, Wirksamkeits-Stichprobe) – Schlussanalyse, Studie 3
Jugendliche ab 13 Jahren
Die Wirksamkeit und Sicherheit von Brexpiprazol bei der Behandlung von pädiatrischen Patienten mit Schizophrenie wurde in einer 6-wöchigen, randomisierten, doppelblinden und placebokontrollierten Studie (Studie 6) untersucht und in einer laufenden langfristigen 24-monatige Open-Label-Studie. An der Kurzzeitstudie nahmen 110 Patienten teil, die randomisiert Brexpiprazol erhielten, 101 Patienten, die aufgrund der Testempfindlichkeit Aripiprazol erhielten, und 104 Patienten, die randomisiert Placebo erhielten. Das Durchschnittsalter betrug 15 Jahre.
In der Kurzzeitstudie (Studie 6) begannen Patienten in der Brexpiprazol-Gruppe die Behandlung mit 0,5 mg einmal täglich an den Tagen 1 bis 4 und dann mit 1 mg täglich an den Tagen 5 bis 7. Die Brexpiprazol-Dosis wurde an den Tagen 8 bis 14 auf 2 mg erhöht. Die Dosis wurde dann entweder bei 2 mg belassen oder von den Tagen 15 bis 21 auf 3 mg einmal täglich erhöht. Nach der Titrationsphase behielten die Patienten entweder die Erhaltungsdosis bei oder erhöhten bzw. verringerten sie um 1 mg auf maximal 4 mg Brexpiprazol täglich.
Der primäre Wirksamkeitsendpunkt wurde als mittlere Veränderung vom Ausgangswert bis Woche 6 in den Gesamtscores der Positive and Negative Syndrome Scale (PANSS) definiert.
Brexpiprazol 2 bis 4 mg/Tag zeigte im Vergleich zu Placebo eine statistisch signifikante Verbesserung der mittleren Veränderung vom Ausgangswert im PANSS-Gesamtscore.
Tabelle 4: Primäre Wirksamkeitsresultate für die 6-Wochen Studien in Schizophrenie in paediatrischen Patienten
|
Studie
|
Behandlungsgruppe
|
N
|
Primärer Wirksamkeitsparameter: PANSS
| |
|
|
|
Mittlerer Ausgangswert (SD)
|
LS mittlere Abweichung vom Ausgangswert (SE)
|
LS mittlere Differenza (95% KI)
|
p-Wert
| |
6
|
Brexpiprazole (2 mg/Tag bis 4 mg/Tag)*
|
110
|
101.06
(14.87)
|
-22.75
(1.49)
|
-5.33
(-9.55, -1.10)
|
0.0136
| |
|
Aripiprazole (10 mg/Tag bis 20 mg/Tag)
|
101
|
101.03
(13.08)
|
-23.95
(1.57)
|
-6.53
(-10.8, -2.21)
|
0.0032
| |
|
Placebo
|
103b
|
102.17
(16.30)
|
-17.42
(1.58)
|
--
|
--
|
SD: standard deviation (Standardabweichung);
SE: standard error (Standardfehler);
LS Mean: Least-Square mean
KI: unadjusted confidence interval (unbereinigtes Konfidenzintervall)
* Behandlung dem Placebo statistisch signifikant überlegen
a Differenz (Brexpiprazol minus Placebo) in kleinsten Quadraten mittlerer Veränderung vom Ausgangswert, in Woche 6
b Die Wirksamkeitsstichprobe umfasst behandelte Probanden, die eine Baseline- und mindestens eine Post-Baseline-Wirksamkeitsbewertung für den PANSS-Gesamtscore aufweisen.
Langzeitstudie bei Jugendlichen ab 13 Jahren mit Schizophrenie
Zwischenanalysen aus Langzeitstudien mit flexiblen Dosen von Brexpiprazol von 1 bis 4 mg/Tag zeigten eine anhaltende Verbesserung der Symptome vom Ausgangswert bis zum 24. Monat im PANSS-Gesamtscore.
Agitiertheit bei Alzheimer-Demenz
Die Wirksamkeit von REXULTI bei der Behandlung der Agitiertheit bei Alzheimer-Demenz (AAD) wurde in zwei 12-wöchigen randomisierten, doppelblinden, placebokontrollierten Studien mit fixer Dosis (Studie 331-12-283 und 331-14-213) gezeigt. Die Studienpatienten mussten eine Diagnose einer wahrscheinlichen Alzheimer-Krankheit nach den Kriterien des National Institute of Neurological and Communicative Diseases and Stroke/Alzheimer's Disease and Related Disorders Association (NINCDS-ADRDA) und einen Mini-Mental-Examination (MMSE)-Score von ≥5 bis ≤22 erfüllen.
Bei der Aufnahme in die Studie mussten die Patienten einen Gesamtscore ≥4 für das Item Agitation/Aggression des NPI/NPI-NH (Neuropsychiatric Inventory-Nursing Home Version) aufweisen. Die Patienten mussten bei der Aufnahme in die Studie so starke agitierte Verhaltensweisen aufweisen, dass nach Ausschluss anderer Ursachen eine Pharmakotherapie gerechtfertigt war.
AAD-Patienten, die in der Vergangenheit auf zwei oder mehr Antipsychotika nicht angesprochen hatten, waren vom klinischen Entwicklungsprogramm REXULTI ausgeschlossen.
An Studie 331-12-283 nahmen 433 Patienten mit einem Durchschnittsalter von 74 Jahren (Bereich: 51 bis 90 Jahre) teil. An Studie 331-14-213 nahmen 345 Patienten mit einem Durchschnittsalter von 74 Jahren (Bereich: 56 bis 90 Jahre) teil.
In Studie 331-12-283 bestanden bei 78 (18%) der mit REXULTI behandelten Patienten und 36 (8%) der mit Placebo behandelten Patienten vor Studienbeginn psychotische Symptome. In Studie 331-14-213 berichteten 44 (19%) der mit REXULTI behandelten Patienten und 21 (18%) der mit Placebo behandelten Patienten psychotische Symptome in der Vorgeschichte.
Der primäre Wirksamkeitsendpunkt in allen Studien war die Änderung der Cohen-Mansfield Agitation Inventory (CMAI)-Scores zwischen der Ausgangsuntersuchung und Woche 12. Der CMAI ist ein von Ärzten bewerteter Fragebogen, der aus 29 Elementen besteht und die Häufigkeit der Manifestationen von Agitiertheit bei älteren Patienten auf der Grundlage der Eingaben des Pflegepersonals bewertet.
Patienten in Studie 331-14-213 mussten zusätzlich zu den allgemeinen Einschlusskriterien zu Studienbeginn die Kriterien für CMAI-Faktor 1 (aggressives Verhalten) erfüllen. Dafür muss eines der folgenden Merkmale vorliegen: ≥1 aggressive Verhaltensweise, die mehrmals pro Woche auftritt, oder ≥2 aggressive Verhaltensweisen, die ein- oder zweimal pro Woche auftreten, oder ≥3 aggressive Verhaltensweisen, die weniger als einmal pro Woche auftreten. Aggressives Verhalten bezieht sich auf Schlagen (einschliesslich sich selbst), Treten, Kratzen, Greifen, Stossen, Verletzen von sich selbst oder anderen, Werfen von Gegenständen, Fluchen oder verbale Aggression, Spucken, Zerreissen von Gegenständen oder Zerstörung von Eigentum, Schreien und Beissen. Zudem mussten die Patienten in Studie 331-14-213 das Kriterium Agitiertheit bei Patienten mit kognitiven Störungen nach der vorläufigen Konsensusdefinition der International Psychogeriatric Association (IPA) (2014) erfüllen.
Die Patienten in Studie 331-12-283 erhielten randomisiert eine feste Dosis von REXULTI 1 mg einmal täglich, 2 mg einmal täglich oder Placebo. In dieser Studie zeigten Patienten, die REXULTI 2 mg einmal täglich erhielten, in Woche 12 eine Verbesserung der CMAI-Scores gegenüber den Patienten, die Placebo erhielten. Die Patienten in Studie 331-14-213 erhielten randomisiert eine feste Dosis von REXULTI 2 mg oder 3 mg einmal täglich (kombinierter Behandlungsarm) oder Placebo. In dieser Studie zeigten Patienten, die REXULTI 2 mg oder 3 mg einmal täglich erhielten, in Woche 12 eine Verbesserung der CMAI-Scores gegenüber den Patienten, die Placebo erhielten.
Wie in Tabelle 5 gezeigt, war die mittlere Veränderung des CMAI-Scores in Woche 12 bei den Patienten, die mit 2 mg/Tag oder 3 mg/Tag REXULTI behandelt worden waren, signifikant grösser als unter Placebo. Unter der Dosis von 1 mg einmal täglich zeigte sich in dieser Patientenpopulation keine signifikant grössere Veränderung des CMAI-Scores gegenüber dem Ausgangswert.
Tabelle 5: Änderung des CMAI-Gesamtscores Ausgangswert bis Woche 12 bei Patienten mit Agitiertheit bei Alzheimer-Demenz in Studie 331-12-283 und Studie 331-14-213
|
Studie
|
Behandlungsgruppe
|
N
|
Mittlerer Ausgangswert
(SD)
|
Veränderung des LS Mean
(SE)
|
Unterschied zwischen Behandlungen†
(95%-KI)
| |
331-12-283
|
REXULTI 1 mg/Tag
|
134
|
70,5 (16,0)
|
-17,6 (1,3)
|
0,2 (-3,4, 3,9)
| |
REXULTI 2 mg/Tag‡
|
138
|
71,0 (16,6)
|
-21.6 (1.3)
|
-3,8 (-7,4, -0,2)
| |
Placebo
|
131
|
72,2 (17,9)
|
-17,8 (1,3)
|
—
| |
331-14-213*
|
REXULTI 2 mg/Tag oder 3 mg/Tag‡
|
225
|
80,6 (16,6)
|
-22,6 (1,1)
|
-5.3 (-8,8, -1,9)
| |
Placebo
|
116
|
79,2 (17,5)
|
-17,3 (1,4)
|
—
|
SD: Standardabweichung, SE: Standardfehler, LS Mean: angepasster Mittelwert (least-square mean), KI: nicht adjustiertes Konfidenzintervall
† Differenz (Arzneimittel minus Placebo) zwischen den Veränderungen der angepassten Mittelwerte seit dem Ausgangswert
‡ Dosierungen statistisch signifikant überlegen gegenüber Placebo
* Angereicherte Wirksamkeitsprobe (AAD-Probanden mit einem Minimum an aggressivem Verhalten, definiert als CMAI-Faktor-1-Kriterium).
Die Untersuchung des primären Endpunkts in Populationsuntergruppen (nach Alter, ethnischer Abstammung oder Geschlecht) ergab keine Unterschiede bezüglich des Ansprechens zwischen diesen Gruppen.
Für die Studie 331-14-213 wurde eine Post-hoc-Reaktionsanalyse durchgeführt, bei der eine klinisch bedeutsame Veränderung (Meaningful Within Patient Change (MWPC)) auf Brexpiprazol als eine Änderung des CMAI-Gesamtscores um mindestens 20 Punkte vom Ausgangswert bis Woche 12 definiert wurde. Der Anteil der Patienten mit einem MWPC betrug 57 % bei den mit REXULTI behandelten Patienten und 37 % bei den mit Placebo behandelten Patienten.
Langzeitdaten
Schizophrenie
Die Langzeitstudie (Studie 331-10-232) war eine randomisierte Placebo-kontrollierte doppelblinde Studie zur Messung der Wirksamkeit, Sicherheit und Verträglichkeit von REXULTI 1–4 mg/Tag als Erhaltungstherapie bei Erwachsenen mit Schizophrenie. Die Brexpiprazol-Dosierung konnte in dieser Studie nach Ermessen des Prüfers angepasst werden. Die häufigste Modaldosis für stabilisierte Patienten, die randomisiert Brexpiprazol zugeordnet waren, betrug 4 mg/Tag (64 Patienten, 66%), gefolgt von 3 mg/Tag (25 Patienten, 25,8%), 2 mg/Tag (7 Patienten, 7,2%) und 1 Patient (1%) erhielt eine Modaldosis von 1 mg/Tag.
Agitierheit bei Alzheimer Demenz
Eine 12-wöchige offene Verlängerungsstudie (Studie 331-201-00182) zur Bewertung der Sicherheit und Verträglichkeit von Brexpiprazol bei Patienten mit Unruhe im Zusammenhang mit Demenz vom Alzheimer-Typ zeigte nach 12-wöchiger Behandlung ähnliche mittlere CMAI-Gesamtwerte bei Patienten, die zuvor in der vorausgegangenen kontrollierten Studie mit Brexpiprazol oder Placebo behandelt worden waren. Bei unterschiedlichen mittleren CMAI-Ausgangswerten (vorherige Brexpiprazol Gruppe 57,3 [SD 17,2] gegenüber vorherige Placebo-Gruppe 62,9 [18,1] war die mittlere (SD) Veränderung gegenüber dem Ausgangswert bei den 86 Probanden in der vorherigen Placebogruppe wie zu erwarten grösser (-12,5 [SD 16,6]) als bei den 140 Probanden in der vorherigen Brexpiprazol-Gruppe (−7,1 [SD 12,3]).
PharmakokinetikAbsorption
Nach Verabreichung von REXULTI Tabletten wird Brexpiprazol gut absorbiert, mit Spitzen-Plasmakonzentrationen innerhalb von 4,0 bis 8 Stunden nach Verabreichung einer Einzeldosis. Die absolute orale Bioverfügbarkeit der Tablettenformulierung beträgt 95,1%. Die Steady-State Konzentration von Brexpiprazol wird nach 10–12 Tagen Verabreichung erreicht und sind etwa 4-fach höher als nach der ersten Dosis. REXULTI kann mit oder ohne Nahrung eingenommen werden. Die Einnahme einer Tablette REXULTI 4 mg zusammen mit einer fettreichen Standardmahlzeit hatte keinen signifikanten Effekt auf die Cmax oder AUC von Brexpiprazol. Nach Einzel- und Mehrfach-Dosen von bis zu 8 mg einmal täglich erhöhte sich die Brexpiprazol Exposition (Cmax und AUC) proportional zur verabreichten Dosis. In-vitro Studien gaben keinen Hinweis darauf, dass Brexpiprazol ein Substrat für Efflux-Transporter wie MDR1 (P-gp) und BCRP ist.
Distribution
Das Distributionsvolumen von Brexpiprazol nach intravenöser Verabreichung ist hoch (1,56 ± 0,418 l/kg), was auf eine extravaskuläre Distribution hinweist. Brexpiprazol zeigt im Plasma eine starke Proteinbindung an Serumalbumin (über 99%) und saures α1-Glykoprotein. Seine Proteinbindung wird durch Nieren- oder Leberfunktionsstörungen nicht beeinflusst. Resultate aus in-vitro Studien zeigen keine Beeinflussung der Proteinbindung von Brexpiprazol durch Warfarin, Diazepam oder Digoxin.
Metabolismus
TEX In-vitro Metabolismus Studien mit Brexpiprazol unter Verwendung von rekombinantem humanem Cytochrom P450 (CYP1A1, 1A2, 2A6, 2B6, 2C8, 2C9, 2C19, 2D6, 2E1 und 3A4) zeigen, dass Brexpiprazol hauptsächlich durch CYP3A4 und CYP2D6 metabolisiert wird.
In-vivo wird Brexpiprazol in erster Linie von den Enzymen CYP3A4 und CYP2D6 metabolisiert. Nach ein- und mehrmaliger Anwendung, sind Brexpiprazol und ein Hauptmetabolit, DM-3411, die vorherrschenden Arzneimittelanteile im systemischen Kreislauf. DM-3411 macht im Steady State 23,1–47,7% der Brexpiprazol Exposition (AUC) im Plasma aus. Allerdings zeigten präklinische in-vivo Studien, dass bei klinisch relevanten Plasmaexpositionen von Brexpiprazol die Exposition von DM-3411 im Hirn unter der Nachweisgrenze lag. Daher trägt DM-3411 wahrscheinlich nicht zur therapeutischen Wirkung von Brexpiprazol bei.
Elimination
Nach einer oralen Einzeldosis von [14C]-markiertem Brexpiprazol wurden etwa 24,6% der verabreichten Radioaktivität im Urin und 46% in den Fäzes nachgewiesen. Weniger als 1% unverändertes Brexpiprazol wurde im Urin ausgeschieden und etwa 14% der oralen Dosis wurden unverändert in den Fäzes nachgewiesen. Die scheinbare orale Clearance von Brexpiprazol nach Verabreichung einer Tablette täglich beträgt 19,8 (±11,4) ml/h/kg. Nach mehrmaliger Verabreichung von REXULTI einmal täglich beträgt die terminale Eliminationshalbwertszeit von Brexpiprazol 91,4 Stunden und vom Hauptme-taboliten DM-3411 85,7 Stunden.
Kinetik spezieller Patientengruppen
Alter / Geschlecht
Nach Verabreichung einer Einzeldosis REXULTI (2 mg) zeigten gesunde ältere Probanden (über 65 Jahre) eine ähnliche systemische Brexpiprazol Exposition (Cmax und AUC) wie erwachsene Probanden (18–45 Jahre). Weibliche Probanden zeigten eine um etwa 40–50% höhere systemische Brexpiprazol Exposition (Cmax und AUC) als männliche Probanden. Bei der Populationspharmakokinetischen Untersuchung wurden Alter und weibliches Geschlecht als statistisch signifikante Kovariaten ermittelt, die die Pharmakokinetik von Brexpiprazol beeinflussen, aber nicht als klinisch relevant beurteilt werden.
Kinder und Jugendliche
Eine PK-Studie mit Mehrfachdosen (0,5, 1, 2, 3 oder 4 mg/Tag) wurde an 43 pädiatrischen Patienten im Alter von 13 bis 17 Jahren durchgeführt. Eine populationsbasierte PK-Analyse ergab, dass die systemische Verfügbarkeit (Cmax und AUC) von Brexpiprazol bei pädiatrischen Patienten (13 bis 17 Jahre) im gesamten Dosisbereich von 0,5 bis 4 mg mit der bei erwachsenen Patienten vergleichbar war.
Ethnische Zugehörigkeit
Obwohl keine spezielle pharmakokinetische Studie zur Auswirkung der ethnischen Zugehörigkeit auf die Disposition von Brexpiprazol durchgeführt wurde, ergab die Populations-pharmakokinetische Beurteilung keinen Hinweis auf klinisch signifikante Unterschiede aufgrund ethnischer Zugehörigkeit in der Pharmakokinetik von Brexpiprazol.
Langsame CYP2D6 Metabolisierer
Etwa bei 8% der Kaukasier und bei 3–8% der Schwarzen/Afroamerikaner fehlt die Fähigkeit, CYP2D6 Substrate zu metabolisieren. Sie werden als langsame Metabolisierer (poor metabolisers, PM) klassifiziert, im Gegensatz zu den extensiven Metabolisierern (extensive metabolisers, EM). Die Brexiprazol Exposition ist bei CYP2D6 PMs etwa 1,8-fach höher als bei EMs. (siehe Dosierung/Anwendung).
Rauchen
In-vitro-Studien mit humanen Leberenzymen zeigen, dass Brexpiprazol kein Substrat für CYP1A2 ist. Rauchen sollte sich somit nicht auf die Pharmakokinetik von Brexpiprazol auswirken.
Leberfunktionsstörungen
Patienten mit mässigen bis schweren Leberfunktionsstörungen (Child-Pugh-Klassifikation ≥7) wiesen in der Regel eine höhere Brexpiprazol Exposition auf als Patienten mit normaler Leberfunktion. Daher sollte die maximal empfohlene Dosis reduziert werden. Bei Probanden mit Leberfunktionsstörungen unterschiedlicher Grade (Child-Pugh-Klassen A, B und C), stieg die AUC von oralem Brexpiprazol (2 mg Einzeldosis) im Vergleich zu entsprechenden gesunden Probanden um 24% bei leichter und um 60% bei mässiger Leberfunktionsstörung, während sie bei schwerer Leberfunktionsstörung unverändert blieb.
Nierenfunktionsstörungen
Patienten mit Nierenfunktionsstörung (CLcr < 60 ml/min) wiesen eine höhere Brexpiprazol Exposition auf als Patienten mit normaler Nierenfunktion. Daher sollte die maximal empfohlene Dosis reduziert werden. Bei Probanden mit schwerer Nierenfunktionsstörung (CLcr < 30 ml/min) stieg die AUC von oralem Brexpiprazol (2 mg Einzeldosis) im Vergleich zu entsprechenden gesunden Probanden um 69%, während die Cmax unverändert blieb.
Präklinische DatenSicherheitspharmakologie
Bei Ratten induzierte Brexpiprazol in hohen Dosen (ED50 = 20 mg/kg) eine Katalepsie (Tiermodell für das Risiko eines extrapyramidalen Syndroms [EPS]). Dies zeigt, dass Brexpiprazol unter klinischen Bedingungen ein geringes Potenzial zur Auslösung eines EPS aufweist.
Beim Hund und beim Affen wurden ein erniedrigter Blutdruck und eine Verlängerung des QT- und des QTc-Intervalls festgestellt. Die Wirkung auf den Blutdruck wurde auf eine Blockade der α1-Adrenorezeptoren in den peripheren Gefässen zurückgeführt, was mit dem pharmakologischen Profil von Brexpiprazol entspricht. Die QT- und die QTc-Verlängerung hängen möglicherweise mit der pharmakologisch vermittelten Senkung der Körpertemperatur zusammen; allerdings hemmte Brexpiprazol auch den Ionentransport durch den hERG-Kanal (mit einem IC50-Wert von 0,117 µM).
Die wichtigsten klinischen Zeichen zur Festlegung des NOAEL (No-Observed-Adverse-Effect-Level, Dosis ohne beobachtete schädliche Wirkung) in den Studien zur Toxizität bei wiederholter Gabe an Ratten und Affen wurden auf die pharmakologisch bedingte Aktivitätsminderung des Zentralnervensystem (Verminderung der Bewegungen, Tremor, Schläfrigkeit, geschlossene Augen, Hypothermie) zurückgeführt. Sonstige klinische Zeichen wie zum Beispiel bei der Ratte beobachtete klonische Krampfanfälle, Hyperaktivität und Aggressivität waren nur von kurzer Dauer.
Wie bei anderen Antipsychotika ist die therapeutische Breite, gemessen anhand der AUC bei Ratten und Affen sowie der Exposition des Menschen bei der maximal empfohlenen Dosis von 4 mg/Tag, relativ gering (Männer/Frauen: 0,1/0,8 und 0,4/0,4 bei Ratten bzw. Affen).
Generell wurden die bei juvenilen Ratten und Hunden beobachteten Wirkungen nach der Gabe von Brexpiprazol auch bei erwachsenen Tieren festgestellt; es reagierten also nicht nur Jungtiere empfindlich auf Brexpiprazol.
Mutagenität
Das mutagene Potenzial von Brexpiprazol wurde im in-vitro Rückmutationstest an Bakterien, im in-vitro Vorwärts-Genmutationstest an Lymphomzellen der Maus, im in-vitro Chromosomen Aberrationstest mit Ovarialzellen des chinesischen Hamsters (CHO), im in-vivo Mikronukleus Test an Ratten sowie im unplanmässigen DNA-Synthese Test an Ratten untersucht. Bei in-vitro-Tests mit Säugetierzellen war Brexpiprazol mutagen und klastogen, aber nur in Dosen, die Zytotoxizität induzierten. In anderen Studien wurde keine Mutagenität oder Genotoxizität beobachtet. Aufgrund der Beweislage wird Brexpiprazol nicht als genotoxisches Risiko für den Menschen beurteilt.
Kanzerogenität
Das lebenslange karzinogene Potenzial von Brexpiprazol wurde in einer zwei-Jahres Studie an ICR Mäusen und Sprague-Dawley Ratten untersucht. Den Mäusen wurde Brexpiprazol während zwei Jahren oral verabreicht (Schlundsonde) in einer Dosierung von 0,75, 2 und 5 mg/kg/Tag (mit der 0,3-3,1-fachen (Männchen) und 0,2-1,1-fachen (Weibchen) Plasma AUC verglichen mit der klinischen Exposition bei der maximal empfohlenen oralen Dosis von 4 mg beim Menschen [MRHD]). Bei den männlichen Tieren nahm die Häufigkeit von Tumoren in keiner Dosisgruppe zu. Bei weiblichen Mäusen stieg die Häufigkeit von Adenokarzinomen der Brustdrüsen, von adenosquamösen Karzinomen sowie von pars-distalis Adenomen der Hypophyse. Den Ratten wurde Brexpiprazol während zwei Jahren oral verabreicht (Schlundsonde) in einer Dosierung von 1, 3 und 10 mg/kg/Tag an männliche Ratten und von 3, 10 und 30 mg/kg/Tag an weibliche Ratten (mit Plasma AUC-Expositionen bis zu 1,0-fach (Männchen) und 4,4-fach (Weibchen) erhöht im Vergleich zur Exposition bei der 4 mg/Tag MRHD). Bei Ratten war Brexpiprazol nicht karzinogen.
Fertilitätsstörungen
Die Auswirkungen von Brexpiprazol auf die Fertilität wurden an weiblichen und männlichen Ratten untersucht. Brexpiprazol wurde weiblichen Ratten vor der Paarung mit unbehandelten Männchen bis zur Empfängnis und Einnistung der Eizelle einmal täglich oral per Schlundsonde verabreicht in Dosen von 0; 0,3; 3 oder 30 mg/kg/Tag mit einer Exposition (Plasma-AUC) bis zum 4,1-fachen der klinischen Exposition bei Anwendung der MRHD). Bei 3 und 30 mg/kg/Tag wurden ein verlängerter Diöstrus und verringerte Fertilität beobachtet, wobei die Exposition (Plasma) um das ≥0,2-fache über der klinischen Exposition bei der MRHD lag. Bei 30 mg/kg/Tag (AUC-Exposition 4,1-fache höher als bei der klinischen Exposition mit MHRD) wurden eine geringfügige Verlängerung der Paarungsphase sowie eine signifikante Zunahme der Verluste vor Einnisten der Eizelle beobachtet. Der no observed adverse effect level für Brexpiprazol war 0,3 mg/kg/Tag (das 0,7-fache der oralen MRHD von 4 mg für einen 60 kg Patienten, berechnet anhand eines Vergleichs der Körperoberfläche).
An männliche Ratten wurde Brexpiprazol einmal täglich oral per Schlundsonde verabreicht in Dosierungen von 0, 3, 10 oder 100 mg/kg/Tag mit einer bis zu 11,6-fach erhöhten Exposition (Plasma AUC) verglichen mit der klinischen Exposition bei der MRHD. Nach 63 Tagen Verabreichung wurden die behandelten Männchen für maximal 14 Tage mit unbehandelten Weibchen zusammengebracht. Es wurden keine erkennbaren Unterschiede in der Paarungs-dauer oder den Fertilitätsindizes in irgendeiner der mit Brexpiprazol behandelten Gruppen festgestellt.
Entwicklungstoxizität und Laktation
In Studien zur Entwicklungstoxizität war Brexpiprazol nicht teratogen und hatte keine unerwünschten Wirkungen auf die Entwicklung. In diesen Studien erhielten trächtige Ratten und Kaninchen während der Phase der Organogenese Brexpiprazol in Dosen von bis zu 30 mg/kg/Tag wobei die Exposition (Plasma AUC), 4,1-fach bzw. 5,9-fach über der klinischen Exposition bei MRHD betrugen).
In der Embryo-fötalen Entwicklungsstudie an Kaninchen, mit einer zu materneller Toxizität führenden oralen Dosis (150 mg/kg/Tag, 16,5-fache AUC Exposition verglichen zur klinischen Exposition) wurden bei den Föten verringertes Körpergewicht, verzögerte Knochenbildung und gehäuftes Auftreten von Anomalien der Eingeweide und des Skeletts beobachtet.
Wurde Brexpiprazol an trächtige Ratten während der Periode von Organogenese bis zur Laktation verabreicht, erhöhte sich die Zahl der perinatalen Todesfälle der Jungen bei 30 mg/kg/Tag (4,1-fach erhörte Plasma AUC-Exposition verglichen zur klinischen Exposition bei der 4 mg MRHD); die No-Effect-Dosis lag bei 10 mg/kg/Tag (0,8-fache Exposition verglichen zur klinischen Exposition).
Wirkung auf das Serum-Prolaktin
Nach chronischer Verabreichung von Antipsychotika an Nager wurden proliferative und/oder neoplastische Veränderungen der Brustdrüsen und der Hypophyse beobachtet, welche wahrscheinlich durch Prolaktin vermittelt werden. Brexpiprazol zeigte sowohl bei Mäusen wie auch bei Ratten ein Potenzial zur Erhöhung der Serum Prolaktinspiegel. Die Relevanz dieser Daten zu Prolaktin vermittelten endokrinen Tumoren bei Nagern für das Risiko beim Menschen ist nicht bekannt.
Missbrauchspotenzial
Brexpiprazol zeigte weder ein Potenzial zur Erzeugung physischer Abhängigkeit bei Ratten noch eine verstärkende Wirkung bei Rhesusaffen. In einer Studie zum Medikamenten Missbrauchspotenzial bei Ratten, wurden keine Entzugserscheinungen, die eine physische Abhängigkeit nahelegen, nachgewiesen. Brexpiprazol wird kein Potenzial zur Erzeugung physischer Abhängigkeit zugeschrieben.
Sonstige HinweiseHaltbarkeit
Das Arzneimittel darf nur bis zu dem auf der Packung mit «EXP» bezeichneten Datum verwendet werden.
Besondere Lagerungshinweise
Ausser Reichweite von Kindern aufbewahren.
Nicht über 30°C lagern.
In der Originalverpackung aufbewahren.
Zulassungsnummer66475 (Swissmedic)
PackungenREXULTI (Brexpiprazol) Filmtabletten sind erhältlich als:
Filmtabletten 0,5 mg: 7 [B]
Filmtabletten 1 mg: 10, 28 [B]
Filmtabletten 2 mg: 28 [B]
Filmtabletten 3 mg: 28 [B]
Filmtabletten 4 mg: 28 [B]
ZulassungsinhaberinLundbeck (Schweiz) AG, Opfikon
Stand der InformationSeptember 2025
09122025FI
|