Propriétés/EffetsCode ATC
A10BD24
Mécanisme d'action
Steglujan
Steglujan associe deux médicaments antidiabétiques ayant des mécanismes d'action complémentaires pour améliorer le contrôle glycémique chez les patients atteints de diabète de type 2: l'ertugliflozine, un inhibiteur du SGLT2, et le phosphate de sitagliptine, un inhibiteur de la DPP-4.
Ertugliflozine
Le SGLT2 est le principal transporteur chargé de la réabsorption du glucose du filtrat glomérulaire vers la circulation sanguine. L'ertugliflozine est un inhibiteur du SGLT2. Par le biais de l'inhibition du SGLT2, l'ertugliflozine réduit la réabsorption rénale du glucose filtré et diminue le seuil rénal pour le glucose, augmentant ainsi l'excrétion urinaire du glucose.
Phosphate de sitagliptine
Le phosphate de sitagliptine est un inhibiteur puissant et hautement sélectif de l'enzyme dipeptidylpeptidase-4 (DPP-4), actif par voie orale pour le traitement du diabète de type 2. Les inhibiteurs de la DPP-4 sont une classe de substances actives ayant un effet renforçant les incrétines. Par le biais de l'inhibition de l'enzyme DPP-4, la sitagliptine augmente la concentration des deux hormones incrétines actives connues, le GLP-1 (glucagon-like peptide-1) et le GIP (glucose-dependent insulinotropic polypeptide). Les incrétines font partie d'un système endogène impliqué dans la régulation physiologique de l'homéostasie du glucose. Lorsque la glycémie est normale ou élevée, le GLP-1 et le GIP favorisent la synthèse et la libération d'insuline par les cellules bêta du pancréas. De plus, le GLP-1 diminue la sécrétion de glucagon par les cellules alpha du pancréas, ce qui entraîne une réduction de la production hépatique de glucose. Ce mécanisme se distingue du mécanisme connu chez les sulfonylurées; ces dernières provoquent une libération d'insuline y compris en cas de valeurs glycémiques basses, ce qui peut entraîner une hypoglycémie induite par les sulfonylurées chez les patients atteints de diabète de type 2 et les personnes en bonne santé. La sitagliptine est un inhibiteur puissant et hautement sélectif de l'enzyme DDP-4 et n'inhibe pas les enzymes étroitement apparentées DPP-8 ou DPP-9 aux concentrations thérapeutiques. La sitagliptine, par sa structure chimique et son action pharmacologique, se distingue des analogues du GLP-1, de l'insuline, des sulfonylurées ou des méglitinides, des biguanides, des agonistes de PPARγ (agonistes du récepteur gamma activé par les proliférateurs de peroxysomes), des inhibiteurs de l'alpha-glucosidase et des analogues de l'amyline.
Pharmacodynamique
Ertugliflozine
Excrétion urinaire du glucose et volume d'urine
Des augmentations dose-dépendantes de la quantité de glucose excrétée dans l'urine ont été observées chez des sujets sains et chez des patients atteints de diabète de type 2 suite à l'administration de doses uniques et de doses multiples d'ertugliflozine. Une modélisation dose-réponse indique que l'ertugliflozine 5 mg et 15 mg entraîne une excrétion urinaire du glucose (EUG) quasi maximale, avec une EUG renforcée de façon incrémentielle à la dose de 15 mg par rapport à la dose de 5 mg. L'EUG renforcée est maintenue après administration de doses multiples. L'EUG ciblée avec l'ertugliflozine entraîne également une augmentation du volume urinaire.
Électrophysiologie cardiaque
Dans une étude croisée, randomisée, contrôlée par placebo avec un comparateur actif, 42 sujets sains ont reçu une dose orale unique suprathérapeutique de 100 mg d'ertugliflozine (correspondant à 6,7 fois la dose maximale recommandée), de la moxifloxacine et le placebo. À la dose de 100 mg d'ertugliflozine, aucun prolongement de l'intervalle QTc n'a été observé.
Sitagliptine
Généralités
Chez des patients atteints de diabète de type 2, l'administration d'une dose unique orale de sitagliptine a pendant 24 heures un effet inhibiteur sur l'activité enzymatique de la DPP-4, ce qui entraîne une augmentation des taux sanguins de GLP-1 et de GIP actifs d'un facteur allant de 2 à 3, une augmentation des concentrations plasmatiques d'insuline et de peptide C, une baisse des concentrations en glucagon, une baisse de la glycémie à jeun et une baisse de l'excursion glycémique après une dose de glucose administrée par voie orale ou un repas.
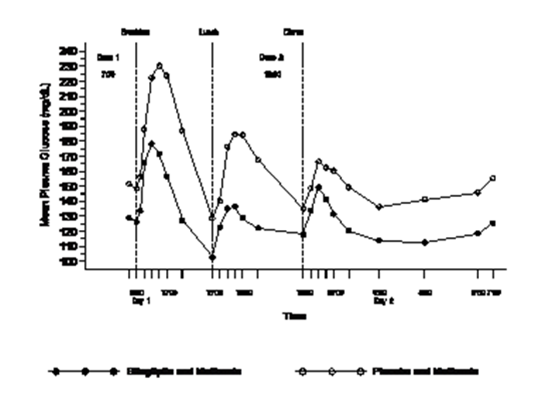
Dans une étude portant sur des patients atteints de diabète de type 2 insuffisamment contrôlé par la metformine en monothérapie, les taux de glycémie observés au cours de la journée chez des patients ayant reçu quotidiennement de la sitagliptine 100 mg (50 mg deux fois par jour) en association avec la metformine étaient significativement plus bas par rapport aux patients ayant reçu le placebo associé à de la metformine (voir figure 1).
Figure 1: Profil de la glycémie plasmatique sur 24 heures après 4 semaines de traitement par la sitagliptine 50 mg deux fois par jour associée à la metformine ou par placebo associé à la metformine
Dans des études cliniques de phase III, d'une durée de 18 à 24 semaines, la fonction des cellules bêta de patients atteints de diabète de type 2 a été notablement améliorée grâce au traitement par 100 mg par jour de sitagliptine. Cet élément a été évalué par plusieurs marqueurs, dont le marqueur HOMA-β (Homeostasis Model Assessment-β), le rapport proinsuline/insuline et des mesures de la sensibilité des cellules bêta du test de tolérance au glucose avec des prélèvements fréquents.
Dans des études de phase II, aucun effet glycémique supplémentaire n'a été atteint avec 50 mg de sitagliptine deux fois par jour en comparaison à la sitagliptine 100 mg une fois par jour.
Dans une étude croisée, réalisée sur 4 périodes de temps, randomisée, contrôlée par placebo, à double insu et en double aveugle, portant sur des sujets adultes sains, les effets sur les concentrations plasmatiques du GLP-1 actif et total et sur le glucose après un repas, après administration simultanée de sitagliptine et de metformine, ont été comparés aux effets après chaque administration biquotidienne de la sitagliptine seule, de la metformine seule ou du placebo. Les concentrations incrémentielles moyennes pondérées du GLP-1 actif quatre heures après le repas étaient, après administration de la sitagliptine seule ou de la metformine seule, environ 2 fois plus élevées en comparaison au placebo. L'effet sur les concentrations du GLP-1 actif après administration simultanée de sitagliptine et de metformine était additif, les concentrations du GLP-1 actif étant environ 4 fois plus élevées en comparaison au placebo. La sitagliptine seule n'a fait qu'augmenter les concentrations de GLP-1 actif, ce qui reflète l'inhibition du DPP-4, alors que la metformine seule a entraîné l'augmentation des concentrations du GLP-1 actif et total dans des proportions similaires. Ces données correspondent aux différents mécanismes impliqués dans l'augmentation des concentrations du GLP-1 actif. Les résultats de l'étude ont par ailleurs montré que la sitagliptine, mais pas la metformine, augmentait les concentrations du GIP actif.
Dans des études portant sur des sujets sains, la sitagliptine n'a pas entraîné de baisse des taux de glycémie ni d'hypoglycémie, ce qui indique que les effets insulinotropes et suppresseurs de glucagon du médicament sont dépendants du glucose.
Effet sur la pression artérielle
Dans une étude croisée, randomisée, contrôlée par placebo portant sur des patients atteints d'hypertension et traités par un ou plusieurs antihypertenseurs (incluant des inhibiteurs de l'enzyme de conversion de l'angiotensine, des antagonistes de l'angiotensine II, des inhibiteurs du canal calcique, des bêtabloquants et des diurétiques), l'administration concomitante de sitagliptine a généralement été bien tolérée. Chez ces patients, la sitagliptine a eu un effet hypotenseur modéré; une dose de 100 mg de sitagliptine par jour a entraîné une baisse de 24 heures de la pression artérielle systolique ambulatoire moyenne d'environ 2 mmHg, en comparaison au placebo. Chez les sujets avec une pression artérielle normale, aucun effet hypotenseur n'a été observé.
Électrophysiologie cardiaque
Dans une étude croisée, randomisée, contrôlée par placebo, 79 sujets sains ont reçu une dose orale unique de 100 mg de sitagliptine, une dose de 800 mg de sitagliptine (correspondant à 8 fois la dose recommandée) ou un placebo. À la dose recommandée de 100 mg, aucun effet n'a été observé sur l'intervalle QTc à la concentration plasmatique maximale ou à tout autre moment de l'étude. Après l'administration de la dose de 800 mg, l'augmentation maximale de la modification moyenne de l'intervalle QTc corrigée par placebo, par rapport à la valeur initiale, 3 heures après l'administration de la dose, était de 8,0 ms. Cette légère augmentation n'a pas été considérée comme cliniquement significative. Lors de l'administration de la dose de 800 mg, les concentrations plasmatiques maximales de sitagliptine étaient environ 11 fois supérieures aux concentrations maximales après une dose de 100 mg.
Chez les patients atteints de diabète de type 2 ayant reçu quotidiennement 100 mg de sitagliptine (N=81) ou 200 mg de sitagliptine (N=63), aucune modification significative de l'intervalle QTc n'a été constatée sur la base des données de l'ECG réalisé au moment où la concentration plasmatique maximale attendue a été atteinte.
Efficacité clinique
Études sur le contrôle glycémique chez des patients atteints de diabète de type 2
L'efficacité et la sécurité de l'ertugliflozine associée à la sitagliptine ont été évaluées dans 3 études cliniques de phase III, multicentriques, randomisées, en double aveugle, contrôlées par placebo et par un comparateur actif et portant sur 1985 patients atteints de diabète de type 2. Ces études comprenaient différents sous-groupes en fonction de la race (Blancs, Noirs, Asiatiques et autres) ou de l'origine ethnique (Hispaniques et personnes appartenant à d'autres ethnies); les patients étaient âgés de 21 à 85 ans.
Le traitement par l'ertugliflozine en association avec la sitagliptine a entraîné, chez des patients atteints de diabète de type 2, une amélioration cliniquement et statistiquement significative de l'HbA1c et de la glycémie plasmatique à jeun par rapport au placebo ou au comparateur actif.
Les patients atteints de diabète de type 2 traités par l'ertugliflozine en association à la sitagliptine ont présenté une amélioration de l'HbA1c similaire au sein des sous-groupes définis en fonction de l'âge, du sexe et de l'origine ethnique.
Étude factorielle avec l'ertugliflozine et la sitagliptine en traitement complémentaire à la metformine
Au total, 1233 patients atteints de diabète de type 2 ont participé à une étude multicentrique, randomisée, en double aveugle, contrôlée par un comparateur actif, d'une durée de 26 semaines, visant à évaluer l'efficacité et la sécurité de 5 mg d'ertugliflozine ou de 15 mg d'ertugliflozine en association avec 100 mg de sitagliptine par rapport aux principes actifs individuels. Des patients atteints de diabète de type 2 dont la glycémie n'était pas contrôlée de façon adéquate par la metformine en monothérapie (≥1500 mg/jour) ont été randomisés dans un des cinq bras de traitement actif: ertugliflozine 5 mg ou 15 mg, sitagliptine 100 mg, ou sitagliptine 100 mg en association avec 5 mg ou 15 mg d'ertugliflozine, une fois par jour en complément du traitement de fond en cours à base de metformine.
À la semaine 26, une amélioration statistiquement significative de l'HbA1c et de la glycémie à jeun a été démontrée sous ertugliflozine en association avec la sitagliptine 100 mg, par rapport aux principes actifs individuels (voir Tableau 5). Plus de patients ont atteint un taux d'HbA1c <7% sous ertugliflozine en association avec la sitagliptine 100 mg que sous traitement par les principes actifs individuels. En outre, le traitement par l'ertugliflozine en association avec la sitagliptine 100 mg a entraîné une réduction statistiquement significative du poids corporel et de la pression artérielle systolique en comparaison à la sitagliptine 100 mg.
Tableau 5: Résultats à la semaine 26 d'une étude factorielle sur l'ertugliflozine et la sitagliptine en traitement complémentaire à la metformine par rapport à chacun des principes actifs individuels*
|
|
Ertugliflozine 5 mg
|
Sitagliptine 100 mg
|
Ertugliflozine 5 mg + sitagliptine 100 mg
| |
HbA1c (%)
|
N = 250
|
N = 247
|
N = 243
| |
Valeur au début de l'étude (moyenne)
|
8,6
|
8,5
|
8,6
| |
Modification par rapport au début de l'étude (moyenne LS†)
|
-1,0
|
-1,1
|
-1,5
| |
Différence par rapport à
la sitagliptine
l'ertugliflozine 5 mg
|
|
|
-0,4‡ (-0,6; -0,3)
-0,5‡ (-0,6; -0,3)
| |
(Moyenne LS†, IC à 95%)
|
|
|
| |
Patients [N (%)] avec HbA1c <7%
|
66 (26,4)
|
81 (32,8)
|
127 (52,3) §
| |
Glycémie à jeun (mg/dl)
|
N = 250
|
N = 247
|
N = 243
| |
Valeur au début de l'étude (moyenne)
|
184,1
|
177,4
|
183,8
| |
Modification par rapport au début de l'étude (moyenne LS†)
|
-35,7
|
-25,6
|
-44,0
| |
Différence par rapport à
la sitagliptine
l'ertugliflozine 5 mg
(Moyenne LS†, IC à 95%)
|
|
|
-18,4‡ (-24,0; -12,8)
-8,2¶ (-13,8; -2,7)
| |
Poids corporel (kg)
|
N = 250
|
N = 247
|
N = 243
| |
Valeur au début de l'étude (moyenne)
|
88,6
|
89,8
|
89,5
| |
Modification par rapport au début de l'étude (moyenne LS†)
|
-2,7
|
-0,7
|
-2,5
| |
Différence par rapport à la sitagliptine
(moyenne LS†, IC à 95%)
|
|
|
-1,8‡ (-2,5; -1,2)
| |
Pression artérielle systolique (mmHg)
|
N = 250
|
N = 247
|
N = 243
| |
Valeur au début de l'étude (moyenne)
|
129,7
|
128,3
|
130,2
| |
Modification par rapport au début de l'étude (moyenne LS†)
|
-3,9
|
-0,7
|
-3,4
| |
Différence par rapport à la sitagliptine
(moyenne LS†, IC à 95%)
|
|
|
-2,8¶ (-4,7; -0,8)
|
* N inclut la totalité des patients randomisés et traités pour lesquels au moins une mesure de la variable d'évaluation était disponible.
† Moyennes des moindres carrés ajustées en fonction du temps, du DFGe initial et de l'interaction temps/traitement.
‡ p <0,001 par rapport au groupe de contrôle.
§ p <0,001 par rapport au groupe de contrôle (sur la base de comparaisons ajustées pour l'odds ratio d'après un modèle de régression logistique utilisant des imputations multiples pour les données manquantes).
¶ p ≤0,005 par rapport au groupe de contrôle.
Ertugliflozine en traitement complémentaire à la metformine et à la sitagliptine
Au total, 463 patients atteints de diabète de type 2 dont la glycémie n'était pas contrôlée de façon adéquate par la metformine (≥1500 mg/jour) et la sitagliptine 100 mg une fois par jour ont participé à un essai multicentrique, randomisé, en double aveugle, contrôlé par placebo, d'une durée de 26 semaines, visant à évaluer l'efficacité et la sécurité de l'ertugliflozine. Les patients ont été randomisés pour recevoir de l'ertugliflozine 5 mg ou 15 mg ou un placebo une fois par jour en complément du traitement de fond en cours par la metformine et la sitagliptine.
À la semaine 26, des améliorations statistiquement significatives de l'HbA1c, de la glycémie à jeun, du poids corporel et de la pression artérielle systolique ont été démontrées sous ertugliflozine par rapport au placebo. En outre, sous ertugliflozine, une plus grande proportion de patients ont atteint un HbA1c <7% par rapport au placebo (voir Tableau 6).
Tableau 6: Résultats à la semaine 26 d'une étude en traitement complémentaire d'ertugliflozine à la metformine et à la sitagliptine*
|
|
Ertugliflozine 5 mg
|
Placebo
| |
HbA1c (%)
|
N = 156
|
N = 153
| |
Valeur au début de l'étude (moyenne)
|
8,1
|
8,0
| |
Modification par rapport au début de l'étude (moyenne LS†)
|
-0,8
|
-0,1
| |
Différence par rapport au placebo (moyenne LS†, IC à 95%)
|
-0,7‡ (-0,9; -0,5)
|
| |
Patients [N (%)] avec HbA1c <7%
|
50 (32,1)§
|
26 (17,0)
| |
Glycémie à jeun (mg/dl)
|
N = 156
|
N = 153
| |
Valeur au début de l'étude (moyenne)
|
167,7
|
169,6
| |
Modification par rapport au début de l'étude (moyenne LS†)
|
-26,9
|
-1,8
| |
Différence par rapport au placebo (moyenne LS†, IC à 95%)
|
-25,2‡ (-32,8; -17,5)
|
| |
Poids corporel (kg)
|
N = 156
|
N = 153
| |
Valeur au début de l'étude (moyenne)
|
87,6
|
86,5
| |
Modification par rapport au début de l'étude (moyenne LS†)
|
-3,3
|
-1,3
| |
Différence par rapport au placebo (moyenne LS†, IC à 95%)
|
-2,0‡ (-2,6; -1,4)
|
| |
Pression artérielle systolique (mmHg)
|
N = 156
|
N = 153
| |
Valeur au début de l'étude (moyenne)
|
132,1
|
130,2
| |
Modification par rapport au début de l'étude (moyenne LS†)
|
-3,8
|
-0,9
| |
Différence par rapport au placebo (moyenne LS†, IC à 95%)
|
-2,9¶ (-5,4; -0,5)
|
|
* N inclut la totalité des patients randomisés et traités pour lesquels au moins une mesure de la variable d'évaluation était disponible.
† Moyennes des moindres carrés ajustées en fonction du temps, du traitement antihyperglycémiant antérieur, du DFGe initial et de l'interaction temps/traitement.
‡ p ≤0,001 par rapport au placebo.
§p <0,001 par rapport au placebo (sur la base de comparaisons ajustées pour l'odds ratio d'après un modèle de régression logistique utilisant des imputations multiples pour les données manquantes).
¶ p <0,05 par rapport au placebo.
Événements cardiovasculaires chez des patients atteints de diabète de type 2 et d'une affection cardiovasculaire établie (étude VERTIS-CV)
Ertugliflozine
L'effet de l'ertugliflozine sur le risque cardiovasculaire chez des patients adultes atteints de diabète de type 2 et d'une maladie cardiovasculaire athérosclérotique stable établie a été évalué dans l'étude VERTIS-CV, une étude multicentrique, multinationale, randomisée, en double aveugle, contrôlée contre placebo, basée sur les événements. L'étude a comparé le risque d'événements cardiovasculaires indésirables graves (Major Adverse Cardiac Event, MACE) entre l'ertugliflozine et le placebo. Les deux groupes ont été traités dans l'étude selon les directives thérapeutiques actuelles.
Au total, 8246 patients ont été randomisés (ertugliflozine 5 mg N=2752, ertugliflozine 15 mg N=2747 ou placebo N=2747) et suivis pendant une durée médiane de 3 ans. Environ 88% de la population de l'étude était caucasienne, 6% asiatique et 3% noire. L'âge moyen était de 64 ans et environ 70% étaient des hommes.
À l'inclusion, tous les patients de l'étude avaient un diabète de type 2 insuffisamment contrôlé (HbA1c ≥7%). La durée moyenne du diabète de type 2 était de 13 ans, l'HbA1c moyenne à l'inclusion était de 8,2% et le DFGe moyen était de 76 ml/min/1,73 m2. À l'inclusion, les patients étaient traités par un (32%) ou plusieurs (67%) antidiabétiques, dont la metformine (76%), l'insuline (47%), les sulfonylurées (41%), les inhibiteurs de la DPP-4 (11%) et les agonistes des récepteurs du GLP-1 (3%).
À l'inclusion, presque tous les patients (99%) avaient une maladie cardiovasculaire athérosclérotique établie, dont: des antécédents documentés de coronaropathie (76%), de maladie cérébrovasculaire (23%) ou d'artériopathie périphérique (19%). Environ 24% des patients avaient des antécédents d'insuffisance cardiaque (IC). À l'inclusion, la pression systolique moyenne était de 133 mmHg, la pression diastolique moyenne était de 77 mmHg, le taux moyen de LDL était de 89 mg/dl et le taux moyen de HDL de 44 mg/dl. À l'inclusion, environ 81% des patients étaient traités par des inhibiteurs du système rénine-angiotensine, 69% par des bêtabloquants, 43% par des diurétiques, 82% par des statines, 4% par l'ézétimibe et 89% par des antiagrégants plaquettaires.
Le critère d'évaluation principal de l'étude VERTIS-CV était le délai jusqu'à la première survenue d'un événement cardiovasculaire indésirable grave (MACE). Un événement cardiovasculaire indésirable grave était défini comme la survenue d'un décès d'origine cardiovasculaire, d'un infarctus du myocarde (IM) non fatal ou d'un accident vasculaire cérébral (AVC) non fatal.
Le taux d'incidence des MACE était semblable chez les patients traités par l'ertugliflozine et chez les patients ayant reçu le placebo. Le hazard ratio estimé des MACE associés à l'ertugliflozine par rapport au placebo était de 0,97 pour un intervalle de confiance de 95,6% (0,85, 1,11). La limite supérieure de cet intervalle de confiance excluait un risque supérieur à 1,3 (Tableau 7). Les résultats pour les doses individuelles de 5 mg et de 15 mg étaient cohérents avec les résultats pour les groupes de doses combinées.
Tableau 7: Analyse des MACE et de ses composants et de l'hospitalisation en cas d'insuffisance cardiaque de l'étude VERTIS-CV*
|
|
Placebo (N=2747)
|
Ertugliflozine (N=5499)
|
| |
Critère d'évaluation†
|
N (%)
|
Taux d'événements (pour 100 personnes-années)
|
N (%)
|
Taux d'événements (pour 100 personnes-années)
|
Hazard ratio vs placebo
(IC) ‡
| |
MACE (décès d'origine CV, IM non fatal ou AVC non fatal)
|
327 (11,9)
|
4,0
|
653 (11,9)
|
3,9
|
0,97
(0,85, 1,11)
| |
IM non fatal
|
148 (5,4)
|
1,6
|
310 (5,6)
|
1,7
|
1,04
(0,86, 1,27)
| |
AVC non fatal
|
78 (2,8)
|
0,8
|
157 (2,9)
|
0,8
|
1,00
(0,76, 1,32)
| |
Décès d'origine CV
|
184 (6,7)
|
1,9
|
341 (6,2)
|
1,8
|
0,92
(0,77, 1,11)
|
N=nombre de patients, IC=intervalle de confiance, CV=cardiovasculaire, IM=infarctus du myocarde, AVC=accident vasculaire cérébral.
* Ensemble d'analyse en intention de traiter.
† Le critère MACE a été évalué chez les sujets qui ont pris au moins une dose du médicament de l'étude et chez les sujets qui ont arrêté le médicament de l'étude avant la fin de celle-ci, les événements survenus plus de 365 jours après la dernière dose du médicament de l'étude ont été censurés. D'autres critères ont été évalués pour tous les sujets randomisés et les événements survenus à tout moment après la première dose du médicament de l'étude jusqu'à la dernière date de contact. Le nombre total de premiers événements a été analysé pour chaque critère d'évaluation.
‡ Pour le critère MACE, un IC de 95,6% est représenté et pour les autres critères d'évaluation, un IC de 95%.
Sitagliptine
Étude TECOS
L'étude TECOS (Trial Evaluating Cardiovascular Outcomes with Sitagliptin) était une étude randomisée réalisée chez 14'671 patients de la population en intention de traiter ayant des valeurs d'HbA1c comprises entre ≥6,5 et 8,0% et présentant une maladie cardiovasculaire avérée. Les patients ont reçu soit 100 mg de sitagliptine par jour (7332) (ou 50 mg par jour lorsque la valeur initiale du DFGe était ≥30 et <50 ml/min/1,73 m2) soit un placebo (7339), en complément de leur traitement habituel avec des valeurs cibles conformes aux normes régionales pour l'HbA1c et les facteurs de risque cardiovasculaire. Les patients présentant un DFGe <30 ml/min/1,73 m2 n'ont pas été inclus dans l'étude. La population étudiée comprenait 2004 patients âgés de ≥75 ans et 3324 patients présentant une insuffisance rénale (DFGe <60 ml/min/1,73 m2).
Les patients du groupe sitagliptine ont reçu moins de médicament antihyperglycémiant que ceux du groupe placebo (Hazard Ratio 0,72; IC à 95%, 0,68-0,77; p ≤0,001). De plus, chez les patients qui n'étaient pas sous insuline au début de l'étude, la mise en place d'un traitement chronique par insuline était moins probable (Hazard-Ratio 0,70; IC à 95%, 0,63-0,79; p <0,001).
Le critère d'évaluation cardiovasculaire primaire était une composition de la première survenue d'un décès d'origine cardiovasculaire, d'un infarctus du myocarde non fatal, d'un accident vasculaire cérébral non fatal ou d'une hospitalisation due à une angine de poitrine instable. Les critères d'évaluation cardiovasculaires secondaires étaient la première survenue d'un décès d'origine cardiovasculaire, d'un infarctus du myocarde non fatal ou d'un accident vasculaire cérébral non fatal; la première survenue des composants individuels de la composition primaire; la mortalité globale; et les hospitalisations pour insuffisance cardiaque.
Après un temps de suivi médian de trois ans, la sitagliptine, ajoutée au traitement habituel chez les patients atteints de diabète de type 2, n'a augmenté ni le risque d'événements cardiovasculaires indésirables majeurs ni le risque d'hospitalisation pour insuffisance cardiaque (voir Tableau 8).
Tableau 8: Taux de résultat pour les événements cardiovasculaires et événements secondaires importants
|
|
Sitagliptine 100 mg
|
Placebo
|
Hazard ratio
(IC à 95%)
|
Valeur de p†
| |
N (%)
|
Taux d'incidence pour 100 patients-années*
|
N (%)
|
Taux d'incidence pour 100 patients-années*
| |
Analyse de la population en intention de traiter
| |
Nombre de patients
|
7332
|
7339
|
0,98 (0,89-1,08)
|
<0,001
| |
Critère d'évaluation primaire composé
(décès d'origine cardiovasculaire, infarctus du myocarde non fatal, accident vasculaire cérébral non fatal ou hospitalisation due à une angine de poitrine instable)
|
839 (11,4)
|
4,1
|
851 (11,6)
|
4,2
| |
Critère d'évaluation secondaire composé
(décès d'origine cardiovasculaire, infarctus du myocarde non fatal ou accident vasculaire cérébral non fatal)
|
745 (10,2)
|
3,6
|
746 (10,2)
|
3,6
|
0,99 (0,89-1,10)
|
<0,001
| |
Événements secondaires
| |
Décès d'origine cardiovasculaire
|
380 (5,2)
|
1,7
|
366 (5,0)
|
1,7
|
1,03 (0,89-1,19)
|
0,711
| |
Tous les infarctus du myocarde (fatals ou non fatals)
|
300 (4,1)
|
1,4
|
316 (4,3)
|
1,5
|
0,95 (0,81-1,11)
|
0,487
| |
Tous les accidents vasculaires cérébraux (fatals ou non fatals)
|
178 (2,4)
|
0,8
|
183 (2,5)
|
0,9
|
0,97 (0,79-1,19)
|
0,760
| |
Hospitalisation due à une angine de poitrine instable
|
116 (1,6)
|
0,5
|
129 (1,8)
|
0,6
|
0,90 (0,70-1,16)
|
0,419
| |
Décès toutes causes confondues
|
547 (7,5)
|
2,5
|
537 (7,3)
|
2,5
|
1,01 (0,90-1,14)
|
0,875
| |
Hospitalisation pour insuffisance cardiaque‡
|
228 (3,1)
|
1,1
|
229 (3,1)
|
1,1
|
1,00 (0,83-1,20)
|
0,983
|
* Le taux d'incidence pour 100 patients-années a été calculé ainsi: 100 × (nombre total de patients avec ≥1 évènement pendant la période d'exposition décisive par nombre total de patients-années de suivi).
† Basé sur un modèle de Cox stratifié par région. Pour les critères d'évaluation composés, les valeurs p correspondent à un test de non-infériorité dans le but de démontrer que le Hazard Ratio est inférieur à 1,3. Pour tous les autres critères d'évaluation, les valeurs p correspondent à un test de la différence des Hazard Rates.
‡ L'analyse des hospitalisations pour insuffisance cardiaque a été ajustée en fonction des antécédents d'insuffisance cardiaque à l'inclusion dans l'étude.
|