CompositionPrincipes actifs
Omalizumab.
Excipients
Poudre: Saccharosum, L-Histidinum, L-Histidini hydrochloridum monohydras, Polysorbatum 20.
Solvant: Aqua ad iniectabilia.
Forme pharmaceutique et quantité de principe actif par unitéPoudre et solvant pour solution injectable.
Poudre: 75 mg resp. 150 mg de lyophilisat blanc à blanc cassé dans une ampoule perforable.
Indications/Possibilités d’emploiAsthme allergique
Xolair est indiqué en association avec d'autres traitements anti-asthmatiques pour améliorer le contrôle de l'asthme chez les adultes et les enfants (à partir de 6 ans) atteints d'asthme allergique persistant sévère (ayant un test cutané positif ou une réactivité in vitro contre un pneumallergène perannuel), et qui, malgré un traitement quotidien par un corticostéroïde inhalé à forte dose et un bêta2agoniste inhalé à longue durée d'action, présentent non seulement une réduction de la fonction pulmonaire (VEMS < 80%) mais aussi des symptômes diurnes fréquents ou des réveils nocturnes et des exacerbations de l'asthme.
Polypes nasaux
Xolair (omalizumab) est indiqué dans le traitement des polypes nasaux chez les adultes (à partir de 18 ans) qui ne répondent pas suffisamment aux corticostéroïdes intranasaux.
Urticaire chronique spontanée (UCS)
Xolair est indiqué, en traitement additionnel chez les adultes et les adolescents (à partir de 12 ans), dans l'urticaire chronique spontanée (UCS) de longue durée*, non contrôlée par les antihistaminiques H1 et pour laquelle aucune autre affection sous-jacente n'a été identifiée dans le cadre d'un examen effectué par un médecin ayant de l'expérience dans ce type d'affections.
(*Les études d'enregistrement ont porté sur des patients atteints d'une UCS évoluant depuis 6 mois à 66 ans, en moyenne depuis 6 ans)
Posologie/Mode d’emploiAsthme allergique
Posologie usuelle
Adultes et enfants à partir de 6 ans
La dose et la fréquence d'administration adaptées de Xolair sont déterminées en fonction du taux sérique initial d'immunoglobulines E (IgE) (UI/ml), mesuré avant le début du traitement, et du poids corporel (kg). Le taux des IgE sériques du patient devra être déterminé avant la mise en route du traitement par l'une des méthodes disponibles commercialement de dosage des IgE sériques totales afin de définir la dose de Xolair à administrer. Sur la base de ces mesures, une dose de 75 à 600 mg de Xolair en 1 à 4 injections pourra être nécessaire lors de chaque administration.
Les patients avec un taux d'IgE inférieur à 76 UI/ml étaient moins susceptibles de tirer bénéfice du traitement (cf. "Pharmacodynamique" ). Les médecins prescripteurs devront s'assurer que les patients chez qui le taux d'IgE est inférieur à 76 UI/ml ont une réactivité significative in vitro (RAST) à un pneumallergène perannuel avant de débuter le traitement.
Voir le tableau 1 pour la table de conversion et les tableaux 2 et 3 pour les tables de détermination de la dose. Pour des doses de 225 ou 375 mg, Xolair 150 mg peut être utilisé en combinaison avec Xolair 75 mg.
Les patients dont le taux initial d'IgE ou le poids corporel en kg est en dehors des valeurs limites figurant dans la table de détermination de la dose (≤20 ou > 150 kg poids corporel) ne doivent pas être traités par Xolair.
Pour plus d'informations sur les modalités de reconstitution de Xolair, cf. "Remarques particulières/Remarques concernant la manipulation" .
Tableau 1: Correspon
dance de la dose
pour chaque administ
ration en nombre
d'ampoules perforabl
es, nombre d'injecti
ons et en volume
total à injecter
Dose (mg) Nombre d'ampoules Nombre d'injections Volume total à
perforables injecter (ml)
75 mga 150 mgb
75 1c 0 1 0.6
150 0 1 1 1.2
225 1c 1 2 1.8
300 0 2 2 2.4
375 1c 2 3 3.0
450 0 3 3 3.6
600 0 4 4 4.8
a 0.6 ml = volume
maximal de produit
par ampoule perforab
le de Xolair 75 mg.
b 1.2 ml = volume
maximal de produit
par ampoule perforab
le de Xolair 150
mg. c ou utiliser
0.6 ml prélevé
d'une ampoule
perforable de
Xolair 150 mg
Afin d'assurer la traçabilité des médicaments biotechnologiques, il convient de documenter pour chaque traitement le nom commercial et le numéro de lot.
Durée du traitement
Une réduction des exacerbations de l'asthme et de la nécessité d'utiliser des médicaments à la demande ainsi qu'une amélioration de la symptomatologie ont été observées dans les études cliniques sur l'asthme allergique au cours des 16 premières semaines du traitement. Après 16 semaines de traitement par Xolair, le médecin doit réévaluer l'efficacité du traitement avant de poursuivre les injections. La décision de continuer le traitement par Xolair doit se baser sur l'observation d'une amélioration sensible du contrôle général de l'asthme (cf. "Pharmacodynamique" ).
Un arrêt du traitement entraîne généralement un retour à des taux élevés d'IgE libres et aux symptômes associés.
Ajustement de la posologie
Les taux d'IgE totales sont élevés au cours du traitement et le restent jusqu'à une année après l'arrêt du traitement. Par conséquent, un nouveau dosage du taux d'IgE au cours du traitement par Xolair ne peut pas être utilisé pour déterminer de nouveau les doses de Xolair à administrer. Après une interruption de traitement de moins d'une année, la dose à administrer doit être déterminée sur la base du taux d'IgE sériques mesuré lors de la détermination de la dose initiale (cf. "Propriétés/Effets" ). Les taux d'IgE totales ne devraient être mesurés afin de déterminer la dose à administrer que si l'interruption de traitement est d'une année ou plus.
La posologie devra être ajustée en cas de modification significative du poids corporel (cf. tableaux 2 et 3).
Tableau 2: ADMINISTR
ATION TOUTES LES 4
SEMAINES. Asthme
allergique et
polypes nasaux.
Doses de Xolair (mg
par dose) administré
es par injection
sous-cutanée toutes
les 4 semaines
Poids corporel (kg)
Taux initial d'IgE > 20–25 > 25–30 > 30–40 > 40–50 > 50–60 > 60–70 > 70–80 > 80-90 > 90–125 > 125–150
(UI/ml)
≥30–100 75 75 75 150 150 150 150 150 300 300
> 100–200 150 150 150 300 300 300 300 300 450 600
> 200–300 150 150 225 300 300 450 450 450 600
> 300–400 225 225 300 450 450 450 600 600
> 400–500 225 300 450 450 600 600
> 500–600 300 300 450 600 600
> 600–700 300 450 600 ADMINISTRATION
TOUTES LES 2 SEMAINE
S, VOIR TABLEAU 3
Tableau 3: ADMINISTR
ATION TOUTES LES 2
SEMAINES. Asthme
allergique et
polypes nasaux.
Doses de Xolair (mg
par dose) administré
es par injection
sous-cutanée toutes
les 2 semaines
Poids corporel (kg)
Taux initial d'IgE > 20–25 > 25–30 > 30–40 > 40–50 > 50–60 > 60–70 > 70–80 > 80–90 > 90–125 > 125–150
(UI/ml)
≥30–100 ADMINISTRATION
TOUTES LES 4 SEMAINE
S, VOIR TABLEAU 2
> 100–200
> 200–300 375
> 300–400
> 400–500 375 375
> 500–600 375 Données insuffisante
s pour une recommand
ation de posologie
> 600–700 225 375
La dose maximale recommandée est de 375 mg d'omalizumab toutes les 2 semaines.
Urticaire chronique spontanée (UCS)
Posologie usuelle
La dose initiale recommandée est de 300 mg sous la forme d'une injection sous-cutanée toutes les 4 semaines. La plupart des patients qui répondent au traitement montrent déjà une amélioration dans les 4 semaines qui suivent la première dose. Chez les patients qui ne répondent que partiellement à la première dose, la poursuite du traitement peut donner lieu à une amélioration des symptômes.
L'expérience clinique dans le traitement à long terme au-delà de 6 mois dans cette indication est limitée.
Il est conseillé au médecin traitant de régulièrement réévaluer la nécessité de poursuivre le traitement.
Les données cliniques montrent que certains patients peuvent parvenir à contrôler les symptômes moyennant des injections de Xolair 150 mg toutes les 4 semaines. C'est pourquoi on peut essayer, chez les patients libérés des papules (éruptions urticariennes) et du prurit sous Xolair 300 mg et antihistaminiques H1, de réduire les doses à 150 mg toutes les quatre semaines.
Polypes nasaux (à partir de 18 ans)
Dans les études sur les polypes nasaux menées en vue de l'autorisation, le même schéma posologique dépendant des IgE et du poids que celui décrit pour l'asthme allergique a été examiné chez des patients âgés de 18 ans ou plus ayant un poids corporel de 30 kg à 150 kg pour prouver l'efficacité du traitement. Le schéma posologique recommandé pour le traitement de l'urticaire chronique spontanée n'a pas été examiné pour le traitement de la polypose. Chez les patients atteints de polypes nasaux (à partir de 18 ans et d'un poids corporel de 30 kg à 150 kg), le même schéma posologique dépendant des IgE et du poids que celui utilisé chez les patients ayant de l'asthme allergique est donc recommandé (voir ci-dessus).
Instructions posologiques particulières
Patients présentant des troubles de la fonction hépatique et des troubles de la fonction rénale
Aucune étude sur les effets d'une insuffisance hépatique ou rénale sur la pharmacocinétique de Xolair n'a été effectuée. Comme la clairance de l'omalizumab aux posologies cliniques est déterminée par le système réticulo-endothélial (SRE), il est improbable qu'elle soit altérée par un trouble de la fonction hépatique ou rénale. Bien qu'aucun ajustement posologique ne soit recommandé, Xolair doit être utilisé avec prudence chez ces patients (cf. "Mises en garde et précautions" ).
Patients âgés
Il existe seulement des données limitées sur l'administration de Xolair à des patients âgés de 65 ans et plus, mais aucun élément ne suggère que les patients âgés aient besoin d'une posologie différente de celle utilisée chez les patients adultes plus jeunes.
Enfants et adolescents
Dans le cas de l'asthme allergique, la sécurité et l'efficacité de Xolair n'ont pas été étudiées chez les patients âgés de moins de 6 ans. Son utilisation chez ces patients est par conséquent déconseillée.
Dans le cas des polypes nasaux, la sécurité et l'efficacité n'ont pas été étudiées chez les patients âgés de moins de 18 ans.
Urticaire chronique spontanée
La sécurité et l'efficacité n'ont pas été étudiées chez les patients âgés de moins de 12 ans. L'utilisation de Xolair chez ces patients n'est donc pas recommandée.
Mode d'administration
Uniquement par injection sous-cutanée: ne pas administrer par voie intraveineuse ou intramusculaire.
Les injections sous-cutanées seront faites dans la région deltoïde du bras. En cas d'impossibilité, elles pourront être réalisées dans la cuisse.
Il existe peu d'expérience sur l'auto-injection de Xolair. Aussi, le médicament doit être administré par un personnel médical spécialisé.
Contre-indicationsHypersensibilité au principe actif ou à l'un des excipients.
Mises en garde et précautionsRéactions allergiques, réaction anaphylactique
Lors de l'utilisation d'omalizumab, des réactions allergiques locales ou systémiques, y compris réaction anaphylactique mettant en jeu le pronostic vital et choc anaphylactique, peuvent apparaître. De telles réactions peuvent aussi apparaître après une durée de traitement prolongée. La plupart de ces réactions sont survenues dans les 2 heures suivant la première injection et les injections suivantes de Xolair, mais certaines se sont produites plus de 2 heures après et même plus de 24 heures après l'injection. Par conséquent, des médicaments utilisés pour traiter une réaction anaphylactique devront toujours être disponibles pour une utilisation immédiate après administration de Xolair. Les patients doivent être informés que de telles réactions peuvent survenir et qu'elles nécessitent un traitement médical d'urgence quand des réactions allergiques surviennent. En cas de réactions sévères, le traitement par Xolair doit être arrêté immédiatement (cf. "Effets indésirables" ). Selon l'expérience post-marketing, une réaction anaphylactique et des réactions anaphylactoïdes ont été rapportées aussi bien après la première administration de Xolair qu'après les suivantes. Même si la plupart de ces réactions sont survenues dans les 2 heures suivant l'injection, certaines se sont produites seulement après 2 heures.
Une maladie sérique et des réactions de type maladie sérique, qui sont des réactions allergiques retardées de type III, ont été observées dans de rares cas chez des patients traités par des anticorps monoclonaux humanisés, y compris l'omalizumab. Leur survenue a généralement eu lieu 1 à 5 jours après l'administration de la première injection ou d'une injection ultérieure, mais elles se sont également produites après une longue durée de traitement. Les symptômes suggérant une maladie sérique comprennent une arthrite/arthralgie, un rash (urticaire ou autres formes), une fièvre et une lymphadénopathie. Les antihistaminiques et les corticostéroïdes peuvent être utiles pour prévenir ou traiter cette pathologie. Il faut indiquer aux patients qu'ils doivent signaler tout symptôme suspect.
Syndrome de Churg-Strauss et syndrome hyperéosinophilique
Les patients atteints d'un asthme allergique sévère peuvent dans de rares occasions présenter un syndrome hyperéosinophilique systémique ou une vascularite granulomateuse allergique à éosinophiles (syndrome de Churg-Strauss, granulomatose éosinophilique avec polyangéite) qui tous deux sont traités habituellement par des corticostéroïdes systémiques.
Immunogénicité
Comme avec tous les anticorps monoclonaux humanisés fabriqués par ADN recombinant, l'apparition d'anticorps dirigés contre l'omalizumab ne peut être exclue.
De faibles titres d'anticorps dirigés contre Xolair ont été détectés chez environ 1/1843 (< 0.1%) patients traités par Xolair. Ces données reflètent le pourcentage de patients dont les résultats au test ELISA visant à détecter des anticorps anti-Xolair ont été considérés comme positifs et sont fortement dépendants de la sensibilité et de la spécificité du test. En outre, l'incidence des réactions à anticorps positifs, observée dans le test, peut être influencée par différents facteurs tels que la manipulation des échantillons, le moment du prélèvement des échantillons, la prise simultanée d'autres médications et la présence d'autres maladies. La comparaison de l'incidence des anticorps anti-Xolair avec celle d'anticorps dirigés contre d'autres produits peut donc induire en erreur.
Maladies cérébrovasculaires
Dans les essais cliniques contrôlés ayant inclus des adultes et des adolescents de 12 ans et plus, on a rapporté davantage d'événements cérébrovasculaires, y compris des attaques ischémiques transitoires et des accidents vasculaires ischémiques, chez les patients sous Xolair que chez ceux du groupe contrôle (cf. "Effets indésirables" ).
Infestations parasitaires (helminthiases)
Les IgE pourraient être impliquées dans la réponse immunologique à certaines infestations par les helminthes. Dans une étude réalisée au Brésil, des patients ont été traités par l'omalizumab pendant 1 an dans une région à haut risque d'helminthiases. 53% (36/68) des patients traités par l'omalizumab et 42% (29/69) des patients sous placebo ont souffert d'une helminthiase, diagnostiquée par l'examen des selles. Il n'y a pas eu de différence statistiquement significative entre les deux groupes quant aux helminthiases. L'évolution, le degré de sévérité de la maladie et la réponse au traitement de l'infection ont été inchangées. Le taux d'helminthiases dans le programme clinique qui n'était pas conçu pour diagnostiquer de telles maladies, a été inférieur à 1 infection pour 1000 patients. La prudence est cependant de rigueur chez les patients présentant un risque élevé d'helminthiases, spécialement en cas de voyages dans des régions où les helminthiases sont endémiques. Si les patients ne répondent pas au traitement antihelminthique recommandé, l'arrêt de Xolair devra être envisagé.
Cancers
Lors des premiers essais cliniques, il a été observé un déséquilibre du nombre de cancers dans le groupe traité par Xolair par rapport au groupe contrôle. La diversité des types de cancers observés, la durée d'exposition relativement courte et les caractéristiques cliniques de chaque cas rendent improbable une relation de cause à effet. Le taux d'incidence global de cancers observé dans le programme d'études cliniques de Xolair a été comparable à celui rapporté dans la population générale. Des études ultérieures montrent que le risque relatif de cancers sous Xolair n'est pas accru. Cependant, l'ensemble des données actuellement disponibles ne permet pas encore d'exclure complètement l'éventualité d'un léger déséquilibre (cf. "Effets indésirables" ).
Général
Xolair n'est pas indiqué pour le traitement des exacerbations aiguës de l'asthme, du bronchospasme aigu ou de l'état de mal asthmatique.
Xolair n'a pas été étudié chez les patients présentant un syndrome d'hyperimmunoglobulinémie E, une aspergillose broncho-pulmonaire allergique ou en prévention de réactions allergiques.
Xolair n'a pas été suffisamment étudié dans les dermatites atopiques, les rhinites allergiques et les allergies alimentaires.
Le traitement par Xolair n'a pas été étudié chez les patients atteints de maladies auto-immunes ou d'états dus à des complexes immuns et chez ceux présentant une insuffisance rénale ou hépatique préexistante. La prudence est de mise en cas d'administration de Xolair chez ce type de patients.
L'arrêt brutal de la corticothérapie systémique ou inhalée en cas d'asthme allergique ou de polypes nasaux après l'initiation du traitement par Xolair n'est pas recommandé. La diminution de la posologie des corticoïdes doit se faire sous surveillance médicale et, le cas échéant, progressivement.
InteractionsLes enzymes du cytochrome P450, les pompes à efflux et les mécanismes liés à la fixation protéique n'interviennent pas dans la clairance de l'omalizumab; le risque d'interactions médicamenteuses est donc faible. Des études d'interactions spéciales entre Xolair et des médicaments ou des vaccins n'ont pas été effectuées. Du point de vue pharmacologique, aucun risque d'interactions entre l'omalizumab et les médicaments habituellement prescrits dans le traitement de l'asthme, des polypes nasaux ou de l'urticaire chronique spontanée (UCS) n'est attendu.
Asthme allergique
Lors des études cliniques, Xolair a été généralement utilisé en association avec des corticoïdes inhalés ou des corticoïdes oraux, des bêta-agonistes inhalés à courte durée d'action ou à longue durée d'action, des anti-leucotriènes, des théophyllines et des antihistaminiques oraux. Il n'a pas été mis en évidence de modification de la sécurité de Xolair en cas d'administration de ces médicaments antiasthmatiques d'utilisation courante.
L'efficacité du traitement par Xolair administré en association avec une immunothérapie spécifique n'a pas été établie.
Polypes nasaux
Lors des études cliniques, Xolair a été utilisé en association avec un spray nasal de mométasone conformément au plan d'essai. Les autres médicaments concomitants utilisés fréquemment ont inclus d'autres corticostéroïdes administrés par voie intranasale, des bronchodilatateurs, des antihistaminiques, des antagonistes des récepteurs des leucotriènes, des substances adrénergiques/sympathomimétiques et des anesthésiques locaux administrés par voie intranasale. Il n'y a eu aucun signe indiquant une réduction de la sécurité de Xolair en cas d'utilisation concomitante de ces autres médicaments fréquemment utilisés en cas de polypes nasaux.
Urticaire chronique spontanée (UCS)
Dans les essais cliniques sur l'UCS, Xolair a été utilisé en combinaison avec des antihistaminiques (anti-H1, anti-H2) et des antagonistes du récepteur des leucotriènes (LTRA). Dans les essais de phase III Q4881g et Q4882g, tous les patients ont reçu des antihistaminiques H1 en plus de Xolair ou d'un placebo. Dans l'étude de phase III Q4883g, tous les patients ont reçu un ou plusieurs antihistaminiques H1 et/ou H2 et/ou un LTRA en plus de Xolair ou d'un placebo. Rien n'indiquait que la prise concomitante des substances évoquées ci-dessus modifie la sécurité de l'omalizumab par rapport à son profil de sécurité rapporté dans l'asthme allergique. De plus, une analyse de pharmacocinétique de population n'a pas mis en évidence d'influence des antihistaminiques H2 et des LTRA sur la pharmacocinétique de l'omalizumab (cf. "Propriétés/Effets" ).
Grossesse, allaitementGrossesse
Il n'existe pas d'études cliniques bien contrôlées sur Xolair chez les femmes enceintes. Une étude prospective portant sur un registre de grossesse (EXPECT) menée sur 250 femmes enceintes asthmatiques traitées par Xolair a montré que la prévalence des anomalies congénitales majeures était similaire (8.1% vs 8.9%) chez les patientes ayant été traitées par Xolair (EXPECT) et les patientes asthmatiques (asthme modéré à sévère) n'ayant pas reçu Xolair. La proportion des enfants avec un poids à la naissance < 2.5 kg était plus élevé sous Xolair (13.7% vs 9.8%), ce qui peut toutefois également avoir été causé par des différences de degré de gravité de l'asthme. Dans l'ensemble, le risque d'anomalies congénitales ne peut pas non plus être évalué définitivement sur la base de cette étude en raison des limites méthodologiques, dont une conception non randomisée de l'étude et d'éventuelles différences entre la population du registre et le groupe comparatif (cf. "Efficacité clinique" ).
On sait que les molécules d'IgG traversent la barrière placentaire. Les expérimentations animales n'ont révélé aucun signe de toxicité sur la reproduction (cf. "Données précliniques" ).
Risque lié à des maladies pour la mère et/ou l'embryon/le fœtus
Il est prouvé que chez les femmes souffrant d'asthme difficilement ou modérément contrôlé, le risque d'une pré-éclampsie de la mère et d'une naissance prématurée, d'un poids à la naissance faible ainsi que d'une taille corporelle du nouveau-né réduite par rapport à l'âge gestationnel est élevé. Par conséquent, le contrôle de l'asthme doit être étroitement surveillé chez les femmes enceintes et le traitement doit être ajusté, si besoin, afin de maintenir un contrôle optimal.
Allaitement
Bien que la présence d'omalizumab dans le lait maternel après l'administration de Xolair n'ait pas été examinée, les IgG passent dans le lait maternel et il faut donc s'attendre à ce que l'omalizumab soit présent dans le lait maternel. La fréquence d'infections chez les nourrissons qui a été observée lors de l'étude EXPECT a été évaluée comme une mesure indirecte du développement du système immunitaire après exposition pendant la grossesse ou en raison de l'allaitement. La majorité des nourrissons dans la population d'analyse primaire (77.5%, 186/240) ont été allaités.
Des événements indésirables graves (EIG) qui ont été classés comme "infections et infestations" ont été observés chez 11.4% (5/44) des nourrissons non allaités, chez 10.4% (16/154) des nourrissons ayant été exposés à Xolair en raison de l'allaitement et chez 12.5% (4/32) des nourrissons ayant été allaités sans être exposés à Xolair. L'étude présente des limites méthodologiques, dont une conception non randomisée de l'étude.
Les avantages de l'allaitement pour le développement et la santé du nourrisson doivent être évalués en tenant compte de la nécessité clinique de Xolair et d'éventuels effets indésirables de l'omalizumab ou de la maladie sous-jacente de la mère sur l'enfant allaité.
Fertilité
Il n'existe pas de recommandations particulières pour les femmes en âge de procréer.
Effet sur l’aptitude à la conduite et l’utilisation de machinesAucune étude correspondante n'a été effectuée.
Les patients traités par l'omalizumab doivent être avertis que s'ils présentent des étourdissements, de la fatigue, des syncopes ou une somnolence, ils devront s'abstenir de conduire des véhicules ou d'utiliser des machines.
Effets indésirablesFréquences
"Très fréquent" (≥1/10), "fréquent" (≥1/100 à < 1/10), "occasionnel" (≥1/1000 à < 1/100), "rare" (≥1/10 000 à < 1/1000), "très rare" (< 1/10 000).
Asthme allergique
Lors d'études cliniques réalisées chez des adultes et des adolescents de 12 ans et plus, les effets indésirables les plus fréquemment rapportés ont été des céphalées et des réactions au site d'injection, y compris une douleur au site d'injection, un gonflement, un érythème et un prurit. Lors d'études cliniques réalisées chez des patients de 6 ans à moins de 12 ans, les effets indésirables les plus fréquemment rapportés ont été des céphalées, une pyrexie et des douleurs abdominales hautes. La plupart de ces réactions ont été d'intensité légère ou modérée.
Effets indésirables issus des études cliniques sur l'asthme allergique
Infections et infestations
Occasionnel: pharyngite.
Rare: infestations parasitaires.
Affections du système immunitaire
Rare: réactions anaphylactiques et autres réactions allergiques telles que maladie sérique, symptômes pseudo-grippaux tels que fièvre, douleurs articulaires, malaise. Développement d'anticorps anti-thérapeutiques.
Affections du système nerveux
Fréquent: céphalées**.
Occasionnel: étourdissements, somnolence, paresthésies, syncope.
Affections vasculaires
Occasionnel: hypotension orthostatique, bouffées congestives.
Affections respiratoires, thoraciques et médiastinales
Occasionnel: toux, bronchospasme allergique.
Rare: œdème du larynx.
Affections gastro-intestinales
Fréquent: douleurs abdominales hautes*.
Occasionnel: nausées, diarrhées, dyspepsie.
Affections de la peau et du tissu sous-cutané
Occasionnel: urticaire, rash, prurit, photosensibilité.
Rare: angio-œdème.
Troubles généraux et anomalies au site d'administration
Très fréquent: pyrexie*.
Fréquent: douleurs, érythème, prurit, gonflement.
Occasionnel: gain pondéral, fatigue, gonflement au niveau des bras, symptômes grippaux.
L'incidence des effets indésirables était comparable entre le groupe traité et le groupe contrôle.
* chez les enfants de 6 ans à < 12 ans
** très fréquent chez les enfants de 6 ans à < 12 ans
Polypes nasaux
Résumé du profil de sécurité
Les données décrites ci-dessous sont issues de deux études contrôlées contre placebo menées auprès de patients âgés de 18 ans ou plus. Dans ces études, les patients ont reçu soit 150 à 600 mg de Xolair toutes les 2 ou 4 semaines, soit le placebo. Tous les patients ont reçu de la mométasone par voie intranasale comme traitement de fond; le profil de sécurité chez les patients atteints de polypes nasaux correspondait à celui des patients souffrant d'asthme allergique ou d'UCS. Les effets secondaires les plus fréquemment indiqués (> 3%) étant survenus plus fréquemment que dans le traitement par placebo sont représentés dans le tableau 4.
Dans le tableau 4, les effets indésirables qui sont survenus lors des études cliniques dans la population atteinte de polypes nasaux traitée par Xolair et surveillée dans son ensemble sont répertoriés par classes d'organes et en fonction de leur fréquence. Les catégories de fréquence sont définies de la manière suivante: très fréquent (≥1/10), fréquent (≥1/100 à < 1/10), occasionnel (≥1/1000 à < 1/100); rare (≥1/10 000 à < 1/1000) et très rare (< 1/10 000).
Tableau 4: Effets
indésirables issus des
études cliniques menées
auprès de patients
atteints de polypes nasaux
Effets indésirables Essais omalizumab 1 et 2 Catégorie de fréquen
(désignés par le terme portant sur les polypes ce
privilégié selon la nasaux Données cumulées
classification MedDRA)
Placebo N = 130 Omalizumab N = 135
Affections du système
nerveux
Céphalées 7 (5.4%) 11 (8.1%) fréquent
Sensation vertigineuse 1 (0.8%) 4 (3.0%) fréquent
Affections musculosqueletti
ques et du tissu conjonctif
Arthralgie 2 (1.5%) 4 (3.0%) fréquent
Affections gastro-intestina
les
Douleur abdominale 1 (0.8%) 4 (3.0%) fréquent
Troubles généraux et
anomalies au site
d'administration
(Réactions au site 2 (1.5%) 7 (5.2%) fréquent
d'injection, réaction liée
à l'injection, douleurs au
site d'injection)
Urticaire chronique spontanée (UCS)
Résumé du profil de sécurité
La sécurité et la tolérance de l'omalizumab aux doses de 75 mg, 150 mg et 300 mg toutes les 4 semaines ont été étudiées chez 975 patients atteints d'UCS, dont 242 ont reçu un placebo. 733 patients ont été traités par l'omalizumab pendant une période allant jusqu'à 12 semaines et 490 ont été traités pendant une période allant jusqu'à 24 semaines. 175 resp. 412 patients ont reçu les doses recommandées de 150 mg et 300 mg pendant une période allant jusqu'à 12 semaines et 87 resp. 333 patients pendant une période allant jusqu'à 24 semaines.
Les effets indésirables les plus fréquemment rapportés chez les adultes et les adolescents (âgés de 12 ans et plus) dans le cadre des essais cliniques étaient des céphalées et une rhinopharyngite.
Tableau 5: Résumé
tabellaire des
effets indésirables
rapportés au cours
des essais de phase
III cumulés avec
les doses recommandé
es de 150 mg et 300
mg (jour 1 à semaine
12).
Effets indésirables( Essais omalizumab Fréquences
MedDRA) Q4881g, Q4882g et
Q4883g Données
cumulées
Placebo N = 242 150 mg N = 175 300 mg N = 412
Infections et
infestations
Rhinopharyngite 17 (7.0%) 16 (9.1%) 27 (6.6%) Fréquent
Sinusite 5 (2.1%) 2 (1.1%) 20 (4.9%) Fréquent
Infection virale 0 4 (2.3%) 2 (0.5%) Fréquent
des voies aériennes
supérieures
Affections du
système nerveux
Céphalées 7 (2.9%) 21 (12.0%) 25 (6.1%) Très fréquent
Affections musculosq
uelettiques et du
tissu conjonctif
Arthralgies 1 (0.4%) 5 (2.9%) 12 (2.9%) Fréquent
Autres évènements rapportés à un moment ou à un autre durant la période de traitement entre le jour 1 et la semaine 24 (études Q4881g et Q4883g) et remplissant les critères pour des effets indésirables:
Infections et infestations: infections des voies aériennes supérieures (placebo 3.1%, 150 mg 3.4%, 300 mg 5.7%); infection urinaire (placebo 1.8%, 150 mg 4.6%, 300 mg 2.4%).
Affections du système nerveux: céphalées sinusales (placebo 0%, 150 mg 2.3%, 300 mg 0.3%).
Affections musculosquelettiques et du tissu conjonctif: myalgies (placebo 0%, 150 mg 2.3%, 300 mg 0.9%); douleurs des extrémités (placebo 0%, 150 mg 3.4%, 300 mg 0.9%); douleurs musculo-squelettiques (placebo 0%, 150 mg 2.3%, 300 mg 0.9%).
Troubles généraux et anomalies au site d'administration: pyrexie (placebo 1.2%, 150 mg 3.4%, 300 mg 0.9%); les réactions au site d'injection étaient plus fréquentes chez les patients sous omalizumab que chez ceux sous placebo (2.7% sous 300 mg, 0.6% sous 150 mg, 0.8% sous placebo). Ces réactions incluaient: des tuméfactions, des érythèmes, des douleurs, des hématomes, un prurit, des saignements et de l'urticaire.
Dans une étude d'une durée de 48 semaines, 81 patients atteints d'UCS ont reçu 300 mg d'omalizumab toutes les 4 semaines (voir "Efficacité clinique – UCS" ). Le profil de sécurité en cas d'utilisation à long terme était similaire à celui observé dans les études portant sur l'UCS et d'une durée de 24 semaines.
Effets indésirables identifiés après commercialisation
Les annonces spontanées ont permis en premier lieu d'identifier les réactions suivantes:
Système immunitaire: une réaction anaphylactique et des réactions anaphylactoïdes ont été rapportées aussi bien après la première administration qu'après les suivantes (cf. "Mises en garde et précautions" ). Des antécédents d'anaphylaxie non associée à l'omalizumab peuvent constituer un facteur de risque d'anaphylaxie en cas d'utilisation de Xolair. Maladie sérique (cf. "Mises en garde et précautions" ).
Peau: alopécie.
Circulation sanguine et système lymphatique: thrombopénie idiopathique sévère.
Organes respiratoires: syndrome de Churg-Strauss (c.-à-d. granulomatose éosinophilique avec polyangéite).
Système musculo-squelettique: arthralgie, myalgie, tuméfaction articulaire.
Descriptions de certains effets indésirables
Thrombopénie
Lors des études cliniques, il y a eu, chez peu de patients, un nombre de thrombocytes inférieur à la limite inférieure de la valeur normale. Aucune de ces variations n'a été associée à des épisodes hémorragiques ou à une diminution du taux d'hémoglobine. Il n'a pas été observé de diminution persistante des thrombocytes chez l'homme (patients de plus de 6 ans) comme cela avait été observé chez les primates (cf. "Données précliniques" ). Des cas de thrombopénie ont été rapportés après la commercialisation.
Infections parasitaires
Chez des patients allergiques ayant une tendance aux infestations chroniques par les helminthes, une étude contrôlée contre placebo a montré une légère augmentation du taux d'infestation parasitaire dans le groupe traité par l'omalizumab. L'évolution, la sévérité et la réponse au traitement des infestations n'ont pas été modifiées (cf. "Mises en garde et précautions" ).
Cancers
Lors des premiers essais cliniques chez les adultes et les adolescents âgés d'au moins 12 ans, un déséquilibre du nombre de cancers dans le groupe traité par Xolair a été observé par rapport au groupe contrôle. Des cas de cancer sont survenus occasionnellement (< 1/100) aussi bien dans le groupe actif que dans le groupe contrôle. Dans une étude observationnelle ultérieure sur 5 ans ayant comparé 5007 patients traités par Xolair et 2829 patients non traités par Xolair, le risque relatif de cancers sous Xolair n'était pas accru. Le taux d'incidence de cancers primaires pour 1000 années-patients était respectivement de 16.01 (295/18 426 années-patients) et de 19.07 (190/9963 années-patients), ce qui correspond à un rapport des risques de 0.84 (intervalle de confiance à 95%, 0.62–1.13). Dans une analyse prospective des études cliniques randomisées en double aveugle, contrôlées versus placebo et menées chez 4254 patients traités par Xolair et 3178 patients sous placebo, il a été conclu sur la base des taux d'incidence pour 1000 années-patients de 4.14 (14/3382 années-patients) chez les patients sous Xolair et de 4.45 (11/2474 années-patients) chez les patients sous placebo, que le traitement par Xolair n'est pas associé à un risque accru de cancer (rapport des risques de 0.93, intervalle de confiance à 95%, 0.39–2.27). Cependant, l'ensemble des données actuellement disponibles ne permet pas encore d'exclure complètement l'éventualité d'un léger déséquilibre.
Le taux d'incidence global de cancers observé dans l'étude Xolair menée chez des adultes et des adolescents à partir de 12 ans a été comparable à celui rapporté dans la population générale.
Les patients traités par Xolair dans les études de suivi n'ont pas montré de risque relatif de cancer augmenté (cf. "Mises en garde et précautions" ).
Evénements thromboemboliques artériels (ETA)
Dans des études cliniques contrôlées et au cours des analyses intermédiaires d'une étude observationnelle, un déséquilibre numérique des ETA a été observé. Les ETA ont compris les accidents vasculaires cérébraux, les accidents ischémiques transitoires, les infarctus du myocarde, l'angor instable et les décès d'origine cardiovasculaire (y compris les décès de cause inconnue). Dans l'analyse finale de l'étude observationnelle, l'incidence des ETA pour 1000 années-patients était de 7.52 (115/15 286 années-patients) pour les patients traités par Xolair et de 5.12 (51/9963 années-patients) pour les patients du groupe contrôle. Dans une analyse multivariée avec rectification tenant compte des facteurs de risque cardiovasculaires associés au niveau de base, le rapport des risques (hazard ratio) était de 1.32 (intervalle de confiance à 95%, 0.91–1.91).
Dans une analyse prospective des études cliniques regroupées, incluant toutes les études cliniques randomisées en double aveugle, contrôlées contre placebo et d'une durée de 8 semaines ou plus, l'incidence des ETA pour 1000 années-patients était de 2.69 (5/1856 années-patients) pour les patients traités par Xolair et de 2.38 (4/1680 années-patients) pour les patients du groupe placebo (rapport des risques de 1.13, intervalle de confiance à 95%, 0.24–5.71).
Paramètres de laboratoire
Après l'administration de Xolair, les taux sériques d'IgE totales ont augmenté, à cause de la formation de complexes Xolair-IgE (cf. "Pharmacocinétique" , "Posologie/Mode d'emploi" ).
L'annonce d'effets secondaires présumés après l'autorisation est d'une grande importance. Elle permet un suivi continu du rapport bénéfice-risque du médicament. Les professionnels de santé sont tenus de déclarer toute suspicion d'effet secondaire nouveau ou grave via le portail d'annonce en ligne ElViS (Electronic Vigilance System). Vous trouverez des informations à ce sujet sur www.swissmedic.ch
SurdosageLa dose maximale tolérée de Xolair n'est pas établie. Des doses intraveineuses uniques allant jusqu'à 4000 mg ont été administrées à des patients sans manifestation de toxicité dose-limitante. La dose cumulée la plus élevée administrée à un patient a été de 44 000 mg (sur une période de 20 semaines) et cette dose n'a entraîné aucun effet indésirable aigu.
Propriétés/EffetsCode ATC
R03DX05
Mécanisme d'action
L'omalizumab est un anticorps monoclonal humanisé qui se fixe de manière sélective aux immunoglobulines E (IgE) humaines. L'anticorps est une IgG1 kappa obtenue par la fusion d'une région d'origine humaine avec des régions de complémentarité se fixant aux IgE humaines et provenant d'un anticorps murin.
Pharmacodynamique
Patients souffrant d'asthme allergique
En se liant aux IgE libres, l'omalizumab inhibe la liaison des IgE au récepteur FcεRI de haute affinité (récepteur aux IgE de haute affinité). La quantité d'IgE libres pouvant déclencher la chaîne de réactions allergiques est réduite. Le traitement des sujets atopiques par l'omalizumab a entraîné la diminution du nombre de récepteurs FcεRI présents à la surface des basophiles. Par ailleurs, la libération d'histamine in vitro à partir de basophiles isolés chez des patients traités par Xolair a été réduite d'environ 90% après stimulation par un allergène par rapport aux valeurs pré-thérapeutiques.
Le traitement par Xolair entraîne une diminution du nombre d'éosinophiles dans le sang et les tissus ainsi qu'une diminution des médiateurs inflammatoires dont les interleukines (IL-4, IL-5 et IL-13) font également partie.
Dans les études cliniques, les taux sériques d'IgE libres ont subi une diminution dose-dépendante dans l'heure suivant la première dose et se sont maintenus entre les différentes doses.
Lors de l'utilisation des doses recommandées, la réduction moyenne des IgE libres a été supérieure à 96%. Les taux sériques d'IgE totales (c.-à-d. liées et non liées) ont augmenté après la première dose en raison de la formation de complexes omalizumab-IgE. Les complexes omalizumab-IgE ont un taux d'élimination plus lent que les IgE libres. 16 semaines après la première dose, les taux d'IgE totales ont été 5 fois plus élevés que ceux observés lors du traitement préalable, des tests standards ayant été utilisés pour leur mesure. Après l'interruption du traitement par Xolair, l'augmentation des IgE totales et la réduction des IgE libres ont été réversibles, sans qu'un effet rebond des taux d'IgE n'ait été observé après l'élimination de l'omalizumab. Les taux d'IgE totales existant avant le traitement n'ont pas été atteints dans l'année suivant l'interruption du traitement par Xolair.
Les effets de l'omalizumab sur les cellules B porteuses d'IgE et sur la régulation à long terme de la synthèse des IgE spécifiques de l'allergène ne sont pas clairement connus.
Patients atteints de polypes nasaux
Dans les études cliniques menées auprès de patients atteints de polypes nasaux, le traitement par Xolair a entraîné une réduction des taux sériques d'IgE libres et une augmentation des taux sériques d'IgE totales; ces effets sont similaires à ceux observés chez les patients souffrant d'asthme allergique.
Patients avec urticaire chronique spontanée (UCS)
Plusieurs théories sont proposées à propos de l'étiologie de l'UCS. L'une d'entre elles postule l'existence d'un mécanisme auto-immun. Des auto-anticorps anti IgE et contre le récepteur FcεRI de ceux-ci ont été isolés dans le sérum de certains patients atteints d'UCS. Ces auto-anticorps sont capables d'activer des granulocytes basophiles ou des mastocytes avec pour effet une libération d'histamine.
Une hypothèse relative au mécanisme d'action de l'omalizumab dans l'UCS avance une diminution des taux d'anticorps IgE libres dans le sang et par conséquent aussi dans la peau. Ceci induirait une régulation négative des récepteurs de surface des IgE avec diminution de la transmission des signaux descendants par la voie FcεRI et inhibition de l'activation cellulaire et de la réaction inflammatoire. Il en résulterait une diminution de la fréquence et de la sévérité de symptômes de l'UCS. Une autre hypothèse repose sur une diminution des taux d'IgE libre qui aurait pour conséquence une désensibilisation non spécifique rapide des mastocytes cutanés. La régulation négative de la voie FcεRI pourrait favoriser le maintien de cette réaction.
Dans les essais cliniques conduits auprès de patients atteints d'UCS, le traitement d'omalizumab a induit une diminution dose-dépendante des taux d'IgE libre et une augmentation des taux sériques d'IgE totale, comme c'était le cas chez les patients avec asthme allergique. La suppression maximale de l'IgE libre a été observée 3 jours après la dose sous-cutanée initiale. Après des doses multiples une fois toutes les 4 semaines, les taux sériques d'IgE libre mesurés avant l'administration sont restés stables de la semaine 12 à la semaine 24. Les taux sériques d'IgE totale ont augmenté après la dose initiale à la suite de la formation de complexes omalizumab-IgE, dont la vitesse d'élimination est inférieure à celle de l'IgE libre. Après des doses multiples de 75 mg à 300 mg toutes les 4 semaines, les taux d'IgE totale moyens mesurés dans le sérum avant la dose de la semaine 12 étaient deux à trois fois supérieurs à ceux mesurés avant le début du traitement et sont restés stables de la semaine 12 à la semaine 24. À la fin du traitement par Xolair, les taux d'IgE libre ont augmenté au cours d'une période de follow-up de 16 semaines sans traitement, tandis que les taux d'IgE totale ont diminué, tous deux en direction des valeurs observées avant le traitement.
Efficacité clinique
Asthme allergique
Adultes et adolescents (≥12 ans)
L'efficacité et la tolérance de Xolair ont été démontrées dans une étude pivot de 28 semaines, contrôlée contre placebo (étude 5) conduite chez 419 patients atteints d'asthme allergique sévère, âgés de 12 à 79 ans, ayant une réduction de la fonction pulmonaire (volume expiratoire maximal pendant la 1ère seconde: VEMS 40–80% des valeurs prédites) et dont les symptômes de l'asthme étaient mal contrôlés en dépit d'un traitement de > 1000 µg de dipropionate de béclométasone (ou équivalent) et de bêta2-agonistes à longue durée d'action. Les patients éligibles avaient présenté de multiples exacerbations de l'asthme ayant nécessité une corticothérapie systémique, avaient été hospitalisés ou s'étaient présentés dans un service d'urgences en raison d'une exacerbation sévère de l'asthme au cours de l'année précédente malgré un traitement continu par corticothérapie inhalée à fortes doses et de bêta2-agonistes à longue durée d'action. Xolair ou un placebo a été administré par voie sous-cutanée en addition à un traitement par > 1000 μg de dipropionate de béclométasone (ou équivalent) et de bêta2agonistes à longue durée d'action. Les patients ont de plus reçu des traitements de fond par corticoïde oral (22% des patients), théophylline (27%) et anti-leucotriènes (35%). Durant la phase de traitement, les thérapies additives contre l'asthme sont restées inchangées.
Le critère primaire représente le taux d'exacerbations de l'asthme ayant nécessité un traitement aigu par des corticostéroïdes systémiques. L'omalizumab a réduit le taux d'exacerbations de l'asthme de 19% (p = 0.153). Les autres évaluations ayant présenté une significativité statistique (p = 0.05) en faveur de Xolair, comprennent la réduction des exacerbations sévères (dans lesquelles la fonction pulmonaire du patient a été réduite à moins de 60% de la meilleure valeur personnelle et des corticostéroïdes systémiques ont été nécessaires), les consultations au service des urgences en relation avec l'asthme (y compris hospitalisation, consultation au service des urgences et consultations médicales non prévues) ainsi que l'amélioration de l'évaluation générale par le médecin de l'efficacité du traitement, de la qualité de vie en rapport avec l'asthme (AQL), des symptômes de l'asthme et de la fonction pulmonaire.
Dans une analyse de sous-groupe, la probabilité de tirer un bénéfice cliniquement significatif du traitement par Xolair a été plus élevée chez les patients avec des taux pré-thérapeutiques d'IgE totales ≥76 UI/ml. Chez ces patients de l'étude 1, Xolair a réduit de 40% (p = 0.002) la fréquence des exacerbations de l'asthme. Par ailleurs, les patients de la population avec des IgE totales ≥76 UI/ml de l'ensemble du programme de Xolair dans l'asthme sévère ont été plus nombreux à présenter des réponses cliniquement significatives (cf. tableau 6).
Quatre autres larges études secondaires contrôlées contre placebo d'une durée de 28 à 52 semaines conduites chez 1722 adultes et adolescents (études 3, 4, 5, 6) ont évalué l'efficacité et la tolérance de Xolair chez des patients atteints d'asthme persistant sévère. La plupart des patients étaient insuffisamment contrôlés mais ils recevaient un traitement antiasthmatique concomitant plus léger que les patients des études 1 ou 2. Les études 3–5 ont utilisé les exacerbations comme critère principal d'évaluation, tandis que l'étude 6 a principalement évalué l'épargne des corticoïdes inhalés.
L'étude 2 a évalué l'efficacité et la tolérance de l'omalizumab dans une population de 312 patients atteints d'asthme allergique sévère présentant des caractéristiques proches de celles de la population de l'étude 1. Le traitement par Xolair dans cette étude en ouvert a entraîné une réduction de 61% de la fréquence des exacerbations de l'asthme, réduction cliniquement significative par rapport au traitement antiasthmatique en cours administré seul.
Dans les études 3, 4 et 5, les patients traités par Xolair ont présenté une réduction de la fréquence des exacerbations de l'asthme respectivement de 37.5% (p = 0.027), 40.3% (p < 0.001) et 57.6% (p < 0.001) par rapport au placebo.
Dans l'étude 6, un nombre significativement supérieur de patients atteints d'asthme allergique sévère ont pu réduire leur dose de fluticasone à ≤500 μg/jour avec Xolair (60.3%) sans détérioration du contrôle de l'asthme, et ce par rapport aux patients du groupe placebo (45.8%, p < 0.05).
Tableau 6: résultats de l'étude
Ensemble de la
population de
l'étude
Xolair (N = 209) Placebo (N = 210)
Exacerbations de l'asthme
Taux par période de 28 semaines 0.74 0.92
% de réduction, valeur de p pour le rapport des taux 19.4%, p = 0.153
Exacerbations sévères de l'asthme
Taux par période de 28 semaines 0.24 0.48
% de réduction, valeur de p pour le rapport des taux 50.1%, p = 0.002
Visites d'urgence
Taux par période de 28 semaines 0.24 0.43
% de réduction, valeur de p pour le rapport des taux 43.9%, p = 0.038
Évaluation globale du médecin
% de répondeurs* 60.5% 42.8%
Valeur de p** < 0.001
Amélioration à l'AQL***
% de patients ≥0.5 d'amélioration 60.8% 47.8%
Valeur de p 0.008
* amélioration marquée ou contrôle complet ** valeur de
p pour la distribution globale de l'évaluation ***
Asthma Quality of Life
Enfants âgés de 6 à < 12 ans
Les principales données concernant la sécurité d'emploi et l'efficacité de Xolair dans le groupe d'âge de 6 ans à < 12 ans sont issues d'une étude multicentrique randomisée, en double aveugle et contrôlée contre placebo (étude 7).
L'étude 7 était une étude contrôlée contre placebo comprenant un sous-groupe spécifique (N = 235) de patients dont le diagnostic correspondait à l'indication actuelle et qui étaient traités par des corticostéroïdes inhalés à forte dose (≥500 μg d'équivalent fluticasone/jour) et des bêtaagonistes de longue durée d'action.
Une exacerbation cliniquement significative était définie selon le jugement clinique de l'investigateur comme une aggravation des symptômes asthmatiques nécessitant le doublement de la dose initiale du corticostéroïde inhalé pendant au moins 3 jours et/ou un traitement d'urgence par des corticostéroïdes systémiques (oraux ou intraveineux) pendant au moins 3 jours.
Dans le sous-groupe spécifique de patients traités par des corticostéroïdes inhalés à forte dose, le groupe omalizumab a présenté une incidence des exacerbations de l'asthme statistiquement inférieure à celle dans le groupe placebo. À la semaine 24, le taux d'incidence chez les patients traités par l'omalizumab a été inférieur de 34% à celui sous placebo (rapport des taux 0.662; p = 0.047). Pendant la seconde période de traitement en double aveugle de 28 semaines, le taux d'incidence chez les patients traités par l'omalizumab a été inférieur de 63% à celui sous placebo (rapport des taux 0.37; p < 0.001).
Pendant la période de traitement en double aveugle de 52 semaines (comprenant la phase de corticothérapie à posologie fixe de 24 semaines et la phase d'adaptation de la posologie des corticoïdes de 28 semaines), les différences des taux d'incidence entre les groupes de traitement ont révélé une diminution des exacerbations de 50% chez les patients traités par l'omalizumab (rapport des taux 0.504; p < 0.001).
Par rapport au groupe placebo, le groupe omalizumab s'est caractérisé à la fin de la période de traitement de 52 semaines par une diminution plus importante de l'utilisation de bêtaagonistes en tant que traitement d'urgence, bien que la différence entre les groupes de traitement n'ait pas été statistiquement significative. En ce qui concerne l'évaluation globale de l'efficacité du traitement à la fin de la période de traitement en double aveugle de 52 semaines et en ce qui concerne le sous-groupe des patients sévères recevant des corticoïdes inhalés à forte dose et des bêta-agonistes à longue durée d'action, la proportion des patients chez lesquels l'efficacité du traitement a été jugée "excellente" a été plus élevée dans le groupe omalizumab que dans le groupe placebo. Les proportions des patients chez lesquels l'efficacité du traitement a été jugée "modérée" ou "faible" ont été plus faibles dans le groupe omalizumab que dans le groupe placebo. Les différences entre les groupes ont été statistiquement significatives (p < 0.001). Aucune différence n'a été constatée entre les groupes omalizumab et placebo en ce qui concerne les évaluations subjectives faites par les patients au sujet de leur qualité de vie.
Polypes nasaux
La sécurité et l'efficacité de Xolair ont été évaluées dans deux études cliniques randomisées, multicentriques, en double aveugle et contrôlées contre placebo (étude 1, N = 138; étude 2, N = 127) réalisée auprès de patients atteints de rhinosinusite chronique et de polypes nasaux. Les patients ont reçu Xolair ou un placebo par voie sous-cutanée toutes les 2 ou 4 semaines, la posologie et la fréquence d'utilisation correspondant aux données des tableaux 7 et 8 (cf. "Posologie/Mode d'emploi" ). Par ailleurs, tous les patients ont reçu tout au long de l'étude un traitement de fond par de la mométasone administrée par voie intranasale. Une opération sino-nasale précédente ou un traitement systémique préalable par des corticostéroïdes n'étaient pas requis pour l'admission dans l'étude. Les participants à l'étude ont reçu Xolair ou un placebo pendant 24 semaines, et une phase de suivi sans traitement d'une durée de 4 semaines a ensuite eu lieu. Les données démographiques et les caractéristiques de référence incluant les comorbidités allergiques sont représentées dans le tableau 7.
Tableau 7: Données démographiques et caractéristiques
de référence dans les études de polypes nasaux
Paramètres Étude 1 des polypes Étude 2 des polypes
nasaux N = 138 nasaux N = 127
Âge moyen en années (SD) 51.0 (13.2) 50.1 (11.9)
% patients de sexe masculin 63.8 65.4
Patients ayant utilisé des corticostéroïdes systémiques 18.8 26.0
l'année précédente (%)
Score NPS endoscopique bilatéral moyen (SD), 6.2 (1.0) 6.3 (0.9)
intervalle: 0–8
Score moyen de congestion nasale (NC)* (SD), 2.4 (0.6) 2.3 (0.7)
intervalle: 0–3
Score olfactif moyen* (SD), intervalle: 0–3 2.7 (0.7) 2.7 (0.7)
Score total SNOT-22 * (SD), intervalle: 0–110 60.1 (17.7) 59.5 (19.3)
Nombre moyen d'éosinophiles dans le sang (cellules/µl) 346.1 (284.1) 334.6 (187.6)
(SD)
IgE totales moyennes en UI/ml (SD) 160.9 (139.6) 190.2 (200.5)
Asthme (%) 53.6 60.6
Léger (%) 37.8 32.5
Modéré (%) 58.1 58.4
Sévère (%) 4.1 9.1
Affection respiratoire exacerbée par l'aspirine (%) 19.6 35.4
Rhinite allergique 43.5 42.5
SD = écart-type; NPS = score de polypes nasaux (nasal
polyps score); SNOT 22 = questionnaire sur le test de
résultats sino-nasaux comportant 22 questions; IgE =
immunoglobulines E; UI = unités internationales.
Dans le NPS, le NCS et les scores olfactifs, de syndrome de rhinorrhée postérieure et d'écoulement nasal ainsi que dans le score SNOT-22, un nombre de points plus élevé indique une gravité accrue de la maladie.
Les co-critères d'évaluation principaux étaient le score bilatéral de polypes nasaux (NPS) et le score quotidien moyen de congestion nasale (NCS), tous deux déterminés à la semaine 24. Le NPS a été déterminé par endoscopie au début de l'étude et à des moments déterminés au préalable (intervalle: 0–4 par narine), et le NPS total a été calculé à partir de ces valeurs (intervalle: 0 = meilleure valeur à 8 = valeur la plus mauvaise). La congestion nasale a été évaluée tous les jours à l'aide de l'échelle NCS (intervalle: 0 = meilleure valeur à 3 = valeur la plus mauvaise). Avant d'être randomisés, les patients devaient présenter un NPS ≥5 et un NCS hebdomadaire moyen > 1 malgré l'utilisation de mométasone par voie intranasale. Dans les deux études, les NPS moyens des deux groupes de traitement au début de l'étude étaient équilibrés.
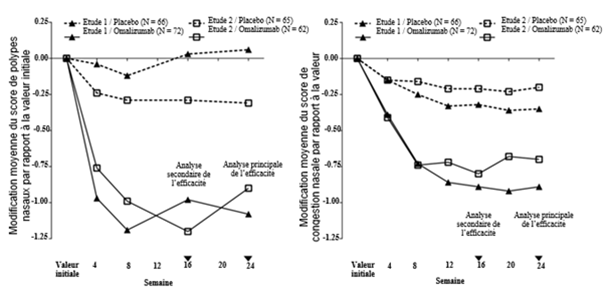
Dans l'étude 1 comme dans l'étude 2 des polypes nasaux, les patients ayant reçu Xolair ont présenté à la semaine 24 une amélioration significativement plus importante d'un point de vue clinique du NPS comme de la moyenne du NCS sur la semaine par rapport à la valeur initiale que les patients ayant reçu le placebo (cf. tableau 8).
Les améliorations les plus importantes du NPS et du NCS dans le groupe traité par Xolair par rapport au groupe placebo ont déjà été observées lors de la première évaluation à la semaine 4, comme le montre la figure 8. À la semaine 4, la différence des moyennes des moindres carrés (LS) pour la modification du NPS par rapport à la valeur initiale dans le groupe traité par Xolair en comparaison avec le groupe placebo était de -0.92 (IC à 95%: -1.37, -0.48) dans l'étude 1 et de -0.52 (IC à 95%: -0.94, -0.11) dans l'étude 2. Pour le NCS, la différence des moyennes des LS pour la modification par rapport à la valeur initiale à la semaine 4 dans le groupe traité par Xolair en comparaison avec le groupe placebo était de -0.25 (IC à 95%: -0.46, -0.04) dans l'étude 1 et de -0.26 (IC à 95%: -0.45, -0.07) dans l'étude 2. Toutefois, les tests statistiques n'étaient pas pré-spécifiés à ce moment-là.
Tableau 8: Modificat
ion du score de
polypes nasaux et
du score moyen de
congestion nasale
sur 7 jours à la
semaine 24 par
rapport à la valeur
initiale dans les
études 1 et 2 des
polypes nasaux
Étude 1 des polypes Étude 2 des polypes
nasaux nasaux
Placebo Xolair Placebo Xolair
N 66 72 65 62
Score de polypes
nasaux
Moyenne de la 6.32 6.19 6.09 6.44
référence
Moyenne des LS pour 0.06 -1.08 -0.31 -0.90
la modification
jusqu'à la semaine
24 par rapport à la
valeur initiale
Différence des -1.14 -0.59
moyennes des LS par
rapport au placebo
IC à 95% de la -1.59, -0.69 -1.05, -0.12
différence
Valeur de p < 0.0001 0.0140
Valeur moyenne sur
7 jours du score
quotidien de congest
ion nasale
Moyenne de la 2.46 2.40 2.29 2.26
référence
Moyenne des LS pour -0.35 -0.89 -0.20 -0.70
la modification
jusqu'à la semaine
24 par rapport à la
valeur initiale
Différence des -0.55 -0.50
moyennes des LS par
rapport au placebo
IC à 95% de la -0.84, -0.25 -0.80, -0.19
différence
Valeur de p 0.0004 0.0017
LS = moindres carrés (détermination de la moyenne selon la méthode des Least Squares = moindres carrés)
Figure 1: Modification moyenne par rapport à la valeur initiale des scores de congestion nasale et de polypes nasaux en fonction du groupe de traitement dans les études 1 et 2 des polypes nasaux
Un critère d'évaluation principal secondaire a été l'évaluation de la modification du score total des symptômes nasaux (total nasal symptom score, TNSS) à la semaine 24 par rapport à la valeur initiale. Le TNSS rapporté par les patients était un score qui correspondait à la somme de 4 scores de symptômes quotidiens équipondérés. Ceux-ci étaient: le NCS, le score olfactif, le score de rhinorrhée postérieure et le score de rhinorrhée antérieure. L'intervalle du TNSS était de 0 = meilleure valeur à 12 = valeur la plus mauvaise. Dans le cadre du traitement par Xolair, il y a eu une amélioration significative du TNSS quotidien moyen par rapport au placebo. La différence des moyennes des LS pour la modification par rapport à la valeur initiale jusqu'à la semaine 24 était de -1.91 points (IC à 95%: -2.85, -0.96; p = 0.0001) dans l'étude 1 et de -2.09 points (IC à 95%: -3.00, -1.18; p < 0.0001) dans l'étude 2.
Dans le cadre du traitement par Xolair, il y a eu une amélioration significative dans le SNOT-22 (Sino-Nasal OutcomeTest) qui combine des questions du domaine des symptômes sino-nasaux, de la psychologie et de la qualité du sommeil. Le SNOT-22 était compris entre 0 et 110 (0 = meilleure valeur, 110 = valeur la plus mauvaise). La différence des moyennes des LS pour la modification du SNOT-22 par rapport à la valeur initiale jusqu'à la semaine 24 dans le cadre du traitement par Xolair, en comparaison avec le placebo, était de -16.12 (IC à 95%: -21.86, -10.38; p < 0.0001) dans l'étude 1 et de -15.04 (IC à 95%: -21.26, -8.82; p < 0.0001) dans l'étude 2.
Dans le cadre du traitement par Xolair, il y a également eu une amélioration significative du score UPSIT (University of Pennsylvania Smell Identification Test) quotidien moyen par rapport au placebo. Le score UPSIT était compris entre 0 et 40 (0 = valeur la plus mauvaise, 40 = meilleure valeur). La différence des moyennes des LS pour la modification par rapport à la valeur initiale jusqu'à la semaine 24 dans le cadre du traitement par Xolair, en comparaison avec le placebo, était de 3.81 points (IC à 95%: 1.38, 6.24; p = 0.0024) dans l'étude 1 et de 3.86 points (IC à 95%: 1.57, 6.15; p = 0.0011) dans l'étude 2.
Dans les deux études, l'effet sur le TNSS et le SNOT-22 a déjà été observé dès la première évaluation à la semaine 4. En outre, l'effet sur le score UPSIT a été observé dans les deux études lors de la première évaluation à la semaine 8.
Des analyses supplémentaires des critères d'évaluation secondaires ont inclus les évaluations du NPS et du NCS à la semaine 16. Dans le cadre du traitement par Xolair, il y a eu une amélioration significative du NPS à la semaine 16 (0 = valeur la plus mauvaise, 8 = meilleure valeur) par rapport au placebo. La différence des moyennes des LS pour la modification par rapport à la valeur initiale jusqu'à la semaine 16 dans le cadre du traitement par Xolair, en comparaison avec le placebo, était de -1.01 (IC à 95%: -1.43, -0.60; p < 0.0001) dans l'étude 1 et de -0.91 (IC à 95%: -1.39, -0.44; p = 0.0002) dans l'étude 2 [XX]. Dans le cadre du traitement par Xolair, il y a eu une amélioration significative du NCS à la semaine 16 (0 = meilleure valeur, 3 = valeur la plus mauvaise) par rapport au placebo. La différence des moyennes des LS pour la modification du NCS quotidien moyen par rapport à la valeur initiale jusqu'à la semaine 16 dans le cadre du traitement par Xolair, en comparaison avec le placebo, était de -0.57 (IC à 95%: -0.83, -0.31; p < 0.0001) dans l'étude 1 et de -0.59 (IC à 95%: -0.87, -0.30; p < 0.0001) dans l'étude 2.
Urticaire chronique spontanée (UCS)
Le programme de développement clinique de phase III dans l'UCS comprenait trois études multicentriques randomisées, en double aveugle, contrôlées contre placebo, en groupes parallèles: Q4881g, Q4882g et Q4883g.
Les observations ont porté sur des adultes et des adolescents (âgés de 12 ans et plus) avec UCS depuis ≥6 mois (de 6 mois à 66 ans, 6 ans en moyenne) et souffrant de poussées récurrentes malgré les antihistaminiques aux doses maximales admises (UAS7 ≥16/42 durant ≥8 jours consécutifs).
Les études Q4881g et Q4882g avaient pour but de tester l'efficacité et la sécurité d'un traitement de 75 mg, 150 mg ou 300 mg de Xolair toutes les 4 semaines durant 24 resp. 12 semaines avec une période de followup sans traitement de 16 semaines chez des patients (12–75 ans) atteints d'une UCS réfractaire malgré l'administration d'antihistaminiques H1.
L'étude Q4883g avait pour objectif de tester l'efficacité et la sécurité de 300 mg de Xolair, administrés toutes les 4 semaines durant 24 semaines avec une période de followup sans traitement de 16 semaines chez des patients (12–75 ans) atteints d'une UCS réfractaire malgré l'administration d'antihistaminiques H1 et/ou H2 et/ou des antagonistes du récepteur des leucotriènes (LTRA).
Tableau 9 Critères d'évaluation
de l'efficacité
Modification de l'Itch Severity Score (ISS, échelle 0–21) hebdomadaire à la Principal critère
semaine 12 versus valeur initiale d'évaluation des
études Q4881g et
Q4882g Critère
d'évaluation secondai
re de l'étude de
sécurité Q4883g
Délai jusqu'à une réponse MID a (diminution de ≥5 points versus valeurs Critères d'évaluation
initiales) du score ISS hebdomadaire jusqu'à la semaine 12 secondaires des
trois essais Q4881g,
Q4882g et Q4883g
Modification du score d'activité de l'urticaire (UAS7 b, échelle 0–42)
mesuré sur une période de 7 jours à la semaine 12 versus valeur initiale
Proportion des patients avec un score d'activité de l'urticaire ≤6 (UAS7b
≤6) mesuré sur 7 jours à la semaine 12
Proportion des patients avec un score d'activité de l'urticaire = 0 (UAS7b
= 0) mesuré sur 7 jours à la semaine 12 c
Modification du score hebdomadaire des papules à la semaine 12 versus
valeur initiale
Modification du score global de l'indice de qualité de vie dermatologique
(DLQI) à la semaine 12 versus valeur initiale
Proportion des patients avec journées sans angio-œdème entre la semaine 4
et la semaine 12 d
a MID: différence minimale significative (Minimally Important Difference) b
UAS7: combinaison de l'intensité du prurit et du nombre de papules; somme
des scores mesurés lors de 7 jours consécutifs c Analyse post-hoc de
l'étude Q4882g d La proportion moyenne des jours sans angio-œdème entre la
semaine 4 et la semaine 12 a été calculée pour l'ensemble de la population
de l'étude, y compris les patients sans symptômes d'angio-œdème.
Dans les essais Q4881g et Q4882g, la dose de 75 mg n'a atteint de manière consistante ni le principal critère d'efficacité (modification de l'Itch Severity Score (ISS) hebdomadaire à la semaine 12 versus valeur initiale) ni plusieurs des critères secondaires. Cette dose a par conséquent été considérée comme non efficace et ces résultats ne sont pas présentés ci-dessous.
Le principal critère d'efficacité, la modification de l'Itch Severity Score hebdomadaire de la semaine 12 versus valeur initiale, a été atteint aussi bien avec la dose de 150 mg qu'avec la dose de 300 mg dans les études Q4881g et Q4882g et il a été atteint avec la dose de 300 mg dans l'étude Q4883g (critère d'évaluation secondaire; cf. Tableau 10).
Tableau 10: Modification de
l'Itch Severity Score
hebdomadaire à la semaine 12
versus valeur initiale, études
Q4881g, Q4882g et Q4883g
(population mITT *)
Placebo Omalizumab150 mg Omalizumab300 mg
Étude Q4881g
N 80 80 81
Moyenne (SD) -3.63 (5.22) -6.66 (6.28) -9.40 (5.73)
Différence des moyennes des LS vs - -2.95 -5.80
placebo1
IC à 95% pour la différence - -4.72,-1.18 -7.49,-4.10
Valeur de p vs placebo2 - 0.0012 < 0.0001
Étude Q4882g
N 79 82 79
Moyenne (SD) -5.14 (5.58) -8.14 (6.44) -9.77 (5.95)
Différence des moyennes des LS vs - -3.04 -4.81
placebo1
IC à 95% pour la différence - -4.85,-1.24 -6.49,-3.13
Valeur de p vs placebo2 - 0.0011 < 0.0001
Étude Q4883g
N 83 - 252
Moyenne (SD) -4.01 (5.87) - -8.55 (6.01)
Différence des moyennes des LS vs - - -4.52
placebo1
IC 95% pour la différence - - -5.97, -3.08
Valeur de p vs placebo2 - - < 0.0001
* Population Intent-to-Treat
modifiée (mITT): englobe tous les
patients randomisés ayant reçu au
moins une dose du médicament à
l'étude. La méthode BOCF
(Baseline Observation Carried
Forward) a été appliquée pour le
calcul des valeurs manquantes. 1
La moyenne des LS a été calculée
à l'aide d'un modèle d'ANCOVA.
Les critères de stratification
étaient la valeur initiale de
l'Itch Severity Score
hebdomadaire (< 13 vs ≥13) et le
poids initial (< 80 kg vs ≥80
kg). 2 Les valeurs de p
proviennent du t-test ANCOVA.
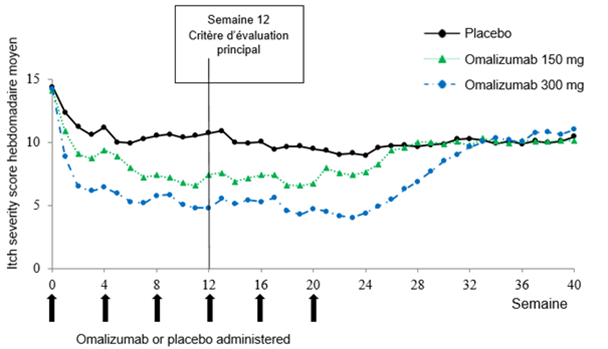
La figure 2 illustre l'évolution temporelle de l'ISS hebdomadaire moyen au cours de l'étude Q4881g. L'Itch Severity Score hebdomadaire moyen a significativement diminué dans les deux groupes de traitement. L'effet maximal a été atteint environ à la semaine 12, puis est resté constant durant la phase de traitement de 24 semaines. Dans les études Q4883g (300 mg durant une période de traitement de 24 semaines) et Q4882g (150 mg ou 300 mg durant une période de traitement de 12 semaines), les résultats étaient semblables à ceux de l'étude Q4881g.
Dans les trois essais (cf. figure 2 pour l'étude Q4881g), l'Itch Severity Score hebdomadaire moyen a augmenté dans les deux groupes de dosage de manière progressive et parallèlement aux symptômes au cours de la phase de follow-up sans traitement de 16 semaines. À l'issue de la phase de follow-up, les valeurs moyennes étaient comparables à celles du groupe placebo, mais inférieures aux valeurs moyennes initiales correspondantes.
Figure 2: Évolution temporelle de l'Itch Severity Score hebdomadaire moyen dans l'étude Q4881g (BOCF, population mITT)
BOCF = Baseline Observation Carried Forward; mITT = population Intent-to-Treat-modifiée
Délai jusqu'à l'apparition d'une réponse MID de l'ISS hebdomadaire jusqu'à la semaine 12
Dans les études Q4881g et Q4882g, le délai médian jusqu'à l'obtention d'une MID de 5 points de l'ISS hebdomadaire était de 2 semaines chez les patients du groupe recevant 150 mg (p = 0.0301 dans l'étude Q4881g; p = 0.0101 dans l'étude Q4882g) et de 1 semaine chez les patients sous 300 mg (p < 0.0001) versus 4 semaines chez les patients des groupes placebo. Des résultats comparables ont été rapportés dans l'étude Q4883g avec un délai médian de 2 semaines jusqu'à l'obtention d'une MID dans le groupe sous 300 mg (p < 0.0001) contre 5 semaines dans le groupe placebo.
Modification de l'UAS7 à la semaine 12 versus valeur initiale
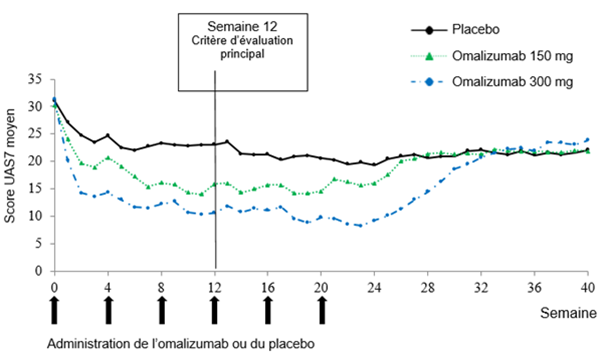
Dans les essais de phase III, les groupes traités par 150 mg et 300 mg d'omalizumab différaient de manière statistiquement significative du groupe placebo en termes de modification moyenne de l'UAS7 à la semaine 12 versus valeur initiale (figure 2 pour l'étude Q4881g). Le seuil de signification statistique (p < 0.0001) a été atteint dans les trois études pour le groupe recevant 300 mg et dans les études Q4881g (p = 0.0008) et Q4882g (p = 0.0001) dans le groupe recevant 150 mg.
La figure 3 représente l'évolution temporelle moyenne de l'UAS7 dans l'étude Q4881g. Celle-ci était marquée par une diminution significative par rapport à la valeur initiale dans les deux groupes de traitement avec un effet maximal aux alentours de la semaine 12. Cet effet est resté constant durant toute la période de traitement de 24 semaines. Dans les études Q4882g (150 mg et 300 mg au cours d'une période de traitement de 12 semaines) et Q4883g (300 mg au cours d'une phase de traitement de 24 semaines), les résultats étaient comparables à ceux observés dans l'étude Q4881g.
Dans les trois études (cf. figure 3 pour l'étude Q4881g), l'UAS7 a progressivement augmenté au cours des 16 semaines de follow-up sans traitement dans les deux groupes sous omalizumab avec une réapparition parallèle des symptômes. À la fin de la phase de followup, les valeurs moyennes étaient comparables à celles sous placebo, mais inférieures aux valeurs initiales correspondantes.
Figure 3: Évolution temporelle moyenne de l'UAS7, étude Q4881g (BOCF, population mITT)
BOCF = Baseline Observation Carried Forward;
mITT = population Intent-to-Treat modifiée;
UAS7 = score d'activité de l'urticaire sur une période de 7 jours
Proportion de patients avec un UAS7 ≤6 à la semaine 12
La proportion des patients ayant un UAS7 ≤6 à la semaine 12 est représentée dans la figure 4. Les taux de réponse étaient tous statistiquement significatifs, se situant entre 52% et 66% (dose de 300 mg; p < 0.0001), resp. entre 40% et 43% (dose de 150 mg; p < 0.001) contre 11–19% dans le groupe placebo.
Figure 4: Proportion des patients avec UAS7 ≤6 à la semaine 12, études Q4881g, Q4882g et Q4883g

Proportion des patients avec un UAS7 = 0 à la semaine 12
La proportion des patients ayant présenté une réponse complète, autrement dit un UAS7 = 0 à la semaine 12 était de 34–44% (dose de 300 mg, statistiquement significatif, tous p < 0.0001), resp. 15–22% (dose de 150 mg) versus 5–9% dans le groupe placebo (figure 5).
Figure 5: Proportion des patients avec un UAS7 = 0 à la semaine 12, études Q4881g, Q4882g et Q4883g

Analyse prospective dans les études Q4881g et Q4883g et analyse post-hoc dans l'étude Q4882g
Modification du score hebdomadaire relatif au nombre de papules à la semaine 12 versus valeur initiale
Dans les trois essais de phase III, le changement moyen du score hebdomadaire relatif au nombre de papules à la semaine 12 versus valeur initiale était statistiquement significatif dans le groupe traité par 300 mg (p < 0.001) avec une diminution du nombre de papules par rapport au placebo(-11.35 dans Q4881g, -11.97 dans Q4882g et -10.46 dans Q4883g versus -4.37, -5.22 resp. -4.49 dans les groupes placebo correspondants). Dans le groupe traité par 150 mg, le changement moyen était de -7.78 (p = 0.0017) dans Q4881g et de -9.75 (p < 0.0001) dans Q4882g.
Proportion des jours sans angio-œdème de la semaine 4 à la semaine 12
Les groupes traités par 300 mg ont bénéficié dans les trois essais de phase III de la plus grande proportion moyenne de jours sans angio-œdème entre les semaines 4 et 12 (91–96%). L'augmentation de la proportion de jours sans angio-œdème était statistiquement significative par rapport au placebo (p < 0.001) (fig. 6). Dans le groupe traité par 150 mg, la proportion moyenne de jours sans angio-œdème pendant le même laps de temps était de 89.6% dans l'étude Q4881g et de 91.6% dans l'étude Q4882g. Les valeurs correspondantes sous placebo étaient de 88.2% et 89.2% dans les deux essais concernés. Les différences entre les groupes sous 150 mg et sous placebo n'ont atteint le seuil de signification statistique dans aucune de ces deux études.
Figure 6: Proportion des jours sans angio-œdème de la semaine 4 à la semaine 12, études Q4881g, Q4882g et Q4883g

Modification du score global de l'indice de la qualité de vie dermatologique (DLQI) à la semaine 12 versus valeur initiale
Dans les trois essais de phase III, le changement moyen du score global DLQI entre la semaine 12 et la valeur initiale était statistiquement significativement supérieur dans le groupe traité par 300 mg (p < 0.001) par rapport à celui du groupe placebo. Dans l'étude Q4882g, le groupe traité par 150 mg d'omalizumab présentait une différence statistiquement significative (p = 0.022) versus placebo (figure 7).
Figure 7: Modification du score global de l'indice de la qualité de vie dermatologique à la semaine 12 versus valeur initiale dans les études Q4881g, Q4882g et Q4883g

Efficacité à l'issue de 24 semaines de traitement
Le tableau 11 présente les résultats après 24 semaines de traitement. Les ordres de grandeur de la réponse sont comparables à ceux observés après 12 semaines de traitement.
Tableau 11: Efficaci
té après 24 semaines
de traitement dans
les études Q4881g
et Q4883g (populatio
n mITT*)
Paramètre de l'étude Semaine Placebo Omalizumab150 mg Omalizumab300 mg
Variation vs ligne
de base du Itch
Severity Score
(BOCF) hebdomadaire,
valeur moyenne
Étude Q4881g Semaine 24 -5.41 -6.47 -9.84**
Étude Q4883g Semaine 24 -4.03 NA -8.60**
Variation vs ligne
de base de l'UAS7
(BOCF), valeur
moyenne
Étude Q4881g Semaine 24 -11.73 -14.21 -22.11**
Étude Q4883g Semaine 24 -8.85 NA -19.15**
Proportion de
patients avec UAS7
≤6, % de patients
Étude Q4881g Semaine 24 25.0 36.3 61.7**
Étude Q4883g Semaine 24 16.9 NA 55.6**
Proportion de
patients avec UAS7
= 0, % de patients
Étude Q4881g Semaine 24 12.5 20.0 48.1**
Étude Q4883g Semaine 24 3.6 NA 42.5**
* Population Intent-
to-Treat modifiée
(mITT): englobe
tous les patients
randomisés ayant
reçu au moins une
dose du médicament
à l'étude ** valeur
de p ≤0.0001 dans
le test statistique
correspondant entre
la substance active
et le placebo NA:
ne s'applique pas
(Not Applicable).
BOCF: Baseline
Observation Carried
Forward
Efficacité après 48 semaines de traitement
Dans une étude d'une durée de 48 semaines, 206 patients atteints d'UCS non contrôlée par un traitement par antihistaminique H1, âgés de 12 à 75 ans, ont participé à une phase de traitement en ouvert par l'omalizumab 300 mg toutes les 4 semaines, d'une durée de 24 semaines, en tant que traitement d'appoint. Les patients qui ont répondu au traitement pendant cette phase en ouvert ont ensuite reçu à l'aveugle de manière aléatoire, l'omalizumab 300 mg (81 patients) ou un placebo (53 patients), toutes les 4 semaines pendant 24 semaines supplémentaires.
Parmi les patients ayant été traités par l'omalizumab pendant un total de 48 semaines, 21% ont présenté une dégradation clinique (valeur UAS7 de 12 ou plus pendant au moins 2 semaines consécutives après la randomisation entre les semaines 24 et 48), par comparaison à 60.4% des patients traités par le placebo à la semaine 48 (La différence est de -39.4%, p <0.0001, IC à 95%: -54.5%, -22.5%).
L'étude prospective portant sur un registre de grossesse (EXPECT)
Une étude prospective portant sur un registre de grossesse menée de 2006 à 2018 aux États-Unis (EXPECT) a porté sur 250 femmes enceintes asthmatiques ayant été traitées par Xolair. 246 d'entre elles avaient été traitées par Xolair au cours du premier trimestre de grossesse et 78.4% (196/250) des femmes avaient été traitées au moins une fois par Xolair au cours de chacun des 3 trimestres de grossesse, la durée totale d'exposition étant de 8.7 mois (médiane). Les résultats de l'étude EXPECT dans les sous-groupes concernés de mères et de nourrissons ont été comparés aux fréquences ajustées en fonction de l'âge dans une cohorte externe de 1153 femmes enceintes asthmatiques (sans traitement par Xolair) ajustée en fonction de la maladie, la cohorte ayant été déterminée à partir de banques de données du domaine de la santé d'habitants de la province canadienne de Québec et désignée comme Quebec External Comparator Cohort (QECC).
Chez les nourrissons de l'étude EXPECT ayant été comparés à des nourrissons de la QECC (n = 223), la prévalence d'anomalies congénitales majeures était de 8.1% et était ainsi comparable à celle des nourrissons de la QECC (8.9%). Dans le cas des grossesses de l'étude EXPECT utilisées pour la comparaison avec la QECC (n = 230), 99.1% ont donné lieu à des naissances vivantes; il en a été de même pour 99.3% des grossesses de la QECC.
Dans une sous-étude d'EXPECT, les taux de plaquettes ont été examinés chez 51 nourrissons nés de mères exposées à Xolair; tous ces taux étaient dans les limites normales.
PharmacocinétiqueAbsorption
Après administration sous-cutanée, l'omalizumab est absorbé avec une biodisponibilité absolue moyenne de 62%. Après l'administration d'une dose sous-cutanée unique chez des patients asthmatiques adultes ou adolescents, l'omalizumab a été absorbé lentement, les concentrations sériques maximales étant atteintes en 7 à 8 jours en moyenne. La pharmacocinétique de l'omalizumab est linéaire aux doses supérieures à 0.5 mg/kg.
Après administration de doses répétées d'omalizumab, les aires sous la courbe de la concentration sérique (entre J0 et J14 à l'état d'équilibre) ont été jusqu'à 6 fois supérieures à celles observées après l'administration de la première dose.
Distribution
In vitro, l'omalizumab forme des complexes à poids moléculaire limité avec les IgE. Il n'a pas été observé, in vitro ou in vivo, de complexes précipitants ni la formation de complexes de poids moléculaire supérieur à 1 000 000 de daltons. Le volume apparent de distribution observé chez les patients après l'administration sous-cutanée a été de 78 ± 32 ml/kg de poids corporel.
Métabolisme
Il n'existe aucune donnée pertinente concernant l'utilisation.
Élimination
L'élimination de l'omalizumab fait intervenir des processus d'élimination des IgG ainsi qu'une élimination par une fixation spécifique et la formation de complexes avec le ligand cible, l'IgE. L'élimination hépatique des IgG fait intervenir une dégradation par le système réticulo-endothélial (SRE) et les cellules endothéliales. Les IgG intacts sont également éliminées dans la bile. Chez les patients asthmatiques, la demi-vie d'élimination sérique de l'omalizumab a été en moyenne de 26 jours, avec une élimination apparente de 2.4 ± 1.1 ml/kg/jour en moyenne.
Par ailleurs, à un poids corporel double correspond à peu près une élimination apparente double.
Patients atteints de polypes nasaux
Les analyses pharmacocinétiques de populations pour l'omalizumab ont indiqué que la pharmacocinétique de l'omalizumab dans le cas des polypes nasaux coïncide avec celle obtenue dans le cas de l'asthme. Des analyses graphiques de covariables ont été réalisées afin d'évaluer les effets de caractéristiques démographiques et d'autres facteurs sur l'exposition à l'omalizumab et la réponse clinique. Ces analyses montrent qu'aucun ajustement de la posologie sur la base de l'âge (18 à 75 ans) ou du sexe n'est nécessaire. Les données disponibles concernant la race et l'appartenance ethnique sont trop limitées pour les patients atteints de polypes nasaux et ne permettent pas d'émettre des affirmations quant à la nécessité éventuelle d'ajuster la posologie.
Linéarité/non-linéarité
La pharmacocinétique de l'omalizumab suit une courbe linéaire pour les doses supérieures à 0.5 mg/kg.
Cinétique pour certains groupes de patients
Les analyses de pharmacocinétique en fonction de l'âge, du sexe, de l'origine ethnique et de l'indice de masse corporelle suggèrent qu'aucun ajustement posologique n'est nécessaire pour ces groupes de patients (6–76 ans).
On ne dispose d'aucune donnée pharmacocinétique ou pharmacodynamique chez les patients insuffisants rénaux ou hépatiques (cf. "Mises en garde et précautions" ).
Âge, provenance ethnique, sexe, poids corporel, indice de masse corporelle, valeur initiale de l'IgE, auto-anticorps anti-FcεRI, médicaments concomitants
Les effets de certaines covariables démographiques et de toute une série d'autres facteurs sur l'exposition à l'omalizumab ont été examinés à l'aide de méthodes de pharmacocinétique de populations. De plus, les effets des covariables ont été testés par l'analyse de la relation entre les concentrations de l'omalizumab et la réponse clinique. Ces analyses suggèrent qu'aucune adaptation des doses n'est requise chez les patients avec UCS en fonction de l'âge (12 à 75 ans), de la provenance ethnique, du sexe, du poids corporel, de l'indice de masse corporelle, de la valeur initiale de l'IgE, des auto-anticorps anti-FcεRI ou de la prise concomitante d'antihistaminiques H2 ou d'antagonistes du récepteur des leucotriènes (LTRA).
Données précliniquesIl n'a pas été mis en évidence de réponse anaphylactique par dégranulation des mastocytes lors de l'administration de Xolair chez les singes Cynomolgus adultes et juvéniles.
Toutes les études d'administration d'omalizumab chez le singe ont montré la présence de complexes d'anticorps IgEomalizumab circulants, sans pour autant qu'une réaction liée aux complexes immuns puisse être détectée dans un organe quelconque (même dans les reins). Les complexes omalizumab-IgE ne peuvent pas se lier au complément ni développer de cytotoxicité dépendante du complément.
Des anticorps dirigés contre l'omalizumab ont été détectés chez certains singes après administration sous-cutanée ou intraveineuse. Après l'administration d'une protéine hétérologue, on pouvait s'attendre à un tel effet. Certains animaux n'ont pas pu être évalués à cause de concentrations sériques d'omalizumab trop élevées, de taux d'IgE élevés, ou les deux à la fois. Pendant toute la durée du traitement lors de l'étude, les animaux ont cependant présenté des concentrations sériques d'omalizumab élevées, et il n'y a pas eu de toxicité manifeste suite à la présence des anticorps antiomalizumab.
Les études de distribution tissulaire menées chez le singe Cynomolgus n'ont pas montré d'absorption spécifique de l'omalizumab marqué à l'l125 par un organe ou un tissu quelconque.
Dans les études réalisées chez la souris et le singe, les complexes omalizumab-IgE ont été éliminés par interaction avec les récepteurs Fcγ dans le SRE, avec des taux de dégradation généralement plus rapides que la clairance des IgG.
Toxicité à long terme (ou toxicité en administration répétée)
Chez les singes (aussi bien adultes que juvéniles), l'administration à long terme de l'omalizumab à des doses allant jusqu'à 250 mg/kg (dose clinique maximale autorisée pour l'asthme ou l'UCS selon les recommandations: 15 mg/kg) a été bien tolérée, à l'exception d'une diminution dose-dépendante des plaquettes chez plusieurs primates ayant une concentration sérique généralement supérieure à la dose maximale d'exposition chez l'homme lors des études cliniques pivots. Les singes juvéniles ont été plus sensibles aux effets sur les thrombocytes que les singes adultes. Par ailleurs, des hémorragies aiguës et des inflammations ont été observées au site d'injection chez le singe Cynomolgus. Ces événements correspondent à une réponse immune localisée en réponse à des administrations répétées d'une protéine hétérologue par voie sous-cutanée.
Carcinogénicité
Aucune étude conventionnelle de carcinogénicité n'a été conduite avec l'omalizumab.
Toxicité pour la reproduction
Dans les études de reproduction conduites chez le singe Cynomolgus, des doses sous-cutanées d'omalizumab allant jusqu'à 75 mg/kg par semaine (rapport d'exposition augmenté d'un facteur 17 environ par rapport à la DMRH sur une période de 4 semaines) n'ont pas provoqué de toxicité maternelle, d'embryotoxicité ou de tératogénicité en cas d'administration pendant toute la durée de l'organogenèse et il n'a pas été mis en évidence d'effets délétères sur la croissance fœtale ou néonatale en cas d'administration pendant la gestation, pendant la mise bas et pendant l'allaitement.
L'excrétion d'omalizumab dans le lait de singe Cynomolgus soumis à des administrations sous-cutanées de 75 mg/kg/semaine a été étudiée. Le taux sérique d'omalizumab chez le nouveau-né après exposition in utero et 28 jours d'allaitement était entre 11 et 94% du taux sérique de la mère. Les taux d'omalizumab retrouvés dans le lait ont représenté 0.15% de la concentration sérique maternelle.
Fertilité
On ne dispose d'aucune donnée concernant l'influence de l'omalizumab sur la fertilité chez l'être humain. Au cours des études non cliniques spécialement conçues pour évaluer la fertilité, y compris des essais concernant l'accouplement, aucun trouble de la fertilité mâle ou femelle n'a été constaté après l'administration de doses répétées d'omalizumab allant jusqu'à 75 mg/kg.
Remarques particulièresIncompatibilités
Xolair ne doit pas être mélangé avec d'autres médicaments ou avec d'autres diluants que de l'eau pour préparations injectables.
Stabilité
Ce médicament ne doit pas être utilisé au-delà de la date figurant après la mention "EXP" sur l'emballage.
Stabilité après ouverture
La préparation ne contient pas de conservateur. La stabilité chimique et physique "in use" du produit reconstitué a été démontrée pendant 8 heures en cas de stockage au réfrigérateur (2–8 °C) et pendant 4 heures à 30 °C. Pour des raisons microbiologiques, la préparation prête à l'emploi doit être utilisée immédiatement après la reconstitution. Si cela n'est pas possible, le délai d'utilisation et les conditions de stockage relèvent de la responsabilité de l'utilisateur mais, de manière générale, l'entreposage ne devrait pas dépasser 8 heures à une température comprise entre 2 et 8 °C et 2 heures à 25 °C, sauf si la dilution/reconstitution se déroule dans des conditions aseptiques contrôlées et validées.
Toute solution reconstituée non utilisée et tout déchet doivent être éliminés conformément à la réglementation en vigueur.
La poudre Xolair 75 mg et 150 mg pour solution injectable est livrée dans une ampoule perforable à usage unique.
Remarques particulières concernant le stockage
Conserver au réfrigérateur (2–8 °C).
Ne pas congeler.
Conserver dans l'emballage d'origine pour le protéger de la lumière (et/ou de l'humidité).
Tenir hors de portée des enfants.
Remarques concernant la manipulation
Il faut habituellement 15–20 minutes pour dissoudre totalement le produit lyophilisé, mais dans certains cas, cela peut prendre plus de temps. Le produit ainsi reconstitué est limpide ou légèrement opaque et il peut se former quelques petites bulles ou un peu de mousse à proximité du bord de l'ampoule perforable. Compte tenu de la légère viscosité du produit reconstitué, il faudra veiller à aspirer tout le produit contenu dans l'ampoule perforable avant d'expulser les bulles d'air ou excédent de solution éventuellement présents dans la seringue afin d'obtenir la dose pleine de 0.6 ml, resp. 1.2 ml.
Préparation de la solution à injecter (reconstitution)
Flacon de 75 mg resp. 150 mg:
1.Prélever 0.9 ml (75 mg), resp. 1.4 ml (150 mg) d'eau pour préparations injectables dans l'ampoule à l'aide d'une seringue munie d'une aiguille de calibre 18.
2.L'ampoule perforable contenant l'omalizumab placé bien droite sur une surface plate, insérer l'aiguille et transférer l'eau pour préparations injectables dans l'ampoule perforable contenant la poudre lyophilisée dans des conditions aseptiques standard, en dirigeant l'eau pour préparations injectables directement sur la poudre.
3.Tout en maintenant l'ampoule perforable bien droite, l'agiter énergiquement par rotation (ne pas secouer) pendant 1 minute environ afin de mouiller la poudre de manière homogène.
4.Pour favoriser la dissolution après l'étape 3, agiter doucement le flacon par rotation pendant 5 à 10 secondes environ toutes les 5 minutes afin de dissoudre les résidus solides éventuels.* A noter que dans certains cas, il faudra plus de 20 minutes pour parvenir à la dissolution complète de la poudre. Dans ce cas, répéter l'étape 4 jusqu'à ce qu'il n'y ait plus de particules gélatineuses visibles dans la solution.Lorsque le produit est complètement dissous, il ne doit y avoir aucune particule gélatineuse visible dans la solution. La présence de petites bulles ou de mousse à proximité du bord de l'ampoule perforable est courante. Le produit reconstitué a un aspect limpide ou légèrement opaque. Ne pas utiliser en présence de corps étrangers.
5.Retourner l'ampoule perforable pendant au moins 15 secondes afin de permettre à la solution de s'écouler vers le bouchon. Prendre une nouvelle seringue de 2 ou 5 ml munie d'une aiguille de calibre 18 et insérer l'aiguille dans l'ampoule perforable retournée. Tout en maintenant le flacon retourné, placer l'extrémité de l'aiguille au niveau le plus bas de la solution dans le flacon pendant l'aspiration de la solution dans la seringue. Avant de retirer l'aiguille du flacon, tirer le piston jusqu'au bout du corps de la seringue pour aspirer toute la solution contenue dans l'ampoule perforable retournée.
6.Remplacer l'aiguille de calibre 18 par une aiguille de calibre 25 pour l'injection sous-cutanée.
7.Expulser l'air, les grosses bulles et tout excédent éventuel de solution afin d'obtenir la dose requise de 0.6 ml (75 mg), resp. 1.2 ml (150 mg). Il est possible qu'une fine couche de petites bulles demeure en haut de la solution dans la seringue. Comme la solution est légèrement visqueuse, l'administration de la solution par injection sous-cutanée peut prendre 5 à 10 secondes. L'ampoule perforable délivre 0.6 ml (75 mg), resp. 1.2 ml (150 mg), de Xolair.Pour administrer une dose de 75 mg, il est également possible de prélever 0.6 ml de solution d'une ampoule perforable de 150 mg dans la seringue et de jeter le reste.
8.Les injections sont administrées par voie sous-cutanée dans la région deltoïde du bras ou dans la cuisse.
Numéro d’autorisation57178 (Swissmedic).
Présentation1 ampoule perforable de 75 mg d'omalizumab avec 1 flacon de solvant de 2 ml. [B]
1 ampoule perforable de 150 mg d'omalizumab avec 1 flacon de solvant de 2 ml. [B]
Titulaire de l’autorisationNovartis Pharma Schweiz AG, Risch; Domicile: 6343 Rotkreuz
Mise à jour de l’informationMars 2025
|