CompositionPrincipes actifs
Tipranavir.
Excipients
Ricinoléate de macrogolglycérol 455 mg, éthanol 7% (v/v), mono- et diglycéride d'acide octanoïque/décanoïque, propylène glycol 73 mg, trométamol, gallate de propyle (E 310).
L'enveloppe de la gélule contient: gélatine, propylène glycol 107,1 mg, «mélange spécial sorbitol-glycérol» (12,6 mg D-sorbitol E 420 et 1,4-sorbitane), dioxyde de titane (E 171) et oxyde de fer rouge (E 172).
L'encre noire contient: propylène glycol, oxyde de fer noir (E 172), phtalate d'acétate de polyvinyle, macrogol.
Indications/Possibilités d’emploiAptivus est toujours administré avec du ritonavir à faible dose et est indiqué dans le traitement antirétroviral associé chez les adultes et les adolescents à partir de 12 ans infectés par le virus VIH-1.
L'administration d'Aptivus est uniquement indiquée chez les patients déjà prétraités plusieurs fois par des antirétroviraux qui sont infectés par des souches de VIH-1 résistantes à de multiples inhibiteurs de la protéase et qui n'ont pas d'autres alternatives thérapeutiques.
Cette indication est basée sur les résultats de deux études de phase III conduites chez des patients adultes prétraités plusieurs fois (ayant déjà reçu en moyenne 12 antirétroviraux) avec virus résistant aux inhibiteurs de la protéase (détails du profil de résistance du VIH des patients en début d'étude, voir la rubrique «Propriétés/Effets») et sur les résultats d'une étude de phase II évaluant la pharmacocinétique, la sécurité d'emploi et l'efficacité d'Aptivus chez des patients adolescents, âgés de 12 à 18 ans. Cette dernière étude a principalement été menée chez des patients prétraités avec réplication actuelle du VIH-1 (voir la rubrique «Propriétés/Effets»).
|
À respecter obligatoirement avant l'initiation d'un traitement par Aptivus/ritonavir:
·Aptivus doit exclusivement être prescrit ou le traitement instauré par des médecins expérimentés dans la thérapie des patients infectés par le VIH.
·L'utilisation d'Aptivus doit, si possible, être guidée par des tests génotypiques adéquats et/ou par les antécédents des différents traitements.
·L'utilisation d'Aptivus associé au ritonavir n'est pas recommandée chez les patients naïfs de traitement infectés par le VIH de type sauvage.
·Il n'existe pas de données d'études démontrant un effet sur la progression clinique de la maladie causée par le VIH-1.
·Des cas d'hépatites cliniques et de défaillance hépatique, dont certains fatals, ont été observés sous Aptivus/ritonavir. Par conséquent, les transaminases doivent être mesurées avant le début de la thérapie et ensuite régulièrement pendant le traitement. Chez les patients présentant des transaminases significativement élevées, une co-infection par une hépatite B ou C, le rapport bénéfice/risque doit être évalué très soigneusement en raison d'une détérioration potentielle de la fonction hépatique (voir «Contre-indications» ainsi que «Mises en garde et précautions»).
·Le potentiel d'interaction très prononcé d'Aptivus/ritonavir avec une multitude de médicaments de différentes classes doit être soigneusement évalué avant l'utilisation de ce produit. Il convient plus particulièrement d'observer la liste des substances formellement contre-indiquées lors d'un traitement par Aptivus/ritonavir (voir «Interactions» et «Contre-indications»).
|
Posologie/Mode d’emploiTraitement associé
Aptivus doit toujours être administré avec du ritonavir à faible dose en tant que «booster» pharmacologique, et en association à au moins deux autres substances antirétrovirales. Avant l'initiation d'un traitement, outre les informations professionnelles d'Aptivus et du ritonavir, il faut donc également consulter celles des autres médicaments antirétroviraux.
Les patients doivent être informés de la nécessité de prendre Aptivus et le ritonavir tous les jours comme prescrit.
Posologie usuelle chez les adultes et adolescents à partir de 12 ans
La dose recommandée d'Aptivus est de 500 mg (2 capsules), co-administrés avec 200 mg de ritonavir, deux fois par jour.
Étant donné la disponibilité actuellement limitée de données sur l'efficacité et la sécurité d'emploi chez les adolescents (voir rubrique «Propriétés/Effets»), une surveillance étroite de la réponse virologique et de la tolérance est particulièrement importante dans ce groupe de patients.
Généralités
Aptivus doit être administré en même temps que le ritonavir à faible dose. Pour améliorer la tolérance du ritonavir, celui-ci devrait être pris avec le repas.
Patients présentant des troubles de la fonction hépatique
Aptivus/ritonavir est formellement contre-indiqué chez les patients présentant une insuffisance hépatique modérée ou sévère (classe Child-Pugh B ou C) (voir «Contre-indications»). En cas d'insuffisance hépatique légère (classe Child-Pugh A), Aptivus/ritonavir peut être utilisé avec la prudence correspondante et une surveillance accrue de la fonction hépatique lorsqu'il n'existe pas d'autre alternative (voir également «Mises en garde et précautions»). Aucun ajustement posologique n'est requis chez les patients présentant une insuffisance hépatique légère.
Patients présentant des troubles de la fonction rénale
La clairance rénale d'Aptivus et du ritonavir est négligeable. Il ne faut donc pas s'attendre à des taux plasmatiques élevés chez les patients insuffisants rénaux et aucun ajustement posologique n'est requis.
Enfants et adolescents
Aucune donnée n'est disponible sur la sécurité et l'efficacité des capsules Aptivus chez les enfants de moins de 12 ans. De plus, un ajustement approprié des doses chez l'enfant de moins de 12 ans ne peut pas être effectué avec Aptivus capsules.
Retard d'administration
Si une dose a été oubliée depuis plus de 5 heures par rapport à l'heure de prise prévue, le patient doit attendre le prochain moment de prise habituelle pour prendre la dose suivante d'Aptivus et de ritonavir. Si l'oubli de la dose date de moins de 5 heures par rapport à l'heure de prise prévue, le patient doit prendre immédiatement la dose oubliée, puis prendre la dose suivante d'Aptivus et de ritonavir à l'heure de prise habituelle.
Contre-indicationsHypersensibilité au principe actif ou à l'un des excipients
Les capsules molles d'Aptivus contenant de petites quantités de sorbitol (12,6 mg par capsule), les patients présentant une rare intolérance héréditaire au fructose ne doivent pas prendre ce médicament.
Aptivus/ritonavir est contre-indiqué chez les patients présentant une insuffisance hépatique modérée ou sévère (classe Child-Pugh B ou C). Aptivus/ritonavir ne doit pas être administré à des patients présentant des taux initiaux d'ASAT ou d'ALAT supérieurs à 5 fois la valeur normale, mais uniquement lorsque ces taux se sont stabilisés en dessous de 5 fois la valeur normale.
Tant le médecin que le patient doivent être attentifs aux signes ou symptômes d'un début d'hépatite. Les médecins doivent surveiller leurs patients sur des symptômes tels que fatigue, malaise, anorexie, nausées, ictère, bilirubinurie, selles acholiques, douleur hépatique ou hépatomégalie.
La rifampicine et Aptivus ne doivent pas être utilisés conjointement, la co-administration pouvant mener à une diminution importante des concentrations de tipranavir susceptible de réduire significativement l'efficacité thérapeutique d'Aptivus (voir «Interactions»).
Les préparations phytothérapeutiques contenant du millepertuis (Hypericum perforatum) ne doivent pas être utilisées pendant le traitement par Aptivus, étant donné le risque de réduction des concentrations plasmatiques et de l'efficacité thérapeutique d'Aptivus (voir «Interactions»).
La co-administration d'Aptivus associé au ritonavir à faible dose et à des principes actifs dont la clairance dépend fortement du CYP3A, et chez lesquels une augmentation des concentrations plasmatiques est associée à des effets secondaires graves et/ou potentiellement mortels, est contre-indiquée.
|
Classe de médicaments/Nom
|
Indications cliniques
| |
Antiarythmiques
p.ex. amiodarone, bépridil, flécaïnide, propafénone, quinidine
|
Contre-indiqués en raison de possibles effets graves et/ou potentiellement mortels, par exemple troubles du rythme cardiaque suite à une augmentation de la concentration plasmatique de l'antiarythmique.
| |
Antihistaminiques
p.ex. astémizole, terfénadine
|
Contre-indiqués en raison de possibles effets graves et/ou potentiellement mortels, par exemple troubles du rythme cardiaque.
| |
Antibiotiques
rifampicine
|
Peut entraîner la disparition de la réponse virologique et l'apparition d'une possible résistance au tipranavir ou à la classe des inhibiteurs de la protéase.
| |
Dérivés de l'ergot de seigle
p.ex. dihydroergotamine, ergonovine, ergotamine, méthylergonovine
|
Contre-indiqués en raison de possibles effets graves et/ou potentiellement mortels, par exemple une ergotoxicité aiguë qui se manifeste par un spasme vasculaire périphérique et une ischémie des extrémités ou d'autres tissus.
| |
Stimulants de la motricité gastro-intestinale
p. ex cisapride
|
Contre-indiqués en raison de possibles effets graves et/ou potentiellement mortels, par exemple troubles du rythme cardiaque.
| |
Phytothérapeutique
Millepertuis
|
Peut entraîner la disparition de la réponse virologique et l'apparition d'une possible résistance au tipranavir ou à la classe des inhibiteurs de la protéase.
| |
Inhibiteurs de l'HMG-CoA réductase
lovastatine, simvastatine
|
Effets graves possibles, par exemple risque de myopathie, rhabdomyolyse incluse.
| |
Antipsychotiques
p.ex. pimozide, sertindole, quétiapine
|
Contre-indiqués en raison de possibles effets graves et/ou potentiellement mortels, par exemple troubles du rythme cardiaque.
| |
Sédatifs et hypnotiques oraux
p.ex. midazolam, triazolam
|
Contre-indiqués en raison de possibles effets graves et/ou potentiellement mortels, par exemple sédation prolongée ou augmentée ou dépression respiratoire.
| |
Inhibiteurs de la phosphodiestérase (IPDE-5)
vardénafil
|
Contre-indiqués, étant donné qu'il faut s'attendre à une augmentation importante de la concentration d'inhibiteurs de la PDE5.
| |
Bloqueurs des adrénorécepteurs α 1 pour le traitement de l'hypertension artérielle pulmonaire
alfuzosine, sildénafil
|
Contre-indiqués en raison de la dépendance élevée par rapport à la clairance du CYP3A, étant donné que des niveaux plasmatiques élevés d'alfuzosine et de sildénafil sont associés à des effets indésirables sévères et/ou engageant le pronostic vital (voir «Interactions»).
|
La prudence est de rigueur lors de la co-administration d'Aptivus associé au ritonavir et d'autres médicaments connus pour être des inducteurs du cytochrome 3A4, étant donné la possibilité d'un échec thérapeutique et d'un développement accéléré d'une résistance. Il s'agit notamment de la phénytoïne, du phénobarbital, de la primidone et du topiramate.
En outre, la co-administration d'Aptivus associé au ritonavir à faible dose et de médicaments dont la clairance est fortement dépendante du CYP2D6, tels que les antiarythmiques flécaïnide et propafénone, est contre-indiquée (voir «Interactions»).
Étant donné qu'Aptivus doit toujours être administré avec le ritonavir à faible dose, l'information professionnelle du ritonavir doit être consultée concernant les contre-indications du ritonavir.
L'administration concomitante de colchicine et d'Aptivus associé au ritonavir n'est pas recommandée (voir «Interactions»).
Mises en garde et précautionsAptivus doit être administré avec le ritonavir à faible dose afin d'assurer son effet thérapeutique (voir « Posologie/Mode d'emploi »). Une co-administration incorrecte d'Aptivus et du ritonavir entraînera une diminution des concentrations plasmatiques du tipranavir, pouvant être insuffisante pour atteindre l'effet antiviral souhaité. Les patients doivent en être informés.
Des doses de ritonavir inférieures à 200 mg deux fois par jour chez l'adulte ne sont pas recommandées, en raison d'un risque d'altération du profil d'efficacité de la combinaison.
Aptivus ne constitue pas un traitement curatif de l'infection par le VIH-1 ou du SIDA. Les patients traités par Aptivus ou tout autre médicament antirétroviral peuvent également développer des infections opportunistes et d'autres complications de l'infection à VIH-1.
Les patients doivent être informés du fait que le traitement antirétroviral actuellement disponible n'a pas prouvé sa capacité à prévenir le risque de transmission du VIH à d'autres personnes, par contact sanguin ou sexuel. L'application des mesures de précaution correspondantes doit être poursuivie.
Les capsules molles d'Aptivus contenant de petites quantités de sorbitol (12,6 mg par capsule), les patients présentant une rare intolérance héréditaire au fructose ne doivent pas prendre ce médicament.
Patients âgés
Les études cliniques conduites avec Aptivus n'ont pas inclus un nombre suffisant de patients âgés de 65 ans ou plus pour permettre de déterminer s'ils répondaient autrement au médicament que les patients plus jeunes. D'une manière générale, la prudence et une surveillance étroite s'imposent lors de l'utilisation d'Aptivus chez les patients âgés. Il faut en effet tenir compte du taux supérieur d'insuffisance hépatique, rénale ou cardiaque, des comorbidités et d'autres mesures thérapeutiques.
Maladies hépatiques
Aptivus est contre-indiqué chez les patients atteints d'une insuffisance hépatique modérée ou sévère (classe Child-Pugh B ou C). Les données actuellement disponibles concernant l'administration d'Aptivus associé au ritonavir à faible dose chez les patients également infectés par l'hépatite B ou C sont limitées. Les patients atteints d'hépatite B ou C chronique et traités par une association d'antirétroviraux présentent un risque accru d'effets secondaires hépatiques sévères et potentiellement fatals. Aptivus ne doit être utilisé chez ces patients que si le bénéfice attendu l'emporte sur le risque potentiel et son utilisation implique une surveillance accrue des paramètres cliniques et biologiques. En cas de traitement antiviral concomitant de l'hépatite B ou C, il convient de tenir également compte des informations professionnelles des médicaments concernés. Des troubles de la fonction hépatique sous association médicamenteuse sont plus fréquents chez les patients présentant un dysfonctionnement hépatique préexistant, y compris une hépatite chronique active. Ces patients doivent être suivis selon la norme de soins. Une interruption ou un arrêt du traitement doit être envisagé en cas de signe d'aggravation de la maladie hépatique.
Les patients présentant une insuffisance hépatique légère (classe Child-Pugh A) doivent être étroitement suivis.
Aptivus associé au ritonavir à faible dose a été associé à des cas d'hépatite cliniquement pertinente et de défaillance hépatique, dont certains avec issue fatale. Ces cas sont généralement survenus chez des patients avec stade avancé de l'affection VIH et prenant de nombreux médicaments adjuvantss. Aucune relation de cause à effet n'a pu être établie avec Aptivus associé au ritonavir.
Les patients présentant des signes d'hépatite doivent arrêter le traitement par Aptivus/ritonavir et consulter leur médecin. La prudence est de mise lors de l'administration d'Aptivus/ritonavir à des patients présentant des anomalies des enzymes hépatiques ou un antécédent d'hépatite. Une surveillance accrue des taux d'AST ou d'ALAT est recommandée chez ces patients. Chez des patients présentant, avant initiation du traitement, des taux d'ASAT ou d'ALAT supérieurs à 5 fois la valeur normale supérieure (5xULN), le traitement par Aptivus ne doit pas être initié avant la stabilisation des taux de base d'ASAT/ALAT à des taux ne dépassant pas 5 fois la valeur normale supérieure (<5xULN), à moins que le bénéfice attendu ne l'emporte sur le risque potentiel.
Le traitement par Aptivus doit être arrêté chez les patients présentant une élévation des taux d'ASAT ou d'ALAT supérieure à 10 fois la valeur normale supérieure (> 10xULN) ou présentant des signes d'une hépatite cliniquement pertinente pendant le traitement. Si une autre cause est identifiée (p.ex. une hépatite virale aiguë A, B ou C, une affection de la vésicule biliaire, d'autres médicaments), une reprise du traitement par Aptivus pourra être envisagée lorsque les taux d'ASAT/ALAT du patient seront revenus à leurs valeurs initiales.
Le tipranavir étant essentiellement métabolisé par voie hépatique, Aptivus doit être utilisé avec prudence chez les patients présentant des lésions hépatiques, car la concentration de tipranavir pourrait être augmentée.
Surveillance de la fonction hépatique: les valeurs hépatiques doivent être contrôlées 2, 4 et 8 semaines après le début du traitement, puis toutes les 8 à 12 semaines. Une surveillance accrue (c.-à-d. toutes les 2 semaines durant les 3 premiers mois de traitement, puis tous les mois) est requise lorsqu'Aptivus associé au ritonavir à faible dose est utilisé chez des patients avec taux d'ASAT et d'ALAT accrus, un léger trouble de la fonction hépatique, une hépatite chronique B ou C ou toute autre affection hépatique préexistante.
Patients naïfs de traitement: dans une étude conduite chez des patients adultes naïfs de traitement, 16,2 % (estimation selon Kaplan-Meier) des patients ayant reçu Aptivus/ritonavir (500 mg/200 mg) pendant 48 semaines ont présenté des élévations des taux d'ALAT de grade 3 ou 4. L'utilisation d'Aptivus associé au ritonavir n'est pas recommandée chez les patients naïfs de traitement infectés par le VIH de type sauvage.
Troubles de la fonction rénale
La clairance rénale du tipranavir étant négligeable, une élévation des taux plasmatiques n'est pas attendue chez les insuffisants rénaux.
Hémophilie
Des cas d'augmentation des hémorragies, notamment des hématomes cutanés spontanés et des hémarthroses, ont été rapportés chez des hémophiles de type A et B traités par inhibiteurs de la protéase. Le facteur VIII a également été administré à certains patients. Dans plus de la moitié des cas rapportés, le traitement par inhibiteurs de la protéase a été poursuivi ou repris après son interruption. Une relation causale a été évoquée, bien que le mécanisme d'action ne soit pas encore élucidé. Il convient donc d'informer les patients hémophiles de l'éventualité d'une augmentation des hémorragies.
Hémorragie intracrânienne
Chez certains patients, l'administration d'Aptivus associé au ritonavir à faible dose était associée à des hémorragies intracrâniennes (HIC) fatales et non fatales. Nombre de ces patients présentaient d'autres états cliniques associés ou étaient traités par des co-médications ayant éventuellement provoqué ou ayant contribué à la survenue d'une HIC. Un lien avec le tipranavir ne peut toutefois être exclu dans certains cas. Aucune relation n'a pu être mise en évidence entre des paramètres hématologiques ou de coagulation anormaux chez ces patients, ni en général, ni en rapport avec le développement d'une HIC. C'est pourquoi aucune détermination de routine des paramètres de coagulation n'est actuellement requise chez les patients traités par Aptivus.
De manière analogue, une incidence supérieure du risque d'HIC des patients traités dans les études Aptivus avait déjà été observée chez d'autres patients atteints de VIH à un stade avancé/SIDA. La prudence est de rigueur lors de l'administration d'Aptivus associé au ritonavir à faible dose à des patients ayant un risque d'hémorragie accru suite à un traumatisme, une opération ou un autre état clinique ou à des patients recevant des médicaments augmentant le risque d'hémorragie (p.ex. des anticoagulants ou des inhibiteurs de l'agrégation des thrombocytes) (voir «Effets indésirables»).
Effet sur l'agrégation plaquettaire et la coagulation
Aptivus associé au ritonavir à faible dose doit être utilisé avec prudence chez les patients qui, suite à un traumatisme, une opération ou pour toute autre raison médicale, présentent un risque d'hémorragie accru ou qui reçoivent des médicaments susceptibles d'augmenter le risque d'hémorragie (p.ex. des antiagrégants plaquettaires et des anticoagulants ou des doses élevées de vitamine E).
Chez des rats, l'administration concomitante d'un dérivé de la vitamine E a renforcé les effets du tipranavir sur les hémorragies (voir la rubrique «Données précliniques»). Toutefois, l'analyse de plasma stocké d'adultes ayant reçu des capsules d'Aptivus associé à du ritonavir à faible dose ainsi que de plasma d'enfants et d'adolescents ayant reçu soit Aptivus capsules, soit une solution buvable de tipranavir (contenant un dérivé de la vitamine E) associé à du ritonavir à faible dose a montré l'absence d'effet du tipranavir, associé ou non à la solution buvable contenant de la vitamine E, sur les facteurs de coagulation dépendant de la vitamine K (facteur II et facteur VII), le facteur V, la prothrombine ou sur le temps de thromboplastine partielle activé.
Des études in vitro menées sur le tipranavir ont montré une inhibition de l'agrégation plaquettaire humaine semblable à celle observée chez des patients recevant Aptivus associé au ritonavir à faible dose.
Diabète sucré/Hyperglycémie
Des cas de diabète sucré, d'hyperglycémie ou d'exacerbation d'un diabète préexistant ont été rapportés chez des patients recevant un traitement antirétroviral, y compris par des inhibiteurs de la protéase. Les hyperglycémies étaient partiellement sévères et également partiellement associées à une acidocétose. De nombreux patients présentaient des comorbidités sous-jacentes dont certaines requéraient partiellement un traitement par des principes actifs susceptibles d'induire l'apparition d'un diabète sucré ou d'une hyperglycémie.
Hyperlipidémie
Le traitement par Aptivus associé au ritonavir à faible dose et à d'autres subtances antirétrovirales a entraîné une augmentation des concentrations plasmatiques des triglycérides et du cholestérol total. Les taux de triglycérides et du cholestérol doivent être mesurés avant l'instauration de la thérapie ainsi que pendant le traitement par Aptivus. L'augmentation des lipides liée au traitement doit être prise en charge selon l'évaluation clinique.
Redistribution des graisses
Chez les patients infectés par le VIH, les traitements par association d'antirétroviraux sont associés à une redistribution de la masse grasse corporelle (lipodystrophie). Les conséquences à long terme de ces événements ne sont pas connues actuellement et le mécanisme n'est pas entièrement élucidé. Une relation éventuelle entre lipomatose viscérale et inhibiteurs de la protéase d'une part et lipoatrophie et inhibiteurs nucléosidiques de la transcriptase inverse d'autre part est discutée. Le risque de lipodystrophie accru a été mis en relation avec des facteurs individuels comme un âge plus avancé et, d'autre part, avec des facteurs liés au traitement tels qu'une plus longue durée de traitement antirétroviral avec les anomalies métaboliques qui lui sont associées. L'examen clinique doit comporter la recherche des signes physiques d'une redistribution des graisses. Des mesures des lipides sériques et de la glycémie à jeun doivent être envisagées. Les troubles lipidiques doivent être pris en charge selon l'évaluation clinique (voir «Effets indésirables»).
L'utilisation d'inhibiteurs de la HMG-CoA réductase (lovastatine et simvastatine) est toutefois contre-indiquée en raison des interactions potentiellement dangereuses (voir «Contre-indications»).
Syndrome de reconstitution immune
Chez les patients séropositifs présentant un déficit immunitaire sévère au moment de l'instauration du traitement par association d'antirétroviraux, y compris Aptivus, une réaction inflammatoire à des agents pathogènes opportunistes asymptomatiques ou résiduels peut apparaître et entraîner des manifestations cliniques graves ou une détérioration des symptômes. De telles réactions ont été observées typiquement au cours des premières semaines ou mois suivant l'instauration d'un traitement par association d'antirétroviraux. Des exemples importants sont les rétinites à cytomégalovirus, les infections mycobactériennes disséminées et/ou localisées et les pneumopathies à Pneumocystis jirovecii. Tout symptôme inflammatoire doit être évalué et un traitement doit être instauré, si nécessaire. Une réactivation d'herpès ou de zona a également été observée au cours d'études cliniques avec Aptivus associé au ritonavir à faible dose. Des affections auto-immunes (p.ex. maladie de Basedow) ont également été observées dans le cadre d'une réactivation immunitaire; le délai de survenue varie toutefois et les événements peuvent survenir de nombreux mois après le début du traitement.
Tractus gastro-intestinal
Aptivus capsules molles contient du ricinoléate de macrogolglycérol qui peut entraîner des indigestions et des diarrhées.
Éruptions cutanées
Des éruptions cutanées légères à modérées comprenant des exanthèmes urticariens et maculopapuleux et une photosensibilité ont été rapportés chez des patients traités par Aptivus associé au ritonavir à faible dose. Au cours des études cliniques de phase III, diverses éruptions cutanées ont été observées jusqu'à la 48e semaine chez 15,5% des hommes et 20,5% des femmes traités par Aptivus en association au ritonavir à faible dose. Dans une étude d'interaction sur des femmes saines recevant une dose unique d'éthinylestradiol suivie de l'administration d'Aptivus associé au ritonavir à faible dose, 33% des sujets ont en outre développé une éruption cutanée. Des éruptions cutanées accompagnées d'une douleur articulaire ou d'une raideur, d'une constriction de la gorge ou d'un prurit généralisé ont été rapportées à la fois chez les hommes et chez les femmes traités par Aptivus associé au ritonavir à faible dose.
Dans l'étude clinique sur les enfants et les adolescents, la fréquence des éruptions cutanées (tous grades et toutes causes confondus) survenues au cours des 48 semaines de traitement était supérieure à celle des patients adultes.
Ostéonécrose
Bien que l'étiologie soit considérée comme multifactorielle (incluant l'utilisation de corticoïdes, la consommation d'alcool, une immunosuppression sévère, un indice de masse corporelle élevé), des cas d'ostéonécrose ont notamment été rapportés chez des patients à un stade avancé de l'affection VIH et/ou lors du traitement au long cours par association d'antirétroviraux (TAR). Les patients doivent être informés qu'ils doivent consulter un médecin en cas de troubles articulaires et d'arthralgies, de raideur articulaire ou de difficultés de mouvement.
Fertilité
Aucune étude n'a été effectuée concernant l'effet du tipranavir sur la fertilité humaine. Les études précliniques n'ont pas montré d'effet indésirable sur la fertilité (voir «Données précliniques»).
InteractionsLe profil d'interaction d'Aptivus associé au ritonavir à faible dose est complexe. Se référer à la rubrique «Interactions» pour une description des mécanismes d'interaction avérés et potentiels d'Aptivus.
Inhibiteurs nucléosidiques de la transcriptase inverse: l'utilisation concomitante d'Aptivus associé au ritonavir à faible dose et d'abacavir ou de zidovudine entraîne une réduction significative des concentrations plasmatiques de ces deux inhibiteurs nucléosidiques de la transcriptase inverse (INTI). L'administration concomitante de la zidovudine ou de l'abacavir avec Aptivus associé au ritonavir à faible dose n'est donc pas recommandée, sauf s'il n'existe pas d'autres inhibiteurs nucléosidiques de la transcriptase inverse appropriés pour le traitement du patient (voir «Interactions»).
Inhibiteurs de la protéase: l'utilisation concomitante d'Aptivus associé au ritonavir à faible dose et des inhibiteurs de la protéase amprénavir, atazanavir, lopinavir ou saquinavir (associés au ritonavir à faible dose) mène à une réduction significative des concentrations plasmatiques de ces inhibiteurs de la protéase (voir «Interactions»).
L'utilisation concomitante d'un inhibiteur de la protéase et d'Aptivus associé au ritonavir à faible dose n'est pas recommandée. Les patients traités concomitamment par Aptivus et amprénavir, tous deux associés à du ritonavir à faible dose, peuvent présenter un risque plus important d'élévation des transaminases hépatiques de grade 3 et 4.
Inhibiteurs de l'HMG-CoA réductase: Aptivus associé au ritonavir à faible dose augmente les concentrations plasmatiques d'atorvastatine (voir «Interactions»). Cette combinaison n'est pas recommandée. L'utilisation d'autres inhibiteurs de l'HMG-CoA réductase, tels que la pravastatine, la fluvastatine ou la rosuvastatine, doit être envisagée.
Inhibiteurs de la phosphodiestérase de type 5 (IPDE5): l'administration concomitante d'Aptivus associé au ritonavir à faible dose et de vardénafil est contre-indiquée. Une prudence particulière est de mise lors de la prescription d'autres inhibiteurs de la phosphodiestérase de type 5 (sildénafil ou tadalafil) à des patients traités par l'Aptivus associé au ritonavir à faible dose. Il faut s'attendre à une augmentation importante des concentrations des inhibiteurs de la PDE5 et de leurs effets secondaires associés, notamment hypotension, troubles visuels et priapisme, en cas d'utilisation concomitante d'Aptivus associé au ritonavir à faible dose et d'IPDE5. L'utilisation concomitante d'Aptivus associé au ritonavir à faible dose et du tadalafil a entraîné une exposition plus importante au tadalafil après la première administration d'Aptivus/ritonavir et aucun changement relatif à l'exposition au tadalafil à l'état d'équilibre d'Aptivus/ritonavir. Lorsque le tadalafil est administré au cours des premiers jours du traitement par Aptivus/ritonavir, il convient d'administrer la dose de tadalafil la plus faible.
Après 7 à 10 jours de traitement par Aptivus/ritonavir, la dose de tadalafil peut être augmentée en fonction des besoins cliniques (voir «Interactions»).
Contraceptifs oraux et œstrogènes: l'administration concomitante d'Aptivus associé au ritonavir n'est pas recommandée en raison de la réduction des concentrations plasmatiques d'éthinylestradiol. Des mesures contraceptives alternatives ou supplémentaires sont indiquées lorsque les contraceptifs oraux à base d'œstrogènes sont co-administrés avec Aptivus associé au ritonavir à faible dose (voir «Interactions»). Les patientes recevant un traitement hormonal substitutif à base d'œstrogènes doivent être suivies cliniquement sur des signes d'insuffisance œstrogénique. Les femmes utilisant des œstrogènes peuvent présenter un risque accru d'éruption cutanée non grave.
Analgésiques narcotiques: l'administration concomitante d'une dose unique de méthadone avec Aptivus associé au ritonavir à faible dose a entraîné une réduction d'env. 50% de la concentration de méthadone (ASC et Cmax). Dans un tel cas, les patients doivent faire l'objet d'un suivi attentif quant à un syndrome de sevrage aux opiacés. Une augmentation de la dose de méthadone peut s'imposer. Aptivus associé au ritonavir à faible dose peut provoquer une réduction des concentrations de la mépéridine et une augmentation des concentrations de son métabolite, la normépéridine. Une augmentation de la dose de la mépéridine et son utilisation au long cours avec Aptivus associé au ritonavir à faible dose ne sont pas recommandées, en raison de l'augmentation des concentrations de son métabolite, la normépéridine. L'action de la normépéridine sur le SNC est à la fois analgésique et stimulante (p.ex. convulsions).
Halofantrine, luméfantrine: l'administration concomitante d'halofantrine et de luméfantrine et d'Aptivus associé au ritonavir à faible dose n'est pas recommandée en raison de leur profil métabolique et du risque inhérent d'apparition de torsades de pointes.
Anticonvulsivants: la prudence est de mise lors de la prescription de carbamazépine, de phénobarbital et de phénytoïne. La réduction des concentrations plasmatiques de tipranavir chez les patients prenant ces médicaments peut entraîner une réduction de l'efficacité d'Aptivus.
Disulfiram/métronidazole: les capsules molles d'Aptivus contiennent de l'alcool (7% d'éthanol, correspondant à 100 mg par capsule ou 200 mg par dose) qui peut induire des réactions de type disulfiram en cas d'administration concomitante avec le disulfiram ou d'autres médicaments pouvant déclencher ces réactions (p.ex. métronidazole).
Fluticasone: l'utilisation concomitante d'Aptivus associé au ritonavir à faible dose et de la fluticasone ou d'autres glucocorticoïdes métabolisés par le CYP3A4 n'est pas recommandée, à moins que le bénéfice attendu du traitement ne l'emporte sur le risque d'effet systémique des corticostéroïdes dont le syndrome de Cushing et l'insuffisance surrénalienne (voir «Interactions»).
Midazolam: lorsqu'Aptivus associé au ritonavir est administré avec le midazolam par voie parentérale, un suivi clinique étroit de l'apparition d'une dépression respiratoire et/ou d'une sédation prolongée doit être instauré et un ajustement de la dose envisagé (voir «Interactions»).
Trazodone: l'administration concomitante de trazodone et d'Aptivus associé au ritonavir à faible dose peut entraîner une augmentation des concentrations plasmatiques de trazodone. Des effets indésirables tels que nausées, vertigines, hypotension et syncope ont été observés au cours de cette étude suite à la co administration de trazodone et de ritonavir. L'association ne doit être utilisée qu'avec prudence et une réduction de la dose de trazodone doit être envisagée.
Colchicine: l'administration concomitante de colchicine et d'Aptivus associé au ritonavir à faible dose n'est pas recommandée (voir «Interactions»).
Salmétérol: l'administration concomitante de salmétérol et d'Aptivus/associéritonavir n'est pas recommandée (voir «Interactions»).
Mises en garde relatives aux excipients
Les capsules molles d'Aptivus contiennent 7% (v/v) d'éthanol. Il convient d'en tenir compte chez les femmes enceintes ou allaitantes, les enfants et les groupes à haut risque comme les patients souffrant d'affections hépatiques ou d'épilepsie. L'éthanol pourrait être nocif pour les patients alcooliques.
La prudence est de mise en cas de changement de traitement vers une autre forme pharmaceutique et/ou un autre médicament contenant le même principe actif. Le patient doit alors faire l'objet d'une surveillance adaptée.
Interactions
Le profil d'interaction d'Aptivus associé au ritonavir à faible dose est complexe et requiert une précaution particulière, notamment lors de l'association à d'autres principes actifs antirétroviraux. Le tipranavir associé au ritonavir peut modifier les concentrations plasmatiques d'autres médicaments et les autres médicaments peuvent influencer celles du tipranavir.
Les études d'interaction ont uniquement porté sur des adultes.
Profil métabolique du tipranavir
Le tipranavir est un substrat, inducteur et inhibiteur de l'iso-enzyme CYP3A du système du cytochrome P450. L'administration en association au ritonavir à la dose recommandée (voir «Posologie/Mode d'emploi») entraîne une inhibition de P450 CYP3A. L'administration concomitante d'Aptivus associé au ritonavir à faible dose et de principes actifs principalement métabolisés par le CYP3A peut mener à une modification des concentrations plasmatiques du tipranavir ou de ces autres principes actifs, ce qui peut modifier leurs effets thérapeutiques et leurs effets indésirables (voir tableau et détails des médicaments concernés ci-dessous). Les médicaments particulièrement contre-indiqués en raison de l'importance attendue des interactions avec le tipranavir et de leur potentiel d'effets secondaires graves sont décrits dans cette rubrique et listés parmi les contre-indications.
Une étude phénotypique a été conduite auprès de 16 volontaires sains pour mesurer l'influence de l'administration pendant 10 jours d'Aptivus capsules associé au ritonavir sur l'activité des cytochromes hépatiques CYP1A2 (caféine), CYP2C9 (warfarine), CYP2C19 (oméprazole), CYP2D6 (dextrométhorphane) et sur l'activité des CYP3A4/5 intestinaux et hépatiques (midazolam) et la glycoprotéine P (P-gp) (digoxine). Cette étude a évalué les effets de 500 mg d'Aptivus associés à 200 mg de ritonavir administrés deux fois par jour sous forme de capsule, après la première administration et à l'état d'équilibre.
Il n'y a pas d'effet net sur le CYP2C9 ou sur la P-gp hépatique, ni après la première dose ni à l'état d'équilibre. Après la première dose, on n'a pas observé d'effet net sur le CYP1A2, mais une induction modérée à l'état d'équilibre. Une légère inhibition sur le CYP2C19 a été observée après la première administration et une induction modérée à l'état d'équilibre. Une forte inhibition de l'activité du CYP2D6 et des CYP3A4/5 hépatiques et intestinaux a été observée après la première administration ainsi qu'à l'état d'équilibre. L'activité de la P-gp intestinale était inhibée après la première administration, mais aucun effet net n'a toutefois été observé à l'état d'équilibre.
Des études sur des microsomes hépatiques humains indiquent que le tipranavir est un inhibiteur du CYP1A2, du CYP2C9, du CYP2C19 et du CYP2D6. Puisque le ritonavir est également un inhibiteur du CYP2D6, l'effet global de tipranavir/ritonavir consiste vraisemblablement en une inhibition du CYP2D6. Les données d'une étude préalable suggèrent que l'effet global in vivo du tipranavir/ritonavir sur le CYP1A2, le CYP2C9 et le CYP2C19 serait le suivant: un potentiel inducteur d'APTIVUS/ritonavir sur le CYP1A2 et dans une moindre mesure sur le CYP2C9 et la P-gp après un traitement de plusieurs jours. On ne sait pas si le tipranavir inhibe ou active les glucuronosyltransférases. Des études in vitro ont montré que le tipranavir est non seulement un substrat, mais aussi un inhibiteur de la glycoprotéine P (P-gp).
Il est difficile de prédire l'effet net d'Aptivus associé au ritonavir à faible dose sur la biodisponibilité orale et les concentrations plasmatiques des principes actifs qui sont à la fois substrat du CYP3A et de la P-gp. L'effet net varie en fonction de l'affinité relative pour le CYP3A et la P-gp des médicaments concomitants utilisés ainsi que de l'importance du premier passage intestinal et de l'efflux.
Aptivus est métabolisé par le CYP3A et est un substrat de la P-gp. L'administration concomitante d'Aptivus et de principes actfis inducteurs du CYP3A et/ou de la P-gp peut abaisser les concentrations du tipranavir et réduire son effet thérapeutique (voir la liste et les détails des principes actifs concernés ci-dessous). L'utilisatioin concomitante d'Aptivus et de médicaments inhibant la P-gp peut augmenter les concentrations plasmatiques du tipranavir.
On ne sait pas encore si le tipranavir inhibe ou active les glucuronosyltransférases.
Interactions pharmacocinétiques
Tipranavir après administration avec des produits associés
|
Produit associé
|
Dosage du produit associé
(protocole d'étude)
|
Dosage TPV/r
(protocole d'étude)
|
n
|
PK
|
Relation (IC à 90 %) de la pharmacocinétique du tipranavir avec/sans produit associé (aucun effet = 1,00)
| |
Cmax
|
ASC
|
Cmin
| |
Atorvastatine
|
10 mg (1 dose)
|
500/200 mg 2x/j (14 doses)
|
22
|
↔
|
0,96 (0,86; 1,07)
|
1,08 (1,00; 1,15)
|
1,04 (0,89; 1,22)
| |
Clarithromycine
|
500 mg 2x/j (25 doses)
|
500/200 mg 2x/j.*
|
24(68)
|
↑
|
1,40 (1,24; 1,47)
|
1,66 (1,43; 1,73)
|
2,00 (1,58; 2,47)
| |
Didanosine
|
400 mg (1 dose)
|
500/100 mg 2x/j (27 doses)
|
5
|
↓
|
1,32 (1,09; 1,60)
|
1,08 (0,82; 1,42)
|
0,66 (0,31; 1,43)
| |
Éfavirenz
|
600 mg 1x/j (8 doses)
|
500/100 mg 2x/j.*
|
21(89
|
↓
|
0,79 (0,69; 0,89)
|
0,69 (0,57; 0,83)
|
0,58 (0,36; 0,86)
| |
|
|
750/200 mg 2x/j.*
|
25(100)
|
↔
|
0,97 (0,85; 1,09)
|
1,01 (0,85; 1,18)
|
0,97 (0,69; 1,28)
| |
Éthinylestradiol/noréthindrone
|
0,035/1,0 mg (1 dose)
|
500/100 mg 2x/j (21 doses)
|
21
|
↓
|
1,10 (0,98; 1,24)
|
0,98 (0,88; 1,11)
|
0,73 (0,59; 0,90)
| |
|
|
750/200 mg 2x/j (21 doses)
|
13
|
↔
|
1,01 (0,96; 1,06)
|
0,98 (0,90; 1,07)
|
0,91 (0,69; 1,20)
| |
Fluconazole
|
100 mg 1x/j (12 doses)
|
500/200 mg 2x/j.*
|
20(68)
|
↑
|
1,32 (1,18; 1,47)
|
1,50 (1,29; 1,73)
|
1,69 (1,33; 2,09)
| |
Lopéramide
|
16 mg (1 dose)
|
750/200 mg 2x/j (21 doses)
|
24
|
↓
|
1,03 (0,92; 1,17)
|
0,98 (0,86; 1,12)
|
0,74 (0,62; 0,88)
| |
Rifabutine
|
150 mg (1 dose)
|
500/200 mg 2x/j (15 doses)
|
21
|
↔
|
0,99 (0,93; 1,07)
|
1,00 (0,96; 1,04)
|
1,16 (1,07; 1,27)
| |
Ténofovir
|
300 mg (1 dose)
|
500/100 mg 2x/j (23 doses)
|
22
|
↓
|
0,83 (0,74; 0,94)
|
0,82 (0,75; 0,91)
|
0,79 (0,70; 0,90)
| |
|
|
750/200 mg 2x/j (23 doses)
|
20
|
↔
|
0,89 (0,84; 0,96)
|
0,91 (0,85; 0,97)
|
0,88 (0,78; 1,00)
| |
Zidovudine
|
300 mg (1 dose)
|
500/100 mg 2x/j (23 doses)
|
29
|
↓
|
0,87 (0,80; 0,94)
|
0,82 (0,76; 0,89)
|
0,77 (0,68; 0,87)
| |
|
|
750/200 mg 2x/j (23 doses)
|
25
|
↔
|
1,02 (0,94; 1,10)
|
1,02 (0,92; 1,13)
|
1,07 (0,86; 1,34)
|
* Comparaison à l'état d'équilibre par rapport aux données existantes
Produits associés après administration de tipranavir/ritonavir
|
Produit associé
|
Dosage du produit associé
(protocole d'étude)
|
Dosage TPV/r
(protocole d'étude)
|
n
|
PK
|
Relation (IC à 90 %) de la pharmacocinétique du produit associé avec/sans tipranavir (aucun effet = 1,00)
| |
Cmax
|
ASC
|
Cmin
| |
Amprénavir/
RTV a
|
600/100 mg 2x/j
(27 doses)
|
500/200 mg 2x/j (28 doses)
|
16
|
↓
|
0,61 (0,51; 0,73)d
|
0,56 (0,49; 0,64)d
|
0,45 (0,38; 0,53)d
| |
|
|
|
74
|
↓
|
-
|
-
|
0,44 (0,39; 0,49)e
| |
Abacavir a
|
300 mg 2x/j
(43 doses)
|
250/200 mg 2x/j (42 doses)
|
28
|
↓
|
0,56 (0,48; 0,66)
|
0,56 (0,49; 0,63)
|
-
| |
|
|
750/100 mg 2x/j (42 doses)
|
14
|
↓
|
0,54 (0,47; 0,63)
|
0,64 (0,55; 0,74)
|
-
| |
|
|
1250/100 mg 2x/j (42 doses)
|
11
|
↓
|
0,48 (0,42; 0,53)
|
0,65 (0,55; 0,76)
|
-
| |
Atorvastatine
|
10 mg (1 dose)
|
500/200 mg 2x/j (17 doses)
|
22
|
↑
|
8,61 (7,25; 10,21)
|
9,36 (8,02; 10,94)
|
5,19 (4,21; 6,40)
| |
Orthohydroxy-atorvastatine
|
|
|
21,
12,
17
|
↓
|
0,02 (0,02; 0,03)
|
0,11 (0,08; 0,17)
|
0,07 (0,06; 0,08)
| |
Parahydroxy-atorvastatine
|
|
|
13,
22,
1
|
↓
|
1,04 (0,87; 1,25)
|
0,18 (0,14; 0,24)
|
0,33 (NA)
| |
Clarithromycine
14-OH-clarithromycine
|
500 mg 2x/j (25 doses)
|
500/200 mg 2x/j (15 doses)
|
21
|
↑
|
0,95 (0,83; 1,09)
|
1,19 (1,04; 1,37)
|
1,68 (1,42; 1,98)
| |
|
|
|
21
|
↓
|
0,03 (0,02; 0,04)
|
0,03 (0,02; 0,04)
|
0,05 (0,04; 0,07)
| |
Didanosine b
|
200 mg 2x/j, >60 kg;
|
250/200 mg 2x/j (42 doses)
|
10
|
↓
|
0,57 (0,42; 0,79)
|
0,67 (0,51; 0,88)
|
-
| |
|
125 mg 2x/j, <60 kg;
|
750/100 mg 2x/j (42 doses)
|
8
|
↔
|
0,76 (0,49; 1,17)
|
0,97 (0,64;1,47)
|
-
| |
|
(43 doses)
|
1250/100 mg 2x/j (42 doses)
|
9
|
↔
|
0,77 (0,47, 1,26)
|
0,87 (0,47; 1,65)
|
-
| |
|
400 mg (1 dose)
|
500/100 mg 2x/j (27 doses)
|
5
|
↔
|
0,80 (0,63; 1,02)
|
0,90 (0,72; 1,11)
|
1,17 (0,62; 2,20)
| |
Éfavirenz b
|
600 mg 1x/j (15 doses)
|
500/100 mg 2x/j (15 doses)
|
24
|
↔
|
1,09 (0,99; 1,19)
|
1,04 (0,97; 1,12)
|
1,02 (0,92; 1,12)
| |
|
|
750/200 mg 2x/j (15 doses)
|
22
|
↔
|
1,12 (0,98; 1,28)
|
1,00 (0,93; 1,09)
|
0,94 (0,84; 1,04)
| |
Éthinylestradiol
|
0,035 mg (1 dose)
|
500/100 mg 2x/j (21 doses)
|
21
|
↓
|
0,52 (0,47; 0,57)
|
0,52 (0,48; 0,56)
|
-
| |
|
|
750/200 mg 2x/j (21 doses)
|
13
|
↓
|
0,48 (0,42; 0,57)
|
0,57 (0,54; 0,60)
|
-
| |
Fluconazole
|
200 mg (jour 1) puis 100 mg 1x/j (6 ou 12 doses)
|
500/200 mg 2x/j (2 ou 14 doses)
|
19
|
↔
|
0,97 (0,94; 1,01)
|
0,99 (0,97; 1,02)
|
0,98 (0,94; 1,02)
| |
|
|
|
19
|
↔
|
0,94 (0,91; 0,98)
|
0,92 (0,88; 0,95)
|
0,89 (0,85; 0,92)
| |
Lopinavir/RTVa
|
400/100 mg 2x/j
(27 doses)
|
500/200 mg 2x/j (28 doses)
|
21
|
↓
|
0,53 (0,40; 0,69)d
|
0,45 (0,32; 0,63)d
|
0,30 (0,17; 0,51)d
| |
|
|
|
69
|
↓
|
-
|
-
|
0,48 (0,40; 0,58)e
| |
Lopéramide
N-desméthyl-lopéramide
|
16 mg (1 dose)
|
750/200 mg 2x/j (21 doses)
|
24
|
↓
|
0,39 (0,31; 0,48)
|
0,49 (0,40; 0,61)
|
-
| |
|
|
|
24
|
↓
|
0,21 (0,17; 0,25)
|
0,23 (0,19; 0,27)
|
| |
Lamivudine a
|
150 mg 2x/j (43 doses)
|
250/200 mg 2x/j (42 doses)
|
64
46
|
↔
|
0,96 (0,89; 1,03)
|
0,95 (0,89; 1,02)
|
-
| |
|
|
750/100 mg 2x/j (42 doses)
|
|
↔
|
0,86 (0,78; 0,94)
|
0,96 (0,90; 1,03)
|
-
| |
|
|
1250/100 mg 2x/j (42 doses)
|
35
|
↔
|
0,71 (0,62; 0,81)
|
0,82 (0,66; 1,00)
|
-
| |
Névirapine a
|
200 mg 2x/j (43 doses)
|
250/200 mg 2x/j (42 doses)
|
26
|
↔
|
0,97 (0,90; 1,04)
|
0,97 (0,91; 1,04)
|
0,96 (0,87; 1,05)
| |
|
|
750/100 mg 2x/j (42 doses)
|
22
|
↔
|
0,86 (0,76; 0,97)
|
0,89 (0,78; 1,01)
|
0,93 (0,80; 1,08)
| |
|
|
1250/100 mg 2x/j (42 doses)
|
17
|
↔
|
0,71 (0,62; 0,82)
|
0,76 (0,63; 0,91)
|
0,77 (0,64; 0,92)
| |
Noréthindrone
|
1,0 mg (1 dose)
|
500/100 mg 2x/j (21 doses)
|
21
|
↔
|
1,03 (0,94; 1,13)
|
1,14 (1,06; 1,22)
|
-
| |
|
|
750/200 mg 2x/j (21 doses)
|
13
|
↔
|
1,08 (0,97; 1,20)
|
1,27 (1,13; 1,43)
|
-
| |
Rifabutine
|
150 mg (1 dose)
|
500/200 mg 2x/j (15 doses)
|
20
|
↑
|
1,70 (1,49; 1,94)
|
2,90 (2,59; 3,26)
|
2,14 (1,90; 2,41)
| |
25-O-désacétyle-rifabutine
|
|
|
20
|
↑
|
|
20,71 (17,66;24,28)
|
7,83 (6,70; 9,14)
| |
Rif. + 25-O-désacétyle-rif. c
|
|
|
20
|
↑
|
1,86 (1,63; 2,12)
|
4,33 (3,86; 4,86)
|
2,76 (2,44; 3,12)
| |
Saquinavir/
RTV a
|
600/100 mg 2x/j
(27 doses)
|
500/200 mg 2x/j (28 doses)
|
20
|
↓
|
0,30 (0,23; 0,40)d
|
0,24 (0,19; 0,32)d
|
0,18 (0,13; 0,26)d
| |
|
|
|
68
|
↓
|
-
|
-
|
0,20 (0,16; 0,25)e
| |
Stavudine a
|
40 mg 2x/j, >60 kg;
|
250/200 mg 2x/j (42 doses)
|
26
|
↔
|
0,90 (0,81; 1,02)
|
1,00 (0,91; 1,11)
|
-
| |
|
30 mg 2x/j, <60 kg;
|
750/100 mg 2x/j (42 doses)
|
22
|
↔
|
0,76 (0,66; 0,89)
|
0,84 (0,74; 0,96)
|
-
| |
|
(43 doses)
|
1250/100 mg 2x/j (42 doses)
|
19
|
↔
|
0,74 (0,69; 0,80)
|
0,93 (0,83; 1,05)
|
-
| |
Ténofovir
|
300 mg (1 dose)
|
500/100 mg 2x/j (23 doses)
|
22
20
|
↓
|
0,77 (0,68; 0,87)
|
0,98 (0,91; 1,05)
|
1,07 (0,98; 1,17)
| |
|
|
750/200 mg 2x/j (23 doses)
|
20
|
↓
|
0,62 (0,54; 0,71)
|
1,02 (0,94; 1,10)
|
1,14 (1,01; 1,27)
| |
Zidovudineb
|
300 mg 2x/j (43 doses)
|
250/200 mg 2x/j (42 doses)
|
48
|
↓
|
0,54 (0,47; 0,62)
|
0,58 (0,51; 0,66)
|
-
| |
|
|
750/100 mg 2x/j (42 doses)
|
31
|
↓
|
0,51 (0,44; 0,60)
|
0,64 (0,55; 0,75)
|
-
| |
|
300 mg (1 dose)
|
1250/100 mg 2x/j (42 doses)
|
23
|
↓
|
0,49 (0,40; 0,59)
|
0,69 (0,49; 0,97)
|
-
| |
|
|
500/100 mg 2x/j (23 doses)
|
29
|
↓
|
0,39 (0,33; 0,45)
|
0,57 (0,52; 0,63)
|
0,89 (0,81; 0,99)
| |
|
|
750/200 mg 2x/j (23 doses)
|
25
|
↓↑
|
0,44 (0,36; 0,54)
|
0,67 (0,62; 0,73)
|
1,25 (1,08; 1,44)
| |
Dérivé glucuronidé de la zidovudine
|
|
500/100 mg 2x/j (23 doses)
|
29
|
↑
|
0,82 (0,74; 0,90)
|
1,02 (0,97; 1,06)
|
1,52 (1,34; 1,71)
| |
|
|
750/200 mg 2x/j (23 doses)
|
25
|
↑
|
0,82 (0,73; 0,92)
|
1,09 (1,05; 1,14)
|
1,94 (1,62; 2,31)
|
a Patients VIH+
b Patients VIH+ (TPV/r 250 mg/200 mg, 750mg/200 mg et 1250 mg/100 mg) et volontaires sains (TPV/r 500 mg/100 mg et 750 mg/200 mg)
c Somme normalisée de la rifabutine et du métabolite actif (25-O-désacétyle-rifabutine)
d Analyse pharmacocinétique intense
e Dosages thérapeutiques du médicament 8-16 h après administration
Inhibiteurs de fusion
Enfuvirtide: dans la population étudiée, l'administration concomitante d'enfuvirtide et d'Aptivus associé au ritonavir à faible dose a entraîné une élévation de la concentration plasmatique minimale à l'état d'équilibre du tipranavir d'environ 45%. Des élévations comparables après association à l'enfuvirtide ont également été observées pour les concentrations plasmatiques minimales de lopinavir (23%) et de saquinavir (63%). Le mécanisme de cette interaction est inconnu. Aucun ajustement posologique du tipranavir ou du ritonavir n'est recommandé.
Inhibiteurs du transfert de brins de l'intégrase du VIH
Raltégravir: l'administration multiple d'Aptivus associé au ritonavir a entraîné les modifications suivantes des concentrations sériques du raltégravir: Cmax ↔, ASC0-12 ↔, C12 ↓ 45 % (moyenne géométrique). Les données relatives à l'efficacité issues des études de phase III dans lesquelles Aptivus/ritonavir était administré avec le raltégravir sans ajustement posologique n'ont pas mis en évidence de signes d'efficacité réduite.
Inhibiteurs nucléosidiques de la transcriptase inverse
Abacavir et zidovudine: Aptivus associé au ritonavir à faible dose diminue d'environ 40% l'ASC de l'abacavir, et d'environ 35% l'ASC de la zidovudine. Les taux du dérivé glucuronidé de la zidovudine ne sont pas influencés. La pertinence clinique de ces réductions n'a pas été établie, mais elles pourraient entraîner une diminution de l'efficacité de ces substances antirétrovirales. L'administration concomitante de la zidovudine ou de l'abacavir avec Aptivus associé au ritonavir à faible dose n'est donc pas recommandée, sauf s'il n'existe pas d'autres inhibiteurs nucléosidiques de la transcriptase inverse appropriés pour le traitement du patient. Aucun ajustement de la dose d'abacavir et de zidovudine ne peut être recommandé dans ces cas (voir «Mises en garde et précautions»).
Didanosine: Aptivus associé au ritonavir à faible dose réduit l'ASC de la didanosine. La pertinence clinique de cette baisse de la concentration de didanosine n'a pas encore été établie. Les comprimés gastrorésistants de didanosine doivent être pris au moins 2 heures avant ou après la prise d'Aptivus associé au ritonavir à faible dose afin d'éviter des incompatibilités.
Stavudine et lamivudine: Aptivus associé au ritonavir à faible dose ne provoque pas de modification significative de l'ASC de la stavudine ou de la lamivudine. Aucun ajustement posologique n'est recommandé pour la stavudine et la lamivudine.
Inhibiteurs nucléotidiques de la transcriptase inverse
Ténofovir: Aptivus associé au ritonavir à faible dose ne provoque pas de modification significative des concentrations plasmatiques du ténofovir. Aucun ajustement de la dose n'est recommandé pour le ténofovir.
Inhibiteurs non nucléosidiques de la transcriptase inverse (INNTI)
Névirapine: aucune interaction significative n'a été observée entre Aptivus associé au ritonavir à faible dose et la névirapine. La dose ne doit donc pas être ajustée.
Éfavirenz: éfavirenz 600 mg une fois par jour à l'état d'équilibre associé à Aptivus à l'état d'équilibre associé au ritonavir à faible dose (500/200 mg deux fois par jour) n'a entraîné aucune influence significative sur l'ASC et la Cmax du tipranavir (baisse de 2,9% et de 8,3%) et a entraîné une augmentation du Cp12h (19,2%) non pertinente sur le plan clinique.
Aptivus associé au ritonavir à faible dose n'a pas entraîné d'effet significatif sur l'ASC et la Cmin de l'éfavirenz.
Étravirine
Aptivus/ritonavir a entraîné une diminution de l'ASC de l'étravirine de 76%, ce qui pourrait réduire de manière significative la réponse virologique à l'étravirine. L'administration concomitante d'étravirine et d'Aptivus/ritonavir n'est pas recommandée.
Rilpivirine
L'administration concomitante de rilpivirine et d'Aptivus/ritonavir n'a pas été étudiée. L'administration concomitante de rilpivirine et de darunavir boosté par ritonavir ou de lopinavir a provoqué une augmentation de la concentration plasmatique de rilpivirine; un ajustement posologique n'est cependant pas recommandé. Une surveillance étroite et/ou un ajustement de la dose des deux médicaments sont de mise lorsqu'Aptivus/ritonavir est administré en même temps que la rilpivirine.
Inhibiteurs de la protéase
Amprénavir, atazanavir, lopinavir, saquinavir:
Au cours d'une étude clinique effectuée avec une association d'inhibiteurs de la protéase doublement boostés chez des adultes séropositifs et ayant déjà reçu de multiples traitements, Aptivus associé au ritonavir à faible dose a diminué les Cmin de l'amprénavir, du lopinavir et du saquinavir respectivement de 55%, 70% et 78%. Une baisse de 81% de la Cmin de l'atazanavir a également été observée dans une étude d'interaction chez des volontaires sains. L'administration concomitante d'Aptivus associé au ritonavir à faible dose et de l'amprénavir, l'atazanavir, du lopinavir et du saquinavir respectivement associés au ritonavir à faible dose n'est donc pas recommandée, la pertinence clinique de cette baisse de leurs taux n'ayant pas été établie. Si l'association est néanmoins considérée comme indispensable, aucun ajustement posologique ne peut être recommandé actuellement.
Aucune donnée n'est actuellement disponible sur les éventuelles interactions d'Aptivus associé au ritonavir à faible dose avec des inhibiteurs de la protéase autres que ceux précités. Leur association à Aptivus associé au ritonavir à faible dose n'est donc pas recommandée (voir «Mises en garde et précautions»).
Antagonistes des récepteurs alpha-1 adrénergiques
Alfuzosine: l'administration concomitante de tipranavir et d'alfuzosine entraîne une augmentation des concentrations d'alfuzosine et peut mener à une hypotension.
Anticonvulsivants
La carbamazépine, le phénobarbital et la phénytoïne doivent être utilisés avec prudence en cas d'association à Aptivus/ritonavir. L'utilisation concomitante de carbamazépine 200 mg deux fois par jour a entraîné une augmentation des concentrations plasmatiques de la carbamazépine (d'environ 23% [moyenne arithmétique] de la Cmin de la quantité totale de carbamazépine et d'époxy-10, 11-carbamazépine; toutes deux sont pharmacologiquement actives) ainsi qu'une diminution de la Cmin du tipranavir (d'env. 61% par rapport aux valeurs témoins connues), ce qui peut entraîner une réduction de l'efficacité.
Antipsychotiques: pimozide, sertindole et quétiapine:
L'administration concomitante d'Aptivus/ritonavir et de pimozide, sertindole ou de quétiapine est contre-indiquée en raison de l'inhibition du CYP3A par Aptivus/ritonavir qui peut entraîner des événements graves mettant en jeu le pronostic vital, coma inclus.
Antifongiques
Fluconazole: Aptivus associé au ritonavir à faible dose n'interfère pas de de manière significative avec les paramètres pharmacocinétiques du fluconazole à l'état d'équilibre. Le fluconazole augmente l'ASC et la Cmin du tipranavir de respectivement 56% et 104% (comparaison avec les données historiques). Aucun ajustement de la dose n'est recommandé. Il est déconseillé d'utiliser des doses de fluconazole supérieures à 200 mg/jour.
Itraconazole/kétoconazole: sur la base de considérations théoriques, Aptivus associé au ritonavir à faible dose devrait augmenter les concentrations d'itraconazole ou de kétoconazole. L'itraconazole ou le kétoconazole doivent donc être utilisés avec précaution (l'utilisation de doses supérieures à 200 mg/jour n'est pas recommandée).
Voriconazole: en raison des nombreux systèmes d'isoenzymes CYP impliqués dans le métabolisme du voriconazole, il est difficile de prédire le type d'interaction avec Aptivus associé au ritonavir à faible dose.
Antigoutteux
Colchicine :
Sur la base de considérations théoriques, il faut s'attendre à ce que le tipranavir associé au ritonavir à faible dose augmente la concentration de colchicine. La colchicine est un substrat du CYP3A4 et de la P-gp (transporteur transmembranaire intestinal).
L'administration concomitante de colchicine et d'Aptivus/ritonavir n'est pas recommandée.
Inhibiteurs de protéase anti-VHC:
Bocéprévir:
L'administration concomitante d'Aptivus/ritonavir et de bocéprévir n'a pas été étudiée. Dans une étude pharmacocinétique menée sur des volontaires sains, le bocéprévir a réduit l'exposition du ritonavir, du lopinavir boosté par ritonavir, de l'atazanavir boosté par ritonavir et du darunavir boosté par ritonavir. L'exposition du bocéprévir était réduite de 45%, resp. 32% en administration concomitante avec du lopinavir boosté par ritonavir et du darunavir boosté par ritonavir. Cette interaction entre les médicaments peut, en administration concomitante, réduire l'efficacité des inhibiteurs de protéase anti-VIH et/ou du bocéprévir. Il est donc recommandé de ne pas administrer concomitamment bocéprévir et Aptivus/ritonavir.
Télaprévir:
L'administration concomitante d'Aptivus/ritonavir et de télaprévir n'a pas été étudiée. Dans une étude pharmacocinétique menée sur des volontaires sains, le lopinavir boosté par ritonavir, le fosamprénavir boosté par ritonavir et le darunavir boosté par ritonavir ont réduit l'exposition du télaprévir de 54%, resp. 32% et 35%. L'exposition du fosamprénavir boosté par ritonavir et du darunavir boosté par ritonavir a également été réduite en administration concomitante avec du télaprévir. Cette interaction entre les médicaments peut, en administration concomitante, réduire l'efficacité des inhibiteurs de protéase anti-VIH et/ou du télaprévir. Il est donc recommandé de ne pas aministrer concomitamment télaprévir et Aptivus/ritonavir.
Antagonistes des récepteurs de l'endothéline
Bosentan: compte tenu de l'inhibition du CYP3A4 par Aptivus/ritonavir, il faut s'attendre à ce que les concentrations de bosentan augmentent en cas d'administration concomitante avec le tipranavir et le ritonavir à faible dose. L'étendue de l'augmentation ne peut être estimée actuellement. Une surveillance étroite doit être garantie (p.ex. dans une unité de soins intensifs) si l'administration concomitante de bosentan et de tipranavir/ritonavir semble absolument indispensable.
Inhibiteurs de la HMG-CoA réductase
Simvastatine et lovastatine: le métabolisme des inhibiteurs de la HMG-CoA réductase simvastatine et lovastatine est fortement dépendant du CYP3A. L'utilisation concomitante d'Aptivus associé au ritonavir à faible dose et de simvastatine ou de lovastatine est contre-indiquée en raison du risque accru de myopathies, dont la rhabdomyolyse (voir «Contre-indications»).
Atorvastatine: Aptivus associé au ritonavir à faible dose multiplie par environ 8 à 10 les concentrations plasmatiques d'une dose unique d'atorvastatine et réduit d'environ 85% l' ASC de ses métabolites. L'atorvastatine ne modifie pas significativement l'ASC, la Cmax et la Cmin du tipranavir. L'administration concomitante d'atorvastatine et d'Aptivus/ritonavir n'est pas recommandée. L'utilisation d'autres inhibiteurs de l'HMG-CoA réductase, tels que la pravastatine, la fluvastatine ou la rosuvastatine, doit être envisagée.
Rosuvastatine et pravastatine: l'utilisation concomitante d'Aptivus/associéritonavir et de rosuvastatine augmente l'ASC (37 %) et la Cmax (123%) de la rosuvastatine; c'est pourquoi l'utilisation concomitante doit être instaurée avec la plus faible dose de rosuvastatine (5 mg/jour).
La titration ultérieure jusqu'à la réponse au traitement doit être accompagnée d'une surveillance clinique attentive des effets secondaires associés à la rosuvastatine, comme décrit dans l'information professionnelle de la rosuvastatine.
Sur la base des similarités d'élimination entre la pravastatine et la rosuvastatine, il est également recommandé d'initier le traitement avec la posologie la plus faible de pravastatine (10 mg/jour), accompagnée d'une surveillance clinique attentive des effets secondaires associés à la pravastatine.
Bêta-agonistes inhalés
Salmétérol: l'administration concomitante d'Aptivus/associéritonavir n'est pas recommandée. Cette association peut entraîner une augmentation du risque d'effets secondaires cardiovasculaires associés au salmétérol, y compris un allongement de l'intervalle QT, des palpitations et une tachycardie sinusale.
Inducteurs des isoenzymes CYP
La prudence est de rigueur lors de l'administration concomitante d'Aptivus associéassocié au ritonavir et d'autres médicaments connus pour être des inducteurs du cytochrome 3A4, étant donné la possibilité d'un échec thérapeutique et d'un développement accéléré d'une résistance. Il s'agit notamment de la phénytoïne, du phénobarbital, de la primidone et du topiramate.
Rifampicine: l'utilisation concomitante de rifampicine et d'inhibiteurs de la protéase réduit significativement les concentrations des inhibiteurs de la protéase. L'utilisation concomitante d'Aptivus associéassocié au ritonavir à faible dose et de rifampicine devrait entraîner des concentrations suboptimales de tipranavir, susceptibles d'entraîner la disparition de la réponse virologique et l'apparition possible de résistances au tipranavir. L'utilisation concomitante d'Aptivus et de rifampicine est par conséquent contre-indiquée (voir «Contre-indications»). Le recours à d'autres agents antibiotiques tels que la rifabutine doit être envisagé.
Rifabutine: Aptivus associé au ritonavir à faible dose entraîne une augmentation des concentrations plasmatiques de la rifabutine et de son métabolite actif par un facteur proche de 3 et de 20, respectivement. La rifabutine augmente la Cmin du tipranavir de 16%. Une réduction d'au moins 75% la dose habituelle de la rifabutine de 300 mg/jour est recommandée (c'est-à-dire 150 mg tous les deux jours ou trois fois par semaine). Les patients recevant de la rifabutine conjointement avec Aptivus associé au ritonavir à faible dose doivent être étroitement surveillés sur l'apparition d'effets indésirables associés au traitement par la rifabutine. Une réduction supplémentaire de la posologie peut s'avérer nécessaire.
Millepertuis (Hypericum perforatum): l'utilisation concomitante de préparations phytothérapeutiques à base de millepertuis (Hypericum perforatum) peut réduire les concentrations plasmatiques du tipranavir par l'intermédiaire de l'induction des enzymes métabolisant le médicament. Les préparations phytothérapeutiques contenant du millepertuis ne doivent donc pas être utilisées en même temps qu'Aptivus. Si un patient prend déjà du millepertuis, il convient d'arrêter le millepertuis, de vérifier la charge virale et, si possible, également les concentrations de tipranavir. Les concentrations de tipranavir peuvent augmenter à l'arrêt du millepertuis et la dose d'Aptivus peut devoir être ajustée. L'effet inducteur du millepertuis peut persister au moins 2 semaines après l'arrêt du traitement (voir «Contre-indications»).
Inhibiteurs des isoenzymes CYP
Clarithromycine: Aptivus associé au ritonavir à faible dose augmente l'ASC et la Cmin de la clarithromycine de respectivement 19% et 68% et réduit de plus de 95% l'ASC du métabolite actif 14-hydroxylé. Bien que ces modifications des paramètres de la clarithromycine ne soient pas considérées comme cliniquement significatives, la réduction de l'ASC du métabolite 14-hydroxylé doit être prise en compte pour le traitement des infections à Haemophilus influenzae, dans lesquelles ce métabolite est le plus actif.
La clarithromycine augmente la Cmin du tipranavir de plus de 100%. Cette forte augmentation de la Cmin est peut-être cliniquement significative. Les patients sous clarithromycine à une posologie supérieure à 500 mg deux fois par jour doivent être rigoureusement surveillés quant aux signes de toxicité. En cas d'insuffisance rénale, les ajustements posologiques suivants doivent être envisagés: la dose de clarithromycine doit être réduite de 50% chez les patients dont la CLCR est comprise entre 30 et 60 ml/min et de 75% chez les patients dont la CLCR est <30 ml/min. Aucun ajustement posologique n'est requis chez les patients présentant une fonction rénale normale.
Cobicistat et préparations contenant du cobicistat
Cobicistat: Aptivus/ritonavir ne doit pas être administré concomitamment avec du cobicistat ou des préparations contenant du cobicistat. Le cobicistat inhibe de façon significative les enzymes hépatiques, de même que d'autres voies du métabolisme. Lors de l'administration concomitante, les expositions de tipranavir et de cobicistat sont nettement inférieures par rapport au tipranavir lorsqu'il est boosté par du ritonavir à faible dose.
Inhibiteurs nucléosidiques de l'ADN polymérase
Valaciclovir: l'administration concomitante de valaciclovir, d'Aptivus et de ritonavir n'ayant pas été associée à des effets pharmacocinétiques cliniquement significatifs, aucun ajustement de la dose n'est requis en cas d'administration concomitante.
Autres principes actifs
La co-administration d'Aptivus associé au ritonavir à faible dose et à des principes actifs dont la clairance dépend fortement du CYP3A, et chez lesquelles une augmentation des concentrations plasmatiques est associée à des effets secondaires graves et/ou potentiellement mortels, est contre-indiquée. Ces principes actifs incluent les antiarythmiques (amiodarone, bépridil, quinidine), les antihistaminiques (astémizole, terfénadine), les dérivés de l'ergot de seigle (dihydroergotamine, ergonovine, ergotamine, méthylergonovine), les stimulants de la motricité gastro-intestinale (cisapride), les neuroleptiques (pimozide, sertindole), les sédatifs et hypnotiques (triazolam, midazolam), les inhibiteurs de l'HMG-CoA réductase (simvastatine et lovastatine) et les inhibiteurs de la phosphodiestérase de type 5 (IPDE5) (vardénafil) (voir «Contre-indications»). En outre, l'utilisation concomitante d'Aptivus associé au ritonavir à faible dose et de médicaments dont la dégradation est fortement dépendante du CYP2D6, tels que les antiarythmiques flécaïnide et propafénone, est contre-indiquée (voir «Contre-indications»).
L'utilisation de certains anti-infectieux n'est pas recommandée (halofantrine, luméfantrine) ainsi que divers autres principes actifs (toltérodine) (voir «Mises en garde et précautions»).
Contraceptifs oraux/œstrogènes: Aptivus associé au ritonavir à faible dose diminue de 50% l'ASC et la Cmax de l'éthinylestradiol, mais ne modifie pas significativement le profil pharmacocinétique de la noréthindrone. L'administration concomitante d'Aptivus associé au ritonavir à faible dose n'est pas recommandée. Des mesures contraceptives alternatives ou supplémentaires sont indiquées lorsque les contraceptifs oraux à base d'œstrogènes sont co-administrés avec Aptivus associé au ritonavir à faible dose. Les préservatifs sont recommandés par principe. Les patientes recevant des œstrogènes comme traitement hormonal substitutif doivent être cliniquement suivies quant aux signes d'insuffisance estrogénique (voir «Mises en garde et précautions»).
Midazolam: l'utilisation concomitante d'Aptivus/ritonavir et de midazolam administré par voie orale est contre-indiquée. Le ritonavir est un puissant inhibiteur du CYP3A4 et peut de ce fait perturber les médicaments métabolisés par cette enzyme. Les concentrations d'une dose unique de midazolam administrée par voie intraveineuse avec Aptivus/ritonavir à l'état d'équilibre ont été multipliées par 2,8 (ASC0-24h). L'exposition (ASC0-24h) était 10 fois plus élevée après l'administration orale de midazolam (voir «Contre-indications»).
Lorsqu'Aptivus associé au ritonavir est administré avec le midazolam par voie parentérale, un suivi clinique étroit de l'apparition d'une dépression respiratoire et/ou d'une sédation prolongée doit être instauré et un ajustement de la dose envisagé.
Inhibiteurs de la phosphodiestérase de type 5 (IPDE5): l'administration concomitante d'Aptivus associé au ritonavir à faible dose et de vardénafil est contre-indiquée. Une prudence particulière est de mise lors de la prescription d'autres inhibiteurs de la phosphodiestérase de type 5 (p.ex. sildénafil ou tadalafil) à des patients traités par l'Aptivus associé au ritonavir à faible dose. Il faut s'attendre à une augmentation importante des concentrations des inhibiteurs de la PDE5 en cas d'utilisation concomitante d'Aptivus associé au ritonavir à faible dose et d'IPDE5. Ceci peut mener à une augmentation des effets secondaires associés aux IPDE5, notamment chute de tension, troubles visuels, priapisme et syncopes.
Sildénafil : une posologie sûre et efficace n'a pas été établie en association avec l'utilisation d'Aptivus associé au ritonavir à faible dose. Les événements indésirables associés au sildénafil (dont troubles visuels, chute de tension, érection prolongée et syncope) peuvent être plus fréquents. L'administration concomitante d'Aptivus/ritonavir et de sildénafil pour le traitement de l'hypertension artérielle pulmonaire est contre-indiquée.
L'utilisation concomitante d'Aptivus associé au ritonavir à faible dose et du tadalafil a entraîné une exposition 2,3 fois supérieure au tadalafil après la première administration d'Aptivus/ritonavir et aucun changement relatif à l'exposition au tadalafil à l'état d'équilibre d'Aptivus/ritonavir. Lorsque le tadalafil est administré au cours des premiers jours du traitement par Aptivus/ritonavir, il convient d'administrer la dose de tadalafil la plus faible. L'état d'équilibre du tipranavir et du ritonavir est atteint après 7 à 10 jours de traitement par Aptivus/ritonavir et la dose de tadalafil peut toutefois être augmentée en fonction des besoins cliniques.
Analgésiques narcotiques (méthadone/mépéridine): l'utilisation concomitante d'une dose unique de méthadone avec Aptivus associé au ritonavir à faible dose a entraîné une réduction d'environ 50% de la concentration de méthadone (ASC et Cmax). Dans un tel cas, les patients doivent faire l'objet d'un suivi attentif quant à un syndrome de sevrage aux opiacés. Une augmentation de la dose de méthadone peut s'imposer. Lors de l'utilisation d'Aptivus associé au ritonavir à faible dose, il faut s'attendre à une réduction des concentrations de la mépéridine et une augmentation de la concentration de son métabolite, la normépéridine. Une augmentation de la dose de la mépéridine et son utilisation au long cours avec Aptivus associé au ritonavir à faible dose ne sont pas recommandées en raison de l'augmentation de la concentration de son métabolite, la normépéridine. L'action de la normépéridine sur le SNC est à la fois analgésique et stimulante (p.ex. convulsions).
Buprénorphine/naloxone: l'administration concomitante de buprénorphine/naloxone et d'Aptivus associé au ritonavir n'a entraîné aucune modification de l'effet clinique de la buprénorphine/naloxone. Avec cette association, la Cmin du tipranavir était réduite de 39%. La pertinence clinique de cette modification de la concentration plasmatique du tipranavir est inconnue.
Bupropion: l'utilisation concomitante d'Aptivus associé au ritonavir à faible dose à l'état d'équilibre a entraîné une réduction de la Cmax et de l'ASC du bupropion d'environ 50%. Un suivi clinique attentif est recommandé lors de l'association de ces trois principes actifs. Il est également recommandé de ne pas augmenter la dose de bupropion.
Immunosuppresseurs (ciclosporine, tacrolimus, sirolimus): l'utilisation concomitante d'Aptivus associé au ritonavir à faible dose et de substrats du CYP3A4/5 a révélé une forte inhibition après la première administration ainsi qu'à l'état d'équilibre d'Aptivus/ritonavir. Lors de l'utilisation concomitante d'Aptivus associé au ritonavir à faible dose avec un substrat de la P-gp, on assiste à une inhibition modérée de la P-gp après la première administration d'Aptivus/ritonavir, mais pas à une inhibition de la P-gp à l'état d'équilibre. On peut s'attendre à des effets similaires avec ces immunosuppresseurs, mais l'effet net des effets partiellement antagonistes n'est pas prévisible. La détermination des concentrations sanguines de ces immunosuppresseurs est recommandée en cas de prise concomitante de ces médicaments et d'Aptivus/ritonavir.
Warfarine et autres anticoagulants oraux: l'utilisation concomitante d'Aptivus associé au ritonavir à faible dose et de warfarine a entraîné une augmentation de 18% de l'exposition à la S-warfarine après la première administration d'Aptivus/ritonavir ainsi que de 12% à l'état d'équilibre d'Aptivus/ritonavir, ce qui peut être associé à une augmentation des valeurs de l'INR («International Normalised Ratio») et du risque d'hémorragies.
Un suivi clinique étroit et la mesure de l'INR sont recommandés lorsque ces médicaments sont associés.
Antiacides: lorsqu'Aptivus associé au ritonavir à faible dose était utilisé avec 20 ml d'une suspension buvable antiacide à base d'aluminium ou de magnésium, l'ASC12h, la Cmax et la Cmin du tipranavir étaient réduites de 25-29%. Un intervalle de 2 heures au moins doit être respecté entre la prise d'Aptivus associé au ritonavir à faible dose et celle des antiacides.
Inhibiteurs de la pompe à protons
Oméprazole/ésoméprazole: l'administration d'Aptivus associé au ritonavir à faible dose réduit l'ASC et la Cmax de l'oméprazole de respectivement 71% et 73%. Aucune modification cliniquement pertinente du tipranavir/ritonavir à l'état d'équilibre n'a été observée. Une augmentation de la dose d'oméprazole doit éventuellement être envisagée en cas d'administration concomitante avec Aptivus et le ritonavir.
Antagonistes des récepteurs H2
Aucune donnée n'est disponible à ce jour concernant les antagonistes des récepteurs H2. Des concentrations plasmatiques réduites de tipranavir liées à l'augmentation du pH gastrique peuvent toutefois survenir lorsque ces médicaments sont administrés en même temps qu'Aptivus associé au ritonavir à faible dose. La prudence est donc requise.
Théophylline: il faut s'attendre à ce qu'Aptivus associé au ritonavir à faible dose réduise les concentrations de théophylline. Une augmentation de la dose de théophylline peut s'imposer et une surveillance du traitement doit être envisagée.
Désipramine: il faut s'attendre à ce qu'Aptivus associé au ritonavir à faible dose augmente les concentrations de désipramine. Une réduction de la dose et une surveillance des concentrations de désipramine sont recommandées.
Lopéramide: une étude d'interactions pharmacodynamiques réalisée chez des volontaires sains a démontré que l'utilisation concomitante de lopéramide et d'Aptivus associé au ritonavir à faible dose ne provoque aucune modification cliniquement significative de la réponse respiratoire au CO2. L'analyse pharmacocinétique a montré que l'ASC et la Cmax du lopéramide sont réduites de 51% et 61% respectivement et la Cmin du tipranavir de 26%. La pertinence clinique de ces résultats est inconnue.
Propionate de fluticasone (interaction avec le ritonavir): lors d'une étude clinique où le ritonavir (capsules de 100 mg, deux fois par jour) était co-administré avec du propionate de fluticasone intranasal (50 µg, 4 fois par jour) pendant 7 jours chez des volontaires sains, les taux plasmatiques de propionate de fluticasone ont augmenté de façon significative tandis que les taux de cortisol intrinsèques ont diminué d'env. 86% (intervalle de confiance à 90%: 82-89%). Il faut s'attende à des effets encore plus importants lorsque le propionate de fluticasone est inhalé. Des effets systémiques des corticostéroïdes, incluant le syndrome de Cushing et une suppression surrénalienne, sont survenus chez des patients recevant du ritonavir et du propionate de fluticasone administré par voie inhalée ou intranasale. Ces effets peuvent également se produire avec d'autres corticostéroïdes métabolisés via le CYP3A, p.ex. le budésonide. L'utilisation concomitante d'Aptivus associé au ritonavir à faible dose et de ces glucocorticoïdes n'est donc pas recommandée, à moins que le bénéfice attendu du traitement ne l'emporte sur le risque d'effet systémique des corticostéroïdes (voir «Mises en garde et précautions»). Une réduction de la dose de glucocorticoïdes est à conseiller, avec une surveillance attentive concomitante des effets locaux et systémiques, ou bien le passage à un glucocorticoïde qui n'est pas un substrat du CYP3A4 (p.ex. la béclométhasone). L'arrêt du glucocorticoïde impose une réduction progressive de la dose sur une période prolongée. Les effets d'une exposition systémique élevée à la fluticasone sur les taux plasmatiques de ritonavir ne sont pas connus jusqu'à présent.
Grossesse, allaitementGrossesse
Il n'existe pas de données suffisantes concernant l'emploi d'Aptivus chez la femme enceinte. Après l'exposition à des doses thérapeutiques utilisées chez l'humain, les expérimentations animales ont révélé une toxicité de reproduction (voir «Données précliniques»). Le risque potentiel pour l'être humain n'est pas connu. Aptivus ne doit pas être administré pendant la grossesse, sauf en cas de nécessité absolue. Les études précliniques sur la reproduction n'ont pas révélé d'effet tératogène du tipranavir (voir «Données précliniques»).
Allaitement
Conformément aux recommandations déconseillant aux femmes infectées par le VIH d'allaiter leurs nourrissons, quelles que soient les circonstances, afin d'éviter tout risque de transmission mère-enfant postnatale du virus, les mères doivent arrêter l'allaitement dès qu'elles reçoivent Aptivus.
Effet sur l’aptitude à la conduite et l’utilisation de machinesAucune étude correspondante n'a été effectuée.
Les patients doivent être informés du fait que les capsules molles d'Aptivus contiennent une petite quantité d'alcool (7% d'éthanol, ce qui correspond à 100 mg par capsule ou 200 mg par dose).
Effets indésirablesAptivus associé au ritonavir à faible dose a été associé à des cas de toxicité hépatique significative. Dans les essais cliniques de phase III RESIST, la fréquence des élévations de transaminases était significativement augmentée dans le bras Aptivus/ritonavir par rapport au bras comparateur. Les patients traités par Aptivus associé au ritonavir à faible dose doivent donc être étroitement surveillés (voir «Mises en garde et précautions»). Les données actuellement disponibles sur l'administration d'Aptivus associé au ritonavir à faible dose aux patients atteints d'hépatite B ou C sont limitées. Aptivus associé au ritonavir à faible dose doit donc être administré avec prudence chez les patients également atteints d'hépatite B ou C. Aptivus ne doit être utilisé chez ces patients que si le bénéfice attendu l'emporte sur le risque potentiel et son utilisation implique une surveillance accrue des paramètres cliniques et biologiques.
Description de certains effets indésirables
Adultes
Aptivus associé au ritonavir à faible dose en association à d'autres antirétroviraux a été évalué dans diverses études cliniques, également « en usage compassionnel », chez plus de 6300 patients adultes séropositifs. Dans des études cliniques formelles, plus de 900 adultes, dont 541 dans les essais pivots de phase III RESIST-1 et RESIST-2, ont été traités par 500 mg/200 mg deux fois par jour pendant au moins 48 semaines.
Dans les études RESIST-1 et RESIST-2, les effets indésirables les plus fréquents dans le bras Aptivus/ritonavir étaient diarrhée, nausées, céphalées, fièvre, vomissements, fatigue et douleurs abdominales. Les probabilités de Kaplan-Meier relatives aux effets indésirables à la semaine 48 ayant entraîné l'interruption du traitement étaient de 13,3% chez les patients traités par Aptivus/ritonavir et de 10,8% chez les patients du bras de comparaison.
Les éléments de sécurité clinique suivants (hépatotoxicité, hyperlipidémie, hémorragies, éruptions cutanées) ont soit été observés à une fréquence accrue chez les patients traités par Aptivus/ritonavir par rapport au bras comparateur lors des études cliniques RESIST ou sont typiquement survenus lors du traitement par Aptivus/ritonavir. La pertinence clinique de ces résultats n'a pas encore été entièrement explorée.
Hépatotoxicité
Après 48 semaines de suivi, la fréquence des anomalies au niveau des ALAT et/ou d'ASAT de grade 3 ou 4 était plus élevée chez les patients traités par Aptivus/ritonavir comparée à celle apparue dans le bras comparateur (10,0% et 3,4%, respectivement). Des analyses multivariées ont montré que des taux de base d'ALAT et d'ASAT supérieurs au grade 1 DAIDS ainsi qu'une co-infection avec une hépatite B ou C étaient des facteurs de risque de ces élévations. La plupart des patients ont pu poursuivre le traitement par Aptivus/ritonavir.
Hyperlipidémie
Les élévations de triglycérides de grade 3 ou 4 étaient plus fréquentes dans le bras Aptivus/ritonavir que dans le bras comparateur. Après 48 semaines, ces taux étaient de 25,2% dans le bras Aptivus/ritonavir et de 15,6% dans le bras comparateur. La pertinence clinique de cette observation n'a pas encore été entièrement explorée.
Hémorragies
Le risque d'hémorragies était plus élevé chez les patients des études RESIST recevant Aptivus/ritonavir: à 24 semaines, le risque relatif était de 1,98 (intervalle de confiance à 95%: 1,03-3,80). Après 48 semaines, le risque relatif était de 1,27 (intervalle de confiance: 0,76 - 2,12). Il n'y avait pas de profil reconnaissable pour l'apparition des hémorragies et il n'y avait pas de différence entre les groupes de traitement concernant les paramètres de la coagulation. La pertinence de ces résultats est encore en cours d'évaluation.
Hémorragie intracrânienne
Chez certains patients, la prise d'Aptivus associé au ritonavir à faible dose était associée à des hémorragies intracrâniennes (HIC) fatales et non fatales. Nombre de ces patients présentaient d'autres états cliniques associés ou étaient traités par des co-médications ayant éventuellement provoqué ou ayant participé à la survenue d'une HIC. Un lien avec Aptivus n'a toutefois pas pu être exclu dans certains cas. De manière générale, les patients atteints ne présentaient pas de profil d'anomalies pour les examens de laboratoire (hématologie, coagulation), même pas immédiatement avant la survenue d'une HIC. La détermination de routine des paramètres de la coagulation pendant le traitement par Aptivus n'est donc pas indiquée actuellement.
De manière analogue, une incidence supérieure du risque d'HIC des patients traités dans les études Aptivus avait déjà été observée chez d'autres patients atteints de VIH à un stade avancé/SIDA.
Éruption cutanée
Une étude d'interaction chez les femmes entre Aptivus associé au ritonavir à faible dose et l'éthinylestradiol/noréthindrone a montré une fréquence très élevée d'éruptions cutanées non graves. Dans les études RESIST, le risque d'éruption cutanée était similaire entre les bras Aptivus/ritonavir et comparateur (16,3% vs 12,5%) (voir «Mises en garde et précautions»). Aucun cas de syndrome de Stevens-Johnson ni de nécrolyse épidermique toxique n'a été rapporté dans le programme de développement clinique d'Aptivus.
Les effets secondaires de toute intensité (grades 1 à 4) les plus fréquents survenus dans les essais cliniques de phase III dans le bras Aptivus/ritonavir (n = 749) sont mentionnés ci-après. Ils sont indiqués par classe de système d'organe et par fréquence conformément aux catégories suivantes: très fréquents: > 1/10; fréquents: > 1/100 – < 1/10.
Troubles du métabolisme et de la nutrition:
Fréquents: hypertriglycéridémie, hyperlipidémie
Affections du système nerveux:
Fréquents: céphalées
Affections gastro-intestinales:
Très fréquents: diarrhée (15,1%), nausées (12,6%)
Fréquents: vomissements, flatulence, distension abdominale, douleur abdominale, dyspepsie
Affections de la peau et du tissu sous-cutané:
Fréquents: éruption cutanée
Troubles généraux:
Fréquents: épuisement
Les effets secondaires cliniquement significatifs d'intensité modérée à sévère qui se sont produits chez moins de 1% (< 1/100) des patients adultes ayant reçu la dose d'Aptivus/ritonavir de 500/200 mg (n = 1397) au cours de toutes les études de phase II et III sont listés ci-après. Ils sont indiqués par classe de système d'organe et par fréquence conformément aux catégories suivantes: Occasionnels: ≥1/1000 - < 1/100; rares: < 1/1000.
Affections hématologiques et du système lymphatique:
Occasionnels: anémie, neutropénie, thrombocytopénie
Affections du système immunitaire:
Occasionnels: réactions d'hypersensibilité
Troubles du métabolisme et de la nutrition:
Fréquents: diminution de l'appétit
Occasionnels: perte de poids, diabète sucré, hyperamylasémie, hypercholestérolémie, hyperglycémie
Rares: déshydratation, lipoatrophie du visage (joues creuses)
Affections psychiatriques:
Occasionnels: insomnie, troubles du sommeil
Affections du système nerveux:
Occasionnels: obnubilation, neuropathie périphérique, somnolence, hémorragie intracrânienne*
Affections respiratoires, thoraciques et médiastinales:
Occasionnels: dyspnée
Affections gastro-intestinales:
Occasionnels: reflux gastro-œsophagien, pancréatite
Rares: augmentation de la lipase
Affections hépatobiliaires:
Occasionnels: élévation des enzymes hépatiques (ALAT, ASAT), test anormal de la fonction hépatique (ALAT, ASAT), hépatite toxique, hépatite
Rares: insuffisance hépatique (y compris d'issue fatale), hyperbilirubinémie, stéatose hépatique
Affections de la peau et du tissu sous-cutané:
Occasionnels: prurit, exanthème, lipoatrophie, lipodystrophie acquise, lipohypertrophie
Affections musculo-squelettiques et systémiques:
Occasionnels: spasmes musculaires, myalgie
Affections du rein et des voies urinaires:
Occasionnels: insuffisance rénale
Troubles généraux:
Occasionnels: affection pseudo-grippale, malaise, fièvre
* Cet effet secondaire n'a pas été observé comme effet secondaire en lien possible avec le traitement dans les études correspondantes sur Aptivus. Conformément à la directive européenne relative au RCP, la fréquence estimée est basée sur la limite supérieure de l'intervalle de confiance à 95%, calculée à partir de la totalité des patients traités (3 de 1397, ce qui correspond à «occasionnel»).
Anomalies biologiques:
Les fréquences des anomalies biologiques cliniquement significatives (grade 3 ou 4) rapportées après 48 semaines chez au moins 2% des patients dans les bras Aptivus/ritonavir au cours des études cliniques de phase III (RESIST-1 et RESIST-2) correspondaient à une augmentation des ASAT (6,1%), une augmentation des ALAT (9,6%), une augmentation de l'amylase (6,0%), une augmentation du cholestérol (4,2%), une augmentation des triglycérides (24,9%) et une diminution du nombre de globules blancs (5,7%).
Au cours de la poursuite des études cliniques RESIST-1 et RESIST-2 jusqu'à 96 semaines, le pourcentage de patients présentant une élévation des ALAT et/ou des ASAT de grade 2 - 4 est passé de 26% à la semaine 48 à 29,3% à la semaine 96 avec Aptivus/ritonavir par rapport à une augmentation de 13,7% à la semaine 48 à 14,6% à la semaine 96 pour le comparateur, l'inhibiteur de la protéase/ritonavir. Ces résultats montrent que le risque de développer une augmentation des transaminases est plus faible au cours de la 2e année de traitement qu'au cours de la 1re année. Le pourcentage de patients ayant développé une élévation des ALAT et/ou des ASAT de grade 3-4 est passé de 10,0% à la semaine 48 à 14,7% à la semaine 96. Dans le bras avec le comparateur inhibiteur de la protéase/ritonavir, les valeurs sont passées de 3,4% (semaine 48) à 4,5% (semaine 96).
Les traitements par association d'antirétroviraux, y compris les traitements contenant un inhibiteur de la protéase, sont associés, chez certains patients, à une redistribution de la masse grasse corporelle, incluant une perte du tissu adipeux sous-cutané périphérique, une augmentation de la masse grasse intra-abdominale, une hypertrophie mammaire et une accumulation de la masse grasse au niveau rétro-cervical (bosse de bison). Les inhibiteurs de la protéase sont également associés à des anomalies métaboliques telles qu'hypertriglycéridémie, hypercholestérolémie, résistance à l'insuline et hyperglycémie. Une augmentation des taux de CPK, des myalgies, des myosites et, rarement, une rhabdomyolyse, ont été rapportés avec les inhibiteurs de la protéase, en particulier en association avec les inhibiteurs nucléosidiques de la transcriptase inverse.
Une réaction inflammatoire à des infections opportunistes asymptomatiques ou résiduelles peut se développer (syndrome de reconstitution immune) chez les patients infectés par le VIH présentant un déficit immunitaire sévère au moment de l'instauration du traitement par association d'antirétroviraux (TAR) (voir «Mises en garde et précautions»).
Une réactivation des virus Herpes simplex et de Varicella zoster a été observée au cours des études RESIST.
Des cas d'ostéonécrose ont notamment été rapportés chez des patients présentant des facteurs de risque connus, un stade avancé de la maladie liée au VIH ou traités au long cours par association d'antirétroviraux (TAR). La fréquence d'apparition n'est pas connue (voir «Mises en garde et précautions»).
Adolescents
Les effets indésirables les plus fréquents observés étaient similaires à ceux décrits pour les adultes. Des vomissements, des éruptions cutanées et une pyrexie étaient plus fréquents chez les adolescents que chez les adultes. Les anomalies biologiques les plus fréquentes apparues pendant le traitement étaient les mêmes que celles observées chez les adultes.
Effets secondaires modérés à sévères rapportés le plus fréquemment chez les patients adolescents âgés de 12 à 18 ans (rapportés chez au moins 2 patients, étude 1182.14, analyses sur 48 semaines, Full Analysis Set)
|
Nombre total de patients traités (n)
|
28
| |
Événements [n (%)]
|
| |
Vomissements/nausées
|
3 (10,7)
| |
Nausées
|
2 (7,1)
| |
Douleurs abdominales1
|
2 (7,1)
| |
Éruption cutanée2
|
3 (10,7)
| |
Insomnie
|
2 (7,1)
| |
Augmentation des ALAT
|
4 (14,3)
|
1 Comprend douleurs abdominales (n = 1) et dyspepsie (n = 1)
2 Le terme éruption cutanée comprend au moins un des termes préférentiels suivants: éruption cutanée, exanthème médicamenteux, exanthème maculeux, exanthème papuleux, érythème, exanthème maculo-papuleux, prurit et urticaire.
L'annonce d'effets secondaires présumés après l'autorisation est d'une grande importance. Elle permet un suivi continu du rapport bénéfice-risque du médicament. Les professionnels de santé sont tenus de déclarer toute suspicion d'effet secondaire nouveau ou grave via le portail d'annonce en ligne ElViS (Electronic Vigilance System). Vous trouverez des informations à ce sujet sur www.swissmedic.ch.
SurdosageIl n'y a pas d'antidote connu à Aptivus en cas de surdosage. Le traitement du surdosage doit comporter des mesures de soutien générales, incluant un suivi des signes vitaux et la surveillance de l'état clinique du patient. Si indiquée, l'élimination du tipranavir non absorbé doit être obtenue par vomissement ou lavage gastrique. Du charbon activé peut également être administré pour éliminer le produit non résorbé. Le tipranavir étant fortement lié aux protéines plasmatiques, il est peu probable que la dialyse soit efficace pour une élimination significative de ce produit.
Propriétés/EffetsCode ATC
J05AE09
Mécanisme d'action
Pharmacodynamique
Le virus de l'immunodéficience humaine (VIH-1) code pour une aspartyl protéase essentielle pour le clivage et la maturation du précurseur des protéines virales. Le tipranavir est un inhibiteur non peptidique de la protéase du VIH-1 qui inhibe la réplication virale en empêchant la maturation des particules virales.
Activité antivirale in vitro
Le tipranavir inhibe la réplication de souches de laboratoire de VIH-1 et d'isolats cliniques dans des modèles d'infection de lymphocytes T, avec des concentrations efficaces à 50% (CE50) et à 90% (CE90) comprises respectivement entre 0,03 - 0,07 µM (18 à 42 ng/ml) et 0,07 - 0,18 µM (42 à 108 ng/ml). Le tipranavir a démontré une activité antivirale in vitro contre un large panel d'isolats du VIH-1 du groupe M sous type non-B (A, C, D, F, G, H, CRF01 AE, CRF02 AG, CRF12 BF). Les isolats du groupe O et du VIH-2 ont, in vitro, une sensibilité réduite au tipranavir (avec des CE50 comprises entre 0,164-1 μM et 0,233-0,522 μM, respectivement).
Les études de liaison aux protéines ont montré une réduction de l'activité antivirale du tipranavir d'un facteur de 3,75 en moyenne en présence de sérum humain. Lors d'associations in vitro à d'autres substances antirétrovirales, le tipranavir a montré des effets additifs à antagonistes avec des inhibiteurs de la protéase (amprénavir, atazanavir, indinavir, lopinavir, nelfinavir, ritonavir et saquinavir) et généralement un effet additif avec les INNTI (délavirdine, éfavirenz et névirapine) et les INTI (abacavir, didanosine, emtricitabine, lamivudine, stavudine, ténofovir et zidovudine). Aptivus a eu un effet synergique avec l'inhibiteur de fusion VIH enfuvirtide. L'association in vitro de tipranavir et d'adéfovir ou de ribavirine, utilisée pour le traitement de l'hépatite virale, n'a pas présenté d'antagonisme.
Résistance
L'apparition de résistances in vitro au tipranavir est un phénomène lent et complexe. Dans une étude in vitro de résistance particulière, un isolat de VIH-1 dont la résistance au tipranavir s'était accrue de 87 fois a été sélectionné au bout de 9 mois. Il présentait 10 mutations au niveau de la protéase: L10F, I13V, V32I, L33F, M36I, K45I, I54V/T, A71V, V82L et I84V ainsi qu'une mutation au niveau du site de clivage de la polyprotéine gag CA/P2. Les résultats des modifications liées à la mutagenèse dirigée ont montré qu'il faut au moins 6 mutations sur le gène de la protéase (I13V, V32I, L33F, K45I, V82L, I84V) pour provoquer une résistance au tipranavir multipliée par un facteur supérieur à 10. Le génotype présentant la totalité des 10 mutations a provoqué une résistance au tipranavir multipliée par un facteur 69. Dans des conditions in vitro, il existe une corrélation inverse entre le degré de résistance au tipranavir et la capacité de réplication des virus. Des virus recombinants montrant une sensibilité au tipranavir diminuée d'un facteur ≥3 ont un taux de réplication inférieur à 1% par rapport à celui enregistré dans les mêmes conditions avec le VIH-1 de type sauvage. Des virus résistants au tipranavir, qui ont émergé in vitro de types sauvages du VIH-1, ont présenté une sensibilité réduite aux inhibiteurs de la protéase suivants: amprénavir, atazanavir, indinavir, lopinavir, nelfinavir et ritonavir, mais sont toutefois restés sensibles au saquinavir.
Une série d'analyses de régression multiple par étape portant sur les génotypes déterminés initialement et sous traitement, à partir de toutes les études cliniques, a montré que 16 acides aminés étaient associés à la réduction de la sensibilité au tipranavir et/ou à une réduction de la réponse virologique au bout de 24 semaines: 10V, 13V, 20M/R/V, 33F, 35G, 36I, 43T, 46L, 47V, 54A/M/V, 58E, 69K, 74P, 82L/T, 83D et 84V. Les isolats cliniques présentant une réduction d'un facteur supérieur à 10 de la sensibilité au tipranavir, contenaient plus de huit mutations associées au tipranavir. Lors des essais cliniques de phase II et de phase III, les génotypes isolés au cours du traitement chez 276 patients ont montré que les principales mutations émergentes sous Aptivus concernaient L33F/I/V, V82T/L et I84V. L'association de ces trois mutations est généralement nécessaire pour obtenir une sensibilité réduite. Les mutations en position 82 apparaissent selon deux mécanismes: l'un par la mutation 82A préexistante qui sélectionne la mutation 82T, et l'autre par un virus de type sauvage présentant la mutation 82V qui sélectionne la mutation 82L.
Dans une étude menée auprès de 373 patients non traités préalablement, on a analysé le développement d'une résistance de la protéase chez 17 patients, qui ont présenté un rebond virologique après un schéma thérapeutique incluant Aptivus/ritonavir. Chez ces patients dont le virus ne présentait aucune mutation préalable aux inhibiteurs de la protéase en début d'étude, aucun virus n'a développé de résistance aux inhibiteurs de la protéase.
Chez les 28 enfants et adolescents de l'étude clinique 1182.14 qui ont présenté un échec virologique ou une non-réponse, les substitutions codons-acides aminés étaient comparables à celles observées chez l'adulte. Comme chez l'adulte, la sensibilité réduite au tipranavir chez les enfants et les adolescents était associée à la survenue de mutations.
Résistance croisée
Le tipranavir maintient une activité antivirale significative (résistance 4 fois plus basse) sur la majorité des isolats cliniques du VIH-1 qui, après traitement, ont fait preuve d'une sensibilité diminuée aux inhibiteurs de la protéase actuellement disponibles: amprénavir, atazanavir, indinavir, lopinavir, ritonavir, nelfinavir et saquinavir. Une résistance au tipranavir d'un facteur supérieur à 10 est rare (moins de 2,5% des isolats testés) pour les virus provenant de patients plusieurs fois prétraités ayant déjà reçu plusieurs inhibiteurs de la protéase peptidiques.
Évaluation de l'ECG
Allongement de l'intervalle QT: à des doses thérapeutiques, Aptivus/ritonavir n'a ni prolongé l'intervalle QTc ni induit d'allongement de l'intervalle QT. À des doses thérapeutiques, Aptivus/ritonavir n'a pas entraîné d'effets cliniquement pertinents sur l'ECG. Par conséquent, le tipranavir à des doses thérapeutiques associé au ritonavir à faible dose n'a que faiblement augmenté l'intervalle QTc (3,2 msec, limite supérieure de l'intervalle de confiance à 95%: 5,6 msec), de 8,3 msec, (limite supérieure de l'intervalle de confiance à 95 % = 10,9 msec) à des doses plus élevées (750 mg/200 mg).
Description des études cliniques
Les données cliniques suivantes sont issues des analyses à 48 semaines des essais cliniques en cours (RESIST-1 et RESIST-2), mesurant les effets sur le taux plasmatique d'ARN du VIH et le taux de cellules CD4. Aucun résultat d'études contrôlées évaluant l'effet d'Aptivus sur la progression de la maladie causée par le VIH n'est disponible à ce jour.
Patients adultes prétraités
RESIST-1 et RESIST-2 sont des études en cours, randomisées, ouvertes, multicentriques, conduites chez des patients séropositifs déjà traités par trithérapie antirétrovirale. Ces études évaluent le traitement par Aptivus associé au ritonavir à faible dose (500 mg/200 mg, deux fois par jour), en association avec un traitement de base optimisé (TBO) défini individuellement pour chaque patient sur la base des tests de résistance génotypique et des antécédents. Le traitement comparateur comporte un inhibiteur de la protéase boosté par le ritonavir (IPC/r, également défini individuellement) en association au TBO. L'inhibiteur de la protéase boosté par le ritonavir sélectionné était le saquinavir, l'amprénavir, l'indinavir ou le lopinavir.
Tous les patients avaient déjà reçu au moins deux traitements antirétroviraux contenant des inhibiteurs de la protéase et étaient en échec d'un traitement par inhibiteur de la protéase à l'inclusion dans l'étude.
À l'inclusion, ils devaient présenter au moins une mutation primaire au niveau du gène de la protéase parmi les mutations 30N, 46I, 46L, 48V, 50V, 82A, 82F, 82L, 82T, 84V ou 90M, mais pas plus de deux mutations sur les codons 33, 82, 84 ou 90.
Après la 8e semaine, les patients du bras comparateur présentant les critères définis par le protocole d'une absence de réponse virologique initiale avaient la possibilité d'arrêter le traitement et de passer à Aptivus/ritonavir dans une étude par rotation séparée.
L'analyse primaire des études RESIST regroupées a tenu compte de 1483 patients (Aptivus/ritonavir n=746, IPC/ritonavir n=737). Les groupes de patients avaient une moyenne d'âge de 43 ans (17-80) ou de 42 ans (21-72) respectivement pour Aptivus/ritonavir et IPC/ritonavir. 84% sur Aptivus/ritonavir, resp. 88% sur IPC/ritonavir étaient des hommes, 77%, resp. 74% étaient blancs, 12,6%, resp. 13,3% noirs et 0,7%, resp. 1,2% asiatiques. Dans les bras Aptivus/ritonavir et IPC/ritonavir, les taux médians de CD4 à l'inclusion étaient respectivement de 158 cellules/mm3 et de 166 cellules/mm3 (interquartile ranges [IQRs]: 66-285 ou 53-280 cellules/mm3); les taux plasmatiques médians d'ARN du VIH-1 à l'inclusion étaient de 4,79 log10 copies/ml et de 4,80 log10 copies/ml, respectivement (IQRs: 4,32-5,24 ou 4,25-5,27 log10 copies/ml). La réponse au traitement et les résultats du traitement randomisé à la semaine 48 sont présentés dans le tableau ci-dessous.
Résultats du traitement randomisé de 48 semaines (résultats compilés des études RESIST-1 et RESIST-2 chez les patients prétraités)
|
|
Aptivus/ritonavir (500/200 mg 2x/jour) + TBO
(n = 746)
|
IPC/ritonavir*** + TBO
(n = 737)
| |
Réponse au traitement*
|
34,2 %
|
15,5 %
| |
·initiation d'enfuvirtide
|
60,5 % (n=75/124)
|
22,7 % (n=22/97)
| |
·sans enfuvirtide
|
29,5 % (n=170/576)
|
14,3 % (n=86/602)
| |
Modification médiane de la charge virale du VIH par rapport à la valeur initiale (log10 copies/ml)
|
-0,64
|
-0,22
| |
Charge virale VIH < 400 copies/ml
|
30,3 %
|
13,6 %
| |
Charge virale VIH < 50 copies/ml
|
22,7 %
|
10,2 %
| |
Augmentation médiane du nombre de cellules CD4+ (cellules/mm3)
|
23
|
4
| |
Échec thérapeutique
|
65,8 %
|
84,5 %
| |
Raisons de l'échec thérapeutique
| |
Décès
|
1,6 %
|
0,7 %
| |
Arrêt du médicament à l'étude ou passage à un TBO en raison d'un manque d'efficacité
|
12,5 %
|
45,9 %
| |
Échec virologique
|
23,1 %
|
18,3 %
| |
Réponse virologique non confirmée
|
49,5 %
|
69,9 %
| |
Arrêt à cause d'effets secondaires
|
8,7 %
|
4,7 %
| |
Arrêt pour d'autres raisons**
|
6,0 %
|
9,2 %
|
* Critère composite défini comme patients présentant une baisse confirmée de 1 log10 de la valeur initiale et sans signe d'échec thérapeutique.
** Patients qui ne sont plus venus pour le suivi, ayant enfreint le protocole, retiré leur consentement ou autres raisons.
*** Comparateur inhibiteur de la protéase: LPV/r 400 mg/100 mg deux fois par jour, IDV/r 800 mg/100 mg deux fois par jour, SQV/r 1000 mg/100 mg deux fois par jour ou 800 mg/200 g deux fois par jour, APV/r 600 mg/100 mg deux fois par jour.
Les données des études RESIST ont également démontré que la réponse à 48 semaines au traitement par Aptivus associé au ritonavir à faible dose était meilleure lorsque le traitement de base optimisé (TBO) comportait des principes actifs antirétroviraux génotypiquement efficaces (p.ex. enfuvirtide).
Sur une période de traitement de 96 semaines, le délai médian jusqu'à l'échec thérapeutique était de 115 jours chez les patients traités par Aptivus/ritonavir par rapport à 0 jour dans le bras comparateur inhibiteur de la protéase/ritonavir. Chez les patients avec initiation d'enfuvirtide (défini comme premier traitement par enfuvirtide), le délai médian jusqu'à l'échec thérapeutique était de 587 jours dans le bras Aptivus/ritonavir contre 60 jours chez les patients traités par IPC/ritonavir.
Analyses de la résistance au tipranavir chez les patients prétraités
Les taux de réponse à Aptivus/ritonavir étaient évalués en fonction du génotype et du phénotype pour le tipranavir à l'inclusion. L'évaluation portait sur la relation entre la sensibilité phénotypique au tipranavir au début de l'étude, les mutations associées à la résistance au tipranavir et la réponse au traitement par Aptivus/ritonavir.
Mutations associées à la résistance au tipranavir:
La réponse virologique et thérapeutique au traitement par Aptivus/ritonavir a été évaluée à l'aide d'un score de mutation associée au tipranavir basé sur le génotype au début de l'étude des patients des essais cliniques RESIST-1 et RESIST-2. Ce score (incluant les 16 acides aminés associés à une réduction de la sensibilité au tipranavir et/ou à une réduction de la réponse en termes de charge virale: 10V, 13V, 20M/R/V, 33F, 35G, 36I, 43T, 46L, 47V, 54A/M/V, 58E, 69K, 74P, 82L/T, 83D et 84V) a été appliqué aux séquences de la protéase virale au début de l'étude. Une corrélation a été découverte entre le score de mutations associées au tipranavir et la réponse au traitement par Aptivus/ritonavir à la 2e et 48e semaine.
Après 48 semaines, le pourcentage de patients sur Aptivus associé au ritonavir à faible dose répondant au traitement était supérieur à celui des patients traités par l'inhibiteur de la protéase comparateur/ritonavir pour presque toutes les combinaisons possibles de mutations de résistance génotypique (Tableau 1).
Tableau 1. Pourcentage de patients avec réponse au traitement à la 48e semaine (recul confirmé de la charge virale ≥1 log10 copies/ml par rapport au début de l'étude), en fonction du score de mutations associées au tipranavir à l'inclusion et de l'utilisation d'enfuvirtide chez les patients de RESIST.
|
|
Initiation d'ENF
|
Pas d'initiation d'ENF1
| |
Score de mutations associées au TPV
|
TPV/r
|
IPC/r
|
TPV/r
|
IPC/r
| |
0, 1
|
73%
|
21%
|
53%
|
25%
| |
2
|
61%
|
43%
|
33%
|
17%
| |
3
|
75%
|
23%
|
27%
|
14%
| |
4
|
59%
|
19%
|
23%
|
8%
| |
≥5
|
47%
|
15%
|
13%
|
13%
| |
Tous les patients
|
61%
|
23%
|
29%
|
14%
|
1 Patients sans traitement par ENF et patients prétraités par ENF qui l'ont continué.
Des réductions durables de l'ARN du VIH-1 (Tableau 2) jusqu'à la 48e semaine ont été observées principalement chez des patients recevant Aptivus/ritonavir et initiant également de l'enfuvirtide. Chez les patients traités par Aptivus/ritonavir sans initiation de l'enfuvirtide, la réponse au traitement observée à la 48e semaine était inférieure à celle des patients ayant initié l'enfuvirtide (voir Tableau 2).
Tableau 2. Recul moyen de la charge virale du début de l'étude à la 48e semaine en fonction du score de mutations associées au tipranavir à l'inclusion et de l'utilisation d'enfuvirtide chez les patients de RESIST.
|
|
Initiation d'ENF
|
Pas d'initiation d'ENF1
| |
Score de mutations associées au TPV
|
TPV/r
|
IPC/r
|
TPV/r
|
IPC/r
| |
0, 1
|
-2,3
|
-1,5
|
-1,6
|
-0,6
| |
2
|
-2,1
|
-1,4
|
-1,1
|
-0,6
| |
3
|
-2,4
|
-1,0
|
-0,9
|
-0,5
| |
4
|
-1,7
|
-0,7
|
-0,8
|
-0,3
| |
≥5
|
-1,9
|
-0,6
|
-0,6
|
-0,4
| |
Tous les patients
|
-2,0
|
-1,0
|
-1,0
|
-0,5
|
1 Patients sans traitement par ENF et patients prétraités par ENF qui l'ont continué.
Mutations de la protéase aux positions 33, 82, 84 et 90:
Les mutations sur deux, trois ou plus de ces positions ont entraîné une réduction de la sensibilité à Aptivus/ritonavir; quatre mutations ont entraîné une résistance.
Résistance phénotypique au tipranavir:
L'augmentation du score phénotypique IC50 («Fold change») en début d'étude est corrélée à une réduction de la réponse virologique au tipranavir. Les isolats avec un score phénotypique IC50 compris entre >0 et 3 sont considérés comme sensibles; les isolats avec un score compris entre >3 et 10 ont une sensibilité réduite; les isolats avec un score IC50 >10 sont résistants.
Les conclusions concernant la pertinence de certaines mutations ou de profils de mutation particuliers sont provisoires et susceptibles de modifications sur la base de données additionnelles. Il est recommandé de toujours consulter les banques de données d'interprétation actualisés lors de l'analyse des résultats des tests de résistance.
PharmacocinétiqueAdultes
Pour obtenir des concentrations plasmatiques efficaces de tipranavir avec une prise biquotidienne, il est essentiel d'administrer Aptivus associé au ritonavir à faible dose deux fois par jour (voir «Posologie/Mode d'emploi»). Le ritonavir agit en inhibant l'iso-enzyme CYP3A4 du cytochrome P450 hépatique, la pompe d'efflux P-glycoprotéine (P-gp) intestinale et peut-être également l'iso-enzyme CYP3A4 du cytochrome P450 intestinal. Une étude d'escalade de doses effectuée chez 113 volontaires sains des deux sexes séronégatifs a montré que le ritonavir augmente l'ASC0-12h, la Cmax et la Cmin et réduit la clairance du tipranavir. Aptivus associé au ritonavir à faible dose (500 mg/200 mg deux fois/jour) était associé à une augmentation d'un facteur de 29 de la moyenne géométrique des taux plasmatiques résiduels matinaux à l'état d'équilibre par rapport à Aptivus 500 mg 2 fois par jour sans ritonavir.
Une étude menée chez des patients séropositifs a analysé la pharmacocinétique et la sécurité d'Aptivus/ritonavir 500/200 mg, administré avec ou sans lopinavir, amprénavir ou saquinavir, par rapport à l'administration concomitante de ritonavir 100 mg et de lopinavir, amprénavir ou saquinavir. La concentration systémique moyenne de ritonavir lors de l'administration concomitante de 200 mg de ritonavir et d'Aptivus ou de 100 mg avec d'autres inhibiteurs de la protéase est de 0,59 µg/ml (SQV/r 1000/100 mg), 0,32 µg/ml (LPV/r 400/100 mg), 0,18 µg/ml (APV/r 600/100 mg) ou de 0,25 µg/ml (TPV/r 500/200 mg).
Absorption
La résorption du tipranavir chez l'homme est limitée, bien qu'elle n'ait pas encore été quantifiée de façon absolue. Le tipranavir est un substrat de la P-gp. Les concentrations plasmatiques maximales sont atteintes 1 à 5 heures après prise de la dose et sont dose-dépendantes. En cas de prise répétée, les concentrations plasmatiques du tipranavir sont inférieures à celles anticipées d'après les données obtenues lors d'une prise unique, probablement en raison de l'induction des enzymes hépatiques ou de l'induction des transporteurs. Chez la plupart des patients, l'état d'équilibre est atteint après 7 jours de traitement. Les paramètres pharmacocinétiques à l'état d'équilibre d'Aptivus associé au ritonavir à faible dose sont linéaires.
L'administration concomitante d'Aptivus capsules molles 500 mg et de 200 mg de ritonavir 2 fois par jour pendant 2 à 4 semaines et sans restriction alimentaire a entraîné une concentration plasmatique maximale de tipranavir (Cmax) moyenne de 94,8 ± 22,8 µM chez les femmes (n = 14) et de 77,6 ± 16,6 µM chez les hommes (n = 106). La concentration plasmatique maximale était atteinte environ 3 heures après l'administration. La concentration plasmatique résiduelle moyenne à l'état d'équilibre avant la dose du matin était de 41,6 ± 24,3 µM chez les femmes et de 35,6 ± 16,7 µM chez les hommes. L'ASC du tipranavir après un intervalle de 12 heures suivant l'administration était en moyenne de 851 ± 309 µM*h (CL=1,15 l/h) chez les femmes et de 710 ± 207 µM*h (CL=1,27 l/h) chez les hommes. La demi-vie moyenne était de 5,5 heures (chez les femmes) ou 6,0 heures (chez les hommes).
Courbe de la concentration plasmatique de tipranavir (IC à 95%) en cas d'administration concomitante de ritonavir (500/200 mg 2 fois par jour)
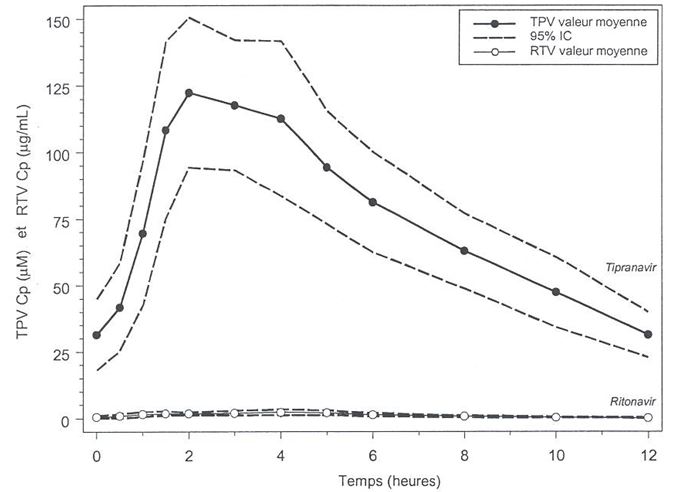
Paramètres pharmacocinétiquesa d'une dose de tipranavir/ritonavir 500/200 mg chez des patients séropositifs en fonction du sexe
|
|
Femmes
(n = 14)
|
Hommes
(n = 106)
| |
Cptrough (µM)
|
41,6 ± 24,3
|
35,6 ± 16,7
| |
Cmax (µM)
|
94,8 ± 22,8
|
77,6 ± 16,6
| |
Tmax (h)
|
2,9
|
3,0
| |
ASC0-12h (µM*h)
|
851 ± 309
|
710 ± 207
| |
CL (l/h)
|
1,15
|
1,27
| |
V (l)
|
7,7
|
10,2
| |
t1/2 (h)
|
5,5
|
6,0
|
a Indication des paramètres pharmacocinétiques en tant que moyenne ± écart-type
Influence de la nourriture sur la résorption orale
Comparée à une prise à jeun, la nourriture (500-682 kcal, 23-25 % de calories provenant des graisses) n'a pas d'influence cliniquement significative sur la Cmax, Cp12h et l'ASC d'Aptivus capsules molles associé au ritonavir à l'état d'équilibre.
Paramètres pharmacocinétiquesa du tipranavir à l'état d'équilibre lors d'une prise orale (tipranavir/ritonavir 500/200 mg deux fois par jour) avec ou sans nourriture
|
|
Tipranavir/ritonavir capsules molles
| |
Cmax (µM)
|
124,7 ± 54,7124,6 ± 60,3
| |
Cp12h (µM)
|
27,6 ± 22,729,8 ± 29,6
| |
ASC0-12h (µM*h)
|
789 ± 395820 ± 479
|
a Indication des paramètres pharmacocinétiques en tant que moyenne ± écart-type
Aptivus associé au ritonavir doit être pris avec un repas, étant donné que le ritonavir est mieux toléré lorsqu'il est pris avec de la nourriture ainsi que l'importance d'une prise concomitante d'Aptivus et de ritonavir (voir «Posologie/Mode d'emploi»).
La résorption d'Aptivus associé au ritonavir à faible dose est réduite en présence d'antiacides (voir «Interactions»).
Distribution
Le tipranavir est fortement lié aux protéines plasmatiques (> 99,9%). D'après les prélèvements sanguins obtenus chez des volontaires sains et des patients VIH-1 positifs ayant reçu Aptivus sans le ritonavir, la fraction plasmatique de tipranavir non lié était comparable dans les deux populations (volontaires sains: 0,015% ±0,006%; sujets séropositifs: 0,019% ± 0,076%). Les concentrations plasmatiques totales de tipranavir de ces échantillons étaient comprises entre 9 et 82 µM. La fraction non liée de tipranavir semblait indépendante de la concentration totale du médicament dans cette plage de concentration.
Aucune étude n'a été effectuée pour déterminer la distribution du tipranavir dans le liquide céphalo-rachidien et le sperme humains.
Métabolisme
Les études de métabolisme in vitro effectuées avec des microsomes de foie humain ont indiqué que l'isoenzyme CYP3A4 constitue l'isoforme CYP prédominante impliquée dans le métabolisme du tipranavir. La clairance du tipranavir était réduite après l'addition de ritonavir, ce qui peut être dû à une réduction de la clairance de premier passage tant digestive qu'hépatique. Le métabolisme du tipranavir est minime en présence du ritonavir à faible dose. Lors d'une étude clinique menée avec du tipranavir marqué au 14C (14C-tipranavir/ritonavir 500 mg/200 mg deux fois par jour), le tipranavir inchangé était prédominant et représentait au moins 98,4% de la radioactivité plasmatique circulante totale 3, 8 et 12 heures après l'administration. Seuls quelques métabolites ont été retrouvés dans le plasma, tous à l'état de traces (≤0,2% de la radioactivité plasmatique). Le tipranavir non métabolisé a également constitué la majorité de la radioactivité retrouvée dans les fèces (79,9% de la radioactivité fécale). Le métabolite fécal le plus abondant, un métabolite hydroxylé du tipranavir, constituait 4,9% de la radioactivité retrouvée dans les selles (soit 3,2% de la dose prise). Dans l'urine, le tipranavir inchangé n'a été retrouvé qu'à l'état de traces (0,5% de la radioactivité urinaire). Un glucuroconjugué du tipranavir était le métabolite urinaire le plus abondant avec 11% de la radioactivité urinaire (soit 0,5% de la dose prise).
Élimination
L'administration de 14C-tipranavir à des sujets (n = 8) ayant reçu l'association Aptivus/ritonavir (500/200 mg) deux fois par jour à l'état d'équilibre a démontré que la majorité de la radioactivité (médiane 82,3%) se retrouvait au niveau des fèces, alors que seule une médiane de 4,4% de la dose radioactive était retrouvée dans l'urine. En outre, la majeure partie de la radioactivité (56%) était excrétée entre 24 et 96 heures après l'administration. La demi-vie moyenne d'élimination efficace de l'association tipranavir/ritonavir (500 mg/200 mg deux fois par jour, avec un repas léger) chez des volontaires sains (n = 67) et des patients adultes séropositifs (n = 120) était approximativement de respectivement 4,8 et 6,0 heures à l'état d'équilibre.
Cinétique pour certains groupes de patients
Les données actuellement disponibles sont limitées et ne permettent pas une analyse définitive. Elles suggèrent toutefois que le profil pharmacocinétique n'est pas modifié chez les personnes âgées et est comparable entre les races. Par contre, l'évaluation des concentrations plasmatiques résiduelles du tipranavir à l'état d'équilibre 10-14 heures après la prise (études RESIST-1 et RESIST-2) montre que les femmes présentent généralement des concentrations de tipranavir plus élevées que les hommes. Après 4 semaines d'utilisation d'Aptivus associé au ritonavir (500 mg/200 mg) deux fois par jour), la concentration plasmatique résiduelle médiane de tipranavir était de 43,9 µM chez les femmes et de 31,1 µM chez les hommes. Cette différence de concentrations ne nécessite toutefois pas d'ajustement de dose.
Enfants et adolescents
Chez les patients pédiatriques de l'étude clinique 1182.14, la concentration plasmatique résiduelle de tipranavir à l'état d'équilibre a été déterminée 10 à 14 heures après l'administration du médicament à l'étude. Les moyennes géométriques des concentrations plasmatiques résiduelles du tipranavir déterminées chez les 50 patients traités par 375 mg/m2/150 mg/m2 deux fois par jour se situaient entre 46,9 et 61,3 μM (entre 3,8 et 73,4 μM en cas de très forte variabilité).
Troubles de la fonction hépatique
Une étude a comparé 9 patients atteints d'insuffisance hépatique légère (classe Child-Pugh A) et 9 témoins. Après l'administration de tipranavir et de ritonavir à dose unique et à doses répétées, les concentrations plasmatiques étaient majorées chez les insuffisants hépatiques, tout en restant néanmoins dans les limites observées au cours des études cliniques. Aucun ajustement de la dose n'est nécessaire en cas d'insuffisance hépatique légère, mais les patients doivent être étroitement surveillés (voir «Posologie/Mode d'emploi», «Contre-indications» ainsi que «Mises en garde et précautions»).
L'influence d'une insuffisance hépatique modérée (classe Child-Pugh B) ou sévère (classe Child-Pugh C) sur la pharmacocinétique après prises multiples de tipranavir ou de ritonavir n'a pas encore été étudiée. Aptivus est contre-indiqué chez les patients atteints d'une insuffisance hépatique modérée ou sévère (voir «Posologie/Mode d'emploi» et «Contre-indications»).
Troubles de la fonction rénale
La pharmacocinétique du tipranavir n'a pas été étudiée chez les patients insuffisants rénaux. La clairance rénale du tipranavir étant négligeable, il ne faut toutefois pas s'attendre à une réduction de la clairance corporelle totale chez les patients présentant une insuffisance rénale.
Données précliniquesPharmacologie de sécurité
Le tipranavir a eu un effet inhibiteur in vitro (IC50 de 2,9 µM dans un environnement sans protéine) sur le canal potassique associé au HERG. Aucune activité concernant un allongement de l'intervalle QT sur la durée du potentiel d'action n'a été observée avec les mêmes concentrations dans des essais in vitro sur des muscles papillaires de cobayes et dans des études ECG in vivo sur des chiens conscients après administration unique.
Paramètres de coagulation
Des études in vitro ont montré que le tipranavir inhibe l'agrégation des plaquettes humaines à des concentrations équivalentes à celles mesurées chez des patients traités par Aptivus/ritonavir.
Dans des études précliniques chez des rats, le traitement par le tipranavir a induit des modifications dose-dépendantes des paramètres de coagulation (augmentation du temps de prothrombine, augmentation du temps de thromboplastine partielle activée et diminution de certains facteurs dépendants de la vitamine K). Chez certains rats, ces modifications ont conduit à des hémorragies dans différents organes et au décès. L'administration concomitante de tipranavir et de vitamine E (en tant que TPGS: succinate de d-alpha-tocophérol polyéthylène glycol 1000) a entraîné une augmentation significative des effets sur les paramètres de coagulation, les événements hémorragiques et les décès. Le mécanisme de ces effets est inconnu. Aucun effet sur les paramètres de coagulation n'a été constaté dans des études précliniques chez des chiens. L'administration concomitante de tipranavir et de vitamine E n'a pas été étudiée chez le chien. L'évaluation clinique des effets sur la coagulation de patients infectés par le VIH-1 n'a révélé aucune influence de l'administration de tipranavir associé au ritonavir sur les paramètres de coagulation.
Toxicité à long terme (ou toxicité en cas d'administration répétée)
Aucune influence négative sur la reproduction ou la fertilité n'a été observée dans une étude menée chez les rats avec le tipranavir à des niveaux d'exposition systémiques (ASC) équivalents à l'exposition chez l'homme à la posologie recommandée en clinique.
Chez les femelles, le tipranavir n'a pas entraîné d'effet tératogène à des niveaux d'exposition systémique équivalents ou même inférieurs aux niveaux d'exposition chez l'homme, à la posologie recommandée en clinique (Aptivus/ritonavir 500 mg/200 mg deux fois par jour).
Une toxicité fœtale (diminution de l'ossification du sternum et du poids corporel) a été observée chez les rats à des niveaux d'exposition au tipranavir équivalents à 0,8 fois l'exposition chez l'homme à la posologie recommandée en clinique. Dans les études de développement prénatal et postnatal chez le rat avec tipranavir, une inhibition de la croissance de la portée a été observée à une posologie maternelle toxique équivalente à environ 0,8 fois l'exposition chez l'homme.
Carcinogénicité
Des essais de carcinogénicité au long cours ont été menés chez la souris et le rat et les résultats ont révélé un potentiel tumorigène, qui est dû à un potentiel d'induction enzymatique hépatique particulièrement important au sein de ces espèces. Ces résultats ne sont donc pas considérés comme cliniquement pertinents. Le tipranavir n'a pas présenté de signes de toxicité génétique dans la batterie de tests in vitro et in vivo.
Remarques particulièresIncompatibilités
Non pertinent.
Stabilité
Le médicament ne doit pas être utilisé au-delà de la date figurant après la mention «EXP» sur le récipient.
Stabilité après ouverture
À utiliser dans les 2 mois après ouverture.
Remarques particulières concernant le stockage
Conserver au réfrigérateur (2-8 °C).
Numéro d’autorisation57330 (Swissmedic).
PrésentationAptivus, 250 mg capsules molles, 120 capsules (A).
Titulaire de l’autorisationBoehringer Ingelheim (Schweiz) GmbH, Bâle.
Mise à jour de l’informationSeptembre 2019.
|