CompositionPrincipes actifs
Furoate de fluticasone
Uméclidinium (sous forme de bromure d'uméclidinium)
Vilantérol (sous forme de trifénatate de vilantérol)
Excipients
Lactose monohydraté (avec de faibles quantités de protéines de lait), stéarate de magnésium.
Forme pharmaceutique et quantité de principe actif par unitéPoudre à inhaler en doses unitaires
Une dose unitaire de Trelegy Ellipta 92/55/22 contient 100 µg de furoate de fluticasone (FF), 62,5 µg d'uméclidinium (correspondant à 74,2 µg de bromure d'uméclidinium) et 25 µg de vilantérol (sous forme de trifénatate de vilantérol). La dose administrée (libérée par l'embout de l'inhalateur Ellipta) est de 92 µg de furoate de fluticasone, 55 µg d'uméclidinium (correspondant à 65,3 µg de bromure d'uméclidinium) et 22 µg de vilantérol.
Un inhalateur Ellipta contient 30 doses unitaires à inhaler.
Indications/Possibilités d’emploiTraitement d'entretien de la bronchopneumopathie chronique obstructive (BPCO) modérée à sévère chez les patients adultes ayant subi ≥1 exacerbation dans les 12 derniers mois et traités de façon non satisfaisante par l'association d'un CSI et d'un LABA ou par l'association d'un LABA et d'un LAMA.
Posologie/Mode d’emploiTrelegy Ellipta est destiné exclusivement à l'inhalation (orale).
Les patients doivent être instruits de se rincer la bouche à l'eau – sans avaler l'eau de rinçage – après l'inhalation. Ces rinçages permettent de prévenir le développement d'une candidose buccale et d'éviter les irritations de la gorge.
Adultes à partir de 18 ans
La dose recommandée et maximale est d'une inhalation de Trelegy 92/55/22 une fois par jour, toujours à la même heure.
Enfants et adolescents
L'utilisation de cette préparation chez les patients de moins de 18 ans n'est pas justifiée en raison de l'indication.
Instructions posologiques particulières
Patients âgés (>65 ans)
Aucun ajustement de la posologie n'est nécessaire chez ce groupe de patients (cf. "Pharmacocinétique" ).
Patients présentant des troubles de la fonction rénale
Aucun ajustement de la posologie n'est nécessaire chez ce groupe de patients (cf. "Pharmacocinétique" ).
Patients présentant des troubles de la fonction hépatique
Aucun ajustement de la posologie n'est nécessaire chez les patients atteints d'insuffisance hépatique. L'uméclidinium n'a pas été évalué chez des patients atteints d'insuffisance hépatique sévère (voir "Mises en garde et précautions" , "Pharmacocinétique" ).
Contre-indicationsTrelegy Ellipta est contre-indiqué chez les patients présentant une hypersensibilité connue à l'un des composants ou une allergie sévère aux protéines du lait (voir "Composition" ).
Mises en garde et précautionsL'utilisation de Trelegy Ellipta n'a pas été évaluée chez les patients asthmatiques et n'est pas recommandée chez ce groupe de patients.
Exacerbations
Trelegy Ellipta est destiné au traitement d'entretien de la bronchopneumopathie chronique obstructive (BPCO). Il ne doit pas être utilisé pour soulager des symptômes aigus, c.-à-d. pour le traitement d'urgence des épisodes aigus de bronchospasme. Les symptômes aigus doivent être traités avec un bronchodilatateur inhalé de courte durée d'action.
Une augmentation de la consommation de bronchodilatateurs de courte durée d'action pour soulager les symptômes indique une détérioration du contrôle de la maladie. Les patients doivent se soumettre à un examen médical.
Les patients ne devraient pas interrompre leur traitement par Trelegy Ellipta sans surveillance médicale, étant donné que l'arrêt peut entraîner la réapparition des symptômes.
Bronchospasme paradoxal
Comme avec d'autres traitements inhalés, un bronchospasme paradoxal avec aggravation immédiate des sifflements respiratoires peut apparaître après l'administration et engager le pronostic vital. Ce phénomène doit être traité immédiatement par l'inhalation d'un bronchodilatateur de courte durée d'action. Dans un tel cas, le patient doit immédiatement interrompre le traitement par Trelegy Ellipta, être soumis à un examen approfondi et recevoir au besoin un traitement alternatif.
Effets cardiovasculaires
Des effets cardiovasculaires tels que des arythmies cardiaques (p.ex. fibrillation auriculaire et tachycardie) peuvent survenir après l'administration d'antagonistes des récepteurs muscariniques ou de sympathomimétiques, y compris d'uméclidinium ou de vilantérol. De plus, on a rapporté sous agonistes bêta-2 adrénergiques tels que Trelegy Ellipta une survenue d'anomalies électrocardiographiques telles qu'un aplatissement de l'onde T, un allongement de l'intervalle QTc et un sous-décalage du segment ST. La signification clinique de ces anomalies reste cependant inconnue. Avant la prescription d'un traitement de fond par un agoniste bêta-2 adrénergique comme Trelegy Ellipta, les patients atteints de BPCO doivent être examinés à la recherche d'éventuelles comorbidités cardiovasculaires. Par mesure de précaution, il est recommandé d'inclure à ces investigations un ECG avec vérification d'un éventuel allongement du QTc.
Hypokaliémie
Le traitement concomitant avec un dérivé de méthylxanthine, des stéroïdes ou un diurétique non épargneur de potassium pourrait renforcer une éventuelle hypokaliémie due aux agonistes bêta-2 adrénergiques.
Patients présentant une insuffisance hépatique
Les patients présentant une insuffisance hépatique modérée ou sévère et traités par Trelegy Ellipta doivent être surveillés afin de détecter l'apparition éventuelle d'effets indésirables systémiques liés au corticostéroïde (voir "Pharmacocinétique" ).
Effets systémiques des corticostéroïdes
L'administration concomitante de Trelegy avec des inhibiteurs puissants du CYP3A4 doit être évitée.
Tous les corticostéroïdes inhalés peuvent provoquer des effets systémiques, surtout lors de la prescription au long cours d'une dose élevée. La probabilité d'une survenue de tels effets reste cependant nettement plus faible qu'avec les corticostéroïdes oraux. Les effets systémiques possibles englobent une suppression de l'axe hypothalamo-hypophyso-surrénalien (HHS), une réduction de la densité minérale osseuse ainsi qu'une cataracte et un glaucome ou des affections oculaires rares telles qu'une choriorétinopathie séreuse centrale (CRSC) qui ont été observées après l'utilisation de corticostéroïdes systémiques ou inhalés.
Comme tous les médicaments contenant un corticoïde, Trelegy Ellipta ne doit pas être administré sans les mesures de précaution qui s'imposent chez des patients atteints d'une tuberculose pulmonaire ou d'une infection chronique ou non traitée.
Troubles visuels
Des troubles visuels peuvent apparaître lors de l'utilisation systémique ou topique de corticostéroïdes. Si un patient se présente avec des symptômes tels qu'une vision floue ou d'autres troubles visuels, on envisagera de l'adresser à un ophtalmologue pour une évaluation des causes possibles; celles-ci incluent entre autres une cataracte, un glaucome ou des maladies rares telles qu'une CRSC qui ont été rapportées après l'utilisation de corticostéroïdes systémiques ou topiques.
Hyperglycémie
Une glycémie accrue a été rapportée chez des patients diabétiques sous corticostéroïdes inhalés. Cet aspect doit être pris en compte lors de la prescription de Trelegy Ellipta à des patients diabétiques.
Effet anticholinergique
En raison de son effet anticholinergique, Trelegy Ellipta doit être utilisé avec précaution chez des patients présentant un glaucome à angle fermé ou une rétention urinaire.
Pneumonies
Des pneumonies (y compris des pneumonies ayant nécessité une hospitalisation) ont été observées chez les patients atteints de bronchopneumopathie chronique obstructive (BPCO) et traités par Trelegy Ellipta, ce qui correspond à l'effet de classe connu des corticostéroïdes inhalés. Dans certains cas, des pneumonies d'issue fatale ont été rapportées lors de l'utilisation de médicaments contenant le corticostéroïde inhalé furoate de fluticasone, y compris Trelegy (voir "Effets indésirables" ). Chez les patients atteints de BPCO, le médecin doit être attentif au développement possible d'une pneumonie, étant donné que les caractéristiques cliniques d'une telle infection coïncident avec les symptômes d'une exacerbation de la BPCO. Chez les patients atteints de BPCO sous médicaments inhalés à base de corticostéroïdes, les facteurs de risque de pneumonie sont notamment le tabagisme, les antécédents de pneumonie, un indice de masse corporelle faible et une BPCO sévère. Ces facteurs doivent être pris en compte lors de la prescription de Trelegy Ellipta et le traitement doit être réévalué lors de l'apparition d'une pneumonie.
InteractionsBêtabloquants
Les bêtabloquants peuvent affaiblir ou supprimer les effets des agonistes bêta-2 adrénergiques tels que le vilantérol. Si un traitement par bêtabloquants s'avère nécessaire, il convient d'envisager l'utilisation de bêtabloquants cardiosélectifs; la prudence est cependant recommandée lors de l'utilisation concomitante de bêtabloquants non sélectifs et sélectifs.
Inhibiteurs du CYP3A4
Aussi bien le furoate de fluticasone que le vilantérol, deux composants de Trelegy Ellipta, sont rapidement métabolisés lors d'un important effet de premier passage hépatique, médié par l'enzyme CYP3A4.
L'administration concomitante avec des inhibiteurs puissants du CYP3A4 (p.ex. kétoconazole, itraconazole, clarithromycine, ritonavir ou médicaments contenant du cobicistat) peut entraîner une exposition accrue aux corticostéroïdes ainsi qu'au vilantérol et, par là même, une augmentation du risque d'effets indésirables, notamment ceux liés aux corticostéroïdes systémiques. L'administration concomitante doit être évitée sauf si le bénéfice attendu l'emporte sur le risque accru d'effets indésirables systémiques liés aux corticostéroïdes; dans ce cas, les patients devront être surveillés afin de détecter d'éventuels effets indésirables systémiques liés aux corticostéroïdes (voir "Pharmacocinétique" ).
L'uméclidinium est un substrat du cytochrome P450 2D6 (CYP2D6). On ne s'attend toutefois à aucune interaction cliniquement significative lors d'une utilisation de Trelegy Ellipta en association avec des inhibiteurs du CYP2D6 ou chez des personnes présentant un déficit en CYP2D6 (métaboliseurs lents) (voir "Pharmacocinétique" ).
Inhibiteurs de la glycoprotéine P
L'uméclidinium et le vilantérol sont des substrats du transporteur glycoprotéine P (P-gp). Les effets du vérapamil (240 mg une fois par jour) – un inhibiteur modéré de la glycoprotéine P (P-gp) – sur la pharmacocinétique de l'uméclidinium et du vilantérol à l'état d'équilibre ont été examinés chez des volontaires sains. Aucun effet du vérapamil sur la Cmax de l'uméclidinium ou du vilantérol n'a été observé. L'AUC de l'uméclidinium a augmenté de 1,4 fois, mais aucun effet n'a été observé sur l'AUC du vilantérol.
Aucune interaction médicamenteuse cliniquement significative n'est toutefois attendue lorsque Trelegy Ellipta est utilisé en même temps que des inhibiteurs de la P-gp.
Médicaments provoquant notoirement un allongement de l'intervalle QTc
Comme pour d'autres agonistes bêta-2 adrénergiques, il existe théoriquement un risque d'interaction pharmacodynamique entre le vilantérol (contenu dans Trelegy Ellipta) et les médicaments connus pour allonger l'intervalle QTc. Une telle interaction pourrait accroître le risque éventuel d'arythmies ventriculaires. Ces médicaments englobent par exemple certains antihistaminiques (p.ex. terfénadine, mizolastine), certains antiarythmiques (p.ex. quinidine), les phénothiazines, l'érythromycine, le ritonavir et les antidépresseurs tricycliques. L'administration supplémentaire de substances sympathomimétiques peut renforcer les effets indésirables cardiovasculaires. Si Trelegy Ellipta est administré à des patients sous inhibiteurs de la MAO ou sous antidépresseurs tricycliques, la prudence est de rigueur parce que les effets des stimulateurs bêta-2 adrénergiques sur le système cardiovasculaire peuvent alors être renforcés.
L'administration en association avec la L-dopa, la L-thyroxine ou l'ocytocine peut avoir une influence négative sur la tolérance cardiaque aux bêta-2 sympathomimétiques.
Hypokaliémie
Le traitement concomitant avec un dérivé de méthylxanthine, des stéroïdes ou un diurétique non épargneur de potassium pourrait renforcer une éventuelle hypokaliémie due aux agonistes bêta-2 adrénergiques.
Autres anticholinergiques de longue durée d'action et agonistes bêta-2 adrénergiques de longue durée d'action
L'administration concomitante de furoate de fluticasone/uméclidinium/vilantérol et d'autres antagonistes des récepteurs muscariniques de longue durée d'action ou d'autres agonistes bêta-2 adrénergiques de longue durée d'action n'a pas été évaluée et n'est pas recommandée, car elle pourrait potentialiser les effets indésirables (voir "Effets indésirables" et "Surdosage" ).
Grossesse, allaitementGrossesse
Il n'existe pas de données suffisantes concernant l'emploi chez la femme enceinte.
Les expérimentations animales ont révélé une toxicité de reproduction après l'administration d'agonistes bêta-2 adrénergiques et de corticostéroïdes (voir "Données précliniques" ).
Trelegy Ellipta ne doit pas être administré pendant la grossesse, sauf en cas de nécessité absolue.
Allaitement
Les données disponibles sur l'excrétion du furoate de fluticasone, de l'uméclidinium, du vilantérol et de leurs métabolites dans le lait maternel sont limitées. D'autres corticostéroïdes, anticholinergiques et agonistes bêta-2 adrénergiques sont toutefois détectables dans le lait maternel.
Un risque pour le nouveau-né/nourrisson allaité ne peut être exclu.
Il faut donc, après avoir soupesé les avantages de l'allaitement pour l'enfant et le bénéfice du traitement pour la mère, choisir d'arrêter soit l'allaitement, soit le traitement par Trelegy Ellipta.
Effet sur l’aptitude à la conduite et l’utilisation de machinesAucune étude correspondante n'a été effectuée sur l'influence de Trelegy Ellipta sur l'aptitude à la conduite et l'utilisation de machines.
Effets indésirablesLe profil de sécurité de Trelegy Ellipta repose sur les données de 3 études cliniques de phase III.
La première étude (CTT116853, FULFIL avec contrôle actif) a inclus des données de sécurité de 911 patients atteints de BPCO ayant reçu Trelegy 92/55/22 une fois par jour pendant jusqu'à 24 semaines, dont 210 patients ayant reçu Trelegy 92/55/22 une fois par jour pendant jusqu'à 52 semaines.
La deuxième étude a inclus des données de sécurité de 527 patients atteints de BPCO ayant reçu Trelegy 92/55/22 une fois par jour pendant jusqu'à 24 semaines et de 528 patients atteints de BPCO ayant reçu l'association furoate de fluticasone/vilantérol 92/22 µg + uméclidinium 55 µg une fois par jour pendant jusqu'à 24 semaines (étude 200812).
La troisième étude (CTT116855, IMPACT avec 2 contrôles actifs) a inclus des données de sécurité de 4151 patients atteints de BPCO ayant reçu Trelegy 92/55/22 une fois par jour pendant jusqu'à 52 semaines.
Lorsque les fréquences des effets indésirables varient selon les études, c'est toujours l'incidence la plus élevée qui est indiquée ci-après.
Les effets indésirables sont classés par systèmes d'organes et par fréquence. Les fréquences sont indiquées par ordre décroissant selon la convention suivante: très fréquents (≥1/10), fréquents (≥1/100 à < 1/10), occasionnels (≥1/1000 à < 1/100), rares (≥1/10 000 à < 1/1000), très rares (< 1/10 000), "Fréquence inconnue" (peut pas être estimée sur la base des données disponibles).
Infections et infestations
Très fréquents: rhinopharyngite (15%)
Fréquents: pneumonie*, infections des voies respiratoires supérieures, bronchite, pharyngite, rhinite, sinusite, grippe, infection virale des voies respiratoires, candidose bucco-pharyngée, infections urinaires
Affections du système nerveux
Fréquents: céphalées
Occasionnels: dysgueusie
Affections cardiaques
Occasionnels: tachyarythmie supraventriculaire, tachycardie, fibrillation auriculaire
Affections respiratoires, thoraciques et médiastinales
Fréquents: toux, douleurs bucco-pharyngées, dysphonie
Affections gastro-intestinales
Fréquents: constipation
Occasionnels: sécheresse buccale
Affections musculosquelettiques et du tissu conjonctif
Fréquents: douleurs articulaires, dorsalgies
Occasionnels: fractures
Description de certains effets indésirables
*Pneumonies (voir "Mises en garde et précautions" )
Chez au total 1810 patients atteints de BPCO avancée (VEMS post-bronchodilatateur moyen à l'inclusion à 45% de la valeur prédite, écart-type ET de 13%), dont 65% avaient présenté une exacerbation de la BPCO modérée à sévère dans l'année précédant l'inclusion dans l'étude (étude CTT 116853), l'incidence des pneumonies était plus élevée chez les patients traités par Trelegy Ellipta (20 patients, 2%) que chez les patients traités par le budésonide/formotérol (7 patients, <1%). Une pneumonie nécessitant une hospitalisation est survenue jusqu'à la semaine 24 chez 1% des patients sous Trelegy Ellipta et chez < 1% des patients sous budésonide/formotérol. Un cas mortel de pneumonie a été rapporté chez un patient traité par Trelegy Ellipta. Dans un sous-groupe de 430 patients traités pendant jusqu'à 52 semaines, l'incidence rapportée des pneumonies a été de 2% aussi bien dans le bras Trelegy que dans le bras budésonide/formotérol.
Dans une étude de 52 semaines, menée chez au total 10 355 patients atteints de BPCO ayant des antécédents connus d'exacerbations modérées à sévères dans les 12 derniers mois (VEMS post-bronchodilatateur moyen à l'inclusion à 46% de la valeur prédite, ET de 15%) (étude CTT116855), l'incidence des pneumonies était de 8% sous furoate de fluticasone/uméclidinium/vilantérol (n = 4151), de 7% sous furoate de fluticasone/vilantérol (n = 4134) et de 5% sous uméclidinium/vilantérol (n = 2070). Une pneumonie d'issue fatale a été observée chez 12 des 4151 patients (3,5 pour 1000 patients-années) sous furoate de fluticasone/uméclidinium/vilantérol, chez 5 des 4134 patients (1,7 pour 1000 patients-années) sous furoate de fluticasone/vilantérol et chez 5 des 2070 patients (2,9 pour 1000 patients-années) sous uméclidinium/vilantérol.
L'incidence des pneumonies sous Trelegy Ellipta est comparable à celle observée sous FF/VI 100/25 dans les études cliniques sur la BPCO.
Données post-marketing:
Affections du système immunitaire
Rares: réactions d'hypersensibilité y compris anaphylaxie, angiœdème, urticaire et exanthème
Troubles du métabolisme et de la nutrition
Fréquence inconnue: hyperglycémie
Affections psychiatriques
Fréquence inconnue: anxiété
Troubles du système nerveux
Fréquence inconnue: tremblements
Affections oculaires
Occasionnels: vue floue, glaucome, douleurs oculaires
Fréquence inconnue: augmentation de la pression intraoculaire
Troubles fonctionnels du cœur
Fréquence inconnue: palpitations
Troubles fonctionnels de l'appareil locomoteur, du tissu conjonctif et des os
Fréquence inconnue: crampes musculaires
Affections du rein et des voies urinaires
Rares: rétention urinaire, dysurie
L'annonce d'effets secondaires présumés après l'autorisation est d'une grande importance. Elle permet un suivi continu du rapport bénéfice-risque du médicament. Les professionnels de santé sont tenus de déclarer toute suspicion d'effet secondaire nouveau ou grave via le portail d'annonce en ligne ElViS (Electronic Vigilance System). Vous trouverez des informations à ce sujet sur www.swissmedic.ch.
SurdosageOn ne dispose pas de données d'études cliniques sur le surdosage de Trelegy Ellipta.
Signes et symptômes
Un surdosage de Trelegy Ellipta peut causer des signes et symptômes correspondant aux effets pharmacologiques des différents composants ou aux effets observés lors d'un surdosage d'autres agonistes bêta-2 adrénergiques ou anticholinergiques, ainsi qu'aux effets de classe connus des corticostéroïdes inhalés (voir "Mises en garde et précautions" et "Pharmacodynamique" ).
Traitement
Aucun traitement spécifique n'est disponible pour le cas d'un surdosage de Trelegy Ellipta. Lors d'un surdosage, le patient doit recevoir le traitement de soutien approprié et être surveillé en conséquence.
Un traitement bêtabloquant cardiosélectif ne doit être envisagé que lors d'un surdosage sévère de vilantérol, causant des symptômes cliniques sérieux et ne répondant pas aux mesures de soutien. Chez les patients ayant des antécédents connus de bronchospasmes, la prudence est de rigueur lors de l'utilisation d'un bêtabloquant cardiosélectif.
La marche à suivre ultérieure dépendra des exigences cliniques ou, le cas échéant, des recommandations du centre d'information toxicologique.
Propriétés/EffetsCode ATC
R03AL08
Mécanisme d'action
Le furoate de fluticasone, l'uméclidinium et le vilantérol sont des représentants de trois classes différentes de médicaments: un corticostéroïde synthétique, un antagoniste des récepteurs muscariniques de longue durée d'action (appelé aussi LAMA ou anticholinergique) et un agoniste bêta-2 adrénergique sélectif de longue durée d'action.
Furoate de fluticasone (FF)
Le FF est un corticostéroïde aux effets anti-inflammatoires puissants. On ignore quel est exactement le mécanisme d'action par lequel le FF influence les symptômes de la BPCO. Il est démontré que les corticostéroïdes ont un large spectre d'action influençant différents types de cellules (p.ex. éosinophiles, macrophages, lymphocytes) et différents médiateurs (p.ex. les cytokines et chimiokines impliquées dans les processus inflammatoires).
Uméclidinium (UMEC)
L'uméclidinium est un antagoniste des récepteurs muscariniques (un anticholinergique) de longue durée d'action. L'uméclidinium exerce une action bronchodilatatrice par inhibition compétitive de la liaison de l'acétylcholine aux récepteurs muscariniques des muscles lisses bronchiques. Il présente in vitro une réversibilité lente au niveau du récepteur muscarinique M3 humain et – dans les modèles précliniques – une durée d'action prolongée in vivo lors d'une administration directement dans les poumons.
Vilantérol (VI)
Le VI est un agoniste bêta-2 adrénergique sélectif de longue durée d'action (LABA).
Les effets pharmacologiques des agonistes bêta-2 adrénergiques tels que le VI reposent au moins partiellement sur la stimulation de l'adénylate cyclase intracellulaire, l'enzyme catalysant la transformation de l'adénosine triphosphate (ATP) en adénosine-3′,5′-monophosphate cyclique (AMPc). Les concentrations accrues d'AMPc provoquent une relaxation de la musculature lisse bronchique et inhibent la libération cellulaire (en particulier par les mastocytes) de médiateurs impliqués dans les réactions d'hypersensibilité immédiate.
Pharmacodynamique
Effets cardiovasculaires
Les effets du furoate de fluticasone/uméclidinium/vilantérol sur l'intervalle QT n'ont pas été examinés dans une étude spécifique approfondie (dite "thorough" ) sur le QT (TQT). Les études TQT menées avec FF/VI et UMEC/VI n'ont pas montré d'effets cliniquement significatifs sur l'intervalle QT aux doses cliniques de FF, UMEC et VI (voir ci-dessous).
Les effets de l'uméclidinium/vilantérol sur l'intervalle QT ont été examinés dans une étude sur le QT, contrôlée contre placebo et contre la moxifloxacine, menée auprès de 103 volontaires sains traités par 125/25 µg ou 500/100 µg d'uméclidinium/vilantérol une fois par jour pendant 10 jours. En comparaison avec le placebo, la différence moyenne maximale de l'allongement de l'intervalle QT (corrigé selon la formule de Fridericia, QTcF) après ajustement par rapport à la valeur initiale était de 4,3 (IC à 90%: 2,2 à 6,4) millisecondes 10 minutes après l'administration de 125/25 µg d'uméclidinium/vilantérol et de 8,2 (IC à 90%: 6,2 à 10,2) millisecondes 30 minutes après l'administration de 500/100 µg d'uméclidinium/vilantérol. À la dose de 125/25 µg d'uméclidinium/vilantérol, aucun effet cliniquement significatif sur l'allongement de l'intervalle QT (corrigé selon la formule de Fridericia) n'a été observé. Par ailleurs, aucun effet cliniquement significatif d'uméclidinium/vilantérol sur le rythme cardiaque n'a été observé sur un enregistrement Holter sur 24 heures, réalisé chez 281 patients traités par 125/25 µg d'uméclidinium/vilantérol une fois par jour pendant 12 mois.
Les effets du furoate de fluticasone/vilantérol sur l'intervalle QT ont été examinés dans une étude croisée en double aveugle, contrôlée contre placebo et traitement actif, menée auprès de 85 volontaires sains ayant reçu plusieurs doses. La différence moyenne maximale (limite supérieure de l'intervalle de confiance à 95%) du QTcF versus placebo après ajustement par rapport à la valeur initiale était respectivement de 4,9 (7,5) millisecondes et 9,6 (12,2) millisecondes 30 minutes après l'administration de 200/25 µg de furoate de fluticasone/vilantérol et de 800/100 µg de furoate de fluticasone/vilantérol. Une élévation dose-dépendante de la fréquence cardiaque a également été observée. La différence moyenne maximale (limite supérieure de l'intervalle de confiance à 95%) de la fréquence cardiaque versus placebo après ajustement par rapport à la valeur initiale était respectivement de 7,8 (9,4) battements/min et 17,1 (18,7) battements/min. 10 minutes après l'administration de 200/25 µg de furoate de fluticasone/vilantérol et de 800/100 µg de furoate de fluticasone/vilantérol.
L'évaluation centralisée des ECG de 911 patients atteints de BPCO et traités par furoate de fluticasone/uméclidinium/vilantérol pendant jusqu'à 24 semaines et d'un sous-groupe de 210 patients exposés pendant jusqu'à 52 semaines n'a montré aucun effet cliniquement significatif sur l'intervalle QTc.
Efficacité clinique
Étude 1
L'efficacité du furoate de fluticasone/uméclidinium/vilantérol (FF/UMEC/VI 100/62,5/25 µg) administré une fois par jour à des patients avec un diagnostic clinique de BPCO a été examinée dans un sous-groupe de patients inclus dans une étude de 24 semaines contrôlée contre traitement actif, avec une extension jusqu'à 52 semaines (CTT116853, FULFIL).
L'administration une fois par jour du FF/UMEC/VI 100/62,5/25 µg a entraîné une amélioration statistiquement significative de la fonction pulmonaire (définie comme la variation du VEMS résiduel à la semaine 24 par rapport à la valeur initiale; co-critère d'évaluation principal) par rapport au budésonide/formotérol (BUD/FOR) 400/12 µg administré deux fois par jour (voir Tableau 2). Les effets bronchodilatateurs du FF/UMEC/VI ont été observés à partir du premier jour de traitement et ont été maintenus pendant les 24 semaines de traitement.
À la semaine 24, le FF/UMEC/VI a entraîné, par rapport au BUD/FOR, une amélioration statistiquement significative de la qualité de vie liée à la santé (HRQoL), mesurée sur le score total du St George's Respiratory Questionnaire (SGRQ) (co-critère d'évaluation principal), sur l'analyse des répondeurs au score SGRQ, sur le score du COPD Assessment Test (CAT), sur l'analyse des répondeurs au score CAT, sur le score des symptômes respiratoires de la BPCO et ses scores annexes (Evaluating Respiratory Symptoms, E-RSTM: BPCO) au cours des semaines 21 à 24, sur le score focal de dyspnée (Transitional Dyspnoea Index, TDI) à la semaine 24, ainsi que sur la consommation de médication de secours, mesurée par le nombre moyen quotidien d'utilisations au cours des semaines 1 à 24 (voir Tableau 1).
Le FF/UMEC/VI a entraîné une réduction statistiquement significative du taux annuel d'exacerbations modérées/sévères (p.ex. nécessitant un traitement par des antibiotiques ou des corticostéroïdes, ou une hospitalisation; extrapolé à partir des données jusqu'à la semaine 24) par rapport au BUD/FOR. Sous FF/UMEC/FI, une réduction du risque d'exacerbation modérée/sévère a été observée par rapport au BUD/FOR (sur la base de l'analyse du délai de survenue de la première exacerbation) (voir Tableau 1).
Tableau 1: principaux critères d'efficacité jusqu'à la semaine 24 (étude CTT116853)
Étude CTT116853 FF/UMEC/VI100/62,5/2 BUD/FOR400/12 µg 2x Comparaison avec
5 µg 1x /j(n = 911) /j (n = 899) BUD/FOR
Différence entreles Rapport entreles
traitements(IC à traitements(IC à
95%)valeur p 95%)valeur p
VEMS résiduel (l) à 0,142 (0,0083) -0,029 (0,0085) 0,171(0,148, 0,194)p -
la semaine 24, <0,001
variation de la
moyenne des moindres
carrés par rapport
à la valeur initiale
(ET) a, b
Score SGRQ total à -6,6 (0,45) -4,3 (0,46) -2,2(-3,5, -1,0)p<0, -
la semaine 24, 001
variation de la
moyenne des moindres
carrés par rapport
à la valeur initiale
(ET) a, c
a Co-critères
d'évaluation princip
aux bDifférence
statistiquement
significative entre
les traitements
FF/UMEC/VI et
BUD/FOR, également
observée aux semaine
s 2, 4 et 12 cDiffér
ence statistiquement
significative
entre les traitement
s FF/UMEC/VI et
BUD/FOR, également
observée à la
semaine 4 Abréviatio
ns: 2x /j = deux
fois par jour; BUD
= budésonide; FOR =
formotérol; IC =
intervalle de
confiance; VEMS =
volume expiratoire
maximal par seconde;
l = litre; µg =
microgramme; n =
nombre de patients
dans la population
en intention de
traiter; 1x /j =
une fois par jour;
ET = écart type;
SGRQ = St George's
Respiratory Question
naire.
Dans un sous-groupe de patients (n = 430), les résultats de la fonction pulmonaire, la qualité de vie liée à la santé (HRQoL), les symptômes et exacerbations jusqu'à 52 semaines de traitement étaient similaires aux résultats obtenus à 24 semaines.
Étude 2 (CTT116855, IMPACT)
L'efficacité à long terme du furoate de fluticasone/uméclidinium/vilantérol (FF/UMEC/VI 100/62,5/25 µg) une fois par jour a été évaluée chez des patients atteints de BPCO avec des antécédents connus d'exacerbations modérées ou sévères dans les 12 mois précédents, dans le cadre d'une étude de 52 semaines contrôlée contre traitement actif, par rapport aux associations fixes de furoate de fluticasone/vilantérol (FF/VI 100/25 µg) et d'uméclidinium/vilantérol (UMEC/VI 62,5/25 µg) (randomisation selon un ratio 2:2:1) (étude CTT116855, IMPACT).
Les patients traités par FF/UMEC/VI ont présenté une réduction statistiquement significative du taux annuel d'exacerbations modérées/sévères sous le traitement (critère d'évaluation principal) par rapport au FF/VI et au UMEC/VI. Les résultats concernant le critère d'efficacité sont présentés dans le Tableau 2.
Tableau 2: principaux critères d'efficacité (étude CTT116855)
FF/UMEC/VI (n = FF/VI (n = 4134) UMEC/VI (n = 2070) FF/UMEC/VI vs FF/VI FF/UMEC/VI vs
4151) UMEC/VI
Taux d'exacerbations
modérées/sévèresa
Exacerbations par an 0,91 1,07 1,21
Réduction des 15% 10; 20 p < 0,001 25% 19; 30 p < 0,001
exacerbations (%)
IC à 95% Valeur p
Délai de survenue
de la première
exacerbation modérée
/sévère
Patients avec un 47% 49% 50%
événement (%)
Réduction du risque 14,8% 9,3; 19,9 p < 16,0% 9,4; 22,1 p <
(%) IC à 95% Valeur 0,001 0,001
p
Taux d'exacerbations
sévères
Exacerbations par an 0,13 0,15 0,19
Réduction des 13% -1; 24 p = 0,064 34% 22; 44 p < 0,001
exacerbations (%)
IC à 95% Valeur p
VEMS résiduel (l)
après 52 semaines
Variation de la 0,094 (0,004) -0,003 (0,004) 0,040 (0,006)
moyenne des MC par
rapport à la valeur
initiale (ET)
Différence entre 0,097 0,085; 0,109 0,054 0,039; 0,069
les traitements IC p < 0,001 p < 0,001
à 95% Valeur p
Score SGRQ total à
la semaine 52
Variation de la -5,5 (0,23) -3,7 (0,24) -3,7 (0,35)
moyenne des MC par
rapport à la valeur
initiale (ET)
Différence entre -1,8 -2,4; -1,1 p < -1,8 -2,6; -1,0 p <
les traitements IC 0,001 0,001
à 95% Valeur p
IC = intervalle de
confiance; VEMS =
volume expiratoire
maximal par seconde;
l = litre; MC =
moindres carrés
(least squared); n
= nombre de patients
dans la population
en intention de
traiter; ET =
erreur type (standar
d error); SGRQ =
St. George's Respira
tory Questionnaire.
a Critère d'évaluati
on principal
Par rapport au FF/VI et au UMEC/VI, les effets du FF/UMEC/VI sur la fonction pulmonaire, rapportés au VEMS résiduel (variation du VEMS résiduel par rapport à la valeur initiale), ont été observés à tous les points de mesure au cours de l'étude de 52 semaines (voir Figure 1).
Figure 1: variation de la moyenne des moindres carrés du VEMS résiduel par rapport à la valeur initiale (l)
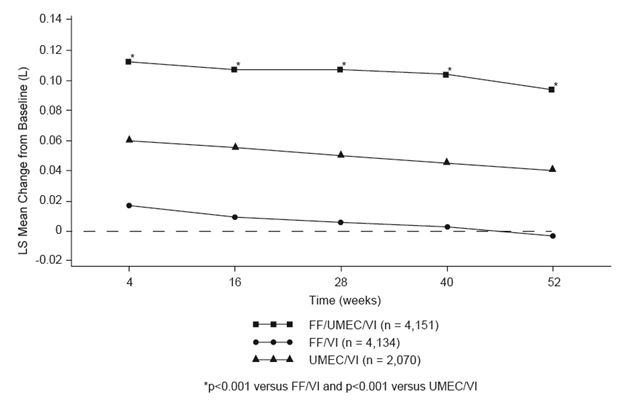
Résultats d'efficacité d'autres études complémentaires
L'étude de non-infériorité 200812, d'une durée de 24 semaines (n = 1055), a comparé l'association furoate de fluticasone/uméclidinium/vilantérol (FF/UMEC/VI 100/62,5/25 µg) administrée une fois par jour avec un inhalateur unique avec l'association furoate de fluticasone/vilantérol (100/25 µg) + uméclidinium (62,5 µg) administrés une fois par jour avec plusieurs inhalateurs, chez des patients ayant des antécédents connus d'exacerbations modérées à sévères au cours des 12 derniers mois. Dans cette étude, le FF/UMEC/VI était non-inférieur au FF/VI + UMEC en ce qui concerne l'amélioration du VEMS résiduel à la semaine 24 par rapport à la valeur initiale. Le seuil prédéfini de non-infériorité était de 50 ml.
Uméclidinium avec furoate de fluticasone/vilantérol
Dans deux études de 12 semaines, contrôlées contre placebo (200109 et 200110), menées auprès de patients adultes avec un diagnostic clinique de BPCO, l'ajout de l'uméclidinium (62,5 µg) au furoate de fluticasone/vilantérol (FF/VI) (100/25 µg) une fois par jour a entraîné des améliorations statistiquement et cliniquement significatives du critère d'évaluation principal, le VEMS résiduel au jour 85 (124 ml [IC à 95%: 93; 154]; p<0,001) dans l'étude 200109 et 122 ml [IC à 95%: 91, 152, p<0,001] dans l'étude 200110), par rapport au placebo plus FF/VI.
Étude de 12 mois avec le furoate de fluticasone/vilantérol
Deux études de 52 semaines, randomisées, en double aveugle et en groupes parallèles (HZC102970 et HZC102871), menées auprès de patients avec un diagnostic clinique de BPCO, ont comparé les taux annuels d'exacerbations modérées à sévères sous traitement par FF/VI et sous traitement par vilantérol, administrés une fois par jour. Les résultats d'une analyse intégrée de ces deux études ont montré que le traitement par FF/VI 100/25 µg une fois par jour a entraîné une réduction de 27% du taux annuel d'exacerbations modérées/sévères de BPCO par rapport au vilantérol (IC à 95%: 16, 37 [p<0,001]). Des réductions du risque d'exacerbations modérées/sévères (basées sur l'analyse du délai de survenue de la première exacerbation) et du taux d'exacerbations nécessitant l'utilisation de corticostéroïdes ont également été observées sous FF/VI 100/25 µg versus vilantérol, administrés une fois par jour.
PharmacocinétiqueLors de l'administration concomitante par inhalation de furoate de fluticasone (FF), d'uméclidinium (UMEC) et de vilantérol (VI) à des volontaires sains par un seul inhalateur, les propriétés pharmacocinétiques de chaque composant étaient similaires à celles observées lorsque chaque principe actif était administré dans l'association furoate de fluticasone/vilantérol ou dans l'association uméclidinium/vilantérol ou lorsque chaque composant était administré en monothérapie.
Les analyses pharmacocinétiques de population avec le FF/UMEC/VI ont été réalisées à partir des données pharmacocinétiques regroupées de 821 patients atteints de BPCO provenant des 3 études de phase III. Les concentrations systémiques (Cmax et AUC à l'état d'équilibre) de FF, UMEC et VI observées après l'administration de FF/UMEC/VI dans un inhalateur (triple association) étaient comprises dans un intervalle similaire à celui observé après l'administration de FF/VI plus UMEC dans deux inhalateurs, après l'administration des doubles associations (FF/VI et UMEC/VI) ainsi qu'après l'administration dans des inhalateurs séparés (FF, UMEC et VI).
Une analyse pharmacocinétique de population a montré une clairance apparente (CL/F) du FF plus élevée lors de l'administration de FF/VI que lors de l'administration de FF/UMEC/VI (1,42, IC à 95% 1,38, 1,45); mais cela est habituellement considéré comme ayant une pertinence clinique minime.
Absorption
Furoate de fluticasone
Après administration par inhalation de furoate de fluticasone/uméclidinium/vilantérol à des volontaires sains, la Cmax du furoate de fluticasone a été atteinte en 15 minutes. La biodisponibilité absolue du furoate de fluticasone lorsqu'il est administré par inhalation avec le vilantérol était en moyenne de 15,2%, principalement en raison de la résorption de la partie inhalée de la dose administrée, l'absorption orale étant négligeable. Après administration répétée par inhalation de furoate de fluticasone/vilantérol, l'état d'équilibre a été atteint en 6 jours, avec une accumulation d'un facteur de 1,6.
Uméclidinium
Après administration par inhalation de furoate de fluticasone/uméclidinium/vilantérol à des volontaires sains, la Cmax de l'uméclidinium a été atteinte en 5 minutes. La biodisponibilité absolue de l'uméclidinium inhalé était en moyenne de 13%, l'absorption orale étant négligeable. Après administration répétée par inhalation d'uméclidinium, l'état d'équilibre a été atteint en 7 à 10 jours, avec une accumulation d'un facteur de 2 au maximum.
Vilantérol
Après administration par inhalation de furoate de fluticasone/uméclidinium/vilantérol à des volontaires sains, la Cmax du vilantérol a été atteinte en 7 minutes. La biodisponibilité absolue du vilantérol administré par inhalation était en moyenne de 27%, l'absorption orale étant négligeable. Après administration répétée par inhalation de furoate de fluticasone/vilantérol, l'état d'équilibre a été atteint en 6 jours, avec une accumulation d'un facteur de 1,5 au maximum.
Distribution
Furoate de fluticasone
Après administration intraveineuse de furoate de fluticasone à des volontaires sains, le volume moyen de distribution était de 661 litres. In vitro, la liaison aux protéines plasmatiques humaines était > 99,6%.
Uméclidinium
Après administration intraveineuse d'uméclidinium à des volontaires sains, le volume moyen de distribution était de 86 litres. In vitro, la liaison aux protéines plasmatiques humaines était en moyenne de 89%.
Vilantérol
Après administration intraveineuse de vilantérol à des volontaires sains, le volume moyen de distribution était de 165 litres. In vitro, la liaison aux protéines plasmatiques humaines était en moyenne de 94%.
Métabolisme
Furoate de fluticasone
Des études in vitro ont montré que le furoate de fluticasone est principalement métabolisé par le CYP3A4 et qu'il est un substrat du transporteur glycoprotéine P (P-gp). Le furoate de fluticasone est principalement métabolisé par hydrolyse du groupe S-fluorométhyl carbothioate en métabolites ayant une activité corticoïde significativement réduite. L'exposition systémique aux métabolites est faible.
Uméclidinium
Des études in vitro ont montré que l'uméclidinium est principalement métabolisé par le CYP2D6 et est un substrat du transporteur P-gp. L'uméclidinium est principalement métabolisé par oxydation (hydroxylation, O-désalkylation), puis par conjugaison (glucuronidation, etc.), en divers métabolites dont l'activité pharmacologique est réduite ou n'a pas été évaluée. L'exposition systémique aux métabolites est faible.
Vilantérol
Des études in vitro ont montré que le vilantérol est principalement métabolisé par le CYP3A4 et est un substrat du transporteur P-gp. Le vilantérol est principalement métabolisé par O-désalkylation, en divers métabolites dont l'activité agoniste bêta-1 et bêta-2 adrénergique est significativement réduite. Dans une étude menée chez l'être humain avec le vilantérol radiomarqué administré par voie orale, les profils métaboliques plasmatiques ont mis en évidence un effet de premier passage important. L'exposition systémique aux métabolites est faible.
Élimination
Furoate de fluticasone
La demi-vie d'élimination plasmatique apparente du furoate de fluticasone administré par inhalation était en moyenne de 24 heures. Après administration intraveineuse, la demi-vie d'élimination était en moyenne de 15,1 heures. La clairance plasmatique après administration intraveineuse était de 65,4 litres/heure. L'excrétion urinaire représentait environ 2% de la dose administrée par voie intraveineuse. Après administration orale, le furoate de fluticasone était éliminé chez l'être humain principalement par métabolisation, les métabolites ayant été excrétés presque exclusivement dans les fèces, avec < 1% de la dose radioactive éliminée dans l'urine.
Uméclidinium
La demi-vie d'élimination plasmatique de l'uméclidinium était en moyenne de 19 heures après administration par inhalation pendant 10 jours. À l'état d'équilibre, 3 à 4% du principe actif étaient éliminés sous forme inchangée dans l'urine. La clairance plasmatique, après administration intraveineuse, était de 151 litres/heure. Après administration intraveineuse, environ 58% de la dose radiomarquée administrée étaient excrétés dans les fèces et environ 22% de la dose radiomarquée administrée étaient excrétés dans l'urine. Après administration intraveineuse, l'élimination de substances dérivées du principe actif dans les selles a permis de conclure à une sécrétion dans la bile. Après administration orale, 92% de la dose radiomarquée administrée étaient essentiellement excrétés dans les fèces. Moins de 1% de la dose administrée par voie orale (1% de la radioactivité retrouvée) était éliminé dans l'urine, ce qui suggère une absorption négligeable après administration orale.
Vilantérol
La demi-vie d'élimination plasmatique du vilantérol était en moyenne de 11 heures après administration par inhalation pendant 10 jours. La clairance plasmatique du vilantérol après administration intraveineuse était de 108 litres/heure. Après administration orale de vilantérol radiomarqué, 70% de la dose radiomarquée ont été excrétés dans l'urine et 30% dans les fèces. Le vilantérol a été principalement éliminé par métabolisation. Les métabolites ont été éliminés dans les fèces et dans l'urine.
Interactions avec d'autres médicaments
Une étude a été effectuée auprès de volontaires sains avec administration répétée de l'association de furoate de fluticasone/vilantérol (200/25 µg) et de kétoconazole (400 mg), un inhibiteur puissant du CYP3A4 et de la P-gp. L'administration concomitante a augmenté l'AUC (0-24) et la Cmax moyennes du furoate de fluticasone de respectivement 36% et 33%. L'augmentation de l'exposition au furoate de fluticasone a été associée à une réduction de 27% de la valeur moyenne pondérée de cortisol sérique de 0 à 24 heures. L'administration concomitante a augmenté l'AUC (0-24) et la Cmax moyennes du vilantérol de respectivement 65% et 22%. L'augmentation de l'exposition systémique au vilantérol n'a pas été associée à une augmentation des effets systémiques typiques des agonistes bêta-2 adrénergiques sur la fréquence cardiaque ou la kaliémie.
Le furoate de fluticasone, l'uméclidinium et le vilantérol sont des substrats de la P-gp. Une étude d'interactions avec administration répétée à des volontaires sains ayant reçu l'uméclidinium/vilantérol ou l'uméclidinium ainsi que du vérapamil (240 mg), un inhibiteur de la P-gp et un inhibiteur modéré du CYP3A4, n'a révélé aucun effet cliniquement significatif sur la pharmacocinétique du vilantérol ou de l'uméclidinium.
L'effet d'un génotype de métaboliseur lent pour le CYP2D6 sur la pharmacocinétique de l'uméclidinium à l'état d'équilibre a été évalué chez des volontaires sains (métaboliseurs normaux pour le CYP2D6 et métaboliseurs lents pour le CYP2D6). Aucune différence cliniquement significative d'exposition systémique à l'uméclidinium (500 µg, ce qui correspond à 8 fois la dose thérapeutique) n'a été observée après administration répétée par inhalation à des métaboliseurs normaux et lents pour le CYP2D6.
Cinétique pour certains groupes de patients
Une analyse pharmacocinétique de population (n = 821) a évalué l'influence de co-variables démographiques (couleur de la peau/origine ethnique, âge, sexe, poids) sur la pharmacocinétique du furoate de fluticasone, de l'uméclidinium et du vilantérol. Les altérations de la fonction rénale et de la fonction hépatique ont été évaluées dans des études séparées.
Patients âgés
Aucun effet cliniquement significatif nécessitant un ajustement posologique n'a été observé en fonction de l'âge.
Troubles de la fonction rénale
Le furoate de fluticasone/uméclidinium/vilantérol n'a pas été évalué chez des patients présentant une insuffisance rénale. Toutefois, des études ont été réalisées avec le furoate de fluticasone/vilantérol et avec l'uméclidinium/vilantérol.
Une étude sur la pharmacologie clinique de FF/VI suggère qu'une insuffisance rénale sévère (clairance de la créatinine < 30 ml/min) n'entraîne pas une exposition significativement accrue au FF ou au VI en comparaison avec les personnes sans insuffisance rénale et qu'elle ne provoque pas d'effets systémiques plus marqués du corticostéroïde ou de l'agoniste bêta-2 adrénergique.
Une étude menée chez des patients atteints d'insuffisance rénale sévère ayant reçu l'uméclidinium/vilantérol n'a pas montré d'augmentation de l'exposition systémique à l'uméclidinium ou au vilantérol (Cmax et AUC). Des études in vitro de liaison aux protéines, réalisées chez des patients atteints d'insuffisance rénale sévère et chez des volontaires sains n'ont montré aucune modification cliniquement significative de la liaison aux protéines plasmatiques.
Les effets de l'hémodialyse n'ont pas été étudiés.
Troubles de la fonction hépatique
Le furoate de fluticasone/uméclidinium/vilantérol n'a pas été évalué chez des patients présentant une insuffisance hépatique. Toutefois, des études ont été réalisées avec le furoate de fluticasone/vilantérol et avec l'uméclidinium/vilantérol.
Après l'administration répétée de FF/VI pendant 7 jours, les personnes souffrant d'insuffisance hépatique (stades Child-Pugh A, B ou C) ont présenté une exposition systémique accrue au FF, pouvant aller, d'après l'AUC(0-24), jusqu'au triple de l'exposition observée chez les personnes sans insuffisance hépatique. Chez les patients présentant une insuffisance hépatique modérée (stade Child-Pugh B), l'augmentation de l'exposition systémique au FF sous FF/VI 200/25 µg était associée à une réduction de 34% en moyenne des concentrations sériques de cortisol par rapport aux personnes sans insuffisance hépatique. Chez les patients présentant une insuffisance hépatique sévère (Child-Pugh C) et ayant reçu la dose faible de FF/VI 100/12,5 µg, aucune réduction du taux sérique de cortisol n'a été observée (augmentation de 10% de la concentration sérique de cortisol).
Après l'administration répétée de FF/VI pendant 7 jours, aucune augmentation significative de l'exposition systémique au VI (Cmax et AUC) n'a été observée chez les personnes présentant une insuffisance hépatique légère, modérée ou sévère (stades Child-Pugh A, B ou C).
Par comparaison avec les sujets sans insuffisance hépatique, aucun effet agoniste bêta-adrénergique systémique (fréquence cardiaque ou taux sériques de potassium) cliniquement significatif n'a été constaté chez les patients présentant une insuffisance hépatique légère à modérée (VI 25 µg) ou sévère (VI 12,5 µg).
Aucune augmentation de l'exposition systémique à l'uméclidinium ou au vilantérol (Cmax et AUC) n'a été constatée chez les patients présentant une insuffisance hépatique modérée. Des études in vitro de liaison aux protéines, réalisées chez des patients présentant une insuffisance hépatique modérée et chez des volontaires sains, n'ont montré aucune modification cliniquement significative de la liaison aux protéines.
L'uméclidinium n'a pas été étudié chez des patients présentant une insuffisance hépatique sévère.
Sexe, poids et IMC des patients
Aucune différence cliniquement significative nécessitant un ajustement posologique n'a été observée en fonction du sexe, du poids ou de l'indice de masse corporelle.
Ethnicité
Chez les patients d'Asie de l'Est (origine japonaise et d'Asie de l'Est) atteints de BPCO (n = 113) et ayant reçu le FF/UMEC/VI, les estimations de l'AUCss du furoate de fluticasone étaient en moyenne de 30% supérieures à celles observées chez les sujets caucasiens. Aucun indice ne laisse toutefois penser que cette augmentation de l'exposition systémique ait des effets cliniquement significatifs sur la concentration sérique de cortisol et l'élimination de cortisol dans l'excrétion urinaire sur 24 heures. Chez les patients atteints de BPCO, aucune influence de l'origine ethnique sur la pharmacocinétique de l'uméclidinium ou du vilantérol n'a été mise en évidence.
Aucune différence cliniquement significative de l'exposition au furoate de fluticasone, à l'uméclidinium et au vilantérol nécessitant un ajustement posologique n'a été observée en fonction de l'origine ethnique.
Autres groupes particuliers de patients
Concernant les autres caractéristiques des patients, une étude menée auprès des métaboliseurs lents pour le CYP2D6 n'a montré aucun effet cliniquement significatif du polymorphisme génétique du CYP2D6 sur l'exposition systémique à l'uméclidinium.
Données précliniquesLes effets pharmacologiques et toxicologiques observés dans les études précliniques sous furoate de fluticasone, uméclidinium ou vilantérol correspondent aux effets caractéristiques des glucocorticoïdes, des antagonistes des récepteurs muscariniques et des agonistes bêta-2 adrénergiques. L'administration concomitante de furoate de fluticasone, d'uméclidinium et de vilantérol à des chiens n'a pas entraîné de nouvelle toxicité significative ni d'exacerbation importante des effets attendus du furoate de fluticasone, de l'uméclidinium et du vilantérol administrés seuls.
Carcinogénicité, mutagénicité
Le FF n'a montré aucune génotoxicité dans une série d'études standard et n'a montré aucune carcinogénicité dans des études sur des rats et des souris avec administration par inhalation pendant la vie entière avec des expositions correspondant – d'après l'AUC – à respectivement 1,4 et 2,9 fois l'exposition humaine à la dose de 100 µg de furoate de fluticasone.
L'uméclidinium n'a montré aucune génotoxicité dans une série d'études standard sur des rats et des souris et n'a montré aucune carcinogénicité dans des études avec administration par inhalation pendant la vie entière avec des expositions correspondant – d'après l'AUC – à ≥ 20 fois (souris) ou ≥ 17 fois (rat) l'exposition clinique humaine à la dose de 62,5 µg d'uméclidinium.
Il ressort des études de génotoxicité que le VI ne présente pas de risques génotoxiques pour l'être humain. En accord avec les résultats obtenus pour d'autres agonistes bêta-2 adrénergiques, les études avec administration de VI par inhalation pendant la vie entière ont révélé des effets prolifératifs sur l'appareil reproducteur chez les rats et les souris femelles ainsi que sur l'hypophyse chez les rats. Aucune augmentation de l'incidence des cancers n'a été observée chez les rats et les souris à des expositions correspondant respectivement – d'après l'AUC – à 0,9 fois et à 22 fois l'exposition clinique humaine à la dose de 25 µg de vilantérol.
Toxicologie de la reproduction
Ni le FF, ni l'UMEC, ni le VI n'ont eu d'effets indésirables sur la fertilité masculine ou féminine chez le rat.
Le FF n'a pas été tératogène chez le rat et le lapin. Aux doses inhalées toxiques pour la mère, il a cependant provoqué des retards du développement chez le rat et des avortements chez le lapin. Aucun effet sur le développement des rats n'a été observé à des expositions correspondant – d'après l'AUC – à 6,6 fois l'exposition humaine à la dose de 100 µg. Le furoate de fluticasone n'a pas eu d'effets indésirables sur le développement prénatal et postnatal chez le rat.
L'uméclidinium n'a pas été tératogène chez le rat ou le lapin. Dans une étude prénatale et postnatale chez le rat, l'administration sous-cutanée d'uméclidinium a provoqué une diminution du gain de poids et de la consommation de nourriture chez les mères ainsi qu'une légère diminution du poids chez la progéniture avant le sevrage, lorsque les mères avaient été traitées à la dose de 180 µg/kg/jour (environ 61 fois l'exposition clinique humaine à la dose de 62,5 µg d'uméclidinium, sur la base de l'AUC).
Le VI n'a pas été tératogène chez le rat. Dans des études d'inhalation chez le lapin, le VI a provoqué des effets similaires à ceux d'autres agonistes bêta-2 adrénergiques (fentes palatines, paupières ouvertes, fusion des sternèbres et déformation par rotation/courbure de membres). Aucun effet n'a été observé lors d'une administration sous-cutanée à des expositions correspondant – d'après l'AUC – à 62 fois l'exposition humaine à la dose de 25 µg de vilantérol.
Le VI n'a pas eu d'effets indésirables sur le développement prénatal et postnatal chez le rat.
Remarques particulièresStabilité
Le médicament ne doit pas être utilisé au-delà de la date figurant après la mention "EXP" sur l'emballage.
Stabilité après ouverture
Durée de conservation après ouverture de la barquette de protection: 6 semaines.
La date d'ouverture de la barquette de protection doit être inscrite sur l'étiquette de l'inhalateur dès que celui-ci est retiré de la barquette de protection.
Remarques particulières concernant le stockage
Ne pas conserver au-dessus de 30°C et le conserver dans son emballage d'origine pour protéger le contenu de l'humidité. En cas de conservation de l'inhalateur au réfrigérateur, le laisser revenir à température ambiante pendant au moins une heure avant de l'utiliser.
Le film protecteur ne doit être retiré que lorsque l'on s'apprête à utiliser l'inhalateur pour la première fois.
Conserver hors de portée des enfants.
Remarques concernant la manipulation de l'inhalateur Ellipta
Voir la notice d'emballage pour un mode d'emploi détaillé de Trelegy Ellipta.
L'inhalateur Ellipta est fourni avec un sachet de dessiccant dans une barquette en film composite. La barquette protège l'inhalateur de l'humidité et ne doit donc être ouverte que lorsque l'on s'apprête à l'utiliser pour la première fois. Une fois que l'emballage a été ouvert, le sachet de dessiccant doit être jeté.
Lorsque l'inhalateur est sorti pour la première fois de son emballage, il est en position "fermé" . Il ne doit être ouvert que lorsque l'on s'apprête à inhaler une dose de médicament.
Si le couvercle de sécurité de l'inhalateur Ellipta est ouvert, puis refermé sans que le médicament soit inhalé, la dose sera perdue. Cette dose restera dans l'inhalateur en toute sécurité, mais ne pourra plus être inhalée. Ainsi, l'administration accidentelle d'une dose trop élevée ou d'une double dose en une seule inhalation est exclue.
Il n'est pas nécessaire de procéder à une vérification du fonctionnement correct ou à une préparation particulière de l'inhalateur Ellipta avant la première utilisation.
Remarque importante
Le compteur de doses indique le nombre de doses encore contenues dans le dispositif. Le compteur indique exactement 30 doses lors de la première utilisation de l'inhalateur. Il retire une unité du compte chaque fois que le couvercle a été ouvert. Lorsqu'il reste moins de 10 doses, la moitié du compteur devient rouge. Après l'inhalation de la dernière dose, la moitié du compteur de dose est rouge et le chiffre 0 apparaît. L'inhalateur est désormais vide. Si le couvercle est à nouveau ouvert, le témoin des doses deviendra entièrement rouge.
Numéro d’autorisation66808 (Swissmedic)
PrésentationTrelegy Ellipta 92/55/22
Emballage de 1 inhalateur Ellipta contenant 30 doses unitaires, B
Emballage de 3 inhalateurs Ellipta contenant 30 doses unitaires, B
Titulaire de l’autorisationGlaxoSmithKline AG
6340 Baar
Mise à jour de l’informationMars 2025
|