CompositionPrincipes actifs
Dose du matin:
Elexacaftor, tezacaftor, ivacaftor
Dose du soir:
Ivacaftor
Excipients
Comprimés pelliculés
Dose du matin:
Noyau du comprimé:
Hypromellose, succinate d'acétate d'hypromellose, laurilsulfate de sodium, croscarmellose sodique, cellulose microcristalline, stéarate de magnésium.
Pelliculage:
Hypromellose, hydroxypropylcellulose, dioxyde de titane, talc, oxyde de fer jaune, oxyde de fer rouge.
Chaque comprimé de 50 mg/25 mg/37,5 mg contient 1,34 mg de sodium.
Chaque comprimé de 100 mg/50 mg/75 mg contient 2,68 mg de sodium.
Dose du soir:
Noyau du comprimé:
Silice colloïdale, croscarmellose sodique, succinate d'acétate d'hypromellose, lactose monohydraté, stéarate de magnésium, cellulose microcristalline, laurilsulfate de sodium.
Pelliculage:
Cire de carnauba, indigotine, macrogol 3350, alcool polyvinylique, talc, dioxyde de titane.
Encre d'impression:
Hydroxyde d'ammonium, oxyde de fer noir, propylène glycol, gommes laques.
Chaque comprimé de 75 mg contient 0,90 mg de sodium et 83,6 mg de lactose monohydraté.
Chaque comprimé de 150 mg contient 1,82 mg de sodium et 167,2 mg de lactose monohydraté.
Granulés en sachet
Dose du matin:
Silice colloïdale, croscarmellose sodique, hypromellose, succinate d'acétate d'hypromellose, lactose monohydraté, stéarate de magnésium, mannitol, laurilsulfate de sodium, sucralose.
Chaque sachet de 80 mg/40 mg/60 mg contient au maximum 2,75 mg de sodium et 188,6 mg de lactose monohydraté.
Chaque sachet de 100 mg/50 mg/75 mg contient au maximum 3,44 mg de sodium et 235,7 mg de lactose monohydraté.
Dose du soir:
Silice colloïdale, croscarmellose sodique, succinate d'acétate d'hypromellose, lactose monohydraté, stéarate de magnésium, mannitol, laurilsulfate de sodium, sucralose.
Chaque sachet de 59,5 mg contient au maximum 1,18 mg de sodium et 87,3 mg de lactose monohydraté.
Chaque sachet de 75 mg contient au maximum 1,49 mg de sodium et 109,8 mg de lactose monohydraté.
Forme pharmaceutique et quantité de principe actif par unitéComprimés pelliculés
Comprimé d'elexacaftor 50 mg/tezacaftor 25 mg/ivacaftor 37,5 mg et comprimé d'ivacaftor 75 mg
Dose du matin:
Chaque comprimé pelliculé de 50 mg/25 mg/37,5 mg contient 50 mg d'elexacaftor, 25 mg de tezacaftor et 37,5 mg d'ivacaftor sous forme d'association fixe.
Comprimé oblong de couleur orange clair, portant la mention "T50" gravée sur une face et uni sur l'autre face (dimensions 6,4 mm × 12,2 mm).
Dose du soir:
Chaque comprimé pelliculé de 75 mg contient 75 mg d'ivacaftor.
Comprimé oblong de couleur bleu clair portant la mention "V 75" imprimée à l'encre noire sur une face et uni sur l'autre face (dimensions 12,7 mm × 6,8 mm).
Comprimé d'elexacaftor 100 mg/tezacaftor 50 mg/ivacaftor 75 mg et comprimé d'ivacaftor 150 mg
Dose du matin:
Chaque comprimé pelliculé de 100 mg/50 mg/75 mg contient 100 mg d'elexacaftor, 50 mg de tezacaftor et 75 mg d'ivacaftor sous forme d'association fixe.
Comprimé oblong de couleur orange, portant la mention "T100" gravée sur une face et uni sur l'autre face (dimensions 7,85 mm × 15,47 mm).
Dose du soir:
Chaque comprimé pelliculé de 150 mg contient 150 mg d'ivacaftor.
Comprimé oblong de couleur bleu clair portant la mention "V 150" imprimée à l'encre noire sur une face et uni sur l'autre face (dimensions 16,5 mm × 8,4 mm).
Granulés en sachet
Tous les granulés sont de couleur blanche à blanc cassé, édulcorés, non aromatisés et mesurent environ 2 mm de diamètre.
Elexacaftor 80 mg/tezacaftor 40 mg/ivacaftor 60 mg et ivacaftor 59,5 mg granulés en sachet.
Dose du matin:
Chaque sachet contient 80 mg d'elexacaftor, 40 mg de tezacaftor et 60 mg d'ivacaftor.
Dose du soir:
Chaque sachet contient 59,5 mg d'ivacaftor.
Elexacaftor 100 mg/tezacaftor 50 mg/ivacaftor 75 mg et ivacaftor 75 mg granulés en sachet
Dose du matin:
Chaque sachet contient 100 mg d'elexacaftor, 50 mg de tezacaftor et 75 mg d'ivacaftor.
Dose du soir:
Chaque sachet contient 75 mg d'ivacaftor.
Indications/Possibilités d’emploiTrikafta est indiqué dans le traitement des patients atteints de mucoviscidose âgés de 2 ans et plus porteurs d'au moins une mutation F508del du gène CFTR (cystic fibrosis transmembrane conductance regulator) ou d'une mutation du gène CFTR ayant montré une réponse selon les données cliniques et/ou in vitro (voir "Propriétés/effets" , tableau 7).
Posologie/Mode d’emploiLa prescription de Trikafta est réservée aux médecins expérimentés dans le traitement de la mucoviscidose. Si le génotype du patient n'est pas connu, un génotypage devra être réalisé pour confirmer la présence d'au moins une mutation F508del ou d'une mutation ayant montré une réponse selon les données cliniques et/ou in vitro.
Posologie usuelle
Adultes, adolescents et enfants âgés de 2 ans et plus
Tableau 1: Recommandations
posologiques pour les
patients âgés de 2 ans et
plus
Âge Poids Dose du matin Dose du soir
2 à < 6 ans 10 à < 14 kg Un sachet d'elexacaftor 80 Un sachet d'ivacafto
mg/tezacaftor 40 mg/ivacaft r 59,5 mg granulés
or 60 mg granulés
2 à < 6 ans ≥14 kg Un sachet d'elexacaftor Un sachet d'ivacafto
100 mg/tezacaftor 50 r 75 mg granulés
mg/ivacaftor 75 mg granulés
6 à < 12 ans < 30 kg Deux comprimés d'elexacafto Un comprimé d'ivacaf
r 50 mg/tezacaftor 25 tor 75 mg
mg/ivacaftor 37,5 mg
6 à < 12 ans ≥30 kg Deux comprimés d'elexacafto Un comprimé d'ivacaf
r 100 mg/tezacaftor 50 tor 150 mg
mg/ivacaftor 75 mg
12 ans et plus - Deux comprimés d'elexacafto Un comprimé d'ivacaf
r 100 mg/tezacaftor 50 tor 150 mg
mg/ivacaftor 75 mg
Les doses du matin et du soir doivent être prises à environ 12 heures d'intervalle avec un repas riche en graisses (voir "Mode d'administration" ).
Prise retardée
S'il s'est écoulé moins de 6 heures depuis l'heure de prise de la dose du matin ou du soir oubliée, le patient doit prendre la dose le plus tôt possible et poursuivre le traitement selon le schéma posologique habituel.
Si un délai de plus de 6 heures s'est écoulé depuis:
l'heure de prise de la dose du matin oubliée, le patient doit prendre la dose oubliée dès que possible et ne doit pas prendre la dose du soir. La dose du matin suivante doit être prise à l'heure habituelle;
l'heure de prise de la dose du soir oubliée, le patient ne doit pas prendre la dose oubliée. La dose du matin suivante doit être prise à l'heure habituelle.
Les doses du matin et du soir ne doivent pas être prises en même temps.
Mode d'administration
Voie orale.
Trikafta doit être pris avec un repas riche en graisses. Les repas ou collations riches en graisses sont ceux qui contiennent du beurre ou de l'huile ou bien des œufs, du beurre de cacahuètes, du fromage, des fruits à coque, du lait entier ou de la viande (voir "Pharmacocinétique" ).
La consommation d'aliments ou boissons contenant du pamplemousse doit être évitée pendant le traitement par Trikafta (voir "Interactions" ).
Comprimés pelliculés
Voie orale. Il convient de préciser aux patients qu'ils doivent avaler les comprimés entiers. Les comprimés ne doivent pas être croqués, cassés ou dissouts avant la prise.
Granulés en sachet
Chaque sachet est à usage unique.
Le contenu de chaque sachet de granulés doit être mélangé avec 5 ml d'aliment semi-liquide ou de liquide adapté à l'âge de l'enfant et ingéré immédiatement en totalité. L'aliment ou le liquide servant à la préparation du mélange doit être à température ambiante ou inférieure. Après le mélange, le médicament reste stable pendant une heure et doit donc être ingéré dans ce délai. Les aliments semi-liquides ou les liquides sont par exemple les compotes de fruits, les purées de légumes, les yaourts, la compote de pommes, l'eau, le lait ou les jus de fruits. Trikafta doit être administré immédiatement après un repas ou une collation riche en graisses.
Instructions posologiques particulières
Patients présentant des troubles de la fonction hépatique
Le traitement n'est pas recommandé chez les patients présentant une insuffisance hépatique modérée (Child-Pugh de classe B).
Chez les patients présentant une insuffisance hépatique modérée, l'utilisation de Trikafta ne doit être envisagée qu'en cas de nécessité médicale absolue et si les bénéfices escomptés prédominent sur les risques.
Il n'a pas été mené d'études chez des patients présentant une insuffisance hépatique sévère (Child-Pugh de classe C). Les patients présentant une insuffisance hépatique sévère ne doivent pas être traités par Trikafta.
Aucune adaptation de la posologie n'est à prévoir chez les patients présentant une insuffisance hépatique légère (Child-Pugh de classe A) (voir "Mises en garde et précautions" , "Effets indésirables" et "Pharmacocinétique" ).
Tableau 2: Recommand
ations en cas
d'utilisation chez
les patients présent
ant une insuffisance
hépatique
Âge Légère (Child-Pugh Modérée (Child-Pugh de classe B) Sévère (Child-Pugh
de classe A) de classe C)
2 à < 6 ans Pas d'adaptation de Utilisation non recommandée. Chez Ne doit pas être
la posologie les patients présentant une utilisé
insuffisance hépatique modérée,
l'utilisation de Trikafta ne doit
être envisagée qu'en cas de
nécessité médicale absolue et si
les bénéfices escomptés
prédominent sur les risques. Dans
ce cas, Trikafta doit être
utilisé avec précaution à une
dose réduite, comme suit: -Jours
1 à 3: un sachet d'elexacaftor/tez
acaftor/ivacaftor granulés chaque
jour -Jour 4: pas de prise -Jours
5 et 6: un sachet d'elexacaftor/te
zacaftor/ivacaftor granules
chaque jour -Jour 7: pas de prise
Le schéma posologique ci-dessus
doit être répété chaque semaine.
La dose du soir d'ivacaftor
granulés ne doit pas être prise.
6 ans et plus Pas d'adaptation de Utilisation non recommandée. Chez Ne doit pas être
la posologie les patients présentant une utilisé
insuffisance hépatique modérée,
l'utilisation de Trikafta ne doit
être envisagée qu'en cas de
nécessité médicale absolue et si
les bénéfices escomptés
prédominent sur les risques. Dans
ce cas, Trikafta doit être
utilisé avec précaution à une
dose réduite, comme suit: -Jour
1: deux comprimés d'elexacaftor/te
zacaftor/ivacaftor le matin -Jour
2: un comprimé d'elexacaftor/tezac
aftor/ivacaftor le matin
Poursuivre ensuite en alternant
les posologies du jour 1 et du
jour 2. La dose du soir
d'ivacaftor comprimés ne doit pas
être prise.
Patients présentant des troubles de la fonction rénale
Aucune adaptation de la posologie n'est recommandée chez les patients présentant une insuffisance rénale légère ou modérée. La prudence est recommandée chez les patients présentant une insuffisance rénale sévère ou en phase terminale (voir "Pharmacocinétique" ).
Administration concomitante avec des inhibiteurs du CYP3A
En cas d'utilisation concomitante d'inhibiteurs modérés du CYP3A (par exemple fluconazole, érythromycine) ou d'inhibiteurs puissants du CYP3A (par exemple kétoconazole, itraconazole, posaconazole, voriconazole, télithromycine et clarithromycine), la posologie doit être réduite selon les recommandations conformément au tableau 3 cidessous (voir "Mises en garde et précautions" et "Interactions" ).
En cas d'utilisation concomitante de ciprofloxacine, aucune adaptation de la posologie n'est recommandée, car aucun effet cliniquement pertinent sur la biodisponibilité de Trikafta n'est attendu (voir "Interactions" ).
Tableau 3: Schéma
posologique en cas
d'administration
concomitante de
Trikafta avec des
inhibiteurs modérés ou
puissants du CYP3A
Âge Inhibiteurs modérés du CYP3A Inhibiteurs puissants du CYP3A
2 à < 6 ans En alternance un jour sur deux: -Un Un sachet d'elexacaftor/tezacaft
sachet d'elexacaftor/tezacaftor/ivacaft or/ivacaftor granulés deux fois
or granulés le premier jour. -Un par semaine, à environ 3 à 4
sachet d'ivacaftor granulés le jours d'intervalle. Pas de
lendemain. Pas de prise de sachet prise de sachet d'ivacaftor
d'ivacaftor granulés le soir. granulés le soir.
6 ans et plus En alternance un jour sur deux: -Deux Deux comprimés d'elexacaftor/tez
comprimés d'elexacaftor/tezacaftor/ivac acaftor/ivacaftor deux fois par
aftor le premier jour. -Un comprimé semaine, à environ 3 à 4 jours
d'ivacaftor le lendemain. Pas de d'intervalle. Pas de prise de
prise de comprimé d'ivacaftor le soir. comprimé d'ivacaftor le soir.
Enfants
La sécurité et l'efficacité de Trikafta n'ont pas encore été établies chez les enfants âgés de moins de 2 ans (voir "Effets indésirables" et "Propriétés/Effets" ).
Patients âgés
Il n'a pas été inclus un nombre suffisant de patients âgés de 65 ans et plus dans les études cliniques de Trikafta pour déterminer si la réponse chez ces patients est différente de la réponse chez les patients plus jeunes.
Contre-indicationsHypersensibilité aux principes actifs ou à l'un des excipients (voir "Composition" ).
Mises en garde et précautionsAtteinte hépatique
Des cas d'insuffisance hépatique entraînant une transplantation du foie ont été rapportés chez des patients avec et sans maladie hépatique avancée préexistante au cours des 6 premiers mois de traitement. Trikafta doit être utilisé avec prudence chez les patients présentant une maladie hépatique avancée préexistante (comme la cirrhose, l'hypertension portale), sous étroite surveillance et uniquement si les bénéfices escomptés prédominent sur les risques (voir "Posologie/Mode d'emploi" , "Effets indésirables" et "Pharmacocinétique" ).
Augmentation des enzymes hépatiques
Des augmentations des transaminases sont fréquentes chez les patients atteints de mucoviscidose et ont été observées chez des patients avec ou sans maladie hépatique préexistante traités par Trikafta. Dans certains cas, ces augmentations, parfois sévères, étaient associées à une augmentation concomitante de la bilirubine totale. Dans les études de phase III, les augmentations des transaminases ont été plus fréquentes dans le groupe Trikafta que dans le groupe placebo. Il est donc recommandé de contrôler les taux de transaminases (ALAT et ASAT) et de bilirubine totale chez tous les patients avant l'instauration du traitement, tous les mois pendant les 6 premiers mois, tous les 3 mois pendant les 6 mois suivants, puis une fois par an. Une surveillance plus fréquente doit être envisagée chez les patients ayant des antécédents d'atteinte hépatique ou d'augmentations des transaminases.
Si un patient présente des signes ou symptômes cliniques indiquant une atteinte hépatique (par exemple jaunisse et/ou urine foncée, nausées ou vomissements inexpliqués, douleur dans la partie supérieure droite de l'abdomen ou anorexie), la prise de Trikafta doit être interrompue et les transaminases sériques et la bilirubine totale doivent être mesurées immédiatement. En cas de taux d'ALAT ou d'ASAT > 5 fois la limite supérieure de la normale [LSN] ou d'ALAT ou d'ASAT > 3 × LSN avec une bilirubine totale > 2 × LSN, le traitement doit être interrompu. Des analyses doivent être effectuées de façon très rapprochée jusqu'à normalisation. Après leur normalisation, la décision d'une éventuelle reprise du traitement doit tenir compte des risques encourus par rapport au bénéfice attendu (voir "Posologie/Mode d'emploi" , "Effets indésirables" et "Pharmacocinétique" ). Les patients qui reprennent leur traitement après une interruption doivent faire l'objet d'une surveillance étroite.
Insuffisance hépatique
Le traitement n'est pas recommandé chez les patients présentant une insuffisance hépatique modérée. Chez les patients présentant une insuffisance hépatique modérée, l'utilisation de Trikafta ne doit être envisagée qu'en cas de nécessité médicale absolue et si les bénéfices escomptés prédominent sur les risques. Dans ce cas, il doit être utilisé avec précaution à une dose réduite (voir tableau 2). Les patients présentant une insuffisance hépatique sévère ne doivent pas être traités par Trikafta (voir "Posologie/Mode d'emploi" , "Effets indésirables" et "Pharmacocinétique" ).
Dépression
Des cas de dépression (incluant idées suicidaires et tentatives de suicide), apparaissant généralement au cours des trois mois suivant l'instauration du traitement, ont été rapportés chez des patients traités par Trikafta et chez des patients ayant des antécédents de troubles psychiatriques. Dans certains cas, une amélioration des symptômes a été observée après une réduction de la dose ou l'arrêt du traitement. Les patients (et aidants) doivent être avertis de la nécessité de surveiller l'apparition d'une humeur dépressive, de pensées suicidaires ou de modifications inhabituelles du comportement et de prendre immédiatement avis auprès du médecin en cas de survenue de ces symptômes.
Interactions avec des médicaments
Inducteurs du CYP3A
L'utilisation concomitante d'inducteurs du CYP3A diminue significativement l'exposition systémique de l'ivacaftor et devrait diminuer les expositions systémiques de l'elexacaftor et du tezacaftor, ce qui peut entraîner une diminution de l'efficacité de Trikafta. Par conséquent, l'administration concomitante avec des inducteurs puissants du CYP3A n'est pas recommandée (voir "Interactions" ).
Inhibiteurs du CYP3A
L'administration concomitante d'inhibiteurs puissants ou modérés du CYP3A augmente les expositions systémiques de l'elexacaftor, du tezacaftor et de l'ivacaftor. La dose de Trikafta doit donc être réduite en cas d'utilisation concomitante d'inhibiteurs puissants ou modérés du CYP3A (voir "Interactions" et tableau 3 à la rubrique "Posologie/Mode d'emploi" ).
Cataractes
Des cas d'opacités du cristallin non congénitales sans répercussion sur la vision ont été rapportés chez des enfants et adolescents recevant des traitements comportant l'ivacaftor. Bien que d'autres facteurs de risque aient été présents dans certains cas (par exemple: corticothérapie, exposition à des rayonnements), un risque possible imputable au traitement par l'ivacaftor ne peut être exclu. Des examens ophtalmologiques avant et pendant le traitement sont recommandés en cas d'instauration du traitement par Trikafta chez des enfants et des adolescents (voir "Données précliniques" ).
Patients greffés
L'elexacaftor/tezacaftor/ivacaftor n'a pas été étudié chez les patients atteints de mucoviscidose ayant reçu une greffe d'organe. Par conséquent, l'utilisation chez les patients greffés n'est pas recommandée. Voir "Interactions" pour des informations sur les interactions avec la ciclosporine ou le tacrolimus.
Lactose
Ce médicament contient du lactose. Les patients présentant une intolérance au galactose, un déficit total en lactase ou un syndrome de malabsorption du glucose et du galactose (maladies héréditaires rares) ne doivent pas prendre ce médicament.
Sodium
Ce médicament contient moins de 1 mmol (23 mg) de sodium par dose journalière, c'est-à-dire qu'il est essentiellement "sans sodium" .
InteractionsInteractions pharmacocinétiques
Effets d'autres médicaments sur Trikafta
Inducteurs du CYP3A
L'elexacaftor, le tezacaftor et l'ivacaftor sont des substrats du CYP3A (l'ivacaftor est un substrat de forte affinité du CYP3A). L'utilisation concomitante d'inducteurs du CYP3A peut diminuer les expositions systémiques et donc entraîner une diminution de l'efficacité de Trikafta. L'administration concomitante d'ivacaftor et de rifampicine, un inducteur puissant du CYP3A, a diminué significativement de 89 % l'aire sous la courbe (ASC) de l'ivacaftor. Une diminution significative de l'exposition systémique de l'elexacaftor et du tezacaftor est également attendue en cas d'administration concomitante avec des inducteurs puissants du CYP3A. Par conséquent, l'association avec des inducteurs puissants du CYP3A n'est pas recommandée (voir "Mises en garde et précautions" ).
Les inducteurs puissants du CYP3A sont par exemple:
rifampicine, rifabutine, phénobarbital, carbamazépine, phénytoïne et millepertuis (Hypericum perforatum).
Inhibiteurs du CYP3A
L'administration concomitante d'itraconazole, un inhibiteur puissant du CYP3A, a augmenté de 2,8 fois l'ASC de l'elexacaftor et de 4 à 4,5 fois l'ASC du tezacaftor. L'administration concomitante d'itraconazole ou de kétoconazole a augmenté de respectivement 15,6 fois et 8,5 fois l'ASC de l'ivacaftor. La posologie de Trikafta doit être réduite en cas d'administration concomitante avec des inhibiteurs puissants du CYP3A (voir "Mises en garde et précautions" et tableau 3 à la rubrique "Posologie/Mode d'emploi" ).
Les inhibiteurs puissants du CYP3A sont par exemple:
kétoconazole, itraconazole, posaconazole et voriconazole;
télithromycine et clarithromycine.
Des modèles de simulations ont indiqué que l'administration concomitante avec des inhibiteurs modérés du CYP3A peut augmenter d'environ 1,9 à 2,3 fois l'ASC de l'elexacaftor et du tezacaftor. L'administration concomitante de fluconazole a augmenté de 2,9 fois l'ASC de l'ivacaftor. La posologie de Trikafta doit être réduite en cas d'administration concomitante avec des inhibiteurs modérés du CYP3A (voir "Mises en garde et précautions" et tableau 3 à la rubrique "Posologie/Mode d'emploi" ).
Les inhibiteurs modérés du CYP3A sont par exemple:
fluconazole;
érythromycine.
L'administration concomitante avec du jus de pamplemousse, qui contient un ou plusieurs composants inhibant modérément le CYP3A, peut augmenter l'exposition de l'elexacaftor, du tezacaftor et de l'ivacaftor. La consommation d'aliments ou de boissons contenant du pamplemousse doit être évitée pendant le traitement par Trikafta (voir "Posologie/Mode d'emploi" ).
Ciprofloxacine
L'utilisation concomitante de ciprofloxacine et de Trikafta n'a pas été étudiée. La ciprofloxacine n'a toutefois montré aucun effet cliniquement pertinent sur la biodisponibilité du tezacaftor ou de l'ivacaftor et aucun effet cliniquement pertinent sur la biodisponibilité de l'elexacaftor n'est attendu. C'est pourquoi, une adaptation de la posologie n'est pas nécessaire lors de l'utilisation concomitante de Trikafta et de ciprofloxacine.
Les effets des médicaments administrés de façon concomitante sur l'exposition de l'elexacaftor, du tezacaftor et/ou de l'ivacaftor sont présentés dans le tableau 4.
Tableau 4: Effet
d'autres médicaments
sur l'elexacaftor,
le tezacaftor et/ou
l'ivacaftor
Dose et schéma Effet sur la PK Rapport des moyennes
d'administration d'ELX, de TEZ et/ou géométriques (IC à
d'IVA 90 % de l'elexacafto
r, du tezacaftor et
de l'ivacaftor Pas
d'effet = 1,0
ASC Cmax
Itraconazole 200 mg TEZ 25 mg 1 × j + ↑ Tezacaftor 4,02(3,71; 4,63) 2,83(2,62; 3,07)
ttes les 12 h le IVA 50 mg 1 × j
jour 1, puis 200 mg
1 × j
↑ Ivacaftor 15,6(13,4; 18,1) 8,60(7,41; 9,98)
Itraconazole 200 mg ELX 20 mg + TEZ 50 ↑ Elexacaftor 2,83 (2,59; 3,10) 1,05(0,977; 1,13)
1 × j mg, dose unique
↑ Tezacaftor 4,51 (3,85; 5,29) 1,48 (1,33; 1,65)
Kétoconazole 400 mg IVA 150 mg, dose ↑ Ivacaftor 8,45 (7,14; 10,0) 2,65 (2,21; 3,18)
1 × j unique
Ciprofloxacine 750 TEZ 50 mg ttes les ↔ Tezacaftor 1,08(1,03; 1,13) 1,05(0,99; 1,11)
mg ttes les 12 h 12 h + IVA 150 mg
ttes les 12 h
↑ Ivacaftor* 1,17(1,06; 1,30) 1,18(1,06; 1,31)
Rifampicine 600 mg IVA 150 mg, dose ↓ Ivacaftor 0,114(0,097; 0,136) 0,200(0,168; 0,239)
1 × j unique
Fluconazole 400 mg, IVA 150 mg ttes les ↑ Ivacaftor 2,95(2,27; 3,82) 2,47(1,93; 3,17)
dose unique le jour 12 h
1, puis 200 mg 1 × j
↑ = augmentation, ↓
= diminution, ↔ =
pas de modification.
IC = intervalle de
confiance; ELX =
elexacaftor; TEZ =
tezacaftor; IVA =
ivacaftor; PK =
pharmacocinétique.
* Effet non clinique
ment significatif.
Effets de Trikafta sur d'autres médicaments
Substrats du CYP2C9
L'ivacaftor peut inhiber le CYP2C9. Par conséquent, la surveillance du rapport normalisé international (INR) est recommandée pendant l'administration concomitante de warfarine avec Trikafta. Les autres médicaments dont l'exposition systémique peut être augmentée par Trikafta sont notamment le glimépiride et le glipizide. Ces médicaments doivent être utilisés avec précaution.
Interactions potentielles avec les transporteurs
L'administration concomitante d'ivacaftor ou de tezacaftor/ivacaftor et de digoxine, un substrat ayant une forte affinité pour la Pgp, a entraîné une augmentation de l'ASC de la digoxine d'un facteur 1,3, ce qui correspond à une inhibition faible de la Pgp par l'ivacaftor. L'administration de Trikafta peut augmenter l'exposition systémique des médicaments substrats de forte affinité de la Pgp, ce qui peut augmenter ou prolonger leur effet thérapeutique ainsi que leurs effets indésirables. La prudence et une surveillance appropriée sont préconisées en cas d'administration concomitante avec la digoxine ou avec d'autres substrats de la Pgp à marge thérapeutique étroite tels que la ciclosporine, l'évérolimus, le sirolimus et le tacrolimus.
L'elexacaftor et M23-ELX inhibent la captation par OATP1B1 et OATP1B3 in vitro. L'association tezacaftor/ivacaftor a augmenté de 1,2 fois l'ASC de la pitavastatine, un substrat d'OATP1B1. L'administration concomitante de Trikafta peut augmenter les expositions systémiques des médicaments qui sont des substrats de ces transporteurs tels que les statines, le glibenclamide, le natéglinide et le répaglinide. La prudence et une surveillance adaptée sont préconisées en cas d'administration concomitante avec des substrats d'OATP1B1 et d'OATP1B3. La bilirubine est un substrat d'OATP1B1 et d'OATP1B3. Dans l'étude 445-102, de légères augmentations du taux moyen de bilirubine totale ont été observées (variation allant jusqu'à 4,0 µmol/l par rapport à la valeur initiale). Cette observation concorde avec l'inhibition in vitro des transporteurs de la bilirubine OATP1B1 et OATP1B3 par l'elexacaftor et M23-ELX.
Contraceptifs hormonaux
Trikafta a été étudié avec l'éthinylestradiol/lévonorgestrel et il n'a pas été mis en évidence d'effet cliniquement pertinent sur les expositions systémiques du contraceptif oral. Trikafta ne devrait pas avoir d'effet sur l'efficacité des contraceptifs oraux.
Les effets de l'elexacaftor, du tezacaftor et/ou de l'ivacaftor sur l'exposition des médicaments administrés de façon concomitante sont présentés dans le tableau 5.
Tableau 5: Effet de
l'elexacaftor, du
tezacaftor et/ou de
l'ivacaftor sur
d'autres médicaments
Dose et schéma Effet sur la PK de Rapport des moyennes
d'administration l'autre médicament géométriques (IC à
90 %) de l'autre
médicament Pas
d'effet = 1,0
ASC Cmax
Midazolam 2 mg, TEZ 100 mg 1 × ↔ Midazolam 1,12(1,01; 1,25) 1,13(1,01; 1,25)
dose orale unique j/IVA 150 mg ttes
les 12 h
Digoxine 0,5 mg, TEZ 100 mg 1 × ↑ Digoxine 1,30(1,17; 1,45) 1,32(1,07; 1,64)
dose unique j/IVA 150 mg ttes
les 12 h
Contraceptifs oraux ELX 200 mg 1 × ↑ Éthinylestradiol* 1,33 (1,20; 1,49) 1,26 (1,14; 1,39)
Éthinylestradiol 30 j/TEZ 100 mg 1 ×
µg/lévonorgestrel j/IVA 150 mg ttes
150 µg 1 × j les 12 h
↑ Lévonorgestrel* 1,23 (1,10; 1,37) 1,10 (0,985; 1,23)
Rosiglitazone 4 mg, IVA 150 mg ttes les ↔ Rosiglitazone 0,975(0,897; 1,06) 0,928(0,858; 1,00)
dose orale unique 12 h
Désipramine 50 mg, IVA 150 mg ttes les ↔ Désipramine 1,04(0,985; 1,10) 1,00(0,939; 1,07)
dose unique 12 h
↑ = augmentation, ↓
= diminution, ↔ =
pas de modification.
IC = intervalle de
confiance; ELX =
elexacaftor; TEZ =
tezacaftor; IVA =
ivacaftor; PK =
pharmacocinétique.
* Effet non clinique
ment significatif.
Grossesse, allaitementGrossesse
Il n'a pas été mené d'études spécifiques et bien contrôlées de Trikafta chez la femme enceinte. Les expérimentations animales menées avec chaque principe actif individuellement n'ont révélé aucune toxicité directe ayant une incidence sur la grossesse, le développement embryonnaire, le développement fœtal et/ou le développement postnatal (voir "Données précliniques" ). Par mesure de précaution, l'utilisation du traitement pendant la grossesse doit être évitée.
Allaitement
Des données limitées montrent une excrétion de l'elexacaftor, du tezacaftor et de l'ivacaftor dans le lait maternel humain. Un risque pour les nouveau-nés/nourrissons ne peut être exclu. Une décision doit être prise soit d'interrompre l'allaitement soit d'interrompre/de s'abstenir du traitement avec Trikafta en prenant en compte le bénéfice de l'allaitement pour l'enfant au regard du bénéfice du traitement pour la femme.
Fertilité
Il n'existe pas de données sur l'effet de l'elexacaftor, du tezacaftor et de l'ivacaftor sur la fertilité humaine. Dans les expérimentations animales, l'elexacaftor et l'ivacaftor ont eu un effet sur la fertilité chez le rat. Dans les expérimentations animales, le tezacaftor n'a pas eu d'effet sur le comportement d'accouplement et sur les paramètres de fertilité (voir "Données précliniques" ).
Effet sur l’aptitude à la conduite et l’utilisation de machinesL'influence de Trikafta sur l'aptitude à la conduite et l'utilisation de machines n'a pas été spécifiquement étudiée.
Effets indésirablesRésumé du profil des effets indésirables
Le profil des effets indésirables de Trikafta est fondé sur les données de 510 patients inclus dans deux études de phase III contrôlées en double aveugle d'une durée de traitement de 24 semaines et 4 semaines (études 445-102 et 445-103). Dans les deux études de phase III contrôlées, 257 patients âgés de 12 ans et plus au total ont reçu au moins une dose de Trikafta.
Dans l'étude 445-102, les pourcentages de patients ayant arrêté prématurément le traitement par le médicament expérimental en raison d'événements indésirables étaient de 1 % dans le groupe traité par Trikafta et de 0 % dans le groupe recevant le placebo.
Les effets indésirables graves survenus plus fréquemment chez les patients traités par Trikafta que chez ceux recevant le placebo étaient des événements de type rash, rapportés chez 3 patients (1,5 %) traités par Trikafta contre 1 patient (0,5 %) recevant le placebo. Les effets indésirables les plus fréquents (≥10 %) chez les patients traités par Trikafta étaient: céphalées (17,3%), diarrhée (12,9%) et infection des voies respiratoires supérieures (11,9%).
Le profil des effets indésirables de Trikafta était généralement similaire dans tous les sousgroupes de patients, y compris dans les analyses en fonction de l'âge, du sexe, du volume expiratoire maximum par seconde (VEMS) exprimé en pourcentage de la valeur théorique lors de l'inclusion ou de la région géographique.
Liste tabulée des effets indésirables
Le tableau 6 présente les effets indésirables observés sous elexacaftor/tezacaftor/ivacaftor en association avec l'ivacaftor, sous tezacaftor/ivacaftor en association avec l'ivacaftor et sous ivacaftor en monothérapie. Les effets indésirables rapportés avec Trikafta sont présentés selon la classification de fréquence MedDRA: très fréquents (≥1/10), fréquents (≥1/100 à <1 /10), occasionnels (≥1/1 000 à < 1/100), rares (≥1/10 000 à < 1/1 000), très rares (< 1/10 000) et inconnue (ne peut être estimée sur la base des données disponibles).
Tableau 6: Effets indésirables observés
lors de l'administration d'elexacaftor/t
ezacaftor/ivacaftor, tezacaftor/ivacafto
r et ivacaftor seul chez les
adolescents > 12 ans et les adultes
Classe de systèmes d'organes selon Effets indésirables Fréquence
MedDRA
Infections et infestations Infection des voies respiratoires Très fréquents
supérieures*, rhinopharyngite
Rhinite*, grippe* Fréquents
Troubles du métabolisme et de la Hypoglycémie* Fréquents
nutrition
Affections psychiatriques Dépression Inconnue
Affections du système nerveux Céphalées*, vertiges* Très fréquents
Affections de l'oreille et du labyrinthe Douleurs d'oreille, troubles de Fréquents
l'oreille, acouphènes, hyperémie du
tympan, troubles de l'équilibre
(troubles vestibulaires)
Oreilles bouchées Occasionnels
Affections respiratoires, thoraciques Douleurs oropharyngées, nez bouché* Très fréquents
et médiastinales
Rhinorrhée*, sinus obstrués, rougeur du Fréquents
pharynx, respiration anormale*
Sibilance* Occasionnels
Affections gastro-intestinales Diarrhée*, douleurs abdominales* Très fréquents
Nausées, douleurs abdominales Fréquents
supérieures*, flatulences*
Affections hépatobiliaires Augmentation des transaminases Très fréquents
Augmentation du taux d'alanine Fréquents
aminotransférase*, augmentation du taux
d'aspartate aminotransférase*
Affections de la peau et du tissu Rash* Très fréquents
sous-cutané
Acné*, prurit* Fréquents
Affections des organes de reproduction Masse dans le sein Fréquents
et du sein
Inflammation du sein, gynécomastie, Occasionnels
affection du mamelon douleur au niveau
du mamelon
Investigations Contamination bactérienne de Très fréquents
l'expectoration
Augmentation de la créatine Fréquents
phosphokinase dans le sang*
Hypertension* Occasionnels
* Effets indésirables observés lors des
études cliniques portant sur
elexacaftor/tezacaftor/ivacaftor en
association avec l'ivacaftor
Les données de sécurité issues des études cidessous concordaient avec les données de sécurité observées dans l'étude 445-102.
-Étude randomisée en double aveugle, contrôlée contre comparateur actif de 4 semaines menée chez 107 patients (étude 445-103).
-Étude de sécurité et d'efficacité en ouvert de 192 semaines (étude 445-105) menée chez des patients qui avaient participé aux études 445-102 ou 445-103.
-Étude randomisée en double aveugle, contrôlée contre comparateur actif de 8 semaines menée chez 258 patients (étude 445-104).
-Étude en ouvert de 24 semaines (étude 445-111) menée chez 75 patients âgés de 2 à moins de 6 ans.
-Une étude en ouvert de 24 semaines a été menée chez 66 patients âgés de 6 à moins de 12 ans (étude 445-106 partie B). Pour plus de détails sur les événements indésirables concernant le foie et la peau, voir ci-dessous.
-Une étude de sécurité et d'efficacité en ouvert en deux parties (partie A et partie B) de 192 semaines (étude 445-107) a été menée chez des patients âgés de 6 ans et plus qui avaient participé à l'étude 445-106, avec une analyse intermédiaire de la partie A portant sur 64 patients effectuée à la semaine 96 (60 patients ont terminé cette partie, 93,8 %). Par la suite, 48 patients (75,0 %) ont été transférés dans la partie B jusqu'à la semaine 192. 39 patients (60,9 %) ont terminé la partie B. Le profil de sécurité était comparable à celui observé dans l'étude 445-106, mais des cataractes/opacités du cristallin ont été observées plus fréquemment à long terme (chez 6 patients, 9,4 %).
-Étude randomisée en double aveugle, contrôlée contre placebo, de 24 semaines (étude 445-124) menée chez 307 patients âgés de 6 ans et plus.
Description de certains effets indésirables
Élévations des transaminases et atteinte hépatique
Dans l'étude 445-102, l'incidence de l'augmentation maximale des taux de transaminases (ALAT ou ASAT) > 8, > 5 ou > 3 × LSN était respectivement de 1,5 %, 2,5 % et 7,9 % chez les patients traités par Trikafta et de 1,0 %, 1,5 % et 5,5 % chez les patients recevant le placebo. L'incidence des augmentations des transaminases était de 10,9 % chez les patients traités par Trikafta et de 4,0 % chez les patients recevant le placebo. Aucun des patients traités par Trikafta n'a arrêté le traitement en raison d'une élévation des transaminases.
Pendant l'étude 445-106 partie B menée chez des patients âgés de 6 ans à moins de 12 ans, l'incidence de l'augmentation maximale des taux de transaminases (ALAT ou ASAT) > 8, > 5 ou >3 × LSN était respectivement de 0,0 %, 1,5 % et 10,6 %. Aucun des patients traités par Trikafta n'a présenté d'augmentation des transaminases > 3 × LSN associée à une augmentation de la bilirubine totale > 2 × LSN ni n'a arrêté le traitement en raison d'une élévation des transaminases. Pour les événements indésirables liés à une augmentation des transaminases, le délai moyen (ET) avant l'apparition du premier événement était de 52,1 (62,2) jours et la durée moyenne (ET) était de 15,3 (9,0) jours (voir "Mises en garde et précautions" ).
Pendant l'étude 445-111 menée chez des patients âgés de 2 à moins de 6 ans, l'incidence de l'augmentation maximale des taux de transaminases (ALAT ou ASAT) > 8, > 5, et >3 × LSN était respectivement de 1,3 %, 2,7 %, et 8,0 %. Aucun des patients traités par Trikafta n'a présenté d'augmentation des transaminases > 3 × LSN associée à une augmentation de la bilirubine totale > 2 × LSN ni n'a arrêté le traitement en raison d'une élévation des transaminases (voir "Mises en garde et précautions" ).
Dans l'étude 445-124 menée chez les patients âgés de 6 ans et plus, l'incidence de l'augmentation maximale des taux de transaminases (ALAT ou ASAT) > 8, > 5 et > 3 × LSN était respectivement de 2,0 %, 2,0 % et 6,3 % chez les patients traités par Trikafta et de 0 % chez les patients ayant reçu le placebo. Aucun des patients traités par Trikafta n'a présenté d'augmentation des transaminases > 3 × LSN associée à une augmentation de la bilirubine totale > 2 × LSN. Un patient a arrêté le traitement en raison d'une élévation des transaminases considérée comme un événement indésirable (voir "Mises en gardes et précautions" ).
Les effets indésirables suivants ont été observés dans le cadre de l'utilisation de Trikafta après l'autorisation.
-Insuffisance hépatique nécessitant une transplantation chez des patients avec ou sans maladie hépatique avancée préexistante (p.ex. cirrhose du foie, hypertension portale) (voir "Posologie/Mode d'emploi" , "Mises en garde et précautions" et "Pharmacocinétique" )
-Atteinte hépatique caractérisée par des augmentations simultanées des taux de transaminases (ALAT et ASAT) et de bilirubine totale chez les patients atteints de mucoviscidose avec ou sans maladie hépatique préexistante (voir "Posologie/Mode d'emploi" , "Mises en garde et précautions" et "Pharmacocinétique" ).
Événements de rash
Dans l'étude 445-102, l'incidence des rashs (par exemple rash, rash prurigineux) était de 10,9 % chez les patients traités par Trikafta et de 6,5 % chez les patients recevant le placebo. Les rashs étaient généralement d'intensité légère à modérée. L'incidence en fonction du sexe était de 5,8 % chez les patients de sexe masculin et de 16,3 % chez les patientes dans le groupe traité par Trikafta et de 4,8 % chez les patients de sexe masculin et 8,3 % chez les patientes recevant le placebo.
Dans l'étude 445-106 partie B portant sur 66 patients âgés de 6 à moins de 12 ans traités par Trikafta, l'incidence des rashs (par exemple rash, rash prurigineux) était de 24,2 % (n=16). Les événements indésirables spécifiques comprenaient l'éruption cutanée n=8 (12,1 %), l'éruption érythémateuse n=3 (4,5 %), l'éruption maculo-papuleuse n=2 (3,0 %), l'éruption papuleuse n=2 (3,0 %), l'exfoliation cutanée n=1 (1,5 %), l'urticaire n=1 (1,5 %). Un patient (1,5 %) a présenté une éruption cutanée qui a conduit à l'arrêt du Trikafta. Les autres patients ont présenté des éruptions cutanées qui ont régressé lors de la poursuite du traitement par Trikafta.
Dans l'étude 445-124, des éruptions cutanées sont survenues chez 55 (26,8 %) des patients du groupe recevant Trikafta et chez 3 (2,9 %) des patients du groupe recevant le placebo. La plupart des éruptions cutanées étaient d'intensité légère à modérée. Un patient (0,5 %) du groupe recevant Trikafta a présenté une éruption cutanée grave conduisant à l'arrêt du traitement. Aucun patient du groupe recevant le placebo n'a présenté d'éruption cutanée conduisant à l'arrêt du traitement.
Un rôle des contraceptifs hormonaux dans la survenue d'un rash ne peut être exclu. L'interruption du traitement par Trikafta et des contraceptifs hormonaux doit être envisagée chez les patientes sous contraceptifs hormonaux qui développent un rash. Après disparition du rash, il convient d'évaluer si la reprise du traitement par Trikafta sans contraceptifs hormonaux est appropriée. Si le rash ne récidive pas, la reprise des contraceptifs hormonaux peut être envisagée.
Augmentation de la créatine kinase sanguine
Dans l'étude 445-102, l'incidence de l'augmentation maximale du taux de créatine kinase > 5 × LSN était de 10,4 % chez les patients traités par Trikafta et de 5,0 % chez les patients recevant le placebo. Aucun des patients traités par Trikafta n'a arrêté le traitement en raison d'une augmentation de la créatine kinase.
Augmentation de la pression artérielle
Dans l'étude 445-102, l'augmentation maximale de la pression artérielle systolique et diastolique moyenne par rapport aux valeurs initiales était de respectivement 3,5 mmHg et 1,9 mmHg chez les patients traités par Trikafta (valeurs initiales: PAS 113 mmHg et PAD 69 mmHg) et de respectivement 0,9 mmHg et 0,5 mmHg chez les patients recevant le placebo (valeurs initiales: PAS 114 mmHg et PAD 70 mmHg).
Les pourcentages de patients ayant eu une pression artérielle systolique > 140 mmHg ou une pression artérielle diastolique > 90 mmHg à deux reprises au moins étaient respectivement de 5,0 % et 3,0 % chez les patients traités par Trikafta contre 3,5 % et 3,5 % chez les patients recevant le placebo.
Étude d'extension sur 192 semaines VX17-445-105
Dans l'étude VX17-445-105 ouverte, non contrôlée et à long terme, 400 patients (78,9 %) porteurs d'une mutation F508del et d'une mutation à fonction minimale et 107 patients (21,1 %) porteurs d'une mutation F508del homozygote ont été suivis pendant une durée allant jusqu'à 192 semaines. À la semaine 192, un patient n'avait jamais reçu le traitement, 356 patients (70,4 %) avaient terminé le traitement et 150 patients (29,6 %) avaient interrompu le traitement prématurément pour différentes raisons. Parmi eux, 13 patients (2,6 %) ont interrompu le traitement en raison d'événements indésirables, y compris 8 patients (1,6 %) en raison d'événements liés au foie et un (0,2 %) patient en raison d'une encéphalopathie hépatique.
Une augmentation du taux d'ALAT ou d'ASAT > 3, > 5 et > 8 × LSN a été constatée respectivement chez 63 (12,5 %), 36 (7,1 %) et 11 patients (2,2 %), dont 2 patients (0,4 %) présentant une augmentation du taux d'ALAT ou d'ASAT > 3 × LSN avec une augmentation concomitante et nouvelle de la bilirubine totale > 2 × LSN, un patient ayant déjà présenté une maladie de Meulengracht.
Des augmentations de la CK ≥2,5-≤5, > 5-≤10 et > 10 × LSN ont été constatées chez 69 (13,6 %), 38 (7,5 %) et 47 patients (9,3 %). Une augmentation de la CK a été considérée comme un événement indésirable chez 72 patients (14,2 %). Trois patients (0,6 %) ont subi une rhabdomyolyse sans atteinte rénale ou myoglobinurie.
Des éruptions cutanées sont survenues chez 89 patients (17,6 %). Un patient (0,2 %) a interrompu le traitement en raison d'une éruption cutanée.
La pression artérielle systolique moyenne a augmenté entre 2,7 et 5,6 mmHg, la pression artérielle diastolique moyenne a augmenté entre 1,5 et 3,6 mmHg. Des événements indésirables liés à l'augmentation de la pression artérielle ont été observés chez 20 patients (4,0 %).
Cinq patients (1,0 %) ont présenté des événements indésirables liés à une cataracte qui n'ont pas entraîné de modification de la posologie.
Étude d'extension sur 192 semaines VX19-445-107
Une étude d'extension en ouvert en deux parties (partie A et partie B) de 192 semaines en cours (étude VX19-445-107) est menée chez des patients âgés de 6 ans et plus qui avaient participé à l'étude 445-106, avec une analyse intermédiaire portant sur 64 patients effectuée à la semaine 96. Au total, 28 patients (43,8 %) étaient homozygotes pour la mutation F508del et 36 patients (56,3 %) étaient porteurs du génotype F508del/MF. Dans la partie A, 61 des 64 patients (95,3 %) ont reçu au moins une dose du médicament expérimental et ont terminé le traitement.
Une augmentation du taux d'ALAT ou d'ASAT > 3, > 5 et > 8 × LSN a été constatée respectivement chez 4 (6,3 %) patients, 1 (1,6 %) patient et aucun patient. Aucun patient n'a présenté d'augmentation du taux d'ALAT ou d'ASAT > 3 × LSN avec une augmentation concomitante et nouvelle de la bilirubine totale > 2 × LSN.
Chez la majorité des patients, le taux de CK est resté dans les valeurs normales. Quatre patients (6,3 %) avaient un taux de CK > 2,5 × LSN; aucun patient n'a eu un taux de CK > 5 × LSN. Aucun patient n'a présenté de rhabdomyolyse.
Des éruptions cutanées sont survenues chez 3 patients (4,7%). Aucun événement d'éruption cutanée n'a entraîné l'interruption ou l'arrêt du traitement par le médicament expérimental.
Il n'a pas été observé d'augmentations cliniquement significatives de la pression artérielle.
Six patients (9,4 %) ont présenté des événements indésirables liés à une cataracte qui n'ont pas entraîné de modification de la posologie. Ces cataractes ou opacités du cristallin n'avaient pas de répercussion sur la vision et n'étaient plus observées à la fin de l'étude chez trois de ces patients.
L'annonce d'effets secondaires présumés après autorisation est importante. Elle permet un suivi continu du rapport bénéficerisque du médicament. Les professionnels de santé sont tenus de déclarer toute suspicion d'effet secondaire nouveau ou grave via le portail d'annonce en ligne ElViS (Electronic Vigilance System). Vous trouverez des informations à ce sujet sur www.swissmedic.ch.
SurdosageTraitement
Il n'existe pas d'antidote spécifique en cas de surdosage de Trikafta. La conduite à tenir en cas de surdosage consiste en mesures de soutien, telles que la surveillance des fonctions vitales et de l'état clinique du patient.
Propriétés/EffetsCode ATC
R07AX32
Mécanisme d'action
L'elexacaftor et le tezacaftor sont des correcteurs de la protéine CFTR qui se lient à des sites différents sur la protéine CFTR. Comparativement à chaque molécule seule, ils ont un effet additif pour faciliter la maturation et le trafic cellulaires de la protéine CFTR afin d'augmenter la quantité de protéines CFTR amenées à la surface cellulaire. L'ivacaftor potentialise la probabilité d'ouverture (ou de régulation) du canal CFTR au niveau de la surface cellulaire.
L'effet combiné de l'elexacaftor, du tezacaftor et de l'ivacaftor est une augmentation de la quantité de protéines CFTR et de leur fonction à la surface cellulaire, entraînant une augmentation de l'activité du canal CFTR, mesurée par le transport des ions chlorures par le canal. Les résultats cliniques concordaient avec les résultats in vitro et indiquent que la présence d'une seule mutation F508del suffit pour obtenir une réponse clinique significative (voir "Efficacité clinique" ).
Test de transport des ions chlorure par le canal CFTR dans des cellules thyroïdiennes de rat Fisher (FRT) exprimant la protéine CFTR mutée
Des études électrophysiologiques réalisée dans une chambre d'Ussing ont évalué la réponse du transport des ions chlorures de la protéine CFTR mutée à l'elexacaftor/tezacaftor/ivacaftor. Un ensemble de lignées cellulaires FRT transfectées avec des mutations individuelles de CFTR a été utilisé à cet effet. L'elexacaftor/tezacaftor/ivacaftor a augmenté le transport des ions chlorures dans les cellules FRT exprimant les mutations CFTR sélectionnées.
Le seuil de réponse du transport de chlorure par le canal CFTR in vitro a été fixé à une augmentation nette d'au moins 10 % par rapport à la valeur normale initiale, car elle permet de prédire un bénéfice clinique ou laisse espérer un bénéfice clinique acceptable. Pour certaines mutations, l'ampleur de la variation nette in vitro par rapport à la valeur initiale du transport des ions chlorures médié par la protéine CFTR n'est pas corrélée à l'ampleur de la réponse clinique.
Le tableau 7 présente les mutations CFTR répondeuses dans les cellules FRT sur la base de la réponse clinique et/ou des données in vitro ou de l'extrapolation, qui indiquent que l'elexacaftor/tezacaftor/ivacaftor augmente le transport des ions chlorures d'au moins 10 % par rapport à la valeur normale initiale. La présence des mutations CFTR listées dans le tableau 7 ne doit pas être utilisée à la place d'un diagnostic de mucoviscidose ni comme seul facteur de prescription.
Tableau 7: liste
des mutations du
gène CFTR répondant
à l'elexacaftor/teza
caftor/ivacaftor
Mutations répondant
à Trikafta sur la
base des données
cliniques
2789+5G→A D1152H L997F P5L R1066H
3272-26A→G F508del L1077P R117C S945L
3849+10kbC→T G85E M1101K R347H T338I
A455E L206W N1303K R347P V232D
Mutations répondant
à Trikafta sur la
base des données in
vitro
3141del9 E588V G970D L165S R117G S589N
546insCTA E822K G1061R L320V R117H S737F
A46D F191V G1069R L346P R117L S912L
A120T F311del G1244E L453S R117P S977F
A234D F311L G1249R L967S R170H S1159F
A349V F508C G1349D L1324P R258G S1159P
A554E F508C;S1251N † H139R L1335P R334L S1251N
A1006E F575Y H199Y L1480P R334Q S1255P
A1067T F1016S H939R M152V R347L T1036N
D110E F1052V H1054D M265R R352Q T1053I
D110H F1074L H1085P M952I R352W V201M
D192G F1099L H1085R M952T R553Q V456A
D443Y G27R H1375P P67L R668C V456F
D443Y;G576A; R668C † G126D I148T P205S R751L V562I
D579G G178E I175V P574H R792G V754M
D614G G178R I336K Q98R R933G V1153E
D836Y G194R I502T Q237E R1070Q V1240G
D924N G194V I601F Q237H R1070W V1293G
D979V G314E I618T Q359R R1162L W361R
D1270N G463V I807M Q1291R R1283M W1098C
E56K G480C I980K R31L R1283S W1282R
E60K G551D I1027T R74Q S13F Y109N
E92K G551S I1139V R74W S341P Y161D
E116K G576A I1269N R74W; D1270N † S364P Y161S
E193K G576A;R668C † I1366N R74W;V201M † S492F Y563N
E403D G622D K1060T R74W;V201M; D1270N † S549N Y1014C
E474K G628R L15P R75Q S549R Y1032C
Mutations répondant
à Trikafta sur la
base d'une extrapola
tion de l'étude
445-124
711+3A→G E831X
† Mutations complexe
s/composées dans
lesquelles un seul
allèle du gène CFTR
présente plusieurs
mutations. Elles
existent indépendamm
ent des mutations
présentes sur
l'autre allèle.
Pharmacodynamique
Effets pharmacodynamiques
Effets sur le taux de chlorure sudoral
Dans l'étude 445-102 (patients porteurs d'une mutation F508del sur un allèle, avec sur le second allèle, une mutation entraînant l'absence de synthèse de la protéine CFTR ou la synthèse d'une protéine CFTR ne répondant pas à l'ivacaftor et au tezacaftor/ivacaftor in vitro [mutation à fonction minimale]), une diminution du taux de chlorure sudoral par rapport à la valeur initiale a été observée à la semaine 4 et s'est maintenue pendant la période de traitement de 24 semaines. La différence de la variation absolue moyenne du taux de chlorure sudoral de l'inclusion jusqu'à la semaine 24 avec Trikafta par rapport au placebo était de -41,8 mmol/l (IC à 95 %: -44,4; -39,3; P < 0,0001).
Dans l'étude 445-103 (patients homozygotes pour la mutation F508del), la différence de la variation absolue moyenne du taux de chlorure sudoral à la semaine 4 par rapport à la valeur initiale avec Trikafta par rapport au traitement par tezacaftor/ivacaftor associé à l'ivacaftor (tezacaftor/ivacaftor) était de -45,1 mmol/l (IC à 95 %: -50,1; -40,1; P < 0,0001).
Dans l'étude 445-104 (patients hétérozygotes pour la mutation F508del et porteurs sur le second allèle d'une mutation d'anomalie de régulation du canal chlorure [Gating] ou d'une mutation associée à une activité résiduelle de la protéine CFTR), après une période de pré-inclusion de 4 semaines pendant laquelle les patients ont reçu un traitement par ivacaftor ou tezacaftor/ivacaftor, la variation absolue moyenne du taux de chlorure jusqu'à la semaine 8 par rapport à la valeur initiale dans le groupe Trikafta était de -22,3 mmol/l (IC à 95 %: -24,5; -20,2; P < 0,0001). La différence entre le groupe Trikafta et le groupe contrôle (groupe ivacaftor ou tezacaftor/ivacaftor) était de -23,1 mmol/l (IC à 95 %: -26,1; -20,1; P < 0,0001).
Dans l'étude 445-106 partie B (ayant inclus des patients âgés de 6 à moins de 12 ans homozygotes pour la mutation F508del ou hétérozygotes pour la mutation F508del et porteurs d'une mutation à fonction minimale), la variation absolue moyenne du taux de chlorure jusqu'à la semaine 24 par rapport à la valeur initiale était de -60,9 mmol/l (IC à 95 %: -63,7; -58,2). Les valeurs mesurées pour le taux de chlorure sudoral ont été relevées aux jours de mesure prévus chez le nombre de patients suivant: inclusion n=62, jour 15 n=56, semaine 4 n=56, semaine 12 n=50, semaine 24 n=28.
Dans l'étude 445-111 (ayant inclus des patients âgés de 2 à moins de 6 ans homozygotes pour la mutation F508del ou hétérozygotes pour la mutation F508del et porteurs d'une mutation à fonction minimale), la variation absolue moyenne du taux de chlorure jusqu'à la semaine 24 par rapport à la valeur initiale était de -57,9 mmol/l (IC à 95 %: -61,3; -54,6).
Dans l'étude 445-124 (patients de 6 ans et plus, porteurs d'une mutation autre que F508del et répondant à l'elexacaftor/tezacaftor/ivacaftor grâce à laquelle ils étaient éligibles dans l'étude [voir tableau 8]), la variation absolue moyenne par rapport à la valeur initiale du taux de chlorure sudoral jusqu'à la semaine 24 était de -28,3 mmol/l comparé au placebo (IC à 95 %: -32,1, -24,5 mmol/l; p<0,0001).
Effets cardiovasculaires
Effet sur l'intervalle QT
À des doses allant jusqu'à 2 fois la dose maximale recommandée d'elexacaftor et jusqu'à 3 fois la dose maximale recommandée de tezacaftor et d'ivacaftor, il n'a pas été observé d'allongement de l'intervalle QT/QTc cliniquement significatif chez des volontaires sains.
Fréquence cardiaque
Dans l'étude 445-102, des diminutions moyennes de la fréquence cardiaque de 3,7 à 5,8 battements par minute (bpm) par rapport à la valeur initiale (76 bpm) ont été observées chez les patients traités par Trikafta.
Efficacité clinique
L'efficacité de Trikafta chez les patients atteints de mucoviscidose a été démontrée statistiquement dans quatre études de phase III contrôlées en double aveugle (études 445-102, 445-103, 445-104 et 445-124). Ces études ont inclus des patients atteints de mucoviscidose porteurs d'au moins une mutation F508del ou l'une des mutations répondant à Trikafta listées dans le tableau 8. Quatre études de phase III en ouvert et non contrôlées (étude 445-105, étude 445-106 partie B, étude 445-111 et étude 445-107) confirment les données d'efficacité. Trikafta a été développé en tant qu'association contenant de l'elexacaftor, du tezacaftor et de l'ivacaftor. Le bénéfice de l'elexacaftor seul et du tezacaftor seul par rapport au traitement en association n'a pas été évalué dans des études cliniques et ces principes actifs ne sont pas disponibles individuellement sous forme de médicaments.
L'étude 445-102 était une étude randomisée en double aveugle, contrôlée contre placebo, d'une durée de 24 semaines conduite chez des patients porteurs d'une mutation F508del sur un allèle, avec sur le second allèle une mutation à fonction minimale entraînant l'absence de synthèse de la protéine CFTR ou la synthèse d'une protéine CFTR ne répondant pas à l'ivacaftor et au tezacaftor/ivacaftor. Au total, 403 patients âgés de 12 ans et plus (âge moyen: 26,2 ans) ont été randomisés pour recevoir Trikafta ou le placebo. Les patients avaient un VEMS allant de 40 % à 90 % de la valeur théorique lors de la sélection. Lors de l'inclusion, le VEMS moyen était de 61,4 % de la valeur théorique (valeurs extrêmes: 32,3 % à 97,1 %).
L'étude 445-103 était une étude randomisée en double aveugle, contrôlée contre comparateur actif, d'une durée de 4 semaines menée chez des patients homozygotes pour la mutation F508del. Au total, 107 patients âgés de 12 ans et plus (âge moyen: 28,4 ans) ont reçu un traitement par tezacaftor/ivacaftor et ivacaftor (tezacaftor/ivacaftor) pendant une période de pré-inclusion en ouvert de 4 semaines, puis ont été randomisés pour recevoir Trikafta ou tezacaftor/ivacaftor pendant une période de traitement en double aveugle de 4 semaines. Les patients avaient un VEMS allant de 40 % à 90 % de la valeur théorique lors de la sélection. Le VEMS moyen lors de l'inclusion, après la période de pré-inclusion sous tezacaftor/ivacaftor, était de 60,9 % de la valeur théorique (valeurs extrêmes: 35,0 % à 89,0%).
L'étude 445-104 était une étude randomisée en double aveugle, contrôlée contre comparateur actif, d'une durée de 8 semaines menée chez des patients hétérozygotes pour la mutation F508del (F) et porteurs sur le second allèle d'une mutation d'anomalie de régulation du canal chlorure (Gating) ou d'une mutation associée à une activité résiduelle de la protéine CFTR (RF). Des patients âgés de 12 ans et plus ayant un VEMS allant de 40 % à 90 % de la valeur théorique lors de la sélection ont reçu un traitement par ivacaftor (patients F/Gating) ou par tezacaftor/ivacaftor (patients F/RF) pendant une période de pré-inclusion en ouvert de 4 semaines. Les patients porteurs du génotype F/R117H ont reçu l'ivacaftor pendant la période de pré-inclusion. Les patients ont ensuite été randomisés dans le groupe Trikafta ou ont poursuivi le traitement modulateur de la protéine CFTR reçu pendant la période de pré-inclusion. Lors de l'inclusion après la période de pré-inclusion, l'âge moyen était de 37,7 ans et le VEMS moyen était de 67,6 % de la valeur théorique (valeurs extrêmes: 29,7 % à 113,5 %).
L'étude 445-106 était une étude en ouvert et non contrôlée en deux parties d'une durée de 24 semaines menée chez 66 patients âgés de 6 à moins de 12 ans (âge moyen à l'inclusion: 9,3 ans) homozygotes pour la mutation F508del ou hétérozygotes pour la mutation F508del et porteurs d'une mutation à fonction minimale. La partie A a évalué la pharmacocinétique et les résultats préliminaires de sécurité, tandis que la partie B a examiné la sécurité, la tolérance, l'efficacité et la pharmacocinétique. Les patients pesant moins de 30 kg lors de l'inclusion (36 patients, 54,5 %) ont reçu deux comprimés d'elexacaftor 50 mg/tezacaftor 25 mg/ivacaftor 37,5 mg le matin et un comprimé d'ivacaftor 75 mg le soir. Les patients pesant 30 kg ou plus lors de l'inclusion (30 patients, 45,5 %) ont reçu deux comprimés d'elexacaftor 100 mg/tezacaftor 50 mg/ivacaftor 75 mg le matin et un comprimé d'ivacaftor 150 mg le soir. Les patients avaient un VEMS ≥40 % de la valeur théorique lors de la sélection (valeur initiale moyenne du VEMS: 88,8 %) (valeurs extrêmes: 39,0 % à 127,1 %) et pesaient au moins 15 kg (critère d'inclusion requis).
L'étude 445-111 était une étude en ouvert de 24 semaines menée chez des patients âgés de 2 à moins de 6 ans (âge moyen à l'inclusion 4,1 ans). Des patients porteurs d'au moins une mutation F508del ou d'une mutation connue pour répondre à l'elexacaftor/tezacaftor/ivacaftor étaient admissibles à l'étude. Au total, 75 patients homozygotes pour la mutation F508del ou hétérozygotes pour la mutation F508del et porteurs d'une mutation à fonction minimale ont été inclus et ont reçu le traitement selon une posologie déterminée en fonction du poids. Les patients pesant de 10 kg à moins de 14 kg lors de l'inclusion recevaient l'elexacaftor 80 mg/tezacaftor 40 mg/ivacaftor 60 mg une fois par jour (qd) le matin et l'ivacaftor 59,5 mg chaque soir. Les patients pesant au moins 14 kg lors de l'inclusion recevaient l'elexacaftor 100 mg qd/tezacaftor 50 mg qd/ivacaftor 75 mg toutes les 12 heures (q12h).
L'étude 445-124 était une étude randomisée, en double aveugle et contrôlée contre placebo, d'une durée de 24 semaines et menée en groupes parallèles chez des patients de 6 ans et plus. Les patients porteurs d'au moins une mutation éligible autre que F508del, qui répondaient à l'elexacaftor/tezacaftor/ivacaftor (voir tableau 8), et sans mutation d'exclusion (une autre mutation répondant à l'elexacaftor/tezacaftor/ivacaftor) étaient éligibles à l'étude.
Au total, 307 patients ont été inclus dans l'étude et ont reçu une posologie adaptée à leur âge et leur poids. Les patients âgés de ≥6 à < 12 ans et pesant < 30 kg au début de l'étude ont reçu l'elexacaftor 100 mg une fois par jour/tezacaftor 50 mg une fois par jour/ivacaftor 75 mg toutes les 12 heures (q12h). Les patients âgés de ≥6 à < 12 ans pesant ≥30 kg au début de l'étude ont reçu un traitement par elexacaftor 200 mg une fois par jour/tezacaftor 100 mg une fois par jour /ivacaftor 150 mg toutes les 12 heures (q12h). Les patients âgés de ≥12 ans au début de l'étude ont reçu l'elexacaftor 200 mg une fois par jour/tezacaftor 100 mg une fois par jour/ivacaftor 150 mg toutes les 12 heures (q12h). Lors de l'analyse préliminaire, les patients présentaient un VEMS ≥40 % et ≤100 % et étaient âgés de 6 ans ou plus. La valeur initiale moyenne du VEMS s'élevait à 67,7 % (plage: 34,0 %, 108,7 %)].
Tableau 8: Mutations
CFTR appropriées
répondant à l'elexac
aftor/tezacaftor/iva
caftor
2789+5G>A D1152H L997F R117C T338I
3272-26A>G G85E M1101K R347H V232D
3849+10kbC>T L1077P P5L R347P
A455E L206W R1066H S945L
Dans les études 445-102, 445-103, 445-104, 445-106, 445-111 et 445-124, les patients ont poursuivi leurs traitements de la mucoviscidose (par exemple bronchodilatateurs, antibiotiques inhalés, dornase alfa et solution de chlorure de sodium hypertonique), mais ont arrêté tous les traitements modulateurs de la protéine CFTR antérieurs, à l'exception des médicaments expérimentaux. Les patients avaient un diagnostic confirmé de mucoviscidose et répondaient aux critères d'inclusion de l'étude.
Dans les études 445-102, 445-103, 445-104, 445-106, 445-111 et 445-124, les patients qui présentaient une infection pulmonaire par des agents pathogènes associés à une dégradation plus rapide de l'état pulmonaire, incluant, mais sans s'y limiter, Burkholderia cenocepacia, Burkholderia dolosa ou Mycobacterium abscessus ou une anomalie d'un paramètre hépatique lors de la sélection (ALAT, ASAT, PA ou GGT ≥3 × LSN ou bilirubine totale ≥2 × LSN) étaient exclus des études. Les patients qui avaient un taux d'ALAT ou d'ASAT ≥2 × LSN étaient également exclus de l'étude 445-111.
Les patients ayant participé aux études 445-102 et 445-103 étaient admissibles pour entrer dans une étude d'extension en ouvert de 192 semaines (étude 445-105). Les patients ayant participé aux études 445-104, 445-106, partie B, 445-111 et 445-124 étaient admissibles pour entrer dans des études d'extension en ouvert.
Étude 445-102
Dans l'étude 445-102, le critère d'évaluation principal était la variation absolue moyenne du VEMS exprimé en pourcentage de la valeur théorique de l'inclusion jusqu'à la semaine 24. Par rapport au placebo, le traitement par Trikafta a entraîné une amélioration statistiquement significative de 14,3 % du VEMS (IC à 95 %: 12,7; 15,8; P < 0,0001) (voir tableau 9). L'amélioration moyenne du VEMS a été observée lors de la première évaluation le jour 15 et a persisté pendant toute la période de traitement de 24 semaines (voir figure 1). Des améliorations du VEMS exprimé en pourcentage de la valeur théorique ont été observées quels que soient l'âge, le VEMS à l'inclusion, le sexe et la région géographique. Au total, 18 patients recevant Trikafta avaient un VEMS < 40 % de la valeur théorique lors de l'inclusion. La sécurité et l'efficacité dans ce sousgroupe concordaient avec celles observées dans la population totale de l'étude. Voir le tableau 9 pour une synthèse des résultats sur le critère d'évaluation principal et les principaux critères secondaires.
Tableau 9: Analyses du critère
d'efficacité principal et des
principaux critères secondaires,
population complète d'analyse
(étude 445-102)
Analyse Statistique Placebo N = 203 Trikafta N = 200
Critère d'efficacité principal
Variation absolue du VEMS de Différence entre S/O S/O -0,4 (0,5) 14,3 (12,7; 15,8) P
l'inclusion jusqu'à la semaine 24 les traitements (IC < 0,0001 13,9 (0,6)
(%) à 95 %) Valeur de P
Variation intragroup
e (ES)
Principaux critères d'efficacité
secondaires
Variation absolue du VEMS à la Différence entre S/O S/O -0,2 (0,6) 13,7 (12,0; 15,3)
semaine 4 par rapport à la valeur les traitements (IC P < 0,0001 13,5
initiale (%) à 95 %) Valeur de P (0,6)
Variation intragroup
e (ES)
Nombre d'exacerbations Nombre d'événements 113 (0,98) S/O S/O 41 (0,37) 0,37
pulmonaires de l'inclusion (taux d'événements (0,25; 0,55) P <
jusqu'à la semaine 24‡ annuel††) Rapport 0,0001
des taux (RR) (IC à
95 %) Valeur de P
Variation absolue du taux de Différence entre S/O S/O -0,4 (0,9) -41,8 (-44,4;
chlorure sudoral par rapport à la les traitements (IC -39,3) P < 0,0001
valeur initiale jusqu'à la à 95 %) Valeur de P -42,2 (0,9)
semaine 24 (mmol/l) Variation intragroup
e (ES)
Variation absolue du score du Différence entre S/O S/O -2,7 (1,0) 20,2 (17,5; 23,0) P
domaine respiratoire CFQ-R de les traitements (IC < 0,0001 17,5 (1,0)
l'inclusion jusqu'à la semaine 24 à 95 %) Valeur de P
(points) Variation intragroup
e (ES)
Variation absolue de l'IMC à la Différence entre S/O S/O 0,09 (0,07) 1,04 (0,85; 1,23) P
semaine 24 par rapport à la les traitements (IC < 0,0001 1,13 (0,07)
valeur initiale (kg/m2) à 95 %) Valeur de P
Variation intragroup
e (ES)
Variation absolue du taux de Différence entre S/O S/O 0,1 (1,0) -41,2 (-44,0;
chlorure sudoral à la semaine 4 les traitements (IC -38,5) P < 0,0001
par rapport à la valeur initiale à 95 %) Valeur de P -41,2 (1,0)
(mmol/l) Variation intragroup
e (ES)
Variation absolue du score du Différence entre S/O S/O -1,9 (1,1) 20,1 (16,9; 23,2) P
domaine respiratoire CFQ-R à la les traitements (IC < 0,0001 18,1 (1,1)
semaine 4 par rapport au score à 95 %) Valeur de P
initial (points) Variation intragroup
e (ES)
VEMS: volume expiratoire maximum
par seconde exprimé en
pourcentage de la valeur
théorique; IC: intervalle de
confiance; ES: erreur standard;
S/O: sans objet; CFQ-R: Cystic
Fibrosis Questionnaire Revised,
(questionnaire révisé spécifique
de la mucoviscidose), IMC: indice
de masse corporelle. ‡ Une
exacerbation pulmonaire était
définie comme une modification de
l'antibiothérapie (intraveineuse,
inhalée ou orale) en raison de la
présence d'au moins 4 des 12
signes/symptômes sino-pulmonaires
prédéfinis. †† Taux annuel
d'événements estimé calculé sur
la base de 48 semaines par an.
Figure 1: Variation absolue du VEMS exprimé en pourcentage de la valeur théorique par rapport à la valeur initiale lors de chaque visite dans l'étude 445-102
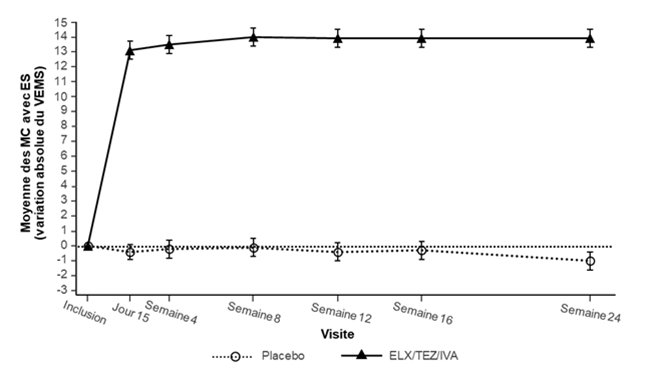
ES: erreur standard; ELX/TEZ/IVA: elexacaftor/tezacaftor/ivacaftor.
Étude 445-103
Dans l'étude 445-103, le critère d'évaluation principal était la variation absolue moyenne du VEMS exprimé en pourcentage de la valeur théorique à la semaine 4 de la période de traitement en double aveugle par rapport à la valeur initiale. Par rapport au traitement par tezacaftor/ivacaftor et ivacaftor (tezacaftor/ivacaftor), le traitement par Trikafta a induit une amélioration statistiquement significative de 10,0 % du VEMS exprimé en pourcentage de la valeur théorique (IC à 95 %: 7,4; 12,6; P < 0,0001) (voir tableau 10). Des améliorations du VEMS exprimé en pourcentage de la valeur théorique ont été observées quels que soient l'âge, le sexe, le VEMS initial et la région géographique. Voir le tableau 10 pour une synthèse des résultats du critère d'évaluation principal et des principaux critères secondaires.$
Tableau 10: Analyses du critère
d’efficacité principal et des
principaux critères secondaires,
population complète d’analyse
(étude 445-103)
Analyse* Statistique Tezacaftor/ ivacafto Trikafta N = 55
r# N = 52
Critère d’efficacité principal
Variation absolue du VEMS à la Différence entre S/O S/O 0,4 (0,9) 10,0 (7,4 ; 12,6) P
semaine 4 par rapport à la valeur les traitements (IC < 0,0001 10,4 (0,9)
initiale (%) à 95 %) Valeur de P
Variation intragroup
e (ES)
Principaux critères d’efficacité
secondaires
Variation absolue du taux de Différence entre S/O S/O 1,7 (1,8) -45,1 (-50,1 ;
chlorure sudoral à la semaine 4 les traitements (IC -40,1) P < 0,0001
par rapport à la valeur initiale à 95 %) Valeur de P -43,4 (1,7)
(mmol/l) Variation intragroup
e (ES)
Variation absolue du score du Différence entre S/O S/O -1,4 (2,0) 17,4 (11,8 ; 23,0)
domaine respiratoire CFQ-R à la les traitements (IC P < 0,0001 16,0
semaine 4 par rapport au score à 95 %) Valeur de P (2,0)
initial (points) Variation intragroup
e (ES)
VEMS : volume expiratoire maximum
par seconde exprimé en
pourcentage de la valeur
théorique ; IC : intervalle de
confiance ; ES : erreur standard
; S/O : sans objet ; CFQ-R :
Cystic Fibrosis Questionnaire
Revised, (questionnaire révisé
spécifique de la mucoviscidose).
* La valeur initiale pour le
critère principal et les
principaux critères secondaires
est définie comme la valeur à la
fin de la période de pré-inclusion
de 4 semaines sous tezacaftor/iva
caftor et ivacaftor. # Traitement
par tezacaftor/ivacaftor et
ivacaftor.
Figure 2: Variation absolue du VEMS exprimé en pourcentage de la valeur théorique par rapport à la valeur initiale lors de chaque visite dans l'étude 445-103
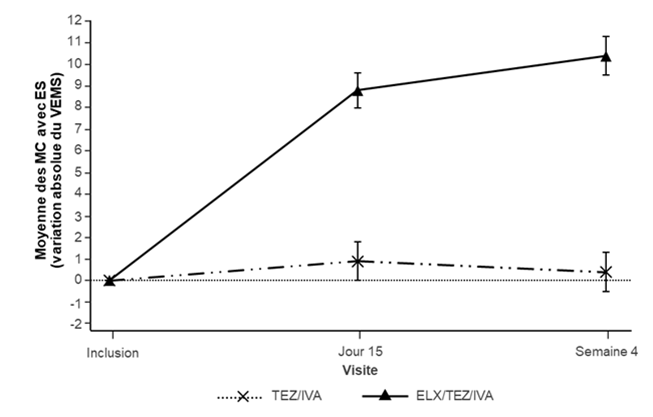
ES: erreur standard; TEZ/IVA: tezacaftor/ivacaftor; ELX/TEZ/IVA: elexacaftor/tezacaftor/ivacaftor
Étude 445-105
L'étude 445-105 était une étude d'extension en ouvert de 192 semaines visant à évaluer la sécurité et l'efficacité du traitement à long terme par Trikafta menée chez les patients qui avaient participé à l'étude 445-102 (N = 399) ou 445-103 (N = 107). Dans cette étude d'extension en ouvert, tous les patients ont reçu Trikafta pendant toute la durée de l'étude.
Dans l'étude 445-105, les patients des groupes contrôle des études principales ont présenté des améliorations des critères d'efficacité qui correspondaient à celles des patients ayant reçu Trikafta dans les études principales. Les patients du bras témoin et les patients ayant reçu le Trikafta dans les études principales ont présenté une amélioration durable du VEMS (voir figure 3 et figure 4) et d'autres critères d'efficacité (voir tableau 11). Les résultats en termes d'IMC et de Zscore d'IMC à la semaine 96 de traitement cumulé (semaine 96 dans l'étude 445-105) concordaient avec ceux observés chez les patients porteurs des génotypes étudiés dans l'étude 445-102.
Figure 3: Variation absolue du VEMS par rapport à la valeur initiale à chaque visite d'étude de l'étude 445-102 et de l'étude 445-105 chez les patients repris de l'étude 445-102*.
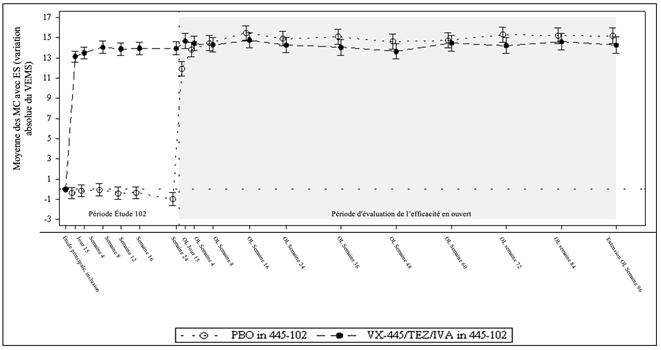
VEMS = volume expiratoire maximum par seconde (VEMS) exprimé en pourcentage de la valeur théorique; moyenne des MC = moyenne calculée par la méthode des moindres carrés; ES = erreur standard; OL = en ouvert.
Figure 4: Variation absolue du VEMS par rapport à la valeur initiale à chaque visite d'étude de l'étude 445-103 et de l'étude 445-105 chez les patients repris de l'étude 445-103.
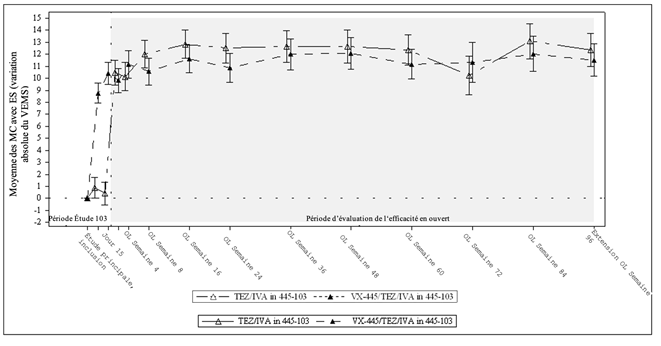
VEMS = volume expiratoire maximum par seconde exprimé en pourcentage de la valeur théorique; moyenne des MC = moyenne calculée par la méthode des moindres carrés; ES = erreur standard; OL = en ouvert.
Tableau 11: Étude
445-105 Analyse en
ouvert des critères
d'efficacité seconda
ires, population
complète d'analyse
(patients porteurs
du génotype F/MF ou
F/F)
Analyse Statistique À la semaine 192 de
l'étude 445-105
PBO dans l'étude ELX/TEZ/IVA dans TEZ/IVA dans l'étude ELX/TEZ/IVA dans
445-102 N = 203 l'étude 445-102 N = 445-103 N = 52 l'étude 445-103 N =
196 55
Variation absoluedu n 136 133 32 36
VEMS parrapport à
la valeurinitiale*
(%)
Moyenne des MC 15,3 13,8 10,9 10,7
IC à 95 % (13,7; 16,8) (12,3; 15,4) (8,2; 13,6) (8,1; 13,3)
Variation absoluedu n 133 128 31 38
taux dechlorure
sudoral par rapport
àla valeur initiale*
(mmol/)
Moyenne des MC -47,0 -45,3 -48,2 -48,2
IC à 95 % (-50,1; -43,9) (-48,5; -42,2) (-55,8; -40,7) (-55,1; -41,3)
Nombrede PExpendant Nombre d'événe-ments 385 71
la périoded'évaluati
on del'efficacité
cumuléede la triplea
ssociation†
Taux annueld'événe-m 0,21 (0,17; 0,25) 0,18 (0,12; 0,25)
entsestimé (IC à95
%)
VEMS = volume
expiratoire maximum
par seconde exprimé
en pourcentage de
la valeur théorique;
PEx = exacerbation
pulmonaire; moyenne
LS = moyenne calculé
e selon la méthode
des moindres carrés;
IC intervalle de
confiance; PBO =
placebo. * Valeur
initiale = valeur
initiale de l'étude
principale. † Pour
les patients randomi
sés dans le groupe
ELX/TEZ/IVA, la
période d'évaluation
de l'efficacité
cumulée de la
triple association
inclut les données
des études principal
es jusqu'à la
semaine 192 de
traitement dans
l'étude 445-105 (N
= 255, dont 4
patients qui ne
sont pas entrés
dans l'étude 445-105
). Pour les patients
randomisés dans le
groupe placebo ou
TEZ/IVA, la période
d'évaluation de
l'efficacité cumulée
de la triple
association inclut
uniquement les
données jusqu'à la
semaine 192 de
traitement dans
l'étude 445-105 (N
= 255).
Étude 445-104
Après une période de pré-inclusion de 4 semaines sous ivacaftor ou tezacaftor/ivacaftor, pour le critère d'évaluation principal, la variation absolue moyenne intragroupe du VEMS jusqu'à la semaine 8 par rapport à la valeur initiale, les résultats ont montré une amélioration statistiquement significative de 3,7 % dans le groupe Trikafta (IC à 95 %: 2,8; 4,6; P < 0,0001) (voir le tableau 12). L'amélioration moyenne du VEMS a été observée lors de la première évaluation le jour 15. Des améliorations globales du VEMS exprimé en pourcentage de la valeur théorique ont été observées quels que soient l'âge, le sexe, le VEMS à l'inclusion, la région géographique ou le groupe de génotype (F/Gating ou F/RF).
Voir le tableau 12 pour une synthèse des résultats sur le critère d'évaluation principal et les critères secondaires dans la population totale de l'étude.
Tableau 12: Analyses du critère
d'efficacité principal et des
principaux critères secondaires,
population complète d'analyse
(étude 445-104)
Analyse* Statistique Groupe contrôle# N Groupe Trikafta N =
= 126 132
Critère principal
Variation absolue du VEMS de Variation intragroup 0,2 (-0,7; 1,1) 3,7 (2,8; 4,6)
l'inclusion jusqu'à la semaine 8 e (IC à 95 %)
(%)
Valeur de P S/O P < 0,0001
Principaux critères secondaires
et autres critères secondaires
Variation absolue du taux de Variation intragroup 0,7 (-1,4; 2,8) -22,3 (-24,5; -20,2)
chlorure sudoral de l'inclusion e (IC à 95 %)
jusqu'à la semaine 8 (mmol/l)
Valeur de P S/O P < 0,0001
Variation absolue du VEMS jusqu'à Différence entre S/O 3,5 (2,2; 4,7)
la semaine 8 par rapport à la les traitements (IC
valeur initiale comparativement à 95 %)
au groupe contrôle (%)
Valeur de P S/O P < 0,0001
Variation absolue du taux de Différence entre S/O -23,1 (-26,1; -20,1)
chlorure sudoral jusqu'à la les traitements (IC
semaine 8 par rapport à la valeur à 95 %)
initiale comparativement au
groupe contrôle (mmol/l)
Valeur de P S/O P < 0,0001
Variation absolue du score du Variation intragroup 1,6 (-0,8; 4,1) 10,3 (8,0; 12,7)
domaine respiratoire CFQ-R de e (IC à 95 %)
l'inclusion jusqu'à la semaine 8
(points) ±
Variation absolue du score du Différence entre S/O 8,7 (5,3; 12,1)
domaine respiratoire CFQ-R les traitements (IC
jusqu'à la semaine 8 par rapport à 95 %)
au score initial comparativement
au groupe contrôle (points) ±
VEMS: volume expiratoire maximum
par seconde exprimé en
pourcentage de la valeur
théorique; IC: intervalle de
confiance; S/O: sans objet;
CFQ-R: Cystic Fibrosis
Questionnaire-Revised,
(questionnaire révisé spécifique
de la mucoviscidose). * La valeur
initiale pour le critère
principal et les critères
secondaires est définie comme la
valeur à la fin de la période de
préinclusion de 4 semaines sous
ivacaftor ou tezacaftor/ivacaftor.
# Groupe ivacaftor ou groupe
tezacaftor/ivacaftor. ± Il n'a
pas été effectué d'ajustement
pour multiplicité des scores du
domaine respiratoire CFQ-R selon
la procédure de tests
hiérarchisés.
Étude 445-106 partie B
Dans l'étude 445-106 partie B, le critère d'évaluation principal était la sécurité et la tolérance sur 24 semaines. Les critères d'évaluation secondaires étaient l'évaluation de l'efficacité et de la pharmacocinétique, y compris la variation absolue du VEMS (1er critère d'évaluation secondaire) et du taux de chlorure sudoral (2e critère d'évaluation secondaire, voir "Pharmacodynamique" ) par rapport à l'inclusion à la semaine 24 et le nombre d'exacerbations pulmonaires de l'inclusion à la semaine 24 comprise. En raison de la réalisation de l'étude 445-106 partie B pendant la pandémie de COVID 19, toutes les mesures n'ont pas pu être effectuées comme prévu initialement. Les mesures des critères d'évaluation secondaires ont été affectées à divers degrés par des mesures non effectuées. Le tableau 13 présente les principaux résultats d'efficacité secondaires dans l'analyse globale sur 24 semaines.
Les valeurs mesurées pour le VEMS ont été relevées les jours de mesure prévus chez le nombre de patients suivant: Inclusion n=62, jour 15 n=51, semaine 4 n=52, semaine 8 n=51, semaine 12 n=43, semaine 16 n=29, semaine 24 n=15.
Les valeurs mesurées pour le taux de chlorure sudoral ont été relevées aux jours de mesure prévus chez le nombre de patients suivant: inclusion n=62, jour 15 n=56, semaine 4 n=56, semaine 12 n=50, semaine 24 n=28.
Tableau 13: Analyses des critères secondaires d'évaluation de l'efficacité,
population complète d'analyse sur 24 semaines (étude 445-106, partie B)
Analyse Variation intragroup
e (IC à 95 %) pour
Trikafta N=66
Variation absolue du VEMS de l'inclusion jusqu'à la semaine 24 (%) 10,2 (7,9; 12,6)
Variation absolue du taux de chlorure sudoral de l'inclusion jusqu'à la -60,9 (-63,7; -58,2)
semaine 24 (mmol/l)
Nombre d'exacerbations pulmonaires jusqu'à la semaine 24‡ 4 (0,12) ††
IC: intervalle de confiance: VEMS: volume expiratoire maximum par seconde en
pourcentage de la valeur théorique. ‡ Une exacerbation pulmonaire était
définie comme une modification de l'antibiothérapie (intraveineuse, inhalée
ou orale) en raison de la présence d'au moins 4 des 12 signes/symptômes
sino-pulmonaires prédéfinis. †† Nombre d'événements et taux annuel
d'événements estimé déterminé sur la base de 48 semaines par an.
Étude 445-107
Une étude d'extension en ouvert en deux parties (partie A et partie B) de 192 semaines visant à évaluer la sécurité et l'efficacité du traitement à long terme par Trikafta a été menée chez des patients qui ont terminé l'étude 445-106. L'analyse de la partie A (96 semaines) portait sur 64 enfants âgés de 6 ans et plus et elle a montré des améliorations maintenues du VEMS et du taux de chlorure sudoral concordant avec les résultats observés dans l'étude 445-106. Par la suite, 48 patients (75,0 %) ont été transférés dans la partie B jusqu'à la semaine 192. 39 patients (60,9 %) ont terminé la partie B, confirmant l'efficacité continue. Les résultats des critères secondaires d'évaluation de l'efficacité des analyses intermédiaire et finale sont présentés dans le tableau 14.
Tableau 14: Analyses des critères
secondaires d'évaluation de
l'efficacité, population complète
d'analyse (N = 64) (étude 445-107)
Analyse Statistique Variation absolue à Variation absolue à
la semaine 96 par la semaine 192 par
rapport à la valeur rapport à la valeur
initiale initiale*
VEMS (%) n 45 27
Moyenne des MC 11,2 9,6
IC à 95 % (8,3; 14,2) (5,4; 13,7)
Taux de chlorure sudoral (mmol/l) n 56 35
Moyenne des MC -62,3 -57,9
IC à 95 % (-65,9; -58,8) (-63,3; -52,5)
Nombre de PEx pendant la période Nombre d'événements 7 11
d'évaluation de l'efficacité
cumulée de la triple association†
Taux annuel d'événements observé 0,04 0,045
VEMS = volume expiratoire maximum
par seconde exprimé en
pourcentage de la valeur
théorique; PEx = exacerbation
pulmonaire; MC = moindres carrés;
IC = intervalle de confiance. MC
= moindres carrés; IC =
intervalle de confiance. * Valeur
initiale = valeur initiale dans
l'étude principale. † La période
d'évaluation de l'efficacité
cumulée de la triple association
inclut les données des 66
patients ayant été inclus et qui
avaient reçu au moins une dose du
traitement dans l'étude
principale (étude 445-106 partie
B) et/ou qui ont reçu au moins
une dose pendant l'étude 445-107.
Étude 445-111
Le profil pharmacocinétique, la sécurité et l'efficacité de Trikafta chez les patients atteints de mucoviscidose âgés de 2 à moins de 6 ans sont étayés par des données d'études de Trikafta menées chez des patients âgés de 12 ans et plus (études 445-102, 445-103 et 445-104), avec des données supplémentaires issues d'une étude de phase III en ouvert de 24 semaines menée chez 75 patients âgés de 2 à moins de 6 ans (étude 445-111).
Dans l'étude 445-111, la sécurité et la tolérance (critère d'évaluation principal) ont été évaluées jusqu'à la semaine 24. Les objectifs secondaires comprenaient une évaluation de la pharmacocinétique et des critères d'efficacité incluant la variation absolue du taux de chlorure sudoral (voir "Pharmacodynamique" ) et de l'ICP2,5 jusqu'à la semaine 24 par rapport aux valeurs initiales. Voir le tableau 15 pour une synthèse des résultats des critères secondaires d'évaluation de l'efficacité.
Tableau 15: Analyses des critères secondaires
d'évaluation de l'efficacité, population complète
d'analyse (étude 445-111)
Analyse Statistique TrikaftaVariation
intra-groupe (IC à
95 %)
Variation absolue du taux de chlorure sudoral jusqu'à N* Moyenne des MC 75 -57,9 (-61,3;
la semaine 24 par rapport à la valeur initiale (mmol/l) (IC à 95 %) -54,6)
Variation absolue de l'ICP2,5 jusqu'à la semaine 24 par N Moyenne des MC 63‡ -0,83 (-1,01;
rapport à la valeur initiale (IC à 95 %) -0,66)
Nombre d'exacerbations pulmonaires jusqu'à la semaine N Nombre d'événement 75 12 (0; 32) ††
24** s (taux annuel
d'événements estimé)
IC: intervalle de confiance; ICP: index de clairance
pulmonaire. * N est le nombre de patients dans la
population complète d'analyse correspondante. ‡ ICP
évalué uniquement chez les patients âgés de 3 ans et
plus lors de la sélection. ** Des définitions
spécifiques à l'âge d'une exacerbation pulmonaire sont
utilisées pour les patients âgés de 2 à 5 ans et de 6
ans et plus. †† Nombre d'événements et taux annuel
d'événements estimé déterminé sur la base de 48
semaines par an.
Étude 445-124
L'efficacité et la sécurité de Trikafta ont été évaluées chez des patients âgés de 6 ans et plus, atteints de mucoviscidose et ne présentant pas la mutation F508del (étude 445-124).
Dans l'étude 445-124, le critère d'évaluation principal de l'efficacité était le VEMS. Les critères d'évaluation secondaires était la variation absolue du taux de chlorure sudoral, le score du domaine respiratoire du questionnaire CFQ-R, les paramètres de croissance (IMC, poids) et le nombre d'exacerbations pulmonaires. Le tableau 16 présente un résumé des critères d'évaluation de l'efficacité principal et secondaires.
Tableau 16: Analyses d'efficacité
principale et secondaires,
population d'analyse complète
(étude 445-124)
Analyse Paramètres statistiq Placebo N = 102 ELX/TEZ/IVA N = 205
ues
Analyse principale
Variation absolue du VEMS à la Différence entre S/O 9,2 (7,2; 11,3)
semaine 24 par rapport à la les traitements (IC
valeur initiale (en pourcentage) à 95 %)
Valeur de p S/O p<0,0001
Variation intragroupe (ES) -0,4 (0,8) 8,9 (0,6)
Analyses secondaires
Variation absolue du taux de Différence entre S/O -28,3 (-32,1, -24,5)
chlorure sudoral à la semaine 24 les traitements (IC
par rapport à la valeur initiale à 95 %)
(mmol/l)
Valeur de p S/O p<0,0001
Variation intragroupe (ES) 0,5 (1,6) -27,8 (1,1)
Variation absolue du score du Différence entre S/O 19,5 (15,5; 23,5)
domaine respiratoire du CFQ-R à les traitements (IC
la semaine 24 par rapport à la à 95 %)
valeur initiale (points)
Valeur de p S/O p<0,0001
Variation intragroupe (ES) -2,0 (1,6) 17,5 (1,2)
Variation absolue de l'IMC à la Différence entre S/O 0,47 (0,24; 0,69)
semaine 24 par rapport à la les traitements (IC
valeur initiale (kg/m2) à 95 %)
Valeur de p S/O p<0,0001
Variation intragroupe (ES) 0,35 (0,09) 0,81 (0,07)
Variation absolue du poids à la Différence entre S/O 1,3 (0,6; 1,9)
semaine 24 par rapport à la les traitements (IC
valeur initiale (kg) à 95 %)
Valeur de p S/O p<0,0001
Variation intragroupe (ES) 1,2 (0,3) 2,4 (0,2)
Nombre de PEx jusqu'à la semaine Rapport des taux S/O 0,28 (0,15; 0,51)
24 incluse (IC à 95 %)
Valeur de p S/O p<0,0001
Nombre d'événements 40 21
Taux annuel d'événements estimé 0,63 0,17
CFQ-R RD: Cystic Fibrosis
Questionnaire-Revised Respiratory
Domain (questionnaire révisé
spécifique de la mucoviscidose,
domaine respiratoire); ELX:
elexacaftor; ES: erreur standard;
IMC: indice de masse corporelle;
IVA: ivacaftor; N: taille de
l'échantillon total; p:
probabilité; PEx: exacerbation
pulmonaire; TEZ: tezacaftor;
VEMS: volume expiratoire maximum
par seconde exprimé en
pourcentage de la valeur
théorique.
Étude CFD-016
L'efficacité de Trikafta chez les patients atteints de mucoviscidose âgés de 6 ans et plus a également été analysée dans une étude rétrospective évaluant les résultats cliniques à partir de données réelles chez des patients atteints de mucoviscidose sans mutation F508del. Les données du registre de patients de l'US Cystic Fibrosis Foundation ont été utilisées à cet effet. Le critère d'évaluation de l'efficacité principal de l'étude CFD-016 était le VEMS. La variation moyenne du VEMS par rapport à la valeur initiale s'élevait à 4,53 % (n = 422; IC à 95 %: 3,50; 5,56).
PharmacocinétiqueLes paramètres pharmacocinétiques de l'elexacaftor, du tezacaftor et de l'ivacaftor sont similaires chez les adultes volontaires sains et les patients atteints de mucoviscidose. Les paramètres pharmacocinétiques de l'elexacaftor, du tezacaftor et de l'ivacaftor chez les patients atteints de mucoviscidose âgés de 12 ans et plus sont présentés dans le tableau 17.
Tableau 17: Paramètres
pharmacocinétiques des composants
de Trikafta
Elexacaftor Tezacaftor Ivacaftor
Informations générales
ASC (ET), µg-h/mla 162 (47,5)b 89,3 (23,2)b 11,7 (4,01)c
Cmax, (ET), µg/mla 9,2 (2,1) 7,7 (1,7) 1,2 (0,3)
Temps jusqu'à l'état d'équilibre, ≤7 jours ≤8 jours ≤3 à 5 jours
jours
Rapport d'accumulation 2,2 2,07 2,4
Absorption
Biodisponibilité absolue 80 % Non déterminée Non déterminée
Tmax médian (valeurs extrêmes), 6 (4 à 12) 3 (2 à 4) 4 (3 à 6)
heures
Effet des aliments Augmentation de 1,9 Pas d'effet clinique Augmentation de 2,5
à 2,5 fois de l'ASC ment significatif à 4 fois de l'exposi
(repas à teneur tion
modérée en graisses)
Distribution
Volume apparent de distribution 53,7 (17,7) 82,0 (22,3) 293 (89,8)
moyen (ET), litresd
Fixation aux protéinese > 99 % environ 99 % environ 99 %
Élimination
Demi-vie effective moyenne (ET), 27,4 (9,31) 25,1 (4,93) 15,0 (3,92)
heuresf
Clairance apparente moyenne (ET), 1,18 (0,29) 0,79 (0,10) 10,2 (3,13)
l/heure
Métabolisme
Voie principale CYP3A4/5 CYP3A4/5 CYP3A4/5
Métabolites actifs M23-ELX M1-TEZ M1-IVA
Activité du métabolite par Similaire Similaire Environ 1/6e de
rapport à la substance mère celle de la substanc
e mère
Excrétiong
Voie principale • Fèces: 87,3 % • Fèces: 72 % (sous • Fèces: 87,8 % •
(principalement forme inchangée ou Urines: 6,6 %
sous forme de sous forme de
métabolites) • M2-TEZ) • Urines:
Urines: 0,23 % 14 % (0,79 % sous
forme inchangée)
a Après administration
d'elexacaftor 200 mg et de
tezacaftor 100 mg une fois par
jour/ivacaftor 150 mg toutes les
12 heures à l'état d'équilibre
chez des patients atteints de
mucoviscidose âgés de 12 ans et
plus. b ASC0-24h. c ASC0-12h. d
Ni l'elexacaftor, ni le
tezacaftor ni l'ivacaftor ne se
fixent de façon préférentielle
dans les hématies humaines. e
L'elexacaftor et le tezacaftor se
fixent principalement à
l'albumine. L'ivacaftor se fixe
principalement à l'albumine, à
l'alpha-1 glycoprotéine acide et
à la gammaglobuline humaine. f
Les demi-vies terminales moyennes
(ET) de l'elexacaftor, du
tezacaftor et de l'ivacaftor sont
respectivement d'environ 24,7
(4,87) heures, 60,3 (15,7) heures
et 13,1 (2,98) heures. g Après
administration de doses
radiomarquées.
Absorption
Voir le tableau 17, "Paramètres pharmacocinétiques des composants de Trikafta" .
Distribution
Voir le tableau 17, "Paramètres pharmacocinétiques des composants de Trikafta" .
Métabolisme
Voir le tableau 17, "Paramètres pharmacocinétiques des composants de Trikafta" .
Élimination
Voir le tableau 17, "Paramètres pharmacocinétiques des composants de Trikafta" .
Cinétique pour certains groupes de patients
Troubles de la fonction hépatique
L'elexacaftor seul ou en association avec le tezacaftor et l'ivacaftor n'a pas été étudié chez les patients présentant une insuffisance hépatique sévère (Child-Pugh de classe C, score de 10 à 15). Après administration répétée d'elexacaftor, de tezacaftor et d'ivacaftor pendant 10 jours, l'ASC de l'elexacaftor était augmentée de 25 % et la Cmax de 12 %, l'ASC du M23elexacaftor était augmentée de 73 % et la Cmax de 70 %, l'ASC de l'elexacaftor et du M23elexacaftor combinés était augmentée de 36 % et la Cmax de 24 %, l'ASC du tezacaftor était augmentée de 20 % mais la Cmax était comparable et l'ASC de l'ivacaftor était augmentée de 50 % et la Cmax de 10 % chez les patients présentant une insuffisance hépatique modérée (Child-Pugh de classe B, score de 7 à 9) comparativement aux volontaires sains appariés pour les données démographiques (voir "Posologie/Mode d'emploi" , "Mises en garde et précautions" et "Effets indésirables" ).
Tezacaftor et ivacaftor
Après administration répétée de tezacaftor et d'ivacaftor pendant 10 jours, l'ASC du tezacaftor était augmentée d'environ 36 % et la Cmax de 10 % et l'ASC de l'ivacaftor était augmentée de 1,5 fois mais la Cmax était comparable chez les patients présentant une insuffisance hépatique modérée comparativement aux volontaires sains appariés pour les données démographiques.
Ivacaftor
Dans une étude menée avec l'ivacaftor seul, la Cmax de l'ivacaftor était comparable mais l'ASC0-∞ était augmentée d'un facteur 2 environ chez les patients présentant une insuffisance hépatique modérée comparativement aux volontaires sains appariés pour les données démographiques.
Troubles de la fonction rénale
L'elexacaftor seul ou en association avec le tezacaftor et l'ivacaftor n'a pas été étudié chez les patients présentant une insuffisance rénale sévère (DFGe < 30 ml/min/1,73 m2) ou une insuffisance rénale en phase terminale.
Dans les études pharmacocinétiques chez l'homme menées avec l'elexacaftor, le tezacaftor et l'ivacaftor, l'élimination urinaire de l'elexacaftor, du tezacaftor et de l'ivacaftor était minime (seulement 0,23 %, 13,7 % [0,79 % sous forme inchangée] et 6,6 % de la radioactivité totale respectivement).
Selon une analyse pharmacocinétique de population, l'exposition de l'elexacaftor était comparable chez les patients présentant une insuffisance rénale légère (N = 75, DFGe de 60 à < 90 ml/min/1,73 m2) et chez les sujets ayant une fonction rénale normale (N = 341, DFGe ≥90 ml/min/1,73 m2).
Une analyse pharmacocinétique de population portant sur 817 patients recevant le tezacaftor seul ou en association avec l'ivacaftor dans les études de phase II ou de phase III a montré que l'insuffisance rénale légère (N = 172, DFGe de 60 à < 90 ml/min/1,73 m2) et l'insuffisance rénale modérée (N = 8, DFGe de 30 à < 60 ml/min/1,73 m2) ne modifiaient pas significativement la clairance du tezacaftor (voir "Posologie/Mode d'emploi" ).
Sexe
Selon l'analyse pharmacocinétique de population, les expositions systémiques de l'elexacaftor, du tezacaftor et de l'ivacaftor sont similaires chez les hommes et les femmes.
Enfants et adolescents âgés de 2 à moins de 18 ans
Les expositions systémiques de l'elexacaftor, du tezacaftor et de l'ivacaftor et celles des métabolites M1-tezacaftor et M23-elexacaftor, déterminées à l'aide d'une analyse pharmacocinétique de population dans les études de phase III, sont présentées par tranche d'âge et par dose administrée dans le tableau 18. Les expositions systémiques de l'elexacaftor, du tezacaftor et de l'ivacaftor chez les patients âgés de 6 à moins de 18 ans se situent dans la fourchette observée chez les patients âgés de 18 ans et plus.
Tableau 18: ASC0-24h
,ss moyenne (ET) de
l'elexacaftor, du
tezacaftor et de
l'ivacaftor par
tranche d'âge
Tranche d'âge/ poids Dose Elexacaftor ASC0-24h M23-elexacaftor Tezacaftor ASC0-24h, M1-tezacaftor Ivacaftor ASC0-12h,s
,ss ( µg-h/ml) ASC0-24h,ss (μg-h/ml ss (µg-h/ml) ASC0-24h,ss (μg-h/ml s (µg-h/ml)
) )
Patients âgés de 2 Elexacaftor 80 mg 128 (24,8) 56,5 (29,4) 87,3 (17,3) 194 (24,8) 11,9 (3,86)
à < 6 ans, < 14 kg une fois par jour/te
(N = 16) zacaftor 40 mg une
fois par jour/ivacaf
tor 60 mg le matin
et ivacaftor 59,5
mg le soir
Patients âgés de 2 Elexacaftor 100 mg 138 (47,0) 59,0 (32,7) 90.2 (27,9) 197 (43,2) 13,0 (6,11)
à < 6 ans, ≥14 kg une fois par jour/te
(N = 59) zacaftor 50 mg une
fois par jour/ivacaf
tor 75 mg toutes
les 12h
Patients âgés de 6 Elexacaftor 100 mg 116 (39,4) 45,4 (25,2) 67,0 (22,3) 153 (36,5) 9,78 (4,50)
à < 12 ans < 30 kg une fois par jour/te
(N = 36) zacaftor 50 mg une
fois par jour/ivacaf
tor 75 mg toutes
les 12 heures
Patients âgés de 6 Elexacaftor 200 mg 195 (59,4) 104 (52) 103 (23,7) 220 (37,5) 17,5 (4,97)
à < 12 ans ≥30 kg une fois par jour/te
(N = 30) zacaftor 100 mg une
fois par jour/ivacaf
tor 150 mg toutes
les 12 heures
Adolescents âgés de Elexacaftor 200 mg 147 (36,8) 58,5 (25,6) 88,8 (21,8) 148 (333) 10,6 (3,35)
12 à < 18 ans (N = une fois par jour/te
72) zacaftor 100 mg une
fois par jour/ivacaf
tor 150 mg toutes
les 12 heures
Adultes âgés de ≥18 Elexacaftor 200 mg 168 (49,9) 64,6 (28,9) 89,5 (23,7) 128 (33,7) 12,1 (4,17)
ans (N = 179) une fois par jour/te
zacaftor 100 mg une
fois par jour/ivacaf
tor 150 mg toutes
les 12 heures
ET: écart-type;
ASCss : aire sous
la courbe de la
concentration par
rapport au temps à
l'état d'équilibre.
Données précliniquesElexacaftor/tezacaftor/ivacaftor
Les études de toxicité en administration répétée réalisées chez le rat et le chien au cours desquelles l'elexacaftor, le tezacaftor et l'ivacaftor ont été administrés en association pour évaluer le potentiel de toxicité additive et/ou synergique n'ont pas montré de toxicités ni d'interactions inattendues.
Il n'a pas été réalisé d'études portant sur la pharmacologie de sécurité, la génotoxicité, la carcinogénicité ou la toxicité sur la reproduction avec Trikafta. Cependant, il existe des études menées avec chaque principe actif individuellement.
Elexacaftor
Les données précliniques issues des études conventionnelles sur la pharmacologie de sécurité, la génotoxicité, et le potentiel cancérogène n'ont pas révélé de risque particulier pour l'homme.
Toxicité en cas d'administration répétée
Dans l'étude de toxicité de 6 mois chez le rat, les principaux organes cibles étaient l'estomac glandulaire (érosion), les testicules et l'épididyme (dégénérescence/atrophie des tubes séminifères, oligospermie/aspermie) et la moelle osseuse (diminution de la cellularité du tissu hématopoïétique). Ces effets ont été observés principalement aux doses non tolérées ≥40 mg/kg/jour chez les mâles et ≥30 mg/kg/jour chez les femelles. L'exposition plasmatique (ASC) chez les animaux à la dose sans effet nocif observé (DSENO) (15 mg/kg/jour) représentait environ 3 fois (mâles) et 11 fois (femelles) l'exposition à la dose maximale recommandée chez l'homme (DMRH). Dans l'étude de toxicité de 9 mois chez le chien, il a été observé chez les animaux ayant reçu l'elexacaftor à la dose de 14 mg/kg/jour (15 fois la DMRH d'après le total des ASC de l'elexacaftor et de son métabolite) une dégénérescence/atrophie bilatérale minime ou légère des tubes séminifères qui ne s'est pas résolue pendant la période de récupération, sans séquelles ultérieures toutefois. La signification éventuelle de ces observations pour l'homme n'est pas connue.
Toxicité sur la reproduction
Chez les mâles à la dose de 75 mg/kg/jour (6 fois la DMRH d'après le total des ASC de l'elexacaftor et de ses métabolites) et chez les femelles à la dose de 35 mg/kg/jour (7 fois la DMRH d'après le total des ASC de l'elexacaftor et de ses métabolites), l'elexacaftor a entraîné une diminution de la fertilité mâle et femelle, de l'accouplement chez les mâles et des indices de fertilité chez les femelles.
L'elexacaftor n'a pas été tératogène à la dose de 40 mg/kg/jour chez le rat et de 125 mg/kg/jour chez le lapin (environ 9 fois et 4 fois respectivement la DMRH d'après le total des ASC de l'elexacaftor et de ses métabolites [chez le rat] et d'après l'ASC de l'elexacaftor [chez le lapin]). Chez le rat, une diminution du poids fœtal moyen a été observée après l'administration de doses ≥25 mg/kg/jour chez les mères (environ 4 fois la DMRH d'après l'ASC). Dans l'étude du développement prénatal et postnatal chez le rat, il n'a pas été observé d'effets indésirables aux doses allant jusqu'à 10 mg/kg/jour (environ 1 fois la DMRH d'après le total des ASC de l'elexacaftor et de ses métabolites). Un passage transplacentaire de l'elexacaftor a été observé chez des rates gestantes.
Toxicité chez les animaux juvéniles
Il n'a pas été observé d'effets indésirables chez de jeunes rats ayant reçu du jour 7 au jour 70 de la période postnatale des doses entraînant une exposition plasmatique représentant environ 3 fois (mâles) et 5 fois (femelles) l'ASC chez les enfants et adolescents (âgés de 12 ans et plus).
Tezacaftor
Les données précliniques issues des études conventionnelles sur la pharmacologie de sécurité, la toxicité en cas d'administration répétée, la génotoxicité et la carcinogénicité n'ont pas révélé de risque particulier pour l'homme.
Toxicité chez les animaux juvéniles
Les études effectuées chez des rats exposés pendant les jours 7 à 35 de la période postnatale ont montré une morbi-mortalité, même à doses faibles. Les effets étaient liés à la dose et généralement plus sévères lorsque l'administration de tezacaftor commençait en début de période postnatale. L'exposition chez le rat du jour 21 au jour 49 de la période postnatale n'a pas entraîné d'effet toxique à la dose la plus élevée, qui représentait environ deux fois l'exposition attendue chez l'homme. Le tezacaftor et son métabolite, M1-TEZ, sont des substrats de la glycoprotéine P. Chez les jeunes rats, l'activité plus faible de la glycoprotéine P dans le cerveau a résulté en des concentrations cérébrales plus élevées du tezacaftor et du M1-TEZ. Ces observations ne sont probablement pas pertinentes pour la population pédiatrique âgée de 2 ans et plus relevant de l'indication, chez laquelle les niveaux d'activité de la glycoprotéine P sont équivalents à ceux observés chez les adultes.
Toxicité sur la reproduction
Chez le rat mâle et femelle, le tezacaftor n'a pas entraîné de toxicité sur la reproduction à la dose de 100 mg/kg/jour, la dose la plus élevée évaluée (environ 3 fois la DMRH d'après le total des ASC du tezacaftor et de M1-TEZ).
Le tezacaftor n'a pas eu d'effet sur la fertilité et les indices des performances de reproduction chez les rats mâles et femelles aux doses allant jusqu'à 100 mg/kg/jour (environ 3 fois la DMRH d'après le total des ASC du tezacaftor et de M1-TEZ).
Chez des rates et lapines gestantes, le tezacaftor n'a pas été tératogène aux doses représentant environ 3 fois et 0,2 fois respectivement l'exposition du tezacaftor chez l'homme à la dose thérapeutique.
Dans une étude du développement prénatal et postnatal, le tezacaftor n'a pas entraîné d'anomalies du développement dans les portées de rates gestantes traitées par voie orale à la dose de 25 mg/kg/jour (environ 1 fois la DMRH d'après le total des ASC du tezacaftor et de M1-TEZ). Aux doses maternotoxiques (≥50 mg/kg/jour), le tezacaftor a entraîné une diminution du poids fœtal, de l'indice de fertilité et a eu des effets sur les cycles œstraux (augmentation de la durée du cycle et diminution du nombre de cycles). À la dose la plus élevée (100 mg/kg/jour), les effets liés au tezacaftor dans les portées étaient une faible survie des petits jusqu'au sevrage, des effets sur le développement avant le sevrage et des retards de maturation sexuelle. Un passage transplacentaire du tezacaftor a été observé chez des rates gestantes.
Ivacaftor
Les données précliniques issues des études conventionnelles sur la pharmacologie de sécurité, la toxicité en cas d'administration répétée, la génotoxicité et la carcinogénicité n'ont pas révélé de risque particulier pour l'homme.
Toxicité sur la reproduction
Chez le rat mâle et femelle, l'ivacaftor a eu des effets sur la fertilité et sur les indices des performances de reproduction aux doses de 200 mg/kg/jour (environ 7 et 5 fois respectivement la DMRH d'après le total des ASC de l'ivacaftor et de ses métabolites). Chez les animaux femelles, l'ivacaftor a entraîné une diminution de l'indice global de fertilité, du nombre de gestations, du nombre de corps jaunes et de sites d'implantation, ainsi que des modifications du cycle œstral. L'ivacaftor a également entraîné une augmentation du nombre de femelles dont les embryons n'étaient pas viables et une diminution du nombre d'embryons viables. De faibles diminutions du poids des vésicules séminales ont été observées chez les mâles. Ces altérations de la fertilité et des performances de reproduction ont été imputées à une toxicité sévère chez les animaux recevant une dose de 200 mg/kg/jour. Il n'a pas été observé d'effets sur la fertilité mâle ou femelle et sur les indices des performances de reproduction après administration de doses ≤100 mg/kg/jour (environ 5 fois et 3 fois respectivement la DMRH d'après le total des ASC de l'ivacaftor et de ses métabolites).
L'ivacaftor n'a pas été tératogène chez le rat après administration de doses de 200 mg/kg/jour et chez le lapin après administration de doses de 100 mg/kg/jour (environ 6 fois et 16 fois respectivement la DMRH d'après le total des ASC de l'ivacaftor et de ses métabolites). Des effets sur le poids fœtal et de faibles augmentations des anomalies fréquentes du développement osseux ont été observés chez le rat aux doses associées à une toxicité significative chez les mères.
Dans l'étude du développement prénatal et postnatal chez des rates gestantes à des doses d'ivacaftor > 100 mg/kg/jour, il a été observé des taux de survie et des indices de lactation représentant respectivement 92 % et 98 % des valeurs chez les animaux témoins, ainsi qu'une réduction du poids des petits. Un passage transplacentaire de l'ivacaftor a été observé chez des rates et des lapines gestantes.
Toxicité chez les animaux juvéniles
Des cataractes ont été observées chez les jeunes rats ayant reçu du jour 7 au jour 35 de la période postnatale des doses d'ivacaftor ≥10 mg/kg/jour (0,2 fois la DMRH d'après l'exposition systémique de l'ivacaftor et de ses métabolites). Ces anomalies n'ont pas été constatées chez les fœtus de rates traitées du 7e au 17e jour de la gestation, ni chez les petits plus ou moins exposés à l'ivacaftor par l'intermédiaire du lait ingéré jusqu'au jour 20 de la période postnatale, ni chez des rats âgés de 7 semaines, ni chez des chiens âgés de 3,5 à 5 mois recevant l'ivacaftor. La signification éventuelle de ces observations pour l'homme n'est pas connue (voir "Mises en garde et précautions" ).
Remarques particulièresStabilité
Le médicament ne doit pas être utilisé audelà de la date figurant après la mention "EXP" sur le récipient.
Remarques particulières concernant le stockage
Ne pas conserver audessus de 30°C.
Conserver hors de vue et de portée des enfants.
Numéro d’autorisation67773, 69212 (Swissmedic)
PrésentationTrikafta comprimés pelliculés
-Comprimé d'elexacaftor 50 mg/tezacaftor 25 mg/ivacaftor 37,5 mg et comprimé d'ivacaftor 75 mg
-Boîte de 84 comprimés (4 pochettes hebdomadaires contenant chacune 14 comprimés pelliculés d'elexacaftor/tezacaftor/ivacaftor 50 mg/25 mg/37,5 mg et 7 comprimés pelliculés d'ivacaftor 75 mg). [A]
-Comprimé d'elexacaftor 100 mg/tezacaftor 50 mg/ivacaftor 75 mg et comprimé d'ivacaftor 150 mg
-Boîte de 84 comprimés (4 pochettes hebdomadaires contenant chacune 14 comprimés pelliculés d'elexacaftor/tezacaftor/ivacaftor 100 mg/50 mg/75 mg et 7 comprimés pelliculés d'ivacaftor 150 mg). [A]
Granulés en sachet
-Granulés en sachet d'elexacaftor 80 mg/tezacaftor 40 mg/ivacaftor 60 mg et granulés en sachet d'ivacaftor 59,5 mg
-Boîte de 56 sachets (4 pochettes hebdomadaires contenant chacune 7 sachets de granulés d'elexacaftor 80 mg/tezacaftor 40 mg/ivacaftor 60 mg et 7 sachets de granulés d'ivacaftor 59,5 mg) [A]
-Granulés en sachet d'elexacaftor 100 mg/tezacaftor 50 mg/ivacaftor 75 mg et granulés en sachet d'ivacaftor 75 mg
-Boîte de 56 sachets (4 pochettes hebdomadaires contenant chacune 7 sachets de granulés d'elexacaftor 100 mg/tezacaftor 50 mg/ivacaftor 75 mg et 7 sachets de granulés d'ivacaftor 75 mg) [A]
Titulaire de l’autorisationVertex Pharmaceuticals (CH) Sàrl
6300 Zoug
Mise à jour de l’informationJuillet 2025
|