ZusammensetzungWirkstoffe
Pasireotidum (ut Pasireotidi Pamoas)
Hilfsstoffe
Durchstechflasche mit Wirkstoff: Poly(D,L-lactide-coglycolide) (50-60:40-50), Poly(D,L-lactide-coglycolide) (50:50)
Fertigspritze mit Lösungsmittel: Mannitol, Natrium-Carmellose entspricht 1.5 mg Natrium Poloxamer 188, Wasser für Injektionszwecke.
Indikationen/Anwendungsmöglichkeiten-Behandlung der Akromegalie bei Patienten, bei denen eine chirurgische Behandlung oder Radiotherapie ungeeignet oder nicht wirksam ist und/oder andere medizinische Therapien nicht den gewünschten Behandlungserfolg erzielt haben.
-Behandlung des Morbus Cushing, wenn alle nicht-medikamentösen Therapiealternativen gemäss geltenden Standards ausgeschöpft sind.
Dosierung/AnwendungSignifor LAR sollte nur von einer geübten medizinischen Fachperson verabreicht werden.
Akromegalie
Die empfohlene Anfangsdosis von Signifor LAR beträgt 40 mg alle 4 Wochen.
Bei Patienten, deren GH- und/oder IGF-1-Spiegel nach 3 Monaten Behandlung mit Signifor LAR 40 mg noch nicht vollständig unter Kontrolle sind, kann die Dosis auf maximal 60 mg erhöht werden.
Morbus Cushing
Anwendung bei Patienten, welche bisher nicht mit Pasireotid behandelt wurden
Die empfohlene Anfangsdosis von Signifor LAR zur Behandlung des Morbus Cushing beträgt 10 mg alle 4 Wochen (28 Tage). Je nach Ansprechen und Verträglichkeit kann die Dosis in Schritten von 10 mg bis auf maximal 40 mg alle 28 Tage gesteigert werden.
Wechsel von subkutan appliziertem Pasireotid
Zu einem Wechsel von subkutan appliziertem Pasireotid auf Signifor LAR bzw. umgekehrt liegen keine Daten vor. Da Signifor (s.c.) und Signifor LAR nicht bioäquivalent sind, können keine Dosierungsempfehlungen gemacht werden (siehe «Warnhinweise / Vorsichtsmassnahmen»). Die Maximaldosis liegt auch in diesem Fall bei 40 mg alle 28 Tage.
Dosisanpassung aufgrund unerwünschter Wirkungen/Interaktionen
Akromegalie: Bei Auftreten (vermuteter) unerwünschter Wirkungen oder einer Überreaktion auf die Behandlung (IGF-1 < untere Grenze des Normbereiches) kann eine Dosisreduktion erforderlich sein. Die Dosis kann dabei in Schritten von 20 mg entweder vorübergehend oder dauerhaft reduziert werden.
Morbus Cushing: Das Auftreten von unerwünschten Wirkungen oder Hinweisen auf einen Hypocortisolismus kann eine Dosisreduktion, eine Unterbrechung der Behandlung oder das Absetzen von Signifor LAR erfordern.
Patienten mit Leberfunktionsstörungen
Bei Patienten mit leicht beeinträchtigter Leberfunktion (Child Pugh A) ist keine Dosisanpassung notwendig. Bei Patienten mit schwerer Leberinsuffizienz (Child Pugh C) ist Signifor LAR kontraindiziert.
Moderate Leberinsuffizienz (Child Pugh B):
Akromegalie: Die empfohlene Anfangsdosis beträgt 20 mg alle 4 Wochen, die Maximaldosis 40 mg alle 4 Wochen (siehe «Pharmakokinetik»).
Morbus-Cushing: Die empfohlene Anfangsdosis beträgt 10 mg alle 4 Wochen, die Maximaldosis 20 mg alle 4 Wochen (s. «Pharmakokinetik»).
Patienten mit Nierenfunktionsstörungen
Bei Patienten mit verminderter Nierenfunktion ist keine Dosisanpassung notwendig (siehe «Pharmakokinetik»).
Ältere Patienten
Daten über die Anwendung von Signifor LAR bei Patienten über 65 Jahre sind limitiert. Es gibt jedoch keine Hinweise darauf, dass bei älteren Patienten eine Dosisanpassung notwendig ist.
Kinder und Jugendliche
Die Sicherheit und Wirksamkeit der Anwendung bei Kindern und Jugendlichen ist nicht untersucht.
Verspätete Dosisgabe
Wenn eine Dosis Signifor LAR versäumt wurde, so sollte die Injektion so bald wie möglich nachgeholt werden. Die nächste Injektion erfolgt dann wieder nach 4 Wochen (d.h. unter Umständen kann sich der vorgesehene Wochentag der Injektion verändern).
Art der Anwendung
Signifor LAR muss als tiefe intramuskuläre Injektion verabreicht werden.
Die Suspension von Signifor LAR darf erst unmittelbar vor der Verabreichung zubereitet werden.
Bei wiederholten intramuskulären Injektionen sollte die Injektionsstelle zwischen dem linken und rechten Musculus glutaeus alterniert werden (siehe «Sonstige Hinweise» / «Hinweise für die Handhabung»).
KontraindikationenÜberempfindlichkeit gegenüber dem Wirkstoff oder einem der Hilfsstoffe.
Schwere Leberinsuffizienz (Child Pugh C).
Warnhinweise und VorsichtsmassnahmenEinleitung und Überwachung der Behandlung mit Signifor sollten nur von Ärzten durchgeführt werden, welche Erfahrung in der Therapie dieser Krankheitsbilder besitzen.
Zur Therapie des Morbus Cushing steht Pasireotid sowohl als Signifor® für die zweimal tägliche subkutane Applikation (in Einzeldosen von 300-900 µg) als auch als Signifor LAR für die einmal monatliche intramuskuläre Anwendung (in Dosen von 10-40 mg) zur Verfügung. Daten zu einem Wechsel zwischen den beiden Darreichungsformen liegen nicht vor. Wird die Therapie von Pasireotid s.c. auf Pasireotid i.m. (oder umgekehrt) umgestellt, so sollte der Patient in den ersten 2-3 Monaten besonders sorgfältig bezüglich seines Ansprechens auf die Therapie sowie bezüglich einer möglichen Verschlechterung des Glucosestoffwechsels überwacht werden.
Hypocortisolismus
Die Behandlung mit Signifor führt zu einer schnellen Unterdrückung der Sekretion von ACTH (Adrenocorticotropes Hormon). Eine schnelle Unterdrückung von ACTH kann zu einem vorübergehenden Hypocortisolismus führen mit den Symptomen Schwäche, Ermüdung, Anorexie, Übelkeit, Erbrechen, Hypotonie, Hyponatriämie oder Hypoglykämie bis hin zur akuten Nebennierenrindeninsuffizienz. In den Studien bei Morbus Cushing wurden Fälle eines Hypocortisolismus insbesondere während der ersten beiden Behandlungsmonate berichtet. Die Patienten sollten daher regelmässig überwacht und über mögliche Symptome eines Hypocortisolismus informiert werden. Falls sich ein Hypocortisolismus manifestiert, kann eine Dosisreduktion bzw. ein Unterbruch oder Abbruch der Therapie mit Signifor LAR und/oder eine temporäre Glukokortikoid-Substitution oder erforderlich sein.
Glukosemetabolismus
Unter Behandlung mit Pasireotid ist mit Veränderungen der Glukoseregulation zu rechnen. Erhöhter Nüchternblutzucker, Hyperglykämien und ein Anstieg des HbA1c sowie weniger häufig Hypoglykämien wurden in klinischen Studien sowohl bei Morbus Cushing als auch bei Akromegalie beobachtet, und bei bis zu 5% der Patienten wurde die Studie aus diesem Grund abgebrochen. Das Auftreten einer Hyperglykämie ist mit einer verminderten Ausschüttung von Insulin und Inkretinhormonen (d.h. Glukagon-ähnlichem Peptid-1 [GLP-1] und Glukoseabhängiges insulinotropes Polypeptid [GIP]) korreliert. Bei prädiabetischer Stoffwechsellage oder manifestem Diabetes mellitus ist der Grad der Glucosedysregulation ausgeprägter. Ein Anstieg von Nüchternglukose und HbA1c wurde dabei insbesondere während der ersten Behandlungsmonate beobachtet, danach kam es zu keinem weiteren Anstieg.
Der Blutzuckerstatus (Nüchternglukose und HbA1c) sollte vor Behandlungsbeginn abgeklärt und während der Behandlung regelmässig überwacht werden. Eine Selbstmessung der Blutglukose und/oder eine Bestimmung des Nüchternblutzuckerspiegels sollte während der ersten 2-3 Behandlungsmonate wöchentlich, danach periodisch in klinisch angemessenen Intervallen sowie wöchentlich während der ersten vier bis sechs Wochen nach einer Dosissteigerung erfolgen.
Nach Absetzen von Pasireotid sinken Nüchternglukose und HbA1c üblicherweise ab, können jedoch gegenüber den Ausgangswerten erhöht bleiben. Der Nüchternblutzucker sollte daher bis drei Wochen, das HbA1c bis drei Monate nach Ende der Behandlung überwacht werden.
Falls eine Hyperglykämie auftritt, ist die prompte Einleitung oder Anpassung einer Therapie der Hyperglykämie mit Inkretinen, Insulinsecretagoga und/oder Insulin angezeigt. Falls die Hyperglykämie trotz geeigneter medizinischer Massnahmen nicht kontrolliert werden kann, soll die Pasireotid-Dosis reduziert oder die Behandlung abgebrochen werden.
Nach der Markteinführung wurde über Fälle einer Ketoazidose unter Behandlung mit Pasireotid berichtet, unabhängig davon, ob bei den Patienten bereits vor Therapiebeginn ein Diabetes vorgelegen hatte. In einigen Fällen lagen prädisponierende Faktoren vor, wie akute Erkrankungen, Infektionen, Erkrankungen des Pankreas (z.B. Pankreasmalignome oder Operationen am Pankreas) oder Alkoholabusus. Alle Patienten mit Symptomen, die auf eine schwere metabolische Azidose hindeuten, sollten auf eine Ketoazidose untersucht werden. Diese Symptome sind häufig unspezifisch und umfassen z.B. übermässigen Durst, Appetitlosigkeit, Bauchschmerzen, Übelkeit, Erbrechen, Dyspnoe, ungewohnte Müdigkeit bzw. Erschöpfung sowie Verwirrtheit. Der Patient muss auf das Risiko (und insbesondere die Risikoerhöhung z.B. durch exzessiven Alkoholkonsum oder längeres Fasten) und die möglichen Symptome einer Ketoazidose hingewiesen werden. Er muss angewiesen werden, bei Auftreten entsprechender Symptome unverzüglich einen Arzt / eine Ärztin zu konsultieren.
Bei Patienten mit schlecht kontrollierter Glykämie (definiert durch HbA1c-Werte >8% unter antidiabetischer Behandlung) sollten Überwachung und Management des Diabetes vor und während der Behandlung mit Signifor LAR intensiviert werden.
In den beiden pivotalen Studien bei Akromegalie waren Frequenz und Schweregrad von unerwünschten Wirkungen im Zusammenhang mit einer Hyperglykämie unter intramuskulär verabreichtem Pasireotid höher als unter dem aktiven Komparator. Akromegaliepatienten, die eine Hyperglykämie entwickelten, schienen im Allgemeinen auf eine antidiabetische Therapie anzusprechen.
Leberfunktionstests
Unter Behandlung mit Pasireotid werden häufig vorübergehende, leichte Erhöhungen der Aminotransferasen beobachtet. In einigen Fällen wurde eine gleichzeitige Erhöhung der ALT auf mehr als 3x ULN (Upper Limit Normal = obere Grenze des Normbereichs) und der Bilirubin-Werte auf mehr als 2x ULN beobachtet.
Eine Kontrolle der Leberfunktion wird vor Beginn der Behandlung mit Signifor LAR, nach den ersten 2-3 Behandlungswochen und anschliessend monatlich während 3 Behandlungsmonaten empfohlen. Danach sollte die Leberfunktion jeweils dann überprüft werden, wenn dies klinisch erforderlich scheint.
Bei Patienten mit erhöhten Transaminasen sollten diese durch eine zweite Leberfunktionsanalyse bestätigt werden. Bei diesen Patienten sollte – ggf. auch nach Absetzen von Signifor LAR – die Leberfunktion häufig kontrolliert werden, bis die Ausgangswerte vor Behandlungsbeginn wieder erreicht sind.
In den folgenden Fällen sollte die Behandlung mit Signifor LAR abgebrochen werden:
bei Auftreten eines Ikterus oder anderer Zeichen einer klinisch relevanten Leberfunktionsstörung
bei anhaltender Erhöhung von AST oder ALT auf ≥5x ULN
bei Anstieg von ALT oder AST auf Werte ≥3x ULN und gleichzeitiger Bilirubin-Erhöhung auf ≥2x ULN
Die Behandlung sollte nicht wiederaufgenommen werden, wenn der Verdacht besteht, dass die Leberfunktionsstörungen mit Signifor LAR in Zusammenhang stehen.
Unerwünschte Wirkungen an der Gallenblase
Eine Cholelithiasis ist eine bekannte unerwünschte Wirkung bei der Langzeit-Behandlung mit Somatostatin-Analoga und wurde in klinischen Studien mit Pasireotid häufig beobachtet. Deshalb wird empfohlen, vor der Behandlung mit Signifor LAR und anschliessend in 6-12monatigen Intervallen eine Sonographie der Gallenblase durchzuführen. Eine Cholelithiasis bei mit Signifor LAR behandelten Patienten ist weitgehend asymptomatisch; symptomatische Konkremente sollten entsprechend gängiger klinischer Praxis behandelt werden. Nach der Markteinführung wurde auch über Fälle einer Cholangitis unter Behandlung mit Signifor LAR berichtet, die in der Mehrzahl als Komplikation einer Cholelithiasis angegeben wurden.
Kardiovaskuläre Ereignisse
Unter Behandlung mit Pasireotid wurden Bradykardien beobachtet. Patienten mit Herzerkrankungen und/oder Risikofaktoren für eine Bradykardie müssen sorgfältig überwacht werden. Hierzu gehören: klinisch relevante Bradykardie in der Anamnese, Mobitz Typ II-Block, Herzinsuffizienz (NYHA Klasse III oder IV), Z.n. Myokardinfarkt, instabile Angina pectoris, ventrikuläre Tachykardie oder Kammerflimmern in der Anamnese. Dosisanpassungen von Arzneimitteln wie Betablockern, Kalziumkanalblockern oder Substanzen, die das Elektrolyt-Gleichgewicht kontrollieren, können erforderlich sein.
In zwei Studien an gesunden Probanden zeigte sich unter Pasireotid eine Verlängerung des QT-Intervalls im EKG (siehe auch «Eigenschaften/Wirkungen»). Die klinische Bedeutung dieser Verlängerung ist unbekannt. Bei 2 von 201 Patienten wurde eine QTcF von >500 ms gemessen. Diese Episoden waren sporadisch, traten ein einziges Mal auf und blieben ohne klinische Konsequenzen. Torsade de pointes wurden nicht beobachtet, weder in diesen Studien, noch in klinischen Studien an Patienten mit Morbus Cushing oder Akromegalie.
Die Phase-III-Studien bei Akromegaliepatienten wiesen zwischen Signifor LAR und den Somatostatin-Analoga, die als aktive Komparatoren eingesetzt wurden, keine klinisch relevanten Unterschiede bezüglich einer QT-Verlängerung auf. Alle QT-bezogenen Ereignisse waren ohne therapeutische Intervention spontan reversibel.
Bei Patienten mit erhöhtem Risiko für eine Verlängerung des QT-Intervalls soll Pasireotid mit Vorsicht angewendet werden, so z.B. bei:
kongenitalem Long-QT-Syndrom
klinisch relevanter Herzerkrankung, wie kürzlichem Myokardinfarkt, kongestiver Herzinsuffizienz, instabiler Angina pectoris oder klinisch relevanter Bradykardie
-Patienten, die Antiarrhythmika oder andere Substanzen anwenden, die dafür bekannt sind, dass sie zu einer QT-Verlängerung führen
-Hypokaliämie und/oder Hypomagnesiämie
Vor Beginn der Behandlung mit Signifor LAR wird empfohlen, ein EKG als Ausgangsbefund zu erstellen. 21 Tage nach Therapiebeginn und immer, wenn es klinisch angebracht ist, ist eine Überwachung hinsichtlich eines möglichen Einflusses auf das QTc-Intervall ratsam.
Eine Hypokaliämie oder Hypomagnesiämie müssen vor der Behandlung mit Signifor LAR korrigiert werden, und die Kalium- und Magnesiumspiegel sollten während der Therapie regelmässig überwacht werden.
Hypophysenhormone
Nach transsphenoidalen Operationen und vor allem nach einer Strahlentherapie der Hypophyse kommt es häufig zu einem Mangel hypophysärer Hormone. Entsprechend vorbehandelte Patienten mit Morbus Cushing oder Akromegalie können daher einen Mangel eines oder mehrerer Hypophysenhormone aufweisen. Da die pharmakologischen Effekte von Pasireotid die Wirkung von Somatostatin imitieren, kann eine Hemmung anderer Hypophysenhormone neben ACTH und GH/IGF-1 nicht ausgeschlossen werden. Deshalb sollte die Hypophysenfunktion (z.B. TSH/freies T4, ACTH, GH/IGF-1) vor Beginn sowie regelmässig während der Behandlung mit Signifor LAR, wie klinisch angemessen, überwacht werden.
Fertilität
Tierexperimentelle Studien mit subkutan verabreichtem Pasireotid haben Auswirkungen auf die weibliche Fertilität gezeigt (s. «Präklinische Daten»). Der Effekt von Pasireotid auf die Fertilität beim Menschen ist unbekannt. Bei der Behandlung von Frauen im gebärfähigen Alter sollte jedoch berücksichtigt werden, dass die weibliche Fertilität reduziert sein könnte.
Anderseits kann die durch die Therapie erreichte Reduktion bzw. Normalisierung des Serumcortisolspiegels bzw. des GH («Growth hormone»)-Spiegels und der IGF-1 («Insulin-like growth factor»)-Konzentration möglicherweise zu einer Verbesserung der Fertilität führen.
Gegebenenfalls sollten Patientinnen im gebärfähigen Alter angewiesen werden, während einer Pasireotid-Behandlung geeignete kontrazeptive Massnahmen anzuwenden (s. «Schwangerschaft/Stillzeit»).
Dieses Arzneimittel enthält weniger als 1 mmol Natrium (23 mg) pro Dosiereinheit, d.h. es ist nahezu „natriumfrei“.
InteraktionenPharmakokinetische Interaktionen
Pasireotid ist moderat an Proteine gebunden, metabolisch stabil und weder Substrat noch Hemmer oder Induktor wichtiger CYP450-Enzyme in der Leber.
Pasireotid scheint ein Substrat des Efflux-Transporters p-Glykoprotein (p-Gp) zu sein. Allerdings ist Pasireotid weder Inhibitor noch Induktor von p-Gp. Pasireotid ist weder Substrat des Efflux-Transporters BCRP (breast cancer resistance protein) noch der Influx- Transporter OCT1 (organic cation transporter 1), OATP (organic anion-transporting polypeptide) 1B1, 1B3 oder 2B1. In klinisch relevanten Konzentrationen wird von Pasireotid keine Hemmung von UGT1A1 (Uridine Diphosphate Glucuronosyltransferase 1A1), des Influx-Transporters OAT1 oder OAT3, von OATP 1B1 oder 1B3, von OCT1 oder OCT2, des Efflux-Transporters Pgp, BCRP, MRP2 (Multi-Drug Resistance Protein 2) oder BSEP (Bile Salt Export Pump) erwartet.
Basierend auf diesen in-vitro-Daten ist das Potential für durch Proteinbindung, Metabolismus und/oder Transporter-vermittelte Interaktionen zwischen Pasireotid und gleichzeitig verabreichten anderen Arzneimitteln in vivo gering.
Pharmakodynamische Interaktionen
Vorsicht ist geboten bei gleichzeitiger Verabreichung von Signifor mit antiarrhythmischen Arzneimitteln oder Wirkstoffen, welche das QT-Intervall verlängern könnten (s. «Warnhinweise und Vorsichtsmassnahmen»).
Wirkung von SIGNIFOR LAR auf andere Arzneimittel
Einige wenige publizierte Daten deuten darauf hin, dass Somatostatin-Analoga durch Unterdrückung der Ausschüttung des Wachstumshormons indirekt die metabolische Clearance von Substanzen reduzieren könnten, die via CYP450-Enzyme metabolisiert werden. Auf der Basis der verfügbaren Daten kann die Möglichkeit, dass Pasireotid eine solche indirekte Wirkung entfalten könnte, nicht ausgeschlossen werden. Vorsicht ist geboten, wenn Pasireotid zusammen mit Arzneimitteln verabreicht wird, die einen niedrigen therapeutischen Index haben und hauptsächlich via CYP3A4 metabolisiert werden.
Bei Hunden führte Pasireotid durch eine verminderte intestinale Absorption von Cyclosporin zu einer Senkung des Cyclosporin-Blutspiegels. Es ist nicht bekannt, ob eine solche Interaktion auch beim Menschen erfolgt. Eine Anpassung der Cyclosporin-Dosis kann nötig sein, wenn Pasireotid und Cyclosporin zusammen verabreicht werden.
Bei gleichzeitiger Gabe von Bromocriptin mit Somatostatin-Analoga kann die Bioverfügbarkeit von Bromocriptin erhöht sein. Die Möglichkeit, dass Pasireotid eine solche Wirkung zeigen könnte, kann nicht ausgeschlossen werden.
Wirkung anderer Arzneimittel auf SIGNIFOR LAR
Der Einfluss eines Pgp-Inhibitors auf die Pharmakokinetik von Pasireotid, verabreicht als Signifor s.c.-Injektion, wurde bei gleichzeitiger Gabe von Verapamil untersucht. Es wurde keine Veränderung der Rate oder des Ausmasses der Pasireotid-Bioverfügbarkeit festgestellt.
Schwangerschaft, StillzeitSchwangerschaft
Es gibt keine adäquaten und gut kontrollierten Studien bei schwangeren Frauen. Studien an Tieren bei s.c.-Verabreichung haben eine Reproduktionstoxizität gezeigt (s. «Präklinische Daten»). Da das potentielle Risiko für den Menschen unbekannt ist, sollte Signifor LAR bei Schwangeren nicht angewendet werden, es sei denn es ist klar notwendig.
Stillzeit
Es ist nicht bekannt, ob Pasireotid in die menschliche Muttermilch übertritt. Untersuchungen mit s.c. appliziertem Pasireotid bei Ratten zeigten einen Übertritt von Pasireotid in die Milch. Da ein Risiko für das zu stillende Kind nicht ausgeschlossen werden kann, sollte während der Anwendung von Signifor LAR nicht gestillt werden.
Wirkung auf die Fahrtüchtigkeit und auf das Bedienen von MaschinenEntsprechende Studien wurden nicht durchgeführt. Unter der Anwendung von Pasireotid wurde jedoch über unerwünschte Wirkungen wie Schwindel sowie selten über Hypoglykämien berichtet, welche die Fahrtüchtigkeit beeinträchtigen könnten. Es sollte daher bekannt sein, wie der Patient auf Signifor LAR reagiert, bevor er ein Fahrzeug steuert oder Maschinen bedient.
Unerwünschte WirkungenDas Sicherheitsprofil ist bei der Behandlung der Indikationen Morbus Cushing und Akromegalie weitgehend ähnlich und ausserdem jenem unter subkutan appliziertem Pasireotid vergleichbar.
Die nachfolgend genannten unerwünschten Wirkungen wurden in klinischen Studien mit Pasireotid gemeldet und sind unter der entsprechenden MedDRA-Organklasse gemäss folgenden Häufigkeitskategorien aufgeführt: sehr häufig (≥1/10); häufig (≥1/100 bis <1/10); gelegentlich (≥1/1‘000 bis <1/100); selten (≥1/10‘000 bis <1/1000); nicht bekannt (basierend überwiegend auf Spontanmeldungen aus der Marktüberwachung, genaue Häufigkeit kann nicht abgeschätzt werden).
Akromegalie
Die Sicherheitsbeurteilung basiert auf 491 Akromegaliepatienten, die Pasireotid in Studien der Phase I bis III erhielten (419 Patienten erhielten Signifor LAR und 72 Signifor s.c.).
Die Sicherheit von Signifor LAR bei Patienten mit aktiver Akromegalie wurde in zwei verblindeten, aktiv kontrollierten Studien untersucht. In der Studie an Patienten, die vorher nicht medikamentös behandelt wurden und bei denen ein chirurgischer Eingriff fehlgeschlagen war oder nicht in Frage kam, waren die häufigsten unerwünschten Wirkungen, die in den Signifor LAR- und Sandostatin LAR-Armen gemeldet wurden, Diarrhoe (33.1% versus 40.6%), Cholelithiasis (30.9% vs. 36.7%), Hyperglykämie (28.1% vs. 7.2%) und Diabetes mellitus (19.7% vs. 3.9%). Unerwünschte Wirkungen vom Schweregrad 3 oder 4 gemäss der allgemeinen Toxizitätskriterien (CTC, Common Toxicity Criteria), die bei mehr als 2% der Patienten im Signifor LAR- oder Sandostatin LAR-Arm gemeldet wurden, waren Diabetes mellitus (4.5% vs. 0%), Diarrhoe (0.6% vs. 2.8%) und Hyperglykämie (2.2% vs. 0.6%). Störungen des Glukosestoffwechsels führten bei knapp 3% der Patienten zu einem vorzeitigen Studienabbruch.
In der Studie an Patienten, bei denen durch eine Therapie mit Somatostatin-Analoga der ersten Generation keine biochemische Kontrolle (GH ≤2.5 Mikrogramm/l und normalisiertes IGF-1) erreicht worden war (bezeichnet als "unzureichend kontrollierte Patienten"), waren die häufigsten unerwünschten Wirkungen, die unter Signifor LAR 40 mg, 60 mg und dem aktiven Komparator beobachtet wurden, Hyperglykämie (33.3%, 29.0% bzw. 6.1%), Diabetes mellitus (19.0%, 25.8% bzw. 4.5%) und Diarrhoe (11.1%, 19.4% bzw. 1.5%). Unerwünschte Wirkungen vom CTC-Grad 3 oder 4, die bei mehr als 2% der Patienten in der Gruppe mit Signifor LAR 40 mg, 60 mg oder in der aktiven Kontrollgruppe gemeldet wurden, waren Hyperglykämie (11.1%, 8.1% bzw. 0%), Diabetes mellitus (0%, 3.2% bzw. 0%) und abdominale Schmerzen (1.6%, 0%, 0%). Störungen des Glukosestoffwechsels führten bei knapp 5% der Patienten zu einem vorzeitigen Studienabbruch.
Erkrankungen des Bluts und Lymphsystems*
Häufig: Anämie
Endokrine Erkrankungen
Häufig: Nebennierenrinden-Insuffizienz**, Cortisol im Serum reduziert
Stoffwechsel- und Ernährungsstörungen
Sehr häufig: Hyperglykämie (28-33%, Diabetes mellitus (19-26%)
Häufig: beeinträchtigte Glukosetoleranz, erhöhtes HbA1c, verminderter Appetit, Blutglukose erhöht
Nicht bekannt: Ketoazidose
Erkrankungen des Nervensystems
Häufig: Schwindel, Kopfschmerzen
Herzerkrankungen
Häufig: Sinusbradykardie***; QT-Verlängerung
Erkrankungen des Gastrointestinaltrakts
Sehr häufig: Diarrhoe (11-33%), Bauchschmerzen (5-13%)
Häufig: Flatulenz, Übelkeit, Erbrechen, Lipase erhöht, Serumamylase erhöht
Nicht bekannt: Steatorrhoe, Stuhlverfärbung
Leber- und Galleerkrankungen ****
Sehr häufig: Cholelithiasis (10-31%)
Häufig: Cholezystitis, Alaninaminotransferase erhöht, Aspartataminotransferase erhöht, Gamma-Glutamyltransferase erhöht
Erkrankungen der Haut und des Unterhautzellgewebes
Sehr häufig: Alopezie (2-16%)
Skelettmuskulatur-, Bindegewebs- und Knochenerkrankungen
Häufig: Kreatinphosphokinase im Blut erhöht
Allgemeine Erkrankungen und Beschwerden am Verabreichungsort
Häufig: Reaktionen an der Injektionsstelle (Schmerzen, Induration, Beschwerden, Hämatom, Pruritus, Schwellung), Müdigkeit
* Über verlängerter Prothrombinzeit wurde mit Signifor bei Morbus-Cushing Patienten berichtet; keine Ereignisse wurden in den Akromegalie Studien durch die Investigators in Zusammenhangt mit der Behandlung mit Signifor LAR gestellt.
**Nebennierenrinden-Insuffizienz schliesst Hypokortisolismus ein
***Sinusbradykardie schliesst Bradykardie ein
**** Über Cholestase wurde mit Signifor in Morbus-Cushing Patienten, jedoch nicht in der Akromegalie-Studie mit Signifor LAR berichtet.
Morbus Cushing
Die nachstehend berichteten Sicherheitsdaten basieren auf einer klinischen Phase III-Studie an n=150 Patienten mit Morbus Cushing, die Anfangsdosen von 10 mg oder 30 mg Signifor LAR erhielten, mit der Möglichkeit zur Auftitration bis zu einer Maximaldosis von 40 mg alle 28 Tage. Die mediane Dauer der Exposition betrug 14.8 Monate (Range 0.9 bis 42.5 Monate) für mit der empfohlenen Anfangsdosis von 10 mg Signifor LAR behandelte Patienten und 12.5 Monate (Range 0.9 bis 45.8 Monate) für mit der Anfangsdosis von 30 mg Signifor LAR behandelte Patienten.
Die am häufigsten berichteten unerwünschten Wirkungen (≥20%), bei denen ein Zusammenhang mit der Behandlung vermutet wurde, waren Hyperglykämie, Diarrhoe, Cholelithiasis und Diabetes mellitus. Störungen des Glukosestoffwechsels führten dabei bei knapp 5% der Patienten zu einem vorzeitigen Studienabbruch.
Bei einem Teil der unerwünschten Wirkungen schienen Häufigkeit und Schweregrad in der Gruppe mit der höheren Anfangsdosis von 30 mg höher zu sein.
Erkrankungen des Blutes und Lymphsystems
Häufig: Anämie
Gelegentlich: Verlängerung der Prothrombinzeit
Endokrinologische Erkrankungen
Häufig: Nebennierenrinden-Insuffizienz
Stoffwechsel- und Ernährungsstörungen
Sehr häufig: Hyperglykämie (47%, Diabetes mellitus (21%)
Häufig: reduzierter Appetit, Erhöhung des HbA1c, eingeschränkte Glukosetoleranz
Erkrankungen des Nervensystems
Häufig: Kopfschmerzen, Schwindel
Herzerkrankungen
Häufig: (Sinus-)Bradykardie
Gelegentlich: QT-Verlängerung
Erkrankungen des Gastrointestinaltraktes
Sehr häufig: Diarrhoe (32%), Übelkeit (15%), Bauchschmerzen (14%)
Häufig: Erbrechen, Erhöhung der Lipase
Gelegentlich: Erhöhung der Amylase
Nicht bekannt: Steatorrhoe, Stuhlverfärbung
Leber- und Gallenerkrankungen
Sehr häufig: Cholelithiasis (31%)
Häufig: Erhöhung von Gamma-GT, ALT oder AST; Cholestase, (akute oder chronische) Cholezystitis
Allgemeine Erkrankungen und Beschwerden am Verabreichungsort
Sehr häufig: Müdigkeit (14%)
Häufig: Reaktionen an der Applikationsstelle (z.B. Schmerzen)
Unerwünschte Wirkungen nach Markteinführung
Die Meldung des Verdachts auf Nebenwirkungen nach der Zulassung ist von grosser Wichtigkeit. Sie ermöglicht eine kontinuierliche Überwachung des Nutzen-Risiko-Verhältnisses des Arzneimittels. Angehörige von Gesundheitsberufen sind aufgefordert, jeden Verdacht einer neuen oder schwerwiegenden Nebenwirkung über das Online-Portal ElViS (Electronic Vigilance System) anzuzeigen. Informationen dazu finden Sie unter www.swissmedic.ch.
ÜberdosierungAnzeichen und Symptome
Im Falle einer Überdosierung ist mit dem vermehrten Auftreten der unter «Unerwünschte Wirkungen» genannten Symptome und Laborveränderungen zu rechnen. In klinischen Studien wurden bei gesunden Probanden Dosen bis zu 2.1 mg zweimal täglich verabreicht; dabei wurde vor allem über Diarrhoe berichtet.
Behandlung
Im Falle einer Überdosierung wird empfohlen, entsprechend dem klinischen Status des Patienten eine geeignete supportive Behandlung einzuleiten, bis die Symptome verschwunden sind.
Eigenschaften/WirkungenATC-Code
H01CB05
Wirkungsmechanismus
Pasireotid (Cyclohexapeptid) ist ein Somatostatin-Analogum und bindet mit hoher Affinität an die humanen Somatostatin-Rezeptoren vom Subtyp SSTR 1, 2, 3 und 5.
Pharmakodynamik
Somatostatin-Rezeptoren werden in zahlreichen Geweben exprimiert, insbesondere in Hypophysentumoren und anderen neuroendokrinen Tumoren, in denen Hormone wie ACTH oder Wachstumshormon im Überschuss sezerniert werden.
Akromegalie: Aufgrund seines breit gefächerten Bindungsprofils bezüglich der Somatostatin-Rezeptoren hat Pasireotid das Potential, sowohl die Rezeptor-Subtypen SSTR2 als auch SSTR5 zu stimulieren, die zur Hemmung der Sekretion von GH und IGF-1 von Bedeutung sind. Daher ist es potenziell wirksamer zur Behandlung von Akromegaliepatienten im Vergleich zu anderen Somatostatin-Analoga.
Morbus Cushing: Pasireotid bindet und aktiviert die hsst-Rezeptoren der corticotropen Zellen in ACTH-produzierenden Adenomen, was zur Hemmung der ACTH-Sekretion führt. Pasireotid weist dabei eine hohe Affinität zu vier der fünf hssts auf, insbesondere zu hsst5.
Sicherheitspharmakodynamik
Glucosestoffwechsel
Die Entwicklung einer Hyperglykämie nach s.c.-Verabreichung von Pasireotid war mit einer signifikanten Abnahme sowohl der Insulinsekretion als auch der Inkretinhormone (d.h. Glukagon-ähnliches Peptid-1 [GLP-1] und glukose-abhängiges insulinotropes Polypeptid [GIP]) korreliert. Pasireotid hatte keinen Einfluss auf die Insulinempfindlichkeit. Bei gesunden Probanden war eine Inkretin-basierte Therapie (GLP-1-Agonisten und DDP-IV-Inhibitoren) zur Behandlung der Pasireotid-assoziierten Hyperglykämie am wirksamsten.
Kardioelektrophysiologie
Der Effekt von subkutan appliziertem Pasireotid auf das QT-Intervall wurde in zwei QT-Studien untersucht. In beiden Studien wurde ein Effekt von Pasireotid auf das QT-Intervall beobachtet. In der Studie mit einer Dosierung von zweimal täglich 1950 µg betrug die maximale mittlere placebo-bereinigte QTcF-Verlängerung 17,5 ms (90%CI: 15.53; 19.38). In der anderen Studie wurde unter Dosen von zweimal täglich 600 µg bzw. 1950 µg eine mittlere, placebo-bereinigte QTcI-Verlängerung von13.19 ms (90%CI: 11.38; 15.01) bzw. 16.12 ms (90%CI: 14.30; 17.95 ms) gemessen. Unter beiden Dosierungen kam es zu einer Reduktion der Pulsfrequenz, mit einer maximalen Abweichung im Vergleich zu Placebo nach 1 Stunde für Pasireotid 600 µg zweimal täglich (–10.39 bpm) und nach 30 Minuten für Pasireotid 1950 µg zweimal täglich (–14.91 bpm).
Die vorhergesagten Peakkonzentrationen für die maximale Signifor LAR-Dosierung von 40 mg bei Patienten mit Morbus Cushing und normaler Leberfunktion sowie von 20 mg bei mässig eingeschränkter Leberfunktion betragen 14 ng/ml bzw. 11.7 ng/ml. Beide Werte sind niedriger als die in den beiden oben genannten Studien beobachteten Peakkonzentrationen.
Die vorhergesagten Peakkonzentrationen für die maximale Signifor LAR-Dosierung von 60 mg bei Akromegaliepatienten mit normaler Leberfunktion und von 40 mg bei Akromegaliepatienten mit moderater Leberinsuffizienz von 25.8 ng/ml bzw. 28.8 ng/ml sind den beobachteten Peakkonzentrationen unter Signifor s.c. 600 Mikrogramm zweimal täglich (24.3 ng/ml) ähnlich und liegen unterhalb der beobachteten Peakkonzentration unter 1'950 Mikrogramm zweimal täglich (80.6 ng/ml).
Die Verlängerung des QT-Intervalls bei Verabreichung von Pasireotid wird nicht über eine Wirkung des hERG-Kaliumkanals vermittelt. Die kardiale Restitution, d.h. die Fähigkeit des Herzens, sich nach jedem vorhergehenden Schlag zu erholen, wurde in einem kontinuierlichen 24-Stunden-EKG untersucht, um die Wirkung von Pasireotid auf die Anfälligkeit für Arrhythmien zu bestimmen. Pasireotid verbesserte signifikant alle Restitutionsparameter in Gegenwart einer QT-Verlängerung. Dies deutet darauf hin, dass die durch Pasireotid vermittelte QT-Verlängerung möglicherweise nicht mit einem erhöhten proarrhythmischen Risiko assoziiert ist. Eine weitergehende quantitative Analyse der T-Wellen-Morphologie zeigte keine Veränderungen, die auf eine beeinträchtigte räumliche Heterogenität der kardialen Repolarisierung während der Pasireotid-Behandlung hindeuten würde.
Klinische Wirksamkeit
Akromegalie
Die Wirkung und Sicherheit von Signifor LAR bei Patienten mit aktiver Akromegalie wurde durch zwei verblindete Studien mit aktiver Kontrolle untersucht. Der primäre Wirksamkeitsendpunkt war definiert als Vergleich des Anteils der Patienten, die eine biochemische Kontrolle (definiert als mittlere GH-Spiegel <2.5 Mikrogramm/l und Normalisierung des geschlechts- und altersadjustierten IGF-1-Wertes) erreichten.
In einer Studie wurden 358 Patienten eingeschlossen, bei welchen entweder eine Adenektomie erfolgt war oder eine Operation nicht in Frage kam (Kontraindikation, fehlende Zustimmung zur Operation). Der primäre Wirksamkeitsendpunkt wurde nach 12 Monaten analysiert. Die Prozentzahl der Patienten, die eine biochemische Kontrolle erreichten, betrug 31.3% und 19.2% für intramuskuläres Pasireotid LAR bzw. Octreotid LAR, (p = 0.007). Eine biochemische Kontrolle wurde in der Studie in beiden Armen früh erreicht (d.h. in Monat 3).
Der Anteil der Patienten mit einer Abnahme des Tumorvolumens um mehr als 20% in Monat 12 betrug für intramuskuläres Pasireotid 80.8% und für Octreotid LAR 77.4%. Bestimmungen der Lebensqualität in Monat 12 zeigten im Vergleich zum Ausgangswert statistisch signifikante Verbesserungen der Scores des körperlichen und des psychologischen Erscheinungsbildes sowie des globalen AcroQol-Scores sowohl unter Signifor LAR als auch unter Octreotid.
Während der Extensions-Phase wurden 74 Patienten weiter mit intramuskulärem Pasireotid und 46 Patienten weiter mit Octreotid LAR behandelt. An Monat 25 erreichten 48.6% der Patienten (36/74) in der intramuskulären Pasireotid-Gruppe und 45.7% (21/46) in der Octreotid LAR-Gruppe eine biochemische Kontrolle. Der prozentuale Anteil an Patienten, welche zum gleichen Zeitpunkt einen mittleren GH-Wert von <2.5 Mikrogramm/l und eine Normalisierung der IGF-1-Werte erreicht hatten, war in beiden Behandlungsarmen vergleichbar. Während der Extensions-Phase ging das Tumorvolumen kontinuierlich zurück, und die Verbesserung der Akromegalie-Symptome blieb zwischen den beiden Behandlungsarmen vergleichbar. Die AcroQolScores blieben während der gesamten Extensions-Phase im intramuskulären Pasireotid-Arm numerisch höher als im Octreotid LAR-Arm.
In einer Studie an vorbehandelten, unzureichend kontrollierten Akromegalie-Patienten (definiert als Patienten mit einer mittleren GH-Konzentration auf einem 5-Punkte-Profil über eine Dauer von 2 Stunden von >2.5 Mikrogramm/l und einem geschlechts- und altersangepassten IGF-1 >1.3 x der oberen Grenze des Normalbereichs (ULN); die Patienten mussten während mindestens 6 Monaten vor der Randomisierung mit der maximal indizierten Dosis von Sandostatin LAR (30 mg) oder Lanreotid ATG (120 mg) behandelt worden sein) wurde entweder Pasireotid 40 mg (n=65) oder 60 mg (n=65) oder die vorhergehende Medikation (n=68) verabreicht. Zwei Drittel der Patienten hatten eine Operation hinter sich. Der mittlere Ausgangswert des GH betrug 17.6 Mikrogramm/l, 12.1 Mikrogramm/l bzw. 9.5 Mikrogramm/l in der Gruppe mit 40 mg, jener mit 60 mg und der aktiven Kontrollgruppe. Die mittleren IGF-1-Werte lagen zu Studienbeginn bei 2.6, 2.8 bzw. 2.9 x ULN.
Der Anteil der Patienten, die eine biochemische Kontrolle in Woche 24 erreichten, belief sich auf 15.4% (p = 0.006) bzw. 20.0% (p- <0.0001) für intramuskuläres Pasireotid 40 mg bzw. 60 mg im Vergleich zu 0% im Kontrollarm (Octreotid LAR , Lanreotid ATG).
Der Anteil der Patienten, die eine Abnahme oder keine Veränderung des Tumorvolumens der Hypophyse in Woche 24 aufwiesen, betrug 81.0% bzw. 70.3% unter intramuskulärem Pasireotid 40 mg bzw. 60 mg und 50.0% in der Kontrollgruppe. Darüber hinaus erzielte ein grösserer Anteil an Patienten unter intramuskulärem Pasireotid (18.5% bzw. 10.8% für 40 mg bzw. 60 mg) eine Abnahme des Tumorvolumens um mindestens 25% als unter dem aktiven Komparator (1.5%). An Woche 24 waren Verbesserungen der Scores des körperlichen und psychologischen Erscheinungsbildes sowie des globalen AcroQol-Scores sowohl in der intramuskulären Pasireotid-Gruppe mit 40 mg als auch in jener mit 60 mg zu erkennen. In der Behandlungsgruppe mit intramuskulärem Pasireotid 40 mg waren diese Veränderungen beim AcroQoL-Subscore für das körperliche Erscheinungsbild gegenüber Baseline statistisch signifikant. In der Gruppe mit intramuskulärem Pasireotid 60 mg waren diese Veränderungen sowohl bei den Subscores für das körperliche und das psychologische Erscheinungsbild als auch für den globalen Score statistisch signifikant. In der Octreotid LAR- und der Lanreotid ATG-Gruppe gab es keine statistisch signifikanten Veränderungen gegenüber Baseline. Die mittlere Verbesserung gegenüber dem Ausgangswert war in der Gruppe mit intramuskulärem Pasireotid 60 mg für alle Scores am grössten. Allerdings waren die Unterschiede in den Veränderungen in Woche 24 im Vergleich zum Ausgangswert zwischen den Behandlungsgruppen nicht statistisch signifikant.
Morbus Cushing
Wirksamkeit und Sicherheit von Signifor LAR wurden in einer multizentrischen, doppelblinden, 1:1 randomisierten Phase III-Studie untersucht an n=150 Patienten mit nach Adenomresektion persistierendem oder rezidivierendem Morbus Cushing sowie Patienten, für welche eine Hypophysen-Operation nicht in Frage kam. Eingeschlossen wurden Patienten, welche ein mittleres freies 24-Stunden-Cortisol im Urin (mUFC) zwischen ≥1.5 und ≤5x ULN aufwiesen. Die Patienten wurden über 12 Monate behandelt und erhielten zunächst entweder 10 mg oder 30 mg Signifor LAR i.m. alle 28 Tage.
Nach vier Monaten Behandlung erhielten Patienten mit einem durchschnittlichen UFC ≤1,5 x ULN weiterhin die Dosis, zu der sie randomisiert worden waren. Bei Patienten, bei welchen das durchschnittliche UFC nach 4 Monaten bei >1,5 x ULN lag, wurde die Dosis von 10 mg auf 30 mg bzw. von 30 mg auf 40 mg erhöht. Falls das UFC weiterhin bei >1x ULN lag, konnte nach 7 und 9 Monaten eine weitere Dosissteigerung erfolgen, wobei die Maximaldosis mit 40 mg festgelegt war. Im Falle einer Unverträglichkeit konnte die Dosierung jederzeit nach unten angepasst werden, wobei die minimal mögliche Dosis bei 5 mg lag.
Primärer Wirksamkeitsendpunkt war der Anteil der Patienten, bei welchen sich nach 7monatiger Behandlung die durchschnittlichen UFC-Werte normalisiert hatten (d.h. UFC ≤ULN; = „Responder“), unabhängig von einer eventuellen Dosissteigerung in Monat 4. Patienten, die vor Monat 4 ausschieden, wurden als Non-Responder gewertet.
Haupt-Sekundärendpunkt war der Anteil der Patienten, die nach 7 Monaten mUFC-Responder waren und bei denen bis zu diesem Zeitpunkt keine Dosiserhöhung erfolgt war. Weitere Sekundärendpunkte waren Veränderungen gegenüber den Ausgangswerten in 24-Stunden-UFC-Test, Plasma-ACTH, Serumcortisolspiegel und klinischen Symptomen des Morbus Cushing sowie gesundheitsbezogene Lebensqualität (gemäss SF-12v2 und CushingQoL). Als Kriterium für die Wirksamkeit war definiert, dass die unter Grenze des 95%-Konfidenzintervalles für die Responserate bei >15% liegen müsste. Dieses Kriterium wurde in beiden Dosisgruppen erfüllt. In der 10 mg-Gruppe lag die Ansprechrate nach 7 Monaten bei 41.9% (95% KI: 30.5-53.9%), in der 30 mg-Gruppe bei 40.8% (95% KI: 29.7-52.7%). Die Reduktion des UFC blieb über den gesamten Behandlungszeitraum erhalten. Anhand des durchschnittlichen Baseline-UFC wurden zwei Strata unterschieden: Bei Patienten mit einem UFC zwischen 1.5 und 2x ULN lag die Ansprechrate in beiden Dosisgruppen bei 52% (95%-KI 31-72%). Bei Patienten mit einem UFC >2x ULN war die Ansprechrate hingegen 37% in der 10 mg-Gruppe (95%-KI 23-52%) und 35% in der 30 mg-Gruppe (95%-KI 22-50%).
Bei 31 von 74 Patienten des 10 mg-Armes sowie bei 28 von 76 Patienten des 30 mg-Armes erfolgte in Monat 4 eine Dosissteigerung. Im Haupt-Sekundärendpunkt (d.h. dem Anteil der Responder ohne vorausgehende Dosissteigerung) fand sich nach 7 Monaten ein Ansprechen in der 10 mg-Gruppe bei 28.4% (95% KI: 18.5-40.1%), in der 30 mg-Gruppe bei 31.6% (95% KI: 21.4-43.3%) der Patienten.
Nach 7 Monaten fand sich in beiden Dosisgruppen eine klinisch relevante Senkung des systolischen und diastolischen Blutdrucks in Rückenlage sowie eine Reduktion des Körpergewichts. Die Veränderungen beider Parameter waren tendenziell bei als mUFC-Responder klassifizierten Patienten stärker. Ähnliche Trends wurden nach 12 Monaten beobachtet.
Nach 7 Monaten zeigte die Mehrzahl der Patienten eine Besserung oder Stabilisierung der klinischen Symptome gegenüber dem Ausgangsbefund. Die Gesichtsrötung besserte sich bei 43.5% der Patienten, und jeweils gut ein Drittel der Patienten zeigte eine Besserung der supraklavikulären und dorsalen Fettpolster. Auch bezüglich Lebensqualität fand sich eine, statistisch allerdings nicht signifikante, Verbesserung.
PharmakokinetikAbsorption
Der Steady-State wird nach Verabreichung von Pasireotid als Signifor LAR innerhalb von 4 Monaten erreicht. Die Pasireotid-Exposition nach wiederholten monatlichen Gaben von Signifor LAR ist annähernd dosisproportional.
Distribution
Pasireotid hat ein Verteilungsvolumen (Vz/F) von >100 l und ist primär im Plasma (91%) zu finden. Die Plasmaproteinbindung ist moderat (88%) und von der Konzentration unabhängig.
Pasireotid ist vermutlich ein Substrat von Pgp, die Liquorgängigkeit ist nicht untersucht.
Metabolismus
Pasireotid erwies sich in Mikrosomen der menschlichen Leber und Niere als metabolisch sehr stabil.
Elimination
Pasireotid wird hauptsächlich unverändert über die Galle eliminiert und nur in geringem Ausmass über die Niere. Die Clearance beträgt 4.5 – 8.5 l/h, die Eliminationshalbwertszeit liegt bei ungefähr 16 Tagen. 55,9 ± 6,63% des radioaktiv markierten Pasireotids wurden während der ersten 10 Tage nach Applikation aufgefunden, wovon 48,3 ± 8,16% der Radioaktivität in den Faezes und 7,63 ± 2,03% im Urin gemessen wurden.
Kinetik spezieller Patientengruppen
Leberfunktionsstörungen
Bei Patienten mit moderater bis schwerer Leberinsuffizienz (Child-Pugh B und C) fand sich nach einer subkutanen Einzeldosis von Pasireotid eine signifikant erhöhte Exposition im Vergleich zu Probanden mit normaler Leberfunktion. In den Gruppen mit moderater bis schwerer Leberinsuffizienz waren die AUCinf um 60% bzw. 79% und die Cmax um 67% bzw. 69% erhöht sowie die CL/F um 37% bzw. 44% reduziert im Vergleich zur Kontrollgruppe.
Nierenfunktionsstörungen
In einer Studie an Patienten mit leicht, mässig oder schwer eingeschränkter Nierenfunktion sowie mit terminaler Niereninsuffizienz unterschied sich die Pharmakokinetik von Pasireotid nach Gabe einer Einzeldosis von 0.9 mg subkutan nicht relevant von jener bei Nierengesunden.
Ältere Patienten
Populationskinetische Analysen deuten darauf hin, dass die Pharmakokinetik von Pasireotid nicht in relevanter Weise durch das Alter beeinflusst wird. Eine spezifische pharmakokinetische Studie bei Patienten ≥65 Jahren liegt jedoch nicht vor.
Kinder und Jugendliche
Die Pharmakokinetik von Pasireotid wurde in dieser Altersgruppe nicht untersucht.
Geschlecht, Ethnizität und Körpergewicht
Populationspharmakokinetische Analysen legen nahe, dass Geschlecht, Körpergewicht und ethnische Zugehörigkeit keinen klinisch relevanten Einfluss auf die pharmakokinetischen Parameter von Signifor LAR haben.
Präklinische DatenNicht-klinische Sicherheitsstudien, die mit subkutan verabreichtem Pasireotid durchgeführt wurden, beinhalteten Sicherheitspharmakologie, Toxizität bei wiederholter Verabreichung, Genotoxizität und Karzinogenität sowie Reproduktions- und Entwicklungstoxizität. Zusätzlich wurden Studien zur Verträglichkeit und Toxizität bei wiederholter Verabreichung mit Pasireotid LAR bei intramuskulärer Verabreichung durchgeführt. Die meisten Befunde der wiederholten Toxizitätsstudien waren reversibel und der Pharmakologie von Pasireotid zuzuschreiben. Die Wirkungen in den präklinischen Studien wurden bei Expositionen beobachtet, die vergleichbar waren oder über der Maximalexposition nach therapeutischen Dosen beim Menschen lagen.
Sicherheitspharmakologie
In sicherheitspharmakologischen Studien (mit Pasireotid als s.c.-Injektion) hatte Pasireotid keine unerwünschten Wirkungen auf respiratorische oder kardiovaskuläre Funktionen. Eine Abnahme der allgemeinen Aktivität und der Verhaltensaktivität wurde bei Mäusen bei einer Dosis von 12 mg/kg beobachtet. Dies ist äquivalent zur etwa 27-fachen geschätzten täglichen Maximaldosis für Pasireotid LAR, bezogen auf die Körperoberfläche.
Mutagenität
Pasireotid erwies sich in zwei in vitro-Genotoxizitätstests (Ames-Test und Chromosomenaberrationstest an peripheren Humanlymphozyten) als nicht genotoxisch. Pasireotid war in einem in vivo-Mikronukleustest an Knochenmark von Ratten bei Dosen bis zu 50 mg/kg nicht genotoxisch. Dies entspricht einer Dosis, die nahezu 224-mal höher ist als die geschätzte Tageshöchstdosis von Pasireotid LAR, bezogen auf die Körperoberfläche.
Karzinogenität
Karzinogenitätsstudien an Ratten und transgenen Mäusen zeigten kein karzinogenes Potenzial.
Reproduktionstoxizität
In Studien zur embryofötalen Entwicklung bei Ratten und Kaninchen erwies sich subkutan injiziertes Pasireotid in materno-toxischen Dosen (entsprechend 10 (Ratte) und 5 (Kaninchen) mg/kg/Tag), die zu einer Exposition (AUC0- 24 h) entsprechend der 106- bzw. 30-fachen Exposition bei der MRHD von Pasireotid LAR führten, als nicht teratogen. Bei Dosen von 10 mg/kg/Tag bei Ratten wurden vermehrt frühe/totale Resorptionen und Gliedmassen in Fehlstellung vorgefunden. Bei Dosen von 5 mg/kg/Tag bei Kaninchen kam es zu vermehrten Aborten, reduziertem Gewicht der Föten und daraus folgenden Veränderungen des Skeletts. Das reduzierte Gewicht der Föten und die resultierende verzögerte Knochenbildung wurden bei einer Dosis von 1 mg/kg/Tag (4.8-fach höhere Exposition als bei der MRHD für Pasireotid LAR) beobachtet. Bei Ratten zeigte Pasireotid in einer prä- und postnatalen Studie bis zu einer Dosis von 10 mg/kg/Tag (45-fache MRHD für Pasireotid LAR, bezogen auf die Körperoberfläche) keine Wirkung auf Wehen und Geburt.
Pasireotid wird in die Milch ausgeschieden. Eine Verzögerung des physiologischen Wachstums der Jungtiere wurde bei einer Dosis von 2 mg/kg/Tag (9-mal höher als die geschätzte Tageshöchstdosis von Signifor LAR, bezogen auf die Körperoberfläche) beobachtet. Nach der Entwöhnung war die Gewichtszunahme der Jungtierratten, die Pasireotid ausgesetzt waren, jener der Kontrolltiere vergleichbar, was eine Reversibilität zeigt.
Pasireotid-Dosen bis zu 10 mg/kg/Tag (45-mal höher als die geschätzte Tagedosis von Signifor LAR, bezogen auf die Körperoberfläche) hatten keinen Einfluss auf die Fertilität männlicher Ratten. Wie aufgrund der Pharmakologie von Pasireotid zu erwarten ist, nahm die Fertilität weiblicher Ratten bei Tagesdosen von 0,1 mg/ kg/Tag (0.5mal die geschätzte Tageshöchstdosis von Pasireotid LAR, bezogen auf die Körperoberfläche) ab, wie eine reduzierte Anzahl von Corpora lutea und Implantationsstellen zeigte. Anormale oder ausbleibende Zyklen wurden bei Dosen von 1 mg/kg/ Tag vorgefunden.
Sonstige HinweiseInkompatibilitäten
Signifor LAR Pulver zur Herstellung einer Injektionssuspension soll als Einzeldosisbehälter benutzt werden und darf nicht mit anderen Arzneimitteln gemischt werden. Daher wurden keine Daten zur Verträglichkeit mit anderen Produkten erhoben.
Haltbarkeit
Das Arzneimittel darf nur bis zu dem auf dem Behälter mit «EXP» bezeichneten Datum verwendet werden.
Besondere Lagerungshinweise
Im Kühlschrank (2-8°C) lagern.
Nicht einfrieren.
Arzneimittel sind für Kinder unerreichbar aufzubewahren.
Hinweise für die Handhabung
Anleitung zur Zubereitung und intramuskulären Injektion von Signifor LAR
AUSSCHLIESSLICH ZUR TIEFEN INTRAMUSKULÄREN INJEKTION
VORSICHT:
Es gibt 2 kritische Schritte bei der Rekonstitution von Signifor LAR. Nicht-Beachtung kann dazu führen, dass das Medikament nicht ordnungsgemäss zum Wirkort gelangt.
Das Injektions-Kit muss auf Raumtemperatur gebracht werden. Nehmen Sie das Injektions-Kit aus dem Kühlschrank und lassen Sie es mindestens 30 Minuten, aber keinesfalls länger als 24 Stunden bei Raumtemperatur stehen, bevor Sie es rekonstituieren.
Nach dem Hinzufügen des Lösungsmittels schütteln Sie die Durchstechflasche leicht in horizontaler Richtung während mindestens 30 Sekunden, bis sich eine einheitliche Suspension gebildet hat.
Inhalt:
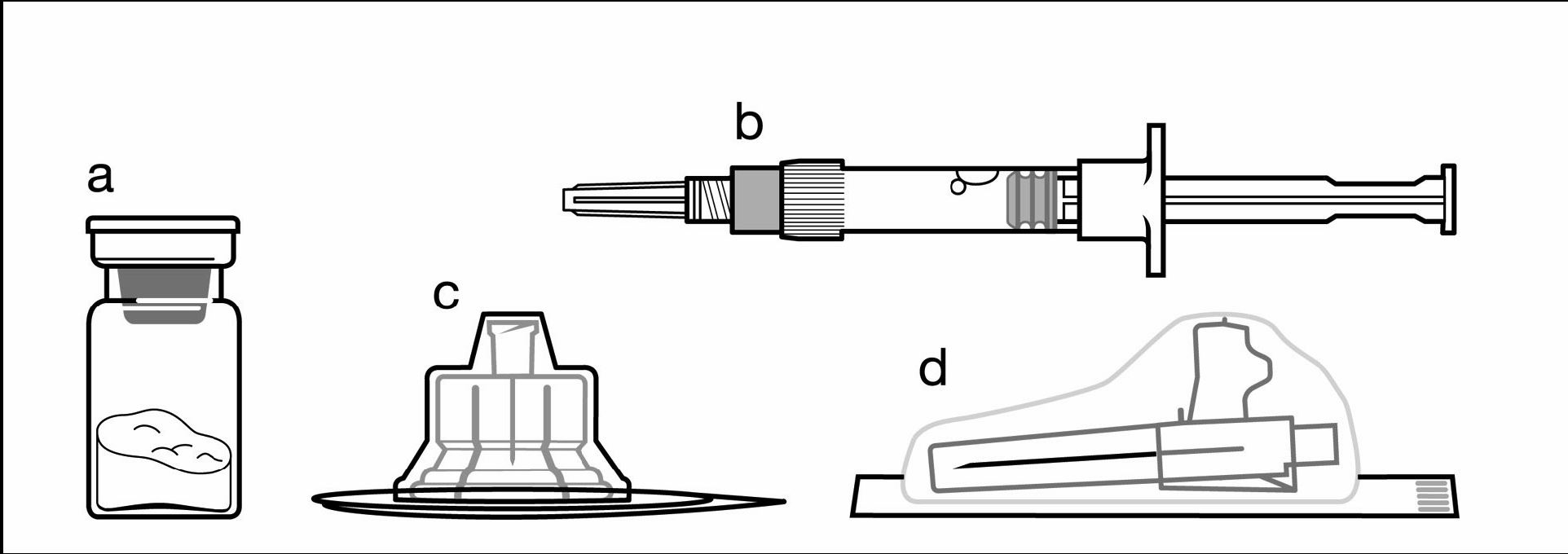
a Eine Durchstechflasche mit Signifor LAR Pulver
b Eine Fertigspritze mit Lösungsmittel zur Rekonstitution
c Ein Adapter für die Durchstechflasche zur Rekonstitution des Arzneimittels
d Eine Sicherheitsinjektionsnadel (20G x 1.5")
Befolgen Sie die untenstehenden Anweisungen sorgfältig, um eine ordnungsgemässe Rekonstitution von Signifor LAR zu gewährleisten, bevor Sie eine tiefe intramuskuläre Injektion vornehmen.
Signifor LAR Suspension darf erst unmittelbar vor der Injektion zubereitet werden.
Signifor LAR sollte nur von geschultem medizinischem Fachpersonal verabreicht werden.
|
Schritt 1
Nehmen Sie das Signifor LAR Injektions-Kit aus der Kühllagerung.
VORSICHT: Es ist ausserordentlich wichtig, dass mit dem Rekonstitutionsvorgang erst begonnen wird, nachdem das Injektions-Kit Raumtemperatur erreicht hat. Lassen Sie das Kit mindestens 30 Minuten, aber keinesfalls länger als 24 Stunden vor der Rekonstitution bei Raumtemperatur stehen.
Anmerkung: Das Injektions-Kit kann, falls erforderlich, wiederholt gekühlt werden.
|
|
| |
Schritt 2
Entfernen Sie die Plastikkappe von der Durchstechflasche und desinfizieren Sie den Gummistopfen mit einem Alkoholtupfer.
|
|
| |
Entfernen Sie die Deckelfolie der Verpackung des Adapters für die Durchstechflasche, aber nehmen Sie den Adapter NICHT aus der Verpackung heraus.
Halten Sie die Verpackung des Adapters und positionieren Sie den Adapter auf der Durchstechflasche. Drücken Sie dann den Adapter vollständig herunter, bis er einschnappt. Dies ist durch ein hörbares "Klicken" erkennbar.
|
|
| |
Heben Sie die Verpackung nun mittels einer vertikalen Bewegung vom Adapter.
|
|
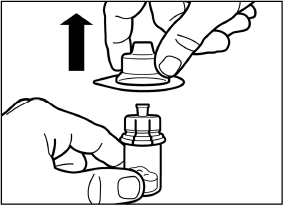
| |
Schritt 3
Entfernen Sie die Verschlusskappe der Fertigspritze mit dem Lösungsmittel und schrauben Sie die Spritze auf den Adapter der Durchstechflasche.
|
|
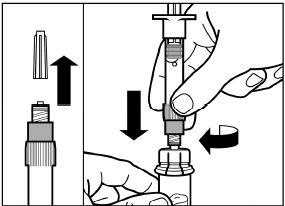
| |
Drücken Sie den Kolben behutsam ganz nach unten, um das gesamte Lösungsmittel in die Durchstechflasche zu injizieren.
|
|
| |
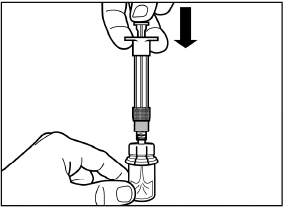
Schritt 4
VORSICHT: Halten Sie den Kolben gedrückt und schütteln Sie die Durchstechflasche während mindestens 30 Sekunden leicht in horizontaler Richtung, bis das Pulver vollständig suspendiert ist. Schütteln Sie nochmals leicht während weiteren 30 Sekunden, wenn das Pulver nicht vollständig suspendiert ist.
|
|
| |
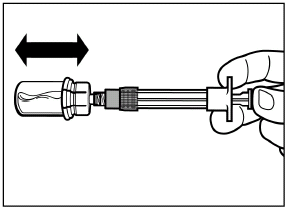
Schritt 5
Drehen Sie Spritze und Durchstechflasche um (so dass die Durchstechflasche oben ist), ziehen Sie den Kolben langsam zurück und ziehen Sie den gesamten Inhalt aus der Durchstechflasche in die Spritze auf.
|
|
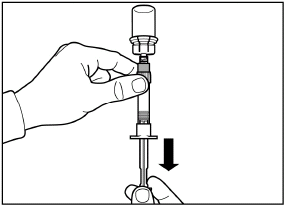
| |
Schrauben Sie die Spritze vom Adapter ab.
|
|
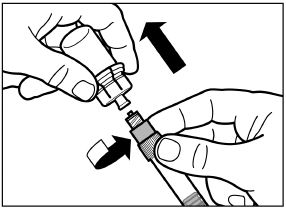
| |
|
| |
|
| |
Schritt 6
Schrauben Sie die Sicherheitsinjektionsnadel auf die Spritze.
|
|

| |
Ziehen Sie die Schutzkappe in gerader Richtung von der Nadel.
Um Sedimentation zu vermeiden, können sie die Spritze vorsichtig schütteln, damit Sie eine einheitliche Suspension beibehalten.
Klopfen Sie mit einem Finger vorsichtig gegen die Spritze, um Luftblasen zu beseitigen, und stossen Sie diese aus der Spritze aus.
Das rekonstituierte Signifor LAR ist nun zur sofortigen Verabreichung bereit.
|
|
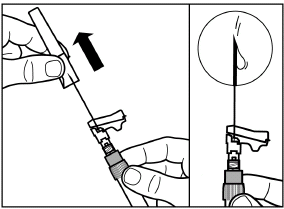
| |
Schritt 7
Signifor LAR darf nur durch tiefe intramuskuläre Injektion verabreicht werden; NIEMALS intravenös.
Desinfizieren Sie die Injektionsstelle mit einem Alkoholtupfer.
Stechen Sie die Nadel vollständig in den linken oder rechten Gesässmuskel (M. glutaeus) in einem Winkel von 90° zur Hautoberfläche.
Ziehen Sie den Kolben langsam zurück (aspirieren), um sicherzustellen, dass kein Blutgefäss angestochen wurde (wenn ein Blutgefäss angestochen wurde, muss eine andere Injektionsstelle gewählt werden).
Drücken Sie den Kolben langsam nach unten, bis die Spritze leer ist. Ziehen Sie die Nadel aus der Injektionsstelle heraus und aktivieren Sie den Nadelschutz (wie in Schritt 8 gezeigt).
|
|
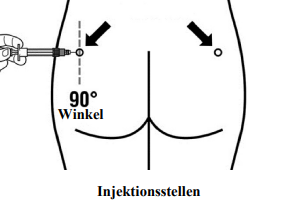
| |
Schritt 8
Aktivieren Sie den Nadelschutz gemäss einer der zwei Vorgehensweisen, die hier beschrieben werden:
·Drücken Sie entweder den klappbaren Abschnitt des Nadelschutzes fest nach unten auf eine harte Oberfläche (Abbildung A)
·oder schieben Sie den klappbaren Abschnitt mit Ihrem Finger nach vorne (Abbildung B).
Ein hörbares "Klicken" bestätigt die korrekte Aktivierung.
Entsorgen Sie die Spritze sofort in einem Sicherheitsbehälter.
|
|

|
Spezielle Vorsichtsmassnahmen für die Entsorgung
Jedes nicht verwendete Produkt oder Abfallmaterial sollte entsprechend den lokalen Anforderungen fachgerecht entsorgt werden.
Zulassungsnummer65148 (Swissmedic)
PackungenSignifor LAR 10 mg Pulver in einer Durchstechflasche, 2ml Lösungsmittel zur Herstellung einer Injektionssuspension in einer Fertigspritze. [A]
Signifor LAR 20 mg Pulver in einer Durchstechflasche, 2ml Lösungsmittel zur Herstellung einer Injektionssuspension in einer Fertigspritze. [A]
Signifor LAR 30 mg Pulver in einer Durchstechflasche, 2ml Lösungsmittel zur Herstellung einer Injektionssuspension in einer Fertigspritze. [A]
Signifor LAR 40 mg Pulver in einer Durchstechflasche, 2ml Lösungsmittel zur Herstellung einer Injektionssuspension in einer Fertigspritze. [A]
Signifor LAR 60 mg Pulver in einer Durchstechflasche, 2ml Lösungsmittel zur Herstellung einer Injektionssuspension in einer Fertigspritze. [A]
ZulassungsinhaberinRecordati AG, 6340 Baar
Stand der InformationFebruar 2025
|