ZusammensetzungWirkstoffe
Fibrinogen vom Menschen.
Hilfsstoffe
Argininhydrochlorid, Isoleucin, Lysinhydrochlorid, Glycin, Natriumcitratdihydrat (E331). Jede Durchstechflasche enthält maximal 69 mg Natrium.
Lösungsmittel: Wasser für Injektionszwecke.
1 Schachtel enthält: eine Durchstechflasche mit Pulver (Glas) + eine Durchstechflasche mit 100 ml Lösungsmittel (Glas) mit einem Stopfen (Halobutyl) und einem Transfersystem, das mit einem Entlüfter mit Sterilfilter ausgestattet ist.
Indikationen/AnwendungsmöglichkeitenBehandlung und Prophylaxe von Blutungen bei Patienten in allen Altersgruppen (Erwachsene, Jugendliche und Kinder) mit kongenitaler Hypo-, Dys- oder Afibrinogenämie mit Blutungsneigung.
Als komplementäre Therapie zur Behandlung von unkontrollierten schweren Blutungen bei erworbener Hypofibrinogenämie.
Dosierung/AnwendungDie Behandlung soll unter Aufsicht eines in der Behandlung von Gerinnungsstörungen erfahrenen Arztes erfolgen.
Dosierung
Dosierung und Dauer der Substitutionstherapie richten sich nach der Schwere der Störung, nach Lokalisation und Ausmass der Blutung sowie nach dem klinischen Zustand des Patienten.
Der (funktionelle) Fibrinogenspiegel im Plasma muss zu Beginn gemessen und regelmässig kontrolliert werden, um die für den Patienten geeignete Dosierung und Häufigkeit der Anwendung zu ermitteln und individuell anzupassen. Der klinische Zustand des Patienten und die anderen Substitutionstherapien sollen regelmässig überwacht werden.
Der normale zirkulierende Fibrinogenspiegel liegt im Durchschnitt bei 1,5 bis 4,5 g/l. Der kritische Fibrinogenspiegel, unterhalb dessen Blutungen auftreten können, liegt bei ungefähr 0,5 bis 1 g/l.
Im Falle eines grösseren chirurgischen Eingriffs ist eine enge Überwachung der Behandlung mit Koagulationstests unbedingt erforderlich.
·Konstitutioneller Mangel
Das Ziel der ersten Injektion besteht im Erreichen eines zirkulierenden Fibrinogenspiegels von über 1 g/l. Dieser Spiegel muss bis zur Kontrolle und Stabilisierung der Homöostase aufrechterhalten werden. Bis zur vollständigen Heilung wird sodann ein Spiegel von über 0,5 g/l empfohlen.
Formel für die Berechnung der Initialdosis:
Zu injizierende Menge (g) = (gewünschter Spiegel (g/l) - Ausgangsspiegel (g/l)) ´ 1 / Wiederfindungsrate (g/l)/(g/kg)* ´ Körpergewicht (kg).
„1 / Wiederfindungsrate“ wird anhand der Wiederfindungsrate des Patienten bestimmt (siehe Rubrik „Pharmakokinetik“) oder, wenn die Wiederfindungsrate unbekannt ist:
-0,053 (g/kg)/(g/l) bei Kindern und Jugendlichen < 40 kg
-0,043 (g/kg)/(g/l) bei Erwachsenen und Jugendlichen ³ 40 kg.
* Beispiel für die Berechnung der Wiederfindungsrate des Patienten und der Dosis
Im Falle eines Patienten von 60 kg mit basal unbestimmbarem Fibrinogenspiegel und bei 1,20 g/l eine Stunde nach Infusion von 0,060 g/kg von Clottafact®:
-Berechnung der Wiederfindungsrate des Patienten:
1,20 (g/l) / 0,060 (g/kg) = 20,0 (g/l)/(g/kg)
-Berechnung der Dosis für eine Erhöhung des Spiegels auf 1,0 g/l:
1,0 g/l x 1 / 20,0 (g/l)/(g/kg) [oder 0,050 (g/kg)/(g/l)] x 60 kg = 3 g
Im Notfall, wenn der Grundspiegel des Fibrinogens nicht verfügbar ist, beträgt die empfohlene intravenös verabreichte Anfangsdosis 0,05 g/kg bei Erwachsenen und Jugendlichen ³ 40 kg und muss bei Kindern und Jugendlichen mit einem Körpergewicht < 40 kg bei 0,06 g/kg liegen.
Kinder und Jugendliche
Die Daten zeigen, dass die Wiederfindungsrate und die Halbwertszeit in vivo bei Kindern und Jugendlichen mit einem Körpergewicht von < 40 kg geringer sind als jene bei Erwachsenen und Jugendlichen mit einem Körpergewicht ³ 40 kg (siehe Rubrik „Pharmakokinetik“). Folglich muss die zu verwendende individuelle Wiederfindungsrate, wenn sie unbekannt ist, anhand des Körpergewichts angepasst werden.
Die nachfolgende Dosierung (Dosen und Häufigkeit der Injektionen) wird dem klinischen Zustand und den Laborwerten des Patienten angepasst.
Die Halbwertszeit von Fibrinogen liegt bei 3 bis 4 Tagen. So ist, wenn kein Verbrauch vorliegt, eine wiederholte Behandlung normalerweise nicht erforderlich. Gemäss der beobachteten Akkumulation bei einer wiederholten prophylaktischen Anwendung müssen Dosierung und Häufigkeit für einen bestimmten Patienten entsprechend den therapeutischen Zielen des praktizierenden Arztes festgelegt werden.
·Erworbener Mangel
Erwachsene:
Generell werden zu Beginn 1 bis 2 g verabreicht und können bei Bedarf wiederholt werden. Bei akuten schweren obstetrischen Blutungen können grössere Mengen (4 bis 8 g) Fibrinogen erforderlich sein.
Im Notfall wird bei akuten Blutungen, wenn die Fibrinogenkonzentration nicht vorliegt, zunächst eine initiale Dosis verabreicht, und die nachfolgenden Dosen werden dann auf Grundlage der zwischenzeitlich vorliegenden Konzentrationen angepasst.
Kinder:
Die Dosierung richtet sich nach dem Körpergewicht und der klinischen Situation und beträgt normalerweise 0,02 bis 0,03 g/kg.
Art der Anwendung
Clottafact® ist ein Pulver, das unmittelbar vor der Anwendung mit Wasser für Injektionszwecke wie in der Rubrik „Sonstige Hinweise“ beschrieben, rekonstituiert werden muss.
Clottafact® wird unmittelbar nach der Rekonstitution als Einzeldosis langsam intravenös injiziert.
In einer stabilen klinischen Situation soll die Geschwindigkeit der Injektion von Clottafact® 4 ml/min nicht überschreiten. Bei akuten schweren Blutungen kann die Injektion mit einer Geschwindigkeit von bis zu 20 ml/min erfolgen.
KontraindikationenÜberempfindlichkeit gegenüber dem Wirkstoff oder einem der Hilfsstoffe gemäss Zusammensetzung.
Warnhinweise und VorsichtsmassnahmenThromboembolie
Es besteht ein Thromboserisiko, wenn Patienten mit einem konstitutionellen oder erworbenen Mangel mit Fibrinogen vom Menschen behandelt werden, insbesondere bei Gabe hoher Dosen oder wiederholter Dosierung. Patienten, die Fibrinogen vom Menschen erhalten, sollen engmaschig auf Zeichen oder Symptome einer Thrombose überwacht werden.
Der mögliche Nutzen einer Therapie mit Fibrinogen vom Menschen soll in folgenden Situationen gegen das Risiko thromboembolischer Komplikationen abgewogen werden: bei Patienten mit anamnestisch bekannter koronarer Herzkrankheit oder Myokardinfarkt; Lebererkrankungen; vor, während und nach Operationen; bei Neugeborenen oder bei Patienten, bei denen das Risiko thromboembolischer Ereignisse oder einer Verbrauchskoagulopathie besteht. Eine engmaschige Überwachung ist erforderlich.
Die erworbene Hypofibrinogenämie geht mit niedrigen Plasmakonzentrationen aller Blutgerinnungsfaktoren (nicht nur Fibrinogen) und –inhibitoren einher. Folglich muss die Verwendung von Blutprodukten, die Blutgerinnungsfaktoren enthalten, in Betracht gezogen werden. Eine sorgfältige Überwachung der Gerinnung ist unbedingt erforderlich.
Bei der Behandlung von akuten schweren Blutungen muss die Verordnung von Clottafact® zusammen mit auf den klinischen und biologischen Zustand des Patienten abgestimmten Reanimationsmassnahmen erfolgen.
Allergische und anaphylaktische Reaktionen
Im Falle einer Allergie mit einer Reaktion vom anaphylaktischen Typ muss die Behandlung sofort unterbrochen und bei einem anaphylaktischen Schock eine geeignete symptomatische Therapie eingeleitet werden.
Natriumspiegel
Clottafact® enthält maximal 69 mg Natrium je Durchstechflasche, entsprechend 3,45 % der von der WHO für einen Erwachsenen empfohlenen maximalen täglichen Natriumaufnahme mit der Nahrung von 2g. Dies ist bei Patienten, die eine strenge kochsalzarme Diät halten müssen, zu beachten.
Übertragbare Erreger
Standardmethoden zur Vermeidung des Risikos einer Übertragung infektiöser Erreger durch Arzneimittel, die aus menschlichem Blut oder Plasma hergestellt werden, umfassen die klinische Auswahl der Spender, die Prüfung jeder einzelnen Spende und jedes Plasmapools auf spezifische Infektionsmarker sowie die Einbeziehung von Herstellungsschritten zur wirksamen Inaktivierung/Elimination von Viren. Dennoch kann das Risiko einer Übertragung von infektiösen Erregern bei der Anwendung von aus menschlichem Blut oder Plasma hergestellten Arzneimitteln nicht vollständig ausgeschlossen werden. Dies gilt auch für bisher unbekannte Viren und andere Typen infektiöser Erreger.
Die getroffenen Massnahmen werden für umhüllte Viren wie das humane Immundefizienz-Virus (HIV), das Hepatitis B-Virus (HBV) und das Hepatitis C-Virus (HCV) als wirksam angesehen.
Für nicht-umhüllte Viren wie das Hepatitis A-Virus (HAV) und Parvovirus B19 könnten die getroffenen Massnahmen nur begrenzt wirksam sein. Eine Infektion mit Parvovirus B19 kann für Foeten und für Patienten mit bestimmten Anämietypen oder Immunmangelzuständen schwerwiegende Folgen haben.
Für Patienten, die regelmässig Fibrinogen erhalten, wird eine geeignete Impfung (Hepatitis A und B) empfohlen.
Immunogenizität
Eine Antikörperbildung kann bei Patienten im Fall einer Substitutionstherapie mit Gerinnungsfaktoren bei anderen konstitutionellen Mangelzuständen beobachtet werden. In Verbindung mit Fibrinogen wurde bislang jedoch über keinen derartigen Fall berichtet.
Kinder und Jugendliche
Die gleichen Warnhinweise und Vorsichtsmassnahmen gelten auch bei Verwendung bei Kindern und Jugendlichen.
InteraktionenInteraktionen von Clottafact® mit anderen Arzneimitteln sind bisher nicht bekannt. Dennoch wird von einer vorherigen Mischung mit anderen Produkten oder Arzneimitteln kategorisch abgeraten.
Schwangerschaft, StillzeitDie Sicherheit der Anwendung von menschlichem Fibrinogen während der Schwangerschaft und Stillzeit wurde bislang nicht in kontrollierten klinischen Studien geprüft. Es wurden keine tierexperimentellen Studien durchgeführt.
Klinische Erfahrungen bei der Behandlung von Geburtskomplikationen weisen jedoch darauf hin, dass keine schädigenden Effekte auf den Verlauf der Schwangerschaft oder auf die foetale oder neonatale Entwicklung zu erwarten sind.
Wirkung auf die Fahrtüchtigkeit und auf das Bedienen von MaschinenClottafact® hat keinen Einfluss auf die Fahrtüchtigkeit oder die Fähigkeit, Maschinen zu bedienen.
Unerwünschte WirkungenTabelle mit unerwünschten Wirkungen
Die in der nachfolgenden Tabelle aufgeführten unerwünschten Wirkungen wurden anhand von klinischen Daten von 47 Patienten mit konstitutionellem Fibrinogenmangel berichtet und stammen aus drei interventionellen klinischen Studien und einer nicht-interventionellen Verträglichkeitsstudie nach der Markteinführung. In diesen Studien wurden 39 unerwünschte Wirkungen bei 14/47 Patienten (29,8%) gemeldet, die insgesamt 631 Infusionen von Clottafact® erhalten haben.
Die nach der Verabreichung von Clottafact® am häufigsten beobachtete Reaktion war Kopfschmerz, der bei 1,4 % der Infusionen (9/631 Infusionen) auftrat. Er war in allen Fällen von leichter bis mittelschwerer Intensität, trat innerhalb von 48 Stunden nach der Infusion auf und klang ohne Folgen ab.
Die Tabelle enthält zudem unerwünschte Wirkungen, die bei 172 Patienten mit erworbenem Fibrinogenmangel berichtet wurden und aus einer interventionellen klinischen Studie und einer nicht-interventionellen Verträglichkeitsstudie nach der Markteinführung stammen. In diesen Studien wurden 5 unerwünschte Wirkungen bei 2/172 Patienten (1,2%) gemeldet, die insgesamt 249 Infusionen von Clottafact® erhalten haben. Im Gesamten wurden 44 unerwünschte Wirkungen bei 16/219 Patienten (7,3%) gemeldet, die insgesamt 880 Infusionen von Clottafact® erhalten haben.
Diese Tabelle wird entsprechend der Klassierung der System Organ Class and Preferred Term Level der MedDRA vorgelegt. Die Häufigkeit wurde pro Infusion nach folgender Klassifikation geschätzt: sehr häufig (³1/10); häufig (³1/100, <1/10); gelegentlich (³1/1.000, <1/100); selten (³1/10.000, <1/1.000); sehr selten (<1/10.000).
Tabelle: Unerwünschte Wirkungen, die bei Erwachsenen sowie bei Kindern und Jugendlichen beobachtet wurden
|
MedDRA-System/Organklasse
|
Unerwünschte Wirkungen
|
Häufigkeit pro Infusion
| |
Störungen des Immunsystems
|
Allergische/anaphylaktische Reaktionen (darunter anaphylaktischer Schock, Blässe, Erbrechen, Husten, Blutdruckabfall, Schüttelfrost, Urtikaria)
|
Gelegentlich
| |
Störungen des Nervensystems
|
Kopfschmerzen
Schwindel
|
Häufig
Gelegentlich
| |
Funktionsstörungen des Ohrs und des Innenohrs
|
Tinnitus
|
Gelegentlich
| |
Funktionsstörungen der Gefässe
|
Thromboembolische Ereignisse (darunter tiefe Venenthrombose, oberflächliche Thrombophlebitis) (siehe Abschnitt „Warnhinweise und Vorsichtsmassnahmen“)
|
Gelegentlich
| |
Atmungsorgane (respiratorische, thorakale und mediastinale Funktionsstörungen)
|
Asthma,
Lungenembolie
|
Gelegentlich
| |
Gastrointestinale Störungen
|
Erbrechen*
|
Gelegentlich
| |
Funktionsstörungen der Haut und des Unterhautzellgewebes
|
Erythematöser Hautausschlag,
|
Gelegentlich
| |
Erythem,
|
| |
Hautreizung,
|
| |
nächtliches Schwitzen
|
| |
Allgemeine Störungen und Reaktionen an der Applikationsstelle
|
Wärmegefühl
|
Gelegentlich
|
* Erbrechen in Verbindung mit Kopfschmerzen
Zur Sicherheit im Hinblick auf übertragbare Erreger siehe Abschnitt „Warnhinweise und Vorsichtsmassnahmen“.
Kinder und Jugendliche:
Unter den 47 Patienten mit konstitutionellem Fibrinogenmangel, die in die Verträglichkeitsanalyse einbezogen wurden, befanden sich 26 Patienten unter 18 Jahren, davon waren 21 Patienten unter 12 Jahre und 10 Patienten unter 6 Jahre alt.
Das globale Verträglichkeitsprofil bei Erwachsenen unterscheidet sich nicht von dem bei Kindern und Jugendlichen.
Unter den 172 Patienten mit erworbenem Fibrinogenmangel, die in die Verträglichkeitsanalyse einbezogen wurden, befanden sich 2 Patienten unter 18 Jahren (zwischen 12 und 17 Jahren).
Das Sicherheitsprofil bei Erwachsenen unterscheidet sich nicht von dem bei Kindern und Jugendlichen.
ÜberdosierungBisher wurde über keinen Fall einer versehentlichen Überdosierung berichtet. Angesichts der physiologischen Eigenschaften von Fibrinogen kann jedoch ein Risiko thromboembolischer Komplikationen nicht ausgeschlossen werden.
Um eine Überdosierung zu vermeiden, ist eine regelmässige Überwachung der Plasmaspiegel von Fibrinogen während der Behandlung angezeigt. Im Fall einer Überdosierung ist die Gefahr einer thromboembolischen Komplikation erhöht.
Eigenschaften/WirkungenATC-Code
B02BB01
Wirkungsmechanismus / Pharmakodynamik
Als Injektion verabreichtes humanes Fibrinogen (Koagulationsfaktor I) wird unter dem Einfluss von Thrombin in Fibrin umgewandelt und bildet in Gegenwart von Faktor-XIII-Aktivität und Kalziumionen ein stabiles dreidimensionales Fibrinnetz, das die Gerinnung sicherstellt.
Die Verabreichung von humanem Fibrinogen löst eine Erhöhung des Plasmaspiegels von Fibrinogen aus und kann die Beeinträchtigung der Gerinnung bei Patienten mit Fibrinogendefizit vorübergehend korrigieren.
Drei klinische, offene, nicht kontrollierte Multicenter-Studien (eine bei Erwachsenen, eine bei Erwachsenen, Jugendlichen und Kindern und eine bei Kindern) haben die klinische Pharmakologie, die Sicherheit und Wirksamkeit von Clottafact® bei kongenitaler Hypofibrinogenämie evaluiert. Zudem wurde nach der Markteinführung eine nichtinterventionelle Verträglichkeitsstudie mit Erwachsenen, Kindern und Jugendlichen durchgeführt. Der „klinische Pharmakologie“-Teil jeder klinischen Studie (siehe Rubrik „Pharmakokinetik“) hat im Gesamten 31 Patienten mit Afibrinogenämie eingeschlossen, die eine auf 0,06 g/kg Clottafact® festgelegte Einmaldosis erhielten. Eine Normalisierung der globalen Koagulationstests (aktivierte partielle Thromboplastinzeit [aPTT] und Prothrombinzeit [PT]) wurde bei Fibrinogenspiegeln von und über 0,5 g/l erreicht. Die Änderung des Medians der maximalen Gerinnselfestigkeit (maximum clot firmness (MCF)) vor der Infusion bis eine Stunde nach der Infusion betrug 6,3 mm bei Patienten mit einem Körpergewicht < 40 kg und 10,0 mm bei Patienten mit einem Körpergewicht von ³ 40 kg.
Klinische Wirksamkeit
Erwachsene
In allen Studien zum kongenitalen Defizit von Fibrinogen waren in den Studien zur Wirksamkeit 19 Patienten ³ 18 Jahre mit einem Median von 30 Jahren (über einen Bereich von 19 – 78 Jahren) entweder zur Behandlung bei Bedarf oder für einen chirurgischen Eingriff eingeschlossen. Davon hatten 18 Patienten eine Afibrinogenämie und ein Patient Dysfibrinogenämie. Clottafact® wurde verabreicht für:
-74 nicht-chirurgische hämorrhagische Episoden bei 12 Patienten (davon 6 grössere Vorfälle bei 3 Patienten)
-24 chirurgische Eingriffe bei 8 Patienten (davon 8 schwere chirurgische Eingriffe bei 5 Patienten)
Die Mehrzahl (94,9%) der Ereignisse (93/98) wurde mit einer Einzeldosis von Clottafact® (0,050 g/kg für hämorrhagische Episoden und 0,055 g/kg für chirurgische Eingriffe) behandelt.
Kinder und Jugendliche
Die Analyse der klinischen Wirksamkeit in Interventionsstudien basiert auf 20 pädiatrischen Patienten von weniger als 18 Jahren (Alter 1 bis 17) mit Afibrinogenämie, die Clottafact® in 80 Fällen entweder zur Behandlung bei Bedarf oder für einen chirurgischen Eingriff erhielten.
14 Patienten wurden mit Clottafact® bei 55 Blutungsepisoden und 15 Patienten bei 25 chirurgischen Eingriffen behandelt. Die Mediandosis pro Infusion betrug 0,064 g/kg bei Blutungen in einer Patientenpopulation mit einem mittleren Körpergewicht von 30 kg und 0,069 g/kg bei chirurgischen Eingriffen in einer Patientenpopulation mit einem mittleren Körpergewicht von 26 kg. Die Mehrzahl (90,0%) der Fälle (72/80) wurde mit einer Einzeldosis von Clottafact® abgeschlossen.
In einer Studie nach Markteinführung erhielten 9 Patienten (einschliesslich 4 Kinder) eine Langzeitprophylaxe über mindestens 12 Monate bei einer Mediandosis von 0,059 g/kg mit einem Medianintervall von 7,6 Tagen zwischen zwei Verabreichungen.
Bei Patientinnen mit einer schweren frühen postpartalen Hämorrhagie (Blutungen bei der Entbindung) induzierte die Gabe von 3 g Clottafact® eine Zunahme der mittels Thromboelastographie gemessenen Gerinnungsfähigkeit des Plasmas. Diese Behandlung ermöglichte eine Blutungskontrolle und einen Verzicht auf invasive Behandlungen (Embolisation oder Ligatur der Uterusarterien, Hysterektomie) in 75% der Fälle.
PharmakokinetikAbsorption / Distribution
Im Plasma beträgt die biologische Halbwertszeit von Fibrinogen 3 bis 4 Tage.
Das Arzneimittel wird intravenös verabreicht und ist im Plasma sofort in einer Konzentration verfügbar, die der verabreichten Dosis entspricht.
In einer klinisch-pharmakologischen Studie wurden 14 erwachsene Patienten und Jugendliche mit einer Afibrinogenämie während 14 Tagen untersucht. Nach der Infusion einer Dosis von 0,06 g/kg von Clottafact® betrug das geometrische Mittel (geometrischer Variationskoeffizient (%)) der Wiederfindungsrate in vivo 93,6% (21). Die maximale Konzentration von Fibrinogen in Aktivität erreichte nach 1 Stunde den Maximalwert von 1,4 g/l (24), worauf eine ungefähr 3- bis 4-tägige Phase langsamen exponentiellen Rückgangs bis zur kritischen Fibrinogenschwelle von 0,5 g/l folgte. Eine nichtkompartementale Analyse ermöglichte die Bestimmung des geometrischen Mittels (geometrischer Variationskoeffizient (%)) der Fläche unter Kurve von 0 bis unendlich von 114 g·h/l (23) und der Halbwertszeit von 69,3 Stunden (22). Das humane Fibrinogen wird weitgehend im vaskulären Kompartiment zurückgehalten: Das geometrische Mittel (geometrischer Variationskoeffizient (%)) des Verteilvolumens betrug 50,7 ml/kg (17).
Metabolismus
keine Angaben
Elimination
Ein pharmakokinetisches Modell der Population, das den allometrischen Ansatz (Körpergewicht) integriert, wurde aufgrund der Daten, die bei 31 Patienten im Alter von 1 bis 48 mit Afibrinogenämie gesammelt wurden, entwickelt: Die aus dem Modell hergeleiteten Parameter werden in Tabelle 2 vorgelegt. Die Kinder und Jugendlichen mit einem Körpergewicht < 40 kg wiesen eine Stunde nach der Injektion eine höhere Clearance, eine kürzere Halbwertszeit und eine schwächere Wiederfindung auf als Jugendliche und Erwachsene mit einem Körpergewicht von ³ 40 kg.
Tabelle 2 : Zusammenfassung der pharmakokinetischen Parameter von Clottafact® (Fibrinogen gemessen in Aktivität) aus der pharmakokinetischen Analyse der Population nach Injektion von 0,06 g/kg und der Wiederfindungsrate eine Stunde nach Injektion, nach Altersgruppen und Körpergewicht
Geometrisches Mittel (geometrischer Variationskoeffizient (%))
|
|
Patienten
< 40 kg
|
Patienten
³ 40 kg
|
Kinder
£ 6 Jahre
|
Kinder
7 bis 12
Jahre
|
Jugendliche
13 bis < 18
Jahre
|
Erwachsene
³ 18 Jahre
| |
Anzahl Patienten
|
12
|
19
|
6
|
8
|
3
|
14
| |
AUC0-∞ (g·h/l)
|
81 (23)
|
133 (26)
|
74 (15)
|
93 (23)
|
144 (30)
|
136 (26)
| |
Cl (ml/h/kg)
|
0,74 (23)
|
0,45 (25)
|
0,81 (15)
|
0,65 (23)
|
0,43 (27)
|
0,44 (25)
| |
t1/2 (h)
|
49,0 (12)
|
66,7 (19)
|
46,6 (10)
|
52,1 (10)
|
64,2 (10)
|
69,3 (20)
| |
MRT (h)
|
70,7 (12)
|
96,2 (13)
|
67,3 (10)
|
75,2 (10)
|
92,6 (10)
|
100,0 (20)
| |
Vss (ml/kg)
|
52,2 (16)
|
43,2 (17)
|
54,4 (10)
|
48,6 (19)
|
39,5 (19)
|
43,8 (18)
| |
|
|
|
|
|
|
| |
Fibrinogenkonzentration nach 1 Stunde (g/l)
|
1,15 (20)
|
1,40 (22)
|
1,12 (14)
|
1,21 (24)
|
1,56 (27)
|
1,38 (23)
| |
Wiederfindungsrate nach 1 Stunde (g/l pro g/kg)
|
19,1 (20)
|
23,3 (21)
|
18,7 (14)
|
20,1 (24)
|
25,4 (24)
|
23,1 (22)
| |
AUC0-∞: Fläche unter der Kurve (Area under the curve) von 0 bis unendlich, Cl: Clearance, t1/2: Halbwertszeit, MRT: Mittlere Verweilzeit (Mean residence time), Vss: Distributionsvolumen bei Steady State
|
Kinetik spezieller Patientengruppen
keine Angaben
Präklinische DatenDer Wirkstoff von Clottafact® ist ein normaler Bestandteil des menschlichen Plasmas und wirkt wie physiologisches Fibrinogen.
Die präklinischen Daten aus Studien zur akuten Toxizität, lokalen Verträglichkeit, subakuten Toxizität, Mutagenität und zum thrombogenen Potenzial lassen keine besonderen Gefahren für den Menschen erkennen.
Es wurden keine Studien zur Reproduktionstoxizität oder Kanzerogenität durchgeführt.
Sonstige HinweiseInkompatibilitäten
Clottafact® darf nicht mit anderen Arzneimitteln gemischt werden und muss auf separatem Injektions-/Infusionsweg verabreicht werden.
Haltbarkeit
Das Arzneimittel darf nur bis zu dem auf der Packung mit „EXP“ bezeichneten Datum verwendet werden.
Besondere Lagerungshinweise
Das Arzneimittel muss nach Rekonstitution sofort verwendet werden und darf nicht aufbewahrt werden.
Nicht über 25°C lagern und ausser Reichweite von Kindern aufbewahren. Den Behälter im Umkarton aufbewahren, um den Inhalt vor Licht zu schützen. Nicht einfrieren.
Hinweise für die Handhabung
Rekonstitution:
Die übliche aseptische Vorgehensweise ist einzuhalten.
|
|
|
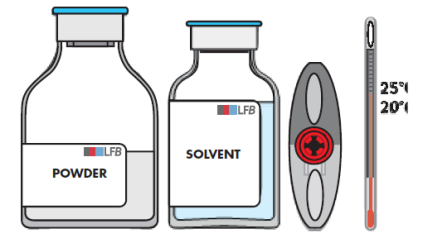
Falls nötig, lassen Sie die beiden Durchstechflaschen (Pulver und Lösungsmittel) Raumtemperatur erreichen.
| |
|
|
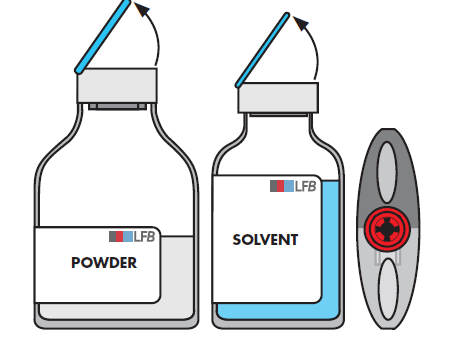
Entfernen Sie die Schutzkappe von der Durchstechflasche mit dem Lösungsmittel und der Durchstechflasche mit dem Pulver.
Desinfizieren Sie die Oberfläche der beiden Stopfen.
| |
|
|
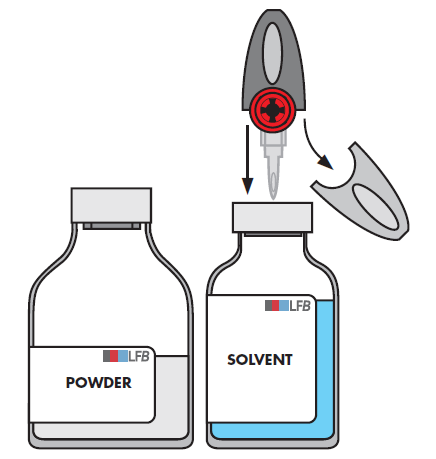
Entfernen Sie den durchsichtigen Schutzdeckel des Transfersystems und stechen Sie den dadurch freigelegten Dorn mit einer Drehbewegung fest in die Mitte des Stopfens der Durchstechflasche mit dem Lösungsmittel ein.
| |
|
|
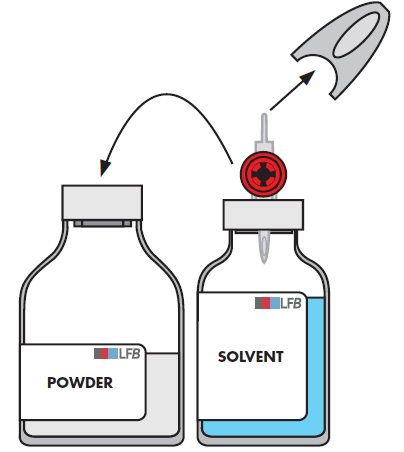
Entfernen Sie den zweiten, grauen Schutzdeckel am anderen Ende des Transfersystems.
Drehen Sie die Durchstechflasche mit dem Lösungsmittel um und stechen Sie mit einer schnellen Bewegung das freigelegte Ende des Dorns in die Mitte des Stopfens der Durchstechflasche mit dem Pulver ein, sodass das Lösungsmittel in das Pulver überführt werden kann.
Achten Sie darauf, dass der Dorn stets mit Lösungsmittel bedeckt ist, damit das Vakuum nicht zu früh aufgehoben wird.
| |
|
|
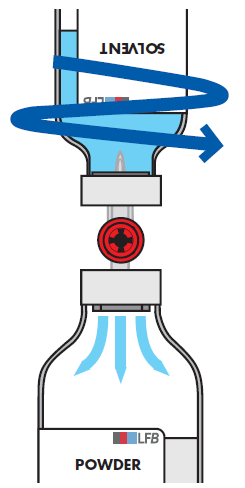
Richten Sie den Lösungsmittelstrahl während des Überleitens durch horizontal kreisende Bewegungen auf die gesamte Pulveroberfläche und gegen die Wand der Durchstechflasche. Achten Sie darauf, dass Sie das gesamte Lösungsmittel in die Durchstechflasche mit dem Pulver überführen.
Nach Abschluss des Übertragungsvorgangs wird das Vakuum automatisch durch sterile Luft aufgehoben, die aus dem Transfersystem eindringt.
| |
|
|
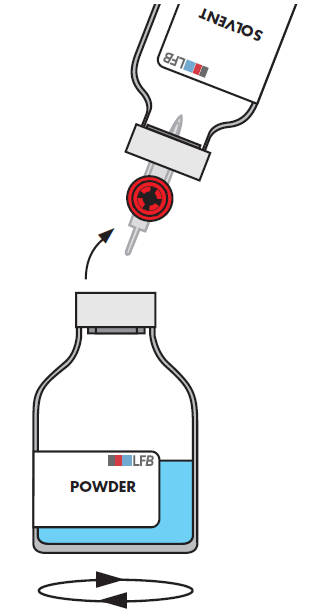
Entfernen Sie die leere Durchstechflasche (Lösungsmittel) zusammen mit dem Transfersystem.
Schwenken Sie die Durchstechflasche mit dem Pulver sanft mit kreisenden Bewegungen, um eine Schaumbildung zu vermeiden, bis sich das Pulver vollständig aufgelöst hat.
|
Das rekonstituierte Produkt muss vor der Verabreichung in Augenschein genommen werden, um sicherzustellen, dass keine Partikel enthalten sind. Die rekonstituierte Lösung erscheint mehr oder weniger opaleszent.
Lösung nicht verwenden, wenn sie trübe erscheint oder Ablagerungen aufweist.
Verabreichung
Clottafact® darf nur intravenös angewendet werden und muss unmittelbar nach der Rekonstitution als Einzeldosis mit einer Injektionsgeschwindigkeit von höchstens 4 ml/min verabreicht werden. Bei starken Blutungen kann die Injektion mit einer Geschwindigkeit von bis zu 20 ml/min erfolgen.
Die rekonstituierte Lösung muss sofort nach Rekonstitution verabreicht werden.
Es wird empfohlen, ein Infusionsset mit einem 15µm-Filter zu verwenden.
Nicht verwendetes Arzneimittel oder Abfallmaterial ist entsprechend zu entsorgen.
Zulassungsnummer66184 (Swissmedic)
PackungenClottafact® Pulver 1,5 g/100 ml Durchstechflasche Lösungsmittel (D 100 ml) 1 [B]
ZulassungsinhaberinOpopharma Vertriebs AG
8153 Rümlang
Stand der InformationFebruar 2023
|