ZusammensetzungWirkstoffe
Inclisiran (als Inclisiran-Natrium)
Hilfsstoffe
Wasser für Injektionszwecke, Natriumhydroxid (zur pH-Einstellung), Phosphorsäure 85% (zur pH-Einstellung).
Darreichungsform und Wirkstoffmenge pro EinheitInjektionslösung in einer Fertigspritze (Injektion).
Die Lösung ist klar, farblos bis blassgelb und im Wesentlichen frei von Partikeln.
Jeder ml enthält Inclisiran-Natrium, das 189 mg Inclisiran entspricht.
Jede Fertigspritze enthält 1,5 ml Lösung mit 284 mg Inclisiran (entspricht 300 mg Inclisiran-Natrium).
Indikationen/AnwendungsmöglichkeitenHypercholesterinämie und gemischte Dyslipidämie
Leqvio ist bei Erwachsenen mit Hypercholesterinämie [einschliesslich heterozygoter familiärer Hypercholesterinämie] oder gemischter Dyslipidämie begleitend zu einer Diät indiziert:
in Kombination mit einer maximal tolerierten Statin-Dosis mit oder ohne andere lipidsenkende Therapien bei Patienten, die eine zusätzliche Low Density Lipoprotein Cholesterin (LDL-C) Senkung benötigen, oder
allein oder in Kombination mit anderen lipidsenkenden Therapien bei Patienten, die statinintolerant sind oder für die Statine kontraindiziert sind.
Die Wirkung von Leqvio auf die kardiovaskuläre Morbidität und Mortalität ist bisher noch nicht bestimmt.
Dosierung/AnwendungÜbliche Dosierung
Hypercholesterinämie und gemischte Dyslipidämie
Die empfohlene Dosierung von Leqvio beträgt 284 mg als einzelne subkutane Injektion zu Behandlungsbeginn, nach 3 Monaten und dann alle 6 Monate.
Verpasste Behandlungen
Wenn eine geplante Behandlung um weniger als 3 Monate verpasst wird, sollte Leqvio verabreicht und der Behandlungsablauf gemäss dem ursprünglichen Zeitplan des Patienten fortgesetzt werden.
Wenn eine geplante Behandlung um mehr als 3 Monate verpasst wird, sollte mit einem neuen Behandlungsplan begonnen werden. Leqvio sollte zu Beginn verabreicht werden, dann erneut nach 3 Monaten und danach alle 6 Monate.
Behandlungsumstellung von monoklonalen Antikörper-PCSK9-Inhibitoren
Leqvio kann unmittelbar nach der letzten Behandlung mit einem monoklonalen Antikörper-PCSK9-Inhibitor verabreicht werden. Um die LDL-C-Senkung beizubehalten, wird empfohlen, Leqvio innerhalb von 2 Wochen nach der letzten Behandlung mit einem monoklonalen Antikörper-PCSK9-Inhibitor zu verabreichen.
Spezielle Dosierungsanweisungen
Patienten mit Leberfunktionsstörungen
Bei Patienten mit einer leichten bis mittelschweren Leberfunktionsstörung ist keine Dosisanpassung erforderlich. Es liegen keine Daten für Patienten mit schwerer Leberfunktionsstörung vor. (Siehe "Pharmakokinetik" ). Bei Patienten mit schwerer Leberfunktionsstörung (Child-Pugh Klasse C) wird die Behandlung mit Leqvio daher nicht empfohlen.
Patienten mit Nierenfunktionsstörungen
Bei Patienten mit leichter, mittelschwerer oder schwerer Nierenfunktionsstörung und bei Patienten mit einer Nierenerkrankung im Endstadium sind keine Dosisanpassungen erforderlich. (Siehe "Pharmakokinetik" ). Es liegen nur begrenzte Erfahrungen mit Leqvio bei Patienten mit schwerer Nierenfunktionsstörung vor. Leqvio sollte bei diesen Patienten mit Vorsicht angewendet werden.
Ältere Patienten
Bei älteren Patienten (≥65 Jahre) ist keine Dosisanpassung erforderlich.
Kinder und Jugendliche
Die Sicherheit und Wirksamkeit von Leqvio bei Kindern und Jugendlichen im Alter von unter 18 Jahren wurde nicht untersucht. Es liegen keine Daten vor.
Art der Anwendung
Subkutane Anwendung.
Leqvio ist zur subkutanen Injektion in den Bauchbereich vorgesehen; alternative Injektionsstellen sind der Oberarm oder Oberschenkel. Das Arzneimittel sollte nicht in Bereiche mit einer aktiven Hauterkrankung oder Verletzung, wie zum Beispiel Sonnenbrand, Hautausschläge, Entzündungen oder Hautinfektionen, injiziert werden.
Jede 284-mg-Dosis wird mit einer einzelnen Fertigspritze verabreicht. Jede Fertigspritze ist nur zum Einmalgebrauch bestimmt.
Leqvio ist für die Verabreichung durch medizinisches Fachpersonal vorgesehen.
KontraindikationenÜberempfindlichkeit gegen den Wirkstoff oder einen der sonstigen Bestandteile.
Warnhinweise und VorsichtsmassnahmenNierenfunktionsstörung
Der Einfluss einer Hämodialyse auf die Pharmakokinetik von Inclisiran wurde nicht untersucht. Da Inclisiran renal eliminiert wird, sollte für mindestens 72 Stunden nach der Verabreichung von Leqvio keine Hämodialyse durchgeführt werden.
Leberfunktionsstörung
Patienten mit schwerer Leberfunktionsstörung (Child-Pugh Klasse C) wurden nicht untersucht (siehe "Pharmakokinetik" ).
Natriumgehalt
Dieses Arzneimittel enthält weniger als 1 mmol Natrium (23 mg) pro Dosis, d.h. es ist nahezu "natriumfrei" .
InteraktionenInclisiran ist kein Substrat von üblichen Arzneistofftransportern, und, obwohl keine in vitro Studien durchgeführt wurden, wird nicht erwartet, dass es ein Substrat von Cytochrom P450 ist. Inclisiran ist kein Inhibitor oder Induktor von Cytochrom-P450-Enzymen (einschliesslich CYP1A2, CYP2B6, CYP2C8, CYP2C9, CYP2C19, CYP2D6 oder CYP3A4/5) oder üblichen Arzneistofftransportern (einschliesslich OAT1, OAT3, OCT1, OCT2, OCT3, OATP1B1, OATP1B3 oder P-gp). Daher ist nicht zu erwarten, dass Leqvio klinisch signifikante Wechselwirkungen mit anderen Arzneimitteln verursacht.
Basierend auf den begrenzten verfügbaren Daten werden keine klinisch relevanten Wechselwirkungen mit Atorvastatin, Rosuvastatin oder anderen Statinen erwartet.
Schwangerschaft, StillzeitSchwangerschaft
Bisher liegen keine oder nur begrenzte Erfahrungen mit der Anwendung von Inclisiran bei schwangeren Frauen vor. Tierexperimentelle Studien ergaben keine Hinweise auf direkte oder indirekte Reproduktionstoxizität (siehe "Präklinische Daten" ). Leqvio sollte während einer Schwangerschaft nicht angewendet werden, es sei denn, der klinische Zustand der Frau erfordert eine Behandlung mit Inclisiran.
Stillzeit
Es ist nicht bekannt, ob Inclisiran in die menschliche Muttermilch übergeht. Die zur Verfügung stehenden pharmakodynamischen/toxikologischen Daten aus tierexperimentellen Studien zeigten, dass Inclisiran in die Milch übergeht (siehe "Präklinische Daten" ). Ein Risiko für Neugeborene/Säuglinge kann nicht ausgeschlossen werden.
Daher muss abgewogen werden, abzustillen oder die Therapie mit Leqvio abzubrechen/auszusetzen, wobei der Nutzen des Stillens für das Kind und der Therapienutzen für die Frau zu berücksichtigen sind.
Fertilität
Es liegen keine Daten zu Effekten von Inclisiran auf die Fertilität beim Menschen vor. Tierexperimentelle Studien zeigten keine Auswirkungen auf die Fertilität bei Expositionswerten, die viel höher waren als bei Patienten, die an klinischen Studien teilnahmen (siehe "Präklinische Daten" ).
Wirkung auf die Fahrtüchtigkeit und auf das Bedienen von MaschinenLeqvio hat keinen oder einen zu vernachlässigenden Einfluss auf die Verkehrstüchtigkeit und die Fähigkeit zum Bedienen von Maschinen.
Unerwünschte WirkungenZusammenfassung des Sicherheitsprofils
Gemäss den Sicherheitsdaten aus den drei zulassungsrelevanten placebokontrollierten Phase-III-Studien traten bei Patienten der Inclisiran-Gruppe und der Placebo-Gruppe therapieassoziierte unerwünschte Ereignisse (TAUE) mit ähnlicher Inzidenz auf. Die Mehrheit der therapieassoziierten unerwünschten Ereignisse war leicht und stand nicht mit Inclisiran oder Placebo in Zusammenhang. Die einzigen Nebenwirkungen, die mit Inclisiran assoziiert wurden, waren in den zulassungsrelevanten Studien unerwünschte Ereignisse an der Injektionsstelle (8.2 %).
Die mit Inclisiran assoziierten Nebenwirkungen leiten sich aus den entsprechenden Berichten aus drei Zulassungsstudien ab, an denen 3'655 Patienten mit atherosklerotischen kardiovaskulären Erkrankungen (atherosclerotic cardiovascular disease, ASCVD, vergleichbaren Risiken (ASCVD-risikoäquivalent) oder familiärer Hypercholesterinämie teilnahmen, die mit Statinen in maximal verträglicher Dosis und Inclisiran oder Placebo behandelt wurden, einschliesslich 1'833 Patienten, die Inclisiran erhielten, und 1'822 Patienten, die bis zu 18 Monate lang Placebo erhielten (durchschnittliche Behandlungsdauer mit Inclisiran von 526 Tagen), und werden unten tabellarisch aufgeführt.
Die Nebenwirkungen sind nach Systemorganklasse aufgeführt. Die Häufigkeitskategorien sind folgendermassen definiert: Sehr häufig (≥1/10); häufig (≥1/100 bis < 1/10); gelegentlich (≥1/1'000 bis < 1/100); selten (≥1/10'000 bis < 1/1'000); sehr selten (< 1/10'000) und unbekannt (Häufigkeit auf Grundlage der vorhandenen Daten nicht abschätzbar).
Allgemeine Erkrankungen und Beschwerden am Verabreichungsort
Häufig: Unerwünschte Ereignisse an der Injektionsstelle1
1Siehe Abschnitt "Beschreibung ausgewählter Nebenwirkungen"
Beschreibung ausgewählter Nebenwirkungen
Unerwünschte Ereignisse an der Injektionsstelle
Unerwünschte Ereignisse an der Injektionsstelle traten bei 8,2 % bzw. 1,8 % der mit Inclisiran und Placebo behandelten Patienten in den Zulassungsstudien auf. Der Anteil der Patienten, welche die Behandlung wegen unerwünschter Ereignisse an der Injektionsstelle bei den mit Inclisiran und den mit Placebo behandelten Patienten abbrachen, betrug 0,2 % bzw. 0,0 %. Alle diese unerwünschten Ereignisse waren leicht oder mässig in ihrem Schweregrad, vorübergehend und heilten ohne Folgeerscheinungen vollständig ab. Die am häufigsten auftretenden unerwünschten Ereignisse an der Injektionsstelle bei Patienten, die mit Inclisiran behandelt wurden, waren Reaktionen an der Injektionsstelle (3,1 %), Schmerzen an der Injektionsstelle (2,2 %), Erythem an der Injektionsstelle (1,6 %) und Hautausschlag an der Injektionsstelle (0,7 %).
Besondere Patientengruppen
Ältere Patienten
Von den 1'833 Patienten, die in den Zulassungsstudien mit Leqvio behandelt wurden, waren 981 (54 %) 65 Jahre oder älter und 239 (13 %) 75 Jahre oder älter. Es wurden keine grundsätzlichen Unterschiede im Hinblick auf die Sicherheit und Wirksamkeit zwischen älteren und jüngeren Patienten beobachtet.
Immunogenität
In den Zulassungsstudien wurden bei 1'830 Patienten Proben auf Antikörper gegen Inclisiran getestet. Ein positives Testergebnis wurde für 1,8 % (33/1.830) der Patienten vor Behandlungsbeginn und 4,9 % (90/1.830) der Patienten während der 18-monatigen Behandlung mit Inclisiran bestätigt. Bei den Patienten, die positiv auf Antikörper gegen Inclisiran getestet wurden, wurden keine klinisch signifikanten Unterschiede bei der klinischen Wirksamkeit, Sicherheit und im pharmakodynamischen Profil von Leqvio beobachtet.
Laborwerte
In den klinischen Phase-III-Studien wurden bei Patienten, die Inclisiran erhielten, häufigere Erhöhungen der hepatischen Transaminasen im Serum zwischen > 1x des Upper limit of normal (ULN) und ≤3x ULN festgestellt (ALT: 19,7 % und AST: 17,2 %) als bei Patienten, die Placebo erhielten (ALT: 13,6 % und AST: 11,1 %). Diese Erhöhungen gingen nicht über die klinisch relevante Schwelle von einer Erhöhung um das 3-Fache der oberen Normgrenze hinaus, waren asymptomatisch und nicht mit Nebenwirkungen oder anderen Anzeichen einer Leberfunktionsstörung verbunden.
Meldung des Verdachts auf Nebenwirkungen
Die Meldung des Verdachts auf Nebenwirkungen nach der Zulassung ist von grosser Wichtigkeit. Sie ermöglicht eine kontinuierliche Überwachung des Nutzen-Risiko-Verhältnisses des Arzneimittels. Angehörige von Gesundheitsberufen sind aufgefordert, jeden Verdacht einer neuen oder schwerwiegenden Nebenwirkung über das Online-Portal ElViS (Electronic Vigilance System) anzuzeigen. Informationen dazu finden Sie unter www.swissmedic.ch.
ÜberdosierungBei gesunden Freiwilligen, die Inclisiran in Dosisstärken bis zum Dreifachen der therapeutischen Dosis erhielten, wurden keine klinisch relevanten unerwünschten Ereignisse beobachtet.
Behandlung
Es gibt keine spezifische Behandlung bei einer Überdosierung von Leqvio. Im Fall einer Überdosierung sollten der Patient symptomatisch behandelt und entsprechende unterstützende Massnahmen ergriffen werden.
Eigenschaften/WirkungenATC-Code
C10AX16.
Wirkungsmechanismus
Inclisiran ist eine cholesterinsenkende kurze doppelsträngige interferierende Ribonukleinsäure, genannt siRNA (small interfering ribonucleid acid), die auf dem Sense-Strang mit einem triantennärem N-Acetylgalactosamin (GalNAc) konjugiert ist, um die Aufnahme in Leberzellen (Hepatozyten) zu erleichtern. In den Hepatozyten nutzt Inclisiran den RNA-Interferenzmechanismus und steuert den katalytischen Abbau der messenger RNA (mRNA) für das Proproteinkonvertase Subtilisin Kexin Typ 9 (PCSK9). Dies erhöht das Recycling des LDL-C-Rezeptors und dessen Expression auf der Oberfläche der Hepatozyten, was die LDL-C-Aufnahme erhöht und die LDL-C-Spiegel im Blut senkt.
Pharmakodynamik
Nach einer einmaligen subkutanen Verabreichung von 284 mg Leqvio zeigte sich innerhalb von 14 Tagen nach der Verabreichung eine LDL-C-Reduktion. 30 bis 60 Tage nach der Verabreichung wurde eine mittlere Reduktion von 49 % - 51 % beim LDL-C beobachtet. An Tag 180 waren die LDL-C-Werte immer noch um ca. 53 % reduziert.
In den Phase-III-Studien waren nach vier Verabreichungen von Leqvio an Tag 1, 90, 270 und 450, LDL-C, Gesamtcholesterin, ApoB, Non-HDL-C und Lp(a) bei Patienten mit Hypercholesterinämie und gemischter Dyslipidämie reduziert.
Kardiale Elektrophysiologie
In einer randomisierten, doppelblinden, placebokontrollierten 3-fach-Crossover-Studie mit aktivem Vergleichspräparat (Moxifloxacin) erhielten 48 gesunde Teilnehmer eine subkutane Behandlung mit 852 mg Inclisiran (das Dreifache der empfohlenen Höchstdosis), Moxifloxacin und Placebo. Bei der supratherapeutischen Dosis von Inclisiran wurde weder eine QTc-Verlängerung noch ein Anstieg eines anderen EKG-Parameters beobachtet.
Klinische Wirksamkeit
Klinische Wirksamkeit bei Hypercholesterinämie und gemischter Dyslipidämie
In klinischen Studien und einigen Veröffentlichungen wird die 284 mg-Dosis von Inclisiran gleichgesetzt mit 300 mg Inclisiran-Natriumsalz oder als solches bezeichnet.
Die Wirksamkeit von Inclisiran wurde in drei Phase-III-Studien bei Patienten mit atherosklerotischen Herz-Kreislauf-Erkrankungen (ASCVD), (koronare Herzkrankheit, zerebrovaskuläre Erkrankung oder periphere arterielle Verschlusskrankheit), ASCVD-Risikoäquivalenten (Diabetes mellitus Typ2, familiäre Hypercholesterinämie oder 10-Jahres-Risiko für ein kardiovaskuläres Ereignis von mindestens 20% gemäß Framingham Risk Score oder einem äquivalenten Risikoscore) und/oder familiärer Hypercholesterinämie (FH) untersucht. Die Patienten nahmen eine maximal verträgliche Dosis eines Statins mit oder ohne anderer lipidmodifizierender Therapie ein und benötigten eine zusätzliche Senkung von LDL-C. Ungefähr 17 % der Patienten waren statinintolerant. Die Patienten erhielten subkutane Injektionen mit 284 mg Leqvio bzw. Placebo an den Tagen 1, 90, 270 und 450. Die Patienten wurden bis Tag 540 nachkontrolliert.
In der gepoolten Analyse der Phase III senkte subkutan verabreichtes Leqvio bereits an Tag 90 den LDL-C-Wert zwischen 50 und 55 % (Abbildung 1), was während der Langzeittherapie beibehalten wurde. Die maximale LDL-C-Reduktion wurde an Tag 150 nach einer zweiten Verabreichung erreicht. Geringe, aber statistisch signifikante erhöhte LDL-C-Reduktionen von bis zu 65 % waren mit niedrigeren LDL-C-Ausgangswerten (ca. < 2 mmol/l [77 mg/dl]), höheren PCSK9-Ausgangswerten und höheren Statindosisstärken und -Intensitäten verbunden.
Die Reduzierung des LDL-C wurde in allen Untergruppen beobachtet, dazu zählen Alter, Hautfarbe, Geschlecht, Region, Body-Mass-Index, Risiko laut US-amerikanischem Cholesterinaufklärungsprogramm NCEP (National Cholesterol Education Program), aktueller Raucherstatus, Risikofaktoren für koronare Baseline-Herzkrankheit (KHK), Familiengeschichte mit frühzeitiger KHK, Glukosetoleranzstatus (d.h. Diabetes mellitus Typ 2, metabolisches Syndrom oder keines von beiden), Bluthochdruck und Triglycerid-Wert bei Baseline.
Inclisiran reduzierte zudem das Non-HDL-C, Apo B, Gesamtcholesterol und Lp(a) bei Patienten mit Hypercholesterinämie und gemischter Dyslipidämie. Es gab keine klinisch signifikanten Veränderungen bei HDL-C und Triglyceriden.
Abbildung 1: Mittlere prozentuale Veränderung des LDL-C-Werts gegenüber dem Ausgangswert bei Patienten mit Hypercholesterinämie bzw. gemischter Dyslipidämie, die mit Leqvio behandelt wurden, im Vergleich zu Placebo (gepoolte Analyse)
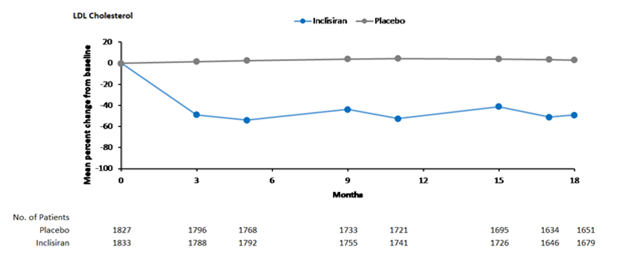
ASCVD und ASCVD-Risikoäquivalente
Zwei Studien wurden mit Patienten mit ASCVD und ASCVD-Risikoäquivalenten durchgeführt (ORION-10 und ORION-11). Die Patienten erhielten eine maximal verträgliche Dosis eines Statins mit oder ohne anderer lipidmodifizierenden Therapie, wie z.B. Ezetimib und benötigten eine zusätzliche Senkung des LDL-C-Werts. Die Patienten erhielten subkutane Injektionen mit 284 mg Leqvio bzw. Placebo an den Tagen 1, 90, 270 und 450. Die co-primären Endpunkte in jeder Studie waren die prozentuale Veränderung des LDL-C-Werts gegenüber dem Ausgangswert bis zu Tag 510 im Vergleich zu Placebo und die zeitbereinigte prozentuale Veränderung des LDL-C-Werts gegenüber dem Ausgangswert nach Tag 90 und bis Tag 540, um die integrierte Wirkung auf die LDL-C-Werte im Laufe der Zeit abzuschätzen. Wichtige sekundäre Endpunkte waren die absolute Veränderung des LCD-C-Werts gegenüber dem Ausgangswert bis zu Tag 510, die zeitbereinigte absolute Veränderung des LDL-C-Werts gegenüber dem Ausgangswert nach Tag 90 und bis Tag 540 und die prozentuale Veränderung gegenüber dem Ausgangswert bis Tag 510 für PCSK9, Gesamtcholesterin, Apo-B und Non-HDL-C. Weitere sekundäre Endpunkte waren das individuelle Ansprechen auf Leqvio und der Anteil der Patienten, die die globalen Lipid-Zielwerte für ihr Risiko für ASCVD erreichen.
ORION-10 war eine multizentrische, doppelblinde, randomisierte, placebokontrollierte Studie über 18 Monate mit 1'561 Patienten mit ASCVD.
Das mittlere Alter betrug zu Beginn 66 Jahre (Altersspanne: 35 bis 90 Jahre), 60 % waren ≥65 Jahre alt, 31 % waren Frauen, 86 % kaukasisch, 13 % afro-amerikanisch, 1 % asiatisch und 14 % waren spanischer oder lateinamerikanischer Herkunft. Der mittlere LDL-C-Ausgangswert betrug 2,7 mmol/l (105 mg/dl). 69 % der Studienteilnehmer nahmen Statine hoher Intensität, 19 % Statine mittlerer Intensität, 1 % Statine niedriger Intensität und 11 % keine Statine. Die am häufigsten verabreichten Statine waren Atorvastatin und Rosuvastatin. Leqvio war in der Studie sicher und gut verträglich, wobei unerwünschte Ereignisse bei nur 2,4 % der mit Leqvio behandelten Patienten gegenüber 2,2 % der mit Placebo behandelten Patienten zum Abbruch der Behandlung führten.
Leqvio reduzierte signifikant die mittlere prozentuale Veränderung des LDL-C-Werts gegenüber dem Ausgangswert bis zu Tag 510 im Vergleich zu Placebo um 52 % (95%-KI: -56 %, -49 %; p < 0,0001) (Tabelle 1 und Abbildung 2).
Leqvio reduzierte zudem signifikant die zeitbereinigte prozentuale Veränderung des LDL-C-Werts gegenüber dem Ausgangswert nach Tag 90 und bis zu Tag 540 um 54 % im Vergleich zu Placebo (95%-KI: -56 %, -51 %; p < 0,0001). Für weitere Ergebnisse siehe Tabelle 1.
Tabelle 1: Mittlere prozentuale Veränderung gegenüber dem Ausgangswert und Differenz zu Placebo bei den Lipidparametern an Tag 510 in der Studie ORION-10
Behandlungsgruppe LDL-C Gesamtcho-lesterin Non-HDL-C Apo B Lp(a)*
Mittlerer Ausgangswe 105 181 134 94 122
rt in mg/dl**
Tag 510 (mittlere
prozentuale Veränder
ung gegenüber dem
Ausgangswert)
Placebo (n=780) 1 0 0 -2 4
Leqvio(n=781) -51 -34 -47 -45 -22
Unterschied zu -52 (-56, -49) -33 (-35, -31) -47 (-50, -44) -43 (-46, -41) -26 (-29, -22)
Placebo (Least-Squar
e-Mittelwert)(95%-KI
)
* An Tag 540; mediane prozentuale Veränderung der Lp(a)-Werte
** Mittlerer Ausgangswert in nmol/l für Lp(a)
Abbildung 2: Mittlere prozentuale Veränderung des LDL-C-Werts gegenüber dem Ausgangswert bei Patienten mit Hypercholesterinämie, gemischter Dyslipidämie und ASCVD, die mit Leqvio behandelt wurden, im Vergleich zu Placebo in der Studie ORION-10

An Tag 510 wurde der LDL-C-Zielwert von < 1,8 mmol/l (70 mg/dl) von 84 % der mit Leqvio behandelten Patienten mit ASCVD im Vergleich zu 18 % der mit Placebo behandelten Patienten erreicht.
Konsistente und statistisch signifikante (p < 0,0001) Reduktionen der prozentualen Veränderung des LDL-C-Wertes gegenüber dem Ausgangswert bis zu Tag 510 und zeitbereinigte prozentuale Veränderungen des LDL-C-Wertes gegenüber dem Ausgangswert nach Tag 90 und bis zu Tag 540 wurden in allen Untergruppen beobachtet, unabhängig von den demographischen Daten und den Merkmalen der Erkrankung bei Baseline (einschliesslich Geschlecht, Alter, Body-Mass-Index, Hautfarbe und Statineinnahme bei Baseline), Komorbiditäten und geographischen Regionen.
ORION-11 war eine internationale, multizentrische, doppelblinde, randomisierte, placebokontrollierte Studie über 18 Monate mit 1'617 Patienten mit ASCVD oder ASCVD-Risikoäquivalenten (ASCVD-Risikoäquivalent wurde definiert als Patienten mit Typ-2-Diabetes mellitus, familiärer Hypercholesterinämie oder einem 10-Jahres-Risiko von 20 % oder mehr für ein kardiovaskuläres Ereignis, das mit dem Framingham-Risiko-Score oder einem gleichwertigen Verfahren beurteilt wurde). Mehr als 75 % der Patienten erhielten eine hochintensive Statin-Hintergrundtherapie, 87 % der Patienten hatten eine ASCVD und 13 % ein vergleichbares Risiko (ASCVD-Risikoäquivalent).
Das mittlere Alter betrug bei Baseline 65 Jahre (Altersspanne: 20 bis 88 Jahre), 55 % waren ≥65 Jahre alt, 28 % waren Frauen, 98 % kaukasisch, 1 % afro-amerikanisch, 1 % asiatisch und 1 % waren spanischer oder lateinamerikanischer Herkunft. Der mittlere LDL-C-Ausgangswert betrug 2,7 mmol/l (105 mg/dl). 78 % der Studienteilnehmer nahmen Statine hoher Intensität, 16 % Statine mittlerer Intensität, 0,4 % Statine niedriger Intensität und 5 % keine Statine. Die am häufigsten verabreichten Statine waren Atorvastatin und Rosuvastatin. Unerwünschte Ereignisse führten bei 2,8 % der mit Leqvio behandelten Patienten gegenüber 2,2 % der mit Placebo behandelten Patienten zum Abbruch der Behandlung.
Leqvio reduzierte signifikant die mittlere prozentuale Veränderung des LDL-C-Werts gegenüber dem Ausgangswert bis zu Tag 510 im Vergleich zu Placebo um 50 % (95%-KI: -53 %, -47 %; p < 0,0001) (Tabelle 2 und Abbildung 3).
Leqvio reduzierte zudem signifikant die zeitbereinigte prozentuale Veränderung des LDL-C-Werts gegenüber dem Ausgangswert nach Tag 90 und bis zum Tag 540 um 49 % im Vergleich zu Placebo (95%-KI: -52 %, -47 %; p < 0,0001). Für weitere Ergebnisse siehe Tabelle 2.
Tabelle 2: Mittlere prozentuale Veränderung gegenüber dem Ausgangswert und Differenz zu Placebo bei den Lipidparametern an Tag 510 in der Studie ORION-11
Behandlungsgruppe LDL-C Gesamtcho-lesterin Non-HDL-C Apo B Lp(a)*
Mittlerer Ausgangswe 105 185 136 96 107
rt in mg/dl**
Tag 510 (mittlere
prozentuale Veränder
ung gegenüber dem
Ausgangswert)
Placebo (n=807) 4 2 2 1 0
Leqvio(n=810) -46 -28 -41 -38 -19
Unterschied zu -50 (-53, -47) -30 (-32, -28) -43 (-46, -41) -39 (-41, -37) -19 (-21, -16)
Placebo (Least-Squar
e-Mittelwert)(95
%-KI)
* An Tag 540; mediane prozentuale Veränderung der Lp(a)-Werte
** Mittlerer Ausgangswert in nmol/l für Lp(a)
Abbildung 3: Mittlere prozentuale Veränderung des LDL-C-Werts gegenüber dem Ausgangswert bei Patienten mit Hypercholesterinämie, gemischter Dyslipidämie und ASCVD/ASCVD-Risikoäquivalenten, die mit Leqvio behandelt wurden, im Vergleich zu Placebo in der Studie ORION-11

An Tag 510 wurde der LDL-C-Zielwert von <1,8 mmol/l (70 mg/dl) von 82 % der mit Leqvio behandelten Patienten mit ASCVD im Vergleich zu 16 % der mit Placebo behandelten Patienten erreicht. Bei Patienten mit einem ASCVD-Risikoäquivalent wurde der LDL-C-Zielwert von < 2,6 mmol/l (100 mg/dl) von 78 % der mit Leqvio behandelten Patienten im Vergleich zu 31 % der mit Placebo behandelten Patienten erreicht.
Konsistente und statistisch signifikante (p < 0,05) Reduktionen der prozentualen Veränderung des LDL-C-Wertes gegenüber dem Ausgangswert bis zum Tag 510 und zeitbereinigte prozentuale Veränderungen des LDL-C-Wertes gegenüber dem Ausgangswert nach Tag 90 und bis zu Tag 540 wurden in allen Untergruppen beobachtet, unabhängig von den demographischen Daten und den Merkmalen der Erkrankung bei Baseline (einschliesslich Geschlecht, Alter, Body-Mass-Index, Hautfarbe und Statineinnahme bei Baseline), Komorbiditäten und geographischen Regionen.
Heterozygotisch familiäre Hypercholesterinämie
ORION-9 war eine internationale, multizentrische, doppelblinde, randomisierte, placebokontrollierte Studie über 18 Monate mit 482 Patienten mit heterozygoter familiärer Hypercholesterinämie (HeFH). Alle Patienten nahmen eine maximal verträgliche Dosis eines Statins mit oder ohne andere lipidmodifizierende Therapie, wie z .B. Ezetimib, ein und benötigten eine zusätzliche Senkung des LDL-C-Werts. Die Diagnose der HeFH wurde entweder durch Genotypisierung oder nach klinischen Kriterien gestellt ( "definitive FH" unter Verwendung der Simon-Broome-Kriterien oder der WHO/Dutch Lipid Network Criteria (DLNC)).
Die co-primären Endpunkte waren die prozentuale Veränderung des LDL-C-Werts gegenüber dem Ausgangswert bis zu Tag 510 im Vergleich zu Placebo und die zeitbereinigte prozentuale Veränderung des LDL-C-Werts gegenüber dem Ausgangswert nach Tag 90 und bis zu Tag 540, um die integrierte Wirkung auf die LDL-C-Werte im Laufe der Zeit abzuschätzen. Wichtige sekundäre Endpunkte waren die absolute Veränderung des LDL-C-Werts gegenüber dem Ausgangswert bis zu Tag 510, die zeitbereinigte absolute Veränderung des LDL-C-Werts gegenüber dem Ausgangswert nach Tag 90 und bis zu Tag 540 und die prozentuale Veränderung gegenüber dem Ausgangswert bis zu Tag 510 für PCSK9, Gesamtcholesterin, Apo-B und Non-HDL-C. Weitere sekundäre Endpunkte waren das individuelle Ansprechen auf Leqvio und der Anteil der Patienten, die die globalen Lipid-Zielwerte für ihr Risiko für ASCVD erreichen.
Das mittlere Alter betrug zu Beginn 55 Jahre (Altersspanne: 21 bis 80 Jahre), 22 % waren ≥65 Jahre alt, 53 % waren Frauen, 94 % kaukasisch, 3 % afro-amerikanisch, 3 % asiatisch und 3 % waren spanischer oder lateinamerikanischer Herkunft. Der mittlere LDL-C-Ausgangswert betrug 4,0 mmol/l (153 mg/dl). Vierundsiebzig Prozent (74 %) der Studienteilnehmer nahmen Statine hoher Intensität, 15 % Statine mittlerer Intensität und 10 % keine Statine. Zweiundfünfzig Prozent (52 %) der Patienten wurden mit Ezetimib behandelt. Die am häufigsten verabreichten Statine waren Atorvastatin und Rosuvastatin. Leqvio war in der Studie sicher und gut verträglich, wobei unerwünschte Ereignisse bei 1 % der mit Leqvio behandelten Patienten gegenüber 0 % der mit Placebo behandelten Patienten zum Abbruch der Behandlung führten.
Leqvio reduzierte signifikant die mittlere prozentuale Veränderung des LDL-C-Werts gegenüber dem Ausgangswert bis zu Tag 510 im Vergleich zu Placebo um 48 % (95%-KI: -54 %, -42 %; p < 0,0001) (Tabelle 3 und Abbildung 4).
Leqvio reduzierte zudem signifikant die zeitbereinigte prozentuale Veränderung des LDL-C-Werts gegenüber dem Ausgangswert nach Tag 90 und bis zu Tag 540 um 44 % im Vergleich zu Placebo (95%-KI: -48 %, -40 %; p < 0,0001). Für weitere Ergebnisse siehe Tabelle 3.
Tabelle 3: Mittlere prozentuale Veränderung gegenüber dem Ausgangswert und Differenz zu Placebo bei den Lipidparametern an Tag 510 in der Studie ORION-9
Behandlungsgruppe LDL-C Gesamtcho-lesterin Non-HDL-C Apo B Lp(a)*
Mittlerer Ausgangswe 153 231 180 124 121
rt in mg/dl**
Tag 510 (mittlere
prozentuale Veränder
ung gegenüber dem
Ausgangswert)
Placebo (n=240) 8 7 7 3 4
Leqvio(n=242) -40 -25 -35 -33 -13
Unterschied zu -48 (-54, -42) -32 (-36, -28) -42 (-47, -37) -36 (-40, -32) -17 (-22, -12)
Placebo (Least-Squar
e-Mittelwert)(95
%-KI)
* An Tag 540; mediane prozentuale Veränderung der Lp(a)-Werte
** Mittlerer Ausgangswert in nmol/l für Lp(a)
Abbildung 4: Mittlere prozentuale Veränderung des LDL-C-Werts gegenüber dem Ausgangswert bei Patienten mit Hypercholesterinämie, gemischter Dyslipidämie und heterozygoter familiärer Hypercholesterinämie, die mit Leqvio behandelt wurden, im Vergleich zu Placebo in der Studie ORION-9

An Tag 510 erreichten 52,5 % der mit Leqvio behandelten Patienten mit ASCVD ihren LDL-C-Zielwert von < 1,8 mmol/l (70 mg/dl) im Vergleich zu 1,4 % der mit Placebo behandelten Patienten mit ASCVD, während 66,9 % der mit Leqvio behandelten Patienten mit ASCVD-Risikoäquivalenten ihren LDL-C-Zielwert von < 2,6 mmol/l (100 mg/dl) im Vergleich zu 8,9 % der mit Placebo behandelten Patienten mit ASCVD-Risikoäquivalenten erreichten.
Konsistente und statistisch signifikante (p < 0,05) Reduktionen der prozentualen Veränderung des LDL-C-Wertes gegenüber dem Ausgangswert bis zum Tag 510 und zeitbereinigte prozentuale Veränderungen des LDL-C-Wertes gegenüber dem Ausgangswert nach Tag 90 und bis zu Tag 540 wurden in allen Untergruppen beobachtet, unabhängig von den demographischen Daten und den Merkmalen der Erkrankung bei Baseline (einschliesslich Geschlecht, Alter, Body-Mass-Index, Hautfarbe und Statineinnahme bei Baseline), Komorbiditäten und geographischen Regionen.
Sicherheit und Wirksamkeit bei pädiatrischen Patienten
Die Sicherheit und Wirksamkeit von Leqvio bei Kindern und Jugendlichen im Alter von unter 18 Jahren wurde nicht etabliert. Es liegen keine Daten vor (siehe "Dosierung/Anwendung" für Informationen zur Anwendung bei Kindern und Jugendlichen).
PharmakokinetikAbsorption
Nach einer einzelnen subkutanen Verabreichung stieg die systemische Exposition gegenüber Inclisiran linear und dosisproportional über einen Bereich von 24 mg bis 756 mg an. Mit dem empfohlenen Dosierungsschema von 284 mg erreichten die Plasmakonzentrationen ca. 4 Stunden nach der Verabreichung ihren Höhepunkt mit einer mittleren Cmax von 509 ng/ml. Die Konzentrationen fielen innerhalb von 48 Stunden nach der Verabreichung auf nicht mehr nachweisbare Werte ab. Die mittlere Fläche unter der Plasmakonzentrations-Zeit-Kurve ab Verabreichung bis unendlich (AUC0-inf) betrug 7'980 ng*h/ml. Die pharmakokinetischen Befunde nach mehrfacher subkutaner Verabreichung von Inclisiran waren ähnlich wie bei der Verabreichung einer Einzeldosis.
Distribution
Basierend auf in vitro Untersuchungen ist Inclisiran bei den relevanten klinischen Plasmakonzentrationen zu 87 % an Plasmaproteine gebunden. Nach einer einzelnen subkutanen Verabreichung von 284 mg Inclisiran bei gesunden Erwachsenen beträgt das scheinbare Verteilungsvolumen rund 500 l. Basierend auf nicht-klinischen Daten wurde gezeigt, dass Inclisiran eine hohe Aufnahme und Selektivität für die Leber, das Zielorgan für die Cholesterinsenkung, aufweist.
Metabolismus
Basierend auf in vitro Untersuchungen, wird Inclisiran hauptsächlich von Nukleasen zu kürzeren inaktiven Nukleotiden unterschiedlicher Länge metabolisiert. Inclisiran ist kein Substrat von üblichen Arzneistofftransportern und, obwohl keine in vitro Studien durchgeführt wurden, wird nicht erwartet, dass es ein Substrat von Cytochrom P450 ist.
Elimination
Die terminale Eliminationshalbwertszeit von Inclisiran beträgt ca. 9 Stunden und bei Mehrfachverabreichung kommt es zu keiner Akkumulation. Sechzehn Prozent (16 %) einer Inclisiran Dosis von 284 mg werden über die Niere ausgeschieden.
Linearität/Nicht Linearität
Nach subkutaner Verabreichung von Inclisiran Einzeldosen zwischen 24 mg und 756 mg wurde eine fast dosisproportionale Erhöhung der Inclisiran-Exposition beobachtet. Es wurden keine Akkumulation und keine zeitabhängigen Veränderungen nach mehrfachen subkutanen Verabreichungen von Inclisiran festgestellt.
Pharmakokinetische/pharmakodynamische Zusammenhänge
Es wurde eine Dissoziation zwischen den pharmakokinetischen Parametern von Inclisiran im Plasma und der pharmakodynamischen Wirkungen auf die Plasma Spiegel von PCSK9 und LDL-C beobachtet. Die beobachtete lange Wirkdauer auf PCSK9 und LDL-C korreliert nicht mit der Eliminationshalbwertszeit von Inclisiran im Plasma von 9 Stunden. Eine maximalen Reduktion von PCSK9 und LDL-C wurden bei einer Dosis von 284 mg beobachtet. höhere Dosen führten nicht zu grösseren Effekten.
Kinetik spezieller Patientengruppen
Leberfunktionsstörungen
Die pharmakokinetische Analyse von Daten aus einer spezifischen Studie zu Leberfunktionsstörung zeigte bei Patienten mit leichter Leberfunktionsstörung (Child Pugh A, n=10) einen Anstieg der Cmax und AUC von Inclisiran auf das 1,1- und 1.3-Fache, im Vergleich zu Patienten mit normaler Leberfunktion (n=12). Trotz der höheren Inclisiran-Plasma-Expositionen war die Reduktion von LDL-C in Patienten mit normaler Leberfunktion und leichter Leberfunktionsstörung ähnlich.
Bei Patienten mit mittlerer Leberfunktionsstörung (Child Pugh B, n=6) waren Cmax und AUC von Inclisiran auf das 2,1- und 2.0- Fache erhöht, die PCSK9-Ausgangswerte deutlich niedriger und die mittlere prozentuale Veränderung der LDL-C-Spiegels gegenüber dem Ausgangswert war bei Patienten mit mittelschwerer Leberfunktionsstörung um 40 % reduziert, gegenüber einer 52 %-igen Reduktion bei Patienten mit normaler Leberfunktion.
Leqvio wurde bei Patienten mit schwerer Leberfunktionsstörung (Child-Pugh-Klasse C) nicht untersucht.
Nierenfunktionsstörungen
Die pharmakokinetische Analyse von Daten aus einer spezifischen Studie zu Nierenfunktionsstörung zeigte einen Anstieg der Cmax und AUC von Inclisiran auf das 2.3-, 2,0- und 3,3-Fache bzw. 1,6-, 1.8- und 2,3-Fache bei Patienten mit leichter, mittlerer oder schwerer Nierenfunktionsstörung im Vergleich zu Patienten mit normaler Nierenfunktion. Maximale prozentuale Veränderungen des LDL-C-Spiegels gegenüber dem Ausgangswert waren 58 %, 35 %, 53 % und 51 % bei Patienten mit normaler Nierenfunktion respektive Patienten mit leichter, mittlerer oder schweren Nierenfunktionsstörung.
Trotz der höheren transienten Plasmaexpositionen über 24 bis 48 Stunden war die Reduktion von LDL-C in allen Nierenfunktionsgruppen ähnlich.
Der Effekt einer Nierenerkrankung im Endstadium oder einer Hämodialyse auf die Pharmakokinetik von Inclisiran wurde nicht untersucht.
Andere spezielle Patientengruppen
Eine pharmakodynamische Populationsanalyse wurde anhand der Daten von 4'328 Patienten durchgeführt. Alter, Körpergewicht, Geschlecht, ethnische Herkunft und Kreatinin-Clearance hatten keinen signifikanten Einfluss auf die Pharmakodynamik von Inclisiran.
Präklinische DatenBasierend auf den konventionellen Studien zu Sicherheitspharmakologie, chronischer Toxizität, Genotoxizität, Kanzerogenität, Reproduktions- und Entwicklungstoxizität lassen die präklinischen Daten keine besonderen Gefahren für den Menschen erkennen.
Langzeittoxizität (bzw. Toxizität bei wiederholter Verabreichung)
In toxikologischen Studien mit wiederholter Verabreichung, die mit Ratten und Affen durchgeführt wurden, wurden die NOAEL Werte (No Observed Adverse Effect Levels) als die höchsten subkutan verabreichten Dosen bestimmt und mit einer Sicherheitsmarge assoziiert, die um ein Vielfaches höher war als die Exposition gegenüber der klinischen Dosis beim Menschen.
Mutagenität
In einer Reihe von Tests, einschliesslich eines Mutagenitätstests mit Bakterienzellen, eines In-vitro-Chromosomenaberrationstests mit humanen peripheren Blutlymphozyten und eines Knochenmark-Mikronukleus-Tests in vivo mit Ratten, wurde kein mutagenes oder klastogenes Potenzial von Inclisiran aufgezeigt.
Karzinogenität
Inclisiran war weder bei Sprague-Dawley-Ratten noch bei TgRasH2-Mäusen, denen Inclisiran in Dosen verabreicht wurde, die deutlich höher lagen als die klinischen Dosen, kanzerogen.
Reproduktionstoxizität
Reproduktionsstudien mit Ratten und Kaninchen ergaben keine Hinweise auf eine Schädigung des Fötus durch Inclisiran bei den höchsten verabreichten Dosen, die zu einer Exposition führten, welche deutlich über der maximalen Exposition beim Menschen lag.
Inclisiran beeinträchtigte weder die Fertilität noch die Fortpflanzungsleistung männlicher und weiblicher Ratten, die vor und während der Trächtigkeit Inclisiran ausgesetzt waren. Die Dosen waren mit systemischen Expositionen verbunden, die um ein Vielfaches höher waren als die Exposition von Menschen gegenüber klinischen Dosen.
Inclisiran wurde in der Milch von laktierenden Ratten festgestellt; es gibt jedoch keine Hinweise auf eine systemische Absorption bei gesäugten neugeborenen Ratten.
Sonstige HinweiseInkompatibilitäten
Da keine Kompatibilitätsstudien durchgeführt wurden, darf dieses Arzneimittel nicht mit anderen Arzneimitteln gemischt werden.
Haltbarkeit
Das Arzneimittel darf nur bis zu dem auf dem Behälter mit "EXP" bezeichneten Datum verwendet werden.
Besondere Lagerungshinweise
Nicht über 25°C lagern. Nicht einfrieren.
Ausser Reichweite von Kindern aufbewahren.
Hinweise für die Handhabung
Leqvio sollte vor der Anwendung visuell kontrolliert werden. Die Lösung muss klar, farblos bis blassgelb und im Wesentlichen frei von Partikeln sein. Wenn die Lösung sichtbare Partikel enthält, darf sie nicht verwendet werden.
Nicht verwendetes Arzneimittel oder Abfallmaterial ist entsprechend den nationalen Anforderungen zu beseitigen.
Zulassungsnummer67836 (Swissmedic)
Packungen1,5 ml Lösung in einer Fertigspritze (Glas TypI) mit Kolbenstopfen (mit FluroTec beschichteter Bromobutyl-Kautschuk), mit Nadel und fester Nadelkappe. [B]
1,5 ml Lösung in einer Fertigspritze (Glas TypI) mit Kolbenstopfen (mit FluroTec beschichteter Bromobutyl-Kautschuk), mit Nadel und fester Nadelkappe, mit Nadelschutz. [B]
ZulassungsinhaberinNovartis Pharma Schweiz AG, Risch; Domizil 6343 Rotkreuz
Stand der InformationJanuar 2023
|