Propriétés/EffetsCode ATC
M09AX07
Mécanisme d'action
Spinraza est un oligonucléotide antisens (AON) spécialement conçu pour traiter la SMA, une maladie neuromusculaire progressive à transmission autosomique récessive, attribuable à des mutations du gène SMN1 (Survival Motor Neuron 1) sur le chromosome 5q. Ces mutations conduisent à une perte de la fonction du gène SMN1 et, par conséquent, à un déficit en protéine SMN. La protéine SMN est également synthétisée par le gène SMN2, mais en quantité beaucoup plus faible. SMA est un terme générique utilisé pour des maladies à l'expression clinique diverse.
L'âge de la primo-apparition de la maladie ainsi que sa sévérité dépendent du nombre de copies disponibles du gène SMN2; un faible nombre de copies du gène SMN2 est associé à une apparition de la maladie à un âge plus précoce et à des symptômes plus prononcés.
Spinraza augmente le pourcentage d'inclusion de l'exon 7 dans les transcrits d'acide ribonucléique messager (ARNm) du gène codant pour la protéine SMN2 en se liant à un site d'inactivation de l'épissage intronique (ISS-N1) présent dans l'intron 7 de l'ARN pré-messager (pré-ARNm) de SMN2. Par l'hybridation, l'oligonucléotide antisens déplace les facteurs d'épissage, qui inhibent normalement l'épissage. Le déplacement de ces facteurs entraîne la rétention de l'exon 7 dans l'ARNm de SMN2 et, lorsque l'ARNm de SMN2 est synthétisé, il peut être traduit en protéine SMN fonctionnelle de pleine longueur.
Pharmacodynamique
Les effets pharmacodynamiques sont en corrélation avec les effets biologiques du nusinersen.
Les échantillons d'autopsie réalisés chez des enfants en bas âge traités ont révélé de plus grandes quantités d'ARNm de SMN2 contenant l'exon 7 dans la moelle épinière thoracique que dans les échantillons prélevés chez des enfants en bas âge atteints de SMA non traités.
Biomarqueur NfL plasmatique
Le NfL plasmatique est un biomarqueur sanguin de l'atteinte axonale et de la dégénérescence neuronale. Dans l'étude SM203, chez les patients atteints de SMA infantile, une réduction de 94 % du taux de NfL plasmatique a été observée entre l'inclusion et le jour 183 dans le groupe traité par 50/28 mg (rapport des moyennes géométriques par rapport à l'inclusion), contre 30 % dans le groupe sous traitement simulé témoin apparié (différence des rapports des moyennes géométriques entre le groupe recevant 50/28 mg et le groupe sous traitement simulé témoin correspondant: 92 %; p < 0,0001). Les taux plasmatiques de NfL ont diminué plus rapidement dans le groupe recevant 50/28 mg que dans le groupe recevant 12 mg, avec une baisse de 88 % (rapport des moyennes géométriques par rapport à l'inclusion) entre l'inclusion et le jour 64, par rapport à une baisse de 77 % dans le groupe recevant 12 mg (différence des rapports des moyennes géométriques entre le groupe recevant 50/28 mg et le groupe recevant 12 mg: 49 %; p = 0,0020).
En conséquence, les taux plasmatiques de NfL ont également diminué plus rapidement chez les participants ayant présenté un début tardif de la maladie dans la partie B pour le groupe recevant 50/28 mg, avec une baisse de 66 % (rapport des moyennes géométriques par rapport à l'inclusion) observée entre l'inclusion et le jour 64, par rapport à une baisse de 42 % dans le groupe recevant 12 mg (différence des rapports des moyennes géométriques entre le groupe recevant 50/28 mg et le groupe recevant 12 mg: 42 %; p = 0,0495) (Figure 1).
Figure 1: Partie B de l'étude SM203, forme infantile de SMA, NfL plasmatique: rapport des moyennes des MC par rapport à l'inclusion (IC à 95 %) après la visite, selon l'analyse ANCOVA utilisant l'IM et des estimations de l'analyse principale du groupe recevant 50/28 mg par rapport au groupe recevant 12 mg et les estimations simulées du groupe recevant 50/28 mg par rapport au groupe sous traitement simulé: en ITT, groupe sous traitement simulé témoin apparié
Efficacité clinique
Résultats en lien avec l'efficacité
L'efficacité de Spinraza a été démontrée dans le cadre de 8 études cliniques menées chez des patients symptomatiques (études CS3B, CS3A, CS4, CS2, CS12, CS7, CS11 et SM203) âgés de 15 jours à 65 ans au moment de la première dose, ainsi que dans une étude clinique réalisée chez des patients présymptomatiques (étude CS5) âgés de 3 à 42 jours au moment de la première dose. Les résultats de ces études concernant l'efficacité corroborent une instauration du traitement aussi rapide que possible après la pose du diagnostic.
Patients symptomatiques sous traitement par 12 mg
Forme infantile
Étude CS3B (ENDEAR)
L'étude CS3B était une étude de phase III randomisée en double aveugle, contrôlée contre traitement simulé et menée chez 121 enfants en bas âge symptomatiques âgés de ≤7 mois, ayant un diagnostic de SMA (apparition des symptômes avant l'âge de 6 mois). Les patients ont été randomisés selon un rapport 2:1 pour recevoir soit Spinraza (selon le schéma posologique autorisé), soit des injections simulées, avec une durée de traitement allant de 6 à 442 jours (médiane: 258 jours).
L'âge médian à l'apparition des signes et symptômes cliniques de SMA était respectivement de 6,5 semaines (intervalle 2 - 18) et de 8 semaines (intervalle 1 - 20) chez les patients traités par Spinraza et chez les patients du groupe de traitement simulé. Dans cette étude, 99 % des patients étaient porteurs de deux copies du gène SMN2 et donc considérés comme très susceptibles de développer une SMA de type I.
À l'inclusion, la valeur totale moyenne des étapes du développement moteur était de 1,37 (intervalle 0 - 6), le résultat médian du score CHOP INTEND de 28 (intervalle 8 - 50,5) et l'amplitude médiane du PAMC de 0,20 (intervalle 0,00 - 0,87) pour le nervus ulnaris et de 0,30 (intervalle 0,00 - 1,50) pour le nervus peroneus. L'âge médian lors de l'administration de la première dose était de 164,5 jours (intervalle 52 - 242) chez les patients traités par Spinraza et de 205 jours (intervalle 30 - 262) chez les patients du groupe de traitement simulé.
Les caractéristiques cliniques initiales étaient très similaires chez les patients traités par Spinraza et les patients du groupe de traitement simulé, à l'exception des événements suivants, observés plus fréquemment chez les patients traités par Spinraza que chez ceux du groupe de traitement simulé: respiration paradoxale (89 % contre 66 %), pneumonie ou symptômes respiratoires (35 % contre 22 %), difficultés de déglutition ou d'alimentation (51 % contre 29 %) et nécessité d'une assistance respiratoire (26 % contre 15 %).
Une analyse intermédiaire planifiée a été réalisée sur la base des résultats de patients soit décédés, soit ayant interrompu l'étude, soit ayant suivi le traitement pendant au moins 183 jours. Au moment de l'analyse intermédiaire, 121 patients suivaient le traitement (Spinraza n = 80, traitement simulé n = 41). Au moment de l'analyse intermédiaire, 78 patients au total avaient suivi le traitement durant au moins 183 jours, étaient décédés ou avaient interrompu l'étude et ont été inclus dans l'ensemble de données provisoires sur l'efficacité pour l'analyse du critère d'évaluation principal (Spinraza n = 51, traitement simulé n = 27). Le critère d'évaluation principal au moment de l'analyse intermédiaire était le pourcentage de répondeurs, c'est-à-dire les patients ayant atteint un degré prédéfini d'amélioration des étapes de développement moteur selon l'échelle du Hammersmith Infant Neurologic Examination (HINE) section 2. Selon cette définition, un patient répondeur était un patient présentant une augmentation d'au moins 2 points [ou un score maximal de 4] de la capacité de gigoter ou une augmentation d'au moins 1 point pour les étapes du développement moteur contrôle de la tête, se retourner, s'asseoir, marcher à quatre pattes, se tenir debout ou marcher. Pour être considérés comme répondeurs, les patients devaient s'améliorer et non pas décliner dans plusieurs catégories des étapes du développement moteur. Les patients décédés ou ayant interrompu l'étude ont été inclus dans l'ensemble de données provisoires sur l'efficacité et considérés comme non répondeurs. Parmi les 78 patients considérés dans l'analyse intermédiaire, le pourcentage de patients correspondant à la définition de répondeur en termes d'étapes du développement moteur était significativement plus élevé sur le plan statistique dans le groupe sous Spinraza (41 %) que dans le groupe de traitement simulé (0 %), avec une différence (IC à 95 %) dans les pourcentages de 41,18 % (18,1 – 61,20) (nusinersen –traitement simulé), p < 0,0001.
Suite aux résultats positifs de l'analyse intermédiaire, l'étude CS3B a été interrompue et les patients ont été inclus dans une étude de prolongation en ouvert (CS11).
Au moment de l'analyse finale, le critère d'évaluation principal était le délai jusqu'au décès ou jusqu'à la mise sous ventilation permanente (ventilation durant ≥16 heures par jour en continu pendant > 21 jours en l'absence d'événement aigu réversible ou de trachéotomie). Des effets statistiquement significatifs ont été observés chez les patients du groupe sous Spinraza, par rapport aux patients du groupe de traitement simulé, s'agissant de la survie sans événement, de la survie globale, du pourcentage de patients correspondant à la définition de répondeur pour les étapes du développement moteur et s'agissant du pourcentage de patients ayant obtenu une amélioration d'au moins 4 points au score CHOP INTEND par rapport à la valeur initiale (Tableau 6).
Une réduction statistiquement significative de 47 % du risque de décès ou de ventilation permanente a été observée dans la population en ITT (p = 0,0046). Une durée médiane jusqu'au décès ou jusqu'à la ventilation permanente n'a pas été atteinte dans le groupe sous Spinraza, tandis qu'elle était de 22,6 semaines dans le groupe de traitement simulé. Une réduction statistiquement significative de 62,8 % du risque de décès a également été observée (p= 0,0041).
Dans l'ensemble de données sur l'efficacité, 18 patients (25 %) du groupe sous Spinraza et 12 patients (32 %) du groupe de traitement simulé ont eu besoin d'une ventilation permanente. Alors qu'aucun patient de l'étude CS3B n'a interrompu la ventilation permanente, 6 (33 %) patients du groupe Spinraza et 0 (0 %) patient du groupe de traitement simulé ont correspondu aux critères déterminants de répondeur moteur définis dans le protocole.
11 patients (61 %) du groupe sous Spinraza et 3 patients (25 %) du groupe de traitement simulé ont atteint au moins 1 point d'amélioration du score total des étapes du développement moteur. Aucun patient (0 %) du groupe sous Spinraza et 3 patients (25 %) du groupe de traitement simulé ont présenté au moins 1 point de détérioration du score total des étapes du développement moteur.
Sur le plan statistique, un pourcentage significativement supérieur (p < 0,0001) des participants de l'ensemble de données sur l'efficacité traités par Spinraza (71 %) a obtenu une amélioration d'au moins 4 points au score CHOP INTEND par rapport au score initial, en comparaison avec les patients recevant le traitement simulé (3 %). De même, 3 % des patients du groupe sous Spinraza et 46 % des patients du groupe sous traitement simulé ont présenté une détérioration du score CHOP INTEND par rapport au score initial.
Tableau 6: Résultats des critères d'évaluation principaux et secondaires lors de l'analyse finale – étude CS3B
Paramètre d'efficacité Patients traités Patients recevant
par Spinraza le traitement simulé
Survie
Survie sans événement2
Nombre de patients décédés ou devant être placés sous 31 (39 %) 28 (68 %)
ventilation permanente
Hazard Ratio (IC à 95 %) 0,53 (0,32 - 0,89)
Valeur de p6 p = 0,0046
Nombre de patients devant être placés sous ventilation 18 (23 %) 13 (32 %)
permanente2
Hazard Ratio (IC à 95 %) 0,66 (0,322 – 1,368)
Valeur de p6 p = 0,1329
Survie globale2
Nombre de patients décédés 13 (16 %) 16 (39 %)
Hazard Ratio (IC à 95 %) 0,37 (0,18 – 0,77)
Valeur de p6 p = 0,0041
Fonction motrice
Étapes du développement moteur3
Pourcentage de patients correspondant aux critères 37 (51 %)1 p < 0 (0 %)
prédéfinis de répondeur en termes d'atteinte des étapes 0,0001
du développement moteur (section 2 du HINE)4,5
Pourcentage au jour 183 30/73 (41 %) 2/37 (5 %)
Pourcentage au jour 302 22/49 (45 %) 0/28 (0 %)
Pourcentage au jour 394 20/37 (54 %) 0/21 (0 %)
Pourcentage présentant une amélioration du score total 49 (67 %) 5 (14 %)
pour les étapes du développement moteur
Pourcentage présentant une détérioration du score total 1 (1 %) 8 (22 %)
pour les étapes du développement moteur
CHOP-INTEND3
Pourcentage présentant une amélioration de 4 points 52 (71 %) p < 0,0001 1 (3 %)
Pourcentage présentant une détérioration de 4 points 2 (3 %) 17 (46 %)
Pourcentage présentant tout niveau d'amélioration 53 (73 %) 1 (3 %)
Pourcentage présentant tout niveau de détérioration 5 (7 %) 18 (49 %)
1 L'étude CS3B a été arrêtée après une analyse statistique positive du critère d'évaluation principal lors de l'analyse intermédiaire (un pourcentage de patients significativement plus élevé sur le plan statistique a correspondu à la définition de répondeur en termes d'étapes du développement moteur dans le groupe sous Spinraza (41 %) que dans le groupe sous traitement simulé (0 %) (p < 0,0001)).
2 Lors de l'analyse finale, la survie sans événement et la survie globale ont été évaluées sur la base de la population en intention de traiter (ITT: Spinraza n = 80; traitement simulé n = 41).
3 Au moment de l'analyse finale, les analyses du score CHOP INTEND et des étapes du développement moteur ont été effectuées sur la base de la population d'analyse de l'efficacité (Spinraza n = 73; traitement simulé n = 37).
4 Évalué lors de la visite du jour 183, du jour 302 ou du jour 394 de l'étude selon la date la plus éloignée de la visite initiale.
5 Selon l'échelle HINE (Hammersmith Infant Neurological Examination) section 2: patient présentant une augmentation ≥2 points [ou un score maximal] de la capacité de pédalage OU une augmentation ≥1 point pour les étapes du développement moteur contrôle de la tête, se retourner, s'asseoir, marcher à quatre pattes, se tenir debout ou marcher ET une amélioration dans plus de catégories d'étapes du développement moteur que de détériorations, défini comme répondeur pour cette analyse principale.
6 Sur la base d'un test des rangs logarithmiques stratifié selon la durée de la maladie.
Sur les 121 patients (80 traités par Spinraza et 41 ayant reçu le traitement simulé témoin) de l'étude CS3B, 89 patients (65 traités par Spinraza et 24 après traitement simulé) ont été inclus dans une étude d'extension en ouvert en cours (étude CS11). Les patients qui avaient reçu un traitement par Spinraza au cours de l'étude CS3B et qui ont été inclus dans l'étude CS11 en prolongation du traitement par Spinraza, ont reçu le médicament pendant une durée allant de 6 jours à 8,3 ans (médiane de 6,7 ans). Les patients randomisés dans le groupe sous traitement simulé au cours de l'étude CS3B et qui ont débuté le traitement par Spinraza dans l'étude CS11 ont reçu le médicament pendant une durée allant de 65 jours à 6,9 ans (médiane de 5,7 ans). Des améliorations de la fonction motrice (Figures 3 et 4) ont été observées aussi bien chez les patients ayant poursuivi le traitement par Spinraza de l'étude CS3B que chez les patients ayant débuté le traitement par Spinraza dans l'étude CS11. Le bénéfice le plus important observé était associé à un début de traitement plus précoce chez les patients. La plupart des patients étaient en vie lors de la dernière visite, après avoir débuté le traitement par Spinraza soit dans l'étude CS3B, soit dans l'étude CS11.
L'âge médian des patients commençant le traitement par Spinraza dans l'étude CS3B était de 5,4 mois (intervalle 1,7 – 14,9 mois). Le délai médian jusqu'au décès ou jusqu'à la ventilation permanente était de 1,4 an après instauration du traitement par Spinraza, en incluant la période d'extension dans l'étude CS11. À la fin de l'étude CS11, 60 des 81 patients (74 %) étaient toujours en vie et 41 des 81 patients (51 %) n'avaient pas été mis sous ventilation permanente selon la définition de l'étude CS11 (ventilation durant ≥16 heures par jour en continu pendant > 21 jours en l'absence d'événement aigu réversible ou de trachéotomie).
Le score total moyen du développement moteur (HINE-2) a augmenté de 5,3 (ET 4,6; n = 53) et le score CHOP INTEND moyen a augmenté de 18,4 (ET 14,7; n = 38) de l'instauration du traitement par Spinraza jusqu'aux périodes de suivi de 394 jours et 6 ans, respectivement.
Chez les patients randomisés dans le groupe sous traitement simulé dans l'étude CS3B et commençant le traitement par Spinraza seulement dans l'étude CS11, l'âge médian était de 17,8 mois (intervalle 10,1 – 23,0 mois). Avant l'instauration du traitement par Spinraza, 12 des 24 patients (50 %) avaient atteint le critère d'efficacité de ventilation permanente de l'étude CS11. Le délai médian jusqu'au décès ou jusqu'à la ventilation permanente était de 2,76 ans après l'instauration du traitement par Spinraza dans l'étude CS11. À la fin de l'étude CS11, 19 des 24 patients (79 %) étaient en vie et 6 patients sur 12 (50 %) étaient en vie sans ventilation permanente. Une amélioration des scores totaux moyens du développement moteur de 1,4 (ET 1,8; n = 12) et du score CHOP INTEND de 11,5 (ET 12,2; n = 10) a été observée entre le début de l'étude et la période de suivi de 394 jours et de 6 ans de l'étude CS11, respectivement.
Étude CS3A
L'étude CS3A était une étude de phase II en ouvert menée chez des patients symptomatiques ayant un diagnostic de SMA. L'âge médian lors de l'apparition des signes et symptômes cliniques était de 56 jours (21 - 254 jours) et les patients étaient porteurs de deux copies (n = 17) ou de trois copies (n = 2) du gène SMN2 (nombre de copies du gène SMN2 inconnu pour un patient). Les patients de cette étude étaient considérés comme très susceptibles de développer une SMA de type I. L'âge médian lors de la première dose était de 162 jours (37 - 223 jours). La durée de participation médiane des patients à l'étude était de 3 ans (62 jours - 3,9 ans).
Le critère d'évaluation principal était le pourcentage de patients présentant une amélioration dans une ou plusieurs catégories d'étapes du développement moteur (selon l'échelle HINE de la section 2: amélioration de ≥2 points [ou un score maximal] de la capacité de gigoter ou de la préhension volontaire OU amélioration de ≥1 point pour les étapes du développement moteur contrôle de la tête, se retourner, s'asseoir, marcher à quatre pattes, se tenir debout ou marcher). Le critère d'évaluation principal a été atteint chez 12 des 20 patients (60 %), avec une amélioration durable du nombre moyen d'étapes du développement moteur atteintes au cours du temps (Figures 3 et 4).
Une amélioration durable du score CHOP INTEND moyen a été observée entre l'inclusion et le jour 1072 (variation moyenne de 21,30). Au total, 11 patients sur 20 (55 %) ont atteint le critère d'évaluation d'augmentation ≥4 points au score CHOP INTEND total lors de leur dernière visite de l'étude avant le gel des données.
Lors de la dernière visite, 11 patients parmi les 20 inclus dans l'étude (55 %) étaient toujours en vie et ne nécessitaient pas de ventilation permanente. Quatre patients remplissaient les critères de ventilation permanente et cinq patients sont décédés pendant l'étude.
Forme d'apparition plus tardive
Étude CS4 (CHERISH)
L'étude CS4 est une étude de phase III randomisée, en double aveugle, contrôlée contre traitement simulé, menée chez 126 enfants symptomatiques présentant une SMA d'apparition plus tardive (apparition des symptômes après l'âge de 6 mois). Les patients ont été randomisés selon un rapport 2:1 pour recevoir soit Spinraza (administration de trois doses de charge puis de doses d'entretien tous les 6 mois) soit le traitement simulé témoin, avec une durée de traitement allant de 324 à 482 jours (médiane de 450 jours). Suite aux résultats positifs de l'analyse intermédiaire, l'étude CS4 a été interrompue et les patients ont été inclus dans l'étude de la phase d'extension en ouvert (CS11).
L'âge médian lors de la sélection était de 3 ans (intervalle compris entre 2 et 9 ans) et l'âge médian lors de l'apparition des signes et symptômes cliniques de SMA était de 11 mois (intervalle compris entre 6 et 20 mois). La majorité des patients (88 %) étaient porteurs de trois copies du gène SMN2 (deux copies chez 8 % des patients, quatre copies chez 2 % et nombre de copies inconnu chez 2 %). Au stade initial, les patients avaient obtenu un score HFMSE moyen de 21,6 et un score RULM moyen de 19,1. Tous les patients avaient atteint l'étape "position assise sans aide" , mais aucun l'étape "marcher sans assistance" . Les patients de cette étude étaient considérés comme très susceptibles de développer une SMA de type II ou III.
Les caractéristiques cliniques initiales étaient généralement comparables, à l'exception d'un déséquilibre dans le pourcentage de patients ayant de temps à autres atteint l'étape "se tenir debout sans aide" (13 % des patients du groupe sous Spinraza contre 29 % des patients du groupe sous traitement simulé) ou "marcher avec une assistance" (24 % des patients du groupe sous Spinraza contre 33 % des patients du groupe sous traitement simulé).
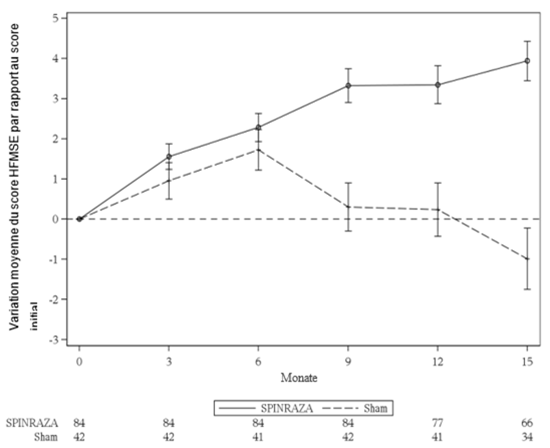
Une analyse intermédiaire a été effectuée lorsque l'évaluation du mois 6 a été réalisée chez tous les patients et que l'évaluation du mois 15 a été réalisée chez au moins 39 patients. Le critère d'évaluation principal examiné au moment de l'analyse intermédiaire était la variation du score HFMSE au mois 15 par rapport au score initial. L'analyse principale a été effectuée sur la population en ITT qui comprenait tous les patients randomisés et ayant reçu au moins 1 dose de Spinraza ou 1 traitement simulé (Spinraza n = 84; traitement simulé n = 42). Les données HFMSE après l'inclusion chez les patients sans visite au mois 15 ont été calculées selon la méthode d'imputation multiple. Par rapport aux patients recevant le traitement simulé, une amélioration statistiquement significative du score HFMSE par rapport au score initial a été observée chez les patients traités par Spinraza (Tableau 7).
Les résultats de l'analyse finale sont conformes aux résultats de l'analyse intermédiaire et montrent, chez les patients traités par Spinraza par rapport aux patients recevant le traitement simulé, une amélioration statistiquement significative du score HFMSE au mois 15 par rapport au score initial (Tableau 7, Figure 2).
Une analyse d'une sous-groupe de patients de la population en ITT qui avaient des valeurs observées au mois 15 a montré des résultats uniformes, statistiquement significatifs (p = 0,0000002). L'analyse statistique a été réalisée selon le modèle ANCOVA et une régression adaptée en fonction de l'âge au moment du screening ainsi que selon le score initial HFMSE. Parmi les patients présentant des valeurs observées au mois 15, une proportion plus élevée de patients traités par Spinraza présentaient une amélioration (73 % contre 41 %) et une proportion plus faible une détérioration (23 % contre 44 %) du score HFMSE total par rapport aux patients recevant le traitement simulé.
Lors de l'analyse finale, tous les critères d'évaluation secondaires, y compris les valeurs fonctionnelles mesurées et les valeurs du développement moteur de l'OMS, ont été testés de manière statistiquement formelle et figurent dans le Tableau 7.
L'amélioration de la fonction motrice a été plus précoce et plus importante lorsque le traitement était instauré rapidement après le diagnostic que lorsque l'instauration du traitement était différée. Cependant, les deux groupes ont obtenu un bénéfice par rapport aux patients recevant le traitement simulé.
Figure 2: Variation moyenne du score HFMSE par rapport au score initial au cours du temps (ITT1) – étude CS41,2
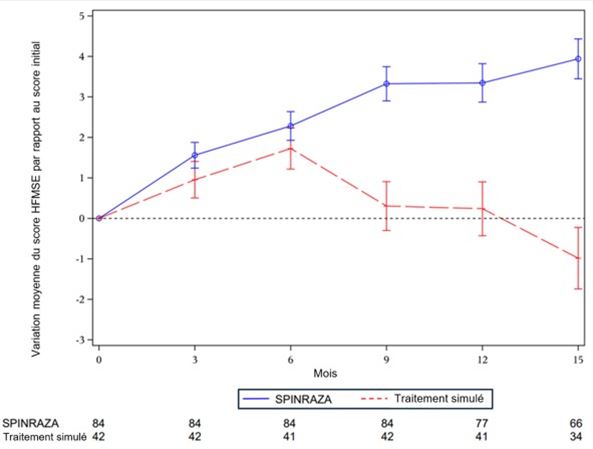
1 Nombre de patients avec une valeur observée à chacun des moments figurant sur l'axe x. Les valeurs moyennes représentées ont été calculées pour la population ITT (Spinraza n = 84, traitement simulé: n = 42) par la méthode d'imputation multiple.
2 Les barres d'erreur indiquent les écarts-types +/-
Tableau 7: Résultats des critères d'évaluation principal et secondaires lors de l'analyse finale -– étude CS41
Patients traités par Patients recevant le
Spinraza traitement simulé
Score HFMSE
Variation du score HFMSE total par rapport à 3,9 (IC à 95 %: 3,0; 4,9) -1,0 (IC à 95 %:
la valeur initiale après 15 mois1,2,3 p = 0,0000001 -2.5, 0.5)
Proportion de patients ayant obtenu une 56,8 % (IC à 95 %: 45,6; 26,3 % (IC à 95 %:
amélioration d'au moins 3 points par rapport à 68,1) p = 0,00065, 6 12.4, 40.2)
la valeur initiale1,4
RULM
Variation moyenne du score RULM total au mois 4,2 (IC à 95 %: 3,4; 5,0) 0,5 (IC à 95 %: -0.6,
15 par rapport au score initial1,2,3 p= 0,00000016 1.6)
Étapes de développement moteur de l'OMS
Pourcentage de patients ayant atteint une 19,7 % (IC à 95 %: 10,9; 5,9 % (IC à 95 %:
nouvelle étape du développement moteur au mois 31,3) p = 0,0811 0.7, 19.7)
154
Nombre atteint moyen de nouvelles étapes du 0,2 (intervalle de -1 à -0,2 (intervalle de
développement moteur2,3,4 2, IC à 95 %: 0,1; 0,3) p -1 à 1, IC à 95 %:
= 0,00016 -0.4, 0.0)3
1 Déterminés sur la base de la population en ITT (Spinraza, n = 84; traitement simulé, n = 42); les données chez les patients sans visite au mois 15 ont été calculées selon la méthode d'imputation multiple.
2 Moyenne des moindres carrés du modèle ANCOVA, où le traitement a été désigné comme l'effet et un ajustement a été réalisé pour chaque patient selon l'âge au moment du screening et selon le score initial.
3 Les valeurs négatives indiquent une détérioration, les valeurs positives une amélioration.
4 Déterminé sur la base des valeurs au mois 15 dans l'ensemble de données sur l'efficacité (Spinraza n = 66; traitement simulé n = 34); analyse reposant sur des valeurs calculées en l'absence de valeurs mesurées.
5 Sur la base d'une régression logistique avec effet thérapeutique et ajustement pour tenir compte de l'âge de chaque patient au moment de la sélection et du score HFMSE à l'inclusion.
6 Valeur nominale de p.
Remarque: nombre de patients en ITT ayant effectué toutes les visites: mois 3, 6, 9 (Spinraza n = 84, traitement simulé n = 42), mois 12 (Spinraza n = 77, traitement simulé n = 41), mois 15 (Spinraza n = 66, traitement simulé n = 34).
125 patients (83 traités par Spinraza et 42 ayant reçu le traitement simulé témoin) ont pris part à l'étude CS4 et ont poursuivi dans l'étude d'extension en ouvert CS11. Les patients randomisés dans le groupe Spinraza dans l'étude CS4 et qui ont été inclus dans l'étude CS11 en prolongation du traitement par Spinraza ont reçu le médicament pendant une durée moyenne de 7,2 ans (1,3 – 8,4 ans). Les patients randomisés dans le groupe sous traitement simulé dans l'étude CS4 et qui ont débuté le traitement par Spinraza dans l'étude CS11 ont reçu le médicament pendant une durée moyenne de 5,8 ans (2,7 – 6,7 ans). De nombreux patients traités par Spinraza ont montré une amélioration de la fonction motrice dans au moins l'un des deux tests de la fonction motrice jusqu'à la visite de suivi à 5,7 ans, le bénéfice le plus important étant observé chez ceux qui avaient débuté le traitement par Spinraza à un âge précoce.
L'âge médian des patients commençant le traitement par Spinraza dans l'étude CS4 était de 4,1 ans (intervalle 2,1 – 9,2 ans). À partir de l'instauration du traitement par Spinraza et en incluant la période d'extension du traitement dans l'étude CS11, le score HFMSE a d'abord augmenté à une valeur moyenne maximale de 4,5 points (ET 6,1; n = 83) après une période d'observation de 1,9 ans, pour ensuite diminuer au cours de l'observation suivante, de telle sorte qu'une augmentation de 1,3 points a été calculée par rapport à l'inclusion (ET 9,4; n = 54) après une période d'observation de 5,7 ans. Quelques patients ont été suivi jusqu'à 7,6 ans, avec une variation de -2,3 points par rapport à l'inclusion (ET 9,6; n = 39). Les scores RULM ont initialement augmenté à une valeur moyenne maximale de 6,4 points (ET 5,6; n = 74) lors de la visite de suivi de 3,9 ans, une valeur qui est restée stable à 6,4 points (ET 6,5; n = 54) jusqu'à la visite de suivi de 5,7 ans. Quelques patients ont été suivi jusqu'à 7,6 ans, avec une variation moyenne de 6,0 points par rapport à l'inclusion (ET 6,8; n = 40).
L'âge moyen des patients randomisés dans le groupe sous traitement simulé dans l'étude CS4 et qui ont débuté le traitement par Spinraza dans l'étude CS11 était de 4,9 ans (3,3 - 9,0 ans). La variation du score HFMSE consistait en une diminution moyenne de 1,3 point (ET 9,3; n = 22) et la variation du score RULM en une augmentation moyenne de 4,2 points (ET 4,4; n = 23) lors de la visite de suivi de 5,7 ans.
L'évolution de la maladie chez des patients non traités d'âge similaire et présentant des caractéristiques cliniques comparables montre une perte progressive de la fonction motrice au cours du temps, avec une diminution moyenne estimée du score HFMSE de 6,6 points sur une période de 5 ans. Au cours de l'évolution à long terme qui a suivi, le score HFMSE a continué de diminuer avec une baisse moyenne estimée de 8,3 points après 7 ans.
Études CS2 et CS12
Ces résultats sont corroborés par deux études en ouvert (études CS2 et CS12). L'analyse a inclus 28 patients qui avaient reçu leur première dose dans le cadre de l'étude CS2 et sont ensuite entrés dans la phase de prolongation (étude CS12). Les patients inclus dans les études étaient âgés de 2 à 15 ans au moment de la première dose. Sur les 28 patients, trois étaient âgés d'au moins 18 ans lors de leur dernière visite dans le cadre de l'étude. Un des 28 patients était porteur de deux copies du gène SMN2, 21 patients étaient porteurs de trois copies et 6 de quatre copies. Les patients de cette étude ont présenté un diagnostic de SMA de type II ou de type III. La valeur HFMSE moyenne au niveau de base était de 21,3 (intervalle 6 - 35) chez les patients avec SMA de type II (n = 11) et de 48,9 (intervalle 20 - 63) chez les patients avec SMA de type III (n = 17).
La valeur moyenne au niveau de base du résultat du test de l'Upper Limb Module (ULM) chez les patients avec SMA de type II était de 11,9 (intervalle 7 - 17) et la valeur moyenne du résultat du test de marche de 6 minutes (6MWT, six-minute walk test) au niveau de base était de 253,3 mètres (intervalle compris entre 0 et 563 mètres) chez les patients ambulatoires de type III (n = 13).
Les patients ont été évalués pendant une période de traitement de 3 ans. Il a été observé une amélioration durable chez les patients atteints de SMA de type II, avec une amélioration moyenne du score HFSME par rapport au score initial après 253 jours de 5,1 (ET de 4,05, n = 11) et après 2,9 ans de 9,1 (ET de 6,61, n = 9). Le nombre moyen de points après 253 jours était de 26,4 (ET de 11,91) et après 2,9 ans de 31,3 (ET de 13,02), sans plateau constaté. Ces résultats sont comparables à la baisse du nombre de points normalement observée au cours du temps chez les patients présentant une forme d'apparition plus tardive de SMA.
Les patients atteints de SMA de type III ont présenté une amélioration moyenne de 1,3 point (ET de 1,87, n = 16) au score HFSME par rapport au score initial après 253 jours et une amélioration de 1,2 point (ET de 4,64, n = 11) après 2,9 ans.
Le test Upper Limb Module a été réalisé chez les patients atteints de SMA de type II et a révélé une amélioration moyenne de 1,9 (ET de 2,68, n = 11) après 253 jours et de 3,5 (ET de 3,32, n = 9) après 2,9 ans. Le score total moyen était de 13,8 (ET de 3,09) après 253 jours et de 15,7 (ET de 1,92) après 2,9 ans.
Le test de marche de 6 minutes (6MWT, six-minute walk test) n'a été réalisé que chez les patients capables de marcher. Chez ces patients, il a été observé après 253 jours une amélioration de 28,6 mètres (ET de 47,22, n = 12) et après 2,9 ans une amélioration de 86,5 mètres (ET de 40,58, n = 8). La distance moyenne parcourue au 6MWT était de 278,5 mètres (ET de 206,46) après 253 jours et de 333,6 mètres (ET de 176,47) après 2,9 ans. Deux patients qui n'étaient jusque-là pas autonomes s'agissant de la marche sans aide (SMA de type III) sont devenus capables de marcher sans assistance, de même qu'un patient qui n'était pas du tout capable de marcher jusque-là (SMA de type II).
Patients atteints de la forme infantile de SMA ou d'une SMA d'apparition plus tardive
Étude CS7 (EMBRACE)
L'étude CS7 est une étude de phase II comportant deux parties; la partie 1 était randomisée, en double aveugle et avec contrôle contre traitement simulé et la partie 2 était une extension effectuée en ouvert. Cette étude a été menée chez des patients symptomatiques atteints d'une forme infantile de SMA chez le nourrisson (≤6 mois) ou d'une SMA d'apparition plus tardive (> 6 mois) et chez des porteurs de 2 ou 3 copies du gène SMN2, qui, en raison de leur âge lors de la sélection, du procédé de sélection ou du nombre de copies du gène SMN2 ne répondaient pas aux critères de participation à l'étude CS3B ou à l'étude CS4. Dans la partie 1 de l'étude, les patients ont été suivis pendant une durée médiane de 302 jours.
Tous les patients traités par Spinraza étaient toujours en vie à la fin de la partie 1 de l'étude, mais un patient du groupe témoin est décédé au jour 289 de l'étude. De plus, aucun des patients du groupe Spinraza ou du groupe de traitement simulé n'a nécessité l'utilisation d'une ventilation permanente. Parmi les 13 patients atteints de SMA infantile, le critère d'efficacité (étapes du développement moteur, échelle HINE section 2) a été atteint chez 7 sur 9 (78 %; IC à 95 %: 45, 94) du groupe Spinraza et chez 0 sur 4 (0 %; IC à 95 %: 0, 60) du groupe de traitement simulé. Parmi les 8 patients atteints d'une SMA d'apparition plus tardive, ce critère a été atteint chez 4 sur 5 (80 %; IC à 95 %: 38, 96) du groupe Spinraza et chez 2 sur 3 (67 %; IC à 95 %: 21, 94) du groupe de traitement simulé.
Nourrissons présymptomatiques
Étude CS5 (NURTURE)
L'étude CS5 est une étude en ouvert menée chez des enfants en bas âge présymptomatiques ayant un diagnostic génétique de SMA, qui ont été inclus dans l'étude à l'âge de 6 semaines ou moins.
En raison de leurs caractéristiques génétiques, les patients de cette étude étaient considérés comme très susceptibles de développer une SMA de type I ou II. L'âge médian au moment de l'administration de la première dose était de 22 jours.
À l'inclusion, le nombre médian des étapes du développement moteur atteintes était de 3 (intervalle compris entre 0 et 7), le score CHOP INTEND total médian était de 50,0 (intervalle compris entre 25 et 60) et l'amplitude médiane du PAMC pour le nervus ulnaris par rapport au moment de l'inclusion était de 2,65 mV (intervalle compris entre 1,00 et 6,7).
L'analyse intermédiaire (gel des données 19 février 2020) a été réalisée au moment où les patients avaient été inclus dans l'étude pendant une durée médiane de 48,3 mois (36,6 - 57,1 mois) et avaient lors de la dernière visite de l'étude un âge médian de 46,0 mois (34,0 - 57,0 mois). Lors de l'analyse intermédiaire, les 25 patients (porteurs de 2 copies du gène SMN2, n = 15; porteurs de 3 copies du gène SMN2, n = 10) étaient tous en vie et sans ventilation permanente. Le critère d'évaluation principal était le délai jusqu'au décès ou jusqu'à une intervention d'assistance respiratoire (définie comme une ventilation invasive ou non invasive durant ≥6 heures par jour en continu pendant ≥7 jours consécutifs OU une trachéostomie) et ce critère n'a pas pu être déterminé en raison d'un trop faible nombre d'événements. Quatre patients (porteurs de 2 copies du gène SMN2) ont nécessité une assistance respiratoire durant > 6 heures par jour pendant ≥7 jours en raison d'une affection aiguë réversible.
Les patients ont atteint des étapes du développement non attendues lors d'une SMA de type I ou de type II et correspondant plutôt à un développement normal. Lors de l'analyse intermédiaire, la totalité des 25 (100 %) patients arrivaient à s'asseoir sans aide et 23 (92 %) patients arrivaient à marcher avec assistance (étapes de développement moteur selon l'échelle de l'OMS) et 22 des 25 patients (88 %) arrivaient à marcher sans assistance. Le score CHOP-INTEND maximal (score de 64) était atteint par 21 (84 %) des patients. Tous les patients étaient capables de téter et de déglutir lors de la dernière visite (jour 778); le score maximal sur l'échelle HINE section 1 a été atteint chez 22 des 25 nourrissons (88 %).
La proportion de patients qui ont développé une SMA cliniquement avérée a été évalué parmi les patients qui avaient atteint la visite du jour 700 lors de l'analyse intermédiaire (n = 25). Les critères de SMA cliniquement avérée définis par le protocole étaient: poids pour l'âge en dessous du 5e percentile de l'OMS, diminution d'au moins 2 percentiles principaux sur la courbe de poids en fonction de l'âge, pose d'une gastrostomie percutanée et/ou incapacité à atteindre une des étapes de développement attendues en fonction de l'âge selon l'OMS (s'asseoir sans aide, se tenir debout avec assistance et marcher à quatre pattes, marcher avec assistance, se tenir debout sans soutien et marcher sans assistance). Au jour 700, 7 patients sur 15 (47 %) porteurs de 2 copies du gène SMN2 et aucun des 10 porteurs de 3 copies du gène SMN2 répondaient aux critères de SMA cliniquement avérée définis par le protocole. Toutefois, ces patients ont présenté une prise de poids et ont atteint des étapes qui ne sont normalement pas atteintes lors d'une SMA de type I. La Figure 3 et la Figure 4 présentent une comparaison du développement moteur chez les patients présentant une SMA infantile symptomatique ou une SMA présymptomatique.
Figure 3: Variation des étapes du développement moteur selon le bilan HINE en fonction des jours d'étude dans le cadre des études CS3B (groupes sous Spinraza et sous traitement simulé), CS3A, CS5 et CS11 (population ITT)

Figure 4: Score CHOP INTEND en fonction du nombre de jours dans l'étude dans le cadre des études CS3B (Spinraza et traitement simulé), CS3A, CS5 et CS11 (population ITT)

Patients symptomatiques sous traitement avec le schéma posologique de 50/28 mg
La partie B de l'étude SM203 était une étude randomisée en double aveugle évaluant la sécurité et l'efficacité du nusinersen 50/28 mg chez des patients non pré-traités atteints de la forme infantile de SMA ou de SMA d'apparition plus tardive. La partie B visait à évaluer l'efficacité chez les patients atteints de la forme infantile de la maladie dans le groupe recevant 50/28 mg par rapport à un groupe sous traitement simulé apparié prédéfini issu de l'étude CS3B. Le schéma posologique à 12 mg dans la partie B de l'étude SM203 a fourni des données probantes, mais la puissance statistique de l'étude n'était pas suffisante pour détecter des différences statistiquement significatives entre les patients randomisés pour recevoir 50/28 mg et ceux randomisés pour recevoir 12 mg de nusinersen. La partie C était une étude ouverte évaluant la sécurité et l'efficacité chez les enfants et les adultes atteints de SMA infantile ou de SMA d'apparition plus tardive qui sont passés d'un schéma posologique de 12 mg à un schéma posologique de 50/28 mg.
Étude SM203, partie B, cohorte déterminante pour l'autorisation atteinte de la forme infantile de la maladie
Les patients atteints de la forme infantile de SMA (porteurs de 2 copies du gène SMN2; apparition des symptômes avant l'âge de 6 mois) ont été randomisés selon un rapport 2:1 et traités selon un schéma posologique de 50/28 mg ou de 12 mg. Sur la base d'analyses prédéfinies, 20 des 37 patients du groupe témoin de l'étude CS3B ont été appariés en fonction de similitudes tant au niveau de la durée de la maladie que du score CHOP-INTEND à l'inclusion. Le critère d'évaluation principal était la variation du score CHOP-INTEND au jour 183 chez les patients atteints de SMA infantile dans le groupe recevant 50/28 mg par rapport au groupe témoin apparié issu de l'étude 1. Par rapport au groupe présentant la forme infantile de SMA dans l'étude CS3B, les patients de l'étude SM203 présentaient une durée plus courte de la maladie (temps écoulé entre l'apparition des symptômes et la sélection) et des scores CHOP-INTEND plus bas à l'inclusion, ce qui indiquait que leur maladie avait une évolution plus rapide et était plus avancée. Une attribution prédéfinie à un sous-groupe du groupe de traitement simulé témoin de l'étude CS3B a permis de minimiser en partie ce déséquilibre. Néanmoins, dans les groupes recevant 50/28 mg et 12 mg de l'étude SM203, la durée moyenne de la maladie (ET) à l'inclusion était toujours plus courte et le score CHOP-INTEND à l'inclusion était plus bas que dans le groupe témoin correspondant (Tableau 8). D'autres caractéristiques démographiques initiales importantes (âge lors de la première dose, âge lors de la sélection, âge lors de l'apparition des symptômes, nombre de copies SMN2 et fonction motrice à l'inclusion) étaient équilibrées entre le groupe recevant 50/28 mg, le groupe recevant 12 mg et le groupe témoin correspondant.
Tableau 8: Caractéristiques initiales des patients participant à la partie B de l'étude SM203
Caractéristiques des patients Spinraza 50/28 mg Spinraza 12 mg (n Traitement simulé
(n = 50) = 25) témoin apparié de
CS3B (n = 20)
Âge médian (fourchette) lors de 18,4 (de 2 à 33) 15,9 (de 3 à 31) 22,2 (de 4 à 34)
la première dose (semaines),
valeur initiale
Âge moyen (ET) au début des 7,5 (5,26) 5,8 (4,44) 8,8 (5,11)
symptômes (semaines), valeur
initiale
Durée moyenne de la maladie (ET), 9,6 (5,29) 9,2 (6,11) 11,1 (4,92)
valeur initiale (temps écoulé
entre l'apparition des symptômes
et la sélection) (semaines)
Score CHOP-INTEND moyen (ET) 20,9 (10,23) 19,9 (9,63) 23,6 (5,84)
(points), valeur initiale
Concentration plasmatique moyenne 304,7 (283,9) 329,4 (175,9) 287,3 (140,4)
(ET) de NfL, valeur initiale
(pg/ml)
La variation moyenne du score CHOP-INTEND entre l'inclusion et le jour 183 était statistiquement plus importante dans le groupe recevant 50/28 mg (amélioration de 15,1 points) que dans le groupe témoin correspondant (aggravation de 11,1 points) (différence entre les moyennes des MC: 26,19 points [IC à 95 %: 20,7; 31,7] p < 0,0001; Figure 5).
La variation du score CHOP-INTEND entre l'inclusion et le jour 302 était numériquement plus élevée dans le groupe recevant 50/28 mg que dans le groupe recevant 12 mg, sur la base de la différence en termes de classement, mais cette différence n'était pas statistiquement significative selon le JRT (différence entre les moyennes des MC en termes de classement (1,00 [IC à 95 %: -9,290; 11,299]; JRT p = 0,8484). La variation des moyennes des MC entre l'inclusion et le jour 302, calculée à partir d'une analyse ANCOVA avec IM, était numériquement plus élevée dans le groupe recevant 12 mg; groupe recevant 50/28 mg (amélioration de 19,6 points), groupe recevant 12 mg (amélioration de 21,6 points; IC à 95 %: 16,5; 22,8) (différence entre les moyennes des MC: -1,94 [7,77; 3,88]). Une proportion statistiquement significative de patients du groupe recevant 50/28 mg correspondait à la définition de répondeur selon le score HINE section 2 (HINE-2) au jour 183 par rapport au groupe témoin correspondant (58 % contre 0 %; p < 0,0001; Tableau 9). Dans une analyse complémentaire, 60 % des patients du groupe recevant 50/28 mg et 44 % des patients du groupe recevant 12 mg correspondaient à la définition de répondeur selon HINE-2 au jour 302. La variation moyenne du score HINE-2 entre l'inclusion et le jour 183 était significativement plus importante dans le groupe recevant 50/28 mg (amélioration de 3,7 points) que dans le groupe témoin correspondant (aggravation de 0,2 points) (différence entre les moyennes des MC: 3,9 (IC à 95 %: 2,5; 5,4; p < 0,0001). La variation du score des étapes du développement moteur HINE-2 entre l'inclusion et le jour 302 était numériquement plus importante dans le groupe recevant 50/28 mg (amélioration de 5,9 points) que dans le groupe recevant 12 mg (amélioration de 5,3 points) (différence entre les moyennes des MC: 0,58 (1,89; 3,04)), mais ces différences n'étaient pas statistiquement significatives.
Tableau 9: Partie B de l'étude SM203 forme infantile: résultats du développement moteur dans le groupe recevant 50/28 mg par rapport au groupe recevant 12 mg et dans le groupe recevant 50/28 mg par rapport au groupe témoin apparié
Paramètres d'efficac Spinraza groupe Spinraza groupe Traitement simulé Différences entre
ité recevant 50/28 mg recevant 12 mg (n = témoin apparié issu les bras (IC à 95 %)
(n = 50) 25) de l'étude CS3B (n
= 20)
Groupe recevant
50/28 mg contre
traitement simulé
témoin apparié
CHOP-INTEND
Moyenne des MC (IC 42,9 (38,7; 47,2) 16,9 (10,1; 23,7) 26,06 (17,94;
à 95 %) pour la 34,17) p < 0,00013
valeur de classement
de la variation
entre l'inclusion
et le jour 183
Variation des 15,1 (12,4; 17,8) -11,1 (-15,9; -6,2) 26,2 (20,7; 31,7)2
valeurs moyennes
des MC (IC à 95 %)
entre l'inclusion
et le jour 1831,2
HINE-2, répondeurs5
Pourcentage de ceux 29 (58%) 0 (0%) 58 % (39,5; 71,8)4
qui ont atteint les p < 0,0001
critères relatifs
au développement
moteur au jour 183
HINE-2, score global
Moyenne des MC (IC 43,1 (39,0; 47,2) 16,5 (9,9; 23,0) 26,67 (18,81;
à 95 %) pour la 34,53) p < 0,00013
valeur de classement
de la variation
entre l'inclusion
et le jour 183
Variation des 3,7 (3,0; 4,4) -0,2 (-1,5; 1,0) 3,94 (2,46; 5,42)2
valeurs moyennes
des MC (IC à 95 %)
entre l'inclusion
et le jour 183 pour
le score global de
HINE-21,2
Groupe recevant
50/28 mg contre
groupe recevant 12
mg
CHOP-INTEND
Moyenne des MC (IC 38,3 (32,7; 44,0) 37,3 (29,1; 45,5) 1,0 (-9,29; 11,30)
à 95 %) pour la p = 0,8483
valeur de classement
de la variation
entre l'inclusion
et le jour 302
Variation des 19,6 (16,5; 22,8) 21,6 (16,6; 26,6) -1,9 (7,8; 3,9)2
valeurs moyennes
des MC (IC à 95 %)
entre l'inclusion
et le jour 3021,2
HINE-2, répondeurs5
Pourcentage de ceux 30 (60%) 11 (44%) 16 % (-7,73; 39,73)
qui ont atteint les p = 0,225
critères relatifs
au développement
moteur au jour 302
HINE-2, score global
Moyenne des MC (IC 40,0 (35,1; 44,9) 33,9 (26,9; 41,0) 6,12 (-2,69; 14,94)
à 95 %) pour la p = 0,1733
valeur de classement
de la variation
entre l'inclusion
et le jour 302
Variation des 5,9 (4,6; 7,2) 5,3 (3,3; 7,4) 0,58 (-1,89; 3,04)
valeurs moyennes
des MC (IC à 95 %)
entre l'inclusion
et le jour 3021,2
1 Utilisation d'ANCOVA et de l'imputation multiple (IM)
2 Différence entre les moyennes des moindres carrés (MC)
3 Test de rang conjoint (Joint Rank Test)
4 Test exact de Fisher
5 Définition des répondeurs dans cette analyse primaire: augmentation du score de ≥2 points [ou score maximal] en termes de capacité à pédaler OU augmentation du score de ≥1 point en termes de critères moteurs (contrôle de la tête, rouler, s'asseoir, ramper, se tenir debout ou marcher) ET amélioration dans un nombre plus élevé de catégories de critères moteurs que d'aggravation).
Figure 5: Partie B: forme infantile de SMA: CHOP-INTEND: variation des moyennes des MC (ET) par rapport à l'inclusion après la visite, analyse ANCOVA avec IM: groupe recevant 50/28 mg et groupe de traitement simulé correspondant.

Les critères d'évaluation secondaires importants dans la cohorte présentant une forme infantile de la maladie dans la partie B de l'étude SM203 étaient les variations du taux plasmatique de NfL par rapport au groupe de traitement simulé correspondant (au jour 183) et au groupe avec la posologie autorisée de 12 mg (au jour 64). Chez les patients atteints de la forme infantile de SMA, les taux plasmatiques moyens de NfL au jour 183 étaient réduits de 94 % dans le groupe recevant 50/28 mg, contre 30 % dans le groupe sous traitement simulé correspondant; différence entre le groupe recevant 50/28 mg et le groupe sous traitement simulé correspondant: 92 %; p < 0,0001. Au jour 64, les taux plasmatiques moyens de NfL étaient réduits de 88 % dans le groupe recevant 50/28 mg, contre 77 % dans le groupe recevant 12 mg; différence entre le groupe recevant 50/28 mg et le groupe recevant 12 mg: 49 %; valeur de p nominale 0,0020.
Tableau 10: Étude SM203 – Partie B forme infantile: rapport entre les valeurs géométriques moyennes de NfL: jour 183: groupe recevant 50/28 mg contre groupe sous traitement simulé correspondant, et jour 64: groupe recevant 50/28 mg contre groupe recevant 12 mg
NfL plasmatique Spinraza 50/28 mg Spinraza 12 mg (n = Traitement simulé Rapports entre les
(n = 50) 25) témoin apparié, moyennes des moindre
patients issus de s carrés des résulta
l'étude CS3B (n = ts pour le bras de
20) l'étude du groupe
de comparaison:
groupe recevant
50/28 mg (IC à 95
%)1
Rapport ajusté des 0,06 0,70 0,08 (0,05; 0,14) p
moyennes géométrique < 0,00013
s, valeur initiale:
jour 183.1,2
Rapport ajusté des 0,12 0,23 0,51 (0,33; 0,78)
moyennes géométrique p < 0,0050†3
s, valeur initiale:
jour 64.1,2
† Valeur de p nominale statistiquement significative
1 Utilisation d'ANCOVA et de l'imputation multiple (IM)
2 Différence entre les moyennes des moindres carrés (MC)
3 Test de rang conjoint (Joint Rank Test)
Dans le groupe recevant 50/28 mg, la réduction du risque de mortalité ou de ventilation permanente était statistiquement significative, avec 68 % (p = 0,0006) par rapport au groupe sous traitement simulé correspondant et 29,9 % (p = 0,2775) par rapport au groupe recevant 12 mg. Le délai médian jusqu'au décès ou à la ventilation permanente n'a pas été atteint dans le groupe recevant 50/28 mg; il était de 24,7 semaines dans le groupe recevant 12 mg et de 19,1 semaines dans le groupe sous traitement simulé apparié. Des observations similaires ont été faites en termes de survie globale (Tableau 11).
Tableau 11: Étude SM203 – Partie B forme infantile: survie sans événement et survie globale: groupe recevant 50/28 mg par rapport au groupe sous traitement simulé correspondant et groupe recevant 50/28 mg par rapport au groupe recevant 12 mg
Paramètres d'efficac Spinraza 50/28 mg Spinraza 12 mg (n = Traitement simulé Hazard Ratio (IC à
ité (n = 50) 25) témoin apparié, 95 %)
patients issus de
l'étude CS3B (n =
20)
Survie
Survie sans événemen 19 (38 %) 12 (48 %) 17 (85 %) 50/28: MS 0,322
t Nombre de (0,158; 0,657) p <
patients décédés ou 0,0006†1
sous ventilation
permanente
50/28:12 0,701
(0,338; 1,452) p =
0,27751
Survie globale 10 (20 %) 6 (24 %) 11 (55 %) 50/28: MS 0,279
Nombre de patients (0,112; 0,696) p <
décédés 0,0012†1
50/28: 12 0,730
(0,264; 2,015) p =
0,48211
† Valeur de p nominale statistiquement significative
1 Valeur de p déterminée par le test du log-rank
Étude M203, partie B, cohorte avec apparition plus tardive de la maladie
24 patients atteints de SMA d'apparition plus tardive (la plupart porteurs de 3 copies du gène SMN2; apparition des symptômes après l'âge de 6 mois) ont été randomisés selon un rapport 2:1 et traités soit avec le schéma posologique de 50/28 mg (n = 16) ou le schéma posologique de 12 mg (n = 8). Les analyses ont été prédéfinies de manière à comparer le schéma posologique de 50/28 mg à des sous-groupes appariés issus de l'étude CS4, y compris un groupe apparié ayant reçu 12 mg (3 doses de charge, suivies de doses d'entretien tous les 6 mois) (n = 32) et un groupe témoin apparié recevant un traitement simulé (n = 16). La puissance statistique des analyses n'était pas suffisante pour détecter des différences significatives entre les groupes de traitement.
Les caractéristiques démographiques initiales du groupe recevant 50/28 mg, du groupe de traitement apparié et du groupe sous traitement simulé apparié étaient globalement équilibrées, à l'exception de l'âge à la première dose. L'âge moyen (ET) lors de la première dose était le suivant: il était de 6,1 (3,0) ans dans le groupe recevant 50/28 mg, de 5,7 (3,0) ans dans le groupe recevant 12 mg, de 5,47 (1,8) ans dans le groupe de traitement correspondant et de 5,13 (1,8) ans dans le groupe sous traitement simulé correspondant.
La variation du score HFMSE entre l'inclusion et le jour 302 (moyenne des MC [IC à 95 %]) était numériquement plus élevée dans le groupe recevant 50/28 mg (3,3 [1,5; 5,0]) que dans le groupe recevant 12 mg (2,6 [0,2; 5,1]). Différence entre les moyennes des MC: 0,63 (-2,5, 3,8; p = 0,70). La variation du score HFMSE entre l'inclusion et le jour 279 était également numériquement plus élevée dans le groupe recevant 50/28 mg que dans le groupe recevant 12 mg correspondant (jour 274) de l'étude CS4 (différence entre les moyennes des MC: 1,7 (-0,3, 3,6); p = 0,095) et le groupe sous traitement simulé correspondant de l'étude CS4 (différence entre les moyennes des MC: 3,2 (0,2; 6,2); p = 0,037). 3,2 (0,2, 6,2); p = 0,037). La variation du score RULM entre l'inclusion et le jour 302 (moyenne des MC [IC à 95 %]) était numériquement plus élevée dans le groupe recevant 50/28 mg (2,5 [0,7, 4,2]) que dans le groupe recevant 12 mg (1,8 [-0,8, 4,4]), mais la différence n'était pas statistiquement significative (p = 0,66). La variation du score RULM entre l'inclusion et le jour 279 était également numériquement plus élevée dans le groupe recevant 50/28 mg que dans le groupe recevant 12 mg correspondant de l'étude CS4 (différence entre les moyennes des MC: 0,5 (-1,0; 1,9; p = 0,55)) et le groupe sous traitement simulé correspondant de l'étude CS4 (différence entre les moyennes des MC: 1,7 (- 0,2, 3,5); p = 0,076).
Étude SM203, partie C
La partie C de l'étude SM203 était une cohorte ouverte comprenant 40 patients âgés de 4 à 65 ans porteurs de 1 à 4 copies du gène SMN2, qui étaient passés d'un traitement de 12 mg à un traitement de 50/28 mg. Les patients ont reçu une dose de 50 mg, suivie de deux doses d'entretien de 28 mg chacune (à 4 mois d'intervalle).
Deux patients (5 %) présentaient la forme infantile, tandis que chez 38 patients (95 %) la maladie s'était déclarée plus tardivement. Au moment où ils ont reçu leur dose de charge de 50 mg, 16 patients étaient âgés de moins de 18 ans et 24 patients de plus de 18 ans. L'âge médian (intervalle) au début des symptômes de SMA était de 24 mois (de 4 à 192). La durée médiane (intervalle) du traitement dans le cadre du schéma thérapeutique à 12 mg de Spinraza était de 3,9 ans (de 1 à 5). À l'inclusion, 21 patients (53 %) étaient capables de faire 15 pas de manière autonome.
Le passage du schéma posologique de 12 mg à celui de 50/28 mg a permis d'observer des améliorations entre l'inclusion et le jour 302 au niveau du score HFMSE (amélioration de 1,8 point [ET 3,99]) et du score RULM (amélioration de 1,2 point [ET 2,14]). Chez les patients adultes (âgés de plus de 18 ans; n = 24), des améliorations ont été observées au jour 302 au niveau du score HFMSE (2,3 points [ET 3,95]) et du score RULM (0,9 point [ET 1,89]) (Figures 6 et 7).
Figure 6: Partie C de l'étude SM203: HFMSE: variation moyenne (ET) du score total après la visite: groupe ITT

Figure 7: Partie C de l'étude SM203: RULM: variation moyenne (ET) du score total après la visite: groupe ITT

Traitement chez les patients adultes atteints de SMA
Dans une analyse intermédiaire, des données d'études cliniques de 7 jeunes patients adultes atteints d'une SMA de type II ou III (âgés de 18,8 à 22,5 ans) dont le traitement a débuté à l'adolescence, qui ont été suivis pendant 5,3 à 6,8 ans dans l'étude CS2/CS12/SHINE, suggèrent une stabilisation ou une amélioration des fonctions motrices chez ces patients, comme l'ont révélé le test de marche de 6 minutes (6MWT) et le score HFMSE.
La partie C de l'étude SM203 a porté sur 24 patients âgés de plus de 18 ans.
Les données de sécurité de la population de patients adultes correspondent au profil de sécurité connu du nusinersen et aux comorbidités associées à la SMA sous-jacente.
Résultats concernant l'utilisation chez l'adulte après la mise sur le marché
Des rapports bibliographiques d'études observationnelles sur l'utilisation du nusinersen 12 mg chez des patients atteints d'une SMA de type II et III, dont le traitement a débuté à l'âge adulte, avec un suivi de 10 à 14 mois, suggèrent une stabilisation ou une amélioration des fonctions motrices (HFMSE, 6MWT, RULM) chez des patients d'âges différents avec une évolution plus ou moins grave de la maladie, alors que chez les patients atteints de SMA non traités, une diminution des fonctions motrices a été rapportée au cours du temps.
|