CompositionPrincipes actifs
Pegcétacoplan
Excipients
Sorbitol (E420), acide acétique glacial (E260), acétate de sodium trihydraté (E262), hydroxyde de sodium (pour ajustement du pH, E524), eau pour préparations injectables.
Contient 41 mg/ml de sorbitol (820 mg/flacon) et au maximum 0,37 mg/ml de sodium (7,4 mg/flacon).
Indications/Possibilités d’emploiAspaveli est indiqué
·En monothérapie pour le traitement de l’hémoglobinurie paroxystique nocturne (HPN) chez les patients adultes qui présentent une anémie hémolytique (voir Posologie/Mode d’emploi et Efficacité clinique).
·Pour le traitement de la glomérulopathie à dépôts de C3 (C3G) ou de la glomérulonéphrite membranoproliférative à médiation par les complexes immuns primaire (IC-MPGN) chez des patients adultes et adolescents (de 12 à 17 ans).
Posologie/Mode d’emploiLe traitement doit être instauré sous la surveillance d’une personne exerçant une profession médicale qui est expérimentée en matière de prise en charge des affections hématologiques ou rénales.
Aspaveli est destiné à être administré par voie sous-cutanée à l’aide d’une pompe à perfusion avec seringue disponible dans le commerce. Une auto-administration est possible. Aspaveli doit être administré dans l’abdomen, les cuisses, les hanches ou le haut des bras.
L’autoperfusion à domicile peut être envisagée pour les patients qui ont bien toléré le traitement dans des centres de traitement expérimentés. Une éventuelle auto-perfusion à domicile doit être décidée après évaluation et sur recommandation du médecin du patient.
L’HPN est une maladie chronique et il est recommandé de poursuivre le traitement par Aspaveli à vie, sauf si l’arrêt de ce dernier est cliniquement indiqué (voir Mises en garde et précautions).
La C3G et l’IC-MPGN primaire sont des maladies chroniques. Il est déconseillé d’arrêter le traitement par ce médicament, sauf si l’arrêt de ce dernier est cliniquement indiqué.
Posologie usuelle
Aspaveli peut être administré par un professionnel de santé, ou par le patient lui-même ou son aidant en suivant les instructions appropriées.
Avant le traitement par Aspaveli:
·Patients déjà vaccinés: il convient de s’assurer que la vaccination contre des bactéries encapsulées telles que Streptococcus pneumoniae, Neisseria meningitidis des types A, C, W, Y et B et Haemophilus influenzae de type B (Hib) a eu lieu dans les deux ans précédant le début du traitement par Aspaveli (voir Mises en garde et précautions).
·Patients non vaccinés auparavant: les vaccins nécessaires doivent être administrés au moins deux semaines avant l’administration de la première dose d’Aspaveli (voir Mises en garde et précautions).
oSi un traitement immédiat par Aspaveli est indiqué, il convient de réaliser les vaccinations nécessaires dès que possible et de prescrire au patient des médicaments antibactériens pour un traitement prophylactique de deux semaines (voir Mises en garde et précautions).
HPN
Patients adultes atteints d’HPN
Aspaveli doit être administré deux fois par semaine sous la forme d’une perfusion sous-cutanée à 1080 mg réalisée à l’aide d’une pompe à perfusion avec seringue disponible dans le commerce, qui permet de délivrer des volumes allant jusqu’à 20 ml. Les deux doses hebdomadaires doivent être administrées le jour 1 et le jour 4 de chaque semaine de traitement (voir Mode d’administration).
Patients atteints d’HPN qui passent d’un inhibiteur de C5 à Aspaveli
·Pendant les quatre premières semaines, Aspaveli doit être administré à raison de deux doses sous-cutanées de 1080 mg par semaine en complément de la dose d’inhibiteur de C5 déjà administrée, afin de limiter le risque d’hémolyse liée à un arrêt brutal du traitement.
·Au-delà des quatre semaines, le patient doit arrêter le traitement par l’inhibiteur de C5 et poursuivre le traitement par Aspaveli en monothérapie.
·Le passage d’inhibiteurs complémentaires autres qu’éculizumab au pegcétacoplan n’a pas été étudié. L’arrêt d’autres inhibiteurs complémentaires avant d’avoir atteint l’état d’équilibre du pegcétacoplan doit être effectué avec prudence (voir Pharmacocinétique).
Ajustement de la posologie en cas d’HPN
·La posologie peut être ajustée à 1080 mg tous les trois jours (c’est-à-dire jour 1, jour 4, jour 7, jour 10, jour 13, etc.) si le patient présente un taux de lactate déshydrogénase (LDH) supérieur à deux fois la limite supérieure de la normale (ULN, upper limit of normal).
·En cas d’augmentation de la dose, le taux de LDH devra être surveillé deux fois par semaine pendant au moins quatre semaines.
C3G et IC-MPGN primaire
Aspaveli est administré deux fois par semaine sous la forme d’une perfusion sous-cutanée réalisée à l’aide d’une pompe à perfusion avec seringue disponible dans le commerce, qui permet de délivrer des volumes allant jusqu’à 20 ml. Les deux doses hebdomadaires doivent être administrées le jour 1 et le jour 4 de chaque semaine de traitement.
Patients adultes atteints de C3G ou d’IC-MPGN primaire
Aspaveli est administré deux fois par semaine sous la forme d’une perfusion sous-cutanée à 1080 mg.
Patients adolescents atteints de C3G ou d’IC-MPGN primaire
Chez les patients adolescents, le schéma posologique est fonction du poids corporel du patient:
|
Poids corporel
|
Première dose
(volume de perfusion)
|
Deuxième dose
(volume de perfusion)
|
Dose d‘entretien
(volume de perfusion)
| |
50 kg et plus
|
1080 mg deux fois par semaine (20 ml)
| |
35 à moins de 50 kg
|
648 mg (12 ml)
|
810 mg (15 ml)
|
810 mg deux fois par semaine (15 ml)
| |
30 à moins de 35 kg
|
540 mg (10 ml)
|
540 mg (10 ml)
|
648 mg deux fois par semaine (12 ml)
|
En cas d’omission d’une dose
Si une dose d’Aspaveli pour le traitement de l’HPN, de la C3G ou de l’IC-MPGN primaire a été oubliée, il convient de l’administrer dès que possible, puis de poursuivre le traitement selon le schéma posologique habituel. N’administrez jamais plus d’une dose le même jour.
Patients atteints de C3G ou d’IC-MPGN primaire après transplantation rénale (récidive de la maladie)
Le diagnostic de récidive de C3G ou d’IC-MPGN primaire doit être établi sur la base d’une biopsie du transplant rénal. Une récidive de C3G ou d’IC-MPGN primaire peut être détectée lors d’une biopsie de routine après la transplantation, autrement, une biopsie doit être réalisée en cas de signes cliniques indiquant une récidive de la maladie. Le traitement par pegcétacoplan peut être commencé, comme dans l’étude APL2-C3G-204 (voir Efficacité clinique), avant l’apparition de signes cliniques, par exemple une baisse du débit de filtration glomérulaire estimé (DFGe) ou une augmentation du ratio protéinurie/créatinurie (uPCR).
Instructions posologiques particulières
Patients présentant des troubles de la fonction hépatique
La sécurité et l’efficacité du pegcétacoplan n’ont pas été étudiées chez des patients qui présentent des troubles de la fonction hépatique; cependant, aucun ajustement posologique n’est préconisé, car l’insuffisance hépatique ne devrait pas avoir d’impact sur la clairance du pegcétacoplan (voir Pharmacocinétique).
Patients présentant des troubles de la fonction rénale
Une insuffisance rénale sévère (clairance de la créatinine < 30 ml/min) n’a eu aucun impact sur la pharmacocinétique du pegcétacoplan; par conséquent, il n’est pas nécessaire d’ajuster la dose de pegcétacoplan chez les patients atteints d’insuffisance rénale. Aucune donnée n’est disponible concernant l’utilisation du pegcétacoplan chez les patients atteints d’insuffisance rénale terminale (IRT) nécessitant une dialyse (voir Pharmacocinétique).
Patients âgés
Bien qu’aucune différence manifeste liée à l’âge n’ait été observée au cours des études cliniques et qu’aucun élément n’indique que des précautions particulières doivent être prises lors du traitement de patients âgés, le nombre de patients âgés de 65 ans et plus n’était pas suffisant pour déterminer s’il existe des différences liées à l’âge.
Enfants et adolescents
La sécurité et l’efficacité du pegcétacoplan chez les enfants atteints d’HPN âgés de 0 à moins de 18 ans ne sont pas établies. Aucune donnée n’est disponible.
La sécurité et l’efficacité du pegcétacoplan chez les enfants atteints de C3G ou d’IC-MPGN primaire âgés de moins de 12 ans ne sont pas établies. Aucune donnée n’est disponible.
Mode d’administration
Aspaveli doit être exclusivement administré par voie sous-cutanée à l’aide d’une pompe à perfusion avec seringue permettant de délivrer des doses d’un volume nominal de 20 ml.
Lors de l’instauration du traitement par Aspaveli, un professionnel de santé qualifié doit former le patient aux techniques de perfusion, à l’utilisation d’une pompe à perfusion avec seringue, à la tenue d’un carnet de suivi du traitement, à l’identification des effets indésirables possibles et aux mesures à prendre le cas échéant.
Aspaveli doit être administré par perfusion sous-cutanée dans l’abdomen, la cuisse ou le haut du bras. Les différents sites de perfusion doivent être espacés d’au moins 7,5 cm. Il convient de changer de site de perfusion à chaque administration et d’éviter toute perfusion dans des zones où la peau est sensible, contusionnée, rouge ou dure ainsi que dans les tatouages, les cicatrices ou les vergetures.
La durée habituelle de la perfusion est d’environ 30 minutes (en cas de perfusion sur deux sites ) ou d’environ 60 minutes (en cas de perfusion sur un site). La perfusion doit débuter dès qu’Aspaveli a été prélevé dans la seringue. L’administration doit être terminée dans les deux heures qui suivent la préparation de la seringue.
Voir Remarques concernant la manipulation et les Instructions sur la notice pour plus d’informations sur la préparation et l’administration du médicament.
Contre-indicationsAspaveli est contre-indiqué chez les patients:
·qui présentent une hypersensibilité au pegcétacoplan ou à l’un des excipients;
·qui présentent une infection non guérie due à une bactérie encapsulée telle que Streptococcus pneumoniae, Neisseria meningitidis et Haemophilus influenzae;
·qui n’ont pas reçu à ce jour de vaccination contre Neisseria meningitidis, Streptococcus pneumoniae et Haemophilus influenzae, à moins qu’ils ne reçoivent un traitement prophylactique par des antibiotiques appropriés jusqu’à deux semaines après la vaccination.
Mises en garde et précautionsInfections graves dues à des bactéries encapsulées
L’administration de pegcétacoplan peut entraîner des infections graves dues à des bactéries encapsulées telles que Streptococcus pneumoniae, Neisseria meningitidis et Haemophilus influenzae. Afin de réduire le risque d’infection, tous les patients doivent être vaccinés contre ces bactéries, conformément aux directives locales en vigueur, au moins deux semaines avant le début du traitement par pegcétacoplan, sauf si le risque d’un report du traitement par pegcétacoplan est supérieur au risque d’infection.
Patients déjà vaccinés auparavant
Avant de démarrer le traitement par pegcétacoplan, il convient de vérifier que les patients déjà vaccinés ont bien reçu des vaccins contre des bactéries encapsulées, dont Streptococcus pneumoniae, Neisseria meningitidis des types A, C, W, Y et B, et Haemophilus influenzae de type B, dans les deux ans précédant le début du traitement par pegcétacoplan.
Patients pas encore vaccinés
Les patients qui ne sont pas encore vaccinés doivent recevoir les vaccins nécessaires au moins deux semaines avant l’administration de la première dose de pegcétacoplan. Si un traitement immédiat est indiqué, les vaccins nécessaires doivent être administrés dès que possible et le patient doit recevoir un traitement par des antibiotiques appropriés jusqu’à deux semaines après la vaccination.
La vaccination pourrait ne pas suffire à prévenir une infection grave. Les recommandations officielles pour l’utilisation appropriée des antibiotiques devront être prises en considération. Tous les patients devront être suivis afin de surveiller l’apparition de signes précoces d’infections dues à des bactéries encapsulées telles que Neisseria meningitidis, Streptococcus pneumoniae et Haemophilus influenzae, être évalués immédiatement en cas de suspicion d’une infection et recevoir un traitement antibiotique approprié si besoin. Les patients doivent être informés des signes et symptômes associés et être invités à consulter immédiatement un médecin, le cas échéant.
Hypersensibilité
Des réactions d’hypersensibilité ont été rapportées. Si une réaction d’hypersensibilité sévère (y compris une anaphylaxie) survient, la perfusion de pegcétacoplan doit être immédiatement arrêtée et un traitement approprié doit être instauré.
Surveillance des manifestations de l’HPN après l’arrêt du traitement par pegcétacoplan
Lors de l’arrêt du traitement par pegcétacoplan chez les patients atteints d’HPN, l’apparition de signes et symptômes d’une hémolyse intravasculaire grave doit être étroitement surveillée. L’hémolyse intravasculaire se manifeste par des taux de LDH élevés accompagnés d’une diminution soudaine de la taille du clone HPN ou du taux d’hémoglobine (Hb), ou par une réapparition de symptômes tels qu’une fatigue, une hémoglobinurie, des douleurs abdominales, une dyspnée, un événement vasculaire indésirable majeur (y compris thrombose), une dysphagie ou une dysfonction érectile. S’il est nécessaire de stopper l’administration de pegcétacoplan, un autre traitement doit être envisagé, car, sans traitement, l’HPN menace le pronostic vital. En cas de survenue d’une hémolyse grave après l’arrêt du traitement, il faut envisager les procédures ou traitements suivants: transfusion sanguine (concentrés érythrocytaires), exsanguino-transfusion, traitement anticoagulant ou corticoïdes. Les patients doivent être étroitement surveillés pendant au moins huit semaines après l’administration de la dernière dose afin de détecter une hémolyse grave et d’autres réactions. Un sevrage lent doit par ailleurs être envisagé.
Contraception chez les femmes en âge de procréer
Il est recommandé aux femmes en âge de procréer d’utiliser des méthodes de contraception efficaces pour éviter une grossesse durant le traitement par le pegcétacoplan et pendant au moins 8 semaines après la dernière dose de pegcétacoplan (voir Grossesse, Allaitement).
Accumulation de polyéthylène glycol (PEG)
Aspaveli est un médicament pegylé. Les effets potentiels à long terme de l’accumulation de PEG dans les reins, dans les plexus choroïdes du cerveau et d’autres organes, sont inconnus (voir Données précliniques). Il est recommandé de réaliser régulièrement des analyses biologiques permettant d’évaluer la fonction rénale.
Matériel éducatif
Tous les médecins qui ont l'intention de prescrire ASPAVELI doivent s'assurer qu'ils ont reçu le matériel éducatif pour les médecins et qu'ils en ont pris connaissance . Les médecins doivent expliquer et discuter des avantages et des risques du traitement par ASPAVELI avec le patient, et lui remettre le dossier d’information du patient et la carte de sécurité du patient. Le patient doit être informé qu'il doit consulter rapidement un médecin s’il présente un quelconque signe ou symptôme d’infection grave ou d’hypersensibilité pendant le traitement par ASPAVELI, en particulier s'il est révélateur d’une infection par des bactéries encapsulées.
Effets sur les analyses biologiques
Des interférences sont possibles entre les réactifs à base de silice utilisés pour les panels d’analyse de la coagulation et le pegcétacoplan, entraînant un allongement artificiel du temps de céphaline activée (TCA). L’utilisation de réactifs à base de silice pour les panels d’analyse de la coagulation devra donc être évitée.
Sorbitol
Ce médicament contient 820 mg de sorbitol par flacon de 20 ml. Les patients présentant une intolérance héréditaire au fructose (IHF) ne doivent pas recevoir ce médicament.
Sodium
Ce médicament contient 7,4 mg de sodium par flacon de 20 ml, c.-à-d. qu’il est essentiellement «sans sodium».
InteractionsAucune étude d’interaction n’a été réalisée. D’après les données in vitro, le risque d’interactions cliniques entre le pegcétacoplan et d’autres médicaments est faible.
Grossesse, AllaitementFemmes en âge de procréer
Il est recommandé aux femmes en âge de procréer d’utiliser des méthodes de contraception efficaces pour éviter une grossesse durant le traitement par le pegcétacoplan et pendant au moins huit semaines après la dernière dose de pegcétacoplan.
Pour les femmes ayant un projet de grossesse, l’utilisation de pegcétacoplan ne doit être envisagée qu’après évaluation du rapport bénéfice/risque (voir Grossesse).
Grossesse
Les données disponibles sur l’utilisation de pegcétacoplan chez la femme enceinte sont limitées, voire inexistantes. Les études effectuées chez l’animal ont mis en évidence une toxicité sur la reproduction (voir Données précliniques).
Le pegcétacoplan ne doit pas être utilisé pendant la grossesse et chez les femmes en âge de procréer n’utilisant pas de contraception, sauf si l’état clinique de la patiente exige un traitement par le pegcétacoplan.
Allaitement
On ne sait pas si le pegcétacoplan passe dans le lait maternel. Une excrétion minime (moins de 1 %, excrétion non significative d’un point de vue pharmacologique) du pegcétacoplan dans le lait maternel a été mise en évidence chez des guenons. Il est improbable que l’exposition soit cliniquement significative chez le nourrisson allaité (voir Données précliniques).
Il est recommandé de ne pas allaiter pendant le traitement par le pegcétacoplan.
Fertilité
Les répercussions du pegcétacoplan sur la fertilité n’ont pas été étudiées chez l’animal. Lors d’études de toxicité menées chez le singe, aucune anomalie microscopique n’a été constatée au niveau des organes reproductifs des mâles et des femelles (voir Données précliniques).
Effet sur l’aptitude à la conduite et l’utilisation de machinesAspaveli n’a aucune influence ou a une influence négligeable sur l’aptitude à la conduite ou l’utilisation de machines.
Effets indésirablesHPN
Résumé du profil de sécurité
Les effets secondaires signalés le plus fréquemment chez les patients atteints d’HPN traités par pegcétacoplan ont été des réactions au site d’injection: érythème au site d’injection, prurit au site d’injection, gonflement au site d’injection, douleur au site d’injection et hématome au site d’injection. D’autres effets indésirables rapportés chez plus de 10 % des patients au cours des études cliniques étaient une infection des voies aériennes supérieures, une diarrhée, une hémolyse, des douleurs abdominales, des céphalées, de la fatigue, de la fièvre, une toux, une infection des voies urinaires, une complication de la vaccination, des douleurs dans les extrémités, une sensation vertigineuse, une arthralgie et une dorsalgie. Les effets indésirables graves signalés le plus fréquemment ont été l’hémolyse et le sepsis.
Liste des effets indésirables
Le tableau 1 présente les effets indésirables observés avec le pegcétacoplan dans les études cliniques et après la mise sur le marché chez des patients atteints d’HPN. Les effets indésirables sont rangés par classe de système d’organes de la classification MedDRA et par fréquence selon la convention suivante: très fréquents (≥ 1/10), fréquents (≥ 1/100, < 1/10), occasionnels (≥ 1/1 000, < 1/100), rares (≥ 1/10 000, < 1/1 000), très rares (< 1/10 000) et fréquence inconnue (ne peut être estimée sur la base des données disponibles).
Au sein de chaque catégorie de fréquence, les effets indésirables sont présentés en respectant un ordre décroissant en termes de sévérité.
Tableau 1 Effets indésirables chez les patients atteints d’HPN observés dans les études cliniques1 et dans la pratique après la mise sur le marché.
|
Classe de système d’organes
|
Fréquence
|
Effet indésirable
| |
Infections et infestations
|
Très fréquents
|
Infection des voies aériennes supérieures
Infection des voies urinaires
| |
Fréquents
|
Sepsis2
COVID-19
Infection gastro-intestinale
Infection fongique
Infection cutanée
Infection buccale
Infection de l’oreille
Infection
Infection des voies aériennes
Infection virale
Infection bactérienne
Infection vaginale
Infection oculaire
| |
Occasionnels
|
Cervicite
Infection de l’aine
Pneumonie
Abcès nasal
Tuberculose
Candidose de l’œsophage
Pneumonie COVID-19
Abcès anal
| |
Affections hématologiques et du système lymphatique
|
Très fréquents
|
Hémolyse
| |
|
Fréquents
|
Thrombopénie
Neutropénie
| |
Affections du système immunitaire
|
Occasionnels
|
Réaction anaphylactique3
Choc anaphylactique3
| |
Troubles du métabolisme et de la nutrition
|
Fréquents
|
Hypokaliémie
| |
Affections du système nerveux
|
Très fréquents
|
Céphalée
Sensation vertigineuse
| |
Affections vasculaires
|
Fréquents
|
Hypertension
| |
Affections respiratoires, thoraciques et médiastinales
|
Très fréquents
|
Toux
| |
Fréquents
|
Dyspnée
Épistaxis
Douleur oropharyngée
Congestion nasale
| |
Affections gastro-intestinales
|
Très fréquents
|
Douleurs abdominales
Diarrhée
| |
Fréquents
|
Nausée
| |
Affections hépatobiliaires
|
Fréquents
|
Alanine aminotransférase augmentée
Bilirubine augmentée
| |
Affections de la peau et du tissu sous-cutané
|
Fréquents
|
Érythème
Rash
Urticaire
| |
Affections musculosquelettiques et du tissu conjonctif
|
Très fréquents
|
Arthralgie
Dorsalgie
Douleurs dans les extrémités
| |
Fréquents
|
Myalgie
Spasmes musculaires
| |
Affections du rein et des voies urinaires
|
Fréquents
|
Insuffisance rénale aiguë
Chromaturie
| |
Troubles généraux et anomalies au site d’administration
|
Très fréquents
|
Érythème au site d’injection
Prurit au site d’injection
Gonflement au site d’injection
Hématome au site d’injection
Fatigue
Fièvre
Douleur au site d’injection
| |
Fréquents
|
Réaction au site d’injection
Induration au site d’injection
| |
Lésions, intoxications et complications d’interventions
|
Très fréquents
|
Complication de la vaccination4
|
1Études APL2-302, APL2-308, APL2-202, APL-2-CP-PNH-204 et APL2-CP0514 chez des patients atteints d’HPN. Lorsque cela est approprié, les termes similaires d’un point de vue médical ont été regroupés sous le même concept médical.
2Le sepsis contient un cas de choc septique.
3Estimé à l’aide des données collectées après la mise sur le marché.
4Les complications vaccinales ont été observées dans le contexte des vaccinations obligatoires.
Description d’effets indésirables spécifiques chez les patients atteints d’HPN
Infections
Compte tenu de son mécanisme d’action, le pegcétacoplan est susceptible de majorer le risque d’infections, en particulier d’infections dues à des bactéries encapsulées telles que Streptococcus pneumoniae, Neisseria meningitidis des types A, C, W, Y et B, et Haemophilus influenzae (voir Mises en garde et précautions). Aucune infection grave due à des bactéries encapsulées n’a été signalée au cours de l’étude APL2-302. Quarante-huit patients ont présenté une infection durant l’étude. Les infections les plus fréquentes chez les patients traités par le pegcétacoplan durant l’étude APL2-302 ont été les infections des voies aériennes supérieures (28 cas, 35 %). La plupart des infections signalées chez les patients traités par le pegcétacoplan durant l’étude APL2-302 étaient sans gravité, et le plus souvent d’intensité légère. Des infections jugées graves ont été signalées chez dix patients, dont l’un est décédé de la COVID-19. Les infections graves les plus fréquentes ont été: sepsis (trois cas) (ayant conduit à l’arrêt du pegcétacoplan chez un patient) et gastroentérite (trois cas); toutes ces infections se sont résolues. Onze patients ont présenté une infection durant l’étude APL2-308. À une exception, toutes les infections signalées étaient légères ou modérément sévères. Un patient présentant une infection a développé un choc septique et est décédé.
Hémolyse
Dix-neuf patients parmi ceux traités par le pegcétacoplan durant l’étude APL2-302 ont présenté une hémolyse. Dans sept cas, l’hémolyse a été considérée comme sévère; dans cinq cas, le pegcétacoplan a dû être arrêté et la dose de pegcétacoplan a été augmentée chez dix patients.
Pendant l’étude APL2-308, trois cas d’hémolyse ont été observés parmi les patients traités par pegcétacoplan. Dans aucun de ces cas, l’hémolyse n’a été considérée comme sévère ou n’a entraîné l’arrêt du pegcétacoplan. La dose de pegcétacoplan a été augmentée chez les trois patients.
Réactions au site d’injection
Des réactions au site d’injection (érythème, gonflement, prurit et douleur, par exemple) ont été rapportées au cours de l’étude APL2-302. Ces réactions étaient d’intensité légère à modérée et n’ont pas nécessité d’interrompre le traitement.
Diarrhée
Des cas de diarrhée, parmi lesquels aucun n’était grave ou n’a abouti à l’interruption du traitement, ont été signalés durant l’étude APL2-302.
Immunogénicité
L’immunogénicité d’Aspaveli a été évaluée à partir de tests d’anticorps anti-médicament (ADA) spécifiques dont l’un permet de mettre spécifiquement en évidence les ADA dirigés contre les composants peptidiques du pegcétacoplan (anticorps anti-peptide du pegcétacoplan) et un deuxième est spécifiquement destiné à la détection des ADA dirigés contre les composants PEG (polyéthylène glycol) du pegcétacoplan (anticorps anti-PEG).
L’incidence des anticorps anti-médicament (ADA induits par le traitement ou amplification d’ADA préexistants) était faible et, lorsque des ADA étaient présents, ils n’ont eu aucun impact notable sur la pharmacocinétique, la pharmacodynamique, l’efficacité ou le profil de sécurité du pegcétacoplan. Sur toute la durée des études APL2-302 et APL-308, trois patients sur 126, ayant reçu du pegcétacoplan, ont développé des anticorps anti-peptide du pegcétacoplan. Les trois patients ont également été testés positifs aux anticorps neutralisants. La survenue d’anticorps neutralisants n’a eu aucun impact évident sur la pharmacocinétique ou l’efficacité clinique. Dix-huit patients sur 126 ont développé des anticorps anti-PEG; dans neuf cas, induits par le traitement. Chez les neuf autres patients, le développement de ces anticorps a été renforcé par le traitement.
C3G et IC-MPGN primaire
Résumé du profil de sécurité
Les effets indésirables les plus souvent signalés par les patients atteints de C3G ou d’IC-MPGN primaire traités par pegcétacoplan ont été des réactions au site de perfusion.
Liste des effets indésirables
Le tableau 2 présente les effets indésirables observés avec le pegcétacoplan dans les études cliniques chez des patients atteints de C3G ou d’IC-MPGN primaire
Les effets indésirables sont rangés par classe de système d’organes de la classification MedDRA et par fréquence selon la convention suivante: très fréquents (≥ 1/10), fréquents (≥ 1/100, < 1/10), occasionnels (≥ 1/1000, < 1/100), rares (≥ 1/10 000, < 1/1000), très rares (< 1/10 000) et fréquence inconnue (ne peut être estimée sur la base des données disponibles). Au sein de chaque catégorie de fréquence, les effets indésirables sont présentés en respectant un ordre décroissant en termes de sévérité.
Tableau 2: effets indésirables chez les patients atteints de C3G ou d’IC-MPGN primaire, observés dans les études cliniques APL2-C3G-310, APL2-C3G-314, APL2-201 et APL2-C3G-204
|
Classe de système d’organes
|
Fréquence
|
Effet indésirable
| |
Affections gastrointestinales
|
Très fréquents
|
Nausée
| |
Affections de la peau et du tissu sous-cutané
|
Fréquents
|
Prurit (ailleurs qu’au site de perfusion)
| |
Affections musculosquelettiques et du tissu conjonctif
|
Fréquents
|
Arthralgie
| |
Troubles généraux et anomalies au site d’administration
|
Très fréquents
|
Pyrexie
Réactions au site de perfusion*
(douleurs, érythème, prurit, gonflements, induration)
|
* Comprend les termes de haut niveau (HLT) réactions au site de perfusion et réactions au site d’injection
Patients transplantés
Chez les patients transplantés atteints de C3G ou d’IC-MPGN primaire (N = 5) inclus dans l’étude APL2 C3G-310, le profil de sécurité correspondait aux résultats généraux de l’étude.
Population pédiatrique
Chez les patients adolescents atteints de C3G ou d’IC-MPGN primaire (N = 28, âgés de 12 à 17 ans), qui ont participé à l’étude APL2 C3G-310, le profil de sécurité correspondait lui aussi aux résultats généraux de l’étude. Les effets indésirables les plus souvent rapportés dans ce groupe de patients étaient les réactions au site de perfusion.
La sécurité du pegcétacoplan n’a pas été étudiée chez les patients pédiatriques âgés de moins de 12 ans.
Immunogénicité
Dans les études cliniques concernant l’HPN ainsi que la C3G et l’IC-MPGN primaire, deux tests différents ont été utilisés pour mettre en évidence la présence d’anticorps anti-peptide du pegcétacoplan (ADA). Le test utilisé pour la C3G ou l’IC-MPGN primaire était plus sensible.
L’incidence des ADA (ADA induits par le traitement ou amplification d’ADA prééxistants) dans l’étude APL2-C3G-310 était de 23,6 % pour les anticorps anti-PEG et de 16,3 % pour les anticorps anti-peptide du pegcétacoplan. Sur la base d’une analyse de la pharmacocinétique et de la pharmacodynamique de la population, les ADA n’avaient pas d’influence clinique significative sur l’efficacité ou les paramètres de pharmacocinétique/pharmacodynamique de la population globale analysée. Cinq patients ont également été testés positifs aux anticorps neutralisants. La survenue d’anticorps neutralisants n’a eu aucun impact évident sur la pharmacocinétique ou l’efficacité clinique. Vingt-neuf patients sur 123 ont développés des anticorps anti-PEG ; chez 14 patients, ils étaient induits par le traitement, chez les 15 autres patients ils étaient renforcés par le traitement. Aucun patient de l’étude APL2-C3G-204 présentant une récidive de la maladie après transplantation n’a développé de réponse ADA positive au peptide du pegcétacoplan ou au PEG (valeur d’ADA induits par le traitement ou valeur d’ADA augmentée par rapport à la valeur précédente). Pendant la période de traitement de 26 semaines contrôlée par placébo de l’étude APL2-C3G-310, aucune influence détectable des ADA sur la sécurité du traitement par pegcétacoplan n’a été observée.
L’annonce d’effets secondaires présumés après l’autorisation est d’une grande importance. Elle permet un suivi continu du rapport bénéfice-risque du médicament. Les professionnels de santé sont tenus de déclarer toute suspicion d’effet secondaire nouveau ou grave via le portail d’annonce en ligne ElViS (Electronic Vigilance System). Vous trouverez des informations à ce sujet sur www.swissmedic.ch.
SurdosageAucun cas de surdosage n’a été rapporté à ce jour.
Propriétés/EffetsCode ATC
L04AJ03
Le pegcétacoplan est une molécule symétrique composée de deux pentadécapeptides identiques attachés par une liaison covalente aux extrémités d’une molécule de polyéthylène glycol (PEG) linéaire. Le poids moléculaire du pegcétacoplan est d’environ 43,5 kilodaltons (kDa). Les unités peptidiques se lient aux compléments C3 et C3b, inhibant ainsi largement la cascade du complément. Le fragment PEG de 40 kDa confère une meilleure solubilité et une durée de vie plus longue dans l’organisme après l’administration du médicament.
Mécanisme d’action
Le pegcétacoplan se lie à la protéine C3 du complément et au fragment C3b résultant de son activation avec une haute affinité, régulant ainsi le clivage de la C3 et la production des effecteurs de l’activation du complément en aval. Dans le cadre de l’HPN, l’hémolyse extravasculaire (HEV) est favorisée par l’opsonisation du fragment C3b, tandis que l’hémolyse intravasculaire (HIV) est médiée par le complexe d’attaque membranaire (CAM) en aval. Le pegcétacoplan régule globalement la cascade du complément grâce à une action proximale sur la formation du C3b et du CAM, contrôlant ainsi les mécanismes qui conduisent à l’HEV et à l’HIV.
Dans le cadre de la C3G et de l’IC-MPGN primaire, il se produit une activation excessive de C3, qui peut se déclencher dans toutes les voies d’activation du complément (alterne, classique et des lectines), avec un dépôt excessif de produits de dégradation de C3 dans les glomérules rénaux. Ceci cause une lésion du parenchyme rénal et une atteinte de la fonction rénale. Le pegcétacoplan cible les effecteurs de l’activation du complément (C3 et C3b) en amont et inhibe ainsi l’activation déclenchée dans toutes les voies du complément (alterne, classique et des lectines). Par l’inhibition de C3, le pegcétacoplan agit directement sur l’activation incorrecte de C3 et influence la maladie sous-jacente, en réduisant le dépôt excessif de produits de dégradation de C3 dans les glomérules. En bloquant C3b, le pegcétacoplan inhibe par ailleurs, par un mécanisme d’action supplémentaire dans la cascade du complément, l’activité de la C3-convertase de la voie alterne d’activation du complément (AP). Ceci permet de réduire davantage le dépôt de produits de dégradation de C3 dans les glomérules.
Pharmacodynamique
HPN
Dans l’étude APL2-302, la concentration sérique moyenne en C3 a augmenté, passant de 0,94 g/l au début de l’étude à 3,83 g/l à la semaine 16 dans le groupe traité par le pegcétacoplan, et restée à ce niveau jusqu’à la semaine 48. Dans l’étude APL2-308, la concentration sérique moyenne en C3 a augmenté, passant d’une valeur à l’inclusion de 0,95 g/l à 3,56 g/l à la semaine 26.
Dans l’étude APL2-302, le pourcentage moyen de globules rouges HPN de types II et III, qui était de 66,80 % à l’inclusion, a augmenté à 93,85 % à la semaine 16 et est resté à ce niveau jusqu’à la semaine 48. Dans l’étude APL2-308, le pourcentage moyen de globules rouges HPN de type II et III, qui était de 42,4 % à l’inclusion, a augmenté à 90,0 % à la semaine 26.
Dans l’étude APL2-302, le pourcentage moyen de globules rouges HPN de types II et III avec dépôt de C3, qui était de 17,73 % à l’inclusion, a diminué pour atteindre 0,20 % à la semaine 16 et est resté à ce niveau jusqu’à la semaine 48. Dans l’étude APL2-308, le pourcentage moyen de globules rouges HPN de type II et III avec dépôt de C3, qui était de 2,85 % à l’inclusion, a diminué pour atteindre 0,09 % à la semaine 26.
C3G et IC-MPGN primaire
Dans l’étude APL2-C3G-310, la concentration sérique moyenne en C3 a augmenté, passant de 0,62 g/l au début de l’étude à 3,71 g/l à la semaine 26 et l’effet a été maintenu jusqu’à la semaine 52 dans le groupe traité par pegcétacoplan. Dans le groupe placébo, la concentration en C3 est restée stable jusqu’à la semaine 26 (0,57 g/l au début de l’étude ; 0,58 g/l à la semaine 26), et a augmenté, après le passage au pegcétacoplan, à 3,59 g/l à la semaine 52.
La concentration sérique moyenne en sC5b-9 a chuté, passant de 902,5 ng/ml au début de l’étude à 290,2 ng/ml à la semaine 26 dans le groupe traité par le pegcétacoplan, et l’effet a été maintenu jusqu’à la semaine 52. Dans le groupe placébo, la concentration en sC5b-9 est restée stable jusqu’à la semaine 26 (768,3 ng/ml au début de l’étude ; 759,9 ng/ml à la semaine 26) et a chuté, après le passage au pegcétacoplan, à 272,9 ng/ml à la semaine 52.
À la semaine 26, le nombre de patients présentant une diminution de l’intensité de coloration C3c lors de la biopsie rénale d’au moins deux ordres de grandeur par rapport à la valeur initiale était de 74,3 % dans le groupe pegcétacoplan, 71,4 % ayant atteint une valeur de coloration nulle, par rapport à 11,8 % des patients présentant une diminution de deux ordres de grandeur et 8,8 % ayant atteint une valeur de coloration nulle dans le groupe placébo.
Dans l’étude APL2-C3G-204, la concentration sérique moyenne en C3 a augmenté chez les patients présentant une récidive de la maladie après transplantation, passant de 0,70 g/l au début de l’étude à 2,80 g/l à la semaine 52, et la concentration sérique moyenne en sC5b-9 a diminué de 525,4 ng/ml au début de l’étude à 151,0 ng/ml à la semaine 52.
Électrophysiologie cardiaque
Aucune étude spécifique n’a été menée pour déterminer si et dans quelle mesure le pegcétacoplan peut retarder la repolarisation cardiaque. Le pegcétacoplan est une structure peptidique pégylée qui ne s’est pas révélée inhiber le canal ionique hERG (human Ether-a-go-go-Gen) lors du test correspondant. L’analyse de la relation concentration-QTc (intervalle QT corrigé en fonction de la fréquence cardiaque) a confirmé que le pegcétacoplan n’avait aucun impact sur la repolarisation cardiaque.
Efficacité clinique
HPN
L’efficacité et la sécurité du pegcétacoplan chez les patients atteints d’HPN ont été évaluées dans deux études contrôlées randomisées ouvertes de phase III : l’étude APL2-302 a été menée chez des patients déjà prétraités par un inhibiteur complémentaire, et l’étude APL2-308 a été menée chez des patients qui n’avaient pas été traités au préalable par un inhibiteur complémentaire. Dans les deux études, la dose de pegcétacoplan était de 1080 mg deux fois par semaine. Si nécessaire, la dose de pegcétacoplan pouvait être ajustée à 1080 mg tous les 3 jours.
Étude chez des patients adultes déjà prétraités par un inhibiteur complémentaire (APL2-302)
L’étude APL2-302 était une étude ouverte, randomisée, comprenant une période contrôlée contre comparateur actif de 16 semaines, suivie d’une période en ouvert de 32 semaines (OLP). Cette étude a été menée chez des patients atteints d’HPN qui étaient traités par éculizumab à dose stable depuis au moins trois mois et présentaient un taux d’hémoglobine < 10,5 g/dl.
Les patients éligibles ont été soumis à une période de préinclusion de quatre semaines durant laquelle on leur a administré du pegcétacoplan par voie sous-cutanée à raison de 1080 mg deux fois par semaine en complément de la dose d’éculizumab qu’ils recevaient déjà. Les patients ont ensuite été randomisés selon un ratio de 1:1 en vue de recevoir soit du pegcétacoplan à raison de 1080 mg deux fois par semaine, soit leur dose actuelle d’éculizumab sur toute la durée de la période contrôlée randomisée de 16 semaines. La randomisation a été stratifiée en fonction du nombre de transfusions de concentrés érythrocytaires reçues dans les 12 mois précédant le jour -28 (< 4 ou ≥ 4) et de la numération plaquettaire lors de la sélection (< 100 000/μl; ≥ 100 000/μl). Les patients arrivés au terme de la période contrôlée randomisée ont été inclus dans la période en ouvert, durant laquelle tous les patients ont reçu du pegcétacoplan pendant jusqu’à 32 semaines (les patients ayant reçu l’éculizumab durant la période contrôlée randomisée ont intégré une période de préinclusion de quatre semaines avant de passer sous pegcétacoplan en monothérapie).
Les patients avaient été vaccinés contre Streptococcus pneumoniae, Neisseria meningitidis des types A, C, W, Y et B, et Haemophilus influenzae de type B (Hib) soit dans les deux ans qui ont précédé le premier jour du traitement, soit dans les deux semaines qui ont suivi le début du traitement par pegcétacoplan. Les patients qui ont été vaccinés après le jour 1 ont reçu un traitement prophylactique par des antibiotiques appropriés jusqu’à deux semaines après la vaccination. De plus, le médecin-investigateur pouvait décider d’administrer une antibiothérapie prophylactique conformément aux directives thérapeutiques locales en vigueur aux patients atteints d’HPN qui étaient traités par un inhibiteur complémentaire. Le pegcétacoplan a été administré en perfusion sous-cutanée. La durée de la perfusion était de 20 à 40 minutes environ.
Les critères d’efficacité principal et secondaires ont été évalués à la semaine 16. Le critère principal d’efficacité était l’évolution du taux Hb entre l’inclusion et la semaine 16 (durant la période contrôlée randomisée). La valeur à l’inclusion a été définie comme la moyenne des mesures obtenues avant l’administration de la première dose de pegcétacoplan. Les transfusions évitées, c-à-d. la part de patients n’ayant pas eu besoin d’une transfusion durant la période contrôlée randomisée, et l’évolution du nombre absolu de réticulocytes, du taux de LDH et du score sur l’échelle FACIT-Fatigue (Functional Assessment of Chronic Illness Therapy – Fatigue) entre l’inclusion et la semaine 16 étaient des critères secondaires d’efficacité importants. Au total, 80 patients ont intégré la période de préinclusion. À la fin de la période de préinclusion, les 80 patients ont tous été randomisés: 41 ont été placés dans le groupe sous pegcétacoplan et 39 dans le groupe sous éculizumab. Les caractéristiques démographiques et les caractéristiques initiales de la maladie étaient globalement équilibrées entre les groupes de traitement (voir Tableau 3). Au total, 38 patients du groupe traité par pegcétacoplan et 39 patients du groupe traité par l’éculizumab sont allés au terme de la période contrôlée randomisée de 16 semaines et ont ensuite participé à la période de traitement en ouvert de 32 semaines. Au total, 12 patients sur 80 traités par pegcétacoplan (15 %) ont arrêté l’étude en raison d’événements indésirables. Chez 15 patients, la posologie a été ajustée à 1080 mg tous les trois jours conformément au protocole de l’étude. Les bénéfices ont été évalués chez douze patients, et chez huit d’entre eux, l’ajustement de la posologie a apporté un bénéfice.
Tableau 3: Données démographiques et caractéristiques initiales des patients dans l’étude APL2-302
|
Paramètres
|
Statistiques
|
Pegcétacoplan (n=41)
|
Éculizumab (n=39)
| |
Âge (ans)
|
Moyenne (ET)
|
50,2 (16,3)
|
47,3 (15,8)
| |
Dose d’éculizumab à l’inclusion
900 mg par voie i.v. toutes les 2 semaines
900 mg par voie i.v. tous les 11 jours
1200 mg par voie i.v. toutes les 2 semaines
1500 mg par voie i.v. toutes les 2 semaines
|
n (%)
n (%)
n (%)
n (%)
|
26 (63,4)
1 (2,4)
12 (29,3)
2 (4,9)
|
29 (74,4)
1 (2,6)
9 (23,1)
0
| |
Femmes
|
n (%)
|
27 (65,9)
|
22 (56,4)
| |
Délai (années) entre le diagnostic d’HPN et le jour -28
|
Moyenne (ET)
|
8,7 (7,4)
|
11,4 (9,7)
| |
Taux Hb (g/dl)
|
Moyenne (ET)
|
8,7 (1,1)
|
8,7 (0,9)
| |
Nombre absolu de réticulocytes (109/l)
|
Moyenne (ET)
|
218 (75,0)
|
216 (69,1)
| |
Taux de LDH (U/l)
|
Moyenne (ET)
|
257,5 (97,6)
|
308,6 (284,8)
| |
Score total FACIT-F*
|
Moyenne (ET)
|
32,2 (11,4)
|
31,6 (12,5)
| |
Nombre de transfusions reçues dans les 12 mois précédant le jour -28
|
Moyenne (ET)
|
6,1 (7,3)
|
6,9 (7,7)
| |
< 4
|
n (%)
|
20 (48,8)
|
16 (41,0)
| |
≥ 4
|
n (%)
|
21 (51,2)
|
23 (59,0)
| |
Numération plaquettaire lors de la sélection (109/l)
|
Moyenne (ET)
|
167 (98,3)
|
147 (68,8)
| |
Numération plaquettaire lors de la sélection <100 000/μl
|
n (%)
|
12 (29,3)
|
9 (23,1)
| |
Numération plaquettaire lors de la sélection ≥100 000/μl
|
n (%)
|
29 (70,7)
|
30 (76,9)
| |
Antécédents d’anémie aplasique
|
n (%)
|
11 (26,8)
|
9 (23,1)
| |
Antécédents de syndrome myélodysplasique
|
n (%)
|
1 (2,4)
|
2 (5,1)
|
* Le score FACIT-Fatigue est mesuré sur une échelle allant de 0 à 52, les valeurs élevées indiquant une fatigue moindre.
Le pegcétacoplan s’est révélé supérieur à l’éculizumab s’agissant du critère d’évaluation principal, à savoir l’évolution du taux d’hémoglobine par rapport à l’inclusion (p < 0,0001). L’évolution médiane ajustée du taux d’hémoglobine par rapport à l’inclusion était de 2,4 g/dl dans le groupe sous pegcétacoplan contre -1,5 g/dl dans le groupe sous éculizumab, soit une augmentation médiane ajustée à la semaine 16 de 3,8 g/dl sous pegcétacoplan par comparaison avec l’éculizumab (figure 1).
Figure 1: Évolution moyenne ajustée (± ET) du taux d’hémoglobine (g/dl) entre l’inclusion et la semaine 16 dans l’étude APL2-302

La non-infériorité s’agissant des critères d’évaluation secondaires importants, à savoir les transfusions évitées et l’évolution du nombre absolu de réticulocytes par rapport à l’inclusion, a été démontrée. Des transfusions ont pu être évitées chez 85 % des patients du groupe sous pegcétacoplan contre chez 15 % des patients du groupe sous éculizumab.
Concernant l’évolution du taux de LDH par rapport à l’inclusion, la non-infériorité n’a pas été prouvée.
En raison de la hiérarchie des tests, l’évolution du score FACIT-Fatigue par rapport à l’inclusion n’a pas été formellement analysée.
Les moyennes ajustées, la différence entre les traitements, les intervalles de confiance et les analyses statistiques réalisées pour les principaux critères d’évaluation secondaires sont présentés à la figure 2.
Figure 2: Analyse des critères d’évaluation secondaires importants dans l’étude APL2-302
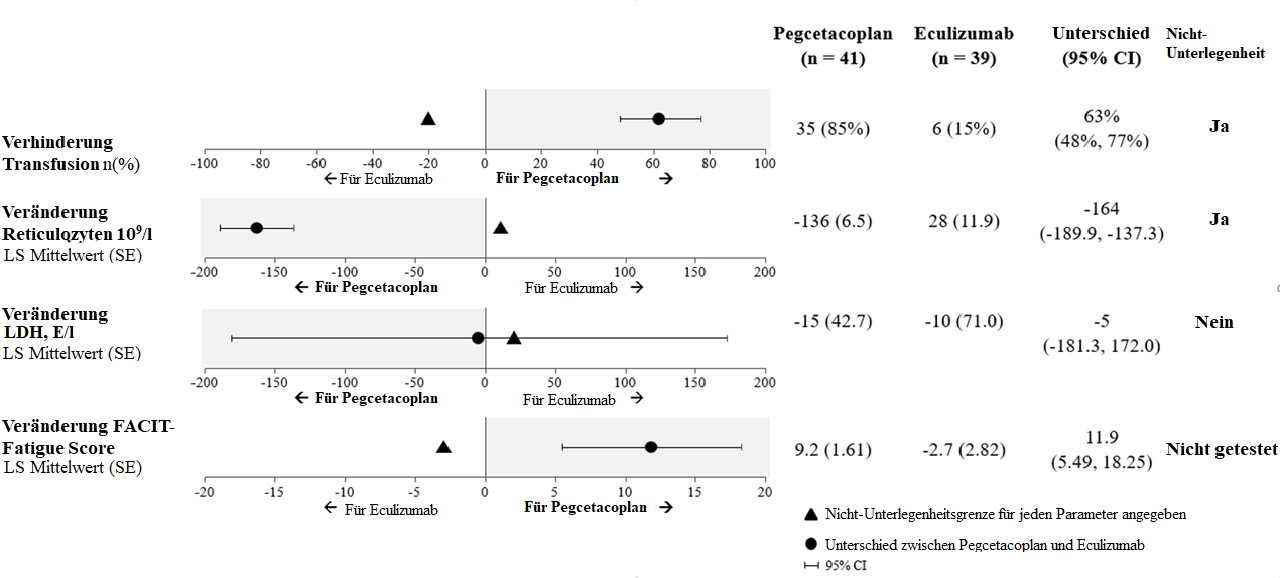
Les résultats ont été cohérents dans toutes les analyses de confirmation du critère principal et des critères secondaires importants, en incluant toutes les données observées, y compris les données post-transfusion.
Les analyses de l’efficacité basées sur les critères d’évaluation principal et secondaires n’ont mis en évidence aucune différence notable entre les patients traités par pegcétacoplan selon le sexe, l’origine ethnique ou l’âge.
Une normalisation du taux Hb a été obtenue à la semaine 16 chez 34 % des patients du groupe sous pegcétacoplan contre 0 % dans le groupe sous éculizumab. Au total, 78 % des patients traités par pegcétacoplan ont vu leur nombre absolu de réticulocytes se normaliser par rapport à 3 % dans le groupe sous éculizumab. Une normalisation du taux de LDH a été obtenue chez 71 % des patients traités par pegcétacoplan contre 15 % dans le groupe sous éculizumab.
Au total, 77 patients ont intégré la période en ouvert de 32 semaines durant laquelle tous les patients ont reçu du pegcétacoplan, ce qui correspond à une exposition totale de 48 semaines au maximum. Globalement, les résultats à 48 semaines concordaient avec ceux enregistrés à la semaine 16 et prouvent que l’efficacité est durable.
Étude chez des patients adultes n’ayant pas reçu de traitement préalable par un inhibiteur complémentaire (APL2-308)
L’étude APL2-308 était une étude ouverte, randomisée, contrôlée, menée chez des patients atteints d’HPN qui n’avaient pas été traités par un inhibiteur complémentaire au cours des trois mois précédant l’étude et qui présentaient un taux Hb inférieur à la limite inférieure de la normale (LLN). Les patients éligibles ont été randomisés selon un ratio de 2:1 et ont reçu du pegcétacoplan ou un traitement de soutien (p. ex. transfusions, corticostéroïdes, supplémentation, notamment en fer, acide folique et vitamine B12), désigné ci-après comme groupe témoin, pendant toute la durée de traitement de 26 semaines.
La randomisation a été stratifiée en fonction du nombre de transfusions de PRBC reçues dans les 12 mois précédant le jour -28 (< 4 ou ≥ 4). À tout moment durant l’étude, un patient affecté au groupe témoin dont le taux Hb était de ≥ 2 g/dl en dessous de la valeur à l’inclusion ou présentant un évènement thromboembolique associé à l’HPN, pouvait passer dans le groupe pegcétacoplan pour le reste de l’étude, conformément au protocole de l’étude.
Au total, 53 patients ont été randomisés : 35 patients dans le groupe pegcétacoplan et 18 patients dans le groupe témoin. Les données démographiques et les caractéristiques de la maladie des patients à l’inclusion étaient globalement équilibrées dans les groupes de traitement. L’âge moyen était de 42,2 ans dans le groupe pegcétacoplan et de 49,1 ans dans le groupe témoin. Le nombre moyen de transfusions de concentrés érythrocytaires reçues dans les 12 mois précédant l’étude préliminaire était de 3,9 dans le groupe pegcétacoplan et de 5,1 dans le groupe témoin. Cinq patients dans chaque groupe (14,3 % dans le groupe pegcétacoplan et 27,8 % dans le groupe témoin) présentaient des antécédents d’anémie aplasique. Les autres valeurs à l’inclusion étaient : valeur moyenne Hb à l’inclusion (groupe pegcétacoplan 9,4 g/dl contre 8,7 g/dl dans le groupe témoin), nombre absolu de réticulocytes (groupe pegcétacoplan 230,2 × 109/l contre 180,3 × 109/l dans le groupe témoin), taux de LDH (groupe pegcétacoplan 2 151,0 E/l contre 1 945,9 E/l dans le groupe témoin) et numération plaquettaire (groupe pegcétacoplan 191,4 × 109/l contre 125,5 × 109/l dans le groupe témoin). 11 patients sur les 18 affectés au groupe témoin sont passés au pegcétacoplan, car leur taux Hb a baissé de ≥ 2 g/dl en dessous de la valeur à l’inclusion. 52 patients randomisés sur 53 (97,8 %) ont reçu un traitement antibiotique prophylactique conformément aux directives de prescription locales.
Les critères d’efficacité principaux et secondaires ont été évalués à la semaine 26. Les deux critères co-principaux d’efficacité étaient la stabilisation du taux Hb, définie comme l’évitement d’une chute de la concentration Hb > 1 g/dl par rapport à l’inclusion sans transfusion, et l’évolution de la concentration de LDH par rapport à l’inclusion.
Dans le groupe traité par pegcétacoplan, 30 patients sur 35 (85,7 %) ont atteint une stabilisation du taux Hb contre 0 patient dans le groupe témoin. L’écart ajusté entre le groupe pegcétacoplan et le groupe témoin était de 73,1 % (IC à 95 % : 57,2 % à 89,0 % ; p < 0,0001).
La variation moyenne (ET) des moindres carrés (LS) de la concentration de LDH à la semaine 26 par rapport à l’inclusion était de -1 870 E/l dans le groupe traité par pegcétacoplan contre -400 E/l dans le groupe témoin (p < 0,0001). La différence entre le groupe pegcétacoplan et le groupe témoin était de 1 470 (IC à 95 % : -2 113 à -827). Les différences de traitement entre le groupe pegcétacoplan et le groupe témoin ont été observées à la semaine 2 et ont perduré jusqu’à la semaine 26 (figure 3). Les concentrations de LDH du groupe témoin sont restées élevées.
Figure 3 : Concentration de LDH (E/l) moyenne (±ET) dans le temps selon le groupe de traitement dans l’étude APL2-308

Pour les critères d’efficacité secondaires importants, réponse de l’Hb sans transfusion, variation du taux Hb et évolution du nombre absolu de réticulocytes, une différence significative entre les traitements a été observée dans le groupe traité par pegcétacoplan par rapport au groupe témoin (tableau 4).
Tableau 4 : analyse des critères secondaires importants dans l’étude APL2-308
|
Paramètre
|
Pegcétacoplan
(N = 35)
|
Groupe témoin
(N = 18)
|
Différence
(IC à 95 %)
Valeur p
| |
Réponse de l’Hb sans transfusiona
(n, %)
|
25 (71 %)
|
1 (6 %)
|
54 % (34 %; 74 %)
p < 0,0001
| |
Variation du taux Hb (g/dl) de l’inclusion à la semaine 26
Moyenne LS (ET)
|
2,9 (0,38)
|
0,3 (0,76)
|
2,7 (1,0 ; 4,4)
| |
Évolution du nombre absolu de réticulocytes (109/l) de l’inclusion à la semaine 26
Moyenne LS (ET)
|
-123 (9,2)
|
-19 (25,2)
|
-104 (-159 ; -49)
|
a La réponse de l’Hb était définie comme l’augmentation de l’hémoglobine de ≥ 1 g/dl de l’inclusion à la semaine 26. IC = intervalle de confiance, LS = moindre carré (least square), ET = erreur type
C3G et IC-MPGN primaire
L’efficacité et la sécurité du pegcétacoplan chez les patients atteints de C3G ou d‘IC-MPGN primaire ont été évaluées dans l’étude contrôlée par placébo, randomisée en double aveugle de phase III APL2-C3G-310 ; étaient inclus des adultes et des adolescents présentant une maladie des reins natifs ou une récidive de C3G ou d’IC-MPGN primaire après une transplantation rénale.
La dose de pegcétacoplan était de 1080 mg deux fois par semaine pour les adultes ou les adolescents d’un poids corporel ≥ 50 kg ou en fonction du poids pour les adolescents dont le poids corporel était inférieur à 50 kg.
Étude chez des patients adultes et adolescents atteints de C3G ou d’IC-MPGN primaire (APL2-C3G-310)
L’étude APL2-C3G-310 était une étude randomisée, en double aveugle, avec une période de traitement contrôlée par placébo de 26 semaines, suivie d’une période de traitement en ouvert de 26 semaines (OLP). Cette étude a été menée chez des adolescents âgés de 12 à 17 ans et des adultes atteints de C3G ou d’IC-MPGN primaire. Ont été inclus des patients présentant une maladie des reins natifs et des patients présentant une récidive de la maladie après transplantation, avec une protéinurie ≥ 1 g/jour et un DFGe ≥ 30 ml/min/1,73 m2. Les patients ont reçu au moins 12 semaines avant la randomisation une dose stable et optimisée pour le traitement de la C3G/IC-MPGN primaire (p. ex. inhibiteurs du SRA, inhibiteurs du cotransporteur sodiumglucose de type 2 [inhibiteur SGLT2], immunosuppreseurs, corticostéroïdes systémiques à une dose maximale équivalente à 20 mg/jour de prednisone).
Les patients en question ont été randomisés selon un ratio de 1:1 en vue de recevoir pendant la phase de traitement de 26 semaines contrôlée par placébo (RCP) le pegcétacoplan ou le placébo deux fois par semaine par voie sous-cutanée. Lors de la randomisation, deux facteurs de stratification ont été appliqués: patients avec une récidive de la maladie après transplantation par rapport aux patients avec une maladie des reins natifs, et patients ayant une biopsie des reins au début de l’étude (soit pendant la sélection soit pendant les 28 semaines avant la randomisation) par rapport aux patients sans biopsie des reins au début de l’étude. Pendant la RCP, les modifications du schéma de traitement initial pour la C3G/l’IC-MPGN primaire ont été limitées au minimum, et entreprises uniquement si ceci était nécessaire au bien-être du patient. Les patients qui avaient conclu la phase RCP sont passés à la phase de traitement en ouvert de 26 semaines (OLP), durant laquelle tous les participants recevaient du pegcétacoplan deux fois par semaine.
124 patients au total ont été randomisés, dont 63 ont reçu du pegcétacoplan et 61 le placébo. Les données démographiques et les caractéristiques de la maladie des patients à l’inclusion étaient réparties de manière globalement équilibrée entre les deux groupes (voir Tableau 5). Au total, 118 patients ont conclu la RCP de 26 semaines, et 114 d’entre eux la phase de traitement OLP par pegcétacoplan (N = 59 pegcétacoplan/pegcétacoplan ; N = 55 placébo/pegcétacoplan).
Tableau 5 : données démographiques initiales et caractéristiques de la maladie des patients de l’étude APL2-C3G-310
|
Paramètre
|
Statistique
|
Pegcétacoplan
(N = 63)
|
Placébo
(N = 61)
| |
Âge (ans)
|
Moyenne (ET)
|
28,2 (17,1)
|
23,6 (14,3)
| |
Adolescents (12–17 ans)
|
n (%)
|
28 (44,4)
|
27 (44,3)
| |
Adultes ≥18 ans
|
n (%)
|
35 (55,6)
|
34 (55,7)
| |
Sexe
Homme
Femme
|
n (%)
n (%)
|
26 (41,3)
37 (58,7)
|
28 (45,9)
33 (54,1)
| |
Maladie à la sélection
|
|
|
| |
C3G
|
n (%)
|
51 (81,0)
|
45 (73,8)
| |
C3GN
|
n (%)
|
45 (71,4)
|
41 (67,2)
| |
DDD
|
n (%)
|
4 (6,3)
|
4 (6,6)
| |
Non définie
|
n (%)
|
2 (3,2)
|
0
| |
IC-MPGN
|
n (%)
|
12 (19,0)
|
16 (26,2)
| |
Délai depuis le diagnostic de C3G/IC-MPGN (années)
|
Moyenne (ET)
|
3,64 (3,47)
|
3,76 (3,62)
| |
Transplantation rénale préalable
|
n (%)
|
5 (7,9)
|
4 (6,6)
| |
Délai depuis la dernière transplantation rénale (années)
|
Moyenne (ET)
|
11,4 (6,7)
|
5,8 (6,4)
| |
Délai depuis la dernière récivide de la maladie après transplantation (années)
|
Moyenne (ET)
|
1,47 (1,49)
|
1,38 (1,64)
| |
uPCR dans l’urine du matin mesurée trois fois à l’inclusion (mg/g)
|
Moyenne (ET)
|
3124 (2408)
|
2541 (2015)
| |
DFGe à l’inclusion (ml/min/1,73 m2)
|
Moyenne (ET)
|
78,5 (34,1)
|
87,2 (37,2)
| |
Coloration C3c dans la biopsie à l’inclusion
|
|
|
| |
3+
|
n (%)
|
51 (81,0)
|
51 (83,6)
| |
2+
|
n (%)
|
12 (19,0)
|
10 (16,4)
| |
Albumine sérique à l’inclusion (g/dl)
|
Moyenne (ET)
|
3,31 (0,61)
|
3,39 (0,70)
| |
C3 sérique à l’inclusion (mg/dl)
|
Moyenne (ET)
|
60,6 (45,7)
|
56,3 (35,6)
| |
Manifestations de la maladie
|
|
|
| |
Œdèmes
|
n (%)
|
45 (71,4)
|
32 (52,5)
| |
Fatigue
|
n (%)
|
16 (25,4)
|
8 (13,1)
| |
Hématurie
|
n (%)
|
37 (58,7)
|
39 (63,9)
| |
Pression artérielle élevée
|
n (%)
|
35 (55,6)
|
29 (47,5)
| |
Syndrome néphrotique
|
n (%)
|
32 (50,8)
|
27 (44,3)
| |
Traitements associés au début de l’étude*
|
|
|
| |
Principes actifs agissant sur le système rénine-angiotensine
|
n (%)
|
59 (93,7)
|
54 (88,5)
| |
Immunsuppresseurs
|
n (%)
|
49 (77,8)
|
45 (73,8)
| |
Glucocorticoïdes
|
n (%)
|
29 (46,0)
|
27 (44,3)
|
* dans un délai de 12 semaines avant le début de l’étude.
C3G = glomérulopathie à dépôts de C3, C3GN = glomérulonéphrite à dépôts de C3, DDD = Dense Deposit Desease (maladie du dépôt dense), IC MPGN = glomérulonéphrite membranoproliférative à médiation par les complexes immuns, FMU = urine du matin, uPCR = ratio protéinure/créatininurie, DFGe = débit de filtration glomérulaire estimé, ET = écart-type
Le critère d’évaluation principal de l’efficacité était le rapport de transformation logarithmique de l’uPCR dans l’urine du matin (FMU) à la semaine 26 par rapport à l’inclusion.
Le pegcétacoplan s’est avéré supérieur au placébo, avec une réduction statistiquement significative de l’uPCR de 68,1 % (IC à 95 %: 57,3 % à 76,2 %, p < 0,0001) par rapport à l’inclusion, contre, pour le groupe placébo, après 26 semaines de traitement (-67,2 % [IC à 95 %: -74,9 % à -57,2 %], c-à-d.+ 2,9 % [IC à 95 %: -8,6 % à 15,9 %] pour le pegcétacoplan par rapport au placébo. Une efficacité similaire a été observée dans les sous-groupes indépendamment de l’âge (adolescents vs adultes), du type de maladie (C3G vs IC-MPGN primaire), du statut de la maladie (native vs récidive après transplantation) et de l’administration simultanée d’immunosuppresseurs/glucocorticoïdes (oui vs non). L’effet du pegcétacoplan sur l’uPCR a été maintenu jusqu’à la semaine 52 (-67,2 % par rapport à l’inclusion). Les patients, qui sont passés du placébo au pegcétacoplan à la semaine 26 (Figure 4) ont présenté une diminution similaire à la semaine 52 (-51,3 %).
Figure 4: Rapport de la moyenne géométrique (IC à 95 %) de l’uCPR dans la FMU par rapport à l’inclusion dans le temps et par groupe de traitement, sur la base du modèle MMRM dans l’étude APL2-C3G-310

Remarque: le rapport de la moyenne géométrique est calculé à partir des moyennes des moindres carrés réexponentialisées
IC = intervalle de confiance, LS = méthode des moindres carrés (Least-Squares), FMU = urine du matin, uPCR = rapport protéinurie/créatinine, MMRM = modèle mixte de mesures répétées
Durant le traitement par pegcétacoplan de 26 semaines, une amélioration statistiquement significative du critère d’évaluation secondaire a été observée : 60,3 % des patients traités par pegcétacoplan ont atteint une réduction ≥ 50 % de l’uPCR, contre 4,9 % dans le groupe placébo (p < 0,0001).
Le traitement par pegcétacoplan de 26 semaines a également donné lieu à une part plus élevée de patients atteignant une réduction de deux ordres de grandeur ou plus sur une échelle de 0 à 3 dans l’intensité de la coloration de C3 des reins, 26 patients (74,3 %) sous pégcétacoplan contre 4 patients (11,8 %) sous placébo (valeur nominale p < 0,0001), ce qui indique une modification de la maladie chez les patients traités par pegcetacoplan.
Le traitement par pegcétacoplan de 26 semaines a montré une stabilisation du DFGe avec une modification par rapport à l’inclusion de -1,497 (2,242) sous pegcétacoplan contre -7,808 (1,919) sous placébo (valeur nominale p = 0,0333). L’effet du pegcétacoplan sur le DFGe a été maintenu jusqu’à la semaine 52.
Une efficacité similaire a été observée pour la réduction de la protéinurie ≥ 50 %, une clairance de la coloration de C3 et une stabilisation du DFGe dans tous les sousgroupes concernés à la semaine 26.
Étude chez des adultes présentant une récidive de C3G ou d’IC-MPGN primaire après transplantation (APL2 C3G-204)
L’étude APL2-C3G-204 était une étude en ouvert, randomisée, de phase II, incluant 13 patients adultes atteints d’une récidive de la maladie C3G (N = 10) ou IC-MPGN primaire (N = 3) après transplantation sur une période de 52 semaines.
Au cours des 12 premières semaines de l’étude, 10 patients ont reçu du pegcétacoplan en plus du traitement standard (SOC), et 3 patients uniquement le traitement standard. Tous les patients ont reçu du pegcétacoplan de la semaine 13 à la semaine 52.
Le critère d’évaluation principal, une réduction de l’intensité de la coloration de C3 lors de la biopsie rénale à la semaine 12, a été observé chez 50 % des patients traités par pegcétacoplan (5 patients sur 10, dont 4 ont obtenu un score de coloration nul), et chez 33,3 % des patients du groupe de contrôle (1 patient sur 3, ce dernier obtenant un score de coloration de 1).
Globalement, les modifications et modifications en pourcentage du DFGe étaient faibles par rapport à la valeur à l’inclusion (critère d’évaluation secondaire). Le DFGe moyen (ET) est passé de 52,3 (12,11) ml/min/1,73 m2 au début de l’étude à 57,3 (25,12) ml/min/1,73 m2 à la semaine 52, et le DFGe médian est passé de 50,5 ml/min/1,73 m2 au début de l’étude à 58,5 ml/min/1,73 m2 à la semaine 52. Chez la plupart des patients (9 patients sur 13 [69,2 %]) dans tous les groupes, une stabilisation ou une amélioration du DFGe à la semaine 52 a été atteinte.
PharmacocinétiqueAbsorption
Le pegcétacoplan est administré par voie sous-cutanée et progressivement absorbé dans la circulation générale, avec un Tmax médian compris entre 108 et 144 heures (4,5 à 6,0 jours). Lors de l’administration de 1080 mg deux fois par semaine chez des patients atteints d’HPN, les concentrations sériques ont atteint l’état d’équilibre quatre à six semaines environ après la première dose. Chez les patients ayant suivi un traitement préalable avec un inhibiteur complémentaire (étude APL2-302), la moyenne géométrique (%CV) des concentrations sériques moyennes à l’état d’équilibre chez les patients traités pendant 16 semaines était comprise entre 655 μg/ml (18,6 %) et 706 μg/ml (15,1 %). Chez les patients n’ayant pas suivi de traitement préalable par un inhibiteur complémentaire (APL2-308), la moyenne géométrique (%CV) des concentrations sériques moyennes à l’état d’équilibre était de 744 µg/ml (25,5 %) à la semaine 26 pour une administration deux fois par semaine. Aucune étude formelle n’a été réalisée concernant la biodisponibilité absolue; lors d’une étude transversale visant à comparer l’exposition après l’administration de formulations sous-cutanées et intraveineuses chez des volontaires sains, la biodisponibilité a été estimée à 87 %.
Les concentrations sériques à l’état d’équilibre ont été atteintes chez les patients atteints de C3G ou d’IC-MPGN primaire après administration deux fois par semaine de 1080 mg, environ 4 à 8 semaines après la première dose, et les concentrations thérapeutiques du pegcétacoplan ont été maintenues jusqu’à la semaine 52. Chez les patients de l’étude APL2-C3G-310, les concentrations sériques moyennes à l’état d’équilibre (%CV) jusqu’à la semaine 26 étaient comprises entre 715,8 (31,2 %) et 765,7 (23,2 %) μg/ml et ont été maintenues jusqu’à la semaine 52 entre 670,1 (30,1 %) et 726,6 (30,5 %) μg/ml.
Distribution
Chez les patients atteints d’HPN, le volume de distribution moyen du compartiment central (%CV) est d’environ 3,98 l (32 %).
Chez les patients adultes atteints de C3G ou d’IC-MPGN primaire, le volume de distribution moyen du compartiment central (%CV) est d’environ 4,31 l (32,1 %).
Métabolisme
En raison de sa structure peptidique pégylée, il est probable que le pegcétacoplan soit dégradé en petits peptides, acides aminés et PEG via des voies cataboliques.
Élimination
Après l’administration répétée de doses de pegcétacoplan par voie sous-cutanée chez des patients atteints d’HPN, la clairance (CL) moyenne (CV%) estimée est de 0,015 l/heure (30 %) et la demi-vie d’élimination effective médiane (t1/2) est de 8,6 jours.
Les résultats d’une étude de radio-marquage chez le singe Cynomolgus suggèrent que la voie d’élimination principale du fragment peptidique marqué est l’excrétion urinaire.
La clairance moyenne estimée (CV%) est de 0,012 l/heure (43 %) chez les patients adultes atteints de C3G ou d’IC-MPGN primaire. La demie-vie d’élimination terminale médiane t1/2 est de 10,1 jours chez les patients adultes atteins de C3G ou d’IC-MPGN primaire.
Linéarité/non-linéarité
L’exposition au pegcétacoplan augmente de façon proportionnelle à la dose dans l’intervalle de 45 à 1440 mg.
Cinétique pour certains groupes de patients
Aucun impact sur la pharmacocinétique du pegcétacoplan n’a été identifié chez les patients atteints d’HPN, de C3G ou d’IC-MPGN primaire en fonction de l’âge (12-81 ans), de l’origine ethnique ou du sexe, d’après les résultats de l’analyse pharmacocinétique de population.
On estime que la concentration moyenne à l’état d’équilibre est jusqu’à 20 % plus élevée chez un patient de 50 kg que chez un patient de référence ayant un poids corporel de 70 kg. On estime que la concentration moyenne chez les patients atteints d’HPN pesant 40 kg est plus élevée de 45 %. Les données disponibles concernant le profil de sécurité du pegcétacoplan chez les patients atteints d’HPN pesant moins de 50 kg sont extrêmement limitées.
Troubles de la fonction rénale
Lors d’une étude menée chez huit patients atteints d’insuffisance rénale sévère, définie sur la base d’une clairance de la créatinine (ClCr) inférieure à 30 ml/min selon la formule de Cockcroft-Gault (dont quatre patients présentant des valeurs inférieures à 20 ml/min), l’insuffisance rénale n’a eu aucun effet sur la pharmacocinétique d’une dose unique de 270 mg de pegcétacoplan (voir Posologie/Mode d’emploi). Les données disponibles concernant les patients atteints d’HPN et d’insuffisance rénale qui ont reçu la dose clinique de 1080 mg deux fois par semaine sont extrêmement limitées. Selon une d’une analyse pharmacocinétique de population, le DFGe n’a montré dans une population d’analyse groupée, aucune influence cliniquement significative sur l’exposition au pegcétacoplan. On ne dispose d’aucune donnée clinique concernant l’utilisation du pegcétacoplan chez des patients atteints d’IRT nécessitant une hémodialyse.
Troubles de la fonction hépatique
Aucune étude spécifique n’a été effectuée pour déterminer les répercussions de troubles de la fonction hépatique sur la pharmacocinétique du pegcétacoplan. La biotransformation ayant principalement lieu par catabolisme, des troubles de la fonction hépatique ne devraient pas influer sur la clairance du pegcétacoplan (voir Patients présentant des troubles de la fonction hépatique).
Patients âgés
Selon une analyse pharmacocinétique de population, la clairance apparente (CL/F) était similaire chez les patients âgés et les patients de moins de 65 ans. Aucune différence manifeste liée à l’âge n’a en outre été observée (voir Posologie/Mode d’emploi – Patients âgés). Le nombre de patients âgés était toutefois limité.
Population pédiatrique
Selon une analyse pharmacocinétique de population, le poids corporel des patients adolescents (12–17 ans) a une influence sur la clairance et le volume de distribution. Le schéma posologique pour les adolescents atteints de C3G ou d’IC-MPGN primaire est fonction du poids corporel du patient. Voir Posologie/Mode d’emploi. L’exposition estimée dans le modèle pour les adolescents atteints de C3G ou d’IC-MPGN primaire correspond à l’exposition de référence chez les adultes.
Données précliniquesLes données de toxicologie in vitro et in vivo n’ont pas révélé de toxicité particulièrement préoccupante pour l’être humain. Les effets observés chez l’animal à des niveaux d’exposition similaires à l’exposition clinique sont décrits ci-dessous.
Toxicité en cas d’administration répétée
Des études en administration répétée ont été réalisées chez le lapin et le singe Cynomolgus avec des doses quotidiennes sous-cutanées de pegcétacoplan correspondant à jusqu’à sept fois la dose clinique (1080 mg deux fois par semaine) chez l’être humain. Les altérations histologiques constatées chez les deux espèces incluaient une vacuolisation épithéliale dépendante de la dose et des infiltrations de macrophages vacuolisés dans différents tissus. Ces observations ont été associées à des doses cumulées importantes de PEG à longue chaîne obtenues avec d’autres médicaments pégylés autorisés. Elles n’ont pas eu de conséquences cliniques et n’ont pas été jugées délétères.
Après quatre semaines et neuf mois d’administration quotidienne de pegcétacoplan, une dégénérescence tubulaire rénale minime et non évolutive a été observée au microscope chez les deux espèces à des niveaux d’exposition (Cmax et ASC) inférieurs ou comparables à ceux atteints à la dose clinique chez l’être humain.
Bien qu’aucun signe manifeste de dysfonctionnement rénal n’ait été observé chez l’animal, les implications cliniques et les conséquences fonctionnelles de ces observations ne sont pas connues.
Génotoxicité
Le pegcétacoplan n’a pas montré de potentiel mutagène lors des tests de mutation inverse bactérienne in vitro (test d’Ames), ni de potentiel génotoxique lors d’un test in vitro sur des cellules TK6 humaines ou d’un test du micronoyau in vivo chez la souris.
Carcinogénicité
Aucune étude de carcinogénicité à long terme n’a été réalisée chez l’animal avec le pegcétacoplan.
Toxicité sur la reproduction
Le pegcétacoplan a fait l’objet d’études de reproduction chez le singe Cynomolgus. L’administration sous-cutanée de pegcétacoplan chez des guenons Cynomolgus gravides à la dose de 28 mg/kg/jour (2,9 fois la Cmax à l’état d’équilibre chez l’être humain) durant la période de gestation jusqu’à la mise bas a conduit à une augmentation statistiquement significative des avortements (31,6 %) ou des mortinaissances (21,1 %) par rapport au groupe témoin (5,0 % et 0 % respectivement).
Ces hausses ont été considérées comme liées au pegcétacoplan et défavorables. En raison de l’incidence accrue d’avortements et de mortinaissances à la dose de 28 mg/kg/jour, la DSENO a été établie à 7 mg/kg/jour dans cette étude.
Aucune toxicité maternelle ni aucun effet tératogène n’ont été observés chez la progéniture née à terme. Par ailleurs, aucun effet sur le développement de la progéniture n’a été observé jusqu’à six mois après la naissance. Une exposition systémique au pegcétacoplan a été détectée chez les fœtus de guenons ayant reçu 28 mg/kg/jour de la période de l’organogenèse jusqu’au deuxième trimestre, mais l’exposition était minime (moins de 1 %, exposition non significative d’un point de vue pharmacologique).
Fertilité
Le pegcétacoplan n’étant pharmacologiquement actif que chez l’être humain et les primates non humains, aucune étude spécifique portant sur la fertilité et le développement embryonnaire précoce n’a été menée avec le pegcétacoplan chez des rongeurs. Des analyses microscopiques des organes sexuels masculins et féminins réalisées au cours d’études de toxicité après administration réitérée chez le singe n’ont révélé aucun effet nocif du pegcétacoplan chez les animaux mâles ou femelles.
Lactation
Il a été démontré que l’excrétion de pegcétacoplan dans le lait maternel de guenons était inférieure à 1 %. Par conséquent, la probabilité d’exposition cliniquement significative de nourrissons au pegcétacoplan par le lait maternel est considérée comme minime.
Remarques particulièresIncompatibilités
Non pertinent.
Stabilité
Ce médicament ne doit pas être utilisé au-delà de la date figurant après la mention «EXP» sur l’emballage.
Remarques particulières concernant le stockage
Conserver au réfrigérateur (2-8 °C).
Conserver le récipient dans son emballage d’origine pour le protéger de la lumière.
Conserver hors de portée des enfants.
Remarques concernant la manipulation
Des instructions détaillées pour la préparation et l’administration d’Aspaveli peuvent être retrouvées dans le mode d’emploi figurant dans la notice.
Ne pas utiliser la solution si elle paraît trouble, contient des particules ou a pris une couleur jaune foncé.
Éliminer les flacons partiellement utilisés et les éléments à usage unique conformément à la réglementation locale.
Numéro d’autorisation68674 (Swissmedic)
PrésentationAspaveli est disponible en solution prête à l’emploi en flacons à usage unique.
1 flacon [A]
8 flacons [A]
Titulaire de l’autorisationSwedish Orphan Biovitrum AG, Basel
Mise à jour de l’informationOctobre 2025
|