CompositionPrincipes actifs
Immunoglobuline humaine normale injectable par voie sous-cutanée (IgSC). Protéine plasmatique humaine dont la teneur minimale en immunoglobuline G (IgG) est ≥98%.
Distribution approximative des sous-classes d'IgG: IgG1 69%, IgG2 26%, IgG3 3%, IgG4 2%.
La teneur maximale en immunoglobuline A (IgA) est de 50 microgrammes/ml.
Excipients
L-proline, polysorbate 80, hydroxyde de sodium (correspond à <10 mmol/l de sodium), acide chlorhydrique, eau pour préparations injectables.
Forme pharmaceutique et quantité de principe actif par unitéSolution injectable par voie sous-cutanée.
1 ml de solution contient: 200 mg de protéines plasmatiques humaines dont au moins 98% d'IgG (solution à 20%).
L'osmolalité est d'environ 380 mOsmol/kg.
La solution est claire et de couleur pouvant varier du jaune pâle ou brun clair.
Indications/Possibilités d’emploiTraitement de substitution chez les adultes et les enfants atteints de:
déficits immunitaires primaires (DIP) tels que:
agammaglobulinémie et hypogammaglobulinémie congénitales
déficit immunitaire commun variable
déficit immunitaire combiné sévère et syndrome de Wiskott Aldrich
déficits en sous-classes d'IgG avec infections récurrentes
-Déficits immunitaires secondaires (DIS) chez les patients souffrant d'infections sévères ou à répétition, réfractaires au traitement antimicrobien et présentant soit une incapacité démontrée à produire suffisamment d'anticorps anti-vaccin spécifiques (PSAF*) soit un taux sérique d'IgG < 4g/l.
* PSAF = incapacité à multiplier par au moins 2 le titre d'anticorps IgG en réponse aux antigènes polysaccharidiques du pneumocoque et aux antigènes polypeptidiques vaccinaux (PSAF = proven spécific antibody failure).
Traitement immunomodulateur:
-Hizentra est indiqué dans le traitement des patients atteints de polyradiculonévrite inflammatoire démyélinisante chronique (PIDC) comme traitement d'entretien après stabilisation par des immunoglobulines pour une administration intraveineuse (IgIV).
Posologie/Mode d’emploiPosologie usuelle
La dose et le schéma posologique dépendent de l'indication.
Le traitement doit être initié et surveillé par un professionnel de santé expérimenté dans le traitement des déficits immunitaires ou de la PIDC par des IgG.
Pour assurer la traçabilité des médicaments biologiques, il est recommandé de documenter le nom commercial et le numéro de lot à chaque traitement.
Traitement de substitution chez l'adulte et l'enfant
La dose peut être adaptée individuellement à chaque patient en fonction de l'évolution clinique et des taux sériques résiduels d'IgG. Les posologies suivantes sont données à titre indicatif:
La thérapie de substitution par Hizentra administré par voie sous-cutanée a pour but de maintenir un taux constant d'IgG. La dose initiale est d'au moins 0,2-0,5 g/kg (1,0-2,5 ml/kg) de poids corporel. Elle peut être répartie sur plusieurs jours. La dose mensuelle pour le maintien d'un taux d'IgG stable, est de l'ordre de 0,4-0,8 g/kg (2,0-4,0 ml/kg) de poids corporel et elle est fractionnée en plus petites doses qui sont administrées à intervalles répétés (voir le chapitre "Pharmacocinétique" ).
Pour les patients passant d'une administration intraveineuse à une administration sous-cutanée, la dose administrée auparavant de façon mensuelle est fractionnée en plus petites doses qui sont administrées à intervalles répétés (voir le chapitre "Pharmacocinétique" ).
Les taux résiduels doivent être mesurés et évalués en tenant compte de l'évolution clinique de la maladie chez le patient. Selon l'évolution clinique (par ex. le taux d'infection), un ajustement des doses et/ou de l'intervalle entre les doses peut être envisagé pour atteindre des taux résiduels plus élevés.
Traitement immunomodulateur dans la PIDC
Le traitement par Hizentra est instauré 1 semaine après la dernière perfusion d'IgIV. La dose sous-cutanée initiale recommandée est de 0,4 g/kg de poids corporel par semaine.
Quand un patient est cliniquement stable, la dose hebdomadaire d'Hizentra peut être réduite à un minimum de 0,2 g/kg de poids corporel. En cas de rechute, après réduction de la dose à 0,2 g/kg de poids corporel par semaine, il convient de revenir à une dose sous-cutanée plus élevée de 0,4 g/kg de poids corporel par semaine. Si le patient ne répond pas ou si une rechute survient aux doses plus élevées, il convient de passer aux IgIV à une dose de saturation initiale. En cas de traitement de rattrapage avec des IgIV, un retour au traitement par IgSC ne peut pas être conseillé.
La dose hebdomadaire peut être fractionnée en doses plus petites et administrée à la fréquence souhaitée. Pour une perfusion toutes les deux semaines, doubler la dose d'Hizentra.
L'efficacité d'Hizentra par rapport au placebo a été démontrée dans des études cliniques après un changement d'immunoglobulines intraveineuses. On ne dispose pas de données comparatives directes sur Hizentra par rapport aux IgIV.
Patients âgés
La dose étant administrée en fonction du poids corporel et ajustée en fonction de la réponse clinique, on ne s'attend pas à ce que la dose pour les sujets âgés soit différente de celle chez les personnes âgées de 18 à 65 ans.
Hizentra a fait l'objet d'études cliniques chez 13 patients atteints de DIP âgés de plus de 65 ans. Aucun ajustement de dose spécifique n'a été nécessaire pour atteindre les taux sériques d'IgG souhaités.
Hizentra a été étudié dans le cadre d'essais cliniques chez 61 patients atteints de PIDC âgés de plus de 65 ans. Aucun ajustement de dose spécifique n'a été nécessaire pour obtenir les résultats cliniques souhaités.
Enfants et adolescents
La posologie est calculée, comme chez l'adulte, en fonction du poids corporel et est ajustée selon l'évolution clinique de la maladie lors du traitement de substitution. Hizentra a été évalué chez 68 enfants âgés de 2 à 12 ans et 57 adolescents âgés de 12 à <18 ans atteints de DIP. Aucun ajustement de la dose relatif à la population pédiatrique n'a été nécessaire pour atteindre les taux sériques d'IgG souhaités.
À ce jour, Hizentra n'a pas été évalué dans des études cliniques chez des patients pédiatriques atteints de PIDC.
Mode d'administration
Hizentra doit être administré uniquement par voie sous-cutanée.
Hizentra peut être perfusé dans des sites tels que la paroi abdominale, la cuisse, le bras, et la face latérale de la hanche (voir Figure 1). Si des quantités importantes (>50 ml) doivent être administrées, il est conseillé de répartir les injections sur plusieurs sites. Chez les nourrissons et les enfants, le site d'injection doit être changé tous les 5-15 ml. Il n'y a pas de nombre limité de sites d'injection. Les sites d'injection doivent être espacés au minimum de 5 cm. Plusieurs' dispositifs de perfusion peuvent être utilisés simultanément. La quantité de produit injecté dans un site particulier peut varier. Il faut changer de site de perfusion en cas d'administrations successives.
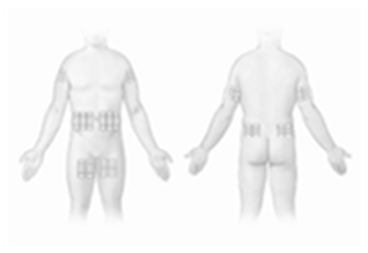
Figure 1: Sites de perfusion possibles pour Hizentra
L'autotraitement à domicile doit être initié et accompagné au début par un professionnel de la santé. Le professionnel de la santé doit avoir l'expérience de la prise en charge de patients autotraités à domicile. Le patient ou le soignant doivent être formés et entraînés à l'utilisation des appareils de perfusion, aux techniques de perfusion, à la tenue du carnet de traitement, à la reconnaissance des effets indésirables graves et aux mesures à prendre en cas de survenue de ces derniers.
Débit de perfusion
Hizentra peut être perfusé:
avec un appareil de perfusion (par ex. une pompe de perfusion)
en perfusion manuelle avec une seringue.
Le débit de perfusion recommandé varie en fonction des besoins individuels de chaque patient.
Perfusion avec un appareil de perfusion:
Traitement de substitution
Le débit de perfusion initial recommandé ne doit pas dépasser 20 ml/heure/site. S'il est bien toléré, le débit de perfusion peut ensuite être augmenté progressivement jusqu'à 100 ml/heure/site.
Polyradiculonévrite inflammatoire démyélinisante chronique (PIDC)
Le débit de perfusion initial recommandé ne doit pas dépasser 20 ml/heure par site de perfusion. S'il est bien toléré, le débit de perfusion peut être progressivement augmenté jusqu'à 50 ml/heure/site de perfusion.
Pour l'administration toutes les deux semaines, le volume nécessaire d'Hizentra peut être administré au besoin en 4 séances maximum, p.ex. une fois le matin et une fois le soir et répété le lendemain ou au cours de la deuxième semaine.
Perfusion manuelle
Le débit de perfusion initial recommandé ne doit pas dépasser 0,5 ml/min par site de perfusion (30 ml/heure par site de perfusion).
S'il est bien toléré, le débit de perfusion peut être progressivement augmenté jusqu'à 2,0 ml/min/site de perfusion (120 ml/heure par site de perfusion) en fonction de l'évaluation par le personnel médical et de la tolérance individuelle du patient.
Il est recommandé d'utiliser des aiguilles d'injection de calibre 24 ou plus (donc avec une gauge plus petite) afin que les patients puissent perfuser manuellement avec un débit plus élevé. L'utilisation d'aiguilles d'injection plus petites (donc avec une gauge plus importante) peut rendre difficile la perfusion d'Hizentra.
On ne peut perfuser manuellement avec une seringue qu'à un seul site de perfusion.
Lorsqu'il est nécessaire d'administrer manuellement un volume supérieur à 20 ml, il est recommandé, en fonction de la dose individuelle à administrer, de perfuser la dose à plusieurs sites. Dans ce cas, une seringue d'Hizentra supplémentaire est nécessaire pour chaque site de perfusion. Si le volume est perfusé à plusieurs sites, il faut utiliser à chaque fois une nouvelle aiguille d'injection stérile pour chaque seringue de perfusion.
Les kits d'administration avec plusieurs aiguilles de perfusion/cathéters ou les systèmes d'administration en Y ne doivent pas être utilisés.
Contre-indicationsHypersensibilité au principe actif ou à l'un des excipients selon la composition (voir aussi le chapitre "Composition" ).
Hyperprolinémie de type I ou II. L'hyperprolinémie est une maladie très rare qui ne touche que quelques familles dans le monde entier.
Hizentra ne doit pas être administré par voie intravasculaire.
Mises en garde et précautionsL'administration accidentelle d'Hizentra par voie intraveineuse peut entraîner un état de choc chez le patient.
Le débit de perfusion recommandé à la rubrique "Posologie/Mode d'emploi: Débit de perfusion" doit être respecté. Lors des premières perfusions accompagnées, les patients doivent être étroitement suivis et surveillés pour détecter tout effet indésirable éventuel survenant au cours de la perfusion et dans les 20 minutes suivant l'administration.
Certains effets indésirables peuvent survenir plus fréquemment chez les patients recevant pour la première fois des immunoglobulines humaines ou, dans de rares cas, lors d'un changement d'immunoglobulines ou lorsque le traitement a été suspendu pendant plus de huit semaines.
Hypersensibilité/réactions allergiques
Les vraies réactions allergiques sont rares. Elles peuvent survenir chez les patients présentant des anticorps anti-IgA. Les patients possédant des anticorps anti-IgA et chez qui l'administration d'IgG par voie sous-cutanée demeure la seule option, doivent être traités par Hizentra uniquement sous étroite surveillance médicale.
Dans de rares cas, l'administration d'immunoglobuline d'origine humaine peut induire une chute soudaine de la pression artérielle associée à une réaction anaphylactique, même chez les patients ayant présenté une bonne tolérance à une administration antérieure d'immunoglobulines.
Les complications potentielles peuvent souvent être évitées en s'assurant que les patients:
ne sont pas hypersensibles aux immunoglobulines d'origine humaine; pour cela on commencera par administrer lentement le produit lors de la première perfusion (≤15 ml/heure/site);
sont étroitement surveillés afin de détecter la survenue de tout symptôme pendant toute la durée de la perfusion et au moins 20 minutes après. Plus particulièrement et ce afin de déceler de potentiels effets indésirables, les patients qui reçoivent pour la première fois de l'immunoglobuline d'origine humaine, qui ont été préalablement traités par une autre préparation d'immunoglobuline, ou pour lesquels un long intervalle de temps s'est écoulé depuis le traitement précédent, doivent être surveillés au cours de la première perfusion et pendant l'heure qui suit.
Toute suspicion de réaction allergique ou anaphylactique exige un arrêt immédiat de l'injection. En cas d'état de choc, les traitements médicaux standards actuels doivent être mis en œuvre.
Événements emboliques et thromboemboliques
Il existe des preuves cliniques d'une association entre l'administration d'immunoglobulines et des événements emboliques/thromboemboliques tels qu'infarctus du myocarde, accident vasculaire cérébral, thrombose veineuse profonde et embolie pulmonaire. La prudence s'impose donc tout particulièrement chez les patients présentant des facteurs de risque préexistants d'événements thrombotiques (tels qu'âge avancé, hypertension, diabète sucré, antécédents de maladie vasculaire ou d'épisodes thrombotiques, risques de thromboses héréditaires ou acquises, immobilité pendant de longues périodes, hypovolémie sévère et maladies augmentant la viscosité du sang). Il convient d'indiquer aux patients quels sont les premiers symptômes des événements thromboemboliques, tels qu'essoufflement, douleur et gonflement d'un membre, déficits neurologiques centraux et douleur thoracique, et de leur conseiller de contacter leur médecin dès qu'apparaissent ces symptômes. Les patients doivent être suffisamment hydratés avant d'utiliser des immunoglobulines.
Syndrome de méningite aseptique (SMA)
Des cas de SMA sont survenus lors de l'utilisation des IGIV ou des IGSC. Généralement, le syndrome survient quelques heures à 2 jours après le traitement par immunoglobulines. Le SMA se caractérise par les signes et symptômes suivants: maux de tête sévères, raideur de la nuque, somnolence, fièvre, photophobie, nausée et vomissement. Les patients présentant des signes et symptômes du SMA doivent faire l'objet d'un examen neurologique approfondi, incluant des examens du liquide céphalo-rachidien (LCR), afin d'écarter toute autre cause de la méningite. L'interruption du traitement par immunoglobulines conduit à la rémission du SMA en l'espace de quelques jours et sans laisser de séquelles.
Complications rénales
Des cas d'effets indésirables rénaux graves ont été rapportés chez des patients sous traitement par immunoglobulines, en particulier dans le cas de préparations contenant du saccharose (Hizentra ne contient pas de saccharose). Il s'agit notamment de: insuffisance rénale aiguë, nécrose tubulaire rénale aiguë, tubulopathie proximale et néphrose osmotique. Les facteurs qui augmentent le risque de complications rénales sont, entre autres, une insuffisance rénale préexistante, un diabète sucré, une hypovolémie, des médicaments concomitants néphrotoxiques, un âge supérieur à 65 ans, une septicémie, une hyperviscosité ou une paraprotéinémie.
Anémie hémolytique
Les produits à base d'immunoglobulines peuvent contenir des anticorps de groupes sanguins pouvant agir comme des hémolysines et induire une liaison des immunoglobulines aux globules rouges. Cela peut induire une réaction antiglobuline directe positive (test de Coombs), et dans de rares cas, une hémolyse. Une anémie hémolytique retardée peut se développer suite au traitement par des immunoglobulines en raison de l'augmentation de la séquestration des globules rouges. Des cas d'anémie hémolytique ont été rapportés.
Information concernant la prévention du risque de transmission virale
Hizentra est préparé à partir de plasma humain. Les mesures habituelles de prévention des infections induites par l'utilisation de médicaments préparés à partir de sang ou de plasma humain comprennent la sélection des donneurs, la recherche des marqueurs spécifiques d'infection sur chaque don et sur les mélanges de plasma ainsi que l'inclusion dans le procédé de fabrication d'étapes efficaces pour l'inactivation/élimination virale (voir également le chapitre "Propriétés/Effets" ). Cependant, lorsque des médicaments préparés à partir de sang ou de plasma humain sont administrés, le risque de transmission d'agents infectieux ne peut pas être totalement exclu. Ceci s'applique également aux virus inconnus ou émergents et aux autres agents pathogènes.
Les mesures prises sont considérées comme efficaces contre les virus à enveloppes tels que le virus de l'immunodéficience humaine (VIH), le virus de l'hépatite B (VHB) et le virus de l'hépatite C (VHC), ainsi que les virus non enveloppés tels que le virus de l'hépatite A (VHA) et le parvovirus B19.
L'expérience clinique montre que la transmission du virus de l'hépatite A ou du parvovirus B19 par des immunoglobulines est inexistante. De plus, on suppose que les anticorps présents contribuent fortement à la sécurité virale.
Il est fortement recommandé d'enregistrer le nom et le numéro du lot du médicament à chaque administration d'Hizentra à un patient, afin de conserver un lien entre le patient et le numéro de lot du médicament.
Hizentra contient moins de 1 mmol de sodium (23 mg) par flacon, ce qui signifie qu'il est pratiquement "sans sodium" .
InteractionsVaccins à virus vivants atténués
L'administration d'immunoglobulines peut limiter, pour une période d'au moins 6 semaines et jusqu'à 3 mois, l'efficacité des vaccins à virus vivants atténués, comme les vaccins contre la rougeole, la rubéole, les oreillons et la varicelle. Après administration de ce médicament, un intervalle de 3 mois doit s'écouler avant une vaccination avec des vaccins constitués de virus vivants atténués. Dans le cas de la rougeole, cette diminution d'efficacité peut persister jusqu'à 1 an. Par conséquent, il est nécessaire de contrôler les taux d'anticorps chez les patients ayant été vaccinés contre la rougeole.
Grossesse, allaitementIl n'existe pas de données cliniques contrôlées concernant l'administration d'Hizentra chez la femme enceinte ou allaitante, c'est pourquoi ce médicament doit être administré avec précaution au cours de la grossesse et de l'allaitement. La longue expérience clinique acquise dans le domaine des immunoglobulines ne suggère néanmoins aucun effet délétère sur le déroulement de la grossesse, sur le fœtus ou sur le nouveau-né.
La poursuite du traitement chez la femme enceinte est importante afin d'assurer la mise en place d'une immunité passive correspondante chez le nouveau-né.
Les immunoglobulines sont excrétées dans le lait maternel et peuvent contribuer à la transmission d'anticorps protecteurs au nouveau-né.
Effet sur l’aptitude à la conduite et l’utilisation de machinesLa capacité à conduire et à utiliser des machines peut être affaiblie par certains effets indésirables associés à Hizentra. Les patients qui présentent des effets indésirables pendant le traitement doivent attendre que ceux-ci disparaissent avant de conduire ou d'utiliser des machines.
Effets indésirablesRésumé du profil de sécurité
Lors de l'administration d'immunoglobulines d'origine humaine, des effets indésirables (EI) de type frissons, céphalée, fièvre, vomissement, réactions allergiques, nausée, arthralgie, hypotension artérielle et lombalgie modérée peuvent survenir occasionnellement.
Dans de rares cas, les immunoglobulines humaines peuvent provoquer une chute brutale de la pression artérielle et, dans des cas isolés, un choc anaphylactique même si le patient n'a pas présenté de réaction d'hypersensibilité lors d'administrations antérieures.
Parmi les réactions locales observées aux sites de perfusion, figurent: gonflement, douleur, rougeur, induration, sensation de chaleur locale, démangeaisons, ecchymose et éruption cutanée.
Effets indésirables (EI) dans des études cliniques et pendant la surveillance post-commercialisation avec Hizentra
Dans sept études cliniques de phase III menées chez des patients atteints de déficit immunitaire primaire DIP (n=231), deux études de phase IV auprès de patients atteints de DIP (n=74), une étude de phase III menée chez des patients atteints de PIDC (n=115) et une étude de prolongation menée auprès de patients atteints de PIDC (n=82) et traités par Hizentra (total n=502), les effets indésirables suivants ont été rapportés et sont présentés par classe de systèmes d'organes (terminologie MedDRA) et par fréquence.
La fréquence par patient a été évaluée selon les critères suivants: très fréquents (≥1/10), fréquents (≥1/100 à <1/10), occasionnels (≥1/1000 à <1/100), rares (≥1/10 000 à <1/1000), très rares (<1/10 000). Pour les EI spontanés post-commercialisation, la fréquence est indiquée sous forme de cas isolés.
Infections et infestations:
Très fréquents: rhinopharyngite (19,3%).
Affections hématologiques et du système lymphatique:
Occasionnels: augmentation de la lactate déshydrogénase sanguine.
Cas isolés: anémie hémolytique**.
Affections du système immunitaire:
Occasionnels: hypersensibilité.
Cas isolés: réactions anaphylactiques.
Affections du système nerveux:
Très fréquents: céphalées (12,5%)
Fréquents: vertiges, migraines
Occasionnels: syndrome de méningite aseptique (SMA)*, tremblements (y compris hyperactivité psychomotrice).
Cas isolés: sensation de brûlure, somnolence.
Affections cardiaques:
Occasionnels: tachycardie (y compris augmentation de la pression artérielle).
Affections vasculaires:
Fréquents: hypertension.
Occasionnels: bouffées de chaleur, augmentation de l'aldolase.
Cas isolés: événements emboliques et thromboemboliques (y compris thrombose, embolie, accident ischémique transitoire, hémiparésie, cécité passagère, accident vasculaire cérébral hémorragique, hémiplégie).
Affections respiratoires, thoraciques et médiastinales:
Fréquents: toux.
Affections gastro-intestinales:
Fréquents: diarrhée, douleurs abdominales, nausées, vomissement.
Affections de la peau et du tissu sous-cutané:
Très fréquents: éruption cutanée (y compris érythème) (10,4%).
Fréquents: prurit, urticaire, dermatite de contact.
Affections musculo-squelettiques et du tissu conjonctif:
Fréquents: douleurs musculo-squelettiques, arthralgie.
Occasionnels: arthrite, spasmes musculaires, faiblesse musculaire, augmentation de la créatine phosphokinase sanguine.
Affections du rein et des voies urinaires:
Occasionnels: hématurie, augmentation de la créatinine.
Cas isolés: insuffisance rénale aiguë**, nécrose tubulaire rénale aiguë** tubulopathie proximale** et néphrose osmotique**.
Troubles généraux et anomalies au site d'administration:
Très fréquents: réactions au site de perfusion (y compris contusion et hématome) (42,2%).
Fréquents: fatigue (y compris sensation de malaise), fièvre (y compris augmentation de la température corporelle), douleurs thoraciques, syndrome grippal, douleurs.
Occasionnels: frissons (y compris hypothermie), perte de poids.
Cas isolés: ulcère au site de perfusion.
* Les symptômes du SMA comprennent: raideur de la nuque, céphalées, pyrexie, photophobie, nausées et vomissement.
** Effets de classe indésirables des immunoglobulines.
Informations sur la sécurité virale: voir le chapitre "Mises en garde et précautions" .
L'annonce d'effets secondaires présumés après l'autorisation est d'une grande importance. Elle permet un suivi continu du rapport bénéfice-risque du médicament. Les professionnels de santé sont tenus de déclarer toute suspicion d'effet secondaire nouveau ou grave via le portail d'annonce en ligne ElViS (Electronic Vigilance System). Vous trouverez des informations à ce sujet sur www.swissmedic.ch.
SurdosageLes conséquences d'un surdosage ne sont pas connues.
Propriétés/EffetsCode ATC
J06BA01
Classe pharmacothérapeutique: antisérums et immunoglobulines: immunoglobulines humaines normales pour administration extravasculaire.
Mécanisme d'action/Propriétés pharmacodynamiques
Hizentra contient un large spectre d'anticorps IgG opsonisants et neutralisants contre de nombreux agents bactériens et viraux.
Hizentra contient les anticorps IgG présents dans la population normale. Hizentra est préparé à partir de pools de plasma provenant d'un minimum de 1000 donneurs. La répartition des sous-classes d'immunoglobulines G correspond approximativement à celle du plasma humain natif.
Des doses suffisantes d'Hizentra peuvent ramener des taux anormalement bas d'anticorps IgG à des valeurs normales et ainsi aider à combattre les infections.
Le mécanisme d'action d'Hizentra chez les patients atteints de PIDC n'est pas entièrement élucidé, mais peut inclure des effets immunomodulateurs.
Efficacité clinique
Déficits immunitaires primaires
La sécurité et l'efficacité d'Hizentra pour le traitement des déficits immunitaires primaires ont été évaluées dans sept études de phase III et deux études de phase IV auprès de patients atteints de DIP.
Dans l'étude européenne pivot prospective, ouverte, en groupe unique et multicentrique, 51 sujets atteints de DIP, âgés de 3 à 60 ans, ont été traités par Hizentra pendant une période allant jusqu'à 41 semaines. La dose moyenne administrée chaque semaine était de 119 mg/kg de poids corporel. Ainsi, il a été possible de maintenir des taux résiduels d'IgG à des concentrations moyennes comprises entre 7,99 et 8,25 g/l tout au long de la durée du traitement. Les sujets ont reçu au total 1831 perfusions d'Hizentra chaque semaine.
40 des patients traités lors de l'étude principale ont été inclus dans l'étude d'extension ultérieure (âge: 4-52 ans). Ces patients ont été traités avec une dose constante d'Hizentra pour une durée de traitement allant jusqu'à 46 mois. Au total, 5405 perfusions hebdomadaires d'Hizentra ont été administrées aux patients.
Durant toute la période de traitement, il a été possible de maintenir des concentrations résiduelles moyennes d'IgG de 7,5-8,5 g/l, confirmant ainsi les résultats de l'étude principale. Le taux d'infections bactériennes aiguës graves (IBA) était de 0,0478 par patient et par an, avec un intervalle de confiance (IC) supérieur à 99% de 0,1252.
Dans l'étude américaine prospective, ouverte, en groupe unique et multicentrique, 49 sujets atteints de déficit immunitaire primaire, âgés de 5 à 72 ans, ont été traités par Hizentra pendant une période allant jusqu'à 15 mois. La dose moyenne administrée chaque semaine était de 228 mg/kg de poids corporel. Ainsi, il a été possible de maintenir des taux résiduels d'IgG à une concentration moyenne de 12,53 g/l tout au long de la durée du traitement. Les sujets ont reçu au total 2264 perfusions d'Hizentra chaque semaine.
Ultérieurement, lors de l'étude d'extension américaine, un total de 21 patients (âge: 5-69 ans) préalablement traités lors de l'étude de base ont été inclus et traités jusqu'à 87 semaines avec un dosage constant. Les patients ont reçu un total de 1735 perfusions hebdomadaires d'Hizentra.
Durant toute la période de traitement, il a été possible de maintenir des concentrations résiduelles moyennes d'IgG de 11,71-12,76 g/l, (11,98 g/l de moyenne générale), confirmant ainsi les résultats de l'étude principale. Chez aucun des patients, le taux résiduel d'IgG n'est descendu en dessous de 5 g/l durant le traitement. Le taux d'IBA était de 0,06 par patient et par an, avec un intervalle de confiance à 99% supérieur de 0,257.
Pour l'évaluation de la sécurité et de la tolérance des vitesses de perfusion élevées utilisées avec des méthodes d'administration manuelles (push) ou avec des pompes, 49 patients atteints de DIP âgés de 2 à 75 ans ont été inclus dans une étude de phase IV multicentrique, non randomisée et en open-label (HiLo) et traités par Hizentra pendant au moins 12 semaines (11 enfants âgés de 2 à < 18 ans, 35 adultes âgés de 18 à 65 ans et 3 patients gériatriques de plus de 65 ans). Dans le premier groupe de patients ayant reçu Hizentra par une méthode manuelle push (n=16), 2 à 7 perfusions ont été administrées par semaine avec des vitesses de 30, 60 et 120 ml/heure/site de perfusion (voir chapitre "Posologie/Mode d'emploi" ).
Dans le deuxième groupe de patients ayant reçu Hizentra avec une pompe de perfusion (n=18), des perfusions hebdomadaires ont été administrées à des vitesses de 25, 50, 75 et 100 ml/heure/site de perfusion. Dans le troisième groupe de patients (n=15), des volumes de perfusion de 25, 40 et 50 ml par site ont été perfusés en plus avec administration par pompe de doses hebdomadaires d'Hizentra. Dans les trois groupes, chaque paramètre de perfusion a été utilisé pendant 4 semaines. Ensuite, les patients ayant bien toléré le traitement (répondeurs) pouvaient passer au paramètre de perfusion suivant plus élevé. Le critère primaire était le pourcentage de patients répondeurs après le passage au paramètre de perfusion plus élevé:
Groupe Paramètre de perfusi Tolérance**
on et taux de
répondeurs* (%)
1. Vitesse de 30 ml/heure 100,0% 60 ml/heure 100,0% 120 ml/heure 87,5% - 1,0
perfusion avec push
manuel
2. Vitesse de 25 ml/heure 77,8% 50 ml/heure 77,8% 75 ml/heure 66,7% 100 ml/ heure 61,1% 1,0 (0,98 – 1,0)
perfusion avec pompe
3. Volumes perfusés 25 ml 86,7% 40 ml 73,3% 50 ml 73,3% - 1,0
avec pompe
* Répondeur: dans le groupe avec perfusion par pompe: patient ayant reçu ≥3 perfusions sur 4 valides pour un paramètre de perfusion; dans le groupe avec push manuel: patient ayant reçu un nombre minimal de perfusions valides (≥60%) pour un paramètre de perfusion.
Une perfusion a été considérée comme valide lorsque ≥95% de la vitesse et du volume prévus pour ≥1 site de perfusion ont été atteints.
** Tolérance: nombre de perfusions sans réaction locale grave divisé par le nombre total de perfusions.
Globalement, le nombre de perfusions sans réaction locale grave par rapport au nombre total de perfusions (tolérance) dans tous les groupes pour tous les paramètres de perfusion a été supérieur ou égal à ≥0,98.
Entre la valeur initiale au jour 1 et à la fin de l'étude, aucune différence cliniquement pertinente n'a été notée en termes de concentrations résiduelles d'IgG sériques chez tous les patients.
Patients âgés
En termes de sécurité et d'efficacité, aucune différence globale n'a été observée entre les patients atteints de DIP âgés de plus de 65 ans et les patients atteints de DIP âgés de 18 à 65 ans. Lors des essais cliniques, Hizentra a été étudié auprès de 13 patients atteints de DIP et âgés de plus de 65 ans.
Pédiatrie
La sécurité et l'efficacité d'Hizentra ont été étudiées chez les patients pédiatriques âgés de 2 à 18 ans.
Hizentra a été évalué chez 68 patients pédiatriques atteints de DIP âgés de 2 à <12 ans et chez 57 patients pédiatriques âgés de 12 à <18 ans.
Aucune différence dans les paramètres pharmacocinétiques, la sécurité et l'efficacité n'a été constatée entre les patients adultes et pédiatriques.
En pédiatrie, aucune posologie spécifique n'a été nécessaire pour atteindre les taux sériques d'IgG souhaités.
Polyradiculonévrite inflammatoire démyélinisante chronique (PIDC)
La sécurité, l'efficacité et la tolérance d'Hizentra chez des patients atteints de PIDC ont été évaluées dans l'étude PATH [Polyneuropathy and Treatment with Hizentra] de phase III, multicentrique, en double aveugle, randomisée, contrôlée contre placebo, en groupes parallèles. 172 participants à l'étude traités préalablement avec des IgIV ont été randomisés dans des groupes recevant Hizentra à raison de 0,2 g/kg de poids corporel par semaine, de 0,4 g/kg de poids corporel ou le placebo. Ces patients ont ensuite été traités pendant 24 semaines. La durée d'exposition moyenne dans le groupe sous 0,2 g/kg de poids corporel était de 118,9 jours et de 129 jours dans le groupe sous 0,4 g/kg de poids corporel (l'exposition maximale dans chaque groupe allait jusqu'à 167 jours et 166 jours respectivement). Les participants à l'étude ont utilisé en général 4 sites de perfusion simultanément (au maximum 8 sites simultanément).
Au total, 57 participants à l'étude ont reçu 1514 perfusions dans le groupe sous placebo, 57 participants à l'étude ont reçu 2007 perfusions dans le groupe sous Hizentra à raison de 0,2 g/kg de poids corporel et 58 participants à l'étude ont reçu 2218 perfusions dans le groupe sous Hizentra à raison de 0,4 g/kg de poids corporel (5739 perfusions au total).
Le critère primaire d'évaluation de l'efficacité était le pourcentage de participants à l'étude qui ont présenté une récidive de la PIDC (définie comme une augmentation de ≥1 point sur l'échelle INCAT ajustée (Inflammatory Neuropathy Cause and Treatment) par rapport à la valeur initiale) ou qui ont été retirés de la phase de traitement par Hizentra pour une autre raison.
Les deux doses d'Hizentra se sont avérées supérieures au placebo pour le critère d'évaluation principal de l'efficacité.
Un pourcentage statistiquement significativement plus faible de patients traités par Hizentra ont présenté une récidive de la PIDC ou ont été retirés de l'étude pour d'autres raisons: 32,8% sous 0,4 g/kg de poids corporel et 38,6% sous 0,2 g/kg de poids corporel, contre 63,2% des participants à l'étude traités par placebo [p <0,001 et p = 0,007 respectivement]. Si les récidives sont considérées séparément, les taux de récidives de PIDC étaient de 19,0% pour la dose d'Hizentra de 0,4 g/kg de poids corporel et de 33,3% pour la dose d'Hizentra de 0,2 g/kg de poids corporel contre 56,1% pour le placebo (p <0,001 et p = 0,012 respectivement). Par conséquent, Hizentra a empêché des récidives pendant une période de traitement allant jusqu'à 24 semaines chez 81% et 67% respectivement des participants à l'étude dans les groupes sous 0,4 g/kg de poids corporel ou sous 0,2 g/kg de poids corporel. Dans le groupe sous placebo, aucune récidive n'est survenue chez 44% des participants à l'étude.
Le délai jusqu'à la récidive de la PIDC a été évalué et les probabilités de récidive de la PIDC correspondantes en fonction des estimations de Kaplan-Meier étaient les suivantes: placebo 58,8%; Hizentra à raison de 0,2 g/kg de poids corporel, 35,0% et Hizentra à raison de 0,4 g/kg de poids corporel, 22,4%.
Les risques relatifs (IC à 95%) pour la dose la plus faible et la plus forte comparée au placebo s'élevaient respectivement à 0,48 (0,27; 0,85) et 0,25 (0,12; 0,49).
La différence observée entre les groupes sous Hizentra à la dose de 0,2 g/kg de poids corporel et de 0,4 g/kg de poids corporel n'était pas cliniquement significative.
Les participants à l'étude sont restés stables en ce qui concerne les résultats d'efficacité (échelle INCAT, force de préhension moyenne et échelle sommaire du Medical Research Council) dans les deux groupes posologiques d'Hizentra comparés au groupe sous placebo, dont les résultats s'étaient dégradés. Les participants à l'étude du groupe sous Hizentra à forte dose sont restés stables sur l'échelle Rasch-built Overall Disability Scale (R-ODS).
Les paramètres électrophysiologiques sont restés stables chez les participants de l'étude dans les deux groupes de dose d'Hizentra.
82 patients de l'étude PIDC issus de l'essai PATH ont été inclus dans l'étude d'extension de phase III, multicentrique et en ouvert, d'une durée de 48 semaines. L'étude d'extension a examiné la sécurité et l'efficacité à long terme du traitement d'entretien par Hizentra avec deux posologies hebdomadaires différentes de 0,2 g/kg et 0,4 g/kg de poids corporel, respectivement. En raison de la conception de l'étude, le même patient pouvait recevoir les deux doses pendant l'étude; 72 patients ont reçu une dose de 0,4 g/kg de poids corporel et 73 patients ont reçu une dose de 0,2 g/kg de poids corporel. La durée moyenne du traitement était de 125,8 jours (intervalle: 1 à 330) dans le groupe de 0,2 g/kg de poids corporel et de 196,1 jours (intervalle: 1 à 330) dans le groupe de 0,4 g/kg de poids corporel.
Les patients qui ont terminé l'étude pivot PATH sans récidive avec une dose de 0,4 g/kg de poids corporel et qui ont initialement reçu cette dose dans l'étude d'extension ont eu un taux de récidive de 5,6% (1 patient sur 18).
Sur l'ensemble des patients ayant reçu la dose de 0,4 g/kg de poids corporel dans l'étude d'extension PATH, 9,7% (7 patients sur 72) ont présenté une récidive.
Les patients qui ont terminé l'étude PATH sans récidive à la dose de 0,2 g/kg de poids corporel et qui ont initialement reçu cette dose dans l'étude de prolongation avaient un taux de récidive de 50% (3 patients sur 6).
Sur l'ensemble des patients ayant reçu 0,2 g/kg de poids corporel dans l'étude de prolongation, 47,9% (35 patients sur 73) ont présenté une récidive. La réduction de la dose de 0,4 g/kg de poids corporel à 0,2 g/kg de poids corporel dans l'étude d'extension sans rechute a été possible chez 67,9% des patients (19 patients sur 28). Tous les patients ayant présenté une récidive se sont rétablis dans les 4 semaines suivant le traitement à la dose de 0,4 g/kg de poids corporel.
La force de préhension, le score cumulatif MRC et le centile R-ODS sont restés stables par rapport à la référence chez les patients qui n'ont pas présenté de récidive dans l'étude d'extension.
Population âgée
Globalement, au niveau de la sécurité et de l'efficacité, aucune différence entre les patients âgés de plus de 65 ans et les patients entre 18 et 65 ans n'a été notée. Dans les études cliniques chez des patients atteints de PIDC, 61 patients traités par Hizentra étaient âgés de plus de 65 ans.
Pédiatrie
À ce jour, le traitement par Hizentra des patients pédiatriques atteints de PIDC n'a pas été étudié dans des études cliniques.
PharmacocinétiqueAbsorption
Après administration par voie sous-cutanée d'Hizentra, les pics plasmatiques d'IgG sont atteints en 2 jours environ.
Distribution
Déficits immunitaires primaires
Les simulations basées sur des modèles pharmacocinétiques de population suggèrent qu'une exposition similaire d'IgG (Cmax, AUC0-14 jours, Cmin, 14 jours) est obtenue si Hizentra est administré toutes les deux semaines en utilisant le double de la dose hebdomadaire.
Ces simulations suggèrent en outre qu'une exposition comparable aux IgG est obtenue lorsque la dose d'entretien hebdomadaire de Hizentra est fractionnée en plusieurs doses (par ex. 2, 3, 5 fois par semaine ou tous les jours).
L'oubli de 2 ou 3 doses journalières dans le cadre d'une administration quotidienne continue se traduit lors des simulations par une baisse du taux d'IgG sérique médian ≤4% par rapport à une administration quotidienne constante. Lorsque les doses oubliées sont administrées ensuite le premier jour de la reprise de l'administration quotidienne en plus de la dose quotidienne normale, le profil de concentration moyenne des IgG se rétablit en 2-3 jours. Cependant, si les doses oubliées ne sont pas remplacées lors de la reprise de l'administration quotidienne des doses, les taux résiduels d'IgG à l'état d'équilibre ne pourront être de nouveau obtenus qu'après une durée de traitement de 5 à 6 semaines.
Patients âgés
Dans l'ensemble, aucune différence n'a été observée dans les paramètres pharmacocinétiques des patients atteints de DIP âgés de plus de 65 ans et de ceux âgés de 18 à 65 ans.
Enfants et adolescents
Aucune différence des réponses pharmacocinétiques n'a été établie entre les participants adultes et pédiatriques de l'étude portant sur la DIP.
Polyradiculonévrite inflammatoire démyélinisante chronique (PIDC)
Dans l'étude PATH, les participants à l'étude (n = 172) ont atteint des taux résiduels durables sur une période de 24 semaines, lorsqu'ils ont reçu des doses hebdomadaires de 0,2 g/kg de poids corporel ou de 0,4 g/kg de poids corporel. Les concentrations résiduelles moyennes (ET) d'IgG après un traitement par Hizentra s'élevaient dans le groupe sous 0,4 g/kg de poids corporel à 20,4 (3,24) g/l et dans le groupe sous 0,2 g/kg de poids corporel à 15,4 (3,06) g/l. Les simulations basées sur des modèles pharmacocinétiques de population dans l'étude PATH suggéraient qu'une exposition similaire aux IgG (Cmax, AUC0-14 jours, Cmin, 14 jours) est obtenue lorsque les participants à l'étude atteints de PIDC reçoivent toutes les deux semaines le double de la dose d'Hizentra. Ces simulations suggèrent en outre que chez la population de patients atteints de PIDC, une exposition comparable aux IgG est par conséquent obtenue lorsque la dose d'entretien hebdomadaire de Hizentra est fractionnée en plusieurs doses plus fréquentes (2 à 7 fois par semaine).
Enfants et adolescents
À ce jour, le traitement par Hizentra des patients pédiatriques de moins de 18 ans atteints de PIDC n'a pas été étudié dans des études cliniques.
Patients âgés
Dans l'ensemble, aucune différence n'a été observée dans les paramètres pharmacocinétiques des patients atteints de PIDC âgés de plus de 65 ans et de ceux âgés de 18 à 65 ans.
Métabolisme
Absence de données.
Élimination
Les IgG et les complexes d'IgG sont dégradés dans les cellules du système réticulo-endothélial.
Données précliniquesLes immunoglobulines sont des constituants normaux du corps humain. La L-proline est un acide aminé physiologique non essentiel.
La sécurité d'Hizentra a été testée dans plusieurs études précliniques, avec une attention particulière pour l'excipient L-proline. Les données précliniques obtenues sur la base des études de pharmacologie de sécurité et de toxicologie n'ont pas révélé de risque particulier pour l'homme.
Remarques particulièresIncompatibilités
Ce médicament ne doit pas être mélangé avec d'autres médicaments.
Influence sur les méthodes de diagnostic
Après injection d'immunoglobulines, l'augmentation transitoire de la concentration de divers anticorps transférés passivement dans le sang des patients peut être responsable de résultats faussement positifs lors des dosages sérologiques.
La transmission passive d'anticorps anti-érythrocytaires, tels que les anticorps anti-A, anti-B ou anti-D, peut interférer avec certains tests sérologiques de recherche d'allo-anticorps contre les globules rouges (par exemple le test de Coombs), le comptage des réticulocytes et le test à l'haptoglobine.
Pour les interactions avec les vaccins à virus vivants atténués, voir le chapitre "Interactions" .
Stabilité
La date limite de conservation d'Hizentra est indiquée sur l'étiquette et l'emballage extérieur sous la mention "EXP" . Le médicament ne doit pas être utilisé au-delà de la date figurant après la mention "EXP" sur l'emballage.
Stabilité après ouverture
Hizentra est destiné à un usage unique. Comme la solution ne contient pas de conservateur, Hizentra doit être utilisé/perfusé dès que possible après ouverture du flacon.
Remarques particulières concernant le stockage
Ne pas conserver au-dessus de 25 °C. Ne pas congeler.
Conserver le flacon dans l'emballage d'origine afin de protéger le contenu de la lumière.
Tenir hors de portée des enfants.
Remarques concernant la manipulation
Hizentra est une solution prête à l'emploi présentée dans des flacons à usage unique.
Le médicament doit être amené à température ambiante ou corporelle avant utilisation.
La solution doit être claire et de couleur jaune pâle ou brun clair. Ne pas utiliser de solution trouble ou contenant des particules.
Tout produit non utilisé ou déchet doit être éliminé conformément à la réglementation en vigueur.
Numéro d’autorisation61547 (Swissmedic).
Présentation1 × 5 ml (1 g) flacon perforable (B)
1 × 10 ml (2 g) flacon perforable (B)
1 × 20 ml (4 g) flacon perforable (B)
1 × 50 ml (10 g) flacon perforable (B)
Titulaire de l’autorisationCSL Behring AG, Berne.
Mise à jour de l’informationOctobre 2022
|