Propriétés/effetsCode ATC
C09XX01
Mécanisme d’action
Le sparsentan est un antagoniste double des récepteurs de l’endothéline et de l’angiotensine.
Il s’agit d’une molécule unique, qui agit en tant qu’antagoniste à forte affinité et à double action à la fois du récepteur de type A de l’endothéline (ETAR) et du récepteur de type 1 de l’angiotensine (AT1R). L’endothéline 1 (via ETAR) et l’angiotensine II (via AT1R) interviennent dans des processus qui conduisent à la progression de la NIgA, tels que les effets hémodynamiques et la prolifération des cellules mésangiales, l’expression et l’activité accrues des médiateurs pro-inflammatoires et profibrotiques, les lésions des podocytes et le stress oxydant. Le sparsentan inhibe l’activation d’ETAR et d’AT1R, réduisant ainsi la protéinurie et ralentissant la progression de la maladie rénale.
Pharmacodynamique
Dans une étude randomisée comprenant un groupe comparateur actif et un groupe placebo et menée auprès de sujets sains, le sparsentan a entraîné un léger allongement de l’intervalle QTcF, avec un effet maximal de 8,8 ms (IC à 90 %: 5,9; 11,8) à une dose de 800 mg et de 8,1 ms (IC à 90 %: 5,2; 11,0) à une dose de 1600 mg. Dans une autre étude menée auprès de sujets sains avec une exposition au sparsentan correspondant à plus de 2 fois la dose maximale recommandée chez l’être humain, aucun allongement significatif de l’intervalle QTcF n’a été observé. L’effet maximal était de 8,3 ms (6,69; 9,90). Il est donc peu probable que le sparsentan ait un effet cliniquement significatif sur l’allongement de l’intervalle QT.
Efficacité clinique
L’efficacité et la sécurité du sparsentan ont été évaluées dans l’étude PROTECT menée auprès de patients atteints d’une NIgA.
L’étude PROTECT est une étude de phase III internationale, multicentrique, randomisée, en double aveugle (110 semaines) et contrôlée contre comparateur actif qui a été menée auprès de patients atteints d’une NIgA. Ont participé à cette étude des patients âgés de ≥ 18 ans, dont 15 patients (8 %) traités par sparsentan âgés de > 65 ans, présentant un DFGe ≥ 30 ml/min/1,73 m2 et une protéinurie totale ≥ 1,0 g/jour. Avant la participation à l’étude, les patients ont reçu la dose maximale tolérée d’un IEC et/ou d’un ARA pendant au moins 3 mois. Le traitement par IEC et/ou par ARA a été arrêté avant l’instauration du sparsentan. Les patients présentant un taux de potassium supérieur à 5,5 mmol/l à l’inclusion ont été exclus.
Au total, 404 patients ont été randomisés et ont reçu soit le sparsentan (n = 202) soit l’irbésartan (n = 202). Le traitement a été instauré à la dose de 200 mg de sparsentan une fois par jour ou de 150 mg d’irbésartan une fois par jour. Après 14 jours, la dose devait être augmentée selon la tolérance jusqu’à la dose recommandée de 400 mg de sparsentan une fois par jour ou de 300 mg d’irbésartan une fois par jour. La tolérance a été définie comme une pression artérielle systolique > 100 mmHg et une pression artérielle diastolique > 60 mmHg après deux semaines et l’absence d’EI (p. ex. aggravation d’un œdème) ou en fonction des résultats d’analyse biologique (p. ex. kaliémie > 5,5 mEq/l [5,5 mmol/l]). Les inhibiteurs du SRAA ou du système endothéline étaient interdits pendant l’étude. D’autres classes d’agents antihypertenseurs étaient autorisées en fonction des besoins, pour atteindre la pression artérielle cible. Un traitement par agents immunosuppresseurs était autorisé pendant l’étude, à la discrétion de l’investigateur.
Les données à l’inclusion en matière de DFGe et de protéinurie étaient comparables entre les groupes de traitement. La population totale présentait un DFGe moyen (ET) de 57 (24) ml/min/1,73 m2 et un rapport protéinurie/créatininurie (UP/C) médian de 1,24 g/g (écart interquartile: 0,83; 1,77). L’âge moyen était de 46 ans (étendue de 18 à 76 ans); 70 % des patients étaient de sexe masculin, 67 % étaient blancs, 28 % étaient asiatiques, 1 % étaient noirs ou afro-américains et 3 % appartenaient à une autre ethnie.
L’analyse principale (intermédiaire) de la protéinurie a été menée 36 semaines après la randomisation d’environ 280 patients, afin de déterminer si l’effet du traitement sur le critère d’évaluation principal de l’efficacité, à savoir la variation du rapport UP/C entre l’inclusion et la semaine 36, était statistiquement significatif. Le critère d’évaluation principal de l’étude, à savoir la variation du rapport UP/C entre l’inclusion et la semaine 36, a été satisfait. La moyenne géométrique du rapport UP/C à la semaine 36 était de 0,62 g/g dans le groupe sparsentan, contre 1,07 g/g dans le groupe irbésartan. La moyenne géométrique des moindres carrés pour la variation en pourcentage du rapport UP/C entre l’inclusion et la semaine 36 était de -49,8 % (intervalle de confiance [IC] à 95 %: -54,98; -43,95) dans le groupe sparsentan, contre -15,1 % (IC à 95 %: -23,72; -5,39) dans le groupe irbésartan (p < 0,0001; Figure 1). Lors de l’analyse finale, le sparsentan a montré un effet antiprotéinurique rapide et durable sur une période de 2 ans, avec une moyenne géométrique pour le rapport UP/C à la semaine 110 de 0,64 g/g dans le groupe sparsentan contre 1,09 g/g dans le groupe irbésartan, ce qui représente une réduction moyenne de 43 % par rapport à l’inclusion (IC à 95 %: -49,75; -34,97), contre seulement 4,4 % avec l’irbésartan (IC à 95 %: -15,84; 8,70). Une amélioration en matière de réduction de la protéinurie a été observée durablement dans le groupe sparsentan après 4 semaines et jusqu’à la semaine 110 (Figure 1).
Figure 1: Évolution en pourcentage du rapport protéinurie/créatininurie par rapport à l’inclusion, par visite (étude PROTECT)
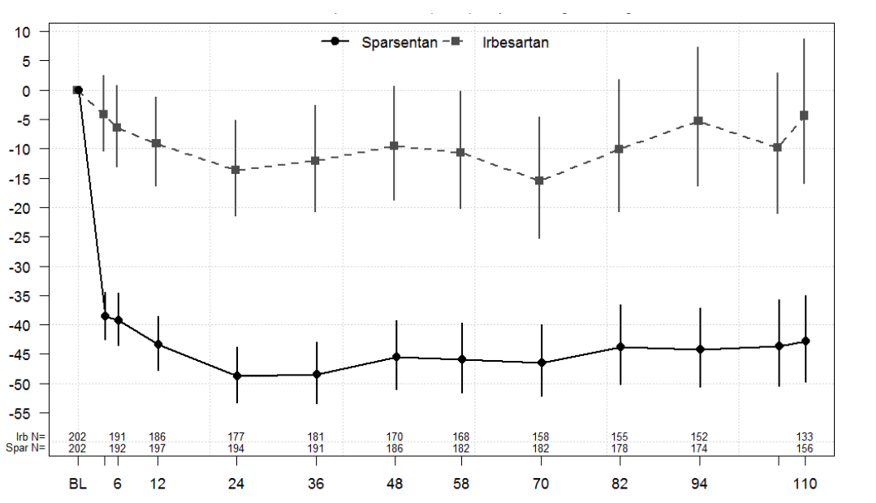
Remarques: la moyenne géométrique des moindres carrés ajustée pour le rapport UP/C par rapport à l’inclusion était basée sur un modèle longitudinal à mesures répétées, stratifié en fonction du DFGe et de la protéinurie à la sélection, avec les valeurs exprimées en pourcentage d’évolution avec leur IC à 95 % respectif. L’analyse inclut les données relatives au rapport UP/C recueillies pendant la phase en double aveugle auprès de tous les patients randomisés ayant reçu au moins une dose de médicament à l’étude. La valeur à l’inclusion a été définie comme la dernière observation non manquante qui a été réalisée avant/pendant l’administration de la première dose.
Abréviations: IC = intervalle de confiance; DFGe = débit de filtration glomérulaire estimé; UP/C = rapport protéinurie/créatininurie.
DFG estimé
Lors de l’analyse confirmatoire, l’amélioration de la pente durable du DFGe à 2 ans (à partir de 6 semaines de traitement) était de 1,1 ml/min/1,73 m2 par an avec le sparsentan par rapport à l’irbésartan (IC à 95 %: -0,07, 2,12; p = 0,037), et l’amélioration correspondante de la pente totale du DFGe à 2 ans (à partir de l’inclusion) était de 1,0 ml/min/1,73 m2 par an (IC à 95 %: -0,03, 1,94; p = 0,058). La variation absolue par rapport à l’inclusion pour le DFGe à 2 ans était de -5,8 ml/min/1,73 m2 (IC à 95 %: -7,38; -4,24) pour le sparsentan, contre -9,5 ml/min/1,73 m2 (IC à 95 %: -11,17; -7,89) pour l’irbésartan.
Informations supplémentaires
Deux études à grande échelle randomisées et contrôlées (ONTARGET [ONgoing Telmisartan Alone and in combination with Ramipril Global Endpoint Trial] et VA NEPHRON-D [The Veterans Affairs Nephropathy in Diabetes]) ont porté sur l’utilisation de l’association d’un IEC et d’un antagoniste des récepteurs de l’angiotensine II. L’étude ONTARGET a été menée auprès de patients présentant des antécédents de maladies cardiovasculaires ou cérébrovasculaires ou de diabète de type 2, et chez lesquels des signes de lésions des organes cibles étaient présents. L’étude VA NEPHRON-D a été menée chez des patients présentant un diabète de type 2 et une néphropathie diabétique. Ces études n’ont pas montré d’effet positif significatif sur les résultats rénaux et/ou cardiovasculaires ni sur la mortalité, tandis qu’un risque accru d’hyperkaliémie, d’insuffisance rénale aiguë et/ou d’hypotension a été observé par rapport à la monothérapie. Compte tenu de leurs propriétés pharmacodynamiques similaires, ces résultats sont aussi pertinents pour les autres IEC et antagonistes des récepteurs de l’angiotensine II. Par conséquent, aucun traitement concomitant par des IEC et des antagonistes des récepteurs de l’angiotensine II ne doit être utilisé chez les patients présentant une néphropathie diabétique. L’étude ALTITUDE (Aliskiren Trial in Type 2 Diabetes Using Cardiovascular and Renal Disease Endpoints) a été conçue pour évaluer les bénéfices de l’ajout d’aliskirène à un traitement standard à base d’IEC ou d’antagoniste des récepteurs de l’angiotensine II chez des patients présentant un diabète de type 2 ainsi qu’une maladie rénale chronique, une maladie cardiovasculaire ou les deux. Il a été mis un terme prématuré à cette étude en raison d’un risque accru d’effets indésirables. Les décès d’origine cardiovasculaire et les accidents vasculaires cérébraux ont été numériquement plus fréquents dans le groupe aliskirène que dans le groupe placebo, et des événements indésirables et des événements indésirables graves d’intérêt (hyperkaliémie, hypotension ou altérations de la fonction rénale) ont également été observés plus fréquemment dans le groupe aliskirène que dans le groupe placebo.
|