ZusammensetzungWirkstoffe
Risdiplamum.
Hilfsstoffe
Pulver zur Herstellung einer Lösung zum Einnehmen
Mannitolum, isomaltum (E953) 2,97 mg/1 ml, acidum tartaricum, natrii benzoas (E211) 0,38 mg/1 ml, macrogolum 6000, sucralosum, acidum ascorbicum, dinatrii edetas, aromatica (Erdbeeraroma) cum maltodextrinum et amylum modificatum (E1450).
80-ml-zubereitete Lösung Evrysdi enthalten 7,2 mg Natrium.
1 ml gebrauchsfertige Lösung enthält 0,09 mg Natrium.
Filmtabletten
Tablettenkern:
Acidum tartaricum, mannitolum, cellulosum microcristallinum, silica colloidalis anhydrica, crospovidonum, natrii stearylis fumaras (entspricht 0,22 mg Natrium), aromatica (Erdbeeraroma) cum maltodextrinum et amylum modificatum (E1450) pro compresso obducto.
Filmüberzug:
Poly(alcohol vinylicus), titanii dioxidum (E171), macrogolum 3350, talcum, ferrum oxydatum flavum (E172).
Darreichungsform und Wirkstoffmenge pro EinheitPulver zur Herstellung einer Lösung zum Einnehmen
Evrysdi 0,75 mg/ml Pulver zur Herstellung einer Lösung zum Einnehmen ist gelblich bis grüngelblich und befindet sich in einer Braunglasflasche. Eine Flasche enthält 60 mg Risdiplam in 2 g Pulver.
Nach Zubereitung der Lösung zum Einnehmen mit gereinigtem Wasser oder Wasser für Injektionszwecke beträgt das Endvolumen 80 ml. 1 ml Lösung enthält 0,75 mg Risdiplam (siehe Rubrik "Sonstige Hinweise" , "Hinweise für die Handhabung" ).
Filmtabletten
Blassgelbe, runde und gewölbte Filmtablette mit der Prägung EVR auf einer Seite.
1 Filmtablette enthält 5 mg Risdiplam.
Indikationen/AnwendungsmöglichkeitenEvrysdi ist für die Behandlung der 5q-assoziierten spinalen Muskelatrophie (SMA) bei pädiatrischen und erwachsenen Patienten indiziert.
Dosierung/AnwendungDie Einleitung und Überwachung der Behandlung mit Evrysdi muss durch Ärzte mit Erfahrung in der Diagnostik und Behandlung von Patienten mit spinaler Muskelatrophie erfolgen.
In das klinische Entwicklungsprogramm wurden keine Patienten mit SMA Typ IV eingeschlossen.
Übliche Dosierung
Pulver zur Herstellung einer Lösung zum Einnehmen
Die Evrysdi-Lösung zum Einnehmen muss vor der Abgabe von einer medizinischen Fachperson, wie z.B. Arzt oder Apotheker bzw. Ärztin oder Apothekerin zubereitet werden.
Es muss sichergestellt werden, dass eine medizinische Fachperson mit dem Patienten bzw. einer Betreuungsperson bespricht, wie die verschriebene tägliche Dosis vorzubereiten und einzunehmen ist, bevor die erste Dosis verabreicht wird (siehe Rubrik "Sonstige Hinweise" , "Hinweise für die Handhabung" ).
Evrysdi-Lösung wird einmal täglich eingenommen bzw. verabreicht, jeden Tag ungefähr zu derselben Tageszeit, mit den in der Packung enthaltenen wiederverwendbaren Spritzen für die orale Verabreichung, mit oder ohne eine Mahlzeit.
Die empfohlene, einmal täglich einzunehmende Dosis Evrysdi zur Behandlung der SMA richtet sich nach dem Alter und Körpergewicht des Patienten (siehe Tabelle 1).
Evrysdi Pulver zur Herstellung einer Lösung zum Einnehmen ist für alle Altersgruppen verfügbar.
Filmtabletten
Neben der Evrysdi-Lösung zum Einnehmen ist für Patienten, denen die 5-mg-Dosis verschrieben wurde, auch eine Tablettenformulierung von Evrysdi verfügbar.
Eine medizinische Fachperson sollte mit dem Patienten bzw. einer Betreuungsperson besprechen, wie die verschriebene tägliche Dosis vorzubereiten und einzunehmen ist, bevor die erste Dosis verabreicht wird (siehe Rubrik "Sonstige Hinweise" , "Hinweise für die Handhabung" ).
Evrysdi Filmtabletten werden einmal täglich eingenommen, jeden Tag ungefähr zu derselben Tageszeit mit oder ohne eine Mahlzeit.
Die empfohlene, einmal täglich einzunehmende Dosis Evrysdi zur Behandlung der SMA richtet sich nach dem Alter und Körpergewicht des Patienten (siehe Tabelle 1).
Tabelle 1: Dosierungsschema nach Alter und Körpergewicht
Altera und Körpergewic Empfohlene Tagesdosi Darreichungsform
ht s
<2 Monate 0,15 mg/kg Pulver zur Herstellung einer Lösung zum Einnehmen
Alter 2 Monate bis <2 0,20 mg/kg
Jahre
Alter ≥2 Jahre (<20 0,25 mg/kg
kg)
Alter ≥2 Jahre (≥20 5 mg Pulver zur Herstellung einer Lösung zum Einnehmen
kg) oder Filmtablette
a Basierend auf dem korrigierten Alter für Frühgeborene.
Der Arzt bzw. die Ärztin sollte die geeignete Darreichungsform je nach der erforderlichen Dosis und den Patientenbedürfnissen verschreiben. Für Patienten, die Schwierigkeiten haben, eine ganze Tablette zu schlucken, kann die Tablette dispergiert oder das Pulver zur Herstellung einer Lösung zum Einnehmen verschrieben werden.
Dosisänderungen müssen unter Aufsicht einer medizinischen Fachperson erfolgen. Die Behandlung mit einer höheren Tagesdosis als 5 mg wurde bisher nicht untersucht. Zu Säuglingen unter 16 Tagen liegen nur sehr begrenzte Sicherheitsdaten aus der Beobachtung nach dem Inverkehrbringen vor, die mit Evrysdi mit der empfohlenen Dosis behandelt wurden. (Siehe Rubrik "Pharmakokinetik" , Kinder und Jugendliche).
Patienten mit Leberfunktionsstörungen
Bei Patienten mit gering- bis mittelgradiger Leberfunktionsstörung ist keine Dosisanpassung erforderlich. Bei Patienten mit schwerer Leberfunktionsstörung ist Evrysdi bisher nicht untersucht worden (siehe Rubrik "Pharmakokinetik" , Kinetik spezieller Patientengruppen).
Patienten mit Nierenfunktionsstörungen
Die Sicherheit und Wirksamkeit von Evrysdi bei Patienten mit Nierenfunktionsstörungen sind bisher nicht untersucht worden. Es ist nicht zu erwarten, dass bei Patienten mit Nierenfunktionsstörungen eine Dosisanpassung erforderlich wird (siehe Rubrik "Pharmakokinetik" , Kinetik spezieller Patientengruppen).
Ältere Patienten
Die Pharmakokinetik (PK) und die Sicherheit von Evrysdi wurden bei Personen bis zu 69 Jahren ohne SMA untersucht. Evrysdi wurde nicht bei SMA-Patienten über 60 Jahren untersucht (siehe Rubrik "Pharmakokinetik" , Ältere Patienten).
Kinder und Jugendliche
Die Anwendung von Evrysdi bei SMA bei Patienten im Alter von 2 Monaten und jünger wird durch pharmakokinetische und Sicherheitsdaten von pädiatrischen Patienten im Alter von 16 Tagen und älter unterstützt (siehe Rubriken "Unerwünschte Wirkungen" , "Klinische Wirksamkeit" und "Pharmakokinetik" ). Es gibt keine klinischen oder pharmakokinetischen Daten in Frühgeborenen sowie in Neugeborenen unter 16 Tagen (siehe Rubrik "Klinische Wirksamkeit" ).
Verspätete Dosisgabe
Evrysdi wird einmal täglich eingenommen, jeden Tag ungefähr zu derselben Tageszeit. Wenn die Einnahme vergessen wird, ist diese so schnell wie möglich nachzuholen, wenn der reguläre Einnahmezeitpunkt weniger als 6 Stunden zurückliegt. Andernfalls ist die vergessene Dosis auszulassen und zum nächsten regulären Einnahmezeitpunkt die normale Dosis einzunehmen.
Wenn eine Dosis nicht vollständig geschluckt wird oder der Patient nach der Einnahme von Evrysdi erbricht, ist keine zusätzliche Dosis zum Ausgleich der unvollständigen Dosis zu verabreichen. Erst am nächsten Tag ist zum nächsten regulären Einnahmezeitpunkt die normale Dosis zu verabreichen.
Art der Anwendung
Pulver zur Herstellung einer Lösung zum Einnehmen
Für die Gabe der täglichen Dosis Evrysdi ist die in der Packung enthaltene wiederverwendbare Spritze für die orale Verabreichung zu verwenden.
Auswahl der zur verordneten Tagesdosis passenden wiederverwendbaren Spritze für die orale Verabreichung:
Tabelle 2: Auswahl der für die verordnete Tagesdosis Evrysdi passenden Spritze für die orale Verabreichung
Spritzengrösse Dosisvolumen Spritzen-Volumenschritte
1 ml 0,3 ml bis 1,0 ml 0,01 ml
6 ml 1,0 ml bis 6,0 ml 0,1 ml
12 ml 6,2 ml bis 6,6 ml 0,2 ml
Bei der Berechnung des Dosisvolumens sind auch die Volumenschritte der Spritze für die orale Verabreichung zu bedenken. Das Dosisvolumen wird auf die nächstliegende Volumenmarkierung der ausgewählten Spritze für die orale Verabreichung auf- oder abgerundet (z.B. 6,30 ml auf 6,4 ml, 3,03 ml auf 3,0 ml und 1,05 ml auf 1,1 ml).
Der Patient soll die Evrysdi-Lösung einnehmen, sobald sie in die wiederverwendbare Spritze für die orale Verabreichung aufgezogen wurde. Wenn der Inhalt der Spritze nicht innerhalb von 5 Minuten eingenommen bzw. verabreicht wird, ist er zu entsorgen (siehe Rubrik "Entsorgung unverbrauchter/nicht mehr haltbarer Arzneimittel" ) und eine neue Dosis vorzubereiten.
Evrysdi ist mit oder ohne Mahlzeit zu verabreichen. Der Patient sollte nach der Einnahme von Evrysdi etwas Wasser trinken, um sicherzustellen, dass das Arzneimittel vollständig heruntergeschluckt wurde. Wenn der Patient nicht in der Lage ist zu schlucken und eine nasogastrale Sonde oder ein Gastrostomiekatheter vorhanden ist, ist Evrysdi über die Sonde bzw. den Schlauch zu verabreichen. Die Sonde/der Schlauch ist nach der Verabreichung von Evrysdi mit Wasser durchzuspülen (siehe Rubrik "Sonstige Hinweise" , "Hinweise für die Handhabung" ).
Filmtabletten
Die Evrysdi Filmtablette soll unzerkaut mit Wasser geschluckt oder in einer kleinen Menge zimmerwarmem, nicht-chloriertem Trinkwasser (z.B. Trinkwasser aus der Flasche) dispergiert eingenommen werden (siehe Rubrik "Sonstige Hinweise, Inkompatibilitäten" und "Hinweise für die Handhabung" ). Die Tabletten dürfen nicht zerkaut, zerteilt oder zerdrückt werden.
Die in nicht-chloriertem Trinkwasser (z.B. Trinkwasser aus der Flasche) dispergierte Evrysdi Filmtablette ist umgehend einzunehmen.
Evrysdi Filmtabletten dürfen ausschliesslich in nicht-chloriertem Trinkwasser (z.B. Trinkwasser aus der Flasche) dispergiert werden.
Die zubereitete Dispersion ist zu verwerfen, wenn sie nicht innerhalb von 10 Minuten nach der Zugabe von Wasser verwendet wird. Die zubereitete Dispersion darf nicht dem Sonnenlicht ausgesetzt werden.
Wenn die zubereitete Dispersion verschüttet wird oder auf die Haut gelangt, den Bereich mit Wasser und Seife waschen.
Evrysdi Filmtabletten sollten nicht über eine transnasale oder eine Gastrostomiesonde verabreicht werden. Falls die Anwendung über eine transnasale oder eine Gastrostomiesonde erforderlich ist, sollte das Evrysdi Pulver zur Herstellung einer Lösung zum Einnehmen verwendet werden.
KontraindikationenEvrysdi ist kontraindiziert bei Patienten mit bekannter Überempfindlichkeit gegenüber Risdiplam oder einem der sonstigen Hilfsstoffe.
Warnhinweise und VorsichtsmassnahmenAllgemeines
In tierexperimentellen Studien wurden retinale Veränderungen, epitheliale Veränderungen insbesondere der Haut sowie des Gastrointestinaltrakts und Hinweise für eine Knochenmarkstoxizität (Veränderungen beim grossen Blutbild) beobachtet. Das Risiko, dass solche Veränderungen auch beim Menschen auftreten können, lässt sich nicht abschliessend beurteilen, da nur begrenzte Langzeitdaten zur Sicherheit vorliegen.
Embryofetale Toxizität
In tierexperimentellen Studien ist embryofetale Toxizität beobachtet worden (siehe Rubrik "Präklinische Daten" ). Fortpflanzungsfähige Patienten sind über die Risiken aufzuklären und müssen für die Dauer der Behandlung und bis mindestens 1 Monat nach der letzten Dosis Evrysdi (Frauen) bzw. 4 Monate nach der letzten Dosis Evrysdi (Männer) zuverlässige Kontrazeption anwenden (siehe Rubrik "Dosierung/Anwendung" ).
Potentielle Auswirkungen auf die männliche Fertilität
Aufgrund der reversiblen Auswirkungen von Evrysdi auf die männliche Fertilität, die in tierexperimentellen Studien beobachtet wurden, dürfen männliche Patienten für die Dauer der Behandlung und in den ersten 4 Monaten nach der letzten Dosis Evrysdi keine Samenspenden abgeben (siehe Rubrik "Pharmakokinetik" , Kinetik spezieller Patientengruppen und Rubrik "Präklinische Daten" ).
Weitere Hinweise
Hautkontakt mit dem Pulver und der gebrauchsfertigen Lösung zum Einnehmen ist zu vermeiden.
Sollte dennoch Arzneimittel (Pulver oder Lösung) auf Ihre Haut gelangen, waschen Sie das Areal mit Wasser und Seife.
Pulver zur Herstellung einer Lösung zum Einnehmen
Dieses Arzneimittel enthält 0,38 mg Natriumbenzoat pro 1 ml. Eine Zunahme des Bilirubingehalts im Blut nach Verdrängung von Albumin kann einen Neugeborenenikterus verstärken und zu einem Kernikterus (nicht-konjugierte Bilirubinablagerungen im Hirngewebe) führen.
Dieses Arzneimittel enthält weniger als 1 mmol Natrium (23 mg) pro Dosis, d.h. es ist nahezu "natriumfrei" .
Dieses Arzneimittel enthält Isomaltitol. Patienten mit der seltenen hereditären Fructose-Intoleranz sollten dieses Arzneimittel nicht anwenden.
Filmtabletten
Dieses Arzneimittel enthält weniger als 1 mmol Natrium (23 mg) pro Dosis, d.h. es ist nahezu "natriumfrei" .
InteraktionenWirkung von Evrysdi auf andere Arzneimittel
In vitro bewirkten Risdiplam und sein zirkulierender Hauptmetabolit M1 keine Induktion von CYP1A2, 2B6, 2C8, 2C9, 2C19 oder 3A4. In vitro bewirkten Risdiplam und M1 keine Inhibition (reversible oder zeitabhängige Inhibition) der untersuchten CYP-Isoenzyme (CYP1A2, 2B6, 2C8, 2C9, 2C19, 2D6) mit Ausnahme von CYP3A. Risdiplam ist ein schwacher Inhibitor von CYP3A. Bei gesunden, erwachsenen Probanden führte die Gabe von Risdiplam einmal täglich über 2 Wochen zu einem leichten Anstieg der Exposition gegenüber Midazolam, einem hochempfindlichen CYP3A-Substrat (11 % höhere AUC, 16 % höhere Cmax). Das Ausmass dieser Interaktion wird nicht als klinisch bedeutsam erachtet, daher ist keine Dosisanpassung für CYP3A-Substrate erforderlich. Der physiologiebasierten Pharmakokinetik-(PBPK-) Modellierung nach zu urteilen, ist bei Kindern und Säuglingen ab 2 Monaten eine vergleichbare Grössenordnung dieses Effekts zu erwarten.
In-vitro-Studien belegen, dass Risdiplam und sein Hauptmetabolit keine signifikanten Inhibitoren von humanem MDR1, Organo-Anion-Transporter-Polypeptiden (OATP) 1B1, OATP1B3 sowie Organische-Anionen-Transportern 1 und 3 (OAT 1 und 3) sind. Risdiplam und sein Hauptmetabolit inhibieren in vitro jedoch den humanen Organische-Kationen-Transporter 2 (OCT2) sowie Multidrug-und-Toxin-Extrusion (MATE) 1 und MATE2-K. In therapeutischen Wirkstoffkonzentrationen sind keine Interaktionen mit OCT2-Substraten zu erwarten. Die Auswirkung der gleichzeitigen Anwendung von Risdiplam auf die Pharmakokinetik von MATE1- und MATE2-K-Substraten beim Menschen ist nicht bekannt. Ausgehend von den In-vitro-Daten könnte Evrysdi die Plasmakonzentrationen von Wirkstoffen erhöhen, die mittels MATE1- oder MATE2-K eliminiert werden, beispielsweise Metformin (siehe Rubrik "Pharmakokinetik" ). Ist eine gemeinsame Anwendung nicht zu vermeiden, sind die Therapie-assoziierten Toxizitäten zu überwachen, und gegebenenfalls ist eine Dosisreduktion des gleichzeitig angewendeten Wirkstoffs in Betracht zu ziehen.
Wirkung anderer Arzneimittel auf Evrysdi
Die Verstoffwechselung von Risdiplam erfolgt primär durch die Flavin-haltigen Monooxygenasen 1 und 3 (FMO1 und FMO3) sowie durch die CYP-Isoenzyme 1A1, 2J2, 3A4 und 3A7. Risdiplam ist kein Substrat des humanen Multidrug-Resistenz-Proteins 1 (MDR1).
Bei gleichzeitiger Anwendung von 200 mg Itraconazol zweimal täglich, einem starken CYP3A-Hemmer, und einer oralen Einzeldosis von 6 mg Risdiplam zeigte sich keine klinisch bedeutsame Auswirkung auf die Pharmakokinetik (PK) von Risdiplam (11 % grössere AUC, 9 % niedrigere Cmax). Bei gleichzeitiger Anwendung von Evrysdi mit einem CYP3A-Inhibitor sind keine Dosisanpassungen erforderlich.
Über den FMO1- und FMO3-Signalweg sind keine Arzneimittelinteraktionen zu erwarten.
Omeprazol, ein Protonenpumpenhemmer, hatte keinen Einfluss auf die Pharmakokinetik von Risdiplam, das als Tablette verabreicht wurde. Die Risdiplam-Tablette kann daher gleichzeitig mit Protonenpumpenhemmer, H2-Rezeptorantagonisten und Antazida verabreicht werden.
Schwangerschaft, StillzeitPräklinischen Studienergebnissen zufolge kann die männliche Fertilität während der Behandlung mit Evrysdi beeinträchtigt sein. In den Fortpflanzungsorganen von Ratten und Affen war eine Degeneration der Spermien und verminderte Anzahl der Spermien zu beobachten (siehe Rubrik "Präklinische Daten" ).
Vor Behandlungsbeginn sind mit Männern, die Evrysdi erhalten sollen, Strategien zur Erhaltung der Fertilität zu besprechen. Männliche Patienten können die Einlagerung von Sperma vor Behandlungsbeginn in Betracht ziehen. Männliche Patienten, die ein Kind zeugen wollen, sollen die Behandlung mit Evrysdi für mindestens 4 Monate unterbrechen (siehe Rubrik "Warnhinweise und Vorsichtsmassnahmen" ).
Den präklinischen Studienergebnissen zufolge sind keine Auswirkungen von Evrysdi auf die weibliche Fertilität zu erwarten (siehe Rubrik "Präklinische Daten" ).
Bei Frauen, die schwanger werden könnten, ist vor Beginn der Behandlung mit Evrysdi der Schwangerschaftsstatus zu untersuchen.
Fortpflanzungsfähige Patienten (männlich und weiblich) sollten die folgenden Anforderungen an die Empfängnisverhütung einhalten:
-Männliche Patienten und ihre Partnerinnen, wenn diese schwanger werden könnten, sollen für die Dauer der Behandlung und mindestens 4 Monate nach der letzten Dosis Evrysdi zuverlässige Kontrazeption anwenden.
-Patientinnen, die schwanger werden könnten, sollen für die Dauer der Behandlung und bis mindestens 1 Monat nach der letzten Dosis Evrysdi zuverlässige Kontrazeption anwenden.
Schwangerschaft
Es liegen keine klinischen Daten mit Anwendung bei Schwangeren vor.
In tierexperimentellen Studien hat Risdiplam embryofetotoxische und teratogene Wirkung gezeigt. Den Ergebnissen der tierexperimentellen Studien nach zu urteilen, passiert Risdiplam die Plazentaschranke und kann den Fetus schädigen (siehe Rubrik "Präklinische Daten" ).
Evrysdi darf während der Schwangerschaft nicht angewendet werden, es sei denn, dies ist eindeutig erforderlich. Wenn eine schwangere Frau mit Evrysdi behandelt werden muss, ist sie nachdrücklich auf die möglichen Risiken für den Fetus hinzuweisen.
Stillzeit
Es ist nicht bekannt, ob Evrysdi in die Muttermilch übertritt. Studien bei Ratten ergaben, dass Risdiplam in die Milch übertritt (siehe Rubrik "Präklinische Daten" ). Da das Potenzial für eine Schädigung des gestillten Säuglings nicht bekannt ist, muss gemeinsam mit dem behandelnden Arzt der Patientin eine Entscheidung getroffen werden. Es wird empfohlen, während der Behandlung mit Evrysdi nicht zu stillen.
Wirkung auf die Fahrtüchtigkeit und auf das Bedienen von MaschinenDie Wirkung von Evrysdi auf die Fahrtüchtigkeit oder die Fähigkeit, Maschinen zu bedienen, wurde nicht in entsprechenden Studien untersucht.
Unerwünschte WirkungenZur Klassifikation der Häufigkeit von unerwünschten Wirkungen wurden die folgenden Definitionen verwendet: Sehr häufig (≥1/10); häufig (≥1/100< 1/10); gelegentlich (≥1/1000< 1/100); selten (≥1/10'000< 1/1000); sehr selten (< 1/10'000); nicht bekannt (auf Grundlage der verfügbaren Daten nicht abschätzbar).
Zusammenfassung des Sicherheitsprofils
Bei Patienten mit infantiler SMA waren die unerwünschten Reaktionen, die im Rahmen der klinischen Studien mit Evrysdi am häufigsten beobachtet wurden Pyrexie (54,8 %), Ausschlag (29,0 %) und Diarrhoe (19,4 %).
Bei Patienten mit einer SMA mit späterem Krankheitsbeginn waren die unerwünschten Reaktionen, die im Rahmen der klinischen Studien mit Evrysdi am häufigsten beobachtet wurden, Pyrexie (21,7 %), Kopfschmerzen (20,0 %), Diarrhoe (16,7 %) und Ausschlag (16,7 %).
Die oben genannten unerwünschten Reaktionen traten ohne erkennbares klinisches oder zeitliches Muster auf und klangen trotz Fortführung der Behandlung mit Evrysdi bei den Patienten mit infantiler SMA und SMA mit späterem Krankheitsbeginn im Allgemeinen wieder ab.
Tabelle 3: Zusammenfassung der unerwünschten Arzneimittelwirkungen bei Patienten mit infantiler SMA und SMA mit späterem Krankheitsbeginn in den klinischen Studien mit Evrysdi
Systemorganklasse Infantile SMA2 (Typ SMA mit späterem Krankheitsbeginn3
1) (Typ 2 und 3)
Erkrankungen des Gastrointestinaltrakts
Diarrhoe Sehr häufig Sehr häufig
Nausea Nicht zutreffend Häufig
Mundgeschwüre und aphthöse Geschwüre Häufig Häufig
Erkrankungen der Haut und des
Unterhautgewebes
Ausschlag1 Sehr häufig Sehr häufig
Erkrankungen des Nervensystems
Kopfschmerzen Nicht zutreffend Sehr häufig
Allgemeine Erkrankungen und Beschwerden
am Verabreichungsort
Pyrexie (einschliesslich Hyperpyrexie) Sehr häufig Sehr häufig
Infektionen und parasitäre Erkrankungen
Infektion des Harntrakts (einschliesslich Häufig Häufig
Zystitis)
Skelettmuskulatur-, Bindegewebs-und
Knochenerkrankungen
Arthralgie Nicht zutreffend Häufig
1 Umfasst Dermatitis, akneiforme Dermatitis, allergische Dermatitis, Erythem, Follikulitis, Ausschlag, erythematöser Ausschlag, makulopapulöser Ausschlag, papulöser Ausschlag.
2 Bei den Patienten mit infantiler SMA (FIREFISH Teil 1 und 2) sind unerwünschte Reaktionen definiert als Ereignisse, die bei ≥2 % der Patienten aufgetreten sind und bei denen ein Kausalzusammenhang mit Evrysdi möglich ist.
3 Bei den Patienten mit SMA mit späterem Krankheitsbeginn (SUNFISH Teil 2) sind unerwünschte Reaktionen definiert als Ereignisse, die im doppelblinden, placebokontrollierten Behandlungsabschnitt bei den mit Evrysdi behandelten Patienten um ≥2 % häufiger aufgetreten sind als bei den Patienten unter Placebo und bei denen ein Kausalzusammenhang mit Evrysdi möglich ist.
Die verfügbaren Sicherheitsdaten sind hinsichtlich der Anzahl der mit Evrysdi exponierten Patienten als auch der Länge der Exposition limitiert. Es kann sein, dass potenzielle, relativ seltene und möglicherweise schwerwiegende unerwünschte Arzneimittelwirkungen (UAWs) im Studienprogramm unerkannt blieben.
Es liegen begrenzte Daten zu Sicherheit von Neugeborenen und Säuglingen mit einer präsymptomatischen SMA aus der Studie RAINBOWFISH vor.
Die RAINBOWFISH-Studie ist eine offene, einarmige Studie, an der 26 Patienten mit präsymptomatischer SMA im Alter zwischen 16 und 41 Tagen (Gewichtsspanne 3,1 bis 5,7kg) bei der ersten Dosis aufgenommen wurden. Bei der primären Analyse betrug die mediane Expositionsdauer 20,4 Monate (Bereich: 10,6 bis 41,9 Monate) (siehe Rubrik "Eigenschaften/Wirkungen" , Klinische Wirksamkeit).
Das Sicherheitsprofil von Evrysdi in präsymptomatischen Patienten in der RAINBOWFISH Studie ist vergleichbar mit dem Sicherheitsprofil in symptomatischen SMA Patienten, welche im Rahmen der klinischen Studien mit Evrysdi behandelt wurden. Es liegen noch keine Langzeitdaten vor.
Sicherheitsprofil bei mit anderen modifizierenden Therapien vorbehandelten SMA-Patienten
Basierend auf der primären Analyse von Probanden der JEWELFISH-Studie deckt sich das Sicherheitsprofil von Evrysdi bei bis zu 59 Monate lang mit Evrysdi behandelten nicht-therapienaiven Patienten in der Studie JEWELFISH mit dem Sicherheitsprofil bei therapienaiven SMA-Patienten, die in den Studien FIREFISH (Teil 1 und Teil 2), SUNFISH (Teil 1 und Teil 2) und RAINBOWFISH mit Evrysdi behandelt wurden. In die JEWELFISH-Studie wurden zuvor mit Nusinersen (n = 76) oder mit Onasemnogene Abeparvovec (n = 14) vorbehandelte Patienten aufgenommen (siehe Rubrik "Eigenschaften/Wirkungen" , Klinische Wirksamkeit).
Nicht klinische Auswirkungen
Die im Abschnitt "Präklinische Daten" beschriebenen nicht-klinischen Auswirkungen auf die Netzhautstruktur, das Epithelgewebe und hämatologische Parameter wurden in klinischen Studien mit SMA bislang nicht beobachtet.
QT-Verlängerung
Eine pharmakokinetische/pharmakodynamische Analyse ergab keine Hinweise auf eine Verlängerung der QTc-Zeit durch Evrysdi bei einem Expositionsniveau im therapeutischen Bereich; für Expositionsniveaus oberhalb des therapeutischen Bereichs liegen keine entsprechenden Daten vor.
Erfahrungen aus der Postmarketingphase
Kutane Vaskulitis wurde nach der Markteinführung festgestellt. Nach dem dauerhaften Absetzen von Evrysdi bildeten sich die Symptome zurück. Inzidenzrate und Häufigkeitskategorie können anhand der verfügbaren Daten nicht geschätzt werden.
Für Neugeborene im Alter von weniger als 16 Tagen liegen nur begrenzte Daten nach Markteinführung vor.
Die Meldung des Verdachts auf Nebenwirkungen nach der Zulassung ist von grosser Wichtigkeit. Sie ermöglicht eine kontinuierliche Überwachung des Nutzen-Risiko-Verhältnisses des Arzneimittels. Angehörige von Gesundheitsberufen sind aufgefordert, jeden Verdacht einer neuen oder schwerwiegenden Nebenwirkung über das Online-Portal ElViS (Electronic Vigilance System) anzuzeigen. Informationen dazu finden Sie unter www.swissmedic.ch.
ÜberdosierungZur Überdosierung von Evrysdi liegen keine Erfahrungen aus den klinischen Studien vor. Es ist kein Antidot für den Fall einer Überdosierung von Evrysdi bekannt. Im Fall einer Überdosierung ist der Patient engmaschig zu überwachen, und supportive Massnahmen sind einzuleiten.
Eigenschaften/WirkungenATC-Code
M09AX10
Wirkungsmechanismus
Risdiplam ist ein Spleissmodifikator der prä-mRNA von SMN2 (survival of motor neuron 2) zur Behandlung von SMA, welche durch einen SMN-Proteinmangel infolge von Mutationen im 5q-Chromosom bedingt ist. Ein Mangel an funktionsfähigem SMN-Protein liegt als pathophysiologischer Mechanismus allen Formen von SMA zugrunde. Risdiplam korrigiert die Spleissung von SMN2 und verschiebt die Balance vom Ausschluss von Exon 7 hin zum Einschluss von Exon 7 in das mRNA-Transkript, sodass grössere Mengen funktionelles und stabiles SMN-Protein gebildet werden. Risdiplam behandelt somit die SMA, indem es die Konzentration funktionellen SMN-Proteins steigert und hoch hält.
Elektrophysiologie des Herzens
Die Wirkung von Risdiplam auf das QTc-Intervall wurde in einer Studie an 47 gesunden erwachsenen Probanden untersucht. Bei der therapeutischen Exposition führte Risdiplam nicht zu einer Verlängerung des QTc-Intervalls.
Pharmakodynamik
Risdiplam verteilt sich gleichmässig auf alle Körperregionen einschliesslich des zentralen Nervensystems (ZNS), da es die Blut-Hirn-Schranke durchdringt, und führt so zur Erhöhung der SMN-Proteinkonzentration im ZNS und im ganzen Körper. Die Konzentrationen von Risdiplam im Plasma und SMN-Protein im Blut spiegeln die Distribution und pharmakodynamischen Wirkungen in Geweben wie Gehirn und Muskeln wieder.
In den klinischen Studien FIREFISH, SUNFISH und JEWELFISH bei Patienten mit infantiler SMA ebenso wie bei Patienten mit späterem SMA-Krankheitsbeginn führte Risdiplam zu einer durchgängigen und anhaltenden Zunahme des SMN-Proteins; im Blut wurde innerhalb von 4 Wochen nach Behandlungsbeginn eine mediane Veränderung gegenüber Behandlungsbeginn auf mehr als das 2-Fache gemessen. Diese Zunahme des SMN-Proteins hatte über den gesamten Behandlungszeitraum von mindestens 24 Monaten Bestand (siehe Rubrik "Eigenschaften/Wirkungen" , Klinische Wirksamkeit).
Klinische Wirksamkeit
Die Wirksamkeit von Evrysdi bei der Behandlung von Patienten mit infantiler SMA (SMA Typ 1) und SMA mit späterem Krankheitsbeginn (SMA Typ 2 und 3) wurde in den beiden pivotalen Studien FIREFISH und SUNFISH untersucht; unterstützend kommen Daten aus der JEWELFISH-Studie hinzu. Die Wirksamkeit von Evrysdi bei der Behandlung von präsymptomatischen SMA-Patienten wurde anhand der RAINBOWFISH-Studie beurteilt.
Patienten mit einer klinischen Diagnose mit SMA Typ 4 wurden nicht in klinischen Studien untersucht.
In klinischen Studien wurde die langfristige Wirksamkeit während einer Behandlungsdauer von mindestens 24 Monaten nachgewiesen. Für längere Zeiträume als zwei Jahre liegen nur begrenzte Daten vor.
Infantile SMA
Studie BP39056 (FIREFISH) ist eine unverblindete, 2-teilige Studie zur Untersuchung der Wirksamkeit, Sicherheit, PK und Pharmakodynamik (PD) von Evrysdi bei symptomatischen Typ-1-SMA-Patienten (alle Patienten hatten eine genetisch bestätigte Erkrankung mit 2 Kopien des SMN2-Gens). Teil 1 von FIREFISH war als Dosisfindungsteil der Studie konzipiert. Im konfirmatorischen Teil 2 der FIREFISH-Studie wurde die Wirksamkeit von Evrysdi in der therapeutischen Dosierung untersucht, die anhand der Ergebnisse von Teil 1 ausgewählt wurde (siehe Rubrik "Dosierung/Anwendung" ). Patienten aus Teil 1 der Studie nahmen nicht an Teil 2 teil.
Insgesamt wurden 62 symptomatische SMA-Typ-1-Patienten in FIREFISH Teil 1 (n = 21) und Teil 2 (n = 41) eingeschlossen, von denen 58 Patienten die therapeutische Dosis erhielten. Das mediane Alter bei der Erstmanifestation der klinischen Zeichen und Symptome war 1,5 Monate (Bereich: 0,9 bis 3,0 Monate). Das mediane Alter bei Einschluss in die Studie betrug 5,6 Monate (Bereich: 2,2 bis 6,9 Monate). Die mediane Zeitspanne zwischen Symptommanifestation und erster Dosis betrug 3,7 Monate (Bereich: 1,0 bis 6,0 Monate). Von diesen Patienten waren 60 % weiblich, 57 % waren kaukasischer und 29 % asiatischer Abstammung. Bei Baseline lag der mediane CHOP-INTEND-Score bei 23 (Bereich: 8 bis 37) und der mediane HINE-2-Score bei 1 (Bereich: 0 bis 5). Die demographischen Baseline-Merkmale und die Baseline-Krankheitsmerkmale der Patienten, die in Teil 1 eingeschlossen wurden, waren mit jenen der Patienten in Teil 2 vergleichbar.
Der primäre Endpunkt der Studie war der Anteil der Patienten, die nach 12-monatiger Behandlung für mindestens 5 Sekunden in Teil 2 ohne Unterstützung sitzen konnten (BSID-III-Grobmotorik-Skala, Item 22). 29 % der Patienten (n = 12/41, 90-%-KI: 17,9 %, 43,1 %, p<0,0001) haben diesen Meilenstein erreicht.
Die wichtigsten Wirksamkeitsendpunkte der mit Evrysdi in FIREFISH behandelten Patienten (gepoolte Daten aus Teil 1 und Teil 2) sind in der Tabelle 4 aufgeführt.
Tabelle 4: Zusammenfassung der wichtigsten Wirksamkeitsendpunkte nach 12 Monaten und nach 24 Monaten (FIREFISH Teil 1 und Teil 2)
Wirksamkeitsendpunkte Monat 12 Monat 24
Anteil Patienten
(90-%-KI)
Motorische Funktion und Entwicklungsmeilensteine n = 58a
BSID-III: Sitzen ohne Unterstützung für mindestens 5 32,8 %(22,6 %, 44,3 %) 60,3 %(48,7 %, 71,2
Sekunden %)
CHOP-INTEND-Score von 40 oder höher 56,9 %(45,3 %, 68,0 %) 74,1 %(63,0 %, 83,3
%)
Anstieg CHOP-INTEND-Score um ≥4 Punkte gegenüber 89,7 %(80,6 %, 95,4 %) 87,9 %(78,5 %, 94,2
Baseline %)
HINE-2: Patienten mit Ansprechen bei motorischen 77,6 %(66,7 %, 86,2 %) 82,8 %(72,5 %, 90,3
Meilensteinenb %)
Überleben und ereignisfreies Überleben n = 62a
Ereignisfreies Überlebenc 87,1 %(78,1 %, 92,6 %) 83,8 %(74,3 %, 90,1
%)
Überleben 91,9 %(83,9 %, 96,1 %) 90,3 %(81,9 %, 94,9
%)
Nahrungsaufnahme n = 58a
Fähigkeit zur oralen Nahrungsaufnahmed 84,5 %(74,5 %, 91,7 %) 82,8 %(72,5 %, 90,3
%)
Abkürzungen: BSID-III: Bayley Scales of Infant and Toddler Development – Third Edition; CHOP-INTEND: Children's Hospital of Philadelphia Infant Test of Neuromuscular Disorders; HINE-2: Module 2 of the Hammersmith Infant Neurological Examination.
a Für das Überleben und das beatmungsfreie Überleben wurden die Daten aller Patienten, die in Teil 1 und Teil 2 eine beliebige Dosis von Risdiplam erhalten hatten (n = 62), gepoolt. Für die Wirksamkeitsendpunkte motorische Funktion und Entwicklungsmeilensteine sowie Nahrungsaufnahme wurden die Daten aller Patienten, die die therapeutische Dosis von Risdiplam erhielten (alle Patienten in Teil 2 und diejenigen in der Hochdosis-Kohorte von Teil 1; n = 58) gepoolt.
b HINE-2-Responder-Definition: Ansprechen ist in dieser Analyse definiert als ≥2 Punkte Steigerung (oder höchstmögliche Punktzahl) beim Strampeln ODER ≥1 Punkt Steigerung bei den motorischen Meilensteinen Kopfkontrolle, Umdrehen, Sitzen, Krabbeln, Stehen oder Gehen UND mehr motorische Meilensteine mit Verbesserung als mit Verschlechterung.
c Ein Ereignis erreicht den Endpunkt der permanenten Beatmung definiert als Tracheostomie oder ≥16 Stunden nicht-invasiver Beatmung pro Tag oder Intubation für >21 aufeinanderfolgende Tage in Abwesenheit, oder folgend der Resolution, eines akuten reversiblen Ereignisses. Vier Patienten erreichten den Endpunkt der permanenten Beatmung vor Monat 24. Diese 4 Patienten erreichten eine Steigerung von mindestens 4 Punkten im CHOP-INTEND-Score im Vergleich zur Baseline.
d Umfasst Patienten, die ausschliesslich oral ernährt wurden (41 Patienten nach 12 und 24 Monaten), und solche mit kombinierter Ernährung (oral und Sonde) (8 Patienten nach 12 Monaten und 7 Patienten nach 24 Monaten).
Im Monat 24 erreichten 40 % (23/58) der Patienten, die die therapeutische Dosis erhielten, freies Sitzen für 30 Sekunden (BSID-III, Item 26). Zudem erreichten die Patienten weitere motorische Meilensteine, die anhand von HINE-2 im Monat 24 bestimmt wurden; 78 % der Patienten konnten sich umdrehen (31 % der Patienten konnten sich auf die Seite drehen, 7 % konnten sich von der Bauchlage in die Rückenlage drehen und 40 % konnten sich von der Rückenlage in die Bauchlage drehen) und 28 % der Patienten konnten stehen (16 % freies Stehen und 12 % Stehen mit Unterstützung).
Der Anteil der Patienten, die ohne permanente Beatmung überlebten (ereignisfreies Überleben), lag bei allen Patienten im Monat 24 bei 84 %. Sechs Säuglinge starben (vier innerhalb der ersten drei Monate nach dem Einschluss in die Studie) und ein weiterer Patient brach die Behandlung ab und verstarb 3,5 Monate später. Vier Patienten benötigten im Monat 24 eine permanente Beatmung.
SMA mit späterem Krankheitsbeginn
Studie BP39055 (SUNFISH) ist eine 2-teilige, multizentrische Studie zur Untersuchung der Wirksamkeit, Sicherheit, PK und PD von Evrysdi bei Patienten mit Typ-2- oder Typ-3-SMA im Alter von 2–25 Jahren. Teil 1 war der Dosisfindungsteil der Studie, Teil 2 der randomisierte, doppelblinde, placebokontrollierte, konfirmatorische Teil. Patienten aus Teil 1 der Studie nahmen nicht an Teil 2 teil.
Der primäre Endpunkt war die Veränderung des MFM32-Scores (Motor Function Measure-32) gegenüber Baseline. Der MFM32 ermöglicht die Beurteilung eines breiten Spektrums motorischer Funktionen bei ganz unterschiedlichen SMA-Patienten. Der MFM32-Gesamtscore wird in Prozent (0 bis 100) des möglichen Höchstscores ausgedrückt, wobei höhere Scores für bessere motorische Funktion stehen. Der MFM32 misst motorische Funktionsfähigkeiten, die für wichtige Alltagstätigkeiten von Bedeutung sind. Kleine Veränderungen der motorischen Funktion können grosse Gewinne bzw. Verluste der Funktionsfähigkeit im Alltag bedeuten.
SUNFISH Teil 2
SUNFISH Teil 2 ist der randomisierte, doppelblinde, placebokontrollierte Teil der SUNFISH-Studie mit 180 nicht-gehfähigen Patienten mit Typ-2- (71 %) oder Typ-3-SMA (29 %). Die Patienten erhielten nach Randomisierung im Verhältnis 2:1 entweder Evrysdi in der therapeutischen Dosierung (siehe Rubrik "Dosierung/Anwendung" ) oder Placebo. Die Randomisierung war nach Altersgruppen stratifiziert (2 bis 5, 6 bis 11, 12 bis 17 und 18 bis 25 Jahre).
Das mediane Alter der Patienten bei Behandlungsbeginn betrug 9,0 Jahre (Bereich: 2 bis 25 Jahre); die mediane Zeitspanne vom Auftreten der ersten SMA-Symptome bis zum Behandlungsbeginn betrug 102,6 Monate (Bereich: 1 bis 275 Monate). Von den 180 in die Studie eingeschlossenen Patienten waren 51 % weiblich, 67 % waren kaukasischer und 19 % asiatischer Abstammung. Bei Baseline lag bei 67 % der Patienten eine Skoliose vor (bei 32 % eine schwere Skoliose). Der mittlere Baseline-MFM32-Score der Patienten betrug 46,1, der mittlere RULM-Score (Revised Upper Limb Module) 20,1. Die demographischen Baseline-Merkmale der Patienten waren insgesamt ausgewogen zwischen der Evrysdi- und der Placebogruppe mit Ausnahme einer Unausgewogenheit bei Patienten mit Skoliose (63,3 % der Patienten in der Evrysdi- und 73,3 % in der Placebogruppe).
Die primäre Analyse in SUNFISH Teil 2, die Veränderung des MFM32-Gesamtscores in Monat 12 gegenüber Baseline, ergab einen klinisch bedeutsamen und statistisch signifikanten Unterschied zwischen den mit Evrysdi behandelten Patienten und denen, die Placebo erhielten. Die Ergebnisse der Primäranalyse und der wichtigsten sekundären Endpunkte sind in Tabelle 5 und Abbildung 1 zusammengefasst.
Tabelle 5: Zusammenfassung der Wirksamkeitsergebnisse bei Patienten mit SMA mit späterem Krankheitsbeginn nach 12-monatiger Behandlung (SUNFISH Teil 2)
Endpunkt Evrysdi (n = 120) Placebo (n = 60)
Primärer Endpunkt
Veränderung des MFM32-Gesamtscores1 nach 12 Monaten vs. 1,36 (0,61; 2,11) -0,19 (-1,22; 0,84)
Baseline LS-Mittelwert (95-%-KI)
Unterschied zu Placebo Schätzwert (95-%-KI) p-Wert2 1,55 (0,30; 2,81)
0,0156
Sekundäre Endpunkte
Anteil Patienten mit Veränderung des 38,3 % (28,9; 47,6) 23,7 % (12,0; 35,4)
MFM32-Gesamtscores1 um 3 oder mehr nach 12 Monaten vs.
Baseline (95-%-KI)
Odds Ratio für das Gesamtansprechen (95-%-KI) 2,35 (1,01; 5,44)
Angepasster (unangepasster) p-Wert3,4 0,0469 (0,0469)
Veränderung des RULM-Gesamtscores5 nach 12 Monaten vs. 1,61 (1,00; 2,22) 0,02 (-0,83; 0,87)
Baseline LS-Mittelwert (95-%-KI)
Unterschied zu Placebo, Schätzwert (95-%-KI) 1,59 (0,55; 2,62)
Angepasster (unangepasster) p-Wert2,4 0,0469 (0,0028)
LS = Least Squares
1 Gemäss der Regel für fehlende Daten beim MFM32 wurden 6 Patienten von der Analyse ausgeschlossen (Evrysdi n = 115; Placebo-Kontrolle n = 59).2 Datenanalyse nach einem gemischten Modell mit wiederholten Messungen mit Baseline-Gesamtscore, Behandlung, Termin, Altersgruppe, Behandlung-nach-Termin und Baseline-Wert-nach-Termin.3 Datenanalyse mittels logistischer Regression für Baseline-Gesamtscore, Behandlung und Altersgruppe.4 Der angepasste p-Wert wurde für die in die hierarchischen Tests einbezogenen Endpunkte ermittelt und auf der Grundlage aller p-Werte von Endpunkten in der hierarchischen Reihenfolge bis zum aktuellen Endpunkt abgeleitet. Unangepasste p-Werte wurden auf 5-%-Signifikanzniveau getestet.5 Gemäss der Regel für fehlende Daten beim RULM wurden 3 Patienten von der Analyse ausgeschlossen (Evrysdi n = 119; Placebo-Kontrolle n = 58).
Nach Abschluss der 12-monatigen Behandlung erhielten 117 Patienten weiterhin Evrysdi. Zum Zeitpunkt der 24-Monats-Analyse zeigten diese während 24 Monaten behandelten Patienten zwischen Monat 12 und Monat 24 eine weitere Verbesserung ihrer motorischen Funktion. Die mittlere Veränderung gegenüber Baseline betrug beim MFM 32 1,83 (95-%-KI: 0,74-2,92) und beim RULM 2,79 (95-%-KI: 1,94-3,64).
Abbildung 1: Mittlere Veränderung (LS) gegenüber Baseline beim MFM32-Gesamtscore über 12 Monate in SUNFISH Teil 2
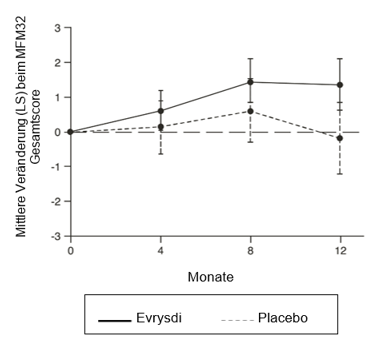
* Fehlerbalken zeichnen das 95%-Konfidenzintervall.
† Der MFM-Gesamtscore wurde gemäß dem Benutzerhandbuch berechnet und als Prozentsatz der maximal möglichen Punktzahl für die Skala ausgedrückt (d.h. die Summe der 32 Itemes Scores geteilt durch 96 und multipliziert mit 100).
SUNFISH Teil 1
Die Wirksamkeit von Evrysdi bei Patienten mit SMA mit späterem Krankheitsbeginn wird auch durch die Ergebnisse von Teil 1 von SUNFISH gestützt, dem Dosisfindungsteil der Studie. In Teil 1 wurden 51 Patienten mit Typ-2- und Typ-3-SMA im Alter von 2 bis 25 Jahren eingeschlossen (darunter 7 gehfähige Patienten). Nach 1-jähriger Behandlung mit der therapeutischen Dosis (der für Teil 2 ausgewählten Dosis) bestand eine klinisch bedeutsame Verbesserung der mittels MFM32 gemessenen motorischen Funktion mit einer mittleren Veränderung gegenüber Baseline um 2,7 Punkte (95-%-KI: 1,5; 3,8). Die Verbesserung beim MFM32 hatte auch nach 2 Jahren unter Evrysdi-Therapie Bestand (mittlere Veränderung um 2,7 Punkte [95-%-KI: 1,2; 4,2]).
In einer explorativen Analyse wurde die mittels MFM beurteilte motorische Funktion zwischen SUNFISH Teil 1 und einer historischen Kohorte mit natürlichem Krankheitsverlauf verglichen (gewichtet nach wichtigen prognostischen Faktoren). Die MFM-Gesamtveränderung gegenüber Baseline nach 1 Jahr und nach 2 Jahren war bei Patienten, die Evrysdi erhielten, grösser als in der natürlichen Verlaufskohorte (nach 1 Jahr: 2,7 Punkte Differenz; p<0,0001; nach 2 Jahren: 4,0 Punkte Differenz; p<0,0001). Die natürliche Verlaufskohorte zeigte eine Abnahme der motorischen Funktion, wie gemäss der natürlichen Progression der SMA zu erwarten ist (mittlere Veränderung nach 1 Jahr: -0,6 Punkte; nach 2 Jahren: -2,0 Punkte).
Präsymptomatische SMA
Die Studie BN40703 (RAINBOWFISH) ist eine offene, einarmige, multizentrische klinische Studie zur Untersuchung der Wirksamkeit, Sicherheit, Pharmakokinetik und Pharmakodynamik von Evrysdi bei Säuglingen ab Geburt bis zum Alter von 6 Wochen (bei der ersten Dosis) mit genetisch bestätigter jedoch noch asymptomatischer SMA.
Die Wirksamkeit bei präsymptomatischen SMA-Patienten wurde in Monat 12 bei 26 Patienten [Intent-to-Treat-(ITT-)Population] untersucht, die mit Evrysdi behandelt wurden. Das mediane Alter dieser Patienten bei der ersten Dosis betrug 25 Tage (Bereich: 16 bis 41 Tage), 62 % waren weiblich und 85 % kaukasischer Abstammung. Acht Patienten, 13 Patienten bzw. 5 Patienten hatten 2, 3 bzw. ≥4 Kopien des SMN2-Gens. Bei Baseline betrug der mediane CHOP-INTEND-Score 51,5 (Bereich: 35,0 bis 62,0), der mediane HINE-2-Score 2,5 (Bereich: 0 bis 6,0) und die mediane Amplitude des muskulären Aktionspotenzials (Compound Muscle Action Potential, CMAP) des Nervus ulnaris 3,6 mV (Bereich: 0,5 mV bis 6,7 mV).
Die primäre Wirksamkeitspopulation (n = 5) umfasste Patienten mit 2 SMN2-Kopien und einer CMAP-Amplitude ≥ 1,5 mV bei Baseline. Bei diesen Patienten betrug der mediane CHOP-INTEND-Score 48,0 (Bereich: 36,0 bis 52,0), der mediane HINE-2-Score 2,0 (Bereich: 1,0 bis 3,0) und die mediane CMAP-Amplitude 2,6 mV (Bereich: 1,6 mV bis 3,8 mV) bei Baseline.
Der primäre Endpunkt war der Anteil der Patienten in der primären Wirksamkeitspopulation, die in Monat 12 mindestens 5 Sekunden lang ohne Unterstützung sitzen konnten (BSID-III Grobmotorik-Skala, Item 22); dieser Meilenstein wurde von einem statistisch signifikanten und klinisch bedeutsamen Anteil der Patienten im Vergleich zum vorgegebenen Leistungskriterium von 5 % erreicht.
Die wichtigsten Wirksamkeitsendpunkte der mit Evrysdi behandelten Patienten sind in den Tabellen 6 und 7 sowie in Abbildung 2 dargestellt.
Tabelle 6: Sitzen gemäss BSID-III-Item 22 für präsymptomatische Patienten in Monat 12
Wirksamkeitsendpunkt Population
Primäre Wirksamkeit Patienten mit 2 ITT (n = 26)
(n = 5) SMN2-Kopiena (n = 8)
Anteil der Patienten, die 80 % (34,3 %, 99,0 87,5 % (52,9 %, 96,2 % (83,0 %,
mindestens 5 Sekunden lang ohne %)p < 0,0001b 99,4 %) 99,8 %)
Unterstützung sitzen (BSID-III,
Item 22); (90-%-KI)
Abkürzungen: BSID-III = Bayley Scales of Infant and Toddler Development – Third Edition; KI = Konfidenzintervall; ITT = Intent-to-Treat.
a Patienten mit 2 SMN2-Kopien hatten bei Baseline eine mediane CMAP-Amplitude von 2,0 (Bereich 0,5 – 3,8).
b Der p-Wert basiert auf einem einseitigen, exakten Binomialtest. Das Ergebnis wird mit einem Grenzwert von 5 % verglichen.
Zusätzlich erreichten 80 % (4/5) der primären Wirksamkeitspopulation, 87,5 % (7/8) der Patienten mit 2SMN2-Kopien und 80,8 % (21/26) der Patienten in der ITT-Population 30 Sekunden lang das Sitzen ohne Unterstützung (BSID-III, Item 26).
Die Patienten in der ITT-Population erreichten auch motorische Meilensteine, gemessen anhand der HINE-2 in Monat 12 (n = 25). In dieser Population konnten 96,0 % der Patienten sitzen [1 Patient (1/8 Patienten mit 2 SMN2-Kopien) erreichte eine stabile Sitzposition und 23 Patienten (6/8, 13/13, 4/4 Patienten mit 2, 3 bzw. ≥4 SMN2-Kopien) konnten sich drehen]. Darüber hinaus konnten 84 % der Patienten stehen; 32 % (n = 8) der Patienten konnten mit Unterstützung stehen (3/8, 3/13 bzw. 2/4 Patienten mit 2, 3 bzw. ≥4 SMN2-Kopien) und 52 % (n = 13) der Patienten konnten ohne Hilfe stehen (1/8, 10/13 bzw. 2/4 der Patienten mit 2, 3 bzw. ≥4 SMN2-Kopien). Darüber hinaus konnten 72 % der Patienten hüpfen, laufen oder gehen; 8 % (n = 2) der Patienten konnten hüpfen (2/8 Patienten mit 2 SMN2-Kopien), 16 % (n = 4) konnten laufen (3/13 bzw. 1/4 Patienten mit 3 bzw. ≥4 SMN2-Kopien) und 48 % (n = 12) konnten unabhängig gehen (1/8, 9/13 bzw. 2/4 Patienten mit 2, 3 bzw. ≥4 SMN2-Kopien). Sieben Patienten wurden in Monat 12 nicht auf Gehfähigkeit getestet.
Tabelle 7: Zusammenfassung der wichtigsten Wirksamkeitsendpunkte für präsymptomatische Patienten in Monat 12
Wirksamkeitsendpunkte ITT-Population (n =
26)
Motorische Funktion
Anteil der Patienten, die im CHOP-INTEND-Score einen Gesamt-Score von 50 oder 92 %a (76,9 %, 98,6
höher erreichen (90-%-KI) %)
Anteil der Patienten, die im CHOP-INTEND-Score einen Gesamt-Score von 60 oder 80 %a (62,5 %, 91,8
höher erreichen (90-%-KI) %)
Nahrungsaufnahme
Anteil der Patienten mit der Fähigkeit zur oralen Nahrungsaufnahme (90-%-KI) 96,2 %b (83,0 %,
99,8 %)
Nutzung des Gesundheitswesens
Anteil der Patienten ohne Krankenhausaufenthaltec; (90-%-KI) 92,3 % (77,7 %,
98,6 %)
Ereignisfreies Überlebend Anteil der Patienten mit ereignisfreiem Überleben 100 % (100 %, 100 %)
(90-%-KI)
Abkürzungen: CHOP-INTEND = Children's Hospital of Philadelphia Infant Test of Neuromuscular Disorders; KI = Konfidenzintervall; ITT = Intent-to-Treat.
a Basierend auf n = 25.
b Ein Patient wurde nicht untersucht.
c Krankenhausaufenthalte umfassen alle Krankenhausaufenthalte, die mindestens zwei Tage dauerten und nicht auf die Studienanforderungen zurückzuführen waren.
d Ein Ereignis bezieht sich auf Tod oder dauerhafte Beatmung; dauerhafte Beatmung ist definiert als Tracheostomie oder ≥16 Stunden nicht-invasive Beatmung pro Tag oder Intubation für > 21 aufeinander folgende Tage bei Abwesenheit von oder nach dem Abklingen eines akuten reversiblen Ereignisses.
Abbildung 2: Mediane CHOP-INTEND-Gesamtpunktzahl nach Besuch und SMN2-Kopienzahl (ITT-Population)
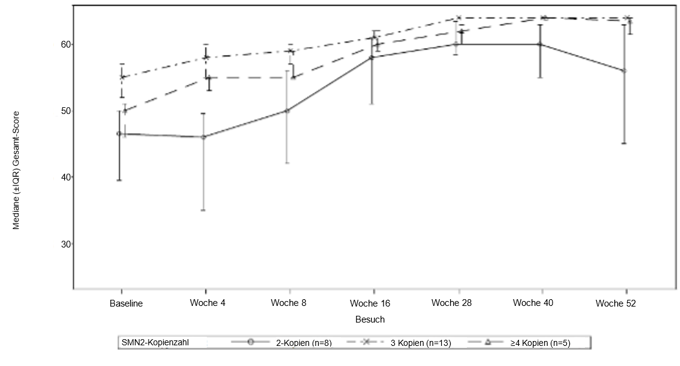
Abkürzungen: IQR – Interquartilbereich; SMN2 = Survival Motor Neuron 2.
Anwendung bei mit anderen modifizierenden Therapien vorbehandelten SMA-Patienten
Studie BP39054 (JEWELFISH) ist eine einarmige, unverblindete Studie zur Untersuchung der Sicherheit, Verträglichkeit, PK und PD von Evrysdi bei Patienten mit infantiler SMA oder SMA mit späterem Krankheitsbeginn im Alter von 6 Monaten bis 60 Jahren, die zuvor andere SMA-Therapien erhalten haben (einschliesslich Nusinersen und Onasemnogene Abeparvovec). Von den 173 Patienten, die Evrysdi erhielten, waren 76 zuvor mit Nusinersen behandelt worden (9 Patienten mit Typ-1-SMA, 43 mit Typ-2-SMA und 24 mit Typ-3-SMA) und 14 Patienten waren zuvor mit Onasemnogen-Abeparvovec behandelt worden (4 Patienten mit Typ-1-SMA und 10 mit Typ-2-SMA). Das mediane Alter der Patienten zu Beginn der Evrysdi-Behandlung betrug 14 Jahre (Bereich 1 - 60 Jahre).
Bei Baseline hatten 83 % der 168 Patienten im Alter von 2 bis 60 Jahren Skoliose (39 % der Patienten hatten eine schwere Skoliose) und 63 % der Patienten hatten einen Score von < 10 Punkten auf der Skala Hammersmith Functional Motor Scale Expanded (HFMSE). In die Studie wurden auch 15 gehfähige Patienten (5 - 46 Jahre) aufgenommen.
Die explorative Wirksamkeit wurde anhand altersgerechter Motorik-Parameter bewertet, einschliesslich der MFM-32- und RULM-Skalen für Patienten im Alter von 2 - 60 Jahren, der BSID-III- und HINE-2-Skalen für Patienten im Alter von unter 2 Jahren und des Sechs-Minuten-Gehtests (6MWT) bei gehfähigen Patienten im Alter von ≥6 Jahren. Bei der Primäranalyse, die nach 24 Behandlungsmonaten durchgeführte wurde, zeigten Patienten im Alter von 2 - 60 Jahren insgesamt eine Stabilisierung der Motorik, gemessen anhand MFM-32 und RULM (n = 137 bzw. n = 133). Patienten unter 2 Jahren (n = 6) konnten motorische Meilensteine wie Kopfkontrolle, selbstständiges Rollen und selbstständiges Sitzen beibehalten oder gewinnen. Die 6MWT-Ergebnisse zeigten eine mittlere Verbesserung von 30,88 Metern (95%-KI: -5,54; 67,29; n = 8). Alle gehfähigen Patienten behielten ihre Gehfähigkeit.
PharmakokinetikDie pharmakokinetischen Parameter von Risdiplam wurden bei gesunden, erwachsenen Probanden sowie bei SMA-Patienten charakterisiert.
Nach Verabreichung von Evrysdi als Lösung zum Einnehmen verlief die PK von Risdiplam zwischen 0,6 und 18 mg etwa linear. Die PK von Risdiplam wird am besten beschrieben mit einem Populations-PK-Modell mit Resorption über drei Transit-Kompartimente, Disposition in zwei Kompartimenten und Elimination mit einer Kinetik erster Ordnung. Ein signifikanter Einfluss von Körpergewicht und Alter auf die PK wurde festgestellt. Die Pharmakokinetik der Tablette (geschluckt oder in Wasser dispergiert) war bioäquivalent zur Lösung zum Einnehmen.
Die geschätzte Exposition (mittlere AUC0-24h) bei Patienten mit infantiler SMA (Alter bei der Aufnahme: 2-7 Monate) unter der therapeutischen Dosis von 0,2 mg/kg einmal täglich betrug 1930 ng·h/ml. Bei präsymptomatischen Säuglingen im Alter von 16 Tage bis <2 Monate betrug die mittlere geschätzte Exposition in der RAINBOWFISH-Studie nach 2 Wochen bei einmal täglicher Gabe von 0,15 mg/kg 2020 ng·h/ml.
Die geschätzte Exposition bei Patienten mit späterem SMA-Krankheitsbeginn (Alter bei der Aufnahme: 2-25 Jahre) in der SUNFISH-Studie (Teil 2) unter der therapeutischen Dosis (0,25 mg/kg einmal täglich bei Patienten mit einem Körpergewicht von <20 kg; 5 mg einmal täglich für Patienten mit einem Körpergewicht von ≥20 kg) betrug 2070 ng·h/ml. Die ermittelte Höchstkonzentration (mittlere Cmax) betrug 194 ng/ml bei 0,2 mg/kg in FIREFISH und 120 ng/ml in SUNFISH Teil 2, und die mittlere geschätzte Höchstkonzentration bei 0,15 mg/kg in RAINBOWFISH betrug 111 ng/ml.
Absorption
Risdiplam wurde im nüchternen Zustand schnell resorbiert, wobei die Plasma-tmax nach der Verabreichung der Lösung, der Tablette oder der dispergierten Tablette zwischen 1 und 5 Stunden lag. Auf der Basis von Daten von 47 gesunden Probanden hatte Nahrung (kalorienreiches Frühstück mit hohem Fettgehalt) keinen wesentlichen Einfluss auf die Exposition gegenüber Risdiplam.
In den klinischen Studien wurde Risdiplam während einer Morgenmahlzeit bzw. nach dem Stillen verabreicht.
Distribution
Die Schätzungen für die populationspharmakokinetischen Parameter betrugen 98 l für das scheinbare zentrale Verteilungsvolumen, 93 l für das periphere Volumen und 0,68 l/h für die interkompartimentale Clearance.
Risdiplam wird vorwiegend durch Serumalbumin gebunden, eine Bindung an Alpha-1-Säure-Glykoprotein findet nicht statt, und die freie Fraktion macht 11 % aus.
Metabolismus
Die Verstoffwechselung von Risdiplam erfolgt primär durch die Flavin-haltigen Monooxygenasen 1 und 3 (FMO1 und FMO3) sowie durch die CYP-Isoenzyme 1A1, 2J2, 3A4 und 3A7. Die Muttersubstanz war die vorwiegend im Plasma gefundene Verbindung; sie machte 83 % der zirkulierenden wirkstoffassoziierten Substanzen aus. Das pharmakologisch inaktive Stoffwechselprodukt M1 wurde als Hauptmetabolit in der Zirkulation identifiziert.
Elimination
Populations-PK-Analysen ergaben eine geschätzte apparente Clearance (Cl/F) von 2,6 l/h für Risdiplam. Die effektive Halbwertszeit von Risdiplam betrug bei SMA-Patienten etwa 50 Stunden.
Etwa 53 % der Dosis (14 % in Form von unverändertem Risdiplam) wurden mit dem Stuhl ausgeschieden und 28 % (8 % als unverändertes Risdiplam) mit dem Urin.
Leberfunktionsstörungen
Eine gering- bis mittelgradige Leberfunktionsstörung hatte keine Auswirkungen auf die PK von Risdiplam. Nach Verabreichung von 5 mg Risdiplam betrug die mittlere Ratio der Cmax und der AUC 0,95 bzw. 0,80 bei geringgradiger (n = 8) sowie 1,20 bzw. 1,08 bei mittelgradiger Leberfunktionsstörung (n = 8), verglichen mit ähnlichen, gesunden Kontrollpersonen (n = 10). Die Sicherheit und PK bei Patienten mit hochgradiger Leberfunktionsstörung sind bisher nicht untersucht worden.
Nierenfunktionsstörungen
Zur PK von Risdiplam bei Patienten mit Nierenfunktionsstörungen sind keine Studien durchgeführt worden. Risdiplam wird zu einem geringen Teil (8 %) als unveränderte Substanz über die Nieren ausgeschieden.
Ältere Patienten
Zur PK von Evrysdi bei SMA-Patienten über 60 Jahren sind keine gesonderten Studien durchgeführt worden. SMA-Patienten bis 60 Jahre wurden in die JEWELFISH-Studie aufgenommen. Bis zu 69-jährige Personen ohne SMA wurden in die klinischen PK-Studien aufgenommen.
Kinder und Jugendliche
Körpergewicht und Alter wurden in der Populations-PK-Analyse als Kovariaten identifiziert. Daher wird die Dosis nach Alter (unter und über 2 Monaten und 2 Jahren) und Gewicht (unter 20 kg) angepasst, um bei Patienten jeden Alters und Körpergewichts eine vergleichbare Exposition zu erzielen. Zu Säuglingen unter 16 Tagen liegen keine pharmakokinetischen Daten vor.
Ethnische Abstammung
Es gab keine Unterschiede in der PK zwischen Personen japanischer und kaukasischer Abstammung.
Präklinische DatenGenotoxizität
Risdiplam war in einem bakteriellen Reverse-Mutations-Assay nicht mutagen. In Säugetierzellen in vitro sowie im Knochenmark von Ratten erhöhte Risdiplam die Zahl mikronukleierter Zellen. Eine Mikronukleus-Induktion im Knochenmark wurde in mehreren Toxizitätsstudien bei Ratten beobachtet, bei adulten wie bei juvenilen Tieren. Die Dosis ohne beobachtete schädliche Wirkungen (NOAEL; no observed adverse effect level) wurde studienübergreifend mit einer Exposition assoziiert, die etwa dem 1,5-Fachen der Exposition beim Menschen unter therapeutischen Dosen entspricht. Die Daten deuten darauf hin, dass dieser Effekt indirekt ist und infolge einer Interferenz von Risdiplam mit dem Zellzyklus von in Teilung begriffenen Zellen entsteht. Derselbe Effekt manifestiert sich auch in anderen Geweben mit hohem Zellumsatz mit Veränderungen in der Haut, im Gastrointestinaltrakt, in männlichen Keimzellen, im Knochenmark sowie als embryonale Toxizität. Risdiplam besitzt kein Potenzial zur direkten Schädigung der DNA.
Kanzerogenität
Derzeit läuft eine 2-jährige Studie zur Karzinogenität bei Ratten. Eine Studie zur Karzinogenität von Risdiplam bei rasH2-transgenen Mäusen ergab keine Hinweise auf ein tumorigenes Potenzial von Risdiplam; die Exposition der Tiere war hierbei bis zu 7-mal so hoch wie beim Menschen unter therapeutischer Dosierung.
Eine 2-jährige Karzinogenitätsstudie an Ratten wurde mit täglichen oralen Dosen von 0,3, 1 und 3 mg/kg Risdiplam durchgeführt. Risdiplam induzierte bei der niedrigen und mittleren Dosis, bei denen die beobachteten Expositionen bei Ratten denen beim Menschen bei der maximal empfohlenen Humandosis (MRHD) von 5 mg entsprachen, keine Tumoren. Statistisch signifikante Anstiege von Tumoren der Präputialdrüse bei männlichen Ratten und von Tumoren der Klitoris bei weiblichen Ratten wurden bei der hohen Dosis beobachtet, die dem 4-Fachen der MRHD entsprach. Da es sich bei beiden um nagerspezifische Organe handelt, haben diese Ergebnisse keine Relevanz für den Menschen.
Reproduktionstoxizität
In Studien mit trächtigen Ratten, die mit Risdiplam behandelt wurden, zeigte sich embryofetale Toxizität mit verringertem fetalem Gewicht und verzögerter Entwicklung. Die NOAEL-Dosis für diesen Effekt betrug etwa das 2-Fache der Exposition, die bei Patienten unter der therapeutischen Dosis Risdiplam erreicht wird. In Studien mit trächtigen Kaninchen traten embryofetale Mortalität und dysmorphogene Effekte bei Expositionen auf, die auch mit mütterlicher Toxizität assoziiert sind. 4 Feten (4 %) aus 4 Würfen (22 %) entwickelten Hydrozephalie. Die NOAEL-Dosis hierfür betrug etwa das 4-Fache der Exposition, die bei Patienten unter der therapeutischen Dosis Risdiplam erreicht wird.
In einer prä- und postnatalen Studie bei Ratten, die täglich Risdiplam erhielten, verursachte Risdiplam eine leichte Verlängerung der Trächtigkeit. Beim Überleben, Wachstum und der funktionellen (verhaltens- oder fortpflanzungsbezogenen) Leistung des Nachwuchses waren keine unerwünschten Wirkungen festzustellen.
Studien bei trächtigen Ratten ergaben, dass Risdiplam die Plazentaschranke passiert und in die Milch übergeht.
Weitere Daten
Die Behandlung mit Risdiplam ist mit Zellzyklusarrest in männlichen Keimzellen bei Ratten und Affen in Verbindung gebracht worden. Dieser Effekt führte zu degenerierten Spermatozyten, zur Degeneration/Nekrose des Samenkanalepithels und zu Oligo-/Aspermie im Nebenhoden. Bei jungen Ratten waren die Effekte bei einer Exposition zu beobachten, wie sie auch bei Patienten unter der therapeutischen Dosis Risdiplam erreicht wird. In einer diesbezüglichen Studie bei Ratten war jedoch keine Beeinträchtigung der männlichen Fertilität zu erkennen. Die Auswirkungen von Risdiplam auf die Spermien hängen wahrscheinlich mit einer Interferenz von Risdiplam mit dem Zellzyklus von in Teilung begriffenen Zellen zusammen und sind stadienspezifisch und reversibel. Bei Ratten und Affen wurden nach Anwendung von Risdiplam keine Auswirkungen auf die weiblichen Fortpflanzungsorgane festgestellt.
Auswirkung auf die Retinastruktur
Die Langzeitanwendung von Risdiplam bei Affen ergab Hinweise auf eine Auswirkung auf die Retina in Form einer Degeneration von Photorezeptoren mit Beginn an der Netzhautperipherie. Nach Beendigung der Behandlung erwiesen sich die Auswirkungen im Retinogramm als partiell reversibel, die Degeneration der Photorezeptoren jedoch war nicht reversibel. Die Effekte wurden mittels optischer Kohärenztomographie (OCT) und Elektroretinographie (ERG) überwacht. Der Effekt trat bei einer Exposition auf, die mehr als das Doppelte der Exposition beim Menschen unter therapeutischer Dosierung betrug.
Auswirkung auf Epithelgewebe
Auswirkungen auf die Histologie der Haut, des Larynx und der Augenlider sowie auf den Gastrointestinaltrakt zeigten sich bei Ratten und Affen, denen Risdiplam verabreicht wurde. Die Veränderungen setzten nach einer Behandlungsdauer von 2 Wochen und länger bei mehr als das 2-Fache der menschlichen Exposition ein. Nach chronischer Anwendung über 39 Wochen bei Affen lag der NOAEL bei einer Exposition von etwa dem 2-Fachen der durchschnittlichen Exposition beim Menschen unter therapeutischer Dosierung.
Auswirkung auf hämatologische Parameter
Im akuten Knochenmarks-Mikronukleus-Test bei Ratten wurde unter der hohen Dosisstufe, die zu einer 15-Fach höheren Exposition als die durchschnittliche Exposition beim Menschen unter therapeutischer Dosierung führte, eine Reduktion des Verhältnisses polychromer (junger) zu normochromer (adulter) Erythrozyten um mehr als 50 % festgestellt, was auf eine erhebliche Knochenmarkstoxizität hindeutet. Bei längerer Behandlung von Ratten über 26 Wochen waren die Expositionsspannen zum NOAEL etwa beim 4-Fachen der durchschnittlichen Exposition beim Menschen unter therapeutischer Dosierung.
Studien bei juvenilen Tieren
Risdiplam wurde bei Ratten und Affen auf Toxizität bei chronischer Anwendung untersucht. Studien mit juvenilen Tieren ergaben bei einer mit der therapeutischen Dosierung beim Menschen vergleichbaren Exposition eine verringerte Nahrungsaufnahme, ein langsameres Wachstum und Anzeichen von Toxizität in den Fortpflanzungsorganen.
Im Hinblick auf die Toxizität, die nach Anwendung von Risdiplam in verschiedenen Organsystemen mit hohem Zellumsatz (Haut, Gastrointestinaltrakt, Knochenmark) auftritt, ergaben die tierexperimentellen Studien keine Hinweise auf Unterschiede in der Empfindlichkeit zwischen juvenilen, adoleszenten und adulten Tieren.
Sonstige HinweisePulver zur Herstellung einer Lösung zum Einnehmen
Inkompatibilitäten
Es wurden keine Inkompatibilitäten zwischen Evrysdi und den empfohlenen wiederverwendbaren Spritzen für die orale Verabreichung beobachtet.
Haltbarkeit
Das Arzneimittel darf nur bis zu dem auf dem Behälter mit "EXP" bezeichneten Datum verwendet werden.
Haltbarkeit nach Anbruch
Die gebrauchsfertige Lösung ist 64 Tage bei Lagerung im Kühlschrank (2-8 °C) haltbar.
Besondere Lagerungshinweise
Pulver:
Den Behälter im Umkarton aufbewahren, um den Inhalt vor Licht und Feuchtigkeit zu schützen.
Nicht über 25 °C lagern.
Gebrauchsfertige Lösung zum Einnehmen:
Den Behälter im Umkarton aufbewahren, um den Inhalt vor Licht zu schützen.
Im Kühlschrank (2-8 °C) lagern.
Falls erforderlich, kann die Patientin/der Patient oder ihr/sein Betreuer die orale Lösung bei Raumtemperatur (unter 40 °C) für eine Gesamtzeit von höchstens 5 Tagen aufbewahren. Die orale Lösung nicht über 40 °C lagern. Die Flasche fest verschlossen halten und immer aufrecht lagern.
Ausser Reichweite von Kindern aufbewahren.
Hinweise für die Handhabung
-Hinweise zur Beachtung vor, während und nach der Zubereitung der Lösung zum Einnehmen: Die Zubereitung der Lösung muss immer durch eine medizinische Fachperson wie z.B. Arzt oder Apotheker bzw. Ärztin oder Apothekerin erfolgen.
-Vermeiden Sie das Inhalieren von Evrysdi-Pulver. Beachten Sie die vor Ort geltenden Vorschriften und verwenden Sie geeignetes Equipment zur Zubereitung der Evrysdi-Lösung.
-Tragen Sie Handschuhe.
-Verwenden Sie das Pulver nicht, wenn das Verfallsdatum überschritten ist. Das Verfallsdatum des Pulvers ist auf dem Etikett der Flasche angegeben.
-Geben Sie die zubereitete Lösung nicht ab, wenn das "Gebrauchsfertige Lösung zum Einnehmen. Nicht mehr verwenden nach" -Datum, das auf dem Flaschenetikett und dem Umkarton steht, nach dem Verfallsdatum des verwendeten Pulvers liegt.
-Vermeiden Sie jeden Kontakt des Arzneimittels mit der Haut. Sollte dennoch Arzneimittel (Pulver oder Lösung) auf Ihre Haut gelangen, waschen Sie das Areal mit Wasser und Seife.
-Verwenden Sie das Arzneimittel nicht, wenn Teile des Packungsinhalts beschädigt sind oder fehlen.
-Verwenden Sie zur Zubereitung der Lösung gereinigtes Wasser oder Wasser für Injektionszwecke.
-Geben Sie keine wiederverwendbaren Spritzen für die orale Verabreichung zusätzlich zu den in der Packung enthaltenen ab.
-Mischen Sie Evrysdi nicht ins Essen oder in Flüssigkeiten (wie z.B. Milch oder Säuglingsmilch).
-Sie dürfen das Evrysdi aus der neuen Flasche nicht mit dem aus der bisher verwendeten Flasche vermischen.
Der Patient bzw. die Betreuungsperson muss vor der Abgabe der zubereiteten Lösung durch eine medizinische Fachperson instruiert werden, wie die verschriebene tägliche Dosis vorzubereiten und einzunehmen ist. (Eine Gebrauchsanleitung für Evrysdi findet sich in der Verpackung).
Zubereitung der Lösung zum Einnehmen
79 ml gereinigtes Wasser oder Wasser für Injektionszwecke werden in die Flasche mit Arzneimittel gegeben.
Der Flaschenadapter wird in den Flaschenhals eingedrückt.
Die Flasche wird nach festem Verschliessen 15 Sekunden lang geschüttelt.
Nach 10 Minuten Warten sollte eine klare Lösung vorliegen. Wenn nicht, wird die Flasche noch einmal gründlich 15 Sekunden lang geschüttelt.
Das "Gebrauchsfertige Lösung zum Einnehmen. Nicht mehr verwenden nach" -Datum 64 Tage nach der Zubereitung der Lösung soll berechnet werden. Der Tag der Zubereitung der Lösung gilt hierbei als Tag 0.
Das berechnete Datum soll auf dem Etikett der Flasche in das dafür vorgesehene Feld bei "Gebrauchsfertige Lösung zum Einnehmen. Nicht mehr verwenden nach (TT.MM.JJJJ)" eingetragen werden und zusätzlich auf das dafür vorgesehene Feld auf dem Umkarton.
Für die nähere Beschreibung liegt eine Anleitung zur Zubereitung für den Arzt oder Apotheker in der Packung bei.
Filmtabletten
Inkompatibilitäten
Risdiplam ist bei Vorhandensein erhöhter Chlorkonzentrationen nicht stabil.
Haltbarkeit
Das Arzneimittel darf nur bis zu dem auf dem Behälter mit "EXP" bezeichneten Datum verwendet werden.
Ausser Reichweite von Kindern aufbewahren.
Den Behälter im Umkarton aufbewahren, um den Inhalt vor Feuchtigkeit zu schützen.
Nicht über 30 °C lagern.
Hinweise für die Handhabung
Zubereitung der Dispersion von Evrysdi Filmtabletten
Der Patient bzw. die Betreuungsperson muss vor der ersten Einnahme des Arzneimittels durch eine medizinische Fachperson instruiert werden, wie die verordnete tägliche Dosis vorzubereiten und einzunehmen ist.
Die Anweisungen zum richtigen Dosierungsschema finden Sie in Rubrik "Dosierung/Anwendung"
Die Evrysdi Filmtablette sollte unzerkaut mit Wasser geschluckt oder in einer kleinen Menge zimmerwarmem, nicht-chloriertem Trinkwasser (z.B. Trinkwasser aus der Flasche) dispergiert eingenommen werden.
Detaillierte Anweisungen zur Herstellung der Dispersion von Evrysdi Filmtabletten in nicht-chloriertem Trinkwasser finden Sie in der Gebrauchsanleitung in der Faltschachtel.
Die Tabletten dürfen nicht zerkaut, zerteilt oder zerdrückt werden.
Bei abgelaufenem Haltbarkeitsdatum oder Beschädigung nicht verwenden (siehe Rubrik "Entsorgung unverbrauchter/nicht mehr haltbarer Arzneimittel" ).
Die in nicht-chloriertem Trinkwasser dispergierte Evrysdi Filmtablette ist umgehend einzunehmen. Evrysdi Filmtabletten sollten ausschliesslich in nicht-chloriertem Trinkwasser dispergiert werden.
Die zubereitete Dispersion ist zu verwerfen, wenn sie nicht innerhalb von 10 Minuten nach der Zugabe von Wasser verwendet wird.
Die zubereitete Dispersion darf nicht dem Sonnenlicht ausgesetzt werden.
Wenn die zubereitete Dispersion von Evrysdi verschüttet wird oder auf die Haut gelangt, den Bereich mit Wasser und Seife waschen.
Evrysdi Filmtabletten sollten nicht über eine transnasale oder eine Gastrostomiesonde verabreicht werden. Falls die Anwendung über eine transnasale oder eine Gastrostomiesonde erforderlich ist, sollte das Evrysdi Pulver zur Herstellung einer Lösung zum Einnehmen verwendet werden.
Entsorgung unverbrauchter/nicht mehr haltbarer Arzneimittel
Die Freisetzung pharmazeutischer Substanzen in die Umwelt muss so gering wie möglich gehalten werden. Arzneimittel dürfen nicht im Abwasser entsorgt werden; die Entsorgung im Haushaltsabfall ist zu vermeiden.
Unverbrauchte/nicht mehr haltbare Arzneimittel sollen von der Abgabestelle (Arzt oder Apotheker) fachgerecht entsorgt werden.
Zulassungsnummer67251, 69919 (Swissmedic).
PackungenPulver zur Herstellung einer Lösung zum Einnehmen
1 Flasche mit Pulver zur Herstellung von 80 ml Lösung zum Einnehmen (0,75 mg/ml Risdiplam) [A].
Die Packung enthält ausserdem 1 Flaschenadapter, 2 wiederverwendbare Spritzen für orale Verabreichung zu 1 ml, 2 wiederverwendbare Spritzen für orale Verabreichung zu 6 ml und 1 wiederverwendbare Spritze für orale Verabreichung zu 12 ml.
Filmtabletten
1 Packung enthält 28 Filmtabletten [A]
ZulassungsinhaberinRoche Pharma (Schweiz) AG, Basel.
Stand der InformationNovember 2025.
Anleitung zur Zubereitung der Lösung (für medizinische Fachpersonen)
Evrysdi 0,75 mg/ml
Pulver zur Herstellung einer Lösung zum Einnehmen
Risdiplam
Die Evrysdi-Lösung zum Einnehmen muss vor der Abgabe von einer medizinischen Fachperson zubereitet werden.
(NUR FÜR MEDIZINISCHE FACHPERSONEN WIE Z.B. ARZT ODER APOTHEKER BZW. ÄRZTIN ODER APOTHEKERIN)
Eine Schachtel EVRYSDI enthält (s. Abbildung A): 1.1 Verschlusskappe 2.1 Abbildung A
Flasche Evrysdi 3.1 wiederverwendbare Spritze für orale Verabreichung à 12 ml
(in Beutel) 4.2 wiederverwendbare Spritzen für orale Verabreichung à 6 ml (in
Beuteln) 5.2 wiederverwendbare Spritzen für orale Verabreichung à 1 ml (in
Beuteln) 6.1 Flaschenadapter zum Eindrücken 7.1 Information für Patientinnen
und Patienten 8.1 Gebrauchsanleitung für Patientinnen und Patienten (nicht
abgebildet) 9.1 Anleitung zur Zubereitung der Lösung für medizinische
Fachpersonen (nicht abgebildet)
Wichtige Hinweise zu Evrysdi -Die Zubereitung der Lösung muss
immer durch eine medizinische Fachperson wie z.B. Arzt oder
Apotheker bzw. Ärztin oder Apothekerin erfolgen. -Vermeiden Sie
das Inhalieren von Evrysdi-Pulver. Beachten Sie die vor Ort
geltenden Vorschriften und verwenden Sie geeignetes Equipment
zur Zubereitung der Evrysdi-Lösung. -Tragen Sie Handschuhe.
-Verwenden Sie das Pulver nicht, wenn das Verfallsdatum
überschritten ist. Das Verfallsdatum des Pulvers ist auf dem
Etikett der Flasche angegeben. -Vermeiden Sie jeden Kontakt des
Arzneimittels mit der Haut. Sollte dennoch Arzneimittel (Pulver
oder Lösung) auf Ihre Haut gelangen, waschen Sie das Areal mit
Wasser und Seife. -Verwenden Sie das Arzneimittel nicht, wenn
Teile des Packungsinhalts beschädigt sind oder fehlen.
-Verwenden Sie zur Zubereitung der Lösung gereinigtes Wasser
oder Wasser für Injektionszwecke. -Geben Sie keine
wiederverwendbaren Spritzen für die orale Verabreichung
zusätzlich zu den in der Packung enthaltenen ab. -Geben Sie die
zubereitete Lösung nicht ab, wenn das "Gebrauchsfertige Lösung
zum Einnehmen. Nicht mehr verwenden nach" -Datum, das auf dem
Flaschenetikett und dem Umkarton steht, nach dem Verfallsdatum
des verwendeten Pulvers liegt. -Der Patient bzw. die
Betreuungsperson muss vor der Abgabe der zubereiteten Lösung
durch eine medizinische Fachperson instruiert werden, wie die
verschriebene tägliche Dosis vorzubereiten und einzunehmen ist.
Wie ist Evrysdi aufzubewahren? -Bewahren Sie das Pulver (das
noch nicht als Lösung zubereitete Arzneimittel) bei
Raumtemperatur unter 25 °C im Umkarton auf, um den Inhalt vor
Licht und Feuchtigkeit zu schützen. -Bewahren Sie die Lösung
(das als Lösung zubereitete Arzneimittel) im Kühlschrank bei 2-8
°C aufrecht stehend im Umkarton auf, um den Inhalt vor Licht zu
schützen. -Falls erforderlich, kann die orale Lösung bei
Raumtemperatur (unter 40 °C) für eine Gesamtzeit von höchstens 5
Tagen aufbewahrt werden. Die orale Lösung nicht über 40 °C
lagern. -Bewahren Sie die Lösung zum Einnehmen in der
Originalflasche, aufrecht stehend und mit fest verschlossener
Kappe auf.
Zubereitung der Lösung
Abbildung B Schritt 1 Klopfen Sie leicht
auf den Flaschenboden, um das
Pulver zu lockern (siehe
Abbildung B).
Abbildung C Schritt 2 Entfernen Sie die
Verschlusskappe, indem Sie sie
nach unten drücken und nach
links (gegen den Uhrzeigersinn)
abschrauben (siehe Abbildung
C). Werfen Sie die Kappe nicht
weg.
Abbildung D Schritt 3 Geben Sie behutsam 79
ml gereinigtes Wasser oder
Wasser für Injektionszwecke in
die Flasche mit dem Arzneimittel
(siehe Abbildung D).
Abbildung E Schritt 4 Halten Sie die auf
dem Tisch stehende Flasche mit
einer Hand fest. Setzen Sie mit
der anderen Hand den
Flaschenadapter zum Eindrücken
in den Flaschenhals ein, indem
Sie ihn herunterdrücken, bis er
vollständig an der Lippe der
Flasche anliegt (siehe
Abbildung E).
Abbildung F Schritt 5 Setzen Sie die
Verschlusskappe wieder auf die
Flasche. Drehen Sie die Kappe
nach rechts (im Uhrzeigersinn),
um die Flasche zu verschliessen.
Stellen Sie sicher, dass die
Flasche fest verschlossen ist,
und schütteln Sie sie gründlich
15 Sekunden lang (siehe
Abbildung F). Warten Sie 10
Minuten. Dann sollte eine klare
Lösung vorliegen. Anschliessend
schütteln Sie die Flasche noch
einmal gründlich 15 Sekunden
lang.
Abbildung G Schritt 6 Berechnen Sie das
"Nicht mehr verwenden nach"
-Datum 64 Tage nach der
Zubereitung der Lösung.
(Anmerkung: Der Tag der
Zubereitung der Lösung gilt
hierbei als Tag 0. Wenn Sie
beispielsweise die Lösung am 1.
April zubereiten, ist das
"Entsorgen am" -Datum der 4.
Juni.). Schreiben Sie das
"Gebrauchsfertige Lösung zum
Einnehmen. Nicht mehr verwenden
nach" -Datum der Lösung auf das
dafür vorgesehene Feld auf dem
Etikett der Flasche (siehe
Abbildung G) und auf dem
Umkarton. Legen Sie die Flasche
zusammen mit den Spritzen (in
Beuteln), der Information für
Patientinnen und Patienten und
der Gebrauchsanleitung wieder
in den Umkarton. Bewahren Sie
alles (Flasche aufrecht
stehend!) im Kühlschrank auf.
|